

Abstract
During development, individual neurons extend highly branched arbors that innervate the surrounding territory, enabling the formation of appropriate synaptic connections. The clustered protocadherins (cPCDH), a family of diverse cell-surface homophilic proteins, provide each neuron with a cell specific identity required for distinguishing between self vs. non-self. While only 52 unique cPcdh isoforms are encoded in the human genome, a combination of stochastic promoter choice and the formation of a protein lattice through engagement of adjacent cPCDH protein cis/trans-tetramers confer the high degree of cellular specificity required for self-recognition. Studies of mice bearing deletions of individual cPcdh gene clusters have identified deficits in circuit formation and behavior. In humans, single nucleotide variants scattered across the cPCDH locus have been identified, which associate with multiple neurodevelopmental disorders, including autism and schizophrenia. To advance our understanding of cPCDH stochastic choice and maintenance, function across cell types, and contribution to neuropsychiatric disease pathogenesis, hiPSC-based models have been developed. Ultimately, integration of human genetic data, biochemical assays, and functional studies is needed to uncover the mechanism underlying neurite repulsion, which has been implicated in neurodevelopmental disorders.
Keywords: protocadherins, homophilic interactions, self-avoidance, neurodevelopmental disorders, stochastic expression, tiling, neural circuits
INTRODUCTION
Functional circuits established among the approximately 80 billion human neurons through trillions of synapses must be established during neuronal development. Failure to establish proper neural networks has been linked to neurodevelopmental and neuropsychiatric disorders1,2. During development, individual neurons extend highly branched neurites that innervate the surrounding territory with minimal overlap. To enable the formation appropriate synaptic connections, neurons have evolved self-avoidance mechanisms to distinguish self from non-self. These mechanisms ensure that neurites from the same neuron avoid one another, allowing extensive arborization without crossing or clumping, while neurites from different neurons can occupy the same field and engage in synaptic interactions3,4. Some neuronal subtypes, such as serotonergic neurons, tile throughout receptive fields in the brain ensuring that their neurites do not overlap sister neurites or neighboring neurites from other serotonergic neurons1,5. In Drosophila, self-avoidance is mediated by the Dscam1 gene, which can generate over 19,008 unique protein isoforms through an extraordinary example of stochastic alternative pre-mRNA splicing, providing the diversity needed for cellular identity6–9. In mammals, a different family of diverse cell-surface receptors, the clustered protocadherins (cPcdh), provide each neuron with the unique cell specific identity required for self-avoidance in the brain10. The protocadherins generate diversity by a remarkable mechanism that combines stochastic promoter choice, nearly random heterodimerization and a novel lattice formation at apposing membranes on the cell surface11.
Consistent with their role in self vs. non-self identification, many studies examining deletion of cPcdh genes in mice have identified deficits in neuronal wiring across several neuronal subtypes in multiple brain regions and altered behaviors1,5,10,12–15. Moreover, large-scale DNA sequencing studies have identified single nucleotide variants (SNVs), scattered across the clustered protocadherin (cPCDH) locus, associated with multiple neurodevelopmental disorders, including ASD and SZ16–18. Here we summarize recent findings related to the regulation and function of clustered protocadherins, examine the contribution of protocadherins to the genetic risk for neurodevelopmental disorders, and highlight new models for functional genetic studies.
ORGANIZATION AND REGULATION OF PROTOCADHERIN GENE EXPRESSION
The clustered protocadherin (cPCDH) locus is organized into three closely linked gene clusters, PCDHα, β, and γ on human chromosome five, together spanning approximately 1 megabase of genomic DNA19. The PCDHα and PCDHγ clusters are more evolutionarily related and contain multiple variable exons that are stochastically chosen20–22, along with two (α) or three (γ) variable exons that are expressed deterministically (PCDH c-types)19,23,24. Each variable exon is then spliced to a 3’ splice site located immediately upstream from three constant exons (Fig. 1A). Each variable exon encodes the extracellular cadherin and transmembrane domains, while the three constant exons encode a common intracellular domain for all of the protein isoforms in the PCDHα or PCDHγ clusters. By contrast, the PCDHβ genes, which are evolutionarily more divergent, consist only of stochastic variable exons, with no constant region encoding an intracellular domain19.
Figure 1:
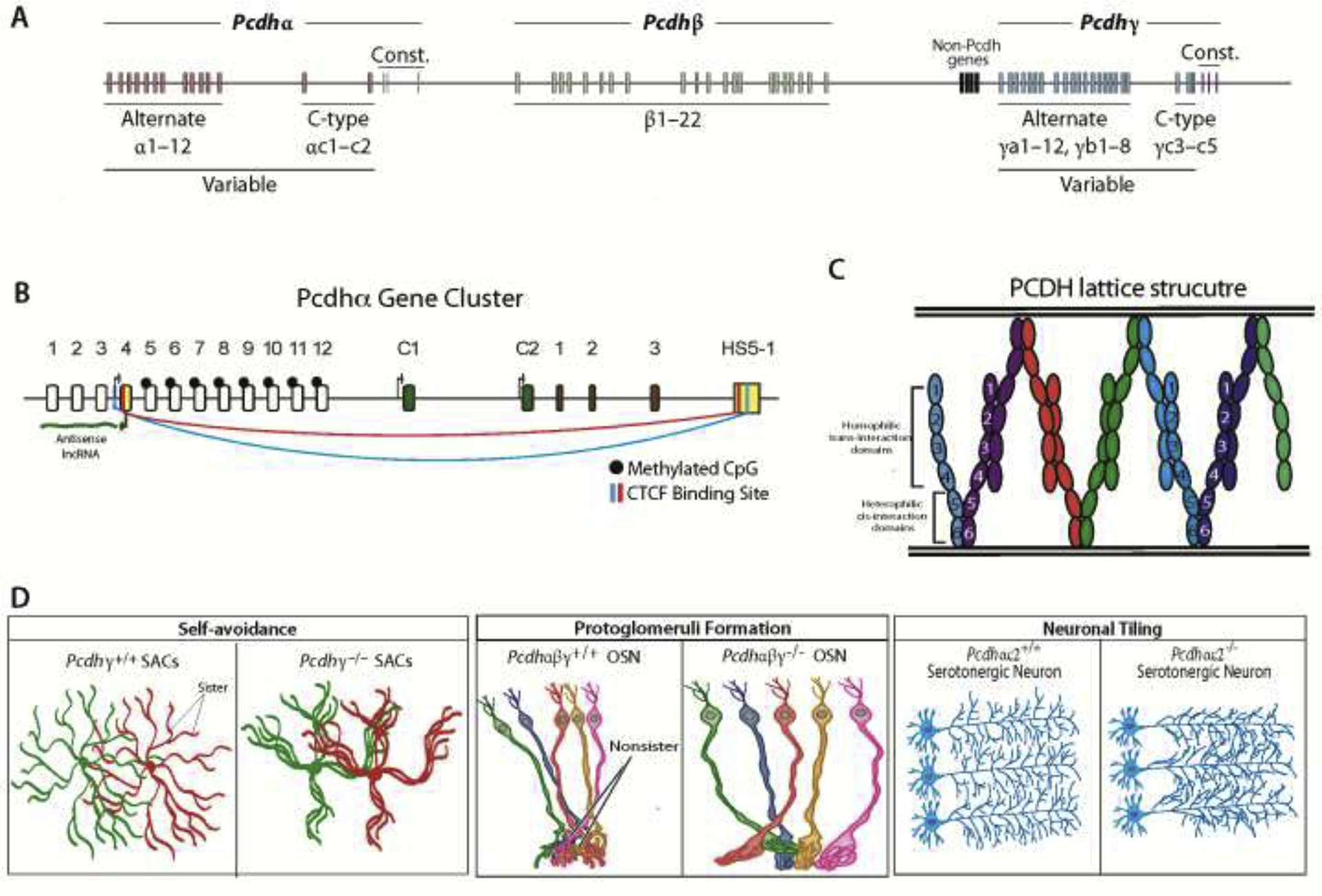
Overview of the protocadherin gene cluster. 1A) Schematic of the cPCDH gene cluster; 1B) Schematic highlighting the mechanism of stochastic choice used to express individual PCDHα isoforms; 1C) Representation of a cPCDH protein lattice formed through homophilic interaction of cis-cPCDH dimers; 1D) Deficits in self-avoidance and neuronal tiling found in mouse cPCDH deletion models demonstrating their role in self vs. non-self identification
The stochastic expression of Pcdhα genes is regulated through long-range DNA looping between individual promoters upstream of each Pcdhα variable exon, and a transcriptional enhancer designated HS5–1, located downstream of the Pcdhα gene cluster25–27. Each Pcdhα variable exon contains two CTCF binding sites, one in the promoter and the other within the protein coding sequence of the exon. These two sites have similar spacing and are in opposite orientation relative to the two CTCF binding sites located in the HS5–1 enhancer25,27. DNA methylation of the promoters appears to play an essential role in stochastic gene activation. The Pcdhα variable gene promoters are highly methylated prior to stochastic activation in ground state and thus do not bind to CTCF23,28. Remarkably, an “antisense” promoter was identified within the protein-coding region of each variable exon, and the stochastic activation of one of these promoters in the Pcdhα gene cluster results in transcription of an antisense polyadenylated lncRNA that extends through the upstream “sense” strand promoter resulting in the demethylation of the sense Pcdhα promoter by a TET demethylase29. This, in turn, permits binding of CTCF to the sense promoter and DNA looping to the HS5–1 enhancer. Consistent with a chromatin scanning mechanism, the demethylated promoter that is most proximal to the HS5–1 enhancer was shown to be the only promoter activated following demethylation, resulting the specific expression of individual cPCDH isoforms (Fig. 1B). In addition to DNA methylation, which was shown to be regulated by DMNT3b28, H3 Lysine 9 trimethylation (H3K9me3) and Smchd1 binding are also critical for the regulation of Pcdhα expression30,31. In contrast, the regulated (as opposed to stochastic) expression of Pcdhαc2 and the stochastic expression of the Pcdhβ and Pcdhγ gene clusters are regulated by an independent, unidentified mechanism, as these genes do not generate an antisense lncRNA and the variable exons do not contain CTCF binding sites.
HOMOPHILIC MATCHING OF PROTOCADHERIN PROTEINS FORMS A PROTEIN LATTICE
While the protocadherin gene cluster encodes only 52 unique isoforms in humans, detailed structural and biochemical studies have demonstrated that complex, heterophilic cis-PCDH dimerization and homophilic trans-PCDH interactions confer the high degree of cellular specificity required proper self-avoidance11,32–36. The cPCDH proteins consist of six extracellular cadherin domains (EC1–6), which are required for specific cPCDH interactions. The crystal structure of a cPCDH cis-dimer and cell aggregation assays established that the EC5 and EC6 domains are required heterophilic cPCDH cis-dimerization, which leads to transport of cPCDH isoforms to the plasma membrane33. While PCDHβ and PCDHγ isoforms engage in dimers with all cPCDH isoforms, PCDHa isoforms (aside from PCDHaC2) must dimerize with either PCDHβ or PCDHγ to be transported to the plasma membrane33.
Systematic in vitro cell aggregation assays in K562 cells, which lack the ability to repel, and thus aggregate, established that all cPCDH proteins (except PCDHαC1 and PCDHγC4) engage in highly specific homophilic trans-interactions. An additional key finding was that the presence of a single mismatched isoform between two cells prevents aggregation in K562 cells expressing multiple Pcdh isoforms32. In contrast to classical cadherin family proteins, the cPCDH dimers on the cell surface engage their matching dimers on opposing membranes through anti-parallel protein-protein interactions (Fig. 1C). These interactions lead to the formation of an extended protein lattice, which was recently visualized by cryo-EM tomagraphy36.
Taken together, these results support a model in which perfect cPCDH isoform matching leads to the formation extended lattice structure on the surface of sister neurites, which triggers the intracellular signaling mechanisms required for contact mediated neurite repulsion. While the intracellular domain of PCDHα and PCDHγ isoforms can bind to receptor tyrosine kinases including RET37 and PYK238, the signaling mechanisms required for cPCDH-mediated self-avoidance remain unknown.
FUNCTIONAL STUDIES IN MOUSE MODELS
The role for cPCDH proteins in self-avoidance was first demonstrated in mouse starburst amacrine cells (SACs), which develop complex radial dendritic arbors that overlap the dendrites of nearby SACs, but do not overlap their own sister dendrites (Fig. 1D, left). Conditional deletion of the Pcdhγ cluster in mice led to a collapse and extensive crossover of sister SAC dendrites10,39. Subsequent studies investigated the role of cPcdh diversity using mature olfactory sensory neurons (OSNs) that project axons to precise locations within the olfactory bulb and converge to form glomeruli. Single-cell RNA-seq studies revealed that OSNs stochastically express distinct sets of alternate isoforms from all three cPcdh clusters and all individual, double and tri-cluster cPcdh gene cluster deletion mice failed to form of normal protoglomeruli and displayed a self-avoidance phenotype (Fig. 1D, middle)15,40. Overexpression of a distinct set of three cPcdh isoforms was unable to rescue glomeruli formation, likely due to inappropriate self-recognition and repulsion caused by the lack of cPCDH isoform diversity15. However, overexpression of the same cPcdh isoforms lacking the intracellular cytoplasmic domain (ICD) formed typical glomeruli structures, indicating that the ICD is required for cPCDH mediated neurite repulsion.
In addition to self-avoidance, cPcdh expression is essential for the proper tiling of serotonergic neurons throughout their target regions in the brain. The spacing between serotonergic axon terminals must be tightly regulated to properly maintain the concentration and distribution of serotonin within target regions. Genetic deletion of the complete Pcdha cluster and only the Pcdha C-type isoforms showed deficits in normal serotonergic tiling across many target regions1,5 and exhibited deficits in cognitive and affective behaviors (Fig. 1D, right)1,5. In addition, translating ribosome affinity purification followed by RNA-seq (TRAP-seq) demonstrated that Pcdhαc2 is the predominant Pcdha isoform expressed in serotonergic neurons, indicating that Pcdhαc2 is required cell autonomously for proper homophilic repulsion and axonal tiling of serotonergic neurons and that these changes in serotonergic circuitry could lead to behavioral deficits seen across multiple neurological disorders.
ASSOCIATION OF PROTOCADHERINS WITH NEURODEVELOPMENTAL DISORDERS
Failure to establish proper neural networks has been linked to neurodevelopmental disorders1,2, including ASD17,41–43 and SZ44–47. These multifaceted disorders are highly heritable and have few effective treatments, most of which are only effective for a subset of patients and/or symptoms48, reflecting the complex genetic etiology. Large-scale sequencing studies have identified hundreds of genes that contribute to the genetic risk of neurodevelopment disorders16,18,49–51. These genes show enrichment for chromatin modifiers, synaptic proteins and cell adhesion molecules, including the protocadherins.
DNA sequencing of family structured cohorts have identified single nucleotide variants (SNVs) across the PCDH gene cluster that associate with ASD. Specifically, 5 SNPs were identified in PCDHα, which showed significant association with ASD in 841 families from the Autism Genetic Resource Exchange17. More recent whole exome sequencing of the Simon’s Simplex Cohort further identified a number of additional de novo SNVs across the cPCDH locus in probands16,52. Additionally, epigenetic dysregulation of cPCDH locus has also been found in Williams-Beuren syndrome, and the reciprocal 7q11.23 duplication syndrome, which are complex neurodevelopmental disorders with ASD behaviors. Many of the genes within the 7q11.23 critical region encode DNA modification enzymes or chromatin remodeling complexes, a gene set significantly associated with ASD53. Consistent with this relationship, MeCP2 was shown to regulate cPCDH promoters54, suggesting a possible convergence between epigenetic modifiers associated with ASD and cPCDH gene regulation.
Early genetic linkage studies first identified an association between chromosome region 5q31, harboring the cPCDH locus, and SZ55,56. More recently, genome wide association studies (GWAS) of over 20,000 SZ cases identified a significant risk locus upstream of the PCDHα cluster18. Examination of DNA looping demonstrated that the SZ risk locus upstream of the PCDHα cluster contained two bundles of DNA interactions in hiPSC-derived neural cells, one bundle interacting with the 3’ end of the PCDHα cluster and the other interacting with the 3’ end of the PCDHγ cluster57. Moreover, dosage of the top SZ GWAS risk SNP within the cPCDH locus is significantly correlated with increased expression of multiple PCDHα genes in the CommonMind dlPFC RNAseq dataset57,58. Furthermore, CRISPR mediated deletion of the region containing the SZ GWAS risk SNP within the cPCDH locus led to significantly decreased expression of PCDHα8 and PCDHα10, suggesting a mechanism in which the SZ-associated SNP impacts DNA looping to PCDHα target genes leading to changes in gene expression.
Genetic associations between cPCDH genes and other neurological disorders have also been reported. SNVs within the PCDHγ cluster were shown to segregate with dyslexia within a multi-incident family. Each of the PCDHγ SNVs fell within the extracellular cadherin domains critical for cPCDH interactions, suggesting a potential impact on cPCDH interactions59. Furthermore, PCDHγC5 expression was shown to be increased in neurons following treatment with B-amyloid and amyloid precursor protein/presenilin-1, suggesting a role for cPCDH isoforms in the progression of Alzheimer’s Disease60. Future work is needed to identify cPCDH variants in larger patient populations and to address the function of cPCDH isoforms across cell types, brain regions and in the context of disease.
Given the genomic complexity and extent of repetitive elements across the cPCDH locus, it is possible that there are both common and rare structural variants, which have not yet been uncovered. While large scale, long read sequencing studies have not yet been completed, some structural variants in cPCDH genes have already been identified. Initially, a 16.7kb deletion was identified as a common structural variant across multiple genetic backgrounds61. Moreover, there were a handful of structural variants (both insertions and deletions) identified through long read sequencing of genomes from 15 individuals62. The presence of these structural variants may make identifying diseases associated SNVs more difficult. Without large-scale, long read sequencing studies of individuals diagnosed with neurodevelopmental disorders, the association both de novo SNVs and structural variants across the cPCDH locus may be under represented.
USING HIPSC MODELS TO UNDERSTAND MECHANISMS OF PCDH FUNCTION
Studies of promoter choice in established cell lines, such as the human neuroblastoma cell line SK-N-SH have shown the promoter choice is maintained through cell division, suggesting that an epigenetic mechanism maintains enhancer/promoter choice, once it is made29. Although mouse models have provided many insights into cPCDH function, new models are beginning to emerge to understand cPCDH expression and relationship to disease. Investigation into the timing and maintenance of cPCDH stochastic choice in hESCs/hiPSCs and in vitro differentiated neurons demonstrated that the expression pattern of cPCDH isoforms in hiPSC-derived neurons could be predicted by the chromatin landscape of the parent hiPSC clone, suggesting stochastic choice of cPCDH isoforms occurs upstream of neural progenitors63. Moreover, evidence for restricted cPCDH isoform selection was found in fetal brains from the Allen BrainSpan collection, but not present in the adult brain samples, providing support for two modes of stochastic choice, one that restricts cPCDH choice during fetal development and a second that expands the repertoire of cPCDH isoforms expressed following an unknown developmental milestone after birth63.
hiPSC models have also been implemented to investigate the contribution of cPCDH to neurological disorders. Recently, hiPSCs derived from major depressive disorder patients classified as either SSRI-remitters or SSRI-non-remitters were differentiated into serotonergic neurons to understand the molecular mechanisms underlying SRRI responsiveness. Serotonergic neurons generated from SSRI-non-remitters displayed deficits in neurite length, morphology, and decreased expression of multiple PCDHα genes64. shRNA mediated knockdown of PCDHα6 and PCDHα8 in control hiPSC-derived serotonergic neurons resulted in increased neurite outgrowth, suggesting PCDHα expression causally contributed to the increased length found in SSRI-non-remitters. In addition, hiPSC-derived cortical interneurons generated from individuals with schizophrenia, showed aberrant cPCDH gene expression and deficits in synaptic density and arborizaiton, which could be rescued by inhibition of protein kinase C, believed to participate in intracellular signaling pathways downstream of cPCDH proteins65. These studies demonstrate the utility of hiPSC-based models to understand cPCDH expression, function and relationship to neurological disorders; however, additional studies are needed to develop the intracellular signaling mechanisms leading to neuronal repulsion and the impact of individual genetic variants across the cPCDH gene clusters.
FUTURE CONSIDERATIONS AND CONCLUSIONS
The mechanisms by which neurite repulsion occurs is fundamental to understanding how neural circuits are established during vertebrate development, and this understanding has important implications for neurodevelopmental and neuropsychiatric disorders1,2. Significant progress has been made in understanding how genomic organization, DNA methylation and chromosome looping leads to the stochastic promoter choice required to generate random sets of Pcdhα isoforms in individual neurons, but we do not yet understand the mechanisms involved in stochastic promoter choice in the Pcdhβ and γ gene clusters66. Structural and biochemical studies along with homophilic binding studies have provided an answer to the question of how a relatively small number of protein isoforms can generate sufficient functional diversity for self-recognition35. However, a key unanswered question is how homophilic engagement of cPCDHs at the cell surface can lead to neurite repulsion. It seems clear that Adam protease and γ-secretase are involved67,68 and the intracellular domains of Pcdhα and γ are required. Biochemical studies have identified protein kinases that associate with cPCDHs38,69, but their role in self-avoidance is not understood. Studies addressing these outstanding questions are likely to continue to provide novel insights into the mechanisms underlying vertebrate neural circuit development and their contribution to neurodevelopmental disorders.
Funding
Funding was received for this work.
All of the sources of funding for the work described in this publication are acknowledged below:
[List funding sources and their role in study design, data analysis, and result interpretation]
NIH grants R01MH108579,R01NS088476, R01MH114817 (T.M.),
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Chen WV et al. Pcdhαc2 is required for axonal tiling and assembly of serotonergic circuitries in mice. Science 356, 406–411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study found that serotonergic neurons predominantly express only the Pcdhαc2 isoform and demonstrated this single cell-type identity is required for axonal tiling and proper serotonergic circuit formation.
- 2.Hirabayashi T ; Yagi T. Protocadherins in Neurological Diseases. Advances in Neurobiology (2014). [DOI] [PubMed] [Google Scholar]
- 3.Grueber WB & Sagasti A Self-avoidance and Tiling: Mechanisms of Dendrite and Axon Spacing. Cold Spring Harbor Perspectives in Biology 2, a001750–a001750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence Zipursky S & Grueber WB The Molecular Basis of Self-Avoidance. Annual Review of Neuroscience 36, 547–568 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Katori S et al. Protocadherin-αC2 is required for diffuse projections of serotonergic axons. Scientific Reports 7, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hattori D et al. Robust discrimination between self and non-self neurites requires thousands of Dscam1 isoforms. Nature 461, 644–648 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wojtowicz WM, Flanagan JJ, Millard SS, Zipursky SL & Clemens JC Alternative splicing of Drosophila Dscam generates axon guidance receptors that exhibit isoform-specific homophilic binding. Cell 118, 619–633 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matthews BJ et al. Dendrite Self-Avoidance Is Controlled by Dscam. Cell 129, 593–604 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Hattori D, Millard SS, Wojtowicz WM & Zipursky SL Dscam-Mediated Cell Recognition Regulates Neural Circuit Formation. Annual Review of Cell and Developmental Biology 24, 597–620 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefebvre JL, Kostadinov D, Chen WV, Maniatis T & Sanes JR Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488, 517–521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinstein R et al. Molecular logic of neuronal self-recognition through protocadherin domain interactions. Cell 163, 629–642 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen WV et al. Functional significance of isoform diversification in the protocadherin gamma gene cluster. Neuron 75, 402–409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasegawa S et al. Clustered Protocadherins Are Required for Building Functional Neural Circuits. Frontiers in Molecular Neuroscience 10, 114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasegawa S et al. Distinct and Cooperative Functions for the Protocadherin-α, -β and -γ Clusters in Neuronal Survival and Axon Targeting. Frontiers in Molecular Neuroscience 9, 1–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mountoufaris G et al. Multicluster Pcdh diversity is required for mouse olfactory neural circuit assembly. Science 356, 411–414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study demonstrated the cooperative function of all three cPCDH gene cluster to provide the diversity required for proper self-avoidance and glomeruli formation of mouse olfactory sensory neurons.
- 16.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anitha A et al. Protocadherin α (PCDHA) as a novel susceptibility gene for autism. Journal of Psychiatry & Neuroscience 38, 192–198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pardinas AF et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nature genetics 50, 381–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Q & Maniatis T A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Esumi S et al. Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nature genetics 37, 171–176 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Hirano K et al. Single-neuron diversity generated by Protocadherin-β cluster in mouse central and peripheral nervous systems. Frontiers in Molecular Neuroscience 5, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaneko R et al. Allelic Gene Regulation of Pcdh- α and Pcdh- γ Clusters Involving Both Monoallelic and Biallelic Expression in Single Purkinje Cells. Journal of Biological Chemistry 281, 30551–30560 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Tasic B et al. Promoter choice determines splice site selection in protocadherin α and γ pre-mRNA splicing. Molecular Cell 10, 21–33 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Wu Q Comparative DNA Sequence Analysis of Mouse and Human Protocadherin Gene Clusters. Genome Research 11, 389–404 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo YY et al. CTCF/cohesin-mediated DNA looping is required for protocadherin promoter choice. Proceedings of the National Academy of Sciences 109, 21081–21086 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kehayova P, Monahan K, Chen W & Maniatis T Regulatory elements required for the activation and repression of the protocadherin- gene cluster. Proceedings of the National Academy of Sciences 108, 17195–17200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monahan K et al. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of Protocadherin- gene expression. Proceedings of the National Academy of Sciences 109, 9125–9130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toyoda S et al. Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 82, 94–108 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Canzio D et al. Antisense lncRNA Transcription Mediates DNA Demethylation to Drive Stochastic Protocadherin α Promoter Choice. Cell 177, 639–653.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study uncovered that stochastic Pcdhα promoter choice occurs following antisense transcription-mediated promoter demethylation, which enables DNA looping between a cPCDH promoter and the downstream HS5–1 enhancer.
- 30.Jiang Y et al. The methyltransferase SETDB1 regulates a large neuron-specific topological chromatin domain. Nature genetics 49, 1239–1250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K et al. Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proceedings of the National Academy of Sciences of the United States of America 112, E3535–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thu CA et al. Single-cell identity generated by combinatorial homophilic interactions between α, β, and γ protocadherins. Cell 158, 1045–1059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodman KM et al. Protocadherin cis -dimer architecture and recognition unit diversity. Proceedings of the National Academy of Sciences 114, E9829–E9837 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodman KM et al. Structural Basis of Diverse Homophilic Recognition by Clustered α- and β-Protocadherins. Neuron 90, 709–723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubinstein R, Goodman KM, Maniatis T, Shapiro L & Honig B Structural origins of clustered protocadherin-mediated neuronal barcoding. Seminars in Cell and Developmental Biology 69, 140–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brasch J et al. Visualization of clustered protocadherin neuronal self-recognition complexes. Nature 569, 280–283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study revealed that cPCDH trans interactions between distinct cis dimers in generate zipper-like lattice structure using cryo-electron tomography. These results provide support for the isoform-mismatch chain-termination model of protocadherin-mediated self-recognition.
- 37.Schalm SS, Ballif BA, Buchanan SM, Phillips GR & Maniatis T Phosphorylation of protocadherin proteins by the receptor tyrosine kinase Ret. Proceedings of the National Academy of Sciences 107, 13894–13899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan L et al. Alpha protocadherins and Pyk2 kinase regulate cortical neuron migration and cytoskeletal dynamics via Rac1 GTPase and WAVE complex in mice. eLife 7, 1–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kostadinov D & Sanes JR Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. eLife 4, 1–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasegawa S et al. Distinct and Cooperative Functions for the Protocadherin-alpha, -beta and -gamma Clusters in Neuronal Survival and Axon Targeting. Frontiers in molecular neuroscience 9, 155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J, Yu S, Fu Y & Li X Synaptic proteins and receptors defects in autism spectrum disorders. Frontiers in cellular neuroscience 8, 276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iossifov I et al. Article De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 74, 285–299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato D et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. The American Journal of Human Genetics 90, 879–887 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barch DM & Ceaser A Cognition in schizophrenia: core psychological and neural mechanisms. Trends in Cognitive Sciences 16, 27–34 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis DA, Glantz LA, Pierri JN & Sweet RA Altered cortical glutamate neurotransmission in schizophrenia: evidence from morphological studies of pyramidal neurons. Annals of the New York Academy of Sciences 1003, 102–12 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Uhlhaas PJ & Singer W Abnormal neural oscillations and synchrony in schizophrenia. Nature Reviews Neuroscience 11, 100–113 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Moghaddam B & Javitt D From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37, 4–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeFilippis M & Wagner KD Treatment of Autism Spectrum Disorder in Children and Adolescents. Psychopharmacology bulletin 46, 18–41 (2016). [PMC free article] [PubMed] [Google Scholar]
- 49.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanders SJ et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ripke S et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krumm N et al. Excess of rare, inherited truncating mutations in autism. Nature Genetics 47, 582–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.El Hajj N, Dittrich M & Haaf T Epigenetic dysregulation of protocadherins in human disease. Seminars in cell & developmental biology 69, 172–182 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Miyake K et al. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: implication for pathogenesis of Rett syndrome. BMC neuroscience 12, 81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwab SG et al. Evidence suggestive of a locus on chromosome 5q31 contributing to susceptibility for schizophrenia in German and Israeli families by multipoint affected sib-pair linkage analysis. Molecular Psychiatry 2, 156–160 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Straub RE, MacLean CJ, O’Neill FA, Walsh D & Kendler KS Support for a possible schizophrenia vulnerability locus in region 5q22–31 in Irish families. Molecular Psychiatry 2, 148–155 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Rajarajan P et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science (New York, N.Y.) 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nature Neuroscience 19, 1442–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naskar T et al. Ancestral Variations of the PCDHG Gene Cluster Predispose to Dyslexia in a Multiplex Family. EBioMedicine 28, 168–179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y et al. Synaptic Adhesion Molecule Pcdh-gammaC5 Mediates Synaptic Dysfunction in Alzheimer’s Disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 37, 9259–9268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noonan JP et al. Extensive linkage disequilibrium, a common 16.7-kilobase deletion, and evidence of balancing selection in the human protocadherin alpha cluster. American journal of human genetics 72, 621–635 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Audano PA et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Almenar-Queralt A et al. Chromatin establishes an immature version of neuronal protocadherin selection during the naive-to-primed conversion of pluripotent stem cells. Nature Genetics 51, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study uncovered a chromatin-based mechanism in which the repertoire of human cPCDH isoforms is limited from establishment of stochastic choice, during the transition from a naive to a primed state, until post-birth where the cPCDH diversity is then expanded.
- 64.Vadodaria KC et al. Altered serotonergic circuitry in SSRI-resistant major depressive disorder patient-derived neurons. Molecular psychiatry 24, 808–818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study generated hiPSC-derived serotonergic neurons from patients classified as SSRI-remitters and SSRI-nonremitters and identified alterations neurite outgrowth and decreased cPCDH expression in SSRI-nonremitters. The alteration in neurite outgrowth was recapitulated by shRNA knockdown of cPCDH genes in control serotonergic neurons.
- 65.Shao Z et al. Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell–derived cortical interneurons from subjects with schizophrenia. Nature Neuroscience (2019). doi: 10.1038/s41593-018-0313-z [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study found that cortical interneurons from schizophrenia patients displayed dysregulated expression of protocadherin genes and showed defects in synaptic density and arborizaiton.
- 66.Canzio D & Maniatis T The generation of a protocadherin cell-surface recognition code for neural circuit assembly. Current Opinion in Neurobiology 59, 213–220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buchanan SM, Schalm SS & Maniatis T Proteolytic processing of protocadherin proteins requires endocytosis. Proceedings of the National Academy of Sciences 107, 17774–17779 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haas IG, Frank M, Veron N & Kemler R Presenilin-dependent processing and nuclear function of gamma-protocadherins. The Journal of biological chemistry 280, 9313–9319 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Suo L, Lu H, Ying G, Capecchi MR & Wu Q Protocadherin clusters and cell adhesion kinase regulate dendrite complexity through Rho GTPase. Journal of molecular cell biology 4, 362–376 (2012). [DOI] [PubMed] [Google Scholar]