

Abstract
While the immune response is likely to play a pivotal role in controlling Kaposi’s sarcoma-associated herpesvirus (KSHV) and preventing disease development, the exact factors responsible for control remain ill-defined. T cell responses are weak and variable, and neutralizing antibodies are more frequently detected in individuals with KS. This suggests a potential role for non-neutralizing antibodies, which to date, have been largely uninvestigated. Antibody-dependent cell cytotoxicity (ADCC) is a common effector function for non-neutralizing antibodies and is known to play a protective role in other herpesvirus infections; yet ADCC has never been investigated in the context of KSHV infection. Here, we provide the first evidence that anti-KSHV antibodies are capable of mediating ADCC responses against infected human cells undergoing lytic reactivation. ADCC activity significantly higher than seronegative controls was detected in 24 of 68 KSHV seropositive individuals tested. However, ADCC responses were not associated with KS development or progression. ADCC activity was also found to be independent of HIV status, sex, age, KSHV antibody titer, and KSHV neutralizing activity. Nevertheless, additional investigations into effector cell function between KS and asymptomatic individuals is needed to determine whether ADCC has a role in preventing KS.
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV), or human herpesvirus-8, is the etiological agent of all forms of Kaposi’s sarcoma (KS), as well as primary effusion lymphoma and multicentric Castleman disease (1–4). Immune dysregulation plays an important role in the transition from KSHV infection to the development of neoplasia, but the specific dysregulation(s) that potentiate this transition remain unclear. KS is more often seen among the elderly (classic KS), HIV-infected (epidemic), or in those undergoing immunosuppressive treatment (iatrogenic). Reconstitution of the immune response via withdrawal or alteration of immunosuppressive medications, or control of HIV with combination anti-retroviral therapy can lead to KS regression (5–7).
The close association between HIV-1 co-infection and KS, the most common AIDS-defining illness, implies a role for cell-mediated immunity. However, T cell responses in both KS patients and asymptomatic KSHV seropositive individuals are weak and variable, suggesting the lack of an immunodominant response associated with control of KSHV infection (8, 9). Similarly, both KS patients and asymptomatic individuals mount Ab responses, but titers and the prevalence of neutralizing Ab are actually higher in KS patients (10, 11). This suggests a potential role for non-neutralizing Ab-mediated effector mechanisms in KSHV control.
Many herpesviruses, including KSHV, downregulate MHC class one expression and modulate innate immune responses (12). KSHV has been demonstrated to alter Ab-independent NK cell cytotoxicity and MHC class I expression, and Ab-independent NK cell functions have been shown to target KSHV infected cells and correlate with control of KSHV infection and disease regression (12–15). Protection against infection and reactivation of herpes simplex virus-1 (HSV-1), HSV-2, and human cytomegalovirus (HCMV) has been associated with Ab-dependent cell cytotoxicity (ADCC), an important effector function of NK cells (16–18). These responses are reviewed by Jenks et al. (18). NK cell activity and ADCC have also been shown to play a role in reduction of infectious mononucleosis and control of KSHV’s closest relative, Epstein-Barr virus (EBV) (19, 20). Despite these associations, KSHV-specific ADCC-mediating Ab have not been investigated to date and their potential role in KS pathogenesis or disease progression is unknown. Here, we investigated whether Ab from KSHV seropositive individuals are capable of mediating ADCC responses against KSHV infected cells. We also determined their prevalence, longitudinal stability, and involvement in KS pathogenesis.
Materials and Methods
Cell Culture
BC3 cells, obtained from ATCC, were cultured in RPMI media (Corning 10-040-CV) supplemented with 20% FBS (Gibco 16000-044) and 1% penicillin/streptomycin (Corning 30-002-CI) and passaged every 2-3 days. BJAB cells, obtained from ATCC, were cultured in RPMI media supplemented with 10% FBS and 1% penicillin/streptomycin and passaged every 2-3 days. L1T2 cells, kindly provided by Dirk Dittmer (University of North Carolina-Chapel Hill) and iSLKBAC16 cells, kindly provided by Jae Jung (University of Southern California) were cultured in DMEM media (Corning10-017-CV) supplemented with 10% FBS and 1% penicillin/streptomycin. iSLKBAC16 cells also received 1μg/mL puromycin (Clontech 631306), 250μg/mL G418 (Thermofisher Scientific 11811023), and 1mg/mL hygromycin B (Thermofisher Scientific 10687010) and were induced with 1μg/mL doxycycline (Sigma Aldrich D9891) and 1mM sodium butyrate (Sigma Aldrich B5887) (21). NK92.05 CD16 176V, a natural killer cell line engineered to express the high affinity FcγRIII graciously provided by Kerry Campbell (Fox Chase Cancer Center) were maintained in αMEM complete media: MEM (Sigma-Aldrich M0644) plus 2.2g/L sodium bicarbonate (Gibco 25080-094), 0.1mM 2-mercaptoethanol (Gibco 31350-010), 2mM L-glutamine (Corning 25-005-CI), 0.2mM myo-inositol (Sigma-Aldrich I5125), 0.02mM folic acid (Sigma-Aldrich F7876), 1% non-essential amino acids (Gibco 11140-050), 1% sodium pyruvate (Gibco 11360-070), 1% penicillin/streptomycin, 12.5% FBS, and 12.5% horse serum (Sigma-Aldrich H1138). NK92.05 CD16 176V cells were passaged every 4 days in the presence of 2.5-5% freshly thawed J558L supernatant (see IL-2 production). J558L Hu-IL-2 cells, also provided by Dr. Kerry Campbell (Fox Chase Cancer Center), were cultured in RPMI media plus 10% FBS, 1% penicillin/streptomycin, 2mM L-glutamine, 1% sodium pyruvate, 0.1mM 2-mercapotethanol, and 1% HEPES (Gibco 25-060-CI).
IL-2 production
The mouse myeloma cell line J558L Hu-IL-2 was used to produce IL-2 as growth supplement for NK cells. J588L cells were thawed and initiated in 10mL culture media (see above) in a T25 flask. The day after thaw, the culture was transferred to a T75 flask and brought to a total volume of 30mL. J558L cells were then expanded every 2-3 days until the desired volume was achieved. Cells were allowed to grow for approximately one week, until the media turned yellow. Supernatant was harvested by centrifuging 3 minutes at 1300rpm. Collected supernatant was filtered through a 0.22μ filter and frozen in 13mL aliquots at −80°C.
Patient plasma
Plasma samples collected for previous KSHV and KS studies conducted in our lab were utilized for ADCC screening (22–25). All samples were obtained with informed consent, and the study was approved by the Institutional Review Board of the University of Nebraska, the University of Zambia Biomedical Research Ethics Committee, Tanzania National Institute for Medical Research, and Ocean Road Cancer Institute. Plasma was heat-inactivated for 60 minutes at 56°C, then centrifuged for 2 minutes at 13000xg to pellet debris. The screening cohort was comprised of 68 individuals, 30 KS patients and 38 KSHV seropositive asymptomatic controls. Of the 30 KS patients, 8 were endemic KS patients and 22 were epidemic KS patients. No differences between endemic and epidemic KS patients were observed and both subtypes are reported as one group. Controls were matched by age, sex, HIV-status, and KSHV Ab titer where possible. 15 additional HIV-negative individuals were included in the control group to ensure sufficient numbers for comparisons between HIV-infected and uninfected individuals. Both cases and controls contained individuals from Zambia and Tanzania. For determination of ADCC Ab stability longitudinally, 10 women from a study designed to examined mother-to-child transmission of KSHV were selected, as this study included regular 3 month follow up visits (25). The 10 women were all HIV-positive and the first two years of samples were examined.
ADCC Calcein release assay
ADCC activity was measured using a 4-hour calcein release assay (26, 27). BC3 and BJAB cells, either unstimulated or 48 hours post PMA (Sigma-Aldrich P8139) stimulation, were labeled with 2μg/mL Calcein-AM (Thermofisher Scientific C3099) at a concentration of 106 cells/mL for 30 minutes at 37°C, 5% CO2. Calcein was then quenched with BC3 or BJAB culture media, cells were washed once with media, and resuspended at a final concentration of 106 cells/mL. 100μL (105) labeled target cells were aliquoted to each well of a 96-well v-bottom plate. Patient plasma (1μL, 1:200 final dilution) was then added to experimental wells and allowed to incubate for 15 minutes while effector cells were prepared. NK92.05 CD16 176V effector cells were resuspend in αMEM complete media at a concentration of 5x106 cells/mL. 100μL (5x105) was aliquoted to all wells except the spontaneous and maximum release control wells. Spontaneous release controls received 100μL of αMEM complete media, while maximum release was achieved through the addition of 100μL of 0.1% Triton-X-100. Plates were spun at 100xg for 2 minutes to increase cell interactions, and incubated at 37°C, 5% CO2 for 4 hours. After incubation, wells were mixed via gentle pipetting, and spun at 400xg for 2 minutes to pellet the cells and debris. 150μL of supernatant was transferred to a black-walled 96-well clear bottom plate and fluorescence was determined with Victor3V plate reader (PerkinElmer).
Samples were tested in quintuplicate and the average of the centroid three was used for all calculations. For longitudinal screening and screening against latent BC3 cells, samples were plated in triplicate and the average of all three was used for analysis. Each plate contained the following controls: target cells only (spontaneous release); target cells and effectors, no plasma (TE); and maximum release. ADCC activity (%ADCC) was determined via the following formula: (Experimental-TE)/(Maximum-spontaneous release)*100 and is presented as the mean and standard deviation from three independent experiments.
Statistical Analysis
A one-way ANOVA with Dunnett post-test was used to identify samples with ADCC activity statistically different from the KSHV seronegative control plasma in both lytic and latent assays. An unpaired t-test was used to assess differences between KS status, HIV status, and sex. Pearson correlation was used to test for a relationship between ADCC activity and age or KSHV neutralization activity, while a Spearman correlation was used for KSHV Ab titer. Ten KS patients were analyzed using a paired T-test to detect differences in ADCC activity pre- and post-treatment. Differences in an individual’s response longitudinally was assessed using a one-way ANOVA. For analysis as discrete variables, Fisher’s Exact was used. Samples with ADCC activity significantly higher than KSHV seronegative controls were considered “positive,” while samples with non-significant differences were deemed “negative.” For KSHV neutralizing activity, samples with greater than 50% neutralization were called “positive,” and those less than 50% were “negative.” All statistical analysis was conducted in GraphPad Prism 8.3.0 (GraphPad Software).
Immunofluorescence Assay
Antibody binding to different target cells lines was assessed using a modified version of our previously established immunofluorescence assay (28). Briefly, plasma samples were diluted 1:200 and incubated with fixed, lytically reactivated target cells. Mouse monoclonal anti-human IgG (American Type Culture Collection CRL-1786) was used as a secondary antibody. Consistent with previous methods, Cy-2 conjugated donkey anti-mouse IgG (Jackson ImmunoResearch 715-225-150) was the detection antibody for L1T2 cells, with 0.004% Evan’s Blue (Sigma Aldrich E2129) serving as a cellular counterstain. As iSLKBAC16 cells express GFP, donkey anti-mouse IgG AlexaFluor647 (Thermofisher Scientific A31571) was used to detect antibody binding at a dilution of 1:300. 300nM DAPI (Thermofisher Scientific D1306) was used to stain nuclei.
Results
To investigate ADCC in the context of KSHV infection, we first compared responses between plasma from KSHV seropositive and seronegative individuals using a calcein-release assay (Figure 1). As an in vitro model for KS tumorigenesis does not exist, the primary effusion B cell lymphoma cell line BC3, which is chronically infected with KSHV, served as target cells. Because ADCC against EBV-infected cells has been shown to differ dramatically between latent and lytically reactivated cells (29), we tested ADCC responses against both unstimulated BC-3 and those 48-hours post-induction with PMA. As shown in Figure 1A, only KSHV seropositive plasma in conjunction with PMA-induced BC3 target cells resulted in a significant increase in killing compared to target and effector only controls (KSHV+ vs TE, black bars, p<0.0001). The relative fluorescence units were then used to calculate %ADCC presented in Figure 1B. To ensure the lack of response in the KSHV seronegative plasma was not specific to a single individual, 12 additional seronegative individuals were tested. No samples showed a response significantly different from Ab-independent controls (Figure 1C). ADCC activity was evaluated at plasma dilutions ranging from 1:50 to 1:10,000 (Figure 1D). KSHV seronegative plasma produced no specific lysis at any dilution (open circles), while KSHV seropositive plasma showed consistently positive responses down to 1:1,000 but no response at 1:10,000 (closed circles). Seven additional KSHV seropositive samples were titered and no evidence of a prozone effect was observed, and samples were low levels responses did not show increased activity with higher plasma dilutions (Supplemental Figure 1). Together these findings demonstrate that a calcein-release assay facilitates identification of polyclonal plasma capable of mediating ADCC responses against BC3 cells undergoing lytic reactivation and can be used to quantify the magnitude and prevalence of their responses.
Figure 1.
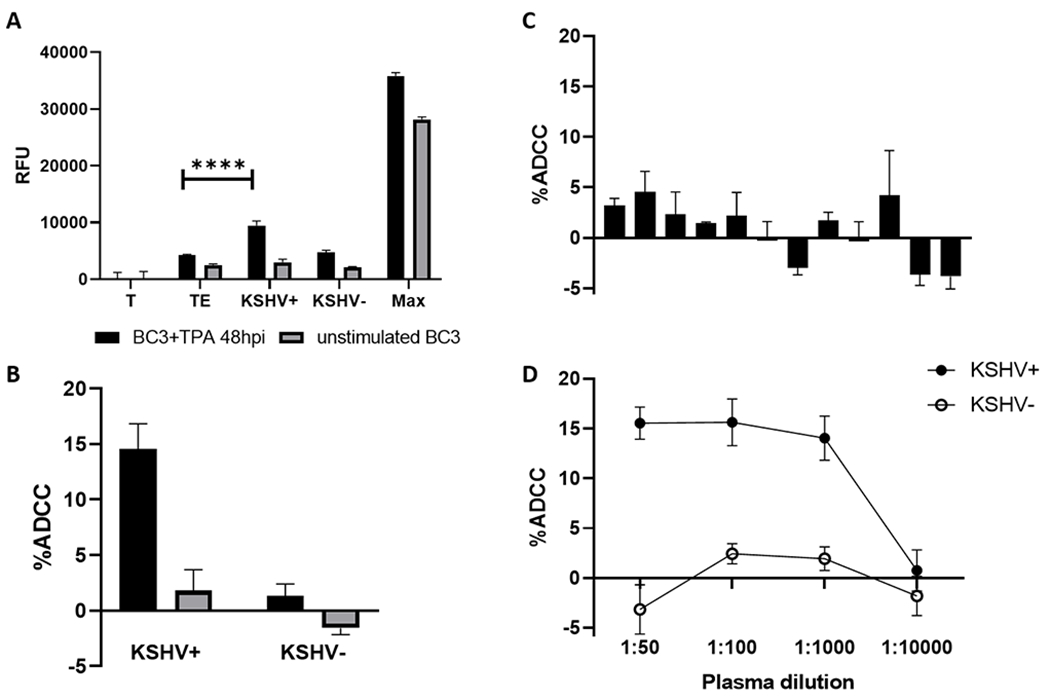
Validation of a KSHV specific ADCC assay. (A) Relative fluorescent units (RFU) of supernatant from wells containing targets only (T), target and effectors (TE), targets and effectors plus KSHV seropositive plasma (KSHV+), targets and effectors plus KSHV seronegative plasma (KSHV−), and target cells plus detergent (Max) for BC3 cell stimulated with PMA (black bars) or unstimulated (gray bars). (B) Percent ADCC was calculated as (Experimental-TE)/(Max-T)*100. (C) ADCC activity for 12 KSHV seronegative individuals. (D) ADCC activity at varying dilutions of KSHV seropositive and seronegative plasma. All data shown is the mean and standard deviation from the centroid three of five experimental replicates. Significant differences denoted: **** p<0.0001
To further confirm the KSHV specificity of the assay, 10 KSHV seropositive samples were screened against the KSHV uninfected B cell lymphoma line BJAB. Figure 2 displays the ADCC activity of each of the samples against both stimulated and unstimulated BC3 (Figure 2A) and BJAB cells (Figure 2B). With the exception of one individual, sample 1, individuals that had detectable ADCC responses against stimulated BC3 cells showed no responses to unstimulated BC3 or BJAB cells. Sample 1 showed significant ADCC responses to all four cell types, indicating an anti-cancer response rather than a virus specific response. Interestingly, sample 8 mounted a significant response against both stimulated and unstimulated BJAB cells but showed no response to BC3 cells (Figure 2). This response was likely against antigens specific to the BJAB cell line, but not lymphomas in general. As latent, unstimulated BC3 cells should not express viral antigen at the cell surface, unstimulated BC3 cells were used as a negative control for the remaining experiments. This allowed for the detected of potential anti-cancer effects as seen in sample 1 without introducing additional cell line specific antigenic variables seen with sample 8.
Figure 2.
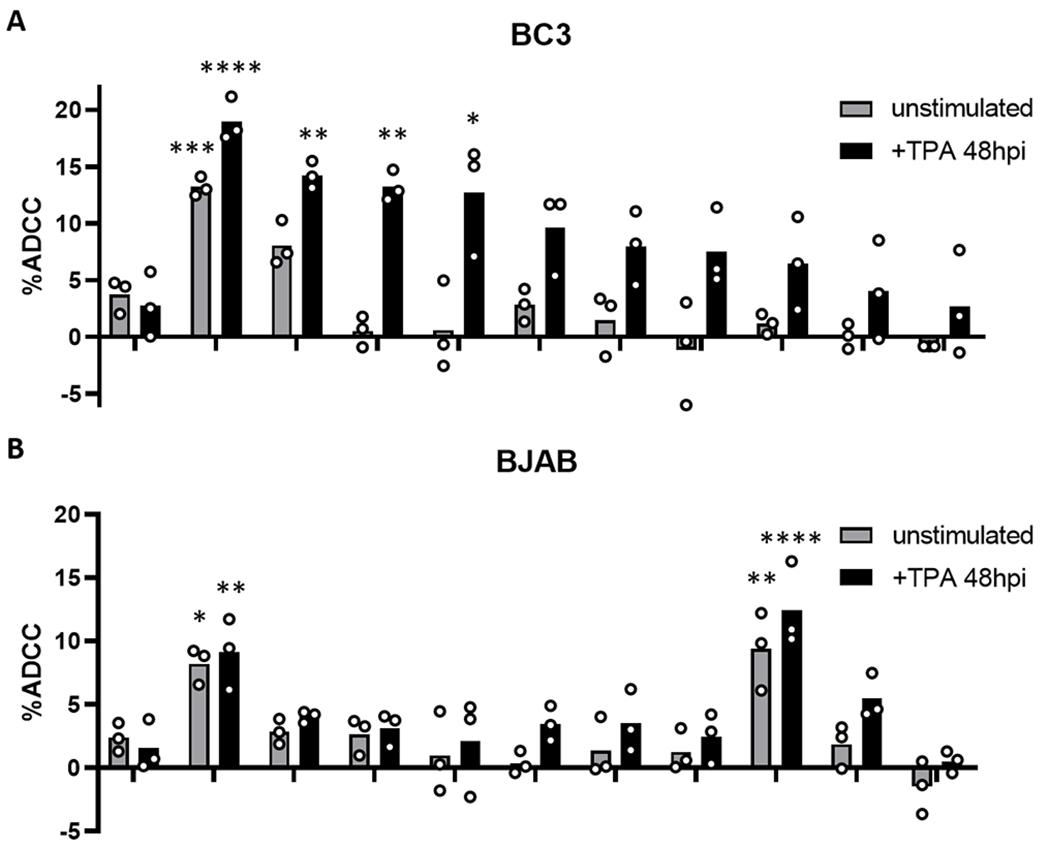
ADCC activity against BC3 and BJAB cells. ADCC activity of KSHV seronegative plasma 10 KSHV seropositive plasma samples against lytically reactivated and unstimulated BC3 cells (A) and BJAB cells (B). Samples with ADCC activity significantly different than their respective seronegative control are indicated with asterisks: * p<0.05; **p<0.01; ***p<0.001; ****p<0.0001
As shown in Figure 3A, plasma from 24 of 68 (35.3%, green bars) KSHV seropositive individuals displayed ADCC activity at levels significantly higher than KSHV seronegative controls when tested against lytically reactivated cells. None of the individuals showed increased ADCC activity against latent cells; however, five samples did show a significant decrease in activity (Figure 3B, red bars). Notably the activity among these samples was also lower than the Ab independent (TE) control. These small reductions in relative killing may be the result of non-specific steric hindrance, where non-KSHV specific Ab bind to either target or effector cells and prevent the formation of cytotoxic immunological synapses. Alternatively, Ab recognition of non-self antigens or anti-cancer responses against either target or effector lymphoma cell lines may inhibit cytotoxicity.
Figure 3.
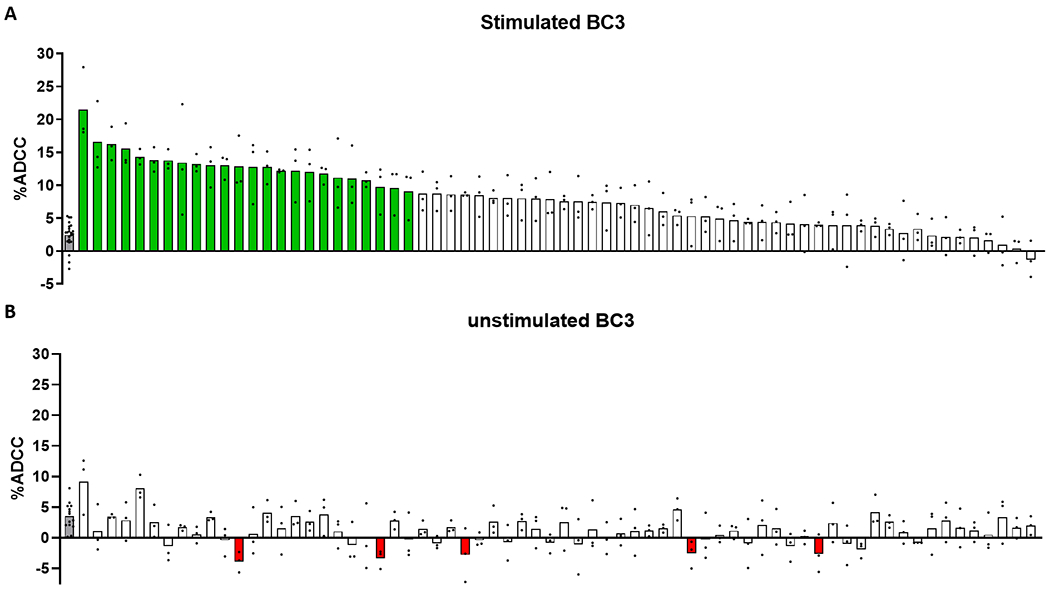
ADCC activity of KSHV seropositive individuals. 68 KSHV seropositive individuals were screened for ADCC activity against lytically reactivated (A) or unstimulated (B) BC3 cells. Gray bars represent KSHV seronegative controls. Data shown is the mean of 3 independent experiments, with the exception of seronegative controls (n=22 in panel A; n=14 in panel B). Each individual value is indicated with a black dot. Samples with activity significantly different (p<0.05, One-way ANOVA with Dunnett’s post-test) from KSHV seronegative controls are highlighted green if the activity is greater than controls and red if the activity is less than controls.
To assess the potential involvement of ADCC in preventing the development of KS, we compared ADCC activity between 30 KS patients and 38 KSHV seropositive asymptomatic controls. As no individuals showed a significant response against latent cells, only ADCC activity against lytic cells was compared. Interestingly, the prevalence of statistically significant ADCC responses was higher among KSHV seropositive controls (16/38, 42.1%) than KS patients (8/30, 26.7%). However, this difference was not significant (Fisher’s Exact, p=0.2113 Supplemental Table 1). There was also no difference between the mean magnitude of ADCC activity between KS patients (7.842%) and controls (7.969%, p=0.9062, Figure 4A). HIV status, sex, age, KSHV Ab titer, and KSHV neutralizing activity were assessed in order to determine what factors, if any, associated with ADCC activity. Magnitude of ADCC activity did not differ by HIV status (p=0.9468) or sex (p=0.3325, Figure 4B,C). Age (Pearson r=−0.08475, p=0.4920), KSHV Ab titer (Spearman r = −0.03576, p=0.8312) and KSHV neutralizing activity (Pearson r =0.05536, p=0.7180) also showed no correlation with ADCC activity (Figure 4D–F). HIV status, sex, and neutralizing activity were also assessed as discrete variables and no significant differences were found (Supplemental Table 1).
Figure 4.
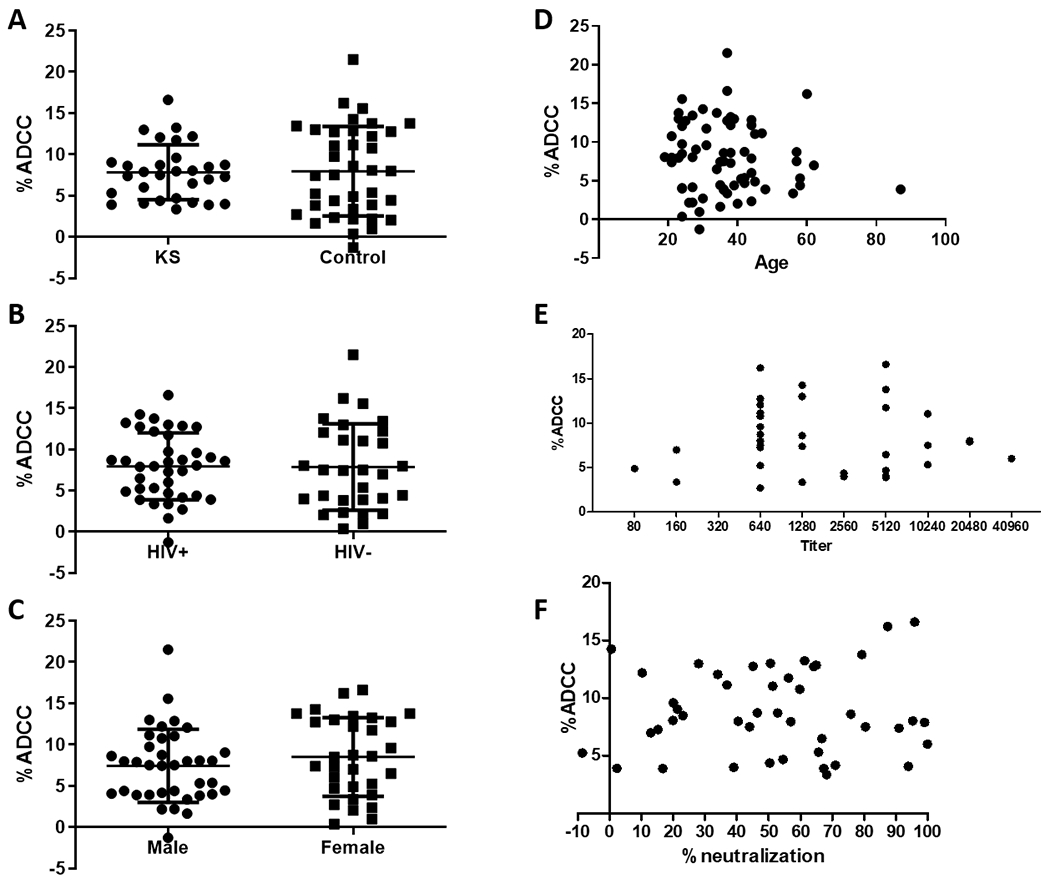
ADCC responses were not found to correlate with any measured variables. ADCC activity was not found to differ by (A) KS status, p=0.9062; (B) HIV status, p=0.9468; (C) sex, p=0.3325. (D) Age, Pearson r=−0.08475, p=0.4920; (E) KSHV Titer, Spearman r = −0.03576, p=0.8312; and (F) KSHV neutralizing activity Pearson r =0.05536, p=0.7180 were not associated with ADCC activity. Each dot represents the mean ADCC from three independent experiments.
Follow up samples taken after chemotherapeutic treatment were available for ten KS patients and were used to determine the potential involvement of ADCC responses in disease progression or control. ADCC activity at pre- and post-treatment time points is shown in Figure 5. No overall change in ADCC activity against either lytic (Figure 5A, p=0.3426) or latent (Figure 5B, p=0.2145) targets was seen post-treatment. Of the ten subjects with follow-up samples, five showed a reduction in the number or size of lesions at follow up (green circles), three presented with new lesions (red squares), and two individuals showed no change in disease status (blue triangles). There was no difference in ADCC activity prior to or after treatment between these treatment response groups. Additionally, no discernable pattern between time points (increasing, decreasing, or constant) was associated with response or non-response.
Figure 5.
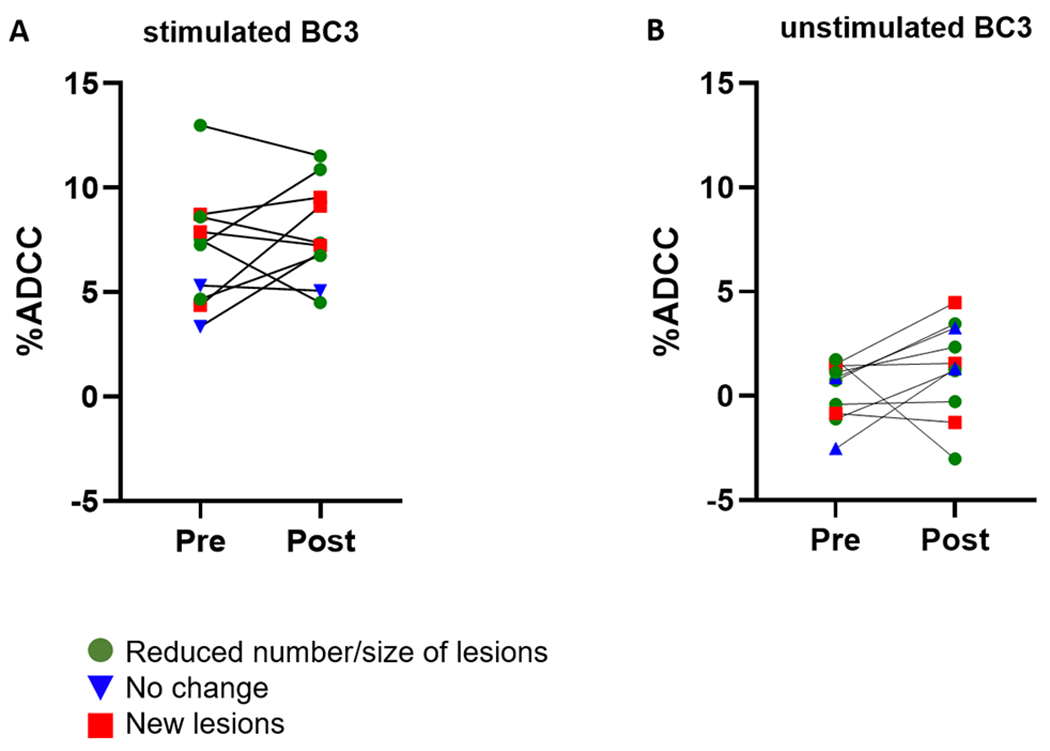
ADCC activity among pre- and post-treatment KS patients. 10 KS patients were tested for ADCC activity against lytically reactivated (A) and unstimulated (B) BC3 cells. Data shown is the mean and standard deviation from 3 independent experiments. Patients are coded by response status at follow up time point. Individuals with a reduced number and/or size of lesions at follow up are represented by green circles, those with no change after treatment are shown in blue triangles, and patients presenting new lesions are depicted with red squares. There was no change in ADCC pre- and post-treatment for the group as a whole, and the magnitude of response or change in response was not found to associate with treatment outcome.
In order to examine the stability of ADCC responses over time, ADCC activity was determined longitudinally for ten different subjects. Two years of samples collected at three-month intervals were examined. All ten were HIV-infected women without KS. ADCC profiles against lytically reactivated BC3 cells for each woman are shown in Figure 6. Four of the women (152M, 594M, 636M, 649M, Figure 6) showed no significant variation in ADCC activity over time, as determined by one-way ANOVA. The remaining 6 showed significant variance between time points. Three were fairly consistent longitudinally with only one or two outliers (159M, 366M, 465M), while the last three showed greater variation (040M, 306M, 470M, Figure 6). Subject 040M appeared to demonstrate a random pattern, with ADCC activity changing at almost every sampling interval, while 306M and 470M trended towards increasing ADCC activity over the two years studied. It should be noted that each woman showed statistically significant ADCC activity at least once. Subject 470M was also unique as this was the only individual in whom ADCC activity against unstimulated, latent BC3 cells was detected. Significant ADCC activity against latent cells was detected at 5 of the 9 time points and likely represents an anti-cancer response as this is the same individual with detectable responses against BJAB cells in Figure 2.
Figure 6.
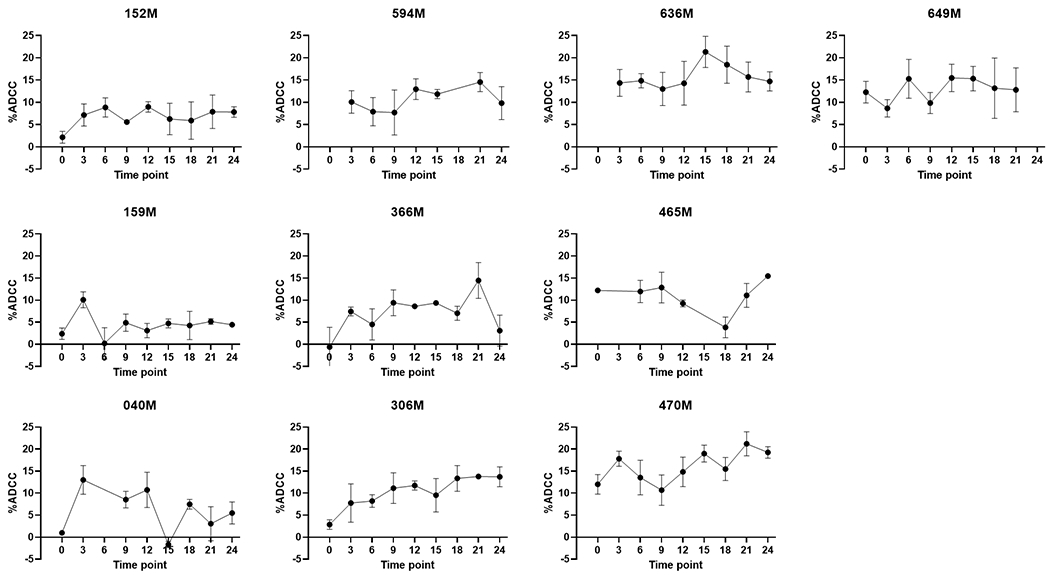
Variation in ADCC activity over time. The longitudinal ADCC activity profiles for 10 individuals are shown. ADCC activity is on the y-axis, while follow-up time point (in months) in on the x-axis. The enrollment sample is represented as time point 0. Each point represents the mean and standard deviation of 3 independent experiments. Time points are without a dot when no sample was available for testing.
Alternative endothelial-like KSHV infected cell lines were also tested for ADCC activity, however, reproducible responses were not detected (data not shown). As endothelial-derived KSHV infected cell lines are notably difficult to infect and induce once infection has been established (30), we hypothesized a lack of antigen/antibody interaction at the cell surface was responsible for the low-level, inconsistent responses observed. Immunofluorescence assays were therefore performed using high titer KSHV seropositive control plasma, strong ADCC mediating plasma (from subject 636M), and KSHV seronegative control plasma to assess antibody binding to stimulated BC3 cells, iSLKBAC16 cells, and L1T2 cells. As shown in Supplemental Figures 2 and 3 antibody binding was only observed on stimulated BC3 cells, confirming a lack of induction and antigen expression on the alternative cell lines.
Discussion
To our knowledge, this is the first study to examine ADCC in the context of KSHV infection and KS pathogenesis. We showed KSHV seropositive individuals have antibodies capable of mediating ADCC; however, only approximately 35% of seropositive individuals mediate significantly more killing than seronegative controls. While exact prevalence of ADCC-mediating antibodies among seropositive individuals is unclear for other herpesviruses, the majority of individuals seropositive against HCMV, HSV-1, and EBV appear to have ADCC-mediating antibodies (20, 31–33). Due to the heterologous criteria used to define positive ADCC responses across the field, it is difficult to make direct comparisons to our data. For example, Nelson et al. stated the majority of HCMV seropositive individuals mediated ADCC responses, though these responses were low-magnitude, and only the average response of all seropositive individuals was tested for significance (31). Stronger ADCC responses were seen in assays using EBV super-infected target cells, where the chronically EBV-infected Raji cell line was infected with an additional EBV strain, and assays where lytic cells were selectively examined via fluorescent markers (29, 33). While we used stringent criteria to identify ADCC positive responses, a majority of KSHV seropositive individuals did demonstrate ADCC activity 2-fold greater than that observed in seronegative controls. Thus, varying assay sensitivity and positive response criteria may account for the seemingly lower prevalence of ADCC responses among KSHV seropositive individuals as compared to other herpesviruses.
Importantly, we observed no relationship between ADCC and KS status or disease progression. Frequency and magnitude of ADCC responses did not differ between KS patients and asymptomatic KSHV seropositive individuals, nor was there a detectable relationship between ADCC activity and treatment outcome in 10 KS patients with follow-up samples. This indicates that for KSHV, ADCC-mediating Abs alone are not likely to be protective against KS disease development. However, both antibodies and effector cells are required to elicit ADCC responses in vivo, and a deficiency in either has been associated with increased disease severity for other herpesviruses. Reductions in ADCC-mediating antibodies compared to healthy seropositive controls have been observed in individuals with infectious mononucleosis (20, 33); however there was no change in ADCC activity prior to the recurrence of disease in Burkitt’s lymphoma patients (34). Individuals with defective ADCC effector cell function show an increase in the severity of HSV symptoms (35). Additionally, both CD16A and IgG1 allotypes are important variables in protection against HSV-1, with the high affinity variant of CD16A (V/V) and IgG1m3 genotypes associated with higher levels of ADCC and clinical repression (17). While we have shown Ab responses do not differ by KS status, it is possible the function of NK and other effector cells may be impaired during disease. Indeed, AIDS-KS patients have been shown to have decreased Ab-independent PBMC cytotoxicity compared to classic KS and healthy blood donors (15). Hyporesponsive PD-1 expressing NK cells have also been reported to be increased in frequency in multiple forms of KS compared to asymptomatic controls (36). Unfortunately, primary PBMCs or NK cells were unavailable for the samples used in this study, therefore an analysis of the effector cell function was not possible at this time. Comparative investigations into effector cell activity between KS and non-KS individuals, specifically with regard to ADCC, is needed before any definitive conclusions can be drawn about the role of ADCC in preventing KS development.
With one exception, only cells undergoing lytic reactivation were targetable by Ab from KSHV seropositive individuals. This finding is supported by similar reports about ADCC against EBV (29, 37). The lack of response against latent cells is unsurprising as limited gene expression during latency results in few or no viral antigens on the cell surface and is a hallmark of all herpesviral latencies. However, cells may be sensitive to ADCC after initial KSHV infection as some lytic genes, including two glycoproteins, are reportedly expressed immediately following infection (38, 39). Additionally, responses targeting lytic cells may still help to maintain control of infection by reducing the viral infectious progeny produced, and thereby limiting dissemination of infection. As lytic reactivation is believed to be a key component in the development of KS (10), ADCC killing of cells undergoing lytic reactivation could also contribute to preventing tumor formation. However, KS tumors are primarily composed of latently infected cells (40–42), thus the lack of response to latent cells suggests natural antibodies are unlikely to target and eliminate KS tumors once formed.
ADCC was not found to relate to HIV status, sex, age, KSHV Ab titer, or neutralizing antibodies. Similarly, Ab titers and neutralization for both HSV and EBV also show no correlation to ADCC responses (33–35). The lack of correlation between ADCC activity and neutralizing activity across herpesviruses implies different antibodies are responsible for ADCC and neutralization. As the Fc-region is most critical for inducing ADCC, some target epitopes could be the same, with differences in class-switching yielding differences in ADCC activity. However, this would not account for the individuals found to have ADCC but no neutralizing activity. Future investigations into the specific proteins and epitopes targeted by both neutralizing Ab and ADCC responses are needed and could provide useful information on antigen priority in designing vaccines and therapeutics.
Over half of the individuals longitudinally examined here had significant variance in ADCC activity. No factors were found to correlate with stability or variability in the response, though all were HIV-infected women. While there was no relationship between strength of ADCC activity and HIV status, we cannot rule-out an effect of HIV on the stability of ADCC responses perhaps through CD4+ T cell depletion or aberrant cytokine signaling. The longitudinal variation matches what we have previously observed with KSHV neutralizing Ab activity, also in HIV-infected women (43). Together, this suggests that the Ab response to KSHV may change qualitatively over time. While a study conducted by Labo et al examined protein specific anti-KSHV Ab, responses were examined at only a single time point (44). Whether individual anti-KSHV Ab responses are consistent longitudinally is unknown and deserves further study.
One limitation to this study is the lack of in vitro models for KS. KSHV infection of endothelial and epithelial cell lines is inefficient, unsustainable, and difficult to reactivate into lytic replication (30). While HUVEC cells have been reported to show increased infection efficiency using recombinant KSHV BAC36, these cells only show robust lytic induction if stimulated in less than 10 days after infection, making independent ADCC replicates from the same infection impossible (30). We hoped that the inducible KSHV infected cell line, iSLKBAC16, which expresses RTA in the presence of doxycycline, would allow for the ADCC assays against both lytic and latent cells (45). However, as seen in Supplemental Figure 2, even the cells stimulated with doxycycline and sodium butyrate were unable to induce sufficient lytic replication to allow for antibody binding at the cell surface and we were unable to detect replicable ADCC against either stimulated or unstimulated iSLKBAC16 cells. Additionally, the original SLK line believed to be a KS cell line was later determined to be contaminated with a renal carcinoma line, further highlighting the lack of good in vitro KS models (46). The L1T2 line was another promising candidate, however this is a heterologous cell line where only a proportion of the cells are infected with KSHV (47, 48). The calcein release method utilized here would be unable to differentiate between the killing of infected and uninfected cells and thus, more intensive, lower throughput methods would be required. In addition, L1T2 cultures were also unable to yield robust lytic reactivation, as seen in Supplemental Figure 3.
Therefore, to avoid the issues with the more KS-like models, we used the primary effusion lymphoma cell line BC3, a naturally occurring KSHV-associated malignancy and the same cell line used to determine KSHV serology (28). As unstimulated cells do not express viral antigens and were not targeted by ADCC mechanisms, we are confident the responses shown here are KSHV specific and not due to non-self or anti-cancer responses. The exception is one individual who showed statistically significant ADCC responses against both lytic and latent BC3 cells, as well as the uninfected BJAB cells, who most likely demonstrated an anti-cancer cell line, rather than KSHV-specific, ADCC response. While we were able to detect KSHV specific responses, the BC3 line may not serve as an accurate representation of KS. Despite the limitations of the current in vitro KS models, future experiments should aim at testing additional cell types, specifically endothelial cells, to determine if ADCC responses are affected.
In summary, we report the first investigation of ADCC in the context of KSHV infection. A portion of KSHV seropositive individuals possess antibodies capable of mediating ADCC responses against KSHV infected cells; however, only cells undergoing lytic reactivation are targeted. While there is no correlation between ADCC-mediating antibodies and KS pathogenesis, further research is needed to determine if effector cell responses are impaired in the context of KS disease development or for other KSHV-associated malignancies.
Supplementary Material
Key points:
KSHV antibodies mediate ADCC responses against lytically reactivated cells.
Over one-third of KSHV seropositive individuals have ADCC responses.
ADCC does not differ between KS patients and asymptomatic controls.
Acknowledgments
The authors would like to acknowledge the individuals in this study for their participation, Dr. Kerry Campbell for generously providing both the NK92.05 CD16 176V cells and the IL-2 producing J558L cell line, and his guidance in culturing the cells, Dr. Dirk Dittmer for providing the L1T2 cells, and Dr. Jae Jung for providing the iSLKBAC16 cells.
1. Funding sources: This study is supported in part by the National Institutes of Health (NIH) under Ruth L. Kirschstein National Research Service Award T32 AI125207 from the National Institute of Allergy and Infectious Diseases, as well as NIH PHS grants: NCI75903, NCI U54CA221204 and D43TW010354 to CW. LKP is a Ruth L. Kirchstein fellow.
2. Abbreviations:
- KSHV
Kaposi’s sarcoma-associated herpesvirus
- KS
Kaposi’s sarcoma
- ADCC
Ab-dependent cell cytotoxicity
- HCMV
Human CMV
References:
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, and Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266: 1865–1869. [DOI] [PubMed] [Google Scholar]
- 2.Moore PS, and Chang Y. 1995. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and those without HIV infection. N Engl J Med 332: 1181–1185. [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, and Knowles DM. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332: 1186–1191. [DOI] [PubMed] [Google Scholar]
- 4.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, and et al. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86: 1276–1280. [PubMed] [Google Scholar]
- 5.Brown EE, Whitby D, Vitale F, Marshall V, Mbisa G, Gamache C, Lauria C, Alberg AJ, Serraino D, Cordiali-Fei P, Messina A, and Goedert JJ. 2006. Virologic, hematologic, and immunologic risk factors for classic Kaposi sarcoma. Cancer 107: 2282–2290. [DOI] [PubMed] [Google Scholar]
- 6.Cesarman E, Damania B, Krown SE, Martin J, Bower M, and Whitby D. 2019. Kaposi sarcoma. Nat Rev Dis Primers 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegel JH, Janis R, Alper JC, Schutte H, Robbins L, and Blaufox MD. 1969. Disseminated visceral Kaposi’s sarcoma. Appearance after human renal homograft operation. JAMA 207: 1493–1496. [PubMed] [Google Scholar]
- 8.Lepone LM, Rappocciolo G, Piazza PA, Campbell DM, Jenkins FJ, and Rinaldo CR. 2017. Regulatory T Cell Effect on CD8(+) T Cell Responses to Human Herpesvirus 8 Infection and Development of Kaposi’s Sarcoma. AIDS Res Hum Retroviruses 33: 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roshan R, Labo N, Trivett M, Miley W, Marshall V, Coren L, Cornejo Castro EM, Perez H, Holdridge B, Davis E, Matus-Nicodemos R, Ayala VI, Sowder R, , Wyvill KM, Aleman K, Fennessey C, Lifson J, Polizzotto MN, Douek D, Keele B, Uldrick TS, Yarchoan R, Ohlen C, Ott D, and Whitby D. 2017. T-cell responses to KSHV infection: a systematic approach. Oncotarget 8: 109402–109416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wakeham K, Johnston WT, Nalwoga A, Webb EL, Mayanja BN, Miley W, Elliott AM, Whitby D, and Newton R. 2015. Trends in Kaposi’s sarcoma-associated Herpesvirus antibodies prior to the development of HIV-associated Kaposi’s sarcoma: a nested case-control study. Int J Cancer 136: 2822–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar P, Kuwa NY, Minhas V, Marimo C, Shea DM, Kankasa C, and Wood C. 2013. Higher levels of neutralizing antibodies against KSHV in KS patients compared to asymptomatic individuals from Zambia. PLoS One 8: e71254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matthews NC, Goodier MR, Robey RC, Bower M, and Gotch FM. 2011. Killing of Kaposi’s sarcoma-associated herpesvirus-infected fibroblasts during latent infection by activated natural killer cells. Eur J Immunol 41: 1958–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupuy S, Lambert M, Zucman D, Choukem SP, Tognarelli S, Pages C, Lebbe C, and Caillat-Zucman S. 2012. Human Herpesvirus 8 (HHV8) sequentially shapes the NK cell repertoire during the course of asymptomatic infection and Kaposi sarcoma. PLoS Pathog 8: e1002486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sirianni MC, Campagna M, Scaramuzzi D, Carbonari M, Toschi E, Bacigalupo I, Monini P, and Ensoli B. 2007. Control of human herpes virus type 8-associated diseases by NK cells. Ann N Y Acad Sci 1096: 37–43. [DOI] [PubMed] [Google Scholar]
- 15.Sirianni MC, Vincenzi L, Topino S, Giovannetti A, Mazzetta F, Libi F, Scaramuzzi D, Andreoni M, Pinter E, Baccarini S, Rezza G, Monini P, and Ensoli B. 2002. NK cell activity controls human herpesvirus 8 latent infection and is restored upon highly active antiretroviral therapy in AIDS patients with regressing Kaposi’s sarcoma. Eur J Immunol 32: 2711–2720. [DOI] [PubMed] [Google Scholar]
- 16.Gorander S, Ekblad M, Bergstrom T, and Liljeqvist JA. 2014. Anti-glycoprotein g antibodies of herpes simplex virus 2 contribute to complete protection after vaccination in mice and induce antibody-dependent cellular cytotoxicity and complement-mediated cytolysis. Viruses 6: 4358–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moraru M, Black LE, Muntasell A, Portero F, Lopez-Botet M, Reyburn HT, Pandey JP, and Vilches C. 2015. NK Cell and Ig Interplay in Defense against Herpes Simplex Virus Type 1: Epistatic Interaction of CD16A and IgG1 Allotypes of Variable Affinities Modulates Antibody-Dependent Cellular Cytotoxicity and Susceptibility to Clinical Reactivation. J Immunol 195: 1676–1684. [DOI] [PubMed] [Google Scholar]
- 18.Jenks JA, Goodwin ML, and Permar SR. 2019. The Roles of Host and Viral Antibody Fc Receptors in Herpes Simplex Virus (HSV) and Human Cytomegalovirus (HCMV) Infections and Immunity. Front Immunol 10: 2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chijioke O, Muller A, Feederle R, Barros MH, Krieg C, Emmel V, Marcenaro E, Leung CS, Antsiferova O, Landtwing V, Bossart W, Moretta A, Hassan R, Boyman O, Niedobitek G, Delecluse HJ, Capaul R, and Munz C. 2013. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep 5: 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jondal M 1976. Antibody-dependent cellular cytotoxicity (ADCC) against Epstein-Barr virus-determined membrane antigens. I. Reactivity in sera from normal persons and from patients with acute infectious mononucleosis. Clin Exp Immunol 25: 1–5. [PMC free article] [PubMed] [Google Scholar]
- 21.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, and Jung JU. 2012. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86: 9708–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lidenge SJ, Tso FY, Ngalamika O, Ngowi JR, Mortazavi Y, Kwon EH, Shea DM, Minhas V, Mwaiselage J, Wood C, and West JT. 2019. Similar Immunological Profiles Between African Endemic and Human Immunodeficiency Virus Type 1-Associated Epidemic Kaposi Sarcoma (KS) Patients Reveal the Primary Role of KS-Associated Herpesvirus in KS Pathogenesis. J Infect Dis 219: 1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olp LN, Shea DM, White MK, Gondwe C, Kankasa C, and Wood C. 2013. Early childhood infection of Kaposi’s sarcoma-associated herpesvirus in Zambian households: a molecular analysis. Int J Cancer 132: 1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minhas V, Crabtree KL, Chao A, Wojcicki JM, Sifuniso AM, Nkonde C, Kankasa C, Mitchell CD, and Wood C. 2011. The Zambia Children’s KS-HHV8 Study: rationale, study design, and study methods. Am J Epidemiol 173: 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olp LN, Minhas V, Gondwe C, Poppe LK, Rogers AM, Kankasa C, West JT, and Wood C. 2016. Longitudinal analysis of the humoral response to Kaposi’s sarcoma-associated herpesvirus after primary infection in children. J Med Virol 88: 1973–1981. [DOI] [PubMed] [Google Scholar]
- 26.Neri S, Mariani E, Meneghetti A, Cattini L, and Facchini A. 2001. Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin Diagn Lab Immunol 8: 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Somanchi SS, McCulley KJ, Somanchi A, Chan LL, and Lee DA. 2015. A Novel Method for Assessment of Natural Killer Cell Cytotoxicity Using Image Cytometry. PLoS One 10: e0141074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crabtree KL, Wojcicki JM, Minhas V, Smith DR, Kankasa C, Mitchell CD, and Wood C. 2014. Risk Factors for Early Childhood Infection of Human Herpesvirus-8 in Zambian Children: The Role of Early Childhood Feeding Practices. Cancer Epidemiol Biomarkers Prev 23: 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez-Montanes M, Alari-Pahissa E, Sintes J, Martinez-Rodriguez JE, Muntasell A, and Lopez-Botet M. 2017. Antibody-Dependent NK Cell Activation Differentially Targets EBV-Infected Cells in Lytic Cycle and Bystander B Lymphocytes Bound to Viral Antigen-Containing Particles. J Immunol 199: 656–665. [DOI] [PubMed] [Google Scholar]
- 30.Gao SJ, Deng JH, and Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J Virol 77: 9738–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson CS, Huffman T, Jenks JA, Cisneros de la Rosa E Xie G, Vandergrift N, Pass RF, Pollara J, and Permar SR. 2018. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc Natl Acad Sci U S A 115: 6267–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shore SL, Nahmias AJ, Starr SE, Wood PA, and McFarlin DE. 1974. Detection of cell-dependent cytotoxic antibody to cells infected with herpes simplex virus. Nature 251: 350–352. [DOI] [PubMed] [Google Scholar]
- 33.Khyatti M, Stefanescu I, Blagdon M, and Menezes J. 1994. Epstein-Barr virus gp350-specific antibody titers and antibody-dependent cellular cytotoxic effector function in different groups of patients: a study using cloned gp350-expressing transfected human T cell targets. J Infect Dis 170: 1439–1447. [DOI] [PubMed] [Google Scholar]
- 34.Jondal M, and Gunven P. 1977. Antibody-dependent cellular cytotoxicity (ADCC) against Epstein-Barr virus-determined antigens. III. Reactivity in sera from patients with Burkitťs lymphoma in relation to tumour development. Clin Exp Immunol 29: 11–15. [PMC free article] [PubMed] [Google Scholar]
- 35.Kohl S 1991. Role of antibody-dependent cellular cytotoxicity in defense against herpes simplex virus infections. Rev Infect Dis 13: 108–114. [DOI] [PubMed] [Google Scholar]
- 36.Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, Dupuy S, Levasseur F, Zucman D, Lebbe C, Sene D, Hivroz C, and Caillat-Zucman S. 2016. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 7: 72961–72977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pappworth IY, Wang EC, and Rowe M. 2007. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol 81: 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, and Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol 78: 3601–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knipe DM, and Howley PM. 2013. Fields virology. Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia, PA. [Google Scholar]
- 40.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, and Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A 96: 4546–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mesri EA, Cesarman E, and Boshoff C. 2010. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer 10: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, and Haase AT. 1997. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71: 715–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poppe LK, Kankasa C, Wood C, and West JT. 2020. Relationships between maternal antibody responses and early childhood infection with KSHV. J Infect Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Labo N, Miley W, Marshall V, Gillette W, Esposito D, Bess M, Turano A, Uldrick T, Polizzotto MN, Wyvill KM, Bagni R, Yarchoan R, and Whitby D. 2014. Heterogeneity and breadth of host antibody response to KSHV infection demonstrated by systematic analysis of the KSHV proteome. PLoS Pathog 10: e1004046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myoung J, and Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sturzl M, Gaus D, Dirks WG, Ganem D, and Jochmann R. 2013. Kaposi’s sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int J Cancer 132: 1954–1958. [DOI] [PubMed] [Google Scholar]
- 47.Roy D, Sin SH, Lucas A, Venkataramanan R, Wang L, Eason A, Chavakula V, Hilton IB, Tamburro KM, Damania B, and Dittmer DP. 2013. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer Res 73: 2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tso FY, West JT, and Wood C. 2019. Reduction of Kaposi’s Sarcoma-Associated Herpesvirus Latency Using CRISPR-Cas9 To Edit the Latency-Associated Nuclear Antigen Gene. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.