

Abstract
Post Translational Modification (PTM) is considered an important biological process with a tremendous impact on the function of proteins in both eukaryotes, and prokaryotes cells. During the past decades, a wide range of PTMs has been identified. Among them, malonylation is a recently identified PTM which plays a vital role in a wide range of biological interactions. Notwithstanding, this modification plays a potential role in energy metabolism in different species including Homo Sapiens. The identification of PTM sites using experimental methods is time-consuming and costly. Hence, there is a demand for introducing fast and cost-effective computational methods. In this study, we propose a new machine learning method, called Mal-Light, to address this problem. To build this model, we extract local evolutionary-based information according to the interaction of neighboring amino acids using a bi-peptide based method. We then use Light Gradient Boosting (LightGBM) as our classifier to predict malonylation sites. Our results demonstrate that Mal-Light is able to significantly improve malonylation site prediction performance compared to previous studies found in the literature. Using Mal-Light we achieve Matthew’s correlation coefficient (MCC) of 0.74 and 0.60, Accuracy of 86.66% and 79.51%, Sensitivity of 78.26% and 67.27%, and Specificity of 95.05% and 91.75%, for Homo Sapiens and Mus Musculus proteins, respectively. Mal-Light is implemented as an online predictor which is publicly available at: (http://brl.uiu.ac.bd/MalLight/)
Index Terms: Cluster Centroid based Majority Under-sampling Technique, Evolutionary Information, Light Gradient Boosting, Lysine Malonylation, Machine Learning, Post Transla tional Modifications
I. Introduction
Post-translational modifications (PTMs) are the key tools for regulating numerous biological processes that are affiliated with the control activities of various cells and diseases [1] – [4]. PTMs are formed after the translation process of proteins from the mRNA sequences when they are elucidated [5], [6]. PTMs are the major components of biological processes for genetic code proliferation and cellular physiology regulation. So far, more than 620 varieties of PTMs [7] have been identified. Lysine is one of the most widely modified residues among the 20 types of natural amino acids through PTM [8]. It has been associated with numerous PTMs including glycation [9], succinylation [10], [11], methylation [12], [13], acetylation [14], and sumoylation [15]. Among them, Lysine malonylation (Kmal) is a recently identified PTM type that is evolutionarily conserved, which is associated with several biological processes in both eukaryotic and prokaryotic cells. Lysine malonylation plays a vital role in a wide range of biological interactions [16]. It has also been found in histones with functions related to gene expression, and chromosome configuration. Thus, identification of malonylation sites can provide detailed insights into the functionality of proteins and their biological interactions. The affluence of malonylated proteins impact on metabolic pathways and notably those adhering to fatty acid metabolism is explained in [17]. In addition, newly identified malonylated sites have been found to be associated with monitoring the conditions in the pathological, and physiological functional structures such as control of appetite and muscle contraction [18], [19].
The foremost techniques for identifying the Kmal sites are experimental methods such as mass spectrometry. However, these methods are costly and time-consuming. In recent years, the identification of PTM sites using a fast and accurate computational method attracted tremendous attention [20]. To identify PTM sites in the protein sequences, various bioinformatics techniques have been suggested [11], [21] – [29]. Among those studies, a five-step rule was proposed in [30], to design an efficient computational predictor for solving these biological problems which have been widely referred and followed in other studies [27], [28], [31], [32], [33]. The following steps comprise: (1) curated the dataset manually or construct the dataset in some valid way to randomly split in both training and testing for the predictor, (2) transforming the biological sequences into numerical values to extracting the feature vector, (3) selecting proper algorithm according to the problem and develop an algorithm to build the predictor, (4) validate the statistical performance matrices and evaluate the predictor enhancement, and (5) design a user-friendly predictor and deploy the method as a web server application publicly. The above-mentioned process is explained in Fig. 1 in the subsequent section.
Fig. 1.
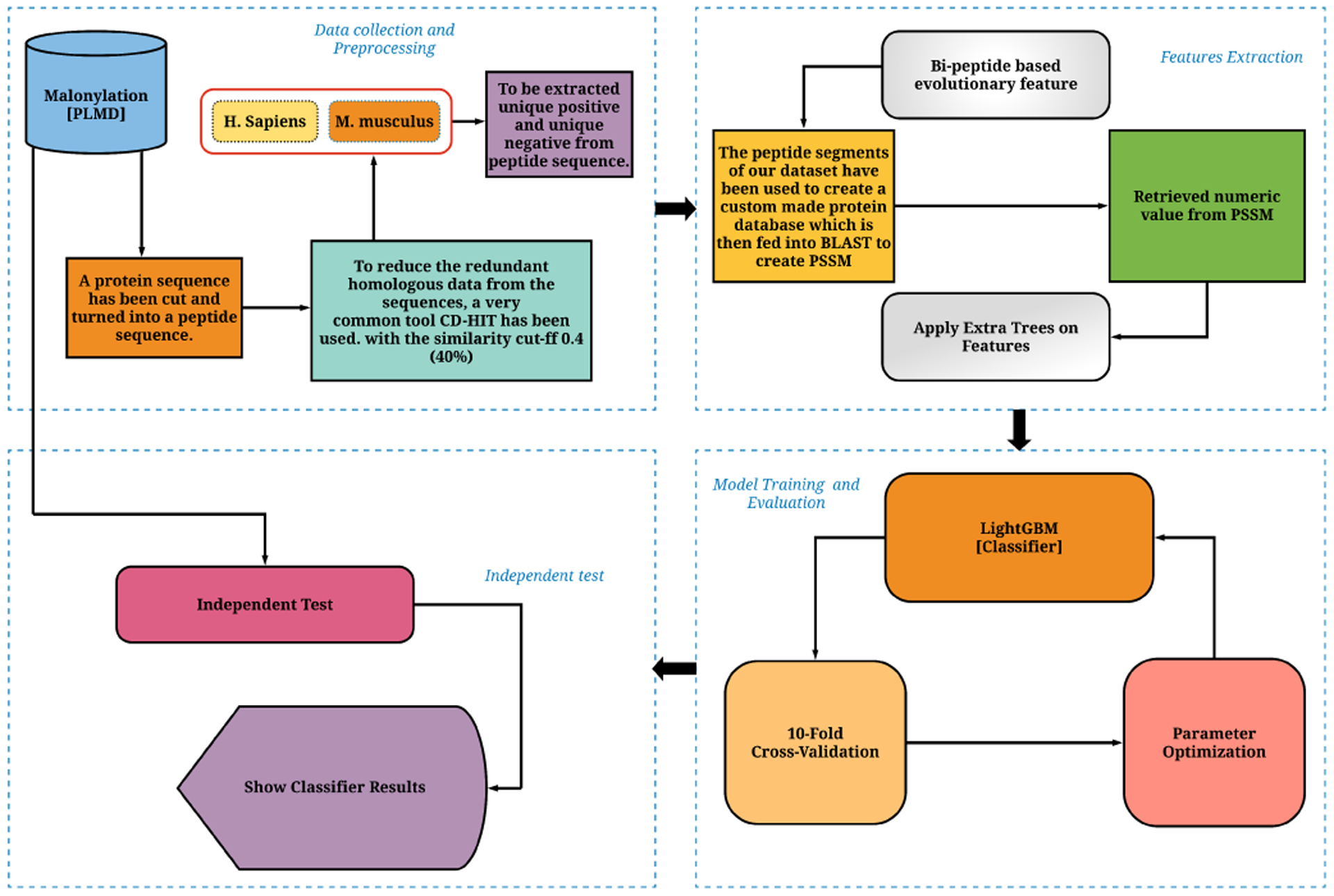
The evaluation and development of Mal-Light that contains in the flowchart strategy. Sequences were yield from a public database and features were generated by our method, named bi-peptide based evolutionary feature extraction approach with the classifier and the classification algorithm was evaluated by using both 10-fold cross-validation and an independent test set
Among computational approaches, predicting the malonylated sites through the Machine Learning (ML) models has attracted the most attention [25], [34] – [40]. The first computational scheme developed by Xu et al. [35], called Mal-Lys, to predict the Kmal sites based on the protein sequences. They extracted three types of features in Mal-Lys based on position-specific amino acid dehydration, sequence order information, and physicochemical properties. They also used maximum relevance minimum redundancy for feature selection task [36]. They also used Support Vector Machine (SVM) as their classifiers to build Mal-Lys. At the same time, Wang et al. [37] manifested an SVM-based classifier, named MaloPred to predict malonylation sites in three different species (Homo sapiens, Mus musculus, and Escherichia coli). In a different study, Xiang et al. [38] trained an SVM model by introducing a new computational method using the Pseudo Amino Acid Composition (PseAAC) scheme to extract features. Their study also validated the diverse pathways and biological processes in several species. In another study, Zhang et al. [39] extracted the characteristics and key patterns from the residue sequences of Kmal sites using 11 different feature encoding methods. Among them, they identified the optimized feature and used Light Gradient Boosting Machine (LightGBM) as their classifier to predict Kmal sites for Homo sapiens, Mus musculus, and Escherichia coli samples.
In a different study, Jianhua et al. [11] developed a new predictor, named pSuc-Lys, by using a feature extraction technique called, PseAAC. To build this model, they combined a vectorized sequence-coupling model into the common form of PseAAC along with using ensemble random forest technique as their classifier. At the same time, Taherzadeh et al. [40] introduced a new machine-learning approach named SPRINT, which is conceived of sequence-based prediction of protein-peptide binding sites directly from protein sequence by using Support Vector Machine. Later on, Taherzadeh et al. [41] also proposed SPRINT-Mal for the Kmal site prediction problem. To build this model, they implicated both sequence-based as well as structural-based features and used SVM as their classifier. They obtained promising results in predicting the malonylation sites for mouse samples. Most recently, Zhe et al. [42] proposed a new SVM base method, entitled CKSAAP FormSite, to solve the class imbalance problem in the prediction of formylation sites prediction task. They have applied a composition of k-spaced amino acid pairs (CKSAAP) feature extraction technique that were utilized to encode each peptide during training.
Despite all the efforts that have been made so far, the Kmal prediction accuracy has still remained limited. In this paper, we propose a new model called Mal-Light, based on the concepts of a bi-peptide based evolutionary feature extraction strategy for enhancing the performance of malonylated sites [43], [44]. We then investigated the performance of 12 different classifiers on our extracted features to identify the best one to build Mal-Light. Among these classifiers, Light Gradient Boosting (LightGBM) obtained the best results. As a result, we use this classifier to build Mal-Light. The above-mentioned process is shown in Fig. 1 and explained in detail in the subsequent section. In fact, our main contribution is to investigate a wide range of models that obtained promising results for different studies but have never been used for Malonylation site prediction problem to enhance the prediction performance. We compared the prediction results of Mal-Light with those of MaloPred [37], and kmal-sp [39]. We obtained Matthew’s correlation coefficient (MCC) of 0.74 and 0.60, Accuracy (ACC) of 86.66% and 79.51%, Sensitivity (SN) of 78.26% and, 67.27%, and Specificity (SP) of 95.05% and 91.75% on our employed independent test set, respectively for the Homo Sapiens (Human) and Mus Musculus (Mouse) samples. Mal-Light obtained promising results by exceeding all the preceding predictors.
II. Materials and Methods
A. Benchmark dataset
For the experimental analysis, we use malonylation data from the Protein Lysine Modification Database (PLMD) [45]. This dataset contains 9,584 malonylation and 677,865 non-malonylation sites in 3,429 proteins belonging to mainly six species. Here we mainly focus on Homo sapiens (5,013 sites in 1,841 proteins) and Mus musculus (4,390 sites in 1,466 proteins) as the number of samples for the remaining species is extremely low. The number of samples belonging to each group and species is shown in Table 1. The responsible residue for the malonylation site is the amino acid lysine (one letter notation of K). For transforming into peptide sequence from protein, the responsible residue is kept in the middle with the window size 2ξ + 1, where ξ is the length of upstream and downstream. In the proposed model, the window size is considered as 21 (length of upstream and downstream is considered as ξ = 10). The optimal window size in the specified range is found by observing the performance of the LightGBM classifier on the features extracted using the amino acid composition technique. For ensuring the uniform length of upstream and downstream, a dummy residue, (X) has been added to any of the ends when required (for n-terminus and c-terminus amino acids that have less than 10 neighboring amino acids at each end). After that, we removed duplicated sites and extracted unique positive and unique negative from peptide sequences of all species as well as Homo sapiens (human) and Mus musculus (mouse). In the next step, to reduce redundant data of homology from the sequences we use CD-HIT [46] which have been widely used for this task. From the peptide sequence, we have found the ratio between positive and negative is quite large. As a result, we merely used CD-HIT [46] over negative sequences only where remaining the positive sequences untouched to avoid losing limited positive samples. If we applied CD-HIT [46] over the positive sequences then the difference ratio between positive and negative sequences more increases. This is why we only apply the CD-HIT [46] over the negative sequence. It reduced the negatives sites with the similarity cut-off 40%. We then generated PSSM for our positive 9,584 and negative 14,972 samples for all the species. Besides, we have a dataset containing 5,013 positive and 12,869 negative samples for human and 4,390 positive and 10,152 negative samples for mouse. To measure the actual effectiveness of our proposed model, we generate independent test data from our original data that is unknown to the training data. In this continuum, we randomly place 90% for the training data and 10% for the independent test data, which is the same for all types of species as well as for human and mouse.
TABLE I.
The total number of sites in different species of protein.
| Species | # of protein | # of sites in protein |
|---|---|---|
| Homo sapiens | 1,841 | 5,013 |
| Mus musculus | 1,466 | 4,390 |
| Saccharopolyspora erythraea | 117 | 175 |
| Saccharomyces cerevisiae | 3 | 3 |
| Sus scrofa | 1 | 2 |
| Escherichia coli | 1 | 1 |
| In Total | 3,429 | 9,584 |
B. Feature extraction
Biological data are usually represented as strings of sequences. Normally strings consist of one-letter notations where each letter represents amino acids for protein and nucleotides for DNA. The string data should have to mutate into numerical values to represent the biological instances through to the classifier. This transformation which is called feature extraction can be accomplished in many ways [44], [47] – [52]. However, the information can not be preserved for all numerical values at the same level that is carried in the letter strings. To maximize the information carried by the string, different feature extraction techniques have been introduced in the literature [43], [44], [47], [48], [50], [51], [53]. Most of these studies introduced sequential-based features extracted from evolutionary-based and structure-based information [43], [44], [50], [52], [53]. Besides, some of these studies incorporate evolutionary-based and physicochemical-based information, simultaneously [47], [51]. Evolutionary-based features are most widely used and provides information on how proteins and peptides evolved or changes through mutation. While structural features provide information on the local structure of the proteins extracted from predicted secondary structure. Similarly, physicochemical-based features are extracted based on different physical, and chemical properties of the amino acids along the protein and peptide sequences. However, in almost all the cases, proposed feature extraction methods failed to extract local discriminatory information based on the interaction of the amino acids along the protein or peptide sequences. Adopting feature extraction techniques that do not preserve important discriminatory information causes low prediction performance in the classification task.
In this study, the sequential evolutionary features were used to represent each malonylated and non-malonylated lysine residue. Their 10 upstream and 10 downstream amino acids were selected to extract features as it obtained the best results compared to other windows sizes. We reflected the missing peptide outspread if a lysine residue did not carry 10 amino acids of upstream or downstream in c-terminus and n-terminus, respectively. This process is shown in detail in Fig. 2. The sequence segment Pξ(⊙) consists of 10 upstream and 10 downstream residues in addition to the central lysine amino acid (K).
Fig. 2.
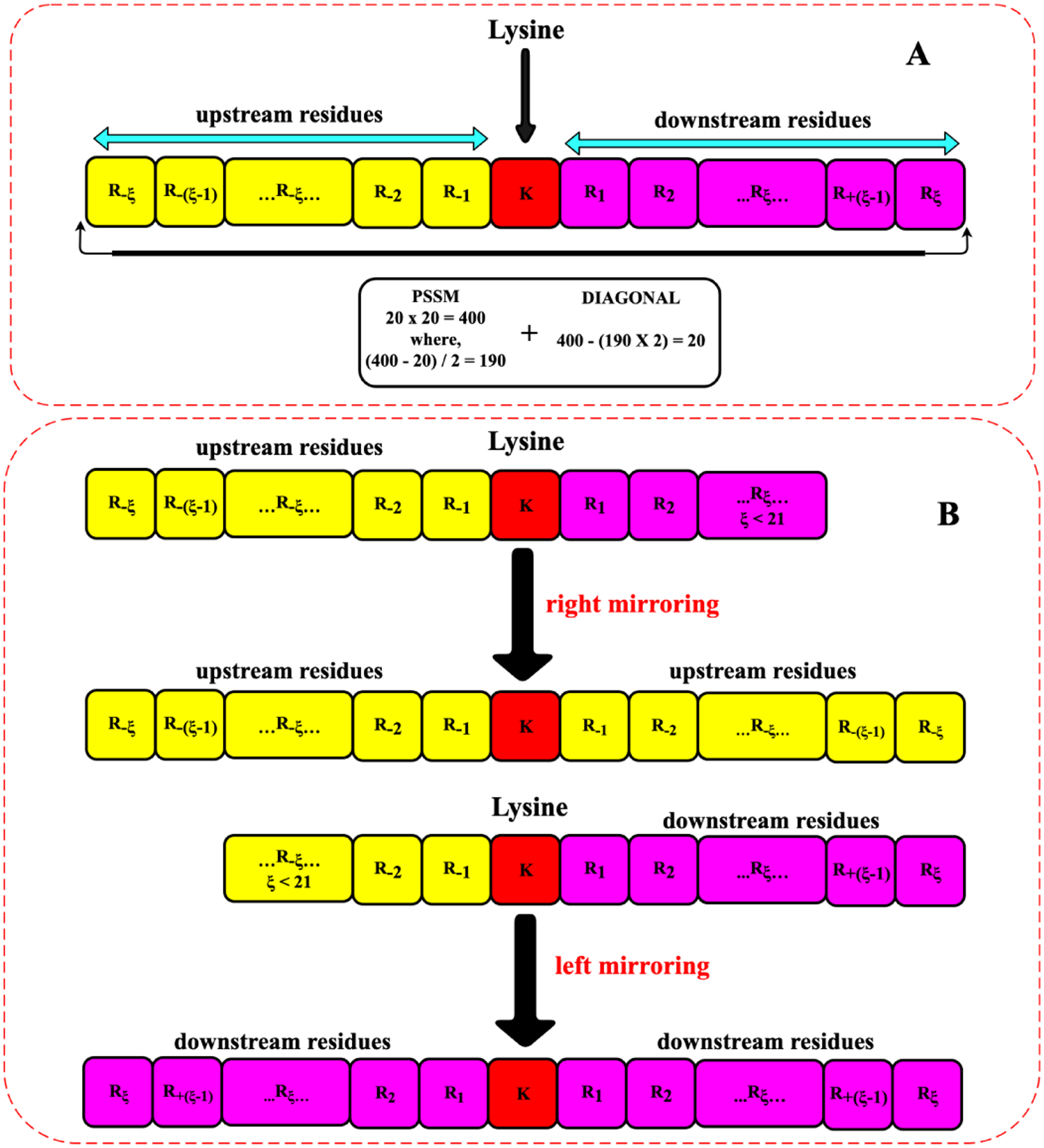
Schematic representation of a lysine residue and its surrounding amino acids. Figure 2.A: lysine residues with both upstream and downstream amino acids with 10 residues. Figure 2.B: Adding dummy residues in n-terminus and c-terminus to complete the window for those amino acids with less than 10 neighboring amino acids on each side.
Here is an example of a peptide sample presented as follows,
| (1) |
Here, ξ is an integer and ⊙ indicates the amino acid lysine (K). Where denotes upstream as R−ξ, and denotes downstream as R+ξ of the peptide sample. Meanwhile, the entire peptide length and a substring of a protein sequence where the sample contains 2ξ + 1 residues. Therefore, each of the peptides specimens befalls below one of two categories, that follows,
| (2) |
The positive malonylation segment symbolizes for and represents the negative malonylation segment where ∈ indicates the association of set principles.
Written the benchmark dataset as follows,
| (3) |
In order that carried malonylated segment, and carried non-malonylated segment, where ∪ is the union operation of set principles.
C. Bi-peptide based evolutionary feature
The bi-peptide based evolutionary concept is the feature extraction technique introduced in the prediction of lysine sites. This technique is a modification of the original sequential evolutionary feature extraction technique that is introduced in [54], [55]. It has been shown as an effective method for feature extraction in similar studies [43], [44], [49], [50]. We extract this feature directly from the Position Specific Scoring Matrix PSSM) which contains important evolutionary information about the interaction of amino acids through mutation. The mutation is the process of sudden alterations, insertions, deletions or rearrangements of amino acids which result in the creation of diverse characteristics for the next generations. This evolution sometimes brings fairly information to nature but also sometimes causes adverse effects. Alignment is the best way to find how similar the peptide sequences are. A widely used tool named BLAST (Basic Local Alignment Search Tool) can be used for finding the alignment of the query sequence against a database. PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) [56] utilizes the concept of BLAST to create the PSSM matrix iteratively based on the cutoff e-value (E) 10−3(0.001).
The following procedure is used to construct a feature vector from a dataset.
-
A peptide query sequence is indicated as P that can be shown as,
(4) PSSM is an L * 20 matrix, where L is the protein length and the 20 columns indicate the amino acids,(5) Here, 20 is points to the diverse 20 amino acids correspond to the alphabetic form, where the length of P denotes as L, and refers to the amino acid responsible residue inclination at the position site ‘i’ that transmute to the amino acid at position site ‘j’ during the evolution process.
-
From Equation (5), the newly created matrix can be derived as—
(6) By means of,(7) Where,(8) Herein, É denotes as the mean and the following equation refers and elucidates by standard deviation,(9) -
The renewed matrix MT M to build 20 * 20 matrix (20 * L * L * 20 = 20 * 20 matrix) is computed by multiplying the main M matrix with its transpose matrix MT resulting in a 20 * 20 matrix. The number of elements in diagonal is 20. So, each triangular matrix consists of (400−20)/2 which is equal to 190 elements. In this study, we only considered the lower triangular matrix along with the diagonal matrix that means, (190 + 20) = 210 as shown below—
(10) The above matrix is then transformed into a vector of 210 elements annotated as Pevo,(11)
D. Addressing Imbalanced dataset issue
After cross-checking the sites from the original sequence, the ratio between the malonylation sites (positive) and the non-malonylation (negative) sites remains largely imbalanced. By comparison, the number of non-malonylation sites is much larger than that of malonylation sites. Due to such proportions, the predictor can be precariously biased towards negative samples. It has been comprehensively studied in machine learning literature that bias-free classification can be difficult to succeed due to the data imbalance in the training data. To address this complexity, a number of balancing strategies have been proposed with regard to the data balance issue [42], [58], [63]. In this case, we can downsample the data, but this can dramatically reduce the number of available samples. Instead of excluding, we use upsample at an early stage as it was done in [60], [64], [65], so that no information for the predictor would be discarded. To handle the class imbalance problem, some studies tried to adjust learning parameters of their model. For example, [42], [60], [62] adjusted the learning parameter for the Support Vector Machine (SVM) classifier to deal with imbalance data. In some other studies, K-Nearest Neighbors (KNN) strategy, and Neighborhood Cleaning Rule (NCR) were adopted to balance the data [59], [61], [63]. In order to calculate their Euclidean distance, they have tuned the value of k with several thresholds in simultaneous iterations.
In this study, to balance our dataset using oversampling with synthetic data construction, the synthetic data must be very similar to the original data. For ensuring the small variation, we took the maximum value of all the feature vectors and found that even if the maximum value is multiplied with the constants 1.0001 or 1.0005, the new value is very much closer to the original value. As multiplying the maximum value results in small variation, multiplying with the other values of feature vectors must generate very small variations of data [61], [62], [63], [66], [67], [68]. This is how we generate our new dataset with a small variation. Therefore, we multiply 1.0001 with 9,584 positive sites (19,168 sites) of all species, 1.0003, and 1.0005 with 5,013 positive sites (15, 039 sites) of Homo sapiens. Besides, we multiply 1.0003 with the 4,390 positive sites (8,780 sites) of the Mus musculus, where the number of negative sites of all species is 14,972, the Homo sapiens has 12,869 negative sites, and the Mus musculus has 10,152 negative sites. Then we use the Cluster Centroid based Majority Under-sampling Technique (CCMUT) [69] to balance the positive and negative sites in the total training data. After applying, the ratio of our positive and negative number of sites in the training data is 1 : 1 (malonylation sites : non-malonylation sites). Note that positive and negative sites for total species and individual species in test data were untouched. In this way, we make sure that our balancing will not impact the generality of our results and we avoid overfitting.
E. Classification Algorithm
To identify the most effective predictor, we have investigated 12 different classifiers that performed outstandingly in numerous biological quandaries [39], [43], [57], [60], [70] – [80]. These classifiers are: Extreme Gradient Boosting (XGBoost) [39], Adaptive Boosting (AdaBoost) [43], Support Vector Machine (SVM) [57], [60], Random Forest (RF) [70], [71], Light Gradient Boosting Machine (LightGBM) [72], [73], Linear Discriminant Analysis (LDA) [74], Quadratic Discriminant Analysis (QDA) [75], Bootstrap Aggregating (Bagging) [76], Decision Tree (DT) [77], Extra-Trees (ET) [78], Gradient Boosting (GB) [79], and Multi-layer Perceptron (MLP) [80], [81]. Finally, we consider the LightGBM [73] as our classifier as it obtained the best results regarding all aspects compared to other classifiers. The comparison of the results with other classifiers is provided in: https://github.com/Wakiloo7/Mal-Light. The Light Gradient Boosting Machine (LightGBM) [72], [73] uses a tree-based learning algorithm which is known as gradient boosting frameworks. Because of its high-speed computation, Light’ titles have been added before GBM. This algorithm uses the minor size of the memory and can handle the large data. It is recommended not to apply LightGBM [73] over small data as it is extremely sensitive because of overfitting. Implementing the LightGBM is straight forward. To implement this powerful algorithm we tune some parameters, such as num leaves, n estimators, and learning rate. In here, num leaves is a base learner maximum tree leaves, n estimators represents the number of base trees, and the third parameter learning rate, is basically the learning rate of boosting. In this study, the optimized values for these parameters are 31, 40, and 0.1 respectively. Alongside this, to fit the method for shrinking or adapting the learning while training, reset parameter callback is used.
F. Performance evaluation metrics
In this study, for the purpose of the computational analysis of our results, we use Accuracy (ACC), sensitivity (SN), specificity (SP), Matthew’s correlation coefficient (MCC), and F1-score(F1). All of the metrics were widely used in the literature [82], [83].
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
In the above equations, the TP indicates the True Positive which notifies how many peptide segments are thoroughly classified as malonylated (positive) sites. TN indicates the True Negative that means how many numbers of non-malonylated (negative) sites are thoroughly classified. Besides, the FP denotes the False Positive which represents the frequencies of non-malonylated (negative) peptide segments that are classified incorrectly as malonylated (positive), and the FN denotes the False Negative, the number of malonylated (positive) sites that were predicted wrongly as non-malonylated (negative). Alongside this, the MCC value is basically regarded as the representative of the total system for the performance. The F1-score is the weighted average or the combination of Precision as PR (also called positive predictive value) and Recall as RE (also known as sensitivity). The FP, and the FN both are taken to calculate this score. However, if anyone has gone through a data imbalance issue, F1-score is coming up with more beneficial information rather than accuracy. The outstanding predictor should be able to perform well in all above mentioned statistical measuring metrics.
III. Results and Discussion
Each proposed predictor aimed at predicting the malonylated sites must have its effectiveness measure to present how well it performs. For the purpose of this study, we examine five statistical performance matrices of Mal-Light namely, accuracy, sensitivity, specificity, F1-score, and Matthew’s correlation coefficient [21] – [23], [49], [82], which has been extensively used in the literature. Mal-Light comprehensive performance for predicting malonylated residues is presented for the above-mentioned five metrics.
A. Analysis of the Results for different species
Here, we report malonylation sites prediction performance for all six species specified in Table 1, and we have collected the dataset from PLMD [45]. As it was explained in the previous section, we applied 12 types of machine learning algorithms on the total and separate species that are trained using 10-fold cross-validation. Among all these algorithms, XGBoost [39], SVM [57], [60], LightGBM [73], GB [79], and MLP [80] obtained the best results. Among these classifiers, LightGBM [73] obtained the best results both for Homo sapiens and Mus musculus species. Whereas all species have been trained by Mal-Light and Homo sapiens well-trained than other species. Our results demonstrate that Mal-Light has the best performance for Homo sapiens, all species (six species), and Mus musculus, respectively in Table 2. To investigate the generality of our model and compare our results with those reported in previous studies, we run Mal-Light on the independent test set as well. Accordingly, we train Mal-Light using training data and use it for the independent test dataset. As shown in Fig. 3, using LightGBM in average obtained better results than other classifiers. Such result is repeated in Fig. 4 for the independent test set which confirms those that are reported in Fig. 3. The consistent results achieved both for 10-fold cross-validation and independent test set demonstrate the generality of using LightGBM as the classifier to build Mal-Light.
TABLE II.
The performance comparison to predict the malonylation sites altogether with six species and separately two species (homo sapiens, mus musculus) trained and tested with 10-fold cross-validation.
| Species | Sensitivity | Specificity | F1-score | ACC | MCC |
|---|---|---|---|---|---|
| Homo sapiens | 71.18% | 98.27% | 0.81 | 83.33% | 0.68 |
| Mus musculus | 66.58% | 92.13% | 0.76 | 79.35% | 0.60 |
| Altogether (six species) | 73.46% | 96.53% | 0.83 | 85.00% | 0.71 |
Fig. 3.
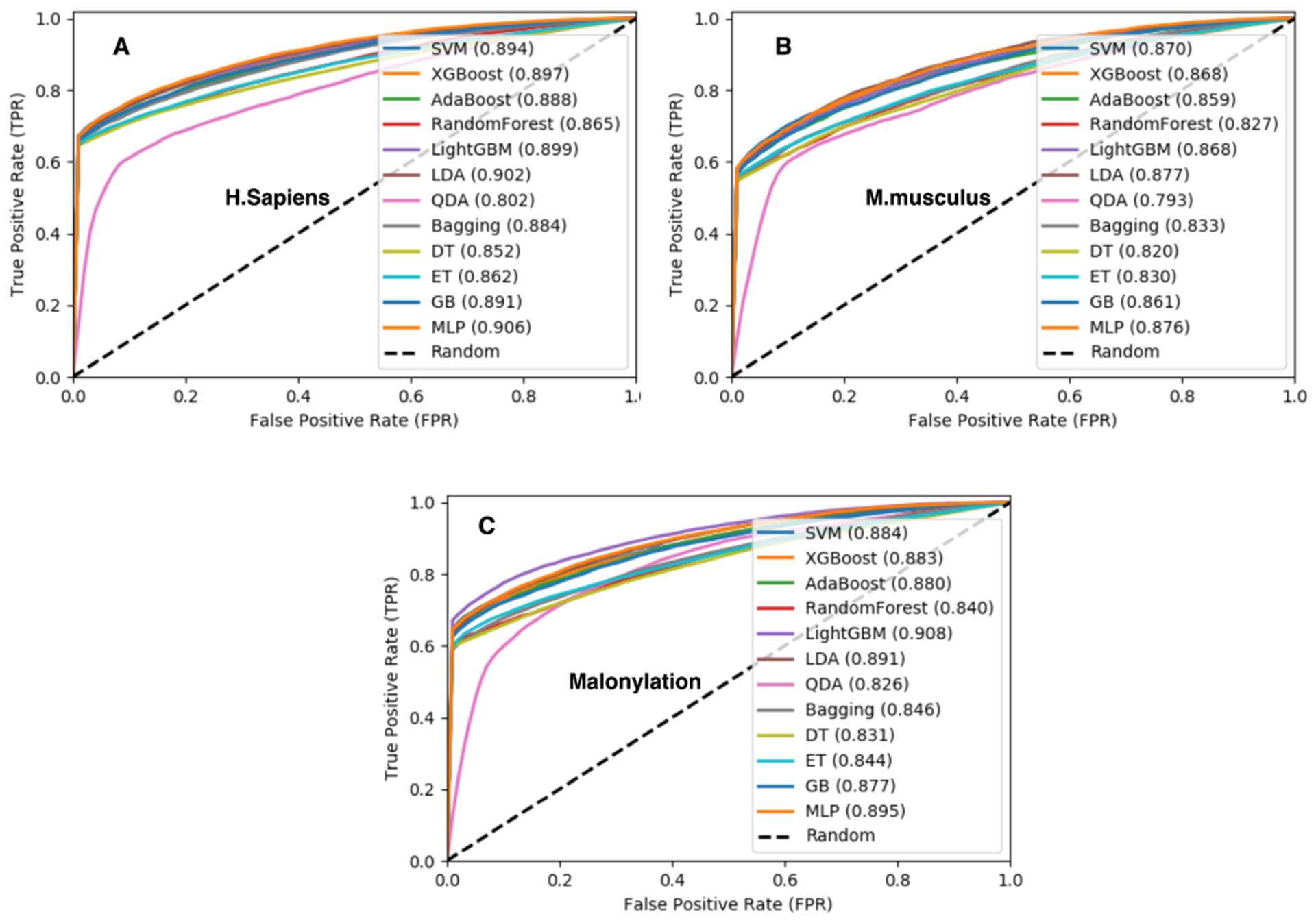
Receiver operator characteristic (ROC) curves Figure (A), (B) and (C) based on the classifiers with 10-fold cross-validation for training the malonylation sites of Homo sapiens, Mus musculus, Altogether (six species), and respectively by using some machine learning algorithms to develop our model for comparing the performances.
Fig. 4.
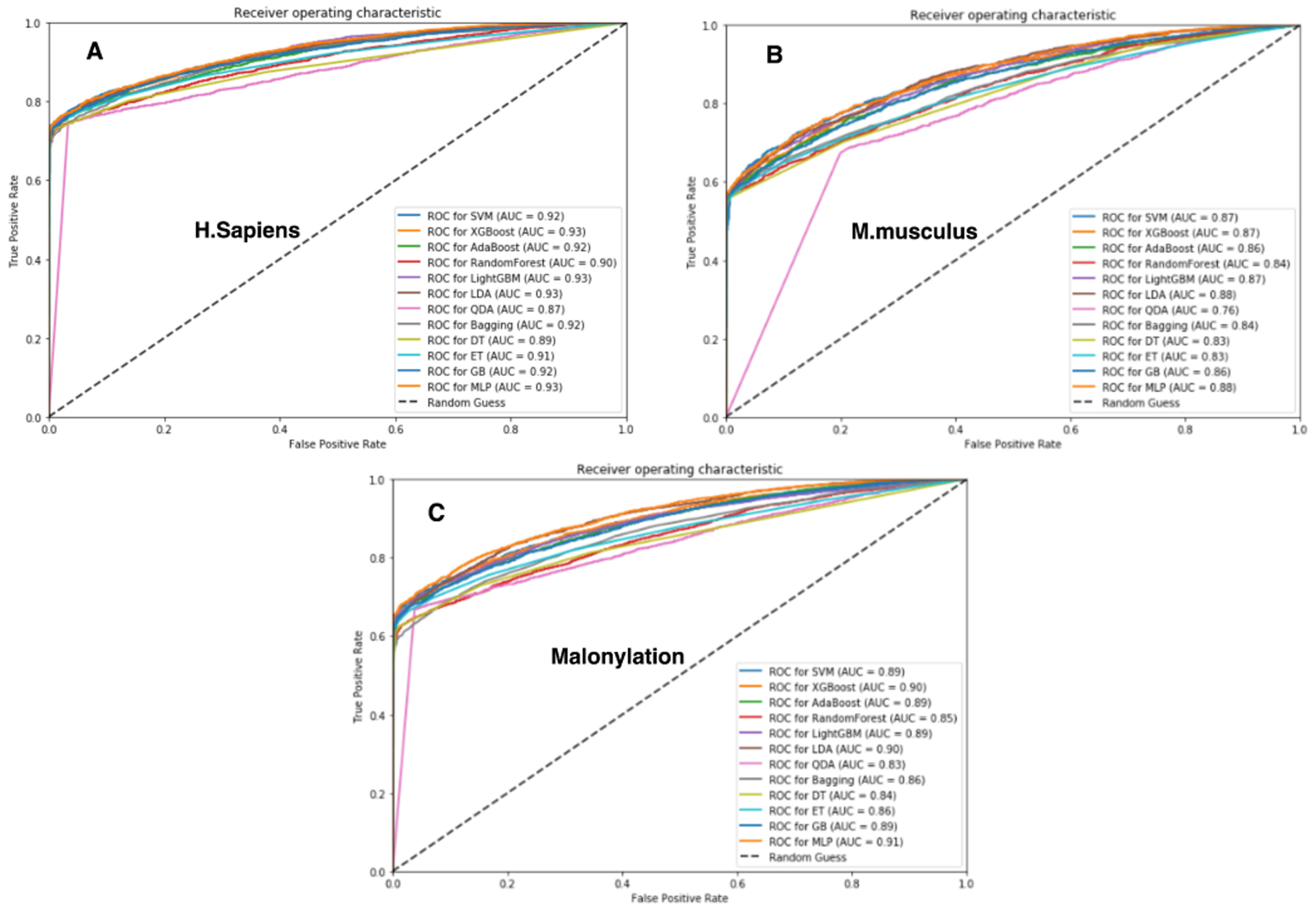
Receiver operator characteristic (ROC) curves Figure (A), (B) and (C) based on the classifiers with the independent test for the malonylation sites of H. sapiens, M. musculus, Altogether (six species), and respectively by using some machine learning algorithms.
B. Performance Comparison with Other Existing Methods
Malonylation has been discovered only a few ages ago. Due to its novelty, to the best of our knowledge, there are only four main tools to predict malonylation sites. These include Mal-Lys [35], which is solely trained on Mus musculus data, SPRINT-Mal [41], only to predict the malonylation sites for Homo sapiens and Mus musculus, MaloPred [37] which designed to predict the malonylation sites for three species (Homo sapiens, Mus musculus, and Escherichia coli), and kmal-sp [39], also designed to predict the malonylation sites for the same three species (Homo sapiens, Mus musculus, and Escherichia coli). Considering our targeted species, we compared Mal-Light with two of those predictors namely, MaloPred [37], kmal-sp [39] which attained the best performance and have online predictors. For the purpose of comparison, we manually transmitted all the peptide sequences to the web servers and retrieved their predictor performance for the measuring assessment. It is worth noting that, MaloPred [37], kmal-sp [39] web servers were pre-trained with some of the corresponding peptides sequences that are utilized in this study for the performance assessment as independent test set. In fact, they used all the data and trained their model and then used 10-fold cross-validation or jackknife cross-validation to evaluate their model. Therefore, their results on the independent test set which is filtered out from the whole data may have been overestimated. In other words, the results reported for those studies on the independent test set are in fact higher than expected. Despite this, our method was able to outperform even those overestimated results.
As a result, we run some of those specific classifiers used in their style over some of those species, as well as run other types of classifier algorithms on our own training data and compare them based on the method and test dataset. Our achieved results compared to MaloPred [37] and kmal-sp [39] for Homo sapiens and Mus musculus are shown in Table 3. Results presented in Table 3, demonstrate that Mal-Light achieves better performance compared to MaloPred [37] and kmal-sp [39]. For example, SP, F1-score, ACC, and MCC prominently enhanced by 12.65%, 0.03, 3.96%, and 0.09 compared to MaloPred for the human samples, respectively. Also, SP, ACC, and MCC are 8.05%, 0.66%, and 0.02 better compared to kmal-sp for the human samples, respectively. In addition, SP increased by 12.05% and 8.05%, respectively for mouse sample compared to MaloPred [37] and kmal-sp [39]. Besides, conducting the T-test demonstrates the statistical significance of the improvement reported in this study compared to those reported in the previous studies (p-value = 0.047). It is also important to note that Mal-Light achieves ACC of 82.36% when predicting malonylation sites for all the data together (consisting of samples belonging to 6 species). These results demonstrate the effectiveness of Mal-Light compared to those previous studies proposed to predict malonylation sites in the literature. We also plot the ROC curve for the 10-fold cross-validation and independent test which is shown in Fig. 3, and Fig. 4, respectively. These figures compare the comprehensive performance between the species. In addition, here we visualize the comparison of Mal-Light with MaloPred and kmal-sp in Fig. 5 that demonstrate the results for each species as well the error bars in bar plots have also been included for better visualization in Fig. 6.
TABLE III.
Performance comparison between our proposed method and malopred [37], kmal-sp [39] for predicting the malonylation sites of the individual species (homo sapiens, mus musculus) and total species (six species) based on the independent test.
| Method | Species | Sensitivity | Specificity | F1-score | ACC | MCC |
|---|---|---|---|---|---|---|
| Mal-Light | Homo sapiens | 78.26% | 95.05% | 0.85 | 86.66% | 0.74 |
| MaloPred [37] | 82.90% | 82.40% | 0.82 | 82.70% | 0.65 | |
| kmal-sp [39] | 84.90% | 87.00% | 0.85 | 86.00% | 0.72 | |
| Mal-Light | Mus musculus | 67.27% | 91.75% | 0.76 | 79.51% | 0.60 |
| MaloPred [37] | 80.60% | 79.70% | 0.80 | 80.20% | 0.60 | |
| kmal-sp [39] | 82.90% | 83.70% | 0.83 | 83.30% | 0.66 | |
| Mal-Light | Altogether (six species) | 68.38% | 96.34% | 0.79 | 82.36% | 0.67 |
| MaloPred [37] | – | – | – | – | – | |
| kmal-sp [39] | – | – | – | – | – |
Note: Due to the insufficient number of Escherichia coli species in our dataset, we did not take this species to in comparision.
Fig. 5.
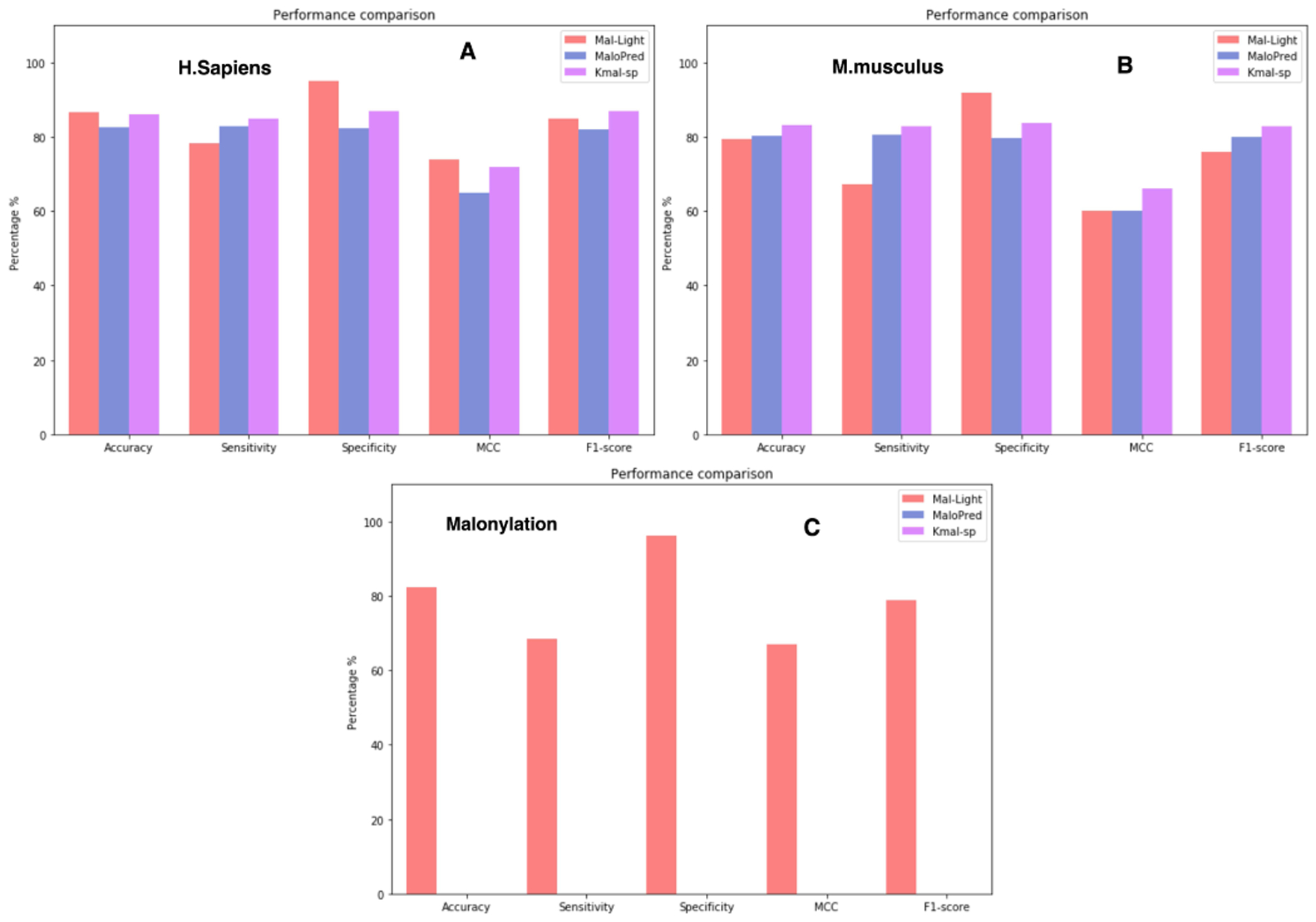
Performance Comparison among MaloPred [37], kmal-sp [39] and our proposed model, Mal-Light.
Fig. 6.
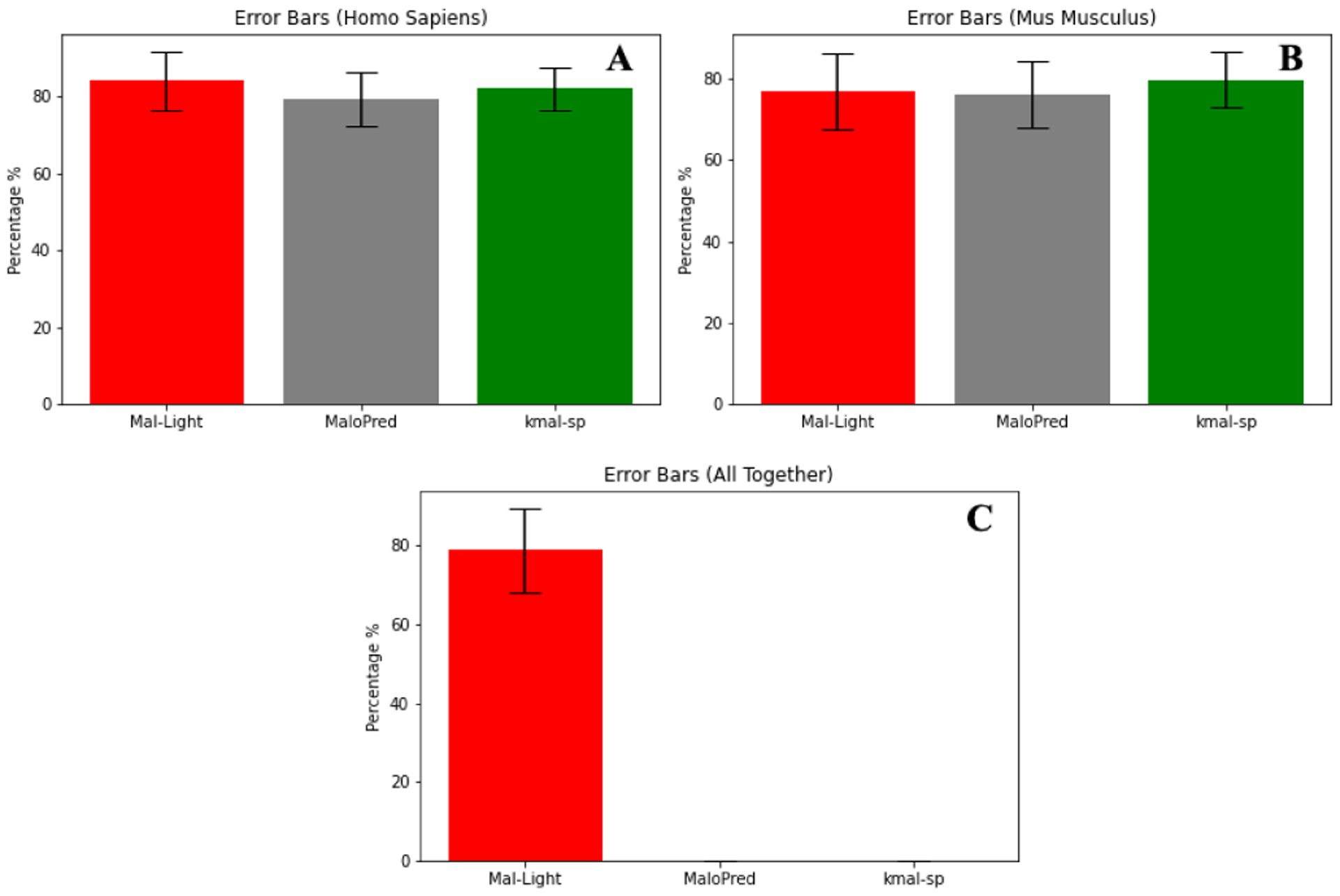
Error bars in a bar plot among our proposed model, Mal-Light and MaloPred [37], kmal-sp [39] in order to H. sapiens, M. musculus, Altogether (six species), respectively.
C. Identifying the most effective features to build Mal-Light
Here we also conduct a comprehensive study to investigate the impact of our extracted features for malonylation sites prediction tasks. To do this, a common approach is to eliminate the combination of features once at a time to show their relative importance in Fig. 7, which shows the impact of the 15 most important features for different species to build Mal-Light. The precision-recall curves for our experiments across all the species that are illustrated in Fig. 8. Besides, we plot the ROC curve for the cross-validation and independent test set shown in Fig. 3, and Fig. 4, which compares the comprehensive performance between the species. Furthermore, we reported a comparison which is shown in Fig. 5 with the corresponding species performance growth in the underneath of the predictor to compare with the same species in MaloPred [37], and kmal-sp [39] and the error bars in bar plots for the important result in Fig. 6.
Fig. 7.
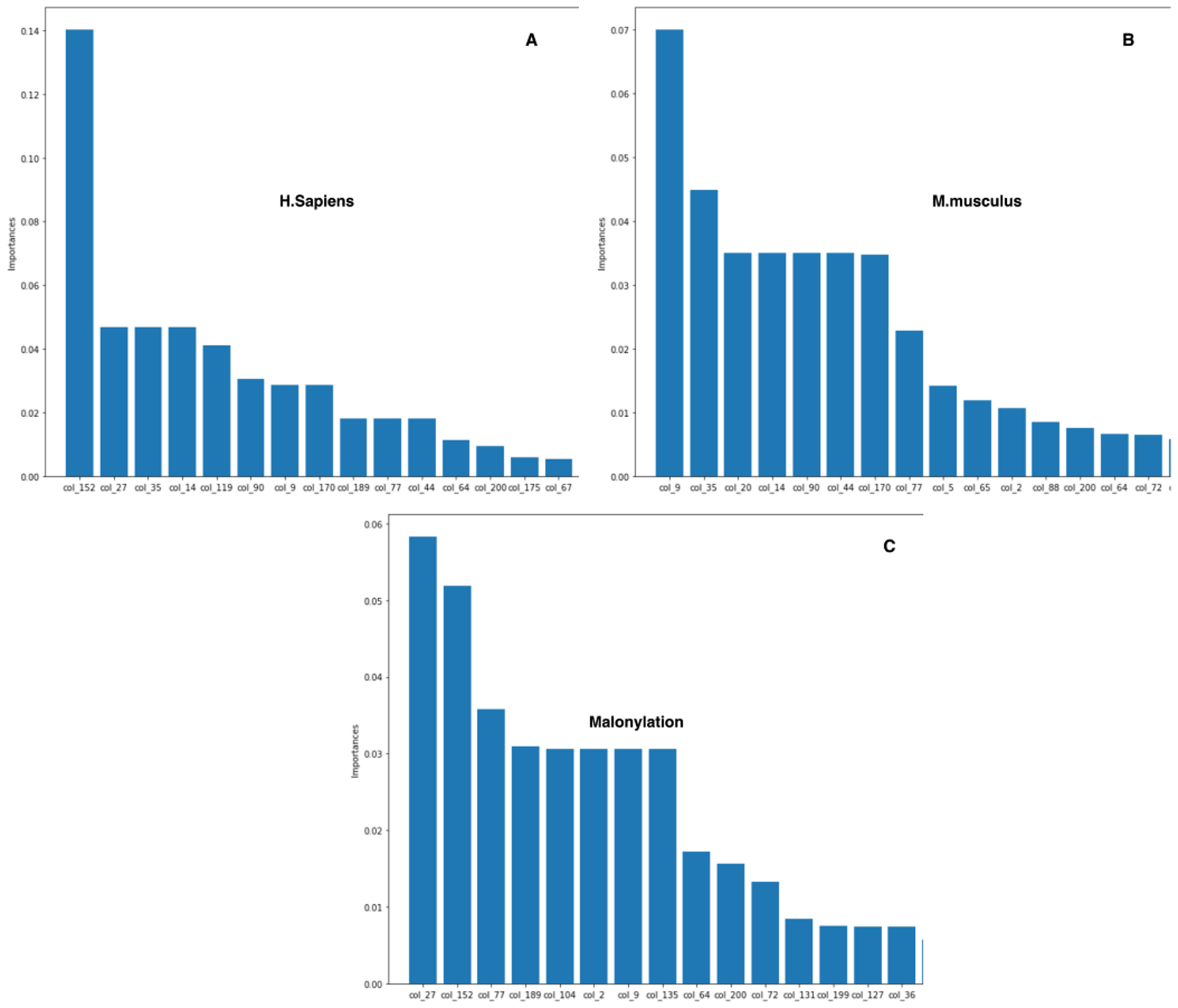
The impacts of the 15 most important features out of the 210 feature vectors for different species, instead of skipping any features in the development of Mal-Light.
Fig. 8.
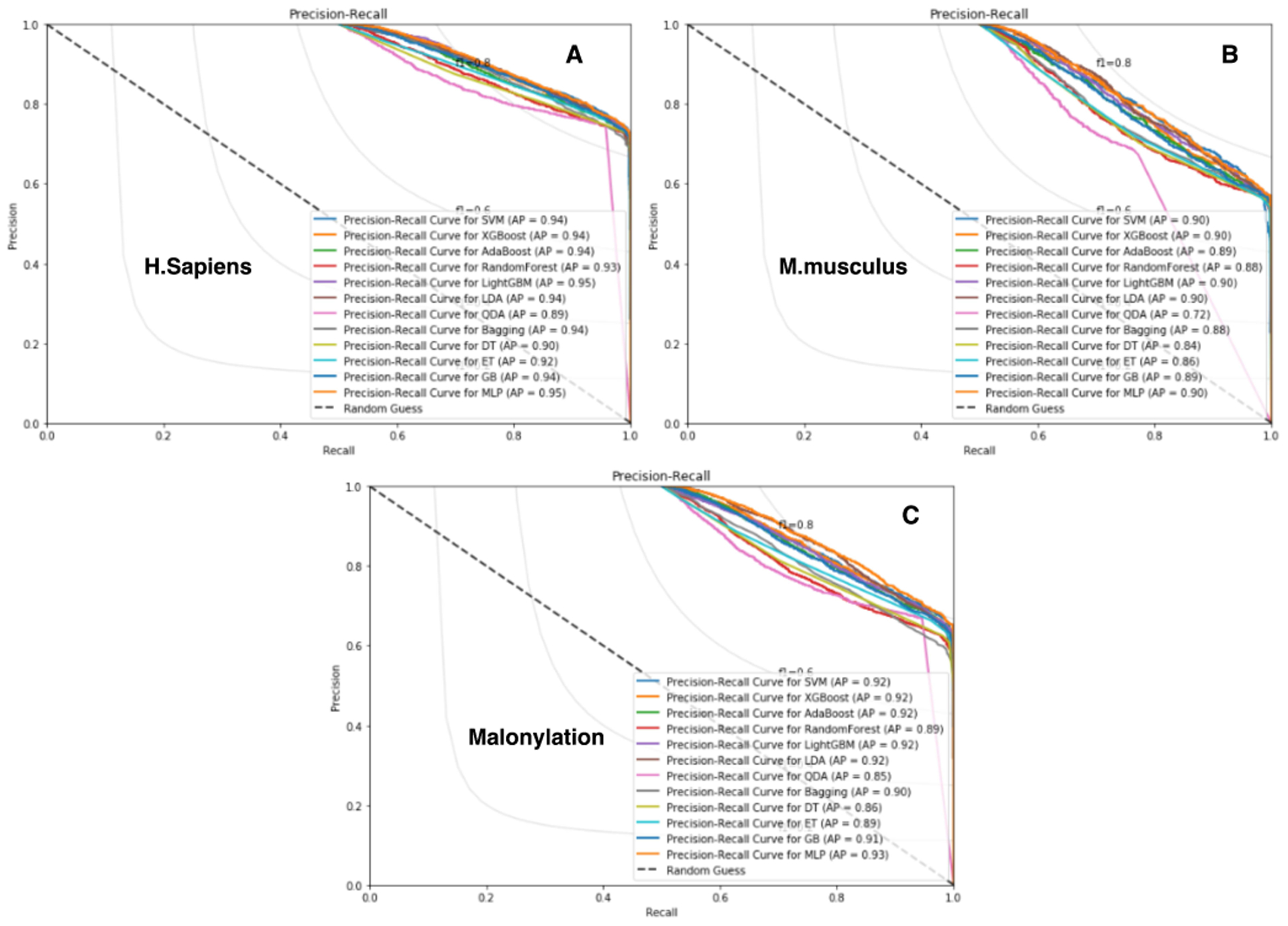
Precision-Recall curves Figure (A), (B) and (C) based on different classifier algorithms for the malonylation sites of Homo sapiens, Mus musculus, Altogether (six species), and respectively.
IV. Conclusion
In this study, we proposed a new predictor named Mal-Light which uses PSSM concepts differently to predict Malonylation sites. Mal-Light incorporates the concept of bi-peptide to extract local features from PSSM. To build our model, we took an oversampling approach with synthetic data construction which was given as the input for the LightGBM classifier for predicting the malonylation site. Our results demonstrate that Mal-Light is able to achieve prominent performance among different species. This also demonstrates that Mal-Light is about to outperform previous studies found in the literature to predict malonylation sites using different evaluation measurements. Our aim is to investigate different window sizes along with different kinds of new evolutionary and structural-based features in our future studies to further enhance the malonylation as one of the most important PTMs. Mal-Light is publicly available as an online malonylation site predictor at: (http://brl.uiu.ac.bd/MalLight/). Also, all our supplementary materials, figures, and their detailed descriptions are available at: (https://github.com/Wakiloo7/Mal-Light).
Acknowledgment
Research reported in this publication was supported by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number U54MD013376. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Research reported in this publication was supported by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number U54MD013376.
Biographies

Md. Wakil Ahmad completed his B.Sc. degrees from United International University, Bangladesh, in 2019. The name of his undergraduate capstone project was machine learning-based, “Automatic Code Review Effectiveness Detection”, under the guidance of Dewan Md. Farid, and the supervision of Chowdhury Rafeed Rahman. At the very end of his capstone project, he published a paper in ICECTE-2019, IEEE Xplore. His main research interests in computational complexity, bioinformatics, computer vision, and machine learning.

Md. Easin Arafat received his B.Sc. degrees from United International University, Bangladesh, in 2019. The title of his undergraduate capstone project was machine learning-based, “Automatic Code Review Effectiveness Detection”, under the guidance and supervision of Dewan Md. Farid, and Chowdhury Rafeed Rahman, respectively. During his undergraduate capstone project, he published a paper in ICECTE-2019, IEEE Xplore. His main research interests are in the field of bioinformatics, machine learning, computational intelligence, and deep learning.

Ghazaleh Taherzadeh received her Bachelor degree from Multimedia University (MMU), Cyberjaya, Malaysia, in 2011, her Master degree in the area of Artificial intelligence from the University of Malaya in 2013, Kuala Lumpur in 2013, and her Ph.D. degree in Bioinformatics from Griffith University, Gold coast, Australia, in 2018. Her research interests include Machine Learning (ML) and Pattern Recognition, Bioinformatics, and other Artificial Intelligence. She has published over 20 scientific articles in these areas. She contributed in reviewing and editing several articles from journals and conferences.

Alok Sharma received the B.Tech. degree from the University of the South Pacific (USP), Suva, Fiji, in 2000 and the M.Eng. degree, and the Ph.D. degree in the area of pattern recognition from Griffith University, Brisbane, Australia, in 2001 and 2006, respectively. He was with the University of Tokyo, Japan (2010–2012), as a Research Fellow (under JSPS). He is a Senior Research Scientist at RIKEN Center for Integrative Medical Sciences, Japan. He also is an Adjunct Professor at the Institute for Integrated and Intelligent Systems (IIIS), Griffith University, Australia, and, a Professor at the USP. He is also a visiting lecturer at Tokyo Medical and Dental University (TMDU), Japan. He participated in various projects carried out in conjunction with Motorola (Sydney), Auslog Pty., Ltd. (Brisbane), CRC Micro Technology (Brisbane), the French Embassy (Suva), and JSPS (Japan). His research interests include artificial intelligence, computer security, human cancer classification, and proteomics. He has published over 120 scientific articles in these areas. He contributed in reviewing and editing several articles from journals and conferences. He is the recipient of Griffith award of academic excellence, Australia, 2002, and the Vice-Chancellor’s Prize for best research output, Fiji, 2013.

Shubhashis Roy Dipta received his B.Sc. degree from Military Institute of Science & Technology, Bangladesh, in 2016. During his undergraduate, he published two papers in the Robotics field (one in IEEE and another one in IEEE Xplore). After that, he is working in the Machine Learning and Bioinformatics field during his Masters at United International University, Bangladesh. His main research interests are machine learning, computational intelligence, and deep learning.

Abdollah (Iman) Dehzangi received his Ph.D. in Bioinformatics and Computational biology from the Griffith University, Brisbane, Australia in 2015. He received his M.S. degree in computer science from the MultiMedia University (MMU), Cyberjaya, Malaysia in 2011 and Bachelor degree in computer engineering from the Shiraz University, Shiraz, Iran in 2007. During his Ph.D., he also served as research scholar at National ICT Australia (NICTA) from 2011 to 2014. After obtaining his Ph.D., he served as research scholar at Griffith University (2014–2015) and later as a postdoctoral research scholar at the University of Iowa (2015–2017). He is currently serving as an assistant professor at the Computer Science Department at Morgan State University (MSU). He is also serving as the M.S. in bioinformatics program director at this institute. His research interests include machine learning, artificial intelligence, and bioinformatics & computational biology in general. He has published over 70 scientific articles in these areas. He also served as reviewer and editor in several journals in these fields.

Swakkar Shatabda has completed his Doctor of Philosophy from Institute for Integrated and Intelligent Systems (IIIS) of Griffith University in October 2014 under the supervision of Professor Abdul Sattar And Dr. Muhammad Abdul Hakim Newton. The title of his Ph.D. thesis was “Local Search Heuristics for Protein Structure Prediction”. During his Ph.D., he published papers in reputed journals like BMC Bioinformatics and foremost conferences like AAAI and ACM-BCB. Prior to his Ph.D., Swakkhar completed his Undergraduate from the Dept of CSE or Bangladesh University of Engineering and Technology (BUET). Swakkhar has been working as an active faculty member at the Dept of CSE of United International University since 2008 and currently serving as Associate Professor and Undergraduate Program Coordinator. His research interests are in the field of AI Search, Optimization, Machine Learning, and Computational Biology. His works are published in reputed journals like the Journal of Theoretical Biology, Scientific Reports, etc.
References
- [1].Westermann S, Weber K, “Post-translational modifications regulate microtubule function,” Nat. Rev. Mol. Cell Biol, vol. 4, no. 12, pp. 938–948, December 2003. DOI: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- [2].Johnson LN, “The regulation of protein phosphorylation,” Biochemical Society Transactions, vol. 37, no. 4, pp. 627–641, July 2009. DOI: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- [3].Gallego M, & Virshup DM, “Post-translational modifications regulate the ticking of the circadian clock,” Nat. Rev. Mol. Cell Biol 8, 139–148, February 2007. DOI: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- [4].Harmel R, Fiedler D, “Features and regulation of non-enzymatic post-translational modifications,” Nat. Chem. Biol, vol.14, pp. 244–252, February 2018. DOI: 10.1038/nchembio.2575. [DOI] [PubMed] [Google Scholar]
- [5].Kouzarides T, “Chromatin modifications and their function,” Cell, vol. 128, no. 4, pp. 693–705, February 2007. DOI: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- [6].Dai C and Gu W, “p53 post-translational modification: Deregulated in tumorigenesis,” Trends Mol. Med, vol. 16, no. 11, pp. 528–536, October 2010. DOI: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Consortium U, “Uniprot: a worldwide hub of protein knowledge,” Nucleic acids research 47, vol. 47, no. D1, pp. D506–D515, January 2019. DOI: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, and Zhao Y, “Lysine succinylation and lysine malonylation in histones,” Molecular & Cellular Proteomics, vol. 11, no. 5, pp. 100–107, March 2012. DOI: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ansari NA, Moinuddin AR, “Glycated Lysine Residues: A Marker for Non-Enzymatic Protein Glycation in Age-Related Diseases,” Disease Markers, vol. 30, no. 6, pp. 317–324, June 2011. DOI: 10.3233/DMA-2011-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang Z, Tan M, Xie Z, Dai L, Chen Y, and Zhao Y, “Identification of lysine succinylation as a new post-translational modification,” Nature Chemical Biology, vol. 7, no. 1, pp. 58–63, December 2011. DOI: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jia J, Liu Z, Xiao X, Liu B, Chou KC, “pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach,” Journal of theoretical biology, vol. 394, pp. 223–230, April 2016. DOI: 10.1016/j.jtbi.2016.01.020. [DOI] [PubMed] [Google Scholar]
- [12].Comb DG, Sarkar N , Pinzino CJ, “The Methylation of Lysine Residues in Protein,” The Journal of Biological Chemistry, vol. 241, no. 8, pp. 1857–62. September 1966. [PubMed] [Google Scholar]
- [13].Martin C, Zhang Y, “The diverse functions of histone lysine methylation,” Nature Reviews Molecular Cell Biology, vol. 6, no. 11, pp. 838–849, November 2005. DOI: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- [14].Drazic A, Myklebust LM, Ree R, Arnesen T, “The world of protein acetylation,” Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics, vol. 1864, no. 10, pp. 1372–401, October 2016. DOI: 10.1016/j.bbapap.2016.06.007. [DOI] [PubMed] [Google Scholar]
- [15].Lamoliatte F, Caron D, Durette C, Mahrouche L, Maroui MA, Lizotte OC et al. , “Large-scale analysis of lysine SUMOylation by SUMO remnant immunoaffinity profiling,” Nature Communications, vol. 5, p. 5409, November 2014. DOI: 10.1038/ncomms6409. [DOI] [PubMed] [Google Scholar]
- [16].Oughtred R et al. , “Biogrid: a resource for studying biological interactions in yeast,” Cold Spring Harb. Protoc, vol. 2016, no. 1, p. pdb– top080754, January 2016. Doi: 10.1101/pdb.top080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Du Y, Cai T, Li T et al. , “Lysine malonylation is elevated in type 2 diabetic mouse models and enriched in metabolic associated proteins,” Mol Cell Proteomics, vol. 14, no. 1, pp. 227–236, January 2015. DOI: 10.1074/mcp.M114.041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Saggerson D, “Malonyl-coa, a key signaling molecule in mammalian cells,” Annu. Rev. Nutr, vol. 28, pp. 253–272, August 2008. DOI: 10.1146/annurev.nutr.28.061807.155434. [DOI] [PubMed] [Google Scholar]
- [19].Nishida Y et al. , “Sirt5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target,” Mol. cell, vol. 59, no. 2, pp. 321–332, July 2015. DOI: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chou KC, “An Unprecedented Revolution in Medicinal Chemistry Driven by the Progress of Biological Science,” Current Topics in Medicinal Chemistry, vol. 17, no. 21, pp. 2337–58, November 2021 DOI: 10.2174/1568026617666170414145508. [DOI] [PubMed] [Google Scholar]
- [21].Qiu WR, Xiao X, Lin WZ, Chou KC, “iMethyl-PseAAC: Identification of Protein Methylation Sites via a Pseudo Amino Acid Composition Approach,” BioMed Research International, vol. 2014, May 2014. DOI: 10.1155/2014/947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xu Y, Wen X, Wen LS, Wu LY, Deng NY, Chou KC, “iNitro-Tyr: Prediction of Nitrotyrosine Sites in Proteins with General Pseudo Amino Acid Composition,” PLoS ONE, vol. 9, no. 8, August 2014. DOI: 10.1371/journal.pone.0105018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Qiu WR, Xiao X, Lin WZ, Chou KC, “iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model,” Journal of Biomolecular Structure and Dynamics, vol. 33, no. 8, pp. 1731–42, August 2015. DOI: 10.1080/07391102.2014.968875. [DOI] [PubMed] [Google Scholar]
- [24].Jia J, Liu Z, Xiao X, Liu B, Chou KC, “iCar-PseCp: identify carbonylation sites in proteins by Monte Carlo sampling and incorporating sequence coupled effects into general PseAAC,” Oncotarget, vol. 7, no. 23, pp. 34558–34570, June 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Qiu WR, Sun BQ, Xiao X, Xu ZC, Chou KC, “iPTM-mLys: identifying multiple lysine PTM sites and their different types,” Bioinformatics, vol. 32, no. 20, pp. 3116–3123, October 2016. DOI: 10.1093/bioinformatics/btw380. [DOI] [PubMed] [Google Scholar]
- [26].Qiu WR, Xiao X, Xu ZC, Chou KC, “iPhos-PseEn: Identifying phosphorylation sites in proteins by fusing different pseudo compo nents into an ensemble classifier,” Oncotarget, vol. 7, no. 32, pp. 51270–51283, August 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feng P, Ding H, Yang H, Chen W, Lin H, Chou KC, “iRNAPseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC,” Molecular Therapy–Nucleic Acids, vol. 7, pp. 155–163, June 2017. DOI: 10.1016/j.omtn.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu LM, Xu Y, Chou KC, “iPGK-PseAAC: Identify Lysine Phosphoglycerylation Sites in Proteins by Incorporating Four Different Tiers of Amino Acid Pairwise Coupling Information into the General PseAAC,” Medicinal Chemistry, vol. 13, no. 6, pp. 552–559, November 2017. DOI: 10.2174/1573406413666170515120507. [DOI] [PubMed] [Google Scholar]
- [29].Qiu WR, Sun BQ, Xiao X, Xu D, Chou KC, “iPhos-PseEvo: Identifying Human Phosphorylated Proteins by Incorporating Evolutionary Information into General PseAAC via Grey System Theory,” Molecular Informatics, vol. 36, no. 5–6, p. 1600010, May 2017. DOI: 10.1002/minf.201600010. [DOI] [PubMed] [Google Scholar]
- [30].Chou KC, “Some remarks on protein attribute prediction and pseudo amino acid composition,” Journal of Theoretical Biology, vol. 273, no. 1, pp. 236–247, March 2011. DOI: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen W, Feng P, Yang H, Ding H, Lin H, Chou KC, “iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences,” Oncotarget, vol. 8, no. 3, pp. 4208–4217, January 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu B, Wang S, Long R, Chou KC, “iRSpot-EL: identify recombination spots with an ensemble learning approach,” Bioinformatics, vol. 33, no. 1, pp. 35–41, January 2017. DOI: 10.1093/bioinformatics/btw539. [DOI] [PubMed] [Google Scholar]
- [33].Liu B, Yang F, Chou KC, “2L-piRNA: A Two-Layer Ensemble Classifier for Identifying Piwi-Interacting RNAs and Their Function,” Molecular Therapy–Nucleic Acids, vol. 7, pp. 267–77, June 2017. DOI: 10.1016/j.omtn.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Du Y, Zhai Z, Li Y, Lu M, Cai T, Zhou B et al. , “Prediction of protein lysine acylation by integrating primary sequence information with multiple functional features,” Journal of proteome research, vol. 15, no. 12, pp. 4234–4244, October 2016. DOI: 10.1021/acs.jproteome.6b00240. [DOI] [PubMed] [Google Scholar]
- [35].Xu Y, Ding Y, Ding J et al. , “Mal-Lys: prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection,” Nat. Publ. Gr, vol. 6, p. 38318, December 2016. DOI: 10.1038/srep38318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Peng H, Long F, Ding C, “Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy,” IEEE Trans Pattern Anal Mach Intell, no. 8, 1226–1238, August 2005. DOI: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- [37].Wang LN, Shi SP, Xu HD, P Wen P, Qiu JD, “Computational prediction of species-specific malonylation sites via enhanced characteristic strategy,” Bioinformatics, vol. 33, no. 10, pp. 1457–1463, December 2016. DOI: 10.1093/bioinformatics/btw755. [DOI] [PubMed] [Google Scholar]
- [38].Xiang Q, Feng K, Liao B et al. , “Prediction of lysine malonylation sites based on pseudo amino acid compositions,” Comb Chem. High Throughput Screen, vol. 20, no. 7, pp. 622–628, November 2017. DOI: 10.2174/1386207320666170314102647. [DOI] [PubMed] [Google Scholar]
- [39].Zhang Y, Xie R, Wang J, Leier A, Marquez-Lago TT, Akutsu T et al. , “Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework,” Briefings in bioinformatics, vol. 5, August 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Taherzadeh G, Yang Y, Zhang T, Liew AWC, Zhou Y. “Sequence-based prediction of protein–peptide binding sites using support vector machine,” Journal of computational chemistry, vol. 37, no. 13, pp. 1223–1229, February 2016. DOI: 10.1002/jcc.24314. [DOI] [PubMed] [Google Scholar]
- [41].Taherzadeh G, Yang Y, Xu H, Xue Y, Liew AWC, Zhou Y, “Predicting lysine-malonylation sites of proteins using sequence and predicted structural features,” Journal of computational chemistry, vol. 39, no. 22, pp. 1757–1763, May 2018. DOI: 10.1002/jcc.25353. [DOI] [PubMed] [Google Scholar]
- [42].Ju Z, Wang SY, “Prediction of lysine formylation sites using the composition of k-spaced amino acid pairs via Chou’s 5-steps rule and general pseudo components,” Genomics, vol. 112, no. 1, pp. 859–866, January 2020. DOI: 10.1016/j.ygeno.2019.05.027. [DOI] [PubMed] [Google Scholar]
- [43].Dehzangi A, López Y, Lal SP, Taherzadeh G, Sattar A, Tsunoda T, & Sharma A, “Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams,” PloS one, vol. 13, no. 2, p. E0191900, February 2018. DOI: 10.1371/journal.pone.0191900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dehzangi A, Paliwal K, Lyons J, Sharma A, & Sattar A, “Enhancing protein fold prediction accuracy using evolutionary and structural features,” In IAPR International Conference on Pattern Recognition in Bioinformatics, pp. 196–207, June 2013. DOI: 10.1007/978-3-642-39159-0_18. [DOI] [Google Scholar]
- [45].Xu H et al. , “PLMD: An updated data resource of protein lysine modifications,” J Genet Genomics, vol. 44, no. 5, pp. 243–250, May 2017. DOI: 10.1016/j.jgg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- [46].Huang Y, Niu B, Gao Y, Fu L, & Li W, “CD-HIT Suite: a web server for clustering and comparing biological sequences,” Bioinformatics, vol. 26, no. 5, pp. 680–682, March 2010. DOI: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dehzangi A, Paliwal K, Sharma A, Dehzangi O, & Sattar A, “A combination of feature extraction methods with an ensemble of different classifiers for protein structural class prediction problem,” IEEE/ACM transactions on computational biology and bioinformatics, vol. 10, no. 3, pp. 564–575, May 2013. DOI: 10.1109/TCBB.2013.65. [DOI] [PubMed] [Google Scholar]
- [48].Dehzangi A, & Karamizadeh S, “Solving protein fold prediction problem using fusion of heterogeneous classifiers,” INFORMATION, vol. 14, no. 11, pp. 3611–3622, November 2011. [Google Scholar]
- [49].Dehzangi A, Heffernan R, Sharma A, Lyons J, Paliwal K, & Sattar A, “Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou’ s general PseAAC,” Journal of theoretical biology, vol. 364, pp. 284–294, January 2015. DOI: 10.1016/j.jtbi.2014.09.029. [DOI] [PubMed] [Google Scholar]
- [50].Chowdhury SY, Shatabda S, & Dehzangi A, “iDNAprot-es: Identification of DNA-binding proteins using evolutionary and structural features,” Scientific reports, vol. 7, no. 1, p. 14938, November 2017. DOI: 10.1038/s41598-017-14945-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dehzangi A, Paliwal K, Sharma A, Lyons J, & Sattar A, “Protein fold recognition using an overlapping segmentation approach and a mixture of feature extraction models,” Australasian Joint Conference on Artificial Intelligence. pp. 32–43, December 2013. DOI: 10.1007/978-3-319-03680-9_4. [DOI] [Google Scholar]
- [52].Dehzangi A, López Y, Lal SP, Taherzadeh G, Michaelson J, Sattar A, & Sharma A, “PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction,” Journal of theoretical biology, vol. 425, pp. 97–102, July 2017. DOI: 10.1016/j.jtbi.2017.05.005. [DOI] [PubMed] [Google Scholar]
- [53].Islam MM, Saha S, Rahman MM, Shatabda S, Farid DM, & Dehzangi A, “iProtGlySS: Identifying protein glycation sites using sequence and structure based features,” Proteins: Structure, Function, and Bioinformatics, vol. 86, no. 7, pp. 777–789, April 2018. DOI: 10.1002/prot.25511. [DOI] [PubMed] [Google Scholar]
- [54].Sharma A, Lyons J, Dehzangi A, & Paliwal KK, “A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition,” Journal of theoretical biology, vol. 320, pp. 41–46, March 2013. DOI: 10.1016/j.jtbi.2012.12.008. [DOI] [PubMed] [Google Scholar]
- [55].Shovan S and Hasan MAM, “Prediction of Lysine Glycation PTM site in Protein using Peptide Sequence Evolution based Features,” International Conference on Electrical, Computer and Communication Engineering (ECCE). IEEE, pp. 1–5, February 2019. DOI: 10.1109/ECACE.2019.8679407. [DOI] [Google Scholar]
- [56].Schaffer A, “Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements,” Nucleic Acids Research, vol. 29, no. 14, pp. 2994–3005, July 2001. DOI: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wu M, Lu P, Yang Y, Liu L, Wang H, Xu Y et al. , “LipoSVM: Prediction of lysine lipoylation in Proteins based on the Support Vector Machine,” Current Genomics, vol. 20, no. 5, pp. 362–370, August 2019. DOI: 10.2174/1389202919666191014092843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jia J, Liu Z, Xiao X, Liu B, Chou KC, “iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset,” Analytical Biochemistry, vol. 497, pp. 48–56, March 2016. DOI: 10.1016/j.ab.2015.12.009. [DOI] [PubMed] [Google Scholar]
- [59].Jia J, Liu Z, Xiao X, Liu B, Chou KC, “iPPBS-Opt: A Sequence-Based Ensemble Classifier for Identifying Protein-Protein Binding Sites by Optimizing Imbalanced Training Datasets,” Molecules, vol. 21, no. 1, p. 95, January 2016. DOI: 10.3390/molecules21010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hasan MAM, Ahmad S, “mLysPTMpred: Multiple Lysine PTM Site Prediction Using Combination of SVM with Resolving Data Imbalance Issue,” Natural Science, vol. 10, no. 09, p. 370, September 2018. DOI: 10.4236/ns.2018.109035. [DOI] [Google Scholar]
- [61].Chandra A, Sharma A, Dehzangi A, Ranganathan S, Jokhan A, Chou KC et al. , “PhoglyStruct: prediction of phosphoglycerylated lysine residues using structural properties of amino acids,” Scientific reports, vol. 8, no. 1, p. 17923, December 2018. DOI: 10.1038/s41598-018-36203-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hasan MAM, Ahmad S, “predSucc-Site: Lysine Succinylation Sites Prediction in Proteins by using Support Vector Machine and Resolving Data Imbalance Issue,” International Journal of Computer Applications, vol. 182, no. 15, p. 8887, September 2018. [Google Scholar]
- [63].López Y, Dehzangi A, Lal SP, Taherzadeh G, Michaelson J, Sattar A et al. , “SucStruct: prediction of succinylated lysine residues by using structural properties of amino acids,” Analytical biochemistry, vol. 527, pp. 24–32. June 2017. DOI: 10.1016/j.ab.2017.03.021. [DOI] [PubMed] [Google Scholar]
- [64].Ahmad MW, Shovan S, Arafat ME, Sifat MHR, Hasan MAM, & Shatabda S, “Improved performance of Lysine Glutarylation PTM using Peptide Evolutionary Features,” in 3rd International Conference on Electrical, Computer & Telecommunication Engineering (ICECTE). IEEE, in press, December 2019. [Google Scholar]
- [65].Bao W, Yang B, Huang DS, Wang D, Liu Q, Chen YH et al. , “IMKPse: Identification of Protein Malonylation Sites by the Key Features Into GeneralPseAAC,” IEEE Access, vol. 7, pp. 54073–54083, March 2019. DOI: 10.1109/ACCESS.2019.2900275. [DOI] [Google Scholar]
- [66].Jia C, Zuo Y, “S-SulfPred: A sensitive predictor to capture S-sulfenylation sites based on a resampling one-sided selection undersampling-synthetic minority oversampling technique,” Journal of theoretical biology, vol. 422, pp. 84–89, June 2017. DOI: 10.1016/j.jtbi.2017.03.031. [DOI] [PubMed] [Google Scholar]
- [67].Sun X, Li J, Gu L, Wang S, Zhang Y, Huang T et al. , “Identifying the Characteristics of the Hypusination Sites Using SMOTE and SVM Algorithmwith Feature Selection,” Current Proteomics, vol. 15, no. 2, pp. 111–118, November 2018. DOI: 10.2174/1570164614666171109120615. [DOI] [Google Scholar]
- [68].AL-barakati HJ, McConnell EW, Hicks LM, Poole LB, Newman RH et al. , “SVM-SulfoSite: A support vector machine based predictor for sulfenylation sites,” Scientific reports, vol. 8, no. 1, p. 11288, July 2018. DOI: 10.1038/s41598-018-29126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhang YP, Zhang LN, and Wang YC, “Cluster-based majority under-sampling approaches for class imbalance learning,” in 2nd IEEE International Conference on Information and Financial Engineering. IEEE, pp. 400–404, September 2010. DOI: 10.1109/ICIFE.2010.5609385. [DOI] [Google Scholar]
- [70].Bao X, Zhao Q, Yang T, Fung YME, Li XD, “A chemical probe for lysine malonylation,” Angewandte Chemie International Edition, vol. 52, no. 18, pp. 4883–4886, March 2013. DOI: 10.1002/anie.201300252. [DOI] [PubMed] [Google Scholar]
- [71].Chen Z, He N, Huang Y, Qin WT, Liu X, Li L, “Integration of a deep learning classifier with a random forest approach for predicting malonylation sites,” Genomics, proteomics & bioinformatics, vol. 16, no. 6, pp. 451–459, December 2018. DOI: 10.1016/j.gpb.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ke G, Meng Q, Finley T, Wang T, Chen W, Ma W et al. , “Lightgbm: A highly efficient gradient boosting decision tree,” Advances in Neural Information Processing Systems, pp. 3146–3154. [Google Scholar]
- [73].Chen C, Zhang Q, Ma Q, Yu B, “LightGBM-PPI: Predicting protein-protein interactions through LightGBM with multi-information fusion,” Chemometrics and Intelligent Laboratory Systems, vol. 191, pp. 54–64, August 2019. DOI: 10.1016/j.chemolab.2019.06.003. [DOI] [Google Scholar]
- [74].Mitteroecker P, Bookstein F, “Linear discrimination, ordination, and the visualization of selection gradients in modern morphometrics,” Evolutionary Biology, vol. 38, no. 1, pp. 100—114, February 2011. DOI: 10.1007/s11692-011-9109-8. [DOI] [Google Scholar]
- [75].Zhang X, Oh C, Riley CP, Buck C, “Current status of computational approaches for protein identification using tandem mass spectra,” Current Proteomics, vol. 4, no. 3, pp. 121–130, November 2007. DOI: 10.2174/157016407783221349. [DOI] [Google Scholar]
- [76].Banerjee S, Basu S, Ghosh D, Nasipuri M, “PhospredRF: Prediction of protein phosphorylation sites using a consensus of random forest classifiers,” International Conference and Workshop on Computing and Communication (IEMCON), IEEE, pp. 1–7, December 2015. DOI: 10.1109/IEMCON.2015.7344514. [DOI] [Google Scholar]
- [77].Shi SP, Sun XY, Qiu JD, Suo SB, Chen X, Huang SY et al. , “The prediction of palmitoylation site locations using a multiple feature extraction method,” Journal of Molecular Graphics and Modelling, vol. 40, pp. 125–130, March 2013. DOI: 10.1016/j.jmgm.2012.12.006. [DOI] [PubMed] [Google Scholar]
- [78].Ismail HD, Newman RH et al. , “RF-Hydroxysite: a random forest based predictor for hydroxylation sites,” Molecular BioSystems, vol. 12, no. 8, pp. 2427–2435, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ai H, Wu R, Zhang L, Wu X, Ma J, Hu H et al. , “pSuc-PseRat: Predicting lysine succinylation in proteins by exploiting the ratios of sequence coupling and properties,” Journal of Computational Biology, vol. 24, no. 10, pp. 1050–1059, October 2017. DOI: 10.1089/cmb.2016.0206. [DOI] [PubMed] [Google Scholar]
- [80].Zahiri J, Mohammad-Noori M, Ebrahimpour R, Saadat S, Bozorgmehr JH, Goldberg T et al. , “LocFuse: human protein–protein interaction prediction via classifier fusion using protein localization information,” Genomics, vol. 104, no. 6, pp. 496–503, December 2014. DOI: 10.1016/j.ygeno.2014.10.006. [DOI] [PubMed] [Google Scholar]
- [81].Sun J, Cao Y, Wang D, Bao W, Chen Y, “K net: Lysine Malonylation Sites Identification with Neural Network,” IEEE Access, vol. 8, pp. 47304–47311, December 2019. DOI: 10.1109/ACCESS.2019.2961941. [DOI] [Google Scholar]
- [82].Jiao Y, Du P, “Performance measures in evaluating machine learning based bioinformatics predictors for classifications,” Quantitative Biology, vol. 4, no. 4, pp. 320–330, December 2016. DOI: 10.1007/s40484-016-0081-2. [DOI] [Google Scholar]
- [83].Chen CY, Tang SL, Chou SCT, “Taxonomy based performance metrics for evaluating taxonomic assignment methods,” BMC bioinformatics, vol. 20, no. 1, p. 310, June 2019. DOI: 10.1186/s12859-019-2896-0. [DOI] [PMC free article] [PubMed] [Google Scholar]