

Abstract
Background.
Sarco(endo)plasmic reticulum calcium ATPase (SERCA) is regulated by oxidative post-translational modifications at cysteine 674 (C674). Since sarcoplasmic reticulum (SR) calcium has been shown to play a critical role in mediating mitochondrial dysfunction in response to reactive oxygen species (ROS), we hypothesized that SERCA oxidation at C674 would modulate the effects of ROS on mitochondrial calcium and mitochondria-dependent apoptosis in cardiac myocytes.
Methods.
Adult rat ventricular myocytes (ARVM) expressing wild-type (WT) SERCA2b or a redox-insensitive mutant in which C674 is replaced by serine (C674S) were exposed to H2O2 (100 μM). Free mitochondrial calcium concentration was measured in ARVM using a genetically-targeted fluorescent probe and SR calcium content was assessed by measuring caffeine-stimulated release. Mice with heterozygous knock-in of the SERCA C674S mutation (SKI) were subjected to chronic ascending aortic constriction (AAC).
Results.
In ARVM expressing WT SERCA, H2O2 caused a 25% increase in mitochondrial calcium concentration that was associated with a 50% decrease in SR calcium content, both of which were prevented by the ryanodine receptor inhibitor tetracaine. In cells expressing the C674S mutant, basal SR calcium content was decreased by 31% and the H2O2-stimulated rise in mitochondrial calcium concentration was attenuated by 40%. In WT cells, H2O2 caused cytochrome c release and apoptosis, both of which were prevented in C674S-expressing cells. In myocytes from SKI mice, basal SERCA activity and SR calcium content were decreased. To test the effect of C674 oxidation on apoptosis in vivo, SKI mice were subjected to chronic AAC. In WT mice, AAC caused myocyte apoptosis, LV dilation and systolic failure – all of which were inhibited in SKI mice.
Conclusions.
Redox activation of SERCA C674 regulates basal SR calcium content thereby mediating the pathologic ROS-stimulated rise in mitochondrial calcium required for myocyte apoptosis and myocardial failure.
Keywords: Sarco(endo)plasmic reticulum calcium ATPase, sarcoplasmic reticulum, mitochondria, calcium, oxidative protein modification, apoptosis, heart failure, cardiac myocyte
Introduction
Reactive oxygen species (ROS) play a central role in pathologic myocardial remodeling and the progression to heart failure 1, 2. Mitochondria are an important source of ROS in the myocardium 3, 4. Among the targets for mitochondrial ROS are proteins that regulate sarcoplasmic reticulum (SR) calcium, including the ryanodine receptor (RyR) 5, 6 and sarco(endo)plasmic reticulum calcium ATPase (SERCA) 7, 8. Appreciation of the close physical association between SR and mitochondria has led to interest in the mechanisms of cross-talk between these organelles. In mice, overexpression of the β2a subunit of the L-type calcium channel leads to mitochondria-mediated apoptosis of cardiac myocytes that is both dependent on SR calcium and prevented by blockade of the RyR 9. Similarly, in mice with post-myocardial infarction ventricular remodeling, SR calcium leak via RyR causes mitochondrial dysfunction and increased ROS production 10. These observations suggest that SR calcium plays a critical role in mediating mitochondrial dysfunction, and conversely, that mitochondrial ROS may promote increased SR calcium leak, thereby creating a maladaptive positive feedback relationship between these closely-associated organelles.
SERCA, the major protein responsible for calcium uptake into the SR, is regulated by multiple mechanisms 11 including oxidative post-translational modification at cysteine 674 (C674): Reversible S-glutathiolation at C674 activates 12, 13 while irreversible S-sulfonylation inhibits enzyme activity 7, 14. In mice with heart failure due to chronic pressure overload, we found that SERCA C674 is oxidized and cardiac myocyte-specific overexpression of catalase, which protected SERCA C674 from oxidation, prevented myocyte apoptosis and the progression to left ventricular (LV) failure 8.
Given the critical role of SERCA in regulating SR calcium content and the susceptibility of SERCA to regulation by ROS, we hypothesized that SERCA oxidation at C674 would mediate the effects of ROS on mitochondrial calcium and mitochondria-dependent apoptosis in cardiac myocytes. Accordingly, adult rat ventricular myocytes (ARVM) expressing wild-type SERCA2b or a redox-insensitive mutant in which C674 is replaced by serine (C674S) were exposed to ROS to induce apoptosis. Mitochondrial calcium was measured using a genetically-targeted calcium indicator and SR calcium content was assessed using Fura-2AM to measure caffeine-stimulated release. Chronic pressure overload by ascending aortic constriction (AAC) causes myocyte apoptosis and oxidation of C674 8. Therefore, to test the role of C674 oxidation in mediating myocyte apoptosis and pathologic LV remodeling in vivo, heterozygous SERCA knock-in (SKI) mice in which 50% of C674 is replaced by serine 15 were subjected to chronic pressure overload by AAC.
Methods
Declaration.
The authors declare that all supporting data are available within the article and its supplement.
Expression of adenoviral constructs in adult rat ventricular myocytes (ARVM).
Calcium-tolerant ARVM were obtained from hearts of male Sprague-Dawley rats (175 to 200 g, Envigo) as we have described 16 by a protocol that was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine. Mitochondrial calcium concentration was measured using a genetically-encoded Cameleon probe targeted specifically to mitochondria and expressed via adenoviral transduction as we have described 17. Adenoviral constructs encoding WT SERCA2b, C674S SERCA2b and β-galactosidase (LacZ) were generated and expressed using methods we have described 13, 15. Cells infected with wild-type (WT) or C674S SERCA (MOI=10) were used for experiments 48 - 72 hours after infection.
Mitochondrial calcium imaging.
ARVM expressing WT or C674S SERCA and the Cameleon probe plated on 10 mm coverslips in 35 mm dishes (MatTek) were imaged at 37°C using an inverted Olympus Spinning Disk confocal microscope in wide-field mode, as we have described 17. After establishment of a stable baseline (t = 0), 250 μL of media with or without H2O2 to yield a final concentration of 100 μM was added and images were obtained every 20 seconds for 10 minutes. Some cells were pre-treated for 20 minutes with tetracaine (Tet; 100 μM, final concentration) to inhibit RyR 18. Ratiometric images were analyzed using NIS Elements software (Nikon) and calculated as the ratio of YFP / CFP emission after background subtraction.
Caffeine-stimulated SR calcium release.
SR content was assessed by measuring cytosolic calcium concentration (Fura-2AM) following the rapid application of caffeine to release SR calcium, as we previously described 14. Briefly, ARVM on 10 mm cover slips in 35 mm plates were loaded with Fura-2AM for 30 minutes at 37°C and washed thoroughly before addition of 250 uL of buffer or buffer containing H2O2 (100 μM, final concentration). Some plates were pre-treated with tetracaine (20 minutes, 100 μM, final concentration). Images were acquired every second for 90 seconds. Caffeine (20 mM, final concentration) was rapidly applied directly to cells after the first image was acquired. Ratiometric data were analyzed using NIS Elements software (Nikon) and the ratio of emissions at 340/380 nm was calculated after background subtraction.
Cytochrome c release.
Cytochrome c release was measured as we have described 19. ARVM grown on 35 mm plates were exposed to H2O2 (100 μM, final concentration) for 6 hours.
Annexin V.
Annexin V and propidium iodide (PI) staining were measured in ARVM plated on 4-well Nunc™ (Lab-Tek) Chamber slides (ThermoFisher) using an Annexin-V-FLUOS staining kit (Millipore-Sigma), as we have described 16. ARVM were incubated for 24 hours (37°C) in media without or with H2O2 (100 μM, final concentration).
SERCA C674S (SKI) mouse.
The SERCA C674S heterozygous knock-in (SKI) mouse was generated on a C57BL/6J background (Ingenious Targeting Laboratory, Inc., Ronkonkoma, NY), as we have described 15. Homozygous knock-in fetuses died in utero just prior to the initial period of vascular development (8–10.5 days), so only heterozygous adult animals that express 50% of the SERCA C674S allele were studied. In SKI mice, total SERCA expression was normal, whereas there was a 50% decrease in biotinylated iodoacetate-labelled cysteines indicative of the C674S mutation 15. The protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine.
Ascending aortic constriction (AAC).
Constriction of the ascending aorta was performed in 10 week-old WT and SKI mice, as we have described 8. Male mice were used to avoid variations in the severity of stenosis related to body size.
Echocardiographic measurements.
Left ventricular (LV) dimensions and function were measured in non-anesthetized mice shortly before surgery and 12 weeks after surgery using an Acuson Sequoia C-256 echocardiograph machine equipped with a 15 MHz linear transducer (model 15L8), as we have described 8. Measurements and analysis were performed by an investigator blinded to experimental group.
Organ weight and histology.
Mice were sacrificed 12 weeks after surgery. Heart, LV with septum, lung and liver were weighed. LV was fixed in 10% buffered formalin, embedded with paraffin and sectioned. Myocyte cross-sectional area and interstitial fibrosis were assessed as we have described 20.
Mouse myocyte contractility and cytosolic calcium.
Cell shortening and cytosolic calcium were measured in myocytes freshly isolated from WT and SKI mice using techniques we have described 21.
Measurement of myocardial apoptosis and caspase activation in myocardium.
Apoptosis was assessed by terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) using an In Situ Cell Death Detection Fluorescein Kit (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s instructions, as we have described 8, 22. The number of apoptotic nuclei was calculated per 10,000 cardiomyocytes. Myocardial cleaved caspase-3 expression was measured by a rabbit anti-active caspase-3 antibody (Abcam, Cambridge, MA) as described 23, 24.
Statistical analysis.
Results are presented as mean ± SEM. Comparisons between experimental groups were performed using an unpaired t-test or by ANOVA for multiple comparisons with a Bonferroni correction. P<0.05 was considered statistically significant.
Results
ROS increases mitochondrial calcium concentration in ARVM.
To examine the effect of exogenous ROS on mitochondrial calcium we exposed ARVM to 100 μM H2O2, a concentration that we previously used to induce apoptosis in these cells 16. H2O2 caused mitochondrial calcium concentration to increase gradually, achieving a mean increase of 25% at 10 minutes (Figure 1A,B). Pre-treatment with tetracaine (100 μM; 20 minutes) to inhibit SR calcium release via RyR decreased the H2O2-stimulated rise in mitochondrial calcium concentration by 85% (Figure 1C), indicating that a major portion of the increase is mediated by SR calcium release.
Figure 1. H2O2 increases mitochondrial calcium concentration in a ryanodine receptor-dependent manner.
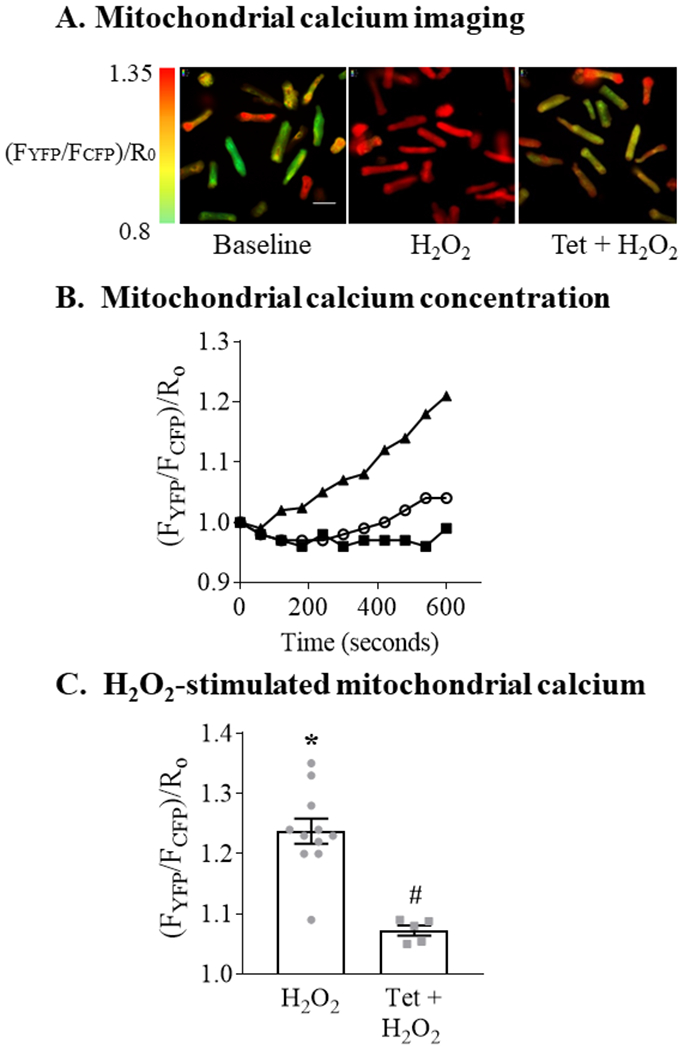
Mitochondrial calcium concentration was measured using a genetically-encoded fluorescent Cameleon indicator targeted to mitochondria. Adult rat ventricular myocytes (ARVM) were exposed to H2O2 (100 μM, final concentration) or control media with or without pretreatment with the ryanodine receptor inhibitor tetracaine (Tet; 100 μM; 20 min). Panel A. Representative images at baseline and 10 minutes after exposure to H2O2 with or without pretreatment with tetracaine. H2O2 was added at time = 0; green represents the lowest mitochondrial calcium concentration and red the highest. Scale bar represents 25 μm. Panel B. Representative experiment depicting mitochondrial calcium concentration over 10 minutes following exposure to H2O2 with (open circles) or without (solid triangles) pretreatment with Tet. Solid squares represent control media. Each time point is the mean of 8-12 cells per condition. Mitochondrial calcium concentration is expressed as the ratio of emission fluorescence (FYFP / FCFP) normalized to the baseline ratio at time = 0 (R0). Panel C. Mean changes in mitochondrial calcium concentration measured 10 minutes after the addition of H2O2 (n = 5 - 11; * = P<0.001 vs. control; # = P <0.001 vs. H2O2; unpaired t-test).
To assess further the role of SR calcium in mediating the ROS-stimulated increase in mitochondrial calcium, we assessed SR calcium content by using Fura-2AM to measure the change in cytosolic calcium concentration due to SR calcium released by a rapid application of caffeine (20 mM). Pretreatment with H2O2 (100 μM; 20 minutes) caused an approximately 50% decrease in caffeine-released SR calcium (Figure 2). Pre-treatment with tetracaine prevented the H2O2-mediated decrease in caffeine-released SR calcium. These findings indicate that the ROS-stimulated increase in mitochondrial calcium concentration is mediated, in large part, by the release of SR calcium via RyR.
Figure 2. H2O2 stimulates SR calcium release via RyR.
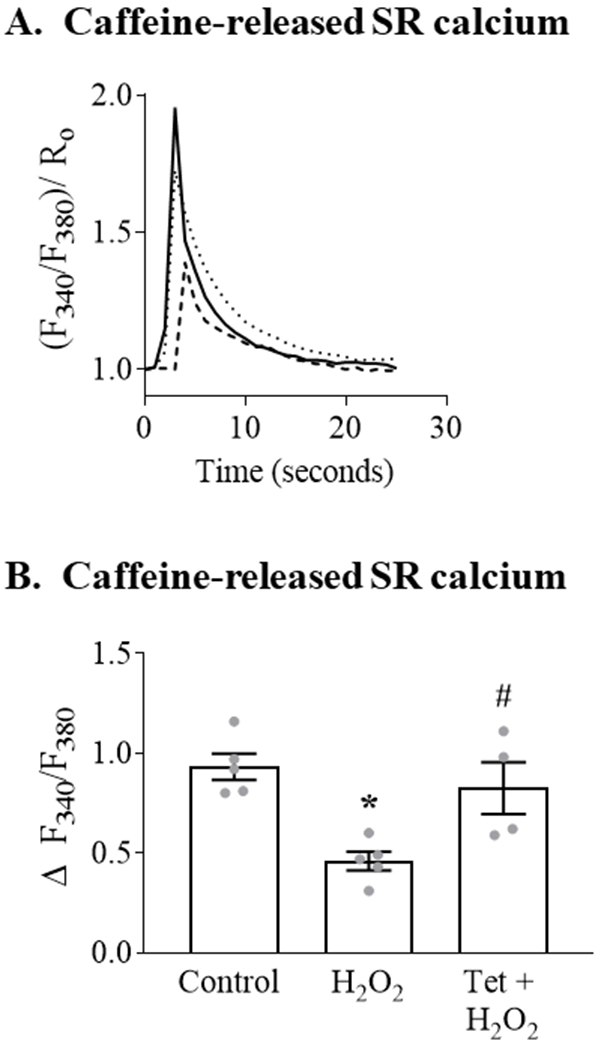
ARVM loaded with the cytosolic calcium indicator Fura 2-AM were incubated with control media (Control) or H2O2 (100 μM; 20 min) with or without Tet pretreatment (100 μM; 20 min) prior to the addition of caffeine (20 mM) to release SR calcium. Panel A. Representative trace showing the caffeine-stimulated increase in cytosolic calcium (F340/F380)/Ro), which is reflective of SR calcium content. Cells were depolarized at t = 0; solid line = control media; dashed line = H2O2; dotted line = Tet + H2O2. Each trace is the mean of 8-12 cells. Panel B. Mean caffeine-stimulated SR calcium release following incubation with H2O2 with or without tetracaine pretreatment (n = 4 - 5, * = P<0.001 vs. Control; # = P< 0.05 vs. H2O2; ANOVA with Bonferroni correction).
The ROS-mediated increase in mitochondrial calcium is attenuated in SERCA C674S-expressing ARVM.
To assess the role of SERCA C674 oxidation in mediating the ROS-stimulated increase in mitochondrial calcium, WT SERCA or the redox-insensitive SERCA C674S mutant were expressed in ARVM. In cells expressing WT SERCA, exposure to H2O2 increased mitochondrial calcium concentration by 20% (Figure 3A). In cells expressing the C674S mutant, basal mitochondrial calcium concentration was decreased by 6% (vs. WT SERCA) and the H2O2-stimulated rise in mitochondrial calcium was attenuated by 40% as compared to cells expressing WT SERCA.
Figure 3. SERCA C674 oxidation modulates basal and H2O2-stimulated mitochondrial calcium concentration and caffeine-stimulated SR calcium release.
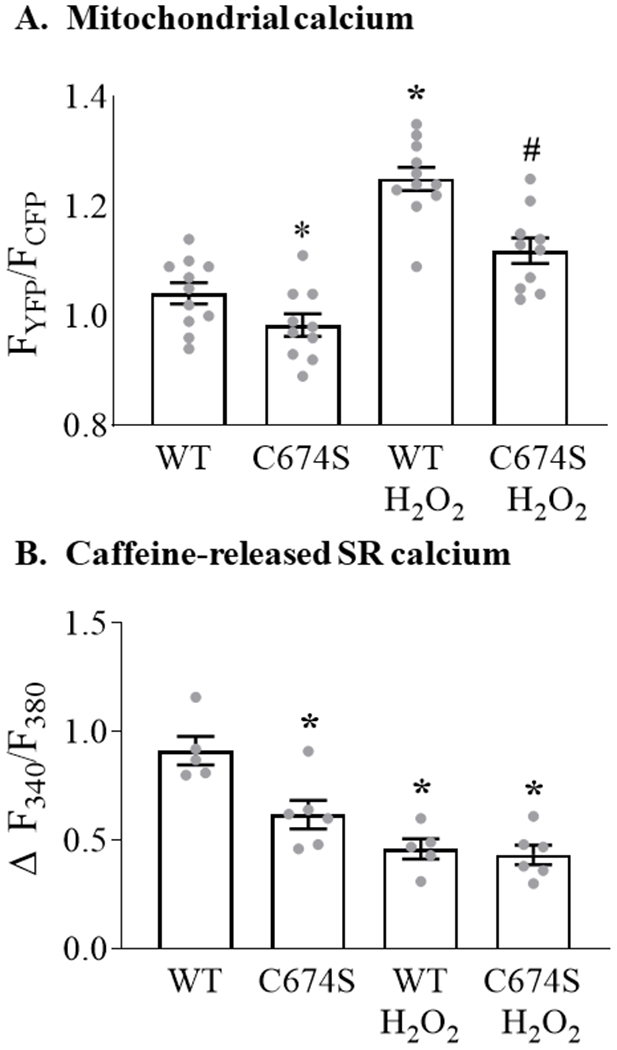
ARVM expressing WT SERCA or the redox-insensitive SERCA C674S mutant were stimulated with H2O2 (100 μM, 10 min) as per Figure 1. Panel A. Mean mitochondrial calcium concentration expressed as the ratio of YFP/CFP fluorescence (n = 10-11; * = P<0.05 vs. WT; # = 0.001 vs. WT / H2O2; ANOVA with Bonferroni correction). Panel B. Change in cytosolic calcium concentration (Fura 2-AM) in response to caffeine in WT and C674S mutant-expressing ARVM with or without pre-incubation with H2O2, expressed as Δ F340/F380 (n = 5 - 6; * = P<0.01 vs. WT; ANOVA with Bonferroni correction).
The observed decreases in basal and H2O2-stimulated mitochondrial calcium in ARVM expressing the C674S mutant raised the possibility that basal SR calcium content is depressed in cells expressing the C674S SERCA mutant. Therefore, SR calcium content was assessed by measuring caffeine-stimulated SR calcium release, as per Figure 2. In cells expressing WT SERCA, H2O2 caused a 49% decrease in caffeine-released SR calcium (Figure 3B). In cells expressing the C674S mutant, basal SR calcium was decreased by 31% vs. WT cells. However, following exposure to H2O2 residual caffeine-released SR calcium was similar in C674S and WT cells. These findings indicate that for cells expressing the SERCA C674S mutant a) the H2O2-stimulated rise in mitochondrial calcium is attenuated, b) SR caffeine-releasable calcium stores are depressed, and c) the magnitude of H2O2-stimulated SR calcium released is diminished.
The SERCA C674S mutation protects ARVM from ROS-induced apoptosis.
In ARVM expressing WT SERCA, exposure to H2O2 caused a 5.5-fold increase in cytochrome c release (Figures 4 A,B). In cells expressing SERCA C674S, the H2O2-stimulated increase was attenuated by 77% vs. WT SERCA.
Figure 4. H2O2-stimulated cytochrome c release and apoptosis are decreased in ARVM expressing the SERCA C674S mutant.
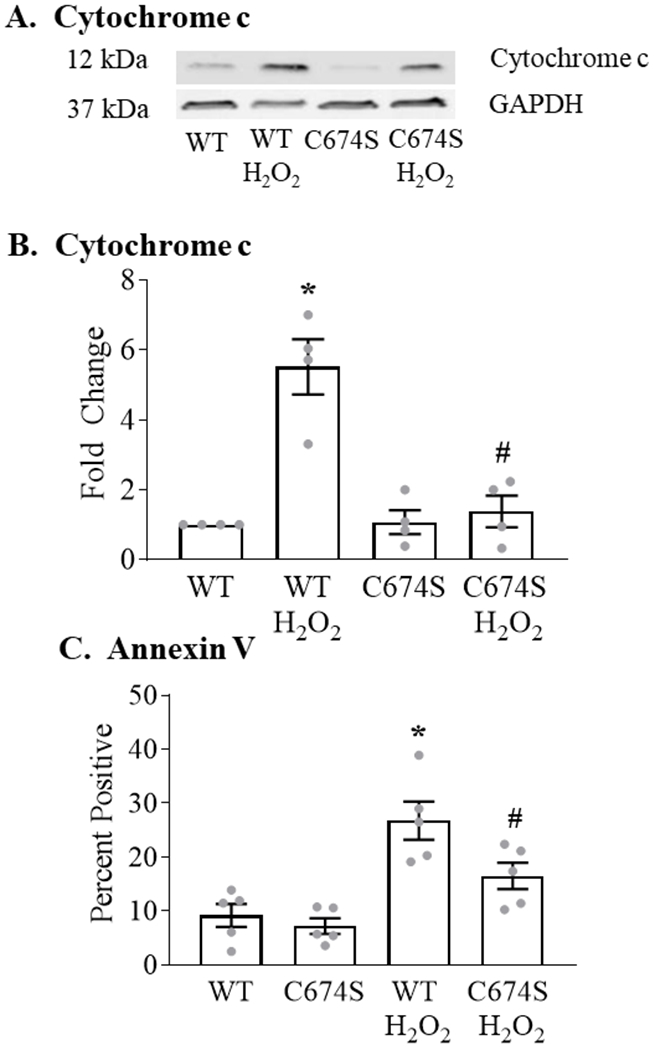
Panel A. Representative Western blot of released cytochrome c measured 6 hours after the addition of H2O2 (100 μM). Panel B. Mean levels of basal and H2O2-stimulated cytochrome c release as per Panel A (n = 4; * = P<0.05 vs. WT; # = 0.05 vs. WT / H2O2; ANOVA with Bonferroni correction). Panel C. Basal and H2O2-stimulated (100 μM; 24 hours) expression of annexin V in ARVM expressing WT or C674S SERCA (n = 4 - 5; * = P<0.01 vs. WT; ANOVA with Bonferroni correction).
We previously showed that annexin V staining, in the absence of an increase in propidium iodide staining, is a marker of ROS-stimulated apoptosis in ARVM 16. In cells expressing WT SERCA, exposure to 100 μM H2O2 for 24 hours caused a 190% increase in annexin V expression (Figure 4C). In C674S-expressing cells, the H2O2-stimulated increase was inhibited by 59%.
Effect of the SERCA C674S mutation on basal cardiac myocyte contractile function and calcium handling (Table I in the Supplement).
To examine further the effects of the SERCA C674S mutation on cardiac myocyte contractile function and calcium handling, ventricular myocytes were isolated from WT mice and heterozygous SERCA knock-in (SKI) mice in which 50% of C674 is replaced by serine 15. In myocytes from SKI mice the extent of basal sarcomere shortening was decreased by 35% vs. cells from WT mice (Figure 5A) and the rate of relaxation, as reflected by myocyte length return velocity, was decreased by 56% (Figure 5B). Likewise, in myocytes from SKI mice the amplitude of the cytosolic calcium transient was decreased by 16% (Figure 5C), due primarily to a decrease in peak systolic calcium (Table I in the Supplement), and the time constant of calcium recovery (Tau), a measure of SERCA activity, was prolonged by 20% (Figure 5D). These findings suggest that the C674S mutation leads to decreases in both SR calcium content (sarcomere shortening, calcium transient) and SERCA activity (relaxation rate, Tau).
Figure 5. Contractile function and calcium transients in freshly isolated cardiac myocytes from WT and SERCA knock-in (SKI) mice in which the C674S mutant replaces 50% of SERCA.
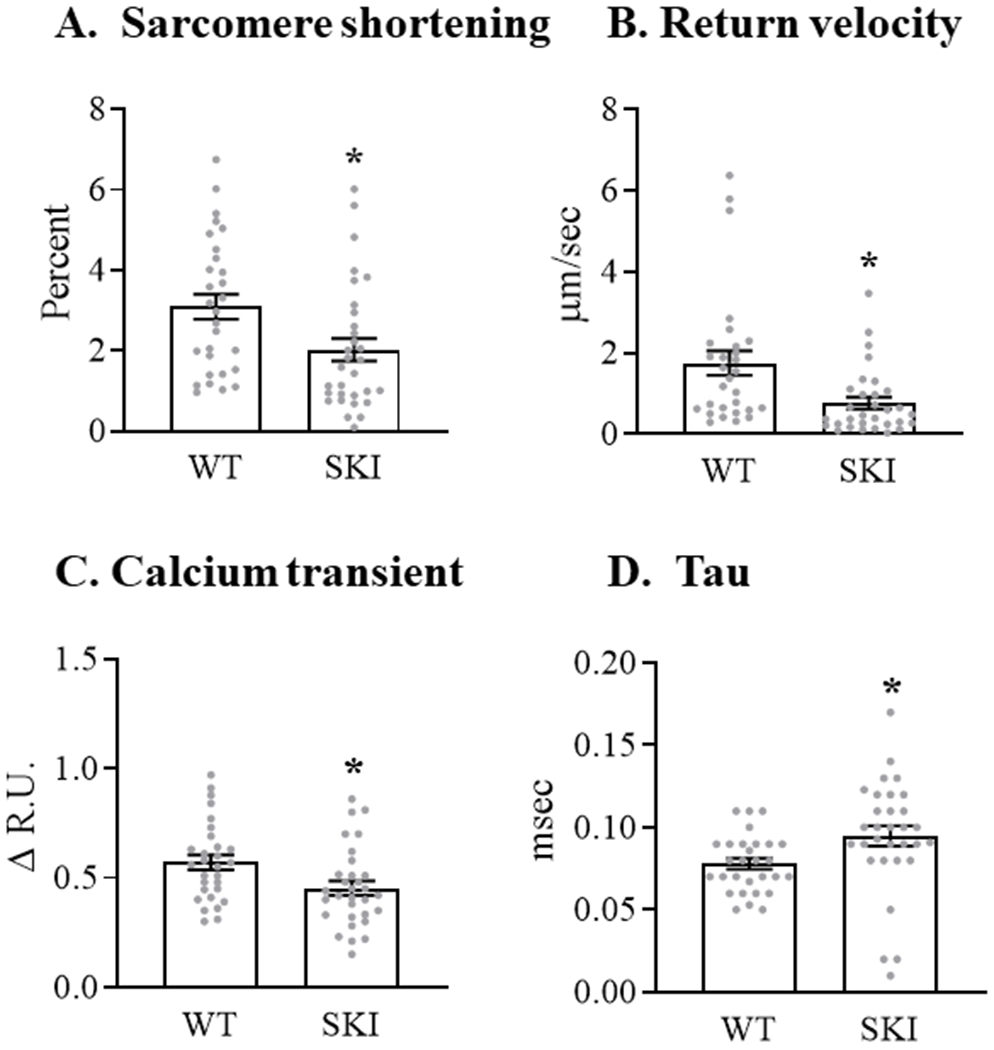
Intracellular calcium concentration was measured at 37°C using Fura-2AM in myocytes paced at 5 Hz. Panel A. Sarcomere shortening. Panel B. Velocity of sarcomere relaxation. Panel C. Amplitude of calcium transient. Panel D. Time constant (Tau) of the resolution of the intracellular calcium transient, a reflection of SERCA activity. For all conditions, data are the means for 29 - 33 myocytes; * = P<0.05 vs. WT; unpaired t-test.
SERCA C674S attenuates LV remodeling and progression to failure in vivo.
To assess the effect of the C674S mutation on the cardiac response to a pathologic oxidative challenge in vivo, SKI mice were subjected to ascending aortic constriction (AAC) for 12 weeks. Heart rate (bpm) was similar in WT (681 ± 11), SKI (651 ± 27), WT-AAC (653 ± 16) and SKI-AAC (668 ± 7) mice (p = ns by ANOVA). In WT mice, LV total wall thickness (septal + posterior wall thickness) measured by echocardiography was increased (Figure 6 A). In SKI mice, the AAC-induced increase in total wall thickness was decreased by approximately 60%. In addition, the AAC-induced increase in heart-to-body weight ratio was attenuated by 76% in SKI mice (Table 1). In WT mice, increases in LV end-diastolic dimension (EDD) (Figure 6 B) and end-systolic dimension (ESD) (Figure 6 C) were associated with a marked decrease in LV fractional shortening (FS) (Figure 6 D) indicative of pathological remodeling. Both LV dilation and the decrease in FS with AAC were prevented in SKI mice. Similarly, the AAC-induced increase in lung-to-body weight ratio, indicative of lung congestion, was abolished in SKI mice (Table 1). Myocyte cross-sectional area was increased in WT mice with AAC, and was attenuated by 82% in SKI mice (Figure I in the Supplement). Likewise, myocardial fibrosis assessed by Masson’s Trichrome staining was increased in WT mice with AAC and was decreased by 94% in SKI mice (Figure II in the Supplement).
Figure 6. LV structure and function in WT and SKI mice subjected to chronic ascending aortic constriction (AAC).
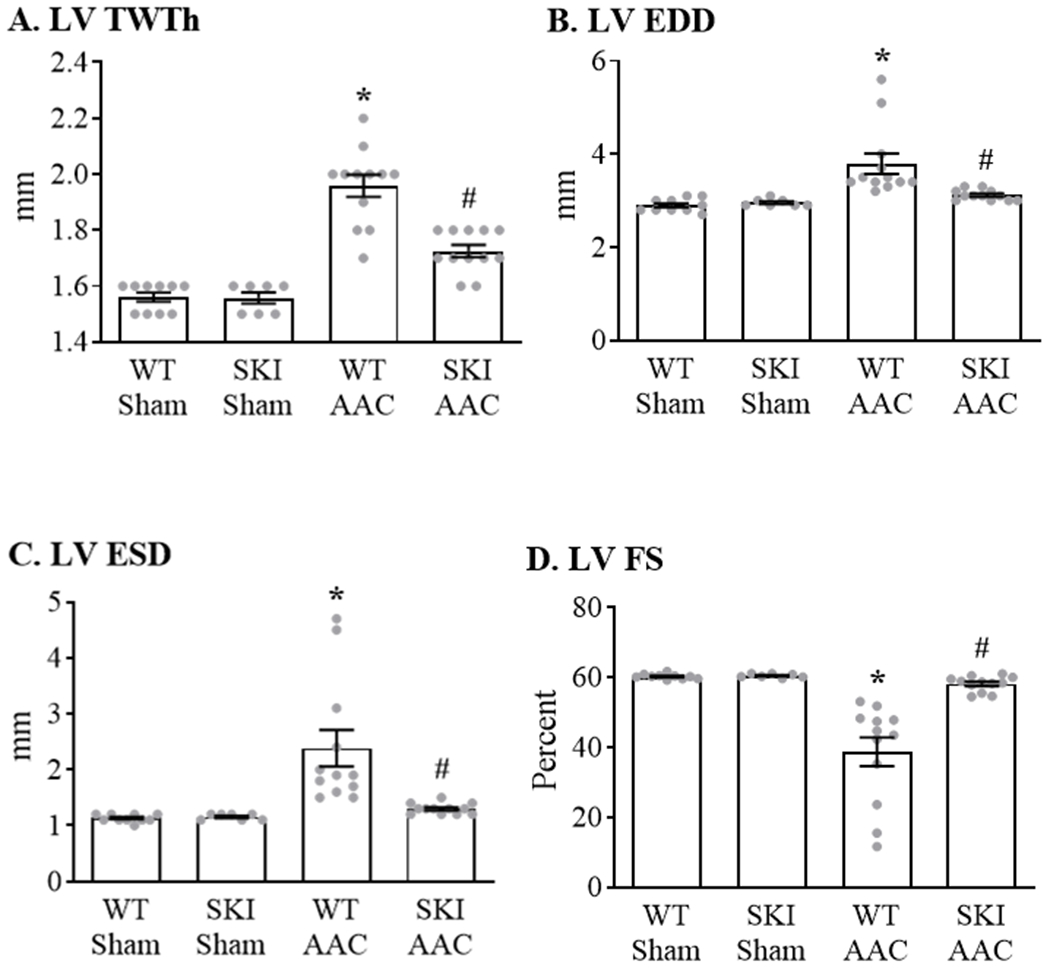
Measurements were made by two-dimensional echocardiography in conscious mice 12 weeks after AAC. Panel A. LV total wall thickness (LV TWTh) calculated as the total of septal and posterior wall thicknesses. Panel B. LV end-diastolic dimension (LV EDD). Panel C. LV end-systolic dimension (LV ESD). Panel D. LV fractional shortening (LV FS). For all panels, n = 7 - 12; * = P<0.02 vs. WT Sham; # = P<0.05 vs. WT AAC; ANOVA with Bonferroni correction.
Table 1.
Body and organ weights
| WT Sham | SKI Sham | WT AAC | SKI AAC | |
|---|---|---|---|---|
| BW (g) | 30.1±1.7 | 32.5±2.4 | 29.3±1.4 | 31.7±1.9 |
| HW (mg) | 135.6±5.6 | 145.7±6.1 | 212.2±16.6* | 169.1±11.3*† |
| HW / BW (mg/g) | 4.59±0.20 | 4.56±0.20 | 7.51±0.72* | 5.37±0.22*† |
| Lung W (mg) | 161.0±4.4 | 154.0±2.1 | 218.3±29.7* | 162.7±3.8† |
| Lung W / BW (mg/g) | 5.54±0.37 | 4.90±0.38 | 8.31±1.25* | 5.36±0.39† |
| Liver W (g) | 1.22±0.15 | 1.33±0.09 | 1.27±0.07 | 1.29±0.09 |
| Liver W / BW (g/g) | 0.041±0.004 | 0.041±0.002 | 0.043±0.002 | 0.041±0.002 |
BW = body weight; HW = heart weight; W = weight; WT = wild-type. SKI = SERCA C674S knock-in mouse; AAC = ascending aortic constriction. Values are mean ± SEM. (n = 7-12;
P < 0.05 vs. WT Sham or SKI Sham
P < 0.05 vs. WT AAC;
ANOVA with Bonferroni correction)
SERCA C674S attenuates myocyte apoptosis and caspase-3 activation in vivo.
Myocardial caspase-3 was cleaved in WT mice subjected to AAC, and the activation was abolished in SKI mice (Figure 7 A; Figure III in the Supplement). Myocyte apoptosis was assessed by measuring TUNEL staining. AAC increased the number of TUNEL-positive myocytes approximately 8-fold in WT mice, whereas the increase was almost completely prevented in SKI mice (Figure 7 B; Figure IV in the Supplement).
Figure 7. Myocyte apoptosis is prevented in SKI mice subjected to chronic AAC.
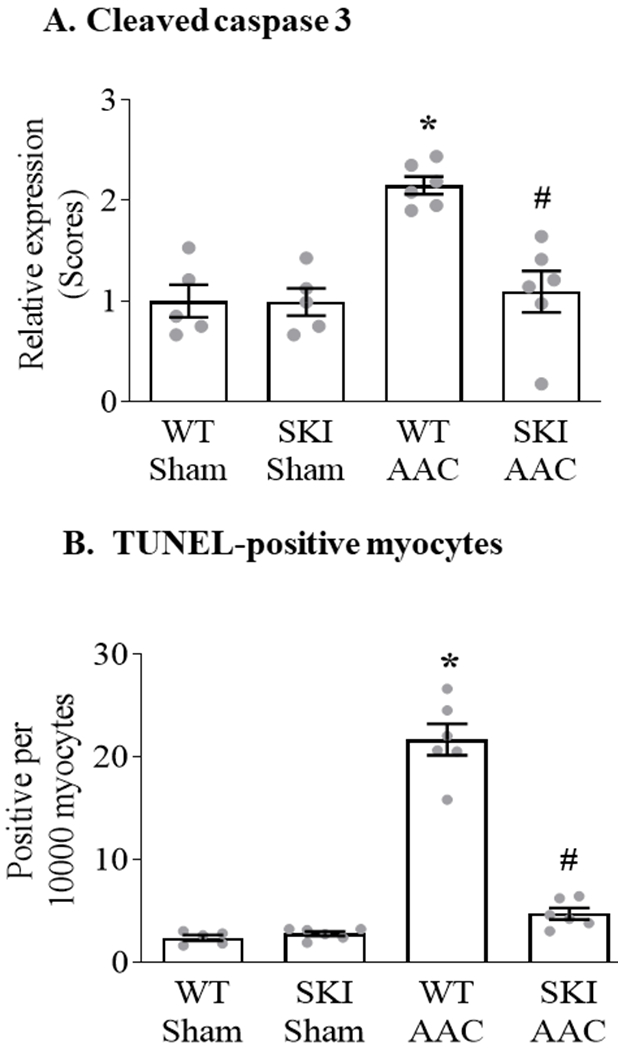
Panel A. Mean change in myocardial cleaved caspase-3 expression 12 weeks after AAC, as assessed by immunohistochemical staining (n = 5 - 6; * = P<0.001 vs. WT Sham; # = P<0.001 vs. WT AAC; ANOVA with Bonferroni correction). Representative images are provided in Figure III in the Supplement. Panel B. Mean number of apoptotic cardiac myocytes per 10,000 cardiac myocytes, as measured by terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) 12 weeks after AAC (n = 5 - 6; * = P<0.001 vs. WT Sham; # = P<0.001 vs. WT AAC; ANOVA with Bonferroni correction). Representative images are provided in Figure IV in the Supplement.
Discussion
Oxidative stress plays a critical role in pathological myocardial remodeling by stimulating multiple cellular events including myocyte apoptosis 1, 2. In mice subjected to chronic pressure overload, we 8 and others 25 have shown that myocyte apoptosis and myocardial failure are prevented by overexpression of the antioxidant catalase. In this model, one of the targets of oxidation is SERCA 8, which plays a dominant role in regulating SR calcium content. Since SR calcium release via the RyR is an important mediator of mitochondrial dysfunction and apoptosis 10, we postulated that oxidation of SERCA plays a role in mitochondrial calcium signaling and apoptosis. To test this possibility in vitro and in vivo we examined an oxidation-resistant mutant of SERCA in cardiac myocytes in culture and in mice subjected to chronic pressure overload. Using a genetically-encoded Cameleon probe targeted to mitochondria 17, we made three novel observations regarding the role of SERCA oxidation in the regulation of SR and mitochondrial calcium in cardiac myocytes. First, under basal in vitro conditions, oxidation of SERCA C674 regulates both SR calcium content and mitochondrial calcium concentration. Second, the regulation of SR calcium content by oxidation of C674 modulates mitochondrial calcium and apoptosis in response to an oxidant challenge. Finally, in mice subjected to chronic hemodynamic overload, SERCA C674 oxidation contributes to SR calcium content and is required for pathological myocardial remodeling.
SR-mitochondrial calcium signaling.
Using a Cameleon probe targeted to mitochondria we observed that exposure to H2O2 caused an increase in mitochondrial calcium that was markedly attenuated by the RyR inhibitor tetracaine, indicating that the majority of the calcium responsible for the increase in mitochondrial calcium was released from the SR via RyR. The increase in mitochondrial calcium presumably occurs due to a local increase in calcium concentration near the mitochondrial calcium uniporter 26. Prior studies have demonstrated that tetracaine-sensitive SR calcium release increases mitochondrial calcium. For example, in adult feline myocytes overexpression of the L-type calcium channel β2a subunit causes SR calcium overload that is associated with increased mitochondrial calcium and apoptosis 9. Similarly, mice expressing a leaky mutant RyR develop mitochondrial calcium overload and activation of the mitochondrial apoptosis pathway 10.
SERCA C674 oxidation regulates SR calcium content and mitochondrial calcium concentration.
Oxidative modification of SERCA can activate or inhibit the enzyme 13, 14. The finding that caffeine-stimulated SR calcium release and mitochondrial calcium concentration were decreased in ARVM expressing redox-insensitive SERCA C674S suggests that under basal conditions, the endogenous level of oxidants sufficiently activates SERCA to maintain the normal SR calcium level. While it is likely that, under the conditions of our study, SERCA cysteines in addition to 674 are oxidatively modified, previous studies in ARVM have shown that the C674S mutant abolishes both oxidative glutathiolation and activation of SERCA, indicating that it is the dominant oxidative modification responsible for SERCA activation 13, 14.
To assess further whether endogenous oxidant levels contribute to SR calcium content, we isolated myocytes from SKI mice in which 50% of SERCA is replaced by the C674S mutant used in the in vitro studies. Myocytes from SKI mice exhibited slowing of myocyte relaxation and prolongation of the time constant for calcium recovery, suggesting a decrease in SERCA activity. Also consistent with the C674S mutant studies in ARVM, sarcomere shortening and the amplitude of the depolarization-stimulated calcium transient were decreased in myocytes from SKI mice, indicating that under physiologic conditions oxidation of SERCA C674 contributes to basal SR calcium content. Despite the observed alterations in calcium handling in myocytes from SKI mice, which are qualitatively similar to those in failing myocytes, LV contractile function as assessed by echocardiography in resting, conscious SKI mice was normal. It is possible that other mechanisms, such as adrenergic tone, are sufficient to maintain adequate SERCA activity under resting conditions, but likely would not support a normal response to an increased work demand.
Mitochondrial calcium and apoptosis are attenuated in C674S-expressing myocytes.
The H2O2-stimulated increases in both SR calcium release and mitochondrial calcium concentration were attenuated in cells expressing the C674S mutant, suggesting that the decrease in basal SR content led to a decrease in the quantity of calcium available for mitochondrial uptake. We previously showed that exposure of ARVM to H2O2 under the conditions used in these studies leads to apoptosis 16. In the present studies, H2O2-stimulated apoptosis, as reflected by cytochrome c release and annexin V expression, was prevented in ARVM expressing the C674S mutant.
Mitochondrial calcium is an essential regulator of mitochondrial function and a well-documented stimulus for cell death in cardiac myocytes 27. In C674S-expressing ARVM, the decrease in SR calcium content available for release in response to an oxidant-stimulated RyR leak is sufficient to attenuate the resulting rise in mitochondrial calcium and myocyte apoptosis. This conclusion complements that of Santulli et al 10 who showed that oxidation of the RyR causes SR calcium leak leading to mitochondrial calcium overload and dysfunction. Our findings indicate that oxidation of SERCA C674 is an important determinant of the quantity of SR calcium that is available for release via the RyR. Taken together, these observations suggest that the coordinated oxidation of SERCA and RyR supports increased SR calcium cycling and a maladaptive increase in mitochondrial calcium leading to apoptotic signaling.
SERCA C674 oxidation regulates pathologic remodeling in mice with chronic pressure overload.
In the SKI mouse, 50% of WT SERCA is replaced by the C674S mutant 15. When subjected to chronic aortic constriction, SKI mice had almost complete prevention of apoptosis and the progression to LV systolic failure. These observations in the SKI mouse indicate that oxidation of SERCA at C674 is necessary for myocyte apoptosis, and thus supports our in vitro observation that SERCA oxidation contributes to SR calcium load and the oxidant-stimulated rise in mitochondrial calcium. By decreasing the SR calcium pool available for release, the C674S mutation in the SKI mouse protects mitochondria from elevated calcium levels. Together, these data are consistent with our prior demonstration that apoptosis and progression to failure are prevented by cardiac myocyte-specific over-expression of peroxisomal catalase, which prevents SERCA oxidation 8.
Limitations.
It should be noted that, in addition to causing SR calcium release via the RyR, H2O2 can activate the mitochondrial calcium uniporter (MCU) directly, which could potentially affect the magnitude of the H2O2-stimulated rise in mitochondrial calcium. However, since MCU activation would likely occur to a similar extent in WT and C674S-expressing cells, it is unlikely to have a qualitative effect on our observations or conclusions. It should also be noted that if there were a change in the sensitivity of the MCU to calcium in C674S mutant, potentially it could modify the magnitude of H2O2-stimulated rise in mitochondrial calcium. Finally, it should be appreciated that SR calcium content was assessed indirectly by measuring the change in cytosolic calcium in response to caffeine, which is subject to potential changes in the uptake of calcium by mitochondria and /or efflux of calcium from the cell.
Implications.
Efforts to treat systolic heart failure have focused on the role of calcium. While an increase in calcium available to the sarcomere may increase myocardial contractile function, alterations in calcium levels in the cardiac myocyte have the potential to promote adverse effects on structure and function. In preclinical studies, the viral expression of SERCA in cardiac myocytes led to improved contractile function 28. However, subsequent studies in patients with severe heart failure were not able to demonstrate a benefit from increased SERCA expression 29. Our findings indicate that, by contributing to SR calcium load, the oxidative activation of SERCA may play a critical role in promoting the adverse effects of chronic hemodynamic overload on pathologic remodeling, and raise the possibility that the expression of a redox-insensitive form of SERCA may be of value in the treatment of heart failure.
Supplementary Material
Clinical Perspective.
What’s New
We tested the hypothesis that sarco(endo)plasmic reticulum calcium ATPase (SERCA), a major regulator of calcium homeostasis in the heart, plays a critical role in mediating mitochondrial calcium and mitochondria-dependent apoptosis.
In adult rat ventricular myocytes expressing an oxidation-resistant mutant of SERCA in which cysteine 674 is replaced by serine (C674S), mitochondrial calcium and the rise in mitochondrial calcium with exposure to an oxidant were decreased, as was apoptotic myocyte death by mitochondrial pathways.
Mice with the same SERCA (C674S) mutation were protected from adverse cardiac remodeling, apoptosis and progression to heart failure following chronic aortic constriction.
What Are the Clinical Implications?
Although the viral expression of SERCA in cardiac myocytes led to improved contractile function in pre-clinical studies, subsequent studies in patients with severe heart failure were not able to demonstrate a benefit from increased SERCA expression.
Our findings indicate that by contributing to sarcoplasmic reticulum calcium load the chronic oxidative activation of SERCA may play a critical role in promoting the adverse effects of hemodynamic overload leading to pathologic remodeling.
These findings illustrate the importance of post-translational modifications of SERCA, and raise the possibility that the expression of a redox-insensitive form of SERCA may be of value in the treatment of heart failure.
Acknowledgements
We would like to acknowledge Dr. Michael Kirber, Director of the Cellular Imaging Core at Boston University School of Medicine, for his expert guidance.
Sources of Funding
This work was supported by NIH grants HL-064750 (WSC), HL-031607 (RAC), F31-HL-144060 (JBG), K08-HL-123744 (MP), R01 DK103750 (MMB), AHA grant 16GRNT27660006 (MMB) and Fellow-to-Faculty Award 15FTF25890062 (IL), and the NHLBI-sponsored Boston University Cardiovascular Proteomics Center (Contract No. N01-HV-28178, RAC and WSC).
Non-standard Abbreviations and Acronyms
- AAC
ascending aortic constriction
- ARVM
adult rat ventricular myocyte
- EDD
end-diastolic dimension
- ESD
end-systolic dimension
- FS
fractional shortening
- LacZ
β-galactosidase
- LV
left ventricular
- MCU
mitochondrial calcium uniporter
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum calcium ATPase
- SKI
SERCA knock-in
- SR
sarcoplasmic reticulum
- Tet
tetracaine
- TWTh
total wall thickness
- WT
wild type
Footnotes
Disclosures
None.
References
- (1).Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol 2002;34:379–388. [DOI] [PubMed] [Google Scholar]
- (2).Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007;49:241–248. [DOI] [PubMed] [Google Scholar]
- (3).Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, Kang YJ, Prolla TA, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011;108:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sverdlov AL, Elezaby A, Qin F, Behring JB, Luptak I, Calamaras TD, Siwik DA, Miller EJ, Liesa M, Shirihai OS, et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J Am Heart Assoc 2016;5:e002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 2008;103:1466–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dridi H, Yehya M, Barsotti R, Reiken S, Angebault C, Jung B, Jaber S, Marks AR, Lacampagne A, Matecki S. Mitochondrial oxidative stress induces leaky ryanodine receptor during mechanical ventilation. Free Radic Biol Med 2020;146:383–391. [DOI] [PubMed] [Google Scholar]
- (7).Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res 2010;107:228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Qin F, Siwik DA, Pimentel DR, Morgan RJ, Biolo A, Tu VH, Kang YJ, Cohen RA, Colucci WS. Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload. Am J Physiol Heart Circ Physiol 2014;306:H1453–H1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res 2005;97:1009–1017. [DOI] [PubMed] [Google Scholar]
- (10).Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A 2015;112:11389–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res 2012;110:1646–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 2004;10:1200–1207. [DOI] [PubMed] [Google Scholar]
- (13).Lancel S, Zhang J, Evangelista A, Trucillo MP, Tong X, Siwik DA, Cohen RA, Colucci WS. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ Res 2009;104:720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kuster GM, Lancel S, Zhang J, Communal C, Trucillo MP, Lim CC, Pfister O, Weinberg EO, Cohen RA, Liao R, et al. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic Biol Med 2010;48:1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Thompson MD, Mei Y, Weisbrod RM, Silver M, Shukla PC, Bolotina VM, Cohen RA, Tong X. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J Biol Chem 2014;289:19907–19916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol 2003;35:615–621. [DOI] [PubMed] [Google Scholar]
- (17).Luptak I, Morgan R, Baka T, Croteau D, Moverman D, Sarnak H, Kirber M, Bachschmid MM, Colucci WS, Pimentel DR. Genetically targeted fluorescent probes reveal dynamic calcium responses to adrenergic signaling in multiple cardiomyocyte compartments. Int J Biochem Cell Biol 2019;114:105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gyorke S, Lukyanenko V, Gyorke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol 1997;500 (Pt 2):297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res 2003;92:136–138. [DOI] [PubMed] [Google Scholar]
- (20).Qin F, Siwik DA, Lancel S, Zhang J, Kuster GM, Luptak I, Wang L, Tong X, Kang YJ, Cohen RA, et al. Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J Am Heart Assoc 2013;2:e000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Morse JC, Huang J, Khona N, Miller EJ, Siwik DA, Colucci WS, Hobai IA. Up-regulation of intracellular calcium handling underlies the recovery of endotoxemic cardiomyopathy in mice. Anesthesiology 2017;126:1125–1138. [DOI] [PubMed] [Google Scholar]
- (22).Qin F, Lennon-Edwards S, Lancel S, Biolo A, Siwik DA, Pimentel DR, Dorn GW, Kang YJ, Colucci WS. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ Heart Fail 2010;3:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 2002;51:1938–1948. [DOI] [PubMed] [Google Scholar]
- (24).Moorjani N, Westaby S, Narula J, Catarino PA, Brittin R, Kemp TJ, Narula N, Sugden PH. Effects of left ventricular volume overload on mitochondrial and death-receptor-mediated apoptotic pathways in the transition to heart failure. Am J Cardiol 2009;103:1261–1268. [DOI] [PubMed] [Google Scholar]
- (25).Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 2012;93:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, et al. Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol Cell 2017;65:1014–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Baumgartner HK, Gerasimenko JV, Thorne C, Ferdek P, Pozzan T, Tepikin AV, Petersen OH, Sutton R, Watson AJ, Gerasimenko OV. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem 2009;284:20796–20803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, et al. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest 1997;100:380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011;124:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.