

Abstract
SARS‐CoV‐2 coronavirus has been recognized the causative agent of the recent and ongoing pandemic. Effective and specific antiviral agents or vaccines are still missing, despite a large plethora of compounds have been proposed and tested worldwide. New compounds are requested urgently and virtual screening can offer fast and robust predictions to investigate. Moreover, natural compounds were shown to exert antiviral effects and can be endowed with limited side effects and wide availability. Our approach consisted in the validation of a docking protocol able to refine the most suitable candidates, within the 31000 natural compounds of the natural product activity and species source (NPASS) library, interacting with the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) spike glycoprotein. After the refinement process two natural compounds, castanospermine and karuquinone B, were shown to be the best‐in‐class derivatives in silico able to target an essential structure of the virus and to act in the early stage of infection.
Keywords: Castanospermine, coronavirus, COVID-19, karuquinone B, library screening, spike glycoprotein
We identified from a database, specialized in natural products, Karuquinone B and Castanospermine as blockers of SARS‐CoV‐2 spike glycoprotein in silico. First, we validated a docking protocol able to refine the most suitable compounds. Then, we studied by molecular dynamics the interactions between the two compounds and the biological target. The two compounds were also characterized in terms of botanical and chemical characteristics, and pharmacokinetic parameters in silico.
1. Introduction
Coronaviruses usually interact with the respiratory tract of humans leading to infections of the related organs and tissues, being the symptoms mild to severe. In the past years, severe acute respiratory syndrome (SARS‐CoV) and Middle East respiratory syndrome (MERS‐CoV) coronaviruses have spread to epidemics with high morbidity and mortality rate.[ 1 , 2 ] Recently, the World Health Organization has also defined as pandemic the COVID‐19 (caused by SARS‐CoV‐2) outbreak following the infection of more than 2 million people. [3] Its genome is characterized by a single strand RNA also acting as a messenger RNA and useful for the synthesis of replication/transcription complex, two proteases and several structural proteins. [4] Exploration of these proteins and their biological interactions by means of an integrated bioinformatics approach have led to the assay of old and new compounds with putative antiviral activity. [5] Furthermore, due to the role exerted by angiotensin‐converting enzyme 2 (ACE2) receptor in lung type 2 alveolar cells for the virus‐cell identification and entry, a detailed structural and comparative analysis of SARS‐CoV‐2 spike glycoproteins (S‐glycoproteins) has been conducted. [6]
The spike glycoprotein of SARS‐CoV‐2 is composed of two subunits (S1 and S2). The former possesses the receptor binding domain (RBD) which has high affinity to ACE2 receptor, whereas S2 stimulates the membrane fusion leading to the virus release into the host cell. Their role and modifications shed new light on the switch of infection from animals to humans, the mortality associated to this new coronavirus and the therapeutic failure of drugs and vaccine licensed for other viral infections. Most importantly, these viral targets were also involved for the development of monoclonal antibodies and vaccines due to their immune stimulating properties.[ 6 , 7 , 8 , 9 , 10 ]
Starting from these premises and keeping in mind that researchers have been screening the antiviral potency and druggability of natural compounds toward different viral proteins,[ 11 , 12 , 13 , 14 ] drug repurposing strategies with in silico design also have recently gained considerable attention on the drugs against the SARS‐CoV‐2.[ 15 , 16 ] Our aim was the identification of naturally occurring molecules able to interact with COVID spike glycoproteins by performing an in silico virtual screening of a large natural product data set (NPASS, natural product activity and species source) containing 31000 compounds belonging to different chemical scaffolds. The fingerprint of glycoprotein‐compound interactions could provide clues on their putative orientations. The novel compounds reported in the study can be further assayed for their antiviral activity for a further development into drugs.
2. Results and discussion
2.1. Active site prediction and virtual screening
Before screening NPASS database, the docking protocol was validated. Since the protein was devoid of any co‐crystallized ligand, we used site map tool available in the Schrödinger suite for the identification of potential binding site to define the binding pocket. Based on the identified site points, a grid box was created which in turn was used for grid generation. The results from sitemap are shown in Table 1 and indicate that the site‐1 was having the highest ‘site score’ and ‘D score’.
Table 1.
Details of the active site of spike protein.
|
Name of Site |
Site score |
D score |
Size |
Volume |
|---|---|---|---|---|
|
Site 1 |
1.061 |
1.091 |
93 |
273.714 |
|
Site 2 |
0.860 |
0.890 |
75 |
158.466 |
|
Site 3 |
0.804 |
0.802 |
54 |
107.359 |
|
Site 4 |
0.777 |
0.783 |
62 |
120.050 |
|
Site 5 |
0.702 |
0.691 |
43 |
150.920 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
We performed the study considering site‐1 as the potential binding site which indicates that these molecules were able to occupy the site as predicted by site map (Figure 1). There was no literature available to validate the same because from the best of our knowledge there is no protein structure for spike protein with a bound ligand available in RCSBPDB website till date.
Figure 1.
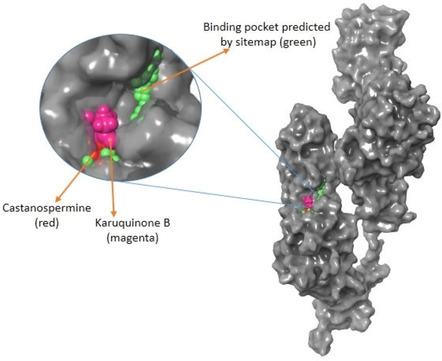
Active site prediction. The entire portion indicated in green ball is the active site predicted by site map and the one shown in magenta and red represents Karuquinone B and Castanospermine, respectively.
In the first attempt, Glide was applied in high‐throughput virtual screen mode, the top 10 % of the compounds were retained; hence about 3100 molecules were picked up after HTVS docking. This procedure allowed to go to the next stage, Glide Single Precision (SP). The criteria set was to pick up the top 10 %. The retrieved hits were narrowed down to 350 molecules and these were retained and docked using Glide Extra Precision (XP) mode keeping the final output to 10 %. Two compounds with better binding affinity were identified based on docking results and visual inspection of ligand. The sequential virtual screening explored in this study is visually described in the flowchart in Figure 2. To further extent our study we performed a structure‐based virtual screening against the same target using a natural product database. The two most active hit compounds NPC472047 (Karuquinone B) and NPC126664 (Castanospermine) were identified from this study.
Figure 2.
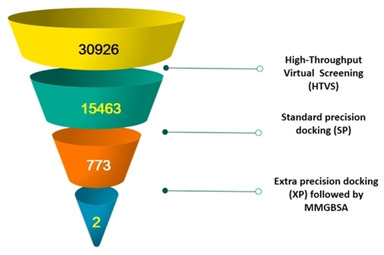
Virtual screening of NPASS database of the current study.
2.2. Binding of CTN and KQB disrupts conformational stability of SARS‐CoV‐2 S‐protein
To understand the inhibitory effects of the two natural compounds on the target protein, we first estimated the Cα RMSD which measures variations in motions of the backbone atoms. Findings revealed that, while atomistic deviation was lower in the unbound S‐protein, the bound proteins exhibited high RMSDs which could indicate the disruptive activities of the compounds on the protein secondary structure. Overall, CTN‐bound S‐protein had the highest mean RMSD, but KQB induced high Cα motions from 35 ns until the end of the simulation time (Figure 3).
Figure 3.
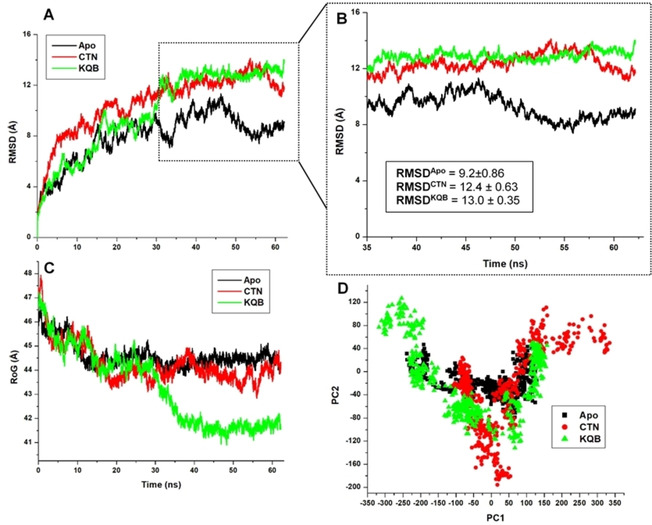
Estimations of structural dynamics and changes that occurred along the MD simulation trajectories among the unbound (black), CTN‐bound (red) and KQB‐bound (green) SARS‐CoV‐2 S‐protein. A. RMSD calculations, while B focuses on the terminal timeframes from 35–65 ns. C. RoG calculations and D. PCA calculations.
Mean RMSD values estimated in Table 2 reveal conformational variations across the three systems. We further investigated the relative structural motions of the proteins using the RoG metrics which compared their structural compactness across the simulation period.
Table 2.
Analysis showing estimations of variations that occurred across the protein structures.
|
Whole structure variations | |||
|---|---|---|---|
|
Structural analyses (Å) |
Apo |
CTN |
KQB |
|
Overall RMSD |
7.9±1.9 |
10.8±2.2 |
10.2±3.2 |
|
RMSD (35‐65 ns) |
9.2±0.86 |
12.4±0.63 |
13.0±0.35 |
|
RoG |
44.4±0.2 |
43.9±0.38 |
43.2±1.57 |
|
RMSF |
3.3±1.5 |
4.0±2.4 |
4.2±2.9 |
|
Ligand binding dynamics | |||
|
RMSD |
|
0.44±0.13 |
0.92±0.17 |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Corroboratively, KQB‐bound S‐protein was highly compact compared to the CTN‐bound and unbound S‐protein. Interestingly, this structural distinctness was more prominent from about 35 ns of the MD trajectory in line with distinct structural motions earlier estimated by the RMSD metrics in the following order: KQB > CTN > Apo. The PCA was also used to capture the motions of the bound and unbound proteins across the conformational phase space as shown in Figure 3. This revealed that while the unbound protein had a stable motion, motions in the bound proteins were more scattered, indicative of a disrupted motion pattern. Per‐residue motion analyses using the RMSF parameter revealed that overall residue fluctuation was the highest in the KQB‐bound system followed by CTN and then by the unbound system which had the least fluctuation (KQB>CTN>Apo).
Notably, structural fluctuation in the KQB‐bound system was the most prominent at regions 300–500 which constitute the S‐protein RBD as widely reported.[ 4 , 5 ] Presumably, conformational perturbations at the RBD as derived by the binding of the compounds could affect interactions with the host hACE2, which could in turn disrupt the viral entry mechanisms (Figure 4). This mechanistic disruption at the RBD could present an allosteric targeting strategy for preventing SARS‐CoV‐2 infectivity as mediated by the S‐protein. Distinct structural variations were also observed at regions 1110–1170 which aligns with the heptad repeat 2 and transmembrane domain.
Figure 4.
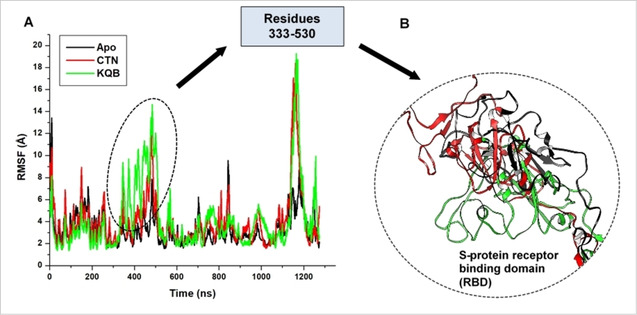
A. Comparative Cα‐RMSF plot showing the degree of fluctuations among individual residues in unbound (black), CTN‐bound (red) and KQB‐bound (green) SARS‐CoV‐2 S‐protein. B. Structural superposition of unbound (black), CTN‐bound (red) and KQB‐bound (green) receptor binding domain of SARS‐CoV‐2 S‐protein.
2.3. Relative ligand binding dynamics and affinities
Furthermore, to better understand the binding activities of the compounds, we investigated their dynamics at the predicted binding site of this target protein. This was essential to understand the relative affinities of the compounds. Firstly, we measured the relative motions of the compounds over the simulation period using the Cα‐RMSD plot. As shown, KQB exhibited higher motions at the target site relative to CTN with mean RMSDs in the KQB > CTN order as presented in Table 2. Visual structural analyses further revealed the differential positioning of the compounds relative to their binding the target site (Figure 5).
Figure 5.
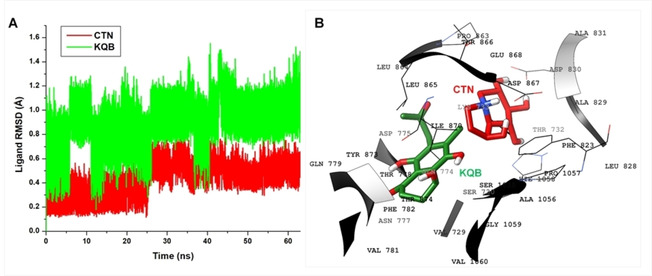
A. Cα‐RMSD plot showing comparative analysis of the motions of CTN (red) and KQB (green) at the predicted target site; B. 3D structural representation of the orientations of CTN (red) and KQB (green) as seen in the ultimate snapshot.
Time‐based interaction analyses were then used to monitor the relative binding patterns of the compounds, which also showed the dynamics of complementary interactions as it occurred with constituent residues of the target site.
Crucial to the binding of CTN was the attractive charge and salt bridge interaction (N−O) that occurred between ASP867 and the N atom of the indolizine ring (Figure 6). This is in addition to the strong H‐bonds (OH–O) mediated by the tetrahydro group of CTN with HIE1058, ASP830, ASP867. We could also deduce that the occurrence of these strong interactions held CTN in place and could account for its relatively lower motion compared to KQB.
Figure 6.
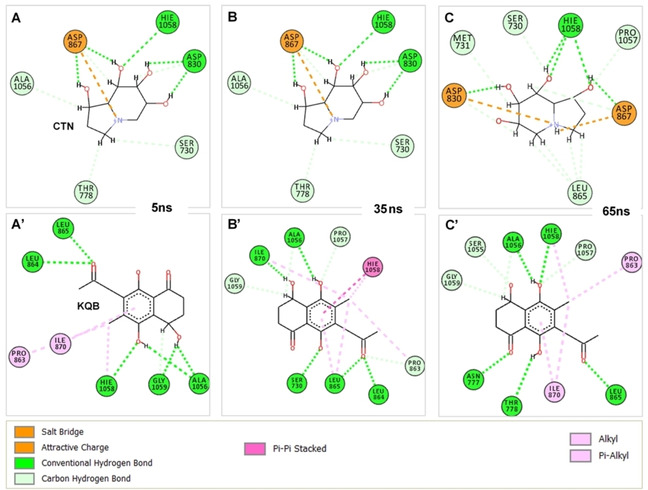
Time‐based interaction dynamics of compounds CTN and KQB at the predicted site of SARS‐CoV‐2 S‐protein. Residues involved in ligand interactions coupled with interaction types are also shown. Panels A‐C show binding dynamics of CTN at 10 ns, 30 ns and 60 ns, while panels A’‐C’ show binding dynamics of KQB at 10 ns, 30 ns and 60 ns.
Non‐conventional H‐bonds (CH−O) were also observed occurring with ALA1056, PRO1057, THR778, LEU865, MET731 and SER730. These interactions were steady across the simulation trajectory. Hydrogen interactions involving S730, N777, T778, LEU864, LEU865, ILE870, ALA1056, HIS1058, GLY1059 and ALA1056, majorly accounted for the high‐affinity binding of KQB at the target site. Moreover, aromatic interactions with the benzene‐1,4‐diol group were observed for ILE870, PRO863 and HIE1058 further contributing to the stability of the compound. Moreover, we measured the binding affinities of the compounds using the MM/PBSA method while per‐residue energy decomposition further showed energy contributions of target site residues to ligand binding. Findings revealed that KQB had a ΔGbind value of −34.80 kcal/mol, while CTN had an estimate of −37.42 kcal/mol (Table 3).
Table 3.
Binding free energy calculations for KQB and CTN.
|
Complexes |
KQB |
CTN |
|---|---|---|
|
ΔEvdw [a] (kcal/mol) |
−40.75±0.19 |
−17.94±0.26 |
|
ΔEele [b] (kcal/mol) |
−16.28±0.34 |
−194.0±0.85 |
|
ΔEGB [c] (kcal/mol) |
27.0±0.23 |
178.1±0.74 |
|
ΔESA [d] (kcal/mol) |
−4.75±0.02 |
−3.6±0.01 |
|
ΔGgas [e] (kcal/mol) |
−57.03±0.38 |
−211.9±0.79 |
|
ΔGsol [f] (kcal/mol) |
22.24±0.22 |
174.5±0.73 |
|
ΔGbind [g] (kcal/mol) |
−34.80±0.26 |
−37.42±0.39 |
[a] ΔEvdW=van der Waals energy; [b] ΔEele=electrostatic energy; [c] ΔEGB=polar desolvation energy; [d] ΔESA non‐polar solvation energy.; [e] ΔGgas=gas phase energy; [f] ΔGsol=solvation energy; [g] ΔGbind=total binding energy.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Presumably, both compounds have similar affinities based on these calculations although higher in CTN which is paradoxical to values obtained from the docking calculations. Also, from our calculations, we could deduce that electrostatic energies majorly contributed to the binding of CTN which could be due to the attractive charges and salt bridge interactions while KQB, on the other hand it was mainly stabilized by van der Waals energies. High ΔEGB for CTN further corroborated that its binding was highly unfavorable in the solvent phase. On the contrary, lower ΔEGB and ΔGsol for KQB suggest its binding contact with the surface region. Taken together, both compounds displayed favorable binding activities at the predicted site of SARS‐CoV‐2 S‐protein.
Energy contributions above −1 kcal/mol were used to establish important residues that can contribute to the binding and stability of CTN and KQB. This is important for future structure‐based drug discovery efforts that aim to achieve the design of highly specific inhibitors of the target protein. From the CTN per‐residue decomposition plot (Figure 7), we observed high electrostatic energy contributions by ASP867, LEU865, ASP830, and ASP775 with respective values of −11.8, −3.9, −8.8 and −4.3 kcal/mol, respectively.
Figure 7.
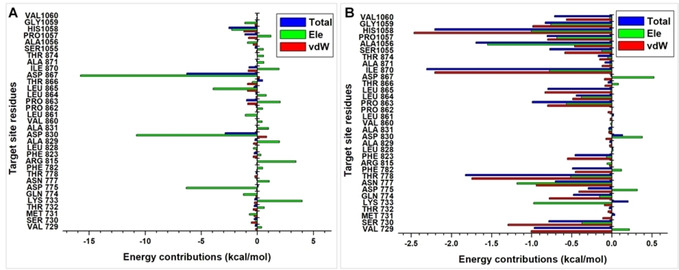
Per‐residue decomposition plots showing energy contributions by constituent residues of the putative S‐protein site. Energy plot for CTN is shown in A, while energy plot for KQB is shown in B.
These energy values further validate the importance of these residues to the binding of CTN. Additional residues above the −1 kcal/mol threshold include HIS1058 (−2.3 kcal/mol), GLN774 (−1.2 kcal/mol), LEU861 (−1.1 kcal/mol), and GLY1059 (−1.1 kcal/mol). ASP830, ASP867, PRO1057 and HIS1058 had total energy values of −2.9, −6.3, −1.1 and −2.5 kcal/mol. All per‐residue vdW energy contributions for CTN at the target site fell below the −1 kcal/mol threshold asides for HIS1058 which had a value of −1.2 kcal/mol. In addition, we observed unfavorable (positive) electrostatic energies for LYS733, ASN777, ARR815, ALA829, ALA831, PRO863, ILE870 and PRO1057, which could be due to steric effects at the protein target site. However, these unfavorable energies were sufficiently compensated by the occurrence of highly negative electrostatic energies.
VAL729, THR778, PRO863, ILE870, ALA1056 and HIS1058 contribute essentially to the binding of KQB with total energy values of −1.0, −1.8, −1.0, −2.3, −1.7 and −2.2 kcal/mol. Moreover, favorable electrostatic energy contributions were observed for LYS733 (−1.0 kcal/mol), ASN777 (‐1.2 kcal/mol), ALA1056 (‐1.6 kcal/mol) and HIS1058 (‐1.0 kcal/mol). High vdW contributions were exhibited by VAL729 (−1.0 kcal/mol), SER730 (−1.3 kcal/mol), THR778 (−1.7 kcal/mol), ILE870 (−2.2 kcal/mol), HIS1058 (−2.5 kcal/mol) and GLY1059 (−1.0 kcal/mol). Unfavorable interactions were highly minimal as they fall below the −1 kcal/mol for contributing residues.
Taken together, the binding of both CTN and KQB were favorable at the putative S‐protein site identified in this study. However, CTN was bound with a higher affinity when compared to KQB due to its ability to form strong attractive charges, salt bridge and H interactions. Nevertheless, the binding of both compounds induced conformational variations in SARS‐CoV‐2 S‐protein which extended to its RBD where interactions with the host hACE2 occur.
2.4. ADME prediction
The physicochemical parameters, and drug likeness of CTN and KQB were evaluated by SwissADME online platform. The compounds have an acceptable pharmacokinetics with satisfied drug‐likeness rules. According to the results (Table 4), both molecules have good/low GI absorption with poor BBB permeation and this agreed with our drug design strategy. [17]
Table 4.
in silico ADME properties for KQB and CTN.
|
Properties |
KQB |
CTN |
|---|---|---|
|
Molecular formula/MW |
C14H16O5/267.2 |
C10H18O3/186.2 |
|
No. H‐bond acceptor |
5 |
3 |
|
No. H‐bond donor |
3 |
3 |
|
LogPO/W |
1.73 |
1.51 |
|
No. rotatable bonds |
2 |
0 |
|
TPSA |
94.83 |
60.69 |
|
LogKP (skin permeation) |
‐7.39 |
−6.96 |
|
Lipinski's rule violation |
No |
No |
|
Bioavailability score |
0.55 |
0.55 |
|
GI absorption |
Yes |
Low |
|
BBB permeation |
No |
No |
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
The online program also shows whether the compounds are suitable for oral administration with the bioavailability radar panel containing physicochemical properties such as flexibility, insolubility, lipophilicity, saturation, size, and polarity and it gives a rapid appraisal of the drug‐likeness of the drug candidates. Both molecules were documented to be within the pink region (optimal ranges) and can be considered endowed with drug‐like characteristics (Figure 8).
Figure 8.
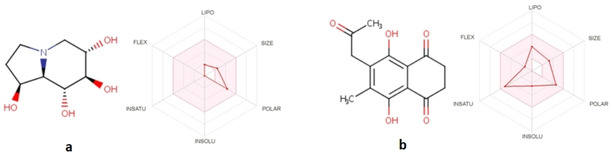
The bioavailability radar of (a) CTN and (b) KQB.
BOILED‐Egg model also predicted the passive human gastrointestinal absorption (HIA) and blood‐brain barrier (BBB) permeation. [18] The BOILED‐Egg construction of CTN and KQB is depicted in Figure 9. The results revealed that KQB has comparatively higher probability of HIA.
Figure 9.
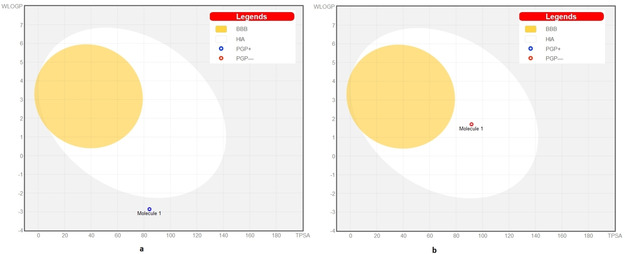
The BOILED‐Egg construction of (a) CTN and (b) KQB.
2.5. Details of the identified compounds
Karuquinone B (4,5,8‐trihydroxy‐6‐methyl‐7‐(2‐oxopropyl)‐3,4‐dihydro‐2H‐naphthalen‐1‐one) is a bioactive dihydronaphthoquinone previously isolated from a culture extract of Fusarium solani, an endophytic organism present on the leaves of Morus alba cultivated in Korea, Japan and China. [19] No information is available regarding its antiviral effects, but only two studies assessed its potential to counteract the glutamate‐mediated cell death in HT22 cells and to exert an antiproliferative action against HUVEC cells. [20] These aspects are important to determine as low the cytotoxicity usually attributed to naphthoquinone compounds.
Conversely and interestingly, castanospermine ((1S,6S,7R,8R,8aR)‐1,2,3,5,6,7,8,8a‐octahydroindolizine‐1,6,7,8‐tetrol), which can be isolated from different plant species (Prunus prostrata, Castanospermum australe, Cassine glauca, Morus bombycis, Richteria pyrethroides) was shown to display antiviral effects against a number of viruses in vitro and in vivo. This indolizidine alkaloid belongs to the class of intestinal α‐glucosidase inhibitors also playing a pivotal role in the inhibition of endoplasmic reticulum (ER)‐resident α‐glucosidases I and II. [21] These enzymes are involved in the trimming of the terminal sugar moieties abundant on the N‐linked glycans at the nascent proteins. This enzymatic reaction is essential for the proper folding and function of the glycoproteins. Fortunately, viral envelope proteins also contain N‐linked glycans, which can be modified, altered or destroyed by these ER α‐glucosidase inhibitors. These biological effects were clearly evident after administration with Castanospermine in the prevention of death in mice infected with flaviviruses and filoviruses, in the suppression of virus multiplication in other infected animals, and in clinical trials against human immunodeficiency, hepatitis C and B, human parainfluenza virus type 3 (HPIV3), Ebola, Zika, and dengue virus.[ 22 , 23 , 24 , 25 , 26 ] This compound seems to be a promising candidate among the large plethora of derivatives tested on SARS‐CoV‐2. [27]
3. Conclusion
In order to suggest new candidates in the struggle against COVID‐19 pandemic, we explored in silico more than 31000 natural compounds for their ability to bind to S‐glycoprotein, thus blocking the early stages of viral infection. Two best‐in‐class compounds, namely Castanospermine and Karuquinone B, were selected on the basis of their binding affinity and pharmacokinetic data. These host function‐targeted, broad‐spectrum antiviral agents would be particularly promising in the management of respiratory tract viral infections and in other medical situations that can be caused by many different enveloped RNA viruses.
Supporting Information Summary
Materials and methods have been carefully described in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are thankful to institute of research and consulting studies at King Khalid University for funding this research through grant no 3‐N‐20/21 and the support of research centre for advanced material science is highly acknowledged.
A. G. Al-Sehemi, F. A. Olotu, S. Dev, M. Pannipara, M. E. Soliman, S. Carradori, B. Mathew, ChemistrySelect 2020, 5, 13309.
Contributor Information
Dr. Simone Carradori, Email: simone.carradori@unich.it.
Dr. Bijo Mathew, Email: bijovilaventgu@gmail.com.
References
- 1. Holshue M. L., DeBolt C., Lindquist S., Lofy K. H., Wiesman J., Bruce H., Spitters C., Ericson K., Wilkerson S., Tural A., Diaz G., Cohn A., Fox L., Patel A., Gerber S. I., Kim L., Tong S., Lu X., Lindstrom S., Pallansch M. A., Weldon W. C., Biggs H. M., Uyeki T. M., PillaI S. K., N. Engl. J. Med. 2020, 382, 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Messina F., Giombini E., Agrati C., Vairo F., Ascoli Bartoli T., Al Moghazi S., Piacentini M., Locatelli F., Kobinger G., Maeurer M., Zumla A., Capobianchi M. R., Lauria F. N., Ippolito G., J. Transl. Med. 2020, 18, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar R., Harilal S., Al-Sehemi A. G., Mathew G. E., Carradori S., Mathew B., Curr. Med. Chem. 2020, doi: 10.2174/0929867327666200702151018. [DOI] [PubMed] [Google Scholar]
- 4. Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K. Y., Wang Q., Zhou H., Yan J., Qi J., Cell 2020, 181, 894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q., Science 2020, 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou D., Tian X., Qi R., Peng C., Zhang W., Glycobiology 2020, doi: 10.1093/glycob/cwaa052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walls A. C., Park Y. J., Tortorici M. A., Wall A., McGuire A. T., Veesler D., Cell 2020, 181, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X., Nature 2020, 581, 215–220. [DOI] [PubMed] [Google Scholar]
- 9. Ahmed S. F., Quadeer A. A., McKay M. R., Viruses 2020, 12, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z., Nat. Commun. 2020, 11, 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gentile D., Patamia V., Scala A., Sciortino M. T., Piperno A., Rescifina A., Mar. Drugs 2020, 18, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Naik B., Gupta N., Ojha R., Singh S., Prajapati V. K., Prusty D., Int. J. Biol. Macromol. 2020, 160, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prasanth D. S. N. B. K., Murahari M., Chandramohan V., Panda S. P., Atmakuri L. R., J. Biomol. Struct. Dyn. 2020, doi: 10.1080/07391102.2020.1779129. [DOI] [PubMed] [Google Scholar]
- 14. Islam R., Parves M. R., Paul A. S., Uddin N., Rahman M. S., Mamun A. A., Hossain M. N., Ali M. A., Halim M. A., J. Biomol. Struct. Dyn. 2020, doi: 10.1080/07391102.2020.1761883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shah B., Modi P., Sagar S. R., Life Sci. 2020, 252, 117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guy R. K., DiPaola R. S., Romanelli F., Dutch R. E., Science 2020, 368, 829–830. [DOI] [PubMed] [Google Scholar]
- 17. Ozkan H., Adem S., ChemistrySelect 2020, 5, 5422–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daina A., Zoe V., ChemMedChem 2016, 11, 1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takemoto K., Kamisuki S., Chia P. T., Kuriyama I., Mizushina Y., Sugawara F., J. Nat. Prod. 2014, 77, 1992–1996. [DOI] [PubMed] [Google Scholar]
- 20. Choi H. G., Song J. H., Park M., Kim S., Kim C. E., Kang K. S., Shim S. H., Biomol. Eng. 2020, 10, 91. [Google Scholar]
- 21. Chang J., Block T. M., Guo J. T., Antiviral Res. 2013, 99, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Norton P. A., Menne S., Sinnathamby G., Betesh L., Cote P. J., Philip R., Mehta A. S., Tennant B. C., Block T. M., Hepatology 2010, 52, 1242–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka Y., Kato J., Kohara M., Galinski M. S., Antiviral Res. 2006, 72, 1–9. [DOI] [PubMed] [Google Scholar]
- 24. Watanabe S., Rathore A. P., Sung C., Lu F., Khoo Y. M., Connolly J., Low J., Ooi E. E., Lee H. S., Vasudevan S. G., Antiviral Res. 2012, 96, 32–35. [DOI] [PubMed] [Google Scholar]
- 25. Bhushan G., Lim L., Bird I., Chothe S. K., Nissly R. H., Kuchipudi S. V., Front. Microbiol. 2020, 11, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dowall S. D., Bewley K., Watson R. J., Vasan S. S., Ghosh C., Konai M. M., Gausdal G., Lorens J. B., Long J., Barclay W., Garcia-Dorival I., Hiscox J., Bosworth A., Taylor I., Easterbrook L., Pitman J., Summers S., Chan-Pensley J., Funnell S., Vipond J., Charlton S., Haldar J., Hewson R., Carroll M. W., Viruses 2016, 8, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carradori S., Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2020, 19, 85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary