

Supplemental Digital Content is available in the text.
Keywords: hemostasis, HNE, inflammation, neutrophils, oxidative stress, thrombosis, tissue factor
Objective:
TF (Tissue factor) plays a key role in hemostasis, but an aberrant expression of TF leads to thrombosis. The objective of the present study is to investigate the effect of 4-hydroxy-2-nonenal (HNE), the most stable and major oxidant produced in various disease conditions, on the release of TF+ microvesicles into the circulation, identify the source of TF+ microvesicles origin, and assess their effect on intravascular coagulation and inflammation.
Approach and Results:
C57BL/6J mice were administered with HNE intraperitoneally, and the release of TF+ microvesicles into circulation was evaluated using coagulation assays and nanoparticle tracking analysis. Various cell-specific markers were used to identify the cellular source of TF+ microvesicles. Vascular permeability was analyzed by the extravasation of Evans blue dye or fluorescein dextran. HNE administration to mice markedly increased the levels of TF+ microvesicles and thrombin generation in the circulation. HNE administration also increased the number of neutrophils in the lungs and elevated the levels of inflammatory cytokines in plasma. Administration of an anti-TF antibody blocked not only HNE-induced thrombin generation but also HNE-induced inflammation. Confocal microscopy and immunoblotting studies showed that HNE does not induce TF expression either in vascular endothelium or circulating monocytes. Microvesicles harvested from HNE-administered mice stained positively with CD248 and α-smooth muscle actin, the markers that are specific to perivascular cells. HNE was found to destabilize endothelial cell barrier integrity.
Conclusions:
HNE promotes the release of TF+ microvesicles from perivascular cells into the circulation. HNE-induced increased TF activity contributes to intravascular coagulation and inflammation.
Highlights.
4-hydroxy-2-nonenal, the most stable and major oxidant produced in various disease conditions, induces the release of TF+ (tissue factor) microvesicles into the circulation.
4-hydroxy-2-nonenal-induced TF+ microvesicles are originated from perivascular cells.
4-hydroxy-2-nonenal induces intravascular coagulation and promotes inflammation.
Blocking of TF activity attenuates 4-hydroxy-2-nonenal-induced intravascular coagulation and inflammation.
See accompanying editorial on page 266
See cover image
TF (Tissue factor) is the principal initiator of blood coagulation in both physiological and pathological conditions.1,2 In health, TF antigen and its associated procoagulant activity are essentially limited to perivascular cells to safeguard the vascular beds and prevent initiation of undesired coagulation in the vascular lumina.3,4 Various pathological conditions induce TF expression in monocytes and endothelium, leading to thrombotic complications.5–8 Studies in the past 2 decades have suggested the presence of low levels of circulating TF in blood.9–11 However, it has been reported that the activity associated with these circulating TF is very low, close to the detection limit, or corresponds to only 5% of the total TF activity in the blood of healthy individuals.11–14 The onset of pathological conditions triggered by an infection or a disease can increase the levels of circulating TF, which can then trigger the activation of coagulation cascade and inflammation.9,11,15–17
It has been suggested that TF+ microvesicles in the circulation are derived from platelets, endothelial cells, leukocytes, vascular smooth muscle cells, atherosclerotic plaques, and others.14,18,19 It has also been proposed that circulating TF+ microvesicles provide an additional source of TF that reinitiates clotting and promotes the growth of thrombus.14,20 Administration of TF+ microvesicles was shown to increase fibrin deposition in a carotid ligation mouse model, suggesting that TF+ microvesicles may contribute to thrombosis.21 Mouse models of endotoxemia were found to have increased levels of TF activity associated with microvesicles.22
Coagulation and inflammation are highly interlinked where excessive coagulation can induce inflammation, while the generation of inflammatory cytokines can ensue coagulation.23 The onset of any one of the processes can result in the activation or amplification of the other, a condition, which unstopped can lead to tissue damage or even multiple organ failure.23 TF plays a central role in circuiting both these phenomena.23 The presence of cancer-derived TF+ microvesicles in the circulation was shown to exacerbate not only the coagulation but also inflammation.24 Aberrant expression of TF was shown to induce lethal inflammatory response during sepsis and blocking TF activity subsided the inflammatory response.25,26 Furthermore, inhibition of the TF-FVIIa (factor VIIa) complex was found to significantly reduce the plasma levels of proinflammatory cytokines, lung inflammation, and edema during septicemia.27,28 These studies suggest that TF-mediated signaling can induce inflammation during septicemia.
A myriad of studies established that several pathological conditions such as bacterial infection, atherosclerosis, diabetes, sepsis, etc induce oxidative stress, which plays a critical role in aggravating morbidity and mortality associated with these diseases.29–34 The above diseases are also known to have high thrombotic risk.35,36 At present, mechanisms by which the oxidative stress increases thrombotic risk are unclear. Oxidative stress induces lipid peroxidation, generating 4-hydroxy-2-nonenal (HNE), a stable and highly reactive aldehyde.37,38 Under normal conditions, the HNE concentration in human blood is about 0.05 to 0.15 µmol/L, but in pathological conditions, HNE concentration is increased to as high as 0.1 to 5 mmol/L in the membranes.39–41 HNE is believed to be involved in various atherosclerotic and cardiovascular diseases.30 Increased generation of HNE was found in the kidney, liver, and other tissues in various murine models of sepsis.29,42 Scavenging of HNE using a general antioxidant, N-acetylcysteine, was shown to completely abrogate the death induced by a lethal dose of lipopolysaccharide.29 N-acetylcysteine treatment also improved end-organ dysfunction.29 Our recent studies showed that HNE enhances TF procoagulant activity on monocytes and promotes microvesicles shedding from fibroblasts and activated endothelial cells.43,44 Therefore, it is conceivable that HNE-induced TF activation or the release of TF+ microvesicles in vivo may lead to intravascular coagulation and inflammation. The present study was performed to investigate the direct effect of HNE on microvesicles shedding and their contribution to activating intravascular coagulation and inflammation.
The data presented in this report provide strong evidence that HNE increases the levels of circulating TF+ microvesicles and induces intravascular coagulation and inflammation. Our data show that HNE-induced intravascular coagulation and inflammation are dependent on TF. More importantly, our data indicate that HNE-induced circulating TF+ microvesicles are originated from perivascular cells and not from intravascular cells.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. See Data Supplement for additional details on Materials and Methods.
Mice
C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) or bred in-house. Eight to 12-week old mice, both sexes weighing between 24 and 30 g, were used in our experiments. About 250 mice were used in the course of the present study. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee and conducted according to the animal welfare guidelines outlined in the Guide for the Care and Use of Laboratory Animals.
Cells
Primary human umbilical vein endothelial cells were purchased from Lonza. Mouse brain endothelial cells (bEnd.3) were obtained from American Type Culture Collection (Manassas, VA). Human umbilical vein endothelial cell and bEnd.3 cells were cultured as described earlier.45 Human peripheral blood mononuclear cells (PBMCs) were isolated from blood from healthy donors by density gradient centrifugation using Ficoll Paque (GE Healthcare, Pittsburg, PA). The Institutional Review Board at the UT Health Science Center at Tyler approved the blood donation protocol, and the participants gave written informed consent.
Animal Treatments
Unless indicated otherwise, mice were treated with saline, HNE, or lipopolysaccharide for 4 hours. The dose of HNE- and lipopolysaccharide-administered to mice was 10 mg/kg, and they were given intraperitoneal in 100 µL volume. When mice were administered with 1H1 anti-murine TF monoclonal antibody or control isotype IgG, one dose (2 mg/kg) was given immediately before HNE administration and the second dose at 2 hours following the HNE administration.
Saphenous Vein Bleeding
The saphenous vein bleeding model has been described in detail in our earlier study.46 The number of hemostatic plugs formed in the 30-minute observation period, average clotting times, and the blood loss from the injury site was determined as described earlier.46
Isolation of Plasma Microvesicles
Mouse blood drawn into citrate anticoagulant was centrifuged at 2500g for 10 minutes in Eppendorf microcentrifuge to obtain plasma. The plasma was recentrifuged at the same speed and duration to remove any cells that might have escaped into plasma in the first centrifugation. The plasma was centrifuged at 21 000g for 1 hour to sediment microvesicles. The microvesicles were resuspended in buffer A (10 mmol/L Hepes, 0.15 mol/L NaCl, 4 mmol/L KCl, and 11 mmol/L glucose, pH 7.5) to the original plasma volume and resedimented by centrifugation at 21 000g for 1 hour. The pellet was suspended in buffer A for further analysis.
Nanoparticle Tracking Analysis
Microvesicles size distribution and concentration were analyzed in Malvern Panalytical NanoSight 300 using nanoparticle tracking analysis software. The analysis parameters were included in the Data Supplement.
Immunoprecipitation of TF+ Microvesicles
Microvesicles harvested from plasma were resuspended in HEPES buffer and incubated with rabbit anti-mTF Ab (10 μg/mL) overnight at 4 °C. The next day, 20 μL of protein A/G beads were added to the suspension and incubated for an additional 2 hours at 4 °C. Protein A/G beads were sedimented by centrifugation, washed 3 times, TF+ microvesicles were eluted with glycine (pH 2.3). The eluate was lysed in SDS-PAGE buffer and subjected to immunoblot analysis.
Coagulation Assays
TF procoagulant activity was measured in a FX activation assay as described earlier.47 To measure TF procoagulant activity in a clotting assay, plasma obtained from saline- or HNE-treated mice was incubated with 1H1 anti-TF antibody (10 µg/mL) or control isotype IgG for 30 minutes at 37 °C, and then, plasma was recalcified. The clot times were recorded using STart coagulizer (Diagnostica Stago). Thrombin: antithrombin (TAT) levels in murine plasma were measured in an ELISA using a commercially available TAT assay kit (Assaypro, St Charles, MO) by following the protocol included in the kit.
Cytokines Levels
Levels of IL (interleukin)-6 and CXCL1/KC (murine IL-8 equivalent) in the plasma were measured using ELISA kits (eBioscience, San Diego, CA, and RayBiotech, Peachtree Corners, GA) as prescribed in the manufacturer’s protocol.
Immunohistochemistry
The processing of lung tissues and immunostaining of the lung tissue sections with Ly6G was described recently.48
Immunofluorescence Confocal Microscopy
Lung tissues were fixed with 4% paraformaldehyde and dehydrated with a 15% sucrose solution followed by 30% sucrose solution until the lung tissues were settled at the bottom. Tissues were then embedded in Tissue-Tek optimal cutting temperature compound (Sakura Finetek, Torrance, CA), and 5-μm thin sections were cut. The sections were fixed in ice-cold acetone for 10 minutes, blocked with Dako antibody diluent solution (Agilent, Santa Clara, CA), and stained overnight at 4 °C with rat anti-mouse CD31 antibody (5 µg/mL), rabbit polyclonal anti-murine TF antibodies (5 µg/mL), and murine anti-human α-smooth muscle actin (α-SMA; 5 µg/mL, cross-reacts with murine α-SMA), followed by Alexa-488-, Alexa-594-, and Alexa-647-conjugated secondary antibodies. The nuclei were stained with DAPI (5 µg/mL). To immunostain cultured endothelial cells, the cells were fixed in 2% paraformaldehyde and stained with antibodies against goat anti-human VE-cadherin (5 µg/mL) and EPCR (endothelial cell protein C receptor) mAb (JRK1500, 5 µg/mL); the nuclei were stained with DAPI. Confocal images were obtained using an LSM 510 confocal system (Carl Zeiss). Immunostained tissue sections or cells were viewed using a Plan-APOCHROMAT 63.3/1.4 NA oil objective lens.
Barrier Permeability Assays
Endothelial cell barrier permeability in vitro and in vivo was analyzed as described in our earlier studies.49,50
Statistical Analysis
Data from both male and female mice were pooled for robust analysis of the data and unbiased interpretation of the findings as we found no discernible differences between males and females in our initial findings on HNE-released TF+-microvesicles procoagulant activity. The values of each parameter within a group were expressed as the mean±SEM. For comparison between 2 groups with normally distributed data, statistical significance between the 2 groups was assessed using the unpaired Student t test. We used D’agostino and Pearson or the Kolmogorov-Smirnov normality tests to determine whether the data are normally distributed. For comparison between groups with non-normally distributed data, nonparametric tests, such as the Mann-Whitney or Wilcoxon signed-rank test, was used to determine statistical significance. Graphpad Prism version 8 was used for statistical analysis and preparation of figures.
Results
HNE Induces the Release of TF+ Microvesicles Into the Circulation
To investigate whether administration of HNE can elicit generation of procoagulant microvesicles, wild-type C57BL/6J mice were injected with saline or HNE (10 mg/kg, IP) and microvesicles from plasma were isolated, and their procoagulant activity was measured in a FX activation assay. As shown in Figure 1A, microvesicles generated in HNE-treated mice activated FX at a significantly higher rate compared with microvesicles isolated from saline-treated animals. Next, we investigated whether the increased FXa (factor Xa) generation associated with microvesicles is TF dependent. To investigate this, microvesicles were first incubated with murine TF-specific monoclonal antibodies (1H1), and then FXa generation was measured. 1H1 antibody significantly reduced the generation of FXa associated with microvesicles, which suggests that the procoagulant activity associated with microvesicles is TF procoagulant activity (Figure 1B). It has been suggested that small quantities of microvesicles are always present in circulation, which increase in response to various disease conditions.14 Therefore, we next investigated whether the increased FXa generation associated with microvesicles is due to the increased number of TF+ microvesicles in the plasma or reflects decryption of TF associated with microvesicles circulating in blood under basal conditions. Nanoparticle tracking analysis studies showed that microvesicles derived from HNE-challenged mice had ≈3-fold higher count compared with saline-administered mice (saline, 1.5×108±4.90×107 versus HNE, 5.0×108±1.03×107) with a significantly higher mean size (saline, 126.1±7.70 nm versus HNE, 159.3±3.10 nm) and mode (saline, 80.0±10.10 nm versus HNE, 135.5±10.02 nm; Figure 1C through 1E). Interestingly, HNE-derived microvesicles exhibited a wider size distribution (Figure 1F). However, >95% of microvesicles fell between 50 and 300 nm (Figure 1C). Overall, the above data suggest that HNE promotes the release of procoagulant TF+ microvesicles into the circulation. However, not all HNE-released microvesicles carry TF. Analysis of microvesicles isolated from HNE-treated mice by fluorescent nanoparticle tracking analysis or flow cytometry for TF expression revealed that only about 7% to 15% microvesicles carry TF. In saline-treated animals, <0.5% microvesicles contain TF (data not shown).
Figure 1.
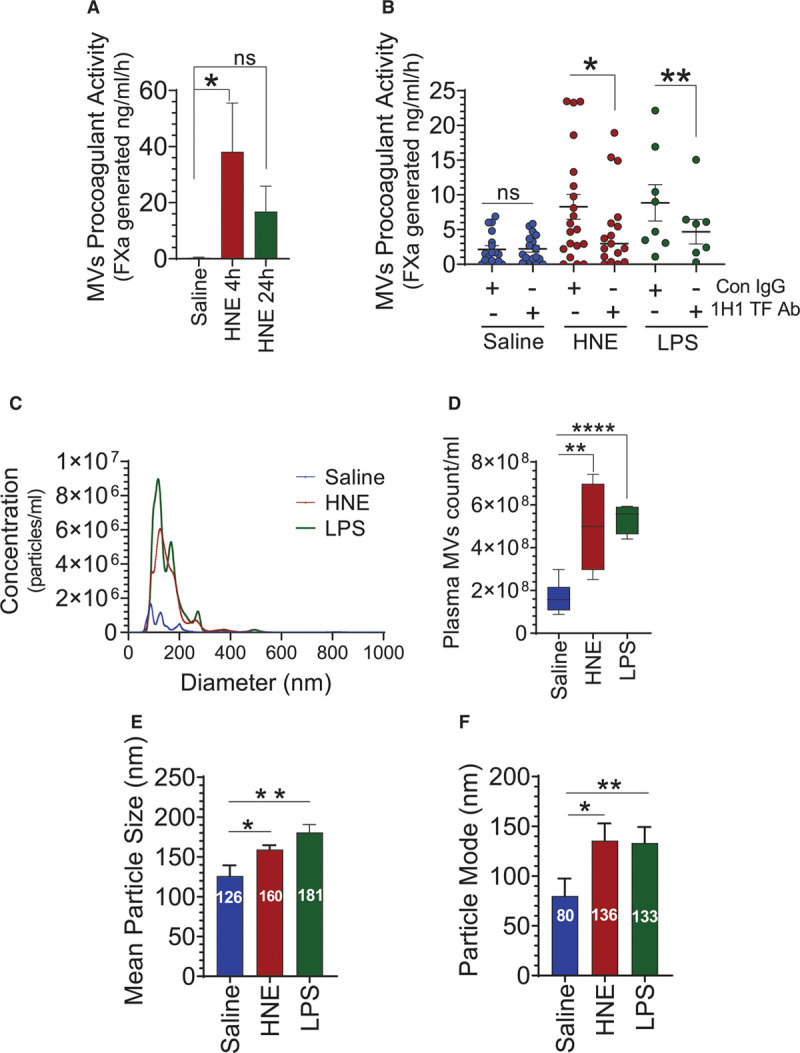
4-hydroxy-2-nonenal (HNE) promotes the release of TF+ (tissue factor) microvesicles. A, C57BL/6J mice were administered with HNE (10 mg/kg, IP) and 24 h following HNE administration, blood was collected via the submandibular vein into citrate anticoagulant. Microvesicles (MVs) were isolated from plasma and their procoagulant activity was measured in FVIIa (factor VIIa) activation of FX. B, MVs isolated from the plasma of saline-, HNE (10 mg/kg)-, or lipopolysaccharide (LPS; 10 mg/kg)-challenged mice (for 4 h) were incubated with control isotype IgG (Con IgG) or 1H1 murine TF mAb (1H1 TF Ab; 10 µg/mL) for 30 min before their procoagulant activity was measured in FX activation assay. C through F, MVs isolated from the plasma of saline-, HNE-, or LPS-administered mice were characterized by nanoparticle tracking analysis using NanoSight NS300. MVs number (C and D) and diameter (C, E, and F) were determined. *P<0.05; **P<0.01; and ****P<0.0001; ns, no statistically significant difference.
HNE Induces Intravascular Coagulation in a TF-Dependent Manner
To test whether HNE-induced microvesicles or other associated factors can promote intravascular coagulation, we measured the plasma levels of thrombin generated in saline- or HNE-challenged mice by measuring TAT levels in plasma. TAT levels were significantly higher in HNE-administered mice compared with control mice (Figure 2A). Next, to investigate the role of TF in HNE-induced thrombin generation, mice were injected with 1H1 murine TF mAb or isotype control IgG along HNE, and the level of thrombin generated was measured. In vitro studies showed that the 1H1 antibody was highly effective in inhibiting murine TF (Figure I in the Data Supplement). As shown in Figure 2B, blockade of TF in vivo with the 1H1 anti-TF antibody significantly impeded the HNE-induced TAT generation. These data indicate that HNE-induced TAT generation is TF dependent. Next, we investigated the clotting times of recalcified plasma from saline- or HNE-administered mice. The data showed that plasma-derived from HNE-challenged mice had a significantly lower clotting time (118.3±10.42 seconds) compared with plasma obtained from saline-treated mice, which did not clot in 300 seconds (maximum duration of the assay). Depletion of microvesicles from the plasma of HNE-challenged mice by relatively low-speed centrifugation (21 000g for 1 hour) or depletion of all extracellular vesicles by high-speed centrifugation (130 000g for 30 minutes) prolonged the plasma clotting time to >300 seconds (Figure 2C). These data suggest that the shortening of clotting times observed in the HNE-derived plasma solely stems from microvesicles-associated procoagulant activity. To determine whether the accelerated clotting time was dependent on TF, we incubated the plasma samples with 1H1 anti-TF antibody or control IgG, and then clotting times were measured. As shown in Figure 2D, the neutralization of TF resulted in prolongation of the clotting time of plasma obtained from HNE-administered mice to over 300 seconds. Since platelets are also important regulators of intravascular coagulation, we investigated whether HNE treatment affected platelet count or volume. Administration of HNE to mice showed no significant difference in the platelet count or mean platelet volume, as revealed by blood analysis studies (Figure 2E and 2F). In additional studies, we wanted to investigate the potential contribution of neutrophil extracellular traps in HNE-induced thrombin generation. However, measurement of myeloperoxidase activity levels in plasma, which is an indicator of neutrophil extracellular traps formation,51 showed no increase in myeloperoxidase activity in the plasma of HNE-challenged mice (Figure 2G). Lipopolysaccharide administration, used as a positive control, markedly elevated myeloperoxidase activity levels in the plasma. These data suggest that HNE-induced thrombin generation is independent of NETosis. Overall, the above data strongly indicate that HNE-induced increased procoagulant activity stems from the increased TF+ microvesicles in the circulation.
Figure 2.
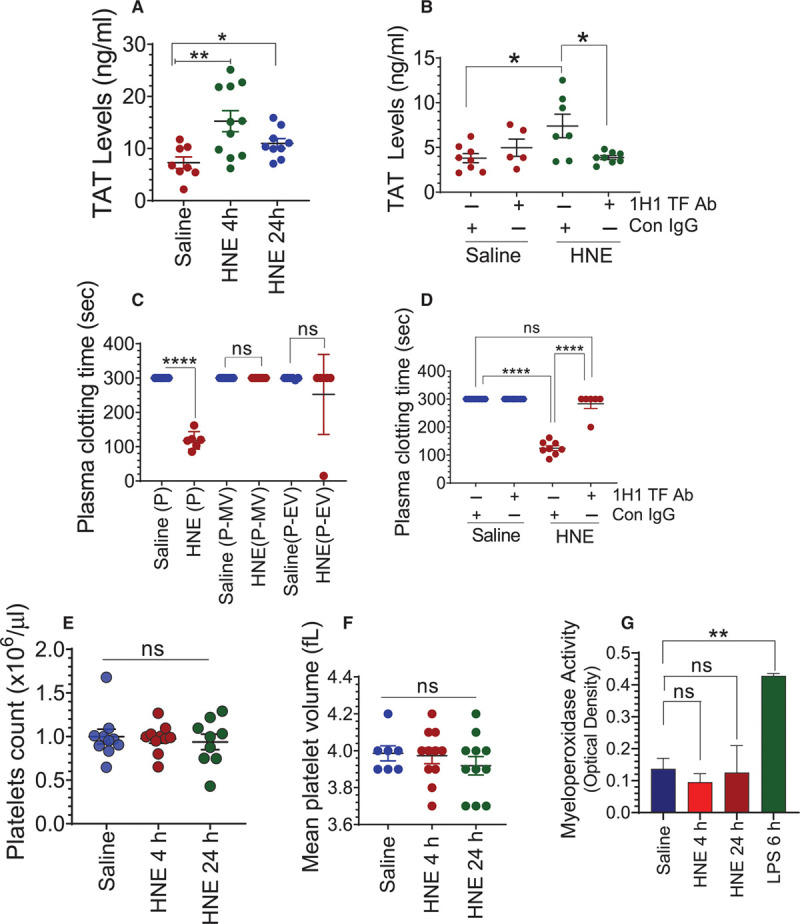
4-hydroxy-2-nonenal (HNE) activates intravascular blood coagulation in a TF (tissue factor)-dependent manner. A, C57BL/6J wild-type mice were administered with saline or HNE (10 mg/kg, IP). Four and 24 h following HNE administration, blood was collected into citrate anticoagulant via submandibular vein puncture. Levels of thrombin-antithrombin (TAT) complexes in the plasma were determined using ELISA. B, Mice were administered with isotype control IgG (Con IgG) or 1H1 murine TF mAb (1H1 TF Ab) immediately before HNE administration and 2 h following HNE administration (2 mg/kg, IP). Four hours following HNE administration, blood was collected from mice, and TAT levels in plasma were measured. C, Plasma (P), plasma depleted of microvesicle (MVs; P-MV) or plasma depleted of all extracellular vesicles (P-EV) of saline- or HNE-treated mice (for 4 h) were recalcified and the clotting times were measured using a semi-automated coagulizer (the maximum assay duration was 300 s). D, Plasma from saline- or HNE-treated mice (for 4 h) were incubated with either control IgG (Con IgG) or 1H1 murine TF mAb (1H1 TF Ab; 10 µg/mL) for 30 min, and then plasma was recalcified to measure the clotting time. E and F, Platelet count and mean platelet volumes were analyzed in an aliquot of blood collected from saline- or HNE-treated mice using HEMAVET. G, Myeloperoxidase activity levels in the plasma obtained from saline-, HNE-, or lipopolysaccharide (LPS)-administered mice. *P<0.05; **P<0.01; and ****P<0.0001; ns, no statistically significant difference.
HNE Shortens Bleeding Time in the Saphenous Vein Injury Model
To investigate whether HNE-induced increased TF procoagulant activity promotes clotting, we evaluated the effect of HNE on the bleeding time in the saphenous vein injury model. As shown in Figure 3A and 3B, the HNE administration significantly reduced the bleeding time and blood loss. Next, we investigated whether the reduced bleeding time and blood loss in HNE-treated mice subjected to the saphenous vein incision is due to HNE-induced increased TF activity. For this, mice were injected with 1H1 anti-TF antibody or control IgG along with HNE, and the average time to clot was recorded. Administration of the anti-TF antibody reversed the reduction in the bleeding time and blood loss observed in HNE-administered mice (Figure 3C and 3D). These data strongly suggest that HNE-induced accelerated clot formation was dependent on TF.
Figure 3.
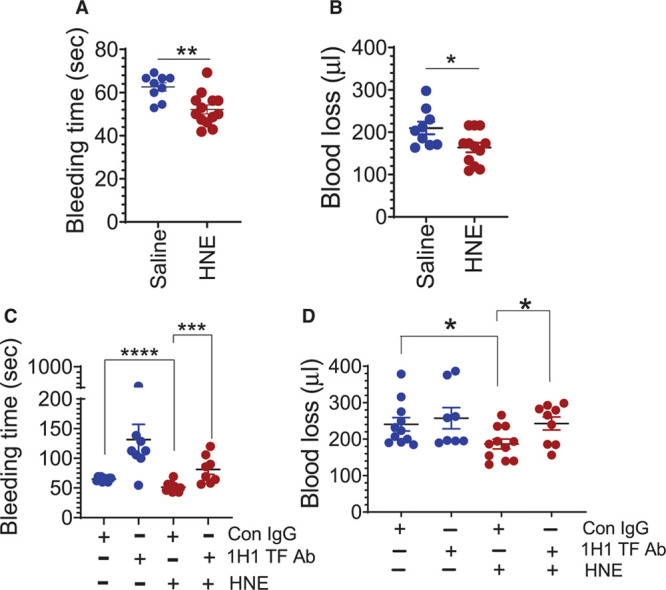
4-hydroxy-2-nonenal (HNE) accelerates coagulation. A and B, C57BL/6J wild-type mice were administered with saline or HNE (10 mg/kg, IP). Four hours after HNE administration, bleeding was initiated by the saphenous vein incision. The average bleeding times were calculated from the number of hemostatic plugs formed in a 30-min bleeding period (A). The volume of blood leaked from the wound site was adsorbed onto Kimwipes for the entire duration of 30 min, and the blood loss was determined by extracting the hemoglobin from wipes and measuring it against known standards (B). C and D, Wild-type mice were administered with control IgG or 1H1 anti-murine TF (tissue factor) mAb (2 mg/kg) just before HNE administration and 2 h after HNE administration. Four hours after HNE administration, mice were subjected to the saphenous vein injury and the bleeding time (C) and the blood loss (D) were determined as described above. *P<0.05; **P<0.01; ***P<0.001; and ****P<0.0001.
HNE-Derived TF+ Microvesicles Elicit Proinflammatory Responses
To investigate the role of HNE-induced TF+ microvesicles on inflammation, we evaluated plasma levels of CXCL1 and IL-6 as markers of inflammation. As shown in Figure 4A and 4B, there was a significant increase in CXCL1 and IL-6 levels in the plasma of HNE-challenged mice. Next, to investigate whether the observed inflammatory response was due to HNE-induced increased TF+ microvesicles, mice were administered with 1H1 anti-TF antibody along HNE. As shown in Figure 4C, inhibition of TF activity significantly attenuated the HNE-induced increase in IL-6 levels. Interestingly, inhibition of TF activity had no significant effect on HNE-induced increased CXCL1 levels (Figure 4D). Analysis of blood showed a significant increase in circulating neutrophils count in 4 hours while circulating monocytes peaked in 24 hours in HNE-administered mice (Figure 4E and 4F). Immunohistochemistry studies showed a robust infiltration of neutrophils in the lungs of HNE-administered mice (Figure 4G). Inhibition of TF activity with 1H1 antibody reduced neutrophil infiltration into the lungs of HNE-administered mice (Figure 4H). Overall, the above data suggest that HNE-induced increase in the proinflammatory cytokine IL-6 and neutrophil infiltration was dependent on TF.
Figure 4.
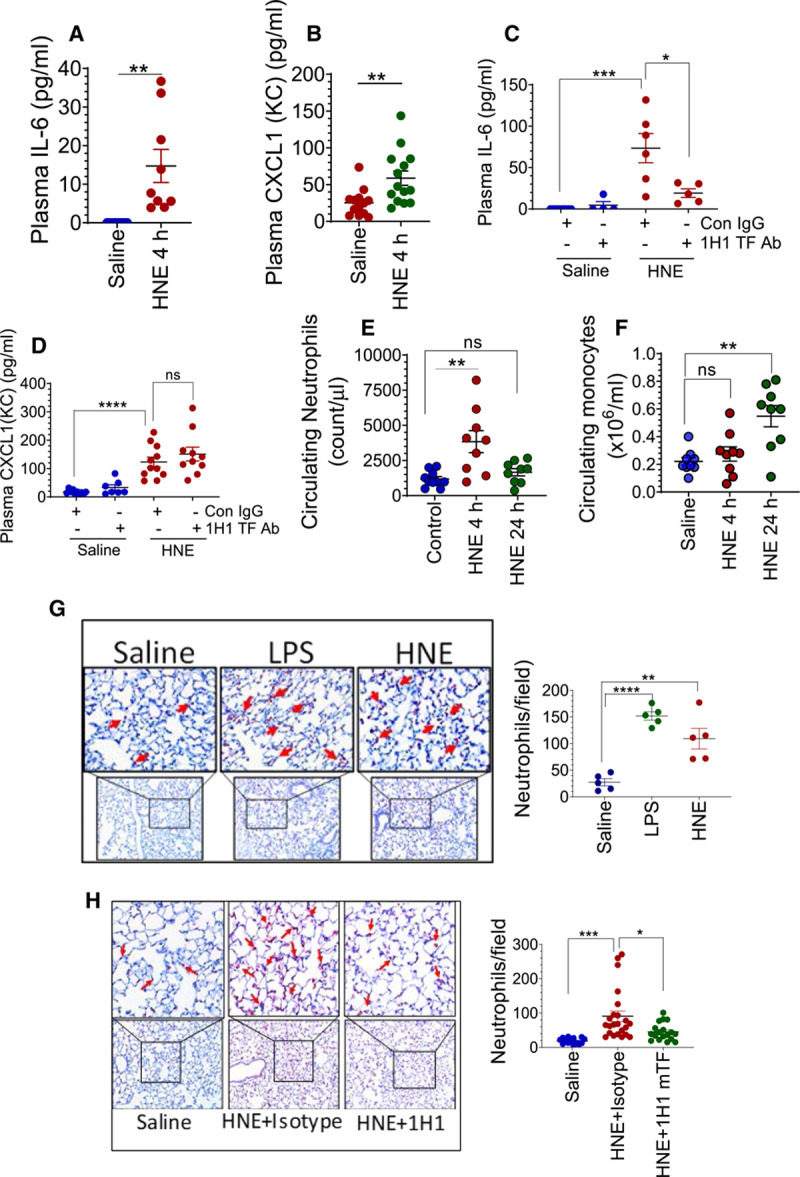
4-hydroxy-2-nonenal (HNE) induces TF (tissue factor)-dependent proinflammatory responses in mice. Cytokines IL (interleukin)-6 (A) and CXCL1 (B) levels in the plasma of saline- or HNE-challenged (for 4 h) wild-type mice. C and D, Effect of murine TF antibody administration on HNE-induced increase in IL-6 (C) and CXCL1 (D) levels in plasma. Mice were administered with control isotype IgG or 1H1 murine TF mAb as described in Figure 2. E and F, HNE induces an increase in neutrophil and monocyte number in circulating blood. Wild-type mice were challenged with saline or HNE. Four and 24 h following HNE administration, the number of neutrophils (E) and monocytes (F) in blood were counted using HEMAVET. G and H, HNE induces neutrophil infiltration into the lungs, and the administration of murine TF antibody attenuates the HNE-induced response. G, Lung tissue sections from saline-, lipopolysaccharide (LPS)-, or HNE-treated mice (for 4 h) were immunostained for neutrophil marker Ly6G to detect neutrophil infiltration (left) and the number neutrophils in each field were counted (right). H, Mice were treated with isotype control IgG or 1H1 anti-murine TF antibody before HNE administration, as described in Figure 2. Lung tissue sections from saline- or HNE-administered mice (for 4 h) were stained for Ly6G (left), and the number of neutrophils in a field were counted (right). *P<0.05; **P<0.01; ***P<0.001; and ****P<0.0001; ns, no statistically significant difference.
HNE Does Not Induce TF Expression in the Endothelium or Circulating Mononuclear Cells
The blood monocytes and the vascular endothelium are among the major sources of TF+ microvesicles generation in the bloodstream.14,52,53 So, to determine whether the endothelium or the blood monocytes are the sources of HNE-induced TF+ microvesicles, we investigated whether HNE induces de novo synthesis of TF in the mouse endothelium or the blood monocytes. Confocal microscopy studies showed that HNE administration in mice did not induce TF expression in endothelial cells (Figure 5A). In contrast, lipopolysaccharide administration induced detectable TF antigen in the endothelium (Figure 5A). There was no signal with isotype IgG control (Figure II in the Data Supplement). In agreement with in vivo data, HNE treatment neither induced TF mRNA nor TF antigen in cultured endothelial cells (Figure 5B and 5C). In controls, cytokine treatment markedly increased both TF mRNA and TF antigen levels in endothelial cells (Figure 5B and 5C). Next, we investigated whether HNE administration in mice induces TF expression in mononuclear cells. We did not detect TF expression in PBMCs derived from HNE-challenged mice by either confocal microscopy (Figure 5D), real-time polymerase chain reaction (Figure 5E), or immunoblot analysis (Figure 5F). We observed similar data in in vitro studies where PBMCs isolated from human blood were treated with HNE (data not shown). In contrast to HNE-administered mice, induction of TF in PBMCs was detectable in lipopolysaccharide-administered mice (Figure 5D through 5F). These observations indicate that HNE does not induce de novo synthesis of TF either in the endothelium or circulating mononuclear cells. Therefore, it is unlikely that the TF+ microvesicles in HNE-administered mice are derived from either endothelial or mononuclear cells.
Figure 5.
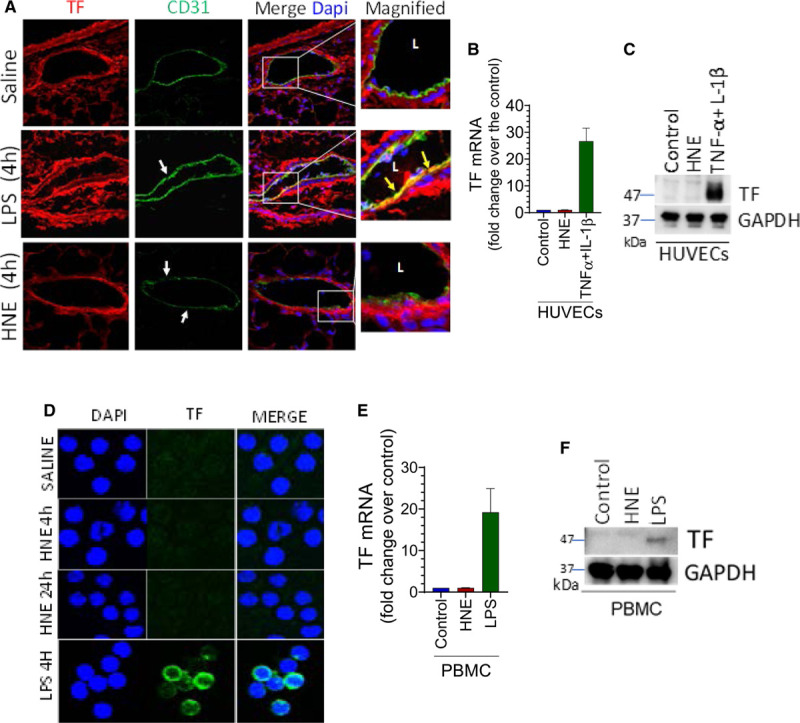
4-hydroxy-2-nonenal (HNE) does not induce TF (tissue factor) expression in vascular cells. A, C57BL/6J mice were injected with saline, HNE (10 mg/kg), or lipopolysaccharide (LPS; 10 mg/kg). After 4 h, the lung tissues were harvested and immunostained with endothelial cell marker CD31 and TF. Images were focused on the immunostaining of blood vessels. Green fluorescence represents CD 31 staining, whereas red fluorescence indicates TF staining. White arrow marks point out endothelial denudation. Yellow arrows on merged images indicate TF staining on the endothelium. L, a lumen of the blood vessel. B and C, Monolayers of human umbilical vein endothelial cells (HUVECs) were treated with saline (control), HNE (40 µmol/L), or TNF (tumor necrosis factor)-α+IL (interleukin)-1β (10 ng/mL, each) for 2 or 4 h to analyze TF mRNA levels by quantitative real-time polymerase chain reaction (RT-PCR; B) or TF protein by western blot analysis (C), respectively. D through F, C57BL/6J mice were treated with saline, HNE (10 mg/kg), or LPS (10 mg/kg). Four hours following HNE or LPS administration, blood was collected, and PBMCs were isolated. TF expression was analyzed by confocal microscopy (D), measuring TF mRNA by quantitative RT-PCR (E), or TF protein by western blot analysis (F). In D, cell nuclei were stained with DAPI. Cells shown are monocytes (other mononuclear cells were much smaller in size than monocytes and were stained weakly with DAPI and negative for TF, and thus not readily visible).
HNE-Induced TF+ Microvesicles Are Derived From Perivascular Cells
To investigate the cellular source of circulating TF+ microvesicles in HNE-administered mice, microvesicles were subjected to immunoblot analysis for the presence of cell type-specific markers. As shown in Figure 6A, HNE-induced microvesicles were stained positively for CD248 and α-SMA, the markers that are specifically expressed by the perivascular cells and not by the endothelium or blood cells.54,55 Furthermore, microvesicles isolated from the plasma of HNE-administered mice lacked CD14, a monocyte marker. CD14 marker was readily detectable in microvesicles isolated from the plasma of lipopolysaccharide-administered mice. VWF (Von Willebrand factor) expression, which was used as a surrogate marker for endothelial cells, was found on all microvesicles, that is, microvesicles derived from saline-, HNE-, or lipopolysaccharide-treated mice. This observation is consistent with the earlier report that suggested a constitutive release of microvesicles into the bloodstream from the endothelial cells.14 To further support our hypothesis that the HNE-induced TF+ microvesicles are derived from perivascular cells, we immunoprecipitated microvesicles isolated from the plasma using a polyclonal anti-murine TF antibody. The analysis of immunoprecipitates showed that microvesicles derived from both HNE- and lipopolysaccharide-treated mice were positive for α-SMA and CD248. However, CD14 was detectable only in microvesicles derived from the plasma of lipopolysaccharide-treated mice but not from the plasma of HNE-challenged mice. These data suggest that HNE, unlike lipopolysaccharide, does not induce TF+ microvesicles from myeloid cells. It has been suggested that brain cells are an important source of TF+ microvesicles.56 So, to determine whether HNE-induced microvesicles are derived from the brain, we probed these microvesicles with markers specific to brain cells. We failed to detect the presence of brain-specific markers,56 such as GFAP (glial fibrillary acidic protein) or Na+/K+ ATPase α3 (Figure 6B).
Figure 6.
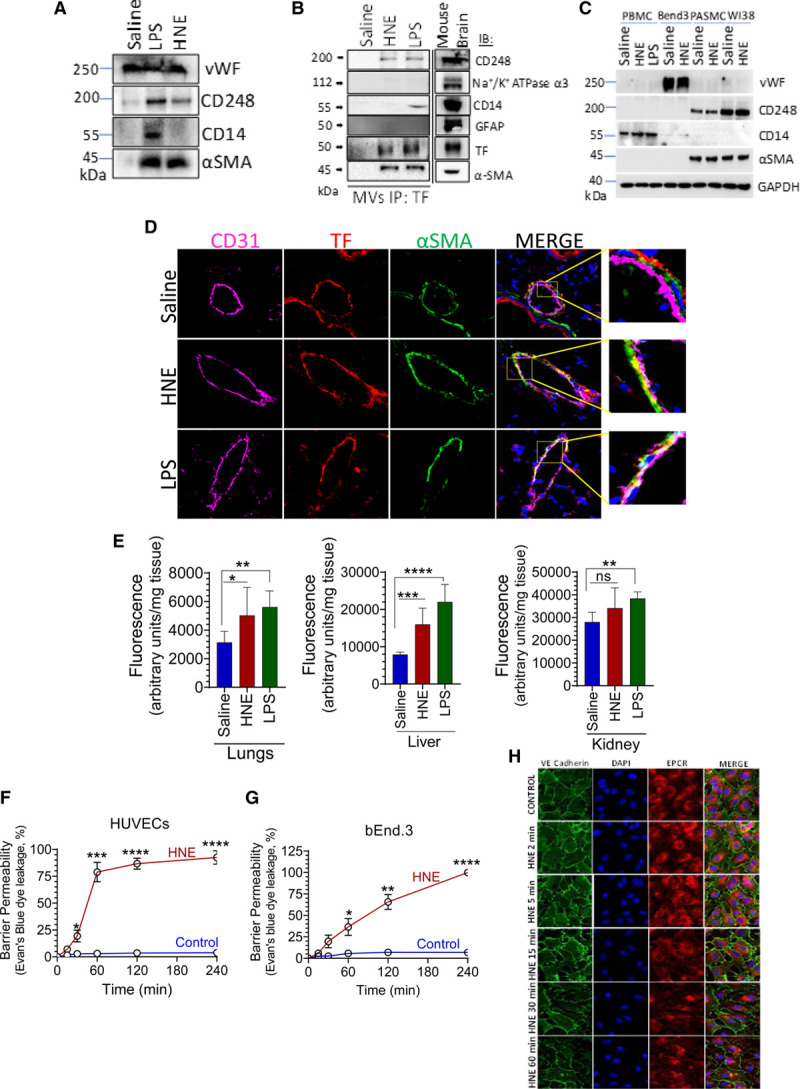
4-hydroxy-2-nonenal (HNE) releases TF+ (tissue factor) microvesicles from perivascular cells and induces vascular barrier disruption. A, C57BL/6J mice were injected with saline, HNE (10 mg/kg), or lipopolysaccharide (LPS; 10 mg/kg). Microvesicle (MVs) harvested from the plasma of saline-, HNE-, or LPS-administered mice were lysed in an equal volume of 1 X SDS lysis buffer and subjected to immunoblot analysis for VWF (Von Willebrand factor), CD248, CD14, or α-SMA. B, MVs harvested from the plasma of saline-, HNE-, or LPS-administered mice were immunoprecipitated with anti-murine TF antibodies, and the immunoprecipitates were analyzed for the presence cell-specific markers by immunoblot analysis. Mouse whole brain lysate was used as a positive control. C, Cell extracts of PBMCs from saline-, HNE-, or LPS-treated mice, bEnd.3 cells, pulmonary artery smooth muscle cells (PASMC), and WI38 fibroblasts treated with or without HNE (20 µmol/L) for 4 h were subjected to immunoblot analysis and probed for VWF, CD248, CD14, or α-SMA. D, Lung tissue sections prepared from the saline-, HNE-, or LPS-treated mice were immunostained for CD31 (Magenta), TF (Red), and α-SMA (Green). A small portion of the merged image was enlarged to show colocalization. E, Three hours following saline-, HNE-, or LPS administration, mice were administered with fluorescein dextran (10 mg/kg) via the tail vein. One hour following fluorescein dextran administration, mice were euthanized, perfused with saline, and tissues were harvested. The extent of fluorescein dextran entered in tissues was determined by measuring the fluorescence intensity of tissue extracts. F and G, Human umbilical vein endothelial cell (HUVEC) or bEnd.3 endothelial cells were cultured in 24-transwell plates for 4 d to form tight confluent monolayers. Cells were treated with HNE (20 µmol/L) for varying periods. In controls, cells were treated for 4 h with a control vehicle. At the end of treatment, Evan blue dye was added to the apical chamber, and the amount of dye leaked into the lower chamber at 10 min was read in a spectrophotometer. H, HUVECs were cultured on coverslips and treated with HNE (20 µmol/L) for varying periods (2–60 min). Following HNE treatment, the cells were washed and fixed with 2% paraformaldehyde. The distribution of VE-cadherin was analyzed by immunostaining the cells with anti-VE-cadherin antibodies, followed by confocal microscopy. EPCR antibodies and DAPI were used to stain the cell surface and nucleus, respectively. *P<0.05; **P<0.01; ***P<0.001; and ****P<0.0001; ns, no statistically significant difference.
To strengthen our data that the presence of CD248 and α-SMA are specific to perivascular cells, we analyzed the PBMCs isolated from HNE- or lipopolysaccharide-administered mice, naïve or HNE-perturbed endothelial cells, and perivascular cells (smooth muscle cells and fibroblasts) for the presence of various cell-specific markers. As shown in Figure 6C, CD248 and α-SMA were present in fibroblasts and smooth muscles and not in PBMCs or endothelial cells. VWF is detectable only in endothelial cells, whereas CD14 was only detectable in PBMCs. Furthermore, immunostaining of mouse lung sections shows that α-SMA staining is restricted to the perivascular region, and TF was colocalized with α-SMA in this region (Figure 6D). These observations strengthen our data that HNE-induced TF+ microvesicles are derived from the perivascular cells and possibly from smooth muscle cells, pericytes, or fibroblasts.
Next, we probed how TF+ microvesicles derived from perivascular cells could enter into circulation. Our earlier studies showed that HNE induces ROS (reactive oxygen species) generation in vitro,44 and the ROS generation could potentially breach endothelial barrier integrity.57,58 Therefore, we investigated whether the HNE administration can disrupt the barrier integrity of the mouse endothelium in vivo. Administration of mice with HNE for 3 hours followed by intravenous injection of the fluorescent dye, fluorescein dextran, showed a significant increase in the leakage of the dye into the lungs and liver (Figure 6E). Similar results were obtained using Evan blue dye (data not shown). Confocal microscopy data depicted in Figure 5A shows partial denudation of the endothelial layer in HNE-administered mice, as evidenced by the ruptured immunostaining of the endothelial protein CD31 (Figure 5A). Additional studies conducted with human and mouse endothelial cells grown in the transwell system to confluency to form tight junctions showed that HNE treatment markedly increased barrier permeability in a time-dependent manner (Figure 6F and 6G). Confocal microscopy studies showed a time-dependent redistribution of the cell adhesion molecule, VE-cadherin, and disruption of the endothelial integrity of confluent human endothelial cell monolayer upon HNE treatment (Figure 6H). Similar effects of HNE was observed in bEnd.3 murine endothelial cells (data not shown).
Discussion
Various cardiovascular diseases and sepsis have been shown to have elevated levels of HNE.29,30 HNE, the most abundant and stable unsaturated aldehyde produced by the oxidation of ω-6 polyunsaturated fatty acids, is a major oxidative stress product.38,59 Our recent studies showed that HNE enhances TF decryption in various cell types, including monocytes, blood-derived macrophages, cytokine-perturbed endothelial cells, and fibroblasts.43,44 HNE was also shown to induce highly procoagulant TF+ microvesicles from fibroblasts but interestingly not from monocytes or macrophages.43 It is unknown at present whether HNE induces TF activation or releases TF+ microvesicles into the circulation in vivo and its impact on coagulation. Data from our present study show that HNE induces the release of TF+ microvesicles from perivascular cells and contributes to intravascular coagulation and inflammation.
Oxidative stress plays a central role in the pathogenesis of many inflammatory diseases, including atherosclerosis, diabetes, and sepsis.29,30 HNE has been shown to be involved in the pathogenesis of the above diseases.30 Wiesel et al29 showed that lipopolysaccharide administration in mice induced the generation of HNE in the liver.29 These studies also showed that antioxidant N-acetylcysteine protected mice from lipopolysaccharide-induced mortality. Increased HNE protein adducts were also found in renal homogenates of cecal ligation and puncture-induced murine sepsis.42 Consistent with these data, we also found the presence of HNE adducts in the lungs, the liver, and the plasma of lipopolysaccharide-administered mice (data not shown). While free radicals are short-lived, HNE is very stable and can persist longer in the system and can diffuse to distant sites from its source of origin.60 Administration of HNE directly to mice, as done in the current study, allows us to investigate the effect of HNE that results from oxidative stress on hemostasis and thrombosis without confounding effects from other mediators generated in the disease.
It has been shown recently that deficiency of antioxidant PON2 (protein paraoxonase-2) causes inflammation and abnormalities in blood coagulation.61 These studies showed that PON2 deficiency in mice provokes endothelial oxidative stress (ROS generation), activates endothelial TF, and impacts the coagulation.61 It has been speculated that increased formation of ROS, exposure of phosphatidylserine, and the lipid peroxidation in PON2 deficiency contribute to endothelial TF procoagulant activity. Our earlier studies showed that HNE induced ROS generation and decrypted TF on cell surfaces.43,44 The present data that show HNE induces TF+ microvesicles and impacts the coagulation is consistent with the conclusion reached in an earlier report with Pon2-deficient mice that oxidative stress impacts the coagulation.61 However, mechanisms by which oxidative stress impacts the coagulation may vary. For example, in the present study, we found no evidence for HNE induction of TF on the vascular endothelium. Similarly, we found no detectable TF antigen in circulating PBMCs of HNE-administered mice. Although it has been suggested that oxidative stress may affect platelet activation and phosphatidylserine exposure,61 our in vivo studies did not show any difference in platelet count or mean platelet volume. A change in the mean platelet volume was shown to be an indicator of platelet activation.62,63 Therefore, it is unlikely that the HNE-induced intravascular coagulation comes from the induction of TF in circulating blood cells or vascular endothelial cells. HNE-induced release of TF+ microvesicles into the circulation appears to be responsible for HNE-induced intravascular coagulation.
Elevated levels of TF bearing microvesicles have been observed in blood samples of various diseases, such as sepsis, sickle cell disease, hyperlipidemia, atherosclerosis, diabetes, and cancer.14 In cancer, circulating TF+ microvesicles were shown to be derived from tumor cells.64 In other disease conditions, the cellular source of circulating TF+ microvesicles was either unknown or shown to be derived from monocytes or endothelial cells.14 Our present data suggest that it is possible that circulating TF+ microvesicles in disease conditions may come from perivascular cells as the microvesicles derived from the HNE-treated mice expressed α-SMA and CD248, markers that are restricted to perivascular cells.54,55 Unlike lipopolysaccharide-derived microvesicles, the HNE-derived microvesicles did not express CD14, a marker for monocytes. These data are consistent with our earlier published data that showed HNE induces the release of TF+ microvesicles from fibroblasts but not from monocytic cells.43 The observation that HNE destabilizes endothelial barrier and induces leaky vasculature explains how TF+ microvesicles from perivascular cells could enter the bloodstream. Although HNE-induced TF+ microvesicles from perivascular cells are likely to be responsible for the increased intravascular coagulation observed in the HNE-treated mice, it is not feasible to completely rule out the contribution of perivascular cell surface-associated TF in this process. It is possible that HNE-induced barrier disruption would also allow the coagulation factors from the bloodstream to come in contact with TF on perivascular cells. At present, it is not possible to selectively deplete circulating microvesicles or inhibit circulating microvesicles TF activity to address the above conundrum. However, it is pertinent to note here that microvesicles isolated from HNE-administered mice were shown to activate FX and reduce the clotting time of plasma in ex vivo in a TF-dependent manner. Furthermore, if thrombin is generated from TF on perivascular cells, thrombin would likely be localized in that compartment rather than in peripheral blood circulation.
Increased coagulation has been associated with increased inflammation and endothelial dysfunction.24 Elevated levels of TF+ microvesicles in the blood have been reported to contribute to inflammation via the protease-activated receptors.24,65 Acute inflammation not only leads to migration of neutrophils and monocytes to tissues but also mobilizes them from bone marrow to bloodstream.66,67 Our study shows that blood from the HNE-administered mice had significantly higher levels of circulating neutrophils and monocytes. More importantly, HNE significantly increased plasma levels of inflammatory mediators IL-6 and CXCL1 and infiltration of neutrophils into the lungs. The observation that blockade of TF using anti-TF antibody markedly attenuate the observed increase in plasma IL-6 levels and neutrophils infiltration into lungs indicate that HNE-induced TF+ microvesicles may be responsible for the inflammation. It is interesting to note here, in contrast to IL-6 levels, the increased levels of CXCL1 observed in HNE-treated mice were not diminished by the administration of the anti-TF antibody. These data suggest HNE induces inflammation in both TF-dependent and TF-independent manner. It may be pertinent to point out here that HNE induced the expression of CXCL1 and not IL-6 in monocytic cells (unpublished data of the authors). It is possible that HNE-induced increase in CXCL1 levels in vivo was the result of direct signaling of HNE, whereas increased IL-6 levels might be due HNE-induced increased TF activity. It would explain why the TF antibody selectively inhibits IL-6 and not CXCL1. Our present data do not address whether TF-FVIIa-induced direct signaling or the signaling induced by the downstream clotting proteases generated by TF-FVIIa-initiated coagulation by TF+ microvesicles is responsible for HNE-induced inflammation, which will be the subject of our future research.
In summary, data of our current study show that HNE induces the release of highly procoagulant TF+ microvesicles that promote thrombin generation and proinflammatory response in vivo (see Figure 7 for schematic presentation). Our data also indicate that the cellular source of TF+ microvesicles is perivascular cells. The breach of endothelial integrity by HNE or other inflammatory agents that would allow the entry of perivascular-derived TF+ microvesicles into the bloodstream could result in prothrombotic events. Our data support the concept that the expression of TF in the intravascular cells is not compulsory for the increased levels of circulating TF+ microvesicles in disease conditions. These data imply that disease conditions that neither induce TF expression nor decrypt TF on intravascular cells could have an impact on intravascular coagulation by increasing vascular permeability and releasing TF+ microvesicles from perivascular cells.
Figure 7.
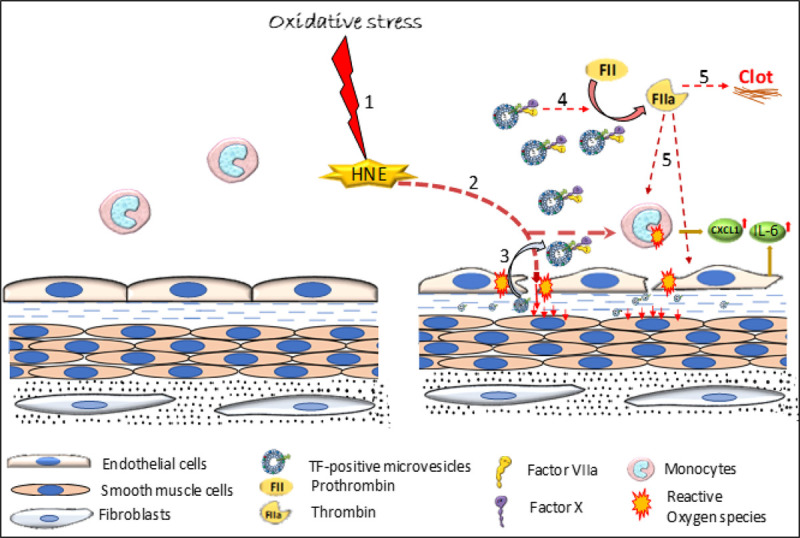
Schematic representation of 4-hydroxy-2-nonenal (HNE)-mediated intravascular coagulation and inflammation. HNE, a lipid peroxidation product that is generated in cardiovascular diseases, sepsis, and other disease conditions, destabilizes endothelial barrier integrity and induces the release of TF+ (tissue factor) microvesicles (MVs) from perivascular cells. Due to the barrier disruption, TF+ MVs from perivascular cells could enter the bloodstream and induce intravascular coagulation and thrombosis. Signaling induced by downstream proteases, such as thrombin, or TF-FVIIa (factor VIIa)-mediated direct signaling in perivascular cells could promote inflammation by inducing inflammatory cytokines and infiltration of neutrophils into tissues. Step 1: Oxidative stress generates HNE; Step 2: HNE induces ROS generation in monocytes and endothelium, and increases vascular permeability; Step 3: Breach in the vasculature leads to leakage of perivascular cells-derived TF+ MVs into the circulation; Step 4: TF+ MVs promote thrombin generation; Step 5: Thrombin induces clot formation and possibly upregulates cytokines expression.
A caveat in our studies is that the bolus injection of HNE into healthy mice may not recapitulate effects that would be seen in a disease state with chronic HNE exposure. Chronic exposure may modulate key receptors and responses differently from that of acute exposure. Furthermore, earlier studies showed that the deletion of TF from perivascular cells had no significant effect on intravascular coagulation or circulating TF+ microvesicles in endotoxemia, atherosclerosis, and ferric chloride injury model systems.68–70 The differences in the data between the present study and the earlier studies on the involvement of perivascular cell-derived TF+ microvesicles in driving intravascular coagulation may be due to differences in the model systems used. As HNE decrypts TF, HNE-released TF+ microvesicles are likely to have higher procoagulant activity than microvesicles released in the other model systems. Further studies are needed to provide the definitive proof that HNE generated in various disease conditions is responsible for aberrant activation of coagulation associated with these diseases. The present data serve as a proof-of-concept for future studies.
Acknowledgments
We thank Dr Daniel Kirchhofer, Genentech Inc, South San Francisco, CA, for providing rat monoclonal antibody against murine TF (1H1) and Dr Charles T. Esmon, Oklahoma Medical Research Foundation, for providing monoclonal antibodies against human EPCR (JRK1500).
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute grants HL124055 and HL107483 to Dr Rao and American Heart Association Post-doctoral Fellowship to S.A. Ansari.
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- FVIIa
- factor VIIa
- FXa
- factor Xa
- GFAP
- glial fibrillary acidic protein
- HNE
- 4-hydroxy-2-nonenal
- IL
- interleukin
- PBMC
- peripheral blood mononuclear cells
- PON2
- protein paraoxonase-2
- TAT
- thrombin: antithrombin
- TF
- tissue factor
- VWF
- Von Willebrand factor
For Sources of Funding and Disclosures, see page 263–264.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315187.
Contributor Information
Shabbir A. Ansari, Email: shabbir.ansari@uthct.edu.
Shiva Keshava, Email: shivakeshava.gaddam@uthct.edu.
Usha R. Pendurthi, Email: usha.pendurthi@uthct.edu.
References
- 1.Rapaport SI, Rao LV. The tissue factor pathway: how it has become a “prima ballerina”. Thromb Haemost. 1995;74:7–17 [PubMed] [Google Scholar]
- 2.Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:709–725. doi: 10.1161/ATVBAHA.117.309846 [DOI] [PubMed] [Google Scholar]
- 3.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–1097 [PMC free article] [PubMed] [Google Scholar]
- 4.Fleck RA, Rao LV, Rapaport SI, Varki N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;59:421–437. doi: 10.1016/0049-3848(90)90148-6 [DOI] [PubMed] [Google Scholar]
- 5.Contrino J, Hair G, Kreutzer DL, Rickles FR. In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med. 1996;2:209–215. doi: 10.1038/nm0296-209 [DOI] [PubMed] [Google Scholar]
- 6.Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. 1983;49:5–7 [PubMed] [Google Scholar]
- 7.Osterud B, Bjorklid E. Tissue factor in blood cells and endothelial cells. Front Biosci (Elite Ed). 2012;4:289–299. doi: 10.2741/376 [DOI] [PubMed] [Google Scholar]
- 8.Drake TA, Cheng J, Chang A, Taylor FB., Jr. Expression of tissue factor, thrombomodulin, and E-selectin in baboons with lethal Escherichia coli sepsis. Am J Pathol. 1993;142:1458–1470 [PMC free article] [PubMed] [Google Scholar]
- 9.Giesen PL, Rauch U, Bohrmann B, Kling D, Roqué M, Fallon JT, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci USA. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berckmans RJ, Nieuwland R, Böing AN, Romijn FP, Hack CE, Sturk A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost. 2001;85:639–646 [PubMed] [Google Scholar]
- 11.Aras O, Shet A, Bach RR, Hysjulien JL, Slungaard A, Hebbel RP, Escolar G, Jilma B, Key NS. Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood. 2004;103:4545–4553. doi: 10.1182/blood-2003-03-0713 [DOI] [PubMed] [Google Scholar]
- 12.Butenas S, Bouchard BA, Brummel-Ziedins KE, Parhami-Seren B, Mann KG. Tissue factor activity in whole blood. Blood. 2005;105:2764–2770. doi: 10.1182/blood-2004-09-3567 [DOI] [PubMed] [Google Scholar]
- 13.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–461. doi: 10.1161/01.ATV.0000202656.53964.04 [DOI] [PubMed] [Google Scholar]
- 14.Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–1297. doi: 10.1161/CIRCRESAHA.110.233056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, Kuypers FA, Bach RR. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91:4216–4223 [PubMed] [Google Scholar]
- 16.Nieuwland R, Berckmans RJ, McGregor S, Böing AN, Romijn FP, Westendorp RG, Hack CE, Sturk A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95:930–935 [PubMed] [Google Scholar]
- 17.Boden G, Rao AK. Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr Diab Rep. 2007;7:223–227. doi: 10.1007/s11892-007-0035-1 [DOI] [PubMed] [Google Scholar]
- 18.Monroe DM, Key NS. The tissue factor-factor VIIa complex: procoagulant activity, regulation, and multitasking. J Thromb Haemost. 2007;5:1097–1105. doi: 10.1111/j.1538-7836.2007.02435.x [DOI] [PubMed] [Google Scholar]
- 19.Müller I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, Zieseniss S, Zahler S, Preissner K, Engelmann B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17:476–478. doi: 10.1096/fj.02-0574fje [DOI] [PubMed] [Google Scholar]
- 20.Hathcock JJ, Nemerson Y. Platelet deposition inhibits tissue factor activity: in vitro clots are impermeable to factor Xa. Blood. 2004;104:123–127. doi: 10.1182/blood-2003-12-4352 [DOI] [PubMed] [Google Scholar]
- 21.Reinhardt C, von Brühl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, Dlugai S, Hess S, Konrad I, Orschiedt L, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest. 2008;118:1110–1122. doi: 10.1172/JCI32376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang JG, Manly D, Kirchhofer D, Pawlinski R, Mackman N. Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J Thromb Haemost. 2009;7:1092–1098. doi: 10.1111/j.1538-7836.2009.03448.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;382 SupplS26–S34. doi: 10.1097/CCM.0b013e3181c98d21 [DOI] [PubMed] [Google Scholar]
- 24.Date K, Ettelaie C, Maraveyas A. Tissue factor-bearing microparticles and inflammation: a potential mechanism for the development of venous thromboembolism in cancer. J Thromb Haemost. 2017;15:2289–2299. doi: 10.1111/jth.13871 [DOI] [PubMed] [Google Scholar]
- 25.Creasey AA, Chang AC, Feigen L, Wün TC, Taylor FB, Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–2860. doi: 10.1172/JCI116529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor FB, Jr, Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R, Blick K, Edgington TS. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock. 1991;33:127–134 [PubMed] [Google Scholar]
- 27.Taylor FB, Chang AC, Peer G, Li A, Ezban M, Hedner U. Active site inhibited factor VIIa (DEGR VIIa) attenuates the coagulant and interleukin-6 and -8, but not tumor necrosis factor, responses of the baboon to LD100 Escherichia coli. Blood. 1998;91:1609–1615 [PubMed] [Google Scholar]
- 28.Miller DL, Welty-Wolf K, Carraway MS, Ezban M, Ghio A, Suliman H, Piantadosi CA. Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 2002;26:650–658. doi: 10.1165/ajrcmb.26.6.4688 [DOI] [PubMed] [Google Scholar]
- 29.Wiesel P, Patel AP, DiFonzo N, Marria PB, Sim CU, Pellacani A, Maemura K, LeBlanc BW, Marino K, Doerschuk CM, et al. Endotoxin-induced mortality is related to increased oxidative stress and end-organ dysfunction, not refractory hypotension, in heme oxygenase-1-deficient mice. Circulation. 2000;102:3015–3022. doi: 10.1161/01.cir.102.24.3015 [DOI] [PubMed] [Google Scholar]
- 30.Dalleau S, Baradat M, Guéraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20:1615–1630. doi: 10.1038/cdd.2013.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057 [DOI] [PubMed] [Google Scholar]
- 32.Kaneto H, Katakami N, Kawamori D, Miyatsuka T, Sakamoto K, Matsuoka TA, Matsuhisa M, Yamasaki Y. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxid Redox Signal. 2007;9:355–366. doi: 10.1089/ars.2006.1465 [DOI] [PubMed] [Google Scholar]
- 33.Negre-Salvayre A, Vieira O, Escargueil-Blanc I, Salvayre R. Oxidized LDL and 4-hydroxynonenal modulate tyrosine kinase receptor activity. Mol Aspects Med. 2003;24:251–261. doi: 10.1016/s0098-2997(03)00020-7 [DOI] [PubMed] [Google Scholar]
- 34.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson FA, Jr, Spencer FA. Risk factors for venous thromboembolism. Circulation. 2003;10723 suppl 1I9–16. doi: 10.1161/01.CIR.0000078469.07362.E6 [DOI] [PubMed] [Google Scholar]
- 36.Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011;9:120–138. doi: 10.2450/2010.0066-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breitzig M, Bhimineni C, Lockey R, Kolliputi N. 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol. 2016;311:C537–C543. doi: 10.1152/ajpcell.00101.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6 [DOI] [PubMed] [Google Scholar]
- 39.Koster JF, Slee RG, Montfoort A, Lang J, Esterbauer H. Comparison of the inactivation of microsomal glucose-6-phosphatase by in situ lipid peroxidation-derived 4-hydroxynonenal and exogenous 4-hydroxynonenal. Free Radic Res Commun. 1986;1:273–287. doi: 10.3109/10715768609051637 [DOI] [PubMed] [Google Scholar]
- 40.Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxid Redox Signal. 2012;17:1590–1609. doi: 10.1089/ars.2011.4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singhal SS, Singh SP, Singhal P, Horne D, Singhal J, Awasthi S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol Appl Pharmacol. 2015;289:361–370. doi: 10.1016/j.taap.2015.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh P, Parajuli N, Mayeux PR, MacMillan-Crow LA. Renal mitochondrial lipid peroxidation during sepsis. J Kidney. 2016;2:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vatsyayan R, Kothari H, Pendurthi UR, Rao LV. 4-Hydroxy-2-nonenal enhances tissue factor activity in human monocytic cells via p38 mitogen-activated protein kinase activation-dependent phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2013;33:1601–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ansari SA, Pendurthi UR, Rao LVM. The lipid peroxidation product 4-hydroxy-2-nonenal induces tissue factor decryption via ROS generation and the thioredoxin system. Blood Adv. 2017;1:2399–2413. doi: 10.1182/bloodadvances.2017010132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen P, Clark CA, Gopalakrishnan R, Hedner U, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa binding to endothelial cell protein C receptor: differences between mouse and human systems. Thromb Haemost. 2012;107:951–961. doi: 10.1160/TH11-09-0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keshava S, Sundaram J, Rajulapati A, Pendurthi UR, Rao LV. Pharmacological concentrations of recombinant factor VIIa restore hemostasis independent of tissue factor in antibody-induced hemophilia mice. J Thromb Haemost. 2016;14:546–550. doi: 10.1111/jth.13244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ansari SA, Pendurthi UR, Sen P, Rao LV. The role of putative phosphatidylserine-interactive residues of tissue factor on its coagulant activity at the cell surface. PLoS One. 2016;11:e0158377 doi: 10.1371/journal.pone.0158377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kondreddy V, Pendurthi UR, Xu X, Griffin JH, Rao LVM. FVIIa (Factor VIIa) Induces biased cytoprotective signaling in mice through the cleavage of PAR (Protease-Activated Receptor)-1 at canonical Arg41 (Arginine41) site. Arterioscler Thromb Vasc Biol. 2020;40:1275–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood. 2011;117:3199–3208. doi: 10.1182/blood-2010-09-310706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magisetty J, Pendurthi UR, Madhunapantula SV, Grandoni J, Rao LVM. Increased accumulation and retention of rhFVIIa (eptacog beta) in knee joints of hemophilia A mice compared to wild-type mice. Thromb Haemost. 2019;119:1283–1294. doi: 10.1055/s-0039-1688907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parker H, Albrett AM, Kettle AJ, Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol. 2012;91:369–376 [DOI] [PubMed] [Google Scholar]
- 52.Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–2683. doi: 10.1182/blood-2003-03-0693 [DOI] [PubMed] [Google Scholar]
- 53.Day SM, Reeve JL, Pedersen B, Farris DM, Myers DD, Im M, Wakefield TW, Mackman N, Fay WP. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105:192–198. doi: 10.1182/blood-2004-06-2225 [DOI] [PubMed] [Google Scholar]
- 54.MacFadyen JR, Haworth O, Roberston D, Hardie D, Webster MT, Morris HR, Panico M, Sutton-Smith M, Dell A, van der Geer P, et al. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005;579:2569–2575. doi: 10.1016/j.febslet.2005.03.071 [DOI] [PubMed] [Google Scholar]
- 55.Hasanov Z, Ruckdeschel T, König C, Mogler C, Kapel SS, Korn C, Spegg C, Eichwald V, Wieland M, Appak S, et al. Endosialin promotes atherosclerosis through phenotypic remodeling of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2017;37:495–505. doi: 10.1161/ATVBAHA.116.308455 [DOI] [PubMed] [Google Scholar]
- 56.Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, Wu X, Zhang Y, Konkle BA, Thiagarajan P, et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015;125:2151–2159. doi: 10.1182/blood-2014-09-598805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monaghan-Benson E, Burridge K. The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J Biol Chem. 2009;284:25602–25611. doi: 10.1074/jbc.M109.009894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, Collard JG, Hordijk PL. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–1846 [DOI] [PubMed] [Google Scholar]
- 59.Catalá A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids. 2009;157:1–11. doi: 10.1016/j.chemphyslip.2008.09.004 [DOI] [PubMed] [Google Scholar]
- 60.Csala M, Kardon T, Legeza B, Lizák B, Mandl J, Margittai É, Puskás F, Száraz P, Szelényi P, Bánhegyi G. On the role of 4-hydroxynonenal in health and disease. Biochim Biophys Acta. 2015;1852:826–838. doi: 10.1016/j.bbadis.2015.01.015 [DOI] [PubMed] [Google Scholar]
- 61.Ebert J, Wilgenbus P, Teiber JF, Jurk K, Schwierczek K, Döhrmann M, Xia N, Li H, Spiecker L, Ruf W, et al. Paraoxonase-2 regulates coagulation activation through endothelial tissue factor. Blood. 2018;131:2161–2172. doi: 10.1182/blood-2017-09-807040 [DOI] [PubMed] [Google Scholar]
- 62.Park Y, Schoene N, Harris W. Mean platelet volume as an indicator of platelet activation: methodological issues. Platelets. 2002;13:301–306. doi: 10.1080/095371002220148332 [DOI] [PubMed] [Google Scholar]
- 63.Ntolios P, Papanas N, Nena E, Boglou P, Koulelidis A, Tzouvelekis A, Xanthoudaki M, Tsigalou C, Froudarakis ME, Bouros D, et al. Mean platelet volume as a surrogate marker for platelet activation in patients with idiopathic pulmonary fibrosis. Clin Appl Thromb Hemost. 2016;22:346–350. doi: 10.1177/1076029615618023 [DOI] [PubMed] [Google Scholar]
- 64.Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood. 2013;122:1873–1880. doi: 10.1182/blood-2013-04-460139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riewald M, Ruf W. Orchestration of coagulation protease signaling by tissue factor. Trends Cardiovasc Med. 2002;12:149–154. doi: 10.1016/s1050-1738(02)00153-6 [DOI] [PubMed] [Google Scholar]
- 66.Strydom N, Rankin SM. Regulation of circulating neutrophil numbers under homeostasis and in disease. J Innate Immun. 2013;5:304–314. doi: 10.1159/000350282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pawlinski R, Mackman N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb Res. 2010;125Suppl 1S70–S73. doi: 10.1016/j.thromres.2010.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leroyer AS, Isobe H, Lesèche G, Castier Y, Wassef M, Mallat Z, Binder BR, Tedgui A, Boulanger CM. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol. 2007;49:772–777. doi: 10.1016/j.jacc.2006.10.053 [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Miller C, Swarthout RF, Rao M, Mackman N, Taubman MB. Vascular smooth muscle-derived tissue factor is critical for arterial thrombosis after ferric chloride-induced injury. Blood. 2009;113:705–713. doi: 10.1182/blood-2007-05-090944 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.