

Abstract
The recent outbreak of coronavirus disease 2019 (COVID‐19), caused by the Severe Acute Respiratory Syndrome Coronavirus‐2 (SARS‐CoV‐2) has resulted in a world‐wide pandemic. Disseminated lung injury with the development of acute respiratory distress syndrome (ARDS) is the main cause of mortality in COVID‐19. Although liver failure does not seem to occur in the absence of pre‐existing liver disease, hepatic involvement in COVID‐19 may correlate with overall disease severity and serve as a prognostic factor for the development of ARDS. The spectrum of liver injury in COVID‐19 may range from direct infection by SARS‐CoV‐2, indirect involvement by systemic inflammation, hypoxic changes, iatrogenic causes such as drugs and ventilation to exacerbation of underlying liver disease. This concise review discusses the potential pathophysiological mechanisms for SARS‐CoV‐2 hepatic tropism as well as acute and possibly long‐term liver injury in COVID‐19.
1. INTRODUCTION
Since December 2019, the outbreak of coronavirus disease 2019 (COVID‐19), caused by the novel Severe Acute Respiratory Syndrome (SARS) Coronavirus (CoV) 2 (SARS‐CoV‐2), has led within a few months to a major global health and economic crisis. As of October 2020, more than 40 million confirmed cases have been reported worldwide, with nearly 1 million deaths, affecting 189 countries. 1 The respiratory tract is considered the main target of SARS‐CoV‐2 infection and a small subset of infected individuals becomes severely ill and may develop acute respiratory distress syndrome (ARDS) with potentially fatal outcome. 2 More recently, systemic features of the disease with the involvement of organs outside the respiratory tract, including the liver and gastrointestinal tract are receiving increasing attention, indicating that COVID‐19 may be considered as a systemic infectious and inflammatory disease. 3 , 4 , 5 , 6 , 7 Although closely related to other Corona virus (CoV) family members SARS‐CoV and MERS‐CoV (Middle East Respiratory Syndrome CoV), infections with the new SARS‐CoV‐2 exhibit a different pathological pattern and the mechanistic link between CoVs‐induced molecular pathophysiological changes and clinical manifestations remains incompletely understood.
Coronaviridae family members, including SARS‐CoV‐2, SARS‐CoV and MERS‐CoV, are enveloped viruses, characterized by a positive single‐stranded RNA genome of about 30Kb. 8 , 9 , 10 The angiotensin‐converting enzyme 2 (ACE2) has been established as the main viral receptor for SARS‐CoV and SARS‐CoV‐2 11 , 12 (Figure 1). Following attachment to the host cell and viral S protein priming by the host transmembrane serine protease 2 (TMPRSS2), 13 SARS‐CoV is internalized by endocytosis and the viral genome is released from the endosome. 14 , 15 In the cytosol, the viral RNA is translated into two polyproteins, pp1a and pp1ab, that are further processed to produce 16 non‐structural proteins (nsp1 to nsp16), 16 the building blocks of the viral replicase–transcriptase complex (RTC). 17 , 18 The full viral genome is then replicated in RTC‐containing vesicles. 19 , 20 In parallel, a set of specific sub‐genomic mRNA is generated 14 for the production of SARS‐CoV structural and accessory proteins, which assemble to form the nucleocapsid and viral envelope at the ER–Golgi intermediate compartment, allowing the subsequent release of mature virions 21 (Figure 1).
Figure 1.
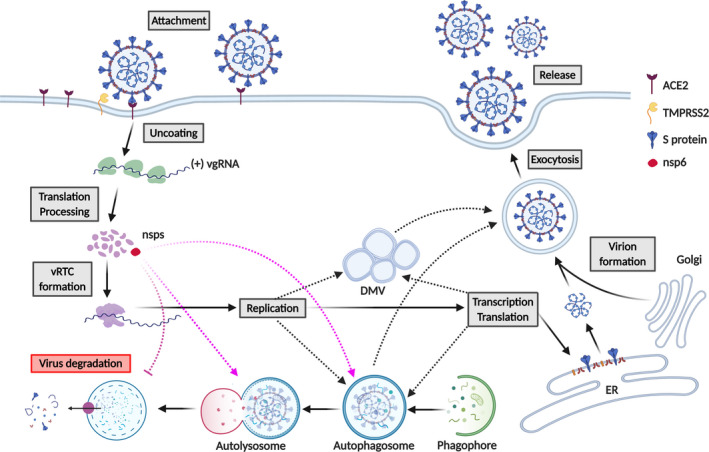
SARS‐CoV‐2 life cycle in host cells. SARS‐CoV‐2 attachment to host cells in liver (eg hepatocytes) may be mediated by the interaction of Spike (S) protein with ACE2. S protein is cleaved by the transmembrane serine protease 2 (TMPRSS2), allowing the cellular entry of the virus. Once uncoated, the viral genome ((+) vgRNA) is released and translated by the ribosome into pp1a and pp1ab (not shown), that are further cleaved into 16 non‐structural proteins (nsps). Following the viral replication/transcription complex (vRTC) assembly, nsp6 (in red) induces autophagosome formation, where viral replication might take place (purple dashed lines). Viral replication might also occur in double‐membrane vesicles (DMV) (black dashed lines). nsp6‐mediated inhibition of autophagosome/lysosome expansion might prevent viral degradation (purple dashed inhibitory line). Newly synthesized viral structural and accessory proteins assemble to form the nucleocapsid and viral envelope at the ER–Golgi intermediate compartment (lower right). Mature virions are then released through the exploitation of the host vesicular system (upper right). DMV and autophagosomes might also be used by the virus for exocytosis and release of mature virions (black dashed lines)
Although COVID‐19 primarily affects the respiratory system, emerging evidence highlights the impact of this viral infection on other organ systems. 3 , 4 , 5 , 22 , 23 The ubiquitous distribution of the main viral entry receptor ACE2 may explain how SARS‐CoV‐2 is able to cause a widespread disease characterized by systemic organ involvement including the intestines, 24 heart, kidneys, pancreas, liver, muscular and nervous system. 11 , 25 , 26 , 27 , 28 In contrast to SARS‐CoV‐2‐induced lung and myocardial injury, the clinical significance of liver involvement has been controversially debated from the very beginning of the COVID‐19 pandemic. 22 , 28 , 29 , 30 , 31 , 32 , 33 However, the scientific progress over the last months has shed more light on several key questions concerning COVID‐19‐associated liver injury. In this review, we will highlight molecular evidence pointing towards a putative hepatic tropism of SARS‐CoV‐2, and further review pathophysiological mechanisms that could explain the hepatic phenotypes associated with COVID‐19.
2. THE SPECTRUM OF LIVER INVOLVEMENT IN COVID‐19
COVID‐19 associated liver injury is defined as any liver damage occurring during disease course and treatment of COVID‐19 patients, with or without pre‐existing liver disease. 4 , 34 , 35 , 36 , 37 , 38 , 39 This includes a broad spectrum of potential pathomechanisms including direct cytotoxicity from active viral replication of SARS‐CoV‐2 in the liver, 40 , 41 immune‐mediated liver damage due to the severe inflammatory response/systemic inflammatory response syndrome (SIRS) in COVID‐19, 42 hypoxic changes induced by respiratory failure, vascular changes due to coagulopathy, endothelitis or cardiac congestion from right heart failure, drug‐induced liver injury and exacerbation of underlying liver disease (Figure 2). The incidence of elevated liver transaminases (ALT and AST) in COVID‐19 patients ranges from 2.5% to 76.3%. 35 , 38 , 43 , 44 In a recent meta‐analysis, the pooled rate for AST and ALT outside the reference range was 20%‐22.5% and 14.6%‐20.1% respectively. 35 , 45 These abnormalities can be accompanied by slightly increased total bilirubin levels in up to 35% of cases. 35 , 38 , 43 , 44 While elevations of cholestatic liver enzymes [alkaline phosphatase (ALP) and gamma glutamyl transferase (γGT)] were initially considered rather rare, 4 , 22 , 23 , 46 recent systemic reviews highlight elevations of ALP and γGT in 6.1% and 21.1% of COVID‐19 patients respectively. 35 , 45 Moreover, a biphasic pattern with initial transaminase elevations followed by cholestatic liver enzymes has been reported, which could reflect SIRS‐induced cholestasis at the hepatocellular/canalicular level or more severe bile duct injury in the later stage of the disease. 47 Although COVID‐19‐associated liver injury has been reported to be mild, it may affect a significant proportion of patients, especially those with a more severe disease course. In the light of the central role of the liver for the production of albumin, acute phase reactants and coagulation factors, hepatic dysfunction may impact on the multisystem manifestations of COVID‐19 such as ARDS, coagulopathy and multiorgan failure. 2 , 3 , 4 , 5 , 6 , 7 , 48 Moreover, the liver is the primary metabolic and detoxifying organ in the human organism, and even a moderate loss of hepatic function could alter the safety profile and therapeutic efficacy of antiviral drugs metabolized in the liver. Hence, it is crucial to understand the causes of COVID‐19‐associated liver injury in more detail.
Figure 2.
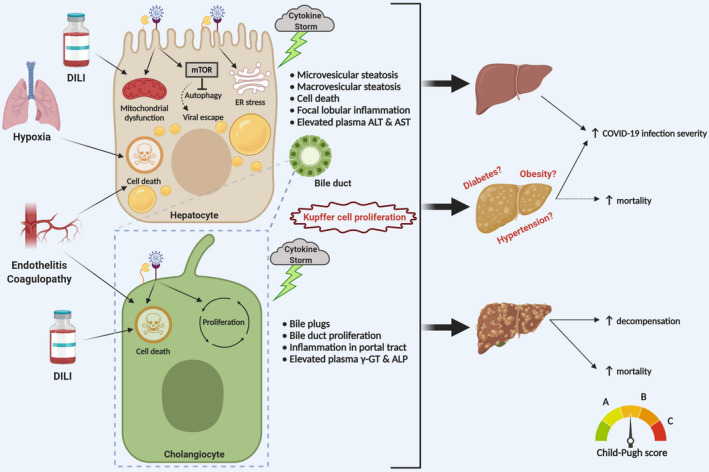
Proposed pathophysiology for liver injury upon SARS‐CoV‐2 infection. COVID‐19‐associated hepatocellular damage is mainly characterized by moderate steatosis, lobular and portal inflammation, apoptotic/necrotic foci and elevation of plasma ALT and AST (upper left panel). Preliminary observations suggest that the injury might be caused by hepatocellular infection with direct cytopathic effects of SARS‐CoV‐2, which could induce mitochondrial dysfunction and ER stress contributing to steatosis. Furthermore, SARS‐CoV‐2 infection might also activate mTOR, which eventually inhibits autophagy (as a mechanism of viral degradation) and facilitates viral escape from the immune system. In addition, cytokine storm, hypoxic conditions due to ARDS and drug‐induced liver injury (DILI) may contribute. COVID‐19‐associated cholangiocellular injury has also been observed and is mainly characterized by bile duct proliferation, occasionally bile plug formation and elevation of plasma γGT and ALP (lower left panel). From a hepatological perspective, COVID‐19‐positive patients may be divided into three categories: patients without pre‐existing chronic liver disease, patients with early stage chronic liver disease and patients with advanced chronic liver disease/cirrhosis. COVID‐19‐associated liver injury may have a more severe outcome in patients with pre‐existing liver disease, such as non‐alcoholic fatty liver disease (NAFLD) and associated metabolic comorbidity. Moreover, COVID‐19 may induce hepatic decompensation with increased mortality in cirrhotic patients (right panel)
So far, systematic information on underlying histopathological alterations is scarce. Hepatic steatosis (in part microvesicular) and Kupffer cell activation appear to be commonly encountered in livers of SARS‐CoV‐2‐infected deceased, together with vascular alterations including derangement of intrahepatic portal vein branches, usually mild lobular and portal inflammation, ductular proliferation and liver cell necrosis. 40 , 46 , 49 , 50 , 51 Of note, examination of liver biopsies from a cohort of 48 deceased COVID‐19 patients revealed extensive luminal thrombosis at the portal and sinusoidal level, together with portal fibrosis accompanied by significant pericyte activation. 51
3. POTENTIAL MOLECULAR MECHANISMS FOR SARS‐COV‐2 TROPISM OF THE LIVER
The presence of SARS‐CoV‐2 viral RNA has recently been demonstrated by qRT‐PCR in liver among various other organs outside the respiratory tract, 52 although the exact cellular site of replication remained unspecified since nucleic acids have been isolated by whole‐tissue homogenization. However, in situ hybridization analyses revealed SARS‐CoV‐2 virions in vessel lumens and endothelial cells of portal veins of COVID‐19 liver specimens. 51 Moreover, electron microscopic analyses on liver samples from two deceased COVID‐19 patients with elevated liver enzymes demonstrated the presence of intact viral particles in the cytoplasm of hepatocytes. 40
Given recent, although still limited, discoveries, 40 , 51 , 52 hepatic tropism for SARS‐CoV‐2 and direct cytopathic effects should be considered as potential mechanism of COVID‐19 associated liver injury, although a classic hepatitic picture has not been reported. 40 , 46 , 49 , 50 , 51 The availability of viral receptors at the host cell surface is a major determinant of viral tropism for a specific tissue. 53 As such, SARS‐CoV‐2 cell entry is mediated by the S protein of the virus, which specifically interacts with host ACE2 and TMPRSS2 (Figure 1). In order to understand whether SARS‐CoV‐2 might be able to infect liver cells, we explored the expression pattern of the human ACE2 and TMPRSS2 proteins using the Human Protein Atlas (data available at https://www.proteinatlas.org/ENSG00000130234‐ACE2/tissue and https://www.proteinatlas.org/ENSG00000184012‐TMPRSS2/tissue). Interestingly, the expression levels of the two proteins is highest in intestine and gall bladder, but it appears to be virtually absent in the liver. These data might be incomplete or lack sensitivity, since in the Human Protein Atlas ACE2 expression also seems to be absent in the lungs, where infection is definitely known to occur. In a recent study, Chai and colleagues applied single‐cell RNAseq to healthy human liver samples and found that ACE2 expression levels in bile duct epithelium (cholangiocytes) is comparable to that of alveolar cells in the lungs, whereas hepatocellular ACE2 expression is low but still detectable. 54 Further confirmation of significant ACE2 and TMPRSS2 expression in liver parenchymal cells comes from bio‐informatics analyses from the single‐cell transcriptome database Single Cell Portal. 55 Interestingly, sinusoidal endothelial cells appear to be ACE2‐negative, in line with previous observations. 56 This finding may be important considering recent reports on endothelitis of large intrahepatic vessels caused by SARS‐CoV‐2 48 , 57 and high ACE2 expression in other endothelia, including central and portal veins, which also can become infected by the virus. 51
Of note, studies in both mice and humans revealed increased hepatic ACE2 expression in hepatocytes upon liver fibrotic/cirrhotic conditions 58 , 59 (and our own unpublished observations). This finding may be of great relevance since pre‐existing liver injury could thereby exacerbate SARS‐CoV‐2 hepatic tropism. Moreover, hypoxia, which is a typical feature in severe COVID‐19 cases, has been shown to be a main regulator of hepatocellular ACE2 expression. 58 This might explain why extra‐pulmonary SARS‐CoV‐2 dissemination is mainly observed in patients manifesting ARDS and other hypoxic conditions. Importantly, inflammatory conditions/diseases in the liver, as shown for other organs, 60 , 61 could also upregulate ACE2 expression. Since drug‐induced liver injury (DILI) may contribute to liver injury in COVID‐19 patients, 62 it might be of interest to explore whether DILI or certain drugs induce hepatic ACE2 over‐expression.
In vitro experiments also showed that the S protein of lineage B beta‐coronaviruses significantly increases the affinity for its receptor when it is pre‐incubated with trypsin, that is when it is proteolytically activated. 63 Since liver epithelial cells express trypsin 64 and a plethora of other serine‐proteases which constantly remodel the extracellular matrix, 65 ACE2 expression required for SARS‐CoV‐2 target and recognition in the liver might be lower than in other tissues with reduced extracellular proteolytic activity. 66 In line with these findings, it has been recently discovered that the S protein of SARS‐CoV‐2 bears a furin‐like proteolytic site never observed before in other coronaviruses of the same lineage. 67 Interestingly, furin is predominantly expressed in organs that have been proposed as permissive for SARS‐CoV‐2 infection, such as salivary glands, kidney, pancreas (data for The Human Protein Atlas, available at https://www.proteinatlas.org/ENSG00000140564‐FURIN/tissue) and the liver. 55
Finally, other factors, as for example ganglioside (GM1), 68 might influence S protein‐ACE2 interaction. Therefore, research should also explore more deeply the S protein‐ACE2 interactome to achieve new molecular and therapeutic insights.
In a recent report, Ou and colleagues tested pseudovirions containing the SARS‐CoV‐2 S protein for their ability to infect different cell lines. Interestingly, HuH7 cells, a hepatocyte cell line, as well as Calu3 cells, a human lung carcinoma cell line, were more efficiently transfected by viral vectors carrying the SARS‐CoV‐2 S protein than control pseudovirions. 69 Moreover, these studies revealed that viral entry might depend on the PIKfyve‐TCP2 endocytotic pathway. A crosscheck in the Human Protein Atlas revealed that both PIKfyve and TPC2 are expressed in liver and gall bladder at comparable levels as in the lung (data available at https://www.proteinatlas.org/ENSG00000115020‐PIKFYVE/tissue and https://www.proteinatlas.org/ENSG00000162341‐TPCN2/tissue), highlighting the potential relevance of this pathway for hepatic tropism, which therefore expands from simple targeting and recognition to support of intracellular viral replication.
In an effort to establish a new and effective functional viromics screening approach aimed at predicting the likelihood of zoonotic events of the known lineage B betacoronaviruses, Letko and colleagues took advantage of HuH7 cells as a permissive model for SARS‐CoV and SARS‐CoV‐2 binding and recognition, 63 further proving SARS‐CoV‐2 tropism for hepatocytes. Of note, HuH7 cells were described as the third most permissive cell line in this study after pulmonary (Calu3) and intestinal (CaCo2) cell models, 63 the latter representing organs with histopathologically proven SARS‐CoV‐2 infection. However, the ability of binding and internalizing viral particles does not necessarily imply that the cell type under investigation is also permissive for effective viral replication. In this regard, both Chu and colleagues and Harcourt et al demonstrated that HuH7 cells support SARS‐CoV‐2 viral replication. 70 , 71 Hepatocyte cell lines are now such an established permissive cell type for SARS‐CoV and SARS‐CoV‐2 infection that HuH7 cells have also been recently used as positive control in SARS‐CoV‐2 immunostainings. 72
Although the above‐reported observations define hepatocytes as putative hosts for SARS‐CoV‐2, it is important to point out that all the data arise from studies in which cancer cell lines have been used. In order to clarify the translational potential of these observations, ACE2 protein expression in HuH7 cells should be compared with that of primary human hepatocytes. Furthermore, future investigations are needed to uncover the molecular changes induced in hepatocytes upon SARS‐CoV‐2 infection.
A reliable source of information comes from recent work by Yang and colleagues, who demonstrated SARS‐CoV‐2 tropism for hepatocytes using organoids obtained from human pluripotent stem cell (hPSC)‐derived hepatocyte and primary adult human hepatocytes. 73 In these systems, pseudovirions expressing SARS‐Cov‐2 S protein were able to infect human hepatocytes, while SARS‐CoV‐2 infection resulted in robust viral replication. 73 Gene expression analyses also showed that SARS‐CoV‐2‐infected primary hepatocytes over‐express pro‐inflammatory cytokines, while downregulating key metabolic processes, as reflected by the inhibition of CYP7A1, CYP2A6, CYP1A2 and CYP2D6 expression. 73
Finally, Wang and colleagues applied electron microscopy imaging to liver samples of two deceased COVID‐19 patients, and identified viral structures in hepatocytes which distinctively resemble SARS‐CoV‐2 virions. 40 This raises the possibility that the histopathological alterations seen in these patients may be caused by direct cytopathic effects of SARS‐CoV‐2 40 although a typical hepatitis pattern appears to be lacking. 40 , 46 , 49 , 50 , 51 However, further studies with larger biopsy/autopsy cohorts and the combined imaging (including immune electron microscopy) may be necessary to confirm these preliminary observations of hepatocellular SARS‐CoV‐2 presence.
Bile duct epithelial cells (cholangiocytes) participate in bile production and flow as well in immune response. 74 Single‐cell sequencing of human long‐term liver ductal organoid cultures showed preservation of ACE2 and TMPRSS2 expression. 75 Following SARS‐CoV‐2 infection, cholangiocytes underwent syncytia formation and the amount of SARS‐CoV‐2 genomic RNA was dramatically increased 24 hours post‐infection. Similar results have been obtained when infecting adult human cholangiocyte organoids with SARS‐CoV‐2. 73 These observations indicate that human liver ductal organoids may be susceptible to SARS‐CoV‐2 infection in vitro and suggest that viral replication could also occur within the bile duct epithelium in vivo. However, despite significantly higher ACE2 expression when compared with hepatocytes, no direct evidence of SARS‐CoV‐2 cholangiocellular infection has been reported so far in COVID‐19 patients. Since bile is primarily produced by hepatocytes and cholangiocytes, and given the continuous and direct contact between biliary fluids and the cholangiocellular apical membrane, identification of SARS‐CoV‐2 viral RNA or proteins in bile could be an indirect proof of SARS‐CoV‐2 cholangiocellular infection. At the moment, only one case report has shown SARS‐CoV‐2 RNA in bile, 76 whereas bile from two other small sample series tested negative. 24 , 49 These discrepancies might rely on the fact that the positive‐tested bile sample has been obtained during surgical resolution of bile duct obstruction, 76 whereas the negatively tested bile was obtained from 48h post‐mortem autopsies. 24 , 49
Tight junctions allow cholangiocytes to act as a protective barrier for parenchymal liver cells from toxic bile components. Viral infection with SARS‐CoV‐2 decreased mRNA expression of cholangiocellular tight junction proteins such as claudin 1 in vitro, 75 implicating reduced barrier function of cholangiocytes. This in turn could cause liver injury through leakage of potentially toxic bile into the periductal space and adjacent liver parenchyma. Of note, expression of the bile acid transporters SLC10A2/ASBT and chloride channel ABCC7/CFTR was significantly down‐regulated by SARS‐CoV‐2 infection. 75 The negative regulation of these hepatobiliary transporters may impair bile acid sensing/signalling by cholangiocytes and bicarbonate secretion, eventually contributing to biliary changes observed in COVID‐19 infection. 49 Furthermore, cholangiocytes infected with SARS‐CoV‐2 virus upregulated inflammatory pathways, depicting the induction of a reactive cholangiocyte phenotype. 73 Future studies will have to explore whether and how SARS‐CoV‐2 may alter secretion of pro‐inflammatory and pro‐fibrogenic cytokines and contribute to the ‘reactive cholangiocyte phenotype’, which could propagate inflammation and fibrosis. 74
Pre‐existing chronic liver diseases seem to be independent risk factors for poor outcome in COVID‐19, and cirrhosis grade has been defined as a predictor of mortality in SARS‐CoV‐2 infected patients 77 (Figure 2). Activation of hepatic stellate cells plays a paramount role in the progression of chronic liver disease as the main cellular source of fibrosis 78 and is induced by pro‐inflammatory and pro‐fibrotic cues, such as Angiotensin II, generated by the catalytic action of ACE as part of the pro‐fibrotic branch of the renin‐angiotensin system. 79 Of note, ACE2 counteracts ACE function by producing the anti‐inflammatory and anti‐fibrotic Angiotensin‐(1‐7) and thereby decreasing the Angiotensin II/Angiotensin‐(1‐7) ratio. 79 However, ACE2 expression has neither been detected in quiescent, nor in fibrogenic/activated hepatic stellate cells. 58 , 80 , 81 , 82 , 83 These findings suggest that these cells may be a rather non‐permissive host for SARS‐CoV‐2. Nevertheless, the pro‐inflammatory milieu generated by direct or indirect COVID‐19‐associated hepatocellular and cholangiocellular injury may pave the way for activation of hepatic stellate cells and consequent induction of fibrosis. This possibility may be even more relevant in patients with underlying CLD, such as NAFLD. Although available data suggest that COVID‐19‐related liver injury is mild and transitory, long‐term follow‐up studies will be necessary to exclude hepatic fibrosis as a potential long‐term consequence of COVID‐19, especially in the presence of pre‐existing liver diseases.
Monocyte‐derived macrophages (MoM) and alveolar macrophages are known to express ACE2, 84 , 85 and there is evidence of alveolar macrophage infection by SARS‐CoV 85 and SARS‐CoV‐2 with detection of viral protein by immunohistochemistry. 24 , 86 However, a histopathologic assessment of ACE2 tissue distribution showed no staining in Kupffer cells and other hepatic immune cells, 56 although Kupffer cell proliferation is typically observed in livers of COVID‐19 diseased. 40 , 49 The recent COVID‐19 pandemic further prompted more in‐detail investigations on ACE2 expression and de novo single‐cell RNAseq analyses, 54 as also in silico evaluations of RNAseq databases 87 , 88 proved that Kupffer cells do not express ACE2. It has to be kept in mind, however, that all the described evidences refer to healthy human liver samples. Therefore, quantification of ACE2 expression in samples obtained from patients with underlying chronic liver disease or acute liver injury may be needed to obtain definitive insights into macrophage ACE2 expression patterns.
Of note, upon liver injury and/or Kupffer cell depletion, MoM can invade the liver and efficiently replenish the hepatic resident macrophage population 89 , 90 , 91 (and reviewed in detail in 92 ). Although in vitro observations proved that MoM does not support efficient replication of SARS‐CoV (and most probably also SARS‐CoV‐2), infected MoM could act as carriers of the pathogen, favouring infection of the ACE2‐expressing cells in the invaded organ. 93 Furthermore, Kupffer cell activation and proliferation are frequently observed as a consequence of systemic inflammation and Kupffer cell activation has been reported in the liver specimen of deceased COVID‐19 patients. 40 , 49 Thus, although Kupffer cells do not express ACE2, monocytic cells might play a key role in SARS‐CoV‐2‐mediated liver injury by propagation of inflammatory stimuli.
4. SARS‐CoV2 AND HEPATIC STEATOSIS
Microvesicular and macrovesicular steatosis have been observed in liver autopsies of COVID‐19 patients who presented with SARS‐CoV‐2 infection as the only risk factor for liver injury, and in some cases, SARS‐CoV‐2 hepatocellular infection has been proven. 40 , 49 Importantly, hepatic lipid accumulation as a result of SARS‐CoV‐2 infection must be differentiated from pre‐existing NAFLD, which has been shown to increase the risk for poor outcome in COVID‐19 patients. 50 Deregulated in host lipid metabolism and mitochondrial activity as a result of potential direct SARS‐CoV‐2 cytopathic effects and/or immunopathology induced by cytokine storm, as well as drug side effects (eg corticosteroids) may be important contributors to the development of hepatic steatosis in COVID‐19 (Figure 2).
Microvesicular steatosis is typically caused by genetic or acquired mitochondrial β‐oxidation defects. 94 Preliminary observations suggest that SARSR‐CoV‐2 affects mitochondrial activity. 95 Furthermore, Wang et al also identified mitochondrial crista abnormalities in liver specimen of COVID‐19 patients. 40 Interestingly, impaired mitochondrial activity has also been implicated in the pathogenesis of NAFLD/NASH. 96 Thus, SARS‐CoV‐2 infection might even worsen the metabolic state and aggravate pre‐existing NAFLD by these mechanisms.
Endoplasmic reticulum (ER) stress is known to induce de novo lipogenesis in hepatocytes. 97 Several studies have implicated SARS‐CoV infection in the induction of ER stress. For instance, significant up‐regulation of ER stress markers glucose‐regulated protein 78 (GRP78) and GRP94 has been observed upon SARS‐CoV infection in several cell lines. 98 , 99 , 100 The coronavirus S protein seems to be a major burden for the host ER and might play a key role in ER stress induction. 98 , 99 Rearrangement of intracellular membranes by extensive depletion of lipid components from the ER during SARS‐CoV‐2 infection may also contribute to ER stress. 20 Moreover, the ER stress‐related PERK‐eIF2‐α pathway is over‐activated upon SARS‐CoV infection in vitro. 101 Finally, electron microscopy examinations, which proved SARS‐CoV‐2 hepatocellular infection, reported a pathological ER dilatation in infected hepatocytes, 40 which most probably will cause ER stress. Collectively, these data could indicate that SARS‐CoV‐2, as other coronaviruses, induces ER stress upon infection, and that the ER stress‐induced de novo lipogenesis could also contribute to the development of steatosis in COVID‐19 patients (Figure 2).
De novo lipogenesis is also induced by the mammalian target of rapamycin (mTOR), 102 which is also the cardinal regulator of autophagy. 103 SARS‐CoV has been previously shown to hijack the autophagy pathway through processes that rely on the viral non‐structural protein 6 (nsp6), highly conserved in SARS‐CoV‐2. 104 , 105 , 106 Furthermore, mTOR hyper‐activation has been observed in MERS‐CoV‐infected HuH7 cells, and inhibition of mTOR signalling pathway by rapamycin inhibits viral replication. 107 Given the recent observations that SARS‐CoV‐2 infection restricts autophagy, 108 it is tempting to speculate that SARS‐CoV‐2, SARS‐CoV and MERS‐CoV share a similar mTOR‐dependent mechanism of infection. Furthermore, significantly increased mTOR activity has been revealed upon IL‐6 stimulation. 109 Thus, SARS‐CoV‐2 infection could lead to a hyper‐activation of hepatic mTOR signalling, via direct infection of hepatic cells, or indirect, cytokine storm‐related systemic IL‐6‐dependent effects, which could contribute to the steatotic phenotype in livers of COVID‐19 patients (Figure 2).
Although disadvantageous for the host, induction of host lipogenesis might be crucial for SARS‐CoV‐2 life cycle. Indeed, enhanced de novo lipogenesis could supply the virus with sufficient amounts of lipids to generate the vesicular systems required for viral replication and exocytosis. mTOR‐mediated promotion of protein synthesis 110 , 111 and inhibition of autophagolysosome formation 112 , 113 may further favour viral replication while preventing viral degradation and ignition of an adequate immune response. Since insulin and glucose signalling positively regulate mTOR activity in the liver, 114 , 115 constitutive mTOR over‐activation in obese and diabetic patients 116 , 117 , 118 could at least in part explain their higher risk for worse outcome of COVID‐19 (Figure 2).
5. SIRS‐INDUCED CHOLESTASIS AND BILE DUCT ALTERATIONS IN COVID‐19
Cholestatic features such as bile duct proliferation, portal inflammatory infiltrates, and in some cases, canalicular/ductular bile plugs have been reported in post‐mortem evaluations on COVID‐19 patients. 49 , 119 The cytokine storm characteristic of the SARS‐CoV‐2‐associated viral sepsis 120 may be a major contributing factor, since cytokines like TNF‐alpha, IL‐1 and IL‐6 can induce hepatocellular cholestasis by down‐regulating hepatobiliary uptake and excretory systems, 121 , 122 resembling the pathomechanisms seen in sepsis‐induced cholestasis. 121 , 122 , 123 , 124 , 125 Further studies will have to explore whether—similar to sepsis—serum bile acids as the most accurate indicators of cholestasis may be relevant prognostic parameters in COVID‐19. 122 , 126 Sustained systemic IL‐6 signalling initiated by SARS‐CoV‐2 infection induces a C/EBPβ‐dependent suppression of albumin synthesis. 127 In addition to hypo‐albuminaemia, cholestasis in SIRS as a result of repressed hepatobiliary excretory function could be viewed as part of the negative acute phase response in COVID‐19.
In addition to hepatocellular features, bile duct changes, such as ductular proliferation have been observed in postmortem studies. 49 Notably, IL‐6 is a strong cholangiocellular mitogen factor 128 and induces a proliferative and pro‐inflammatory phenotype. 74 , 129 Bile ducts from patients with COVID‐19 could therefore be exposed to a ‘triple hit’ from (i) hypoxia from respiratory failure (potentially aggravated by obliteration of the peribiliary arterial plexus through vasculitic/thrombotic changes); (ii) systemic SIRS resulting in a reactive cholangiocyte phenotype or senescence‐associated secretory phenotype, thus actively propagating inflammation as well as fibrosis and (iii) potential viral infection of cholangiocytes themselves. Thus, the hepatobiliary system may become an important target for adverse long‐term hepatic outcomes of COVID‐19. Secondary sclerosing cholangitis of critical ill patients (SSC‐CIP) is a rare but clinically relevant complication in critically ill patients with severe trauma, burn injury, suffering from severe respiratory failure or requiring vasopressor therapy due to hemodynamic instability. 130 , 131 Malperfusion and hypoxia, as well as recurrent inflammatory stimuli, are the main triggers for the destruction of the biliary epithelium in SSC‐CIP, 122 all conditions present in severe COVID‐19 patients.
Therefore, hepatic long‐term follow‐up for COVID‐19 survivors who experienced a severe disease course, such as ARDS with ECMO and prolonged ICU admission might be considered. Early diagnosis is paramount to best manage symptoms and disease progression of SSC‐CIP, which could be counteracted with anti‐cholestatic, cholangio‐protective drugs such as UDCA or more recently norUDCA. 132 , 133 , 134
6. SARS‐COV‐2 AND HYPOXIC HEPATITIS
Causes for hypoxic hepatitis are multifactorial. In general, cardiac failure, sepsis and respiratory failure account for more than 90% of all cases. 135 , 136 , 137 , 138 Additionally, right‐sided heart failure was found to aggravate liver injury by liver congestion as a result of elevated central venous pressure. 122 , 135 , 136 , 137 , 138 , 139 , 140 In cases of long‐lasting hemodynamic and/or respiratory failure, hypoxia results in hepatic cell death, histopathologically defined as centrilobular necrosis. 141
COVID‐19‐associated ARDS remains the most common complication requiring critical care management including invasive ventilation, high levels of positive end‐expiratory pressure (PEEP) and vasoconstrictor therapy in case of hemodynamic instability. 142 , 143 , 144 , 145 These factors may be accompanied by right ventricular dysfunction caused by high pulmonary vascular resistance as a result of hypoxaemia and hypercapnia during ARDS. 146 , 147 Furthermore, COVID‐19 causes a hyper‐coagulate state with a significant incidence of pulmonary thrombotic complications aggravating acute right‐sided heart failure and consequently liver congestion. 148 However, in the majority of cases, SARS‐CoV‐2 associated liver injury was generally mild and did not exceed >5 times the upper reference limit, therefore not fulfilling the diagnostic criteria for hypoxic hepaitis. 35 These findings were also obtained in critically ill patients referred to the ICU, suggesting that even in cases of severe respiratory failure during SARS‐CoV‐2 infection, the adequate oxygen supply to the liver is ensured by compensatory mechanisms. 35 , 36 , 39 , 149 , 150 , 151 , 152 , 153 , 154
7. DRUG‐INDUCED LIVER INJURY
At the beginning of the COVID‐19 outbreak, evidence‐based drug therapy was not available. Over the course of 8 months, multiple studies were performed allowing us to give scientifically valid recommendations for the treatment of SARS‐CoV‐2 infection. In the meantime, various antiviral (remdesivir, lopinavir/ritonavir), antibiotic (macrolids), antimalaria/antirheumatic (hydroxychloroquine), immunomodulating (corticosteroids, tocilizumab) and antipyretic (acetaminophen) drugs have been used in clinical studies or in an off‐label fashion. For most of these drugs (eg ritonavir, remdesivir) a hepatotoxic potential has already been confirmed in in vitro/in vivo experiments and in their respective registration studies. Moreover, corticosteroid therapy, which is now recommended by the WHO in patients with severe SARS‐CoV‐2 infection, 155 is also clearly associated with steatosis or glycogenosis. 156 Recently, the first case of DILI associated with tocilizumab use in a COVID‐19 patient has been reported. 62 Tocilizumab undergoes minimal hepatic metabolism, and the most probable etiology for its hepatotoxic effect is the interference with the IL‐6 pathway, which plays a key role in hepatic regeneration. 157
8. THE GUT‐LIVER AXIS AS THE POTENTIAL ROUTE FOR SARS‐COV‐2 HEPATIC INFECTION
Since SARS‐CoV‐2 infection affects also the gastrointestinal (GI) tract, 158 a significant proportion of COVID‐19 patients experience gastrointestinal symptoms, including diarrhea (2%‐35.6%), nausea (1%‐17.3%) and vomiting (1%‐6.4%). 158 Notably, both SARS‐CoV‐2 RNA and viable virions have been identified in stool samples of infected patients and post‐mortem. 24 , 159 , 160 , 161 , 162 Hepatic and gastrointestinal manifestations appear more frequently in severe forms of COVID‐19 infections. 3 , 4 , 5 , 163 , 164 , 165 Interestingly, a recent study by Jin and colleagues showed that individuals with pre‐existing liver diseases are more susceptible to develop an intestinal phenotype upon SARS‐CoV‐2 infection. 163 SARS‐CoV‐2 is potentially able to infect cells of the gastrointestinal tract, since ileal and colonic enterocytes co‐express ACE2 and TMPRSS2, the central proteins for viral attachment. 166 , 167 , 168 Recently, viral nucleocapsid protein could be demonstrated within enterocytes by immunohistochemistry. 24 The Human Protein Atlas database further corroborates these observations, with intestinal cells exhibiting the highest pattern of ACE2 expression across the whole human cell type repertoire (data available at https://www.proteinatlas.org/ENSG00000130234‐ACE2/tissue). Moreover, human intestinal organoids have been shown to be permissive to SARS‐CoV and SARS‐CoV‐2 infection. 169 Direct gastrointestinal infection has been reported also by biopsy‐proven RNA and nucleocapsid protein detection in gastric, duodenal and rectal epithelia. 160 Interestingly, gastrointestinal symptoms may appear before or even in the absence of manifestations in the respiratory tract. 165 This suggests that the GI tract might be a primary site of COVID‐19 infection, and therefore that oral‐fecal transmission could be an alternative route of infection for SARS‐CoV‐2 (this has been extensively reviewed). 162 , 170
We would like to propose the following putative way of SARS‐CoV‐2 infection through the hepatobiliary system. COVID‐19 intestinal infection might impair the intestinal epithelial and vascular barriers, eventually leading to hepatic translocation of the virus through the portal vein. Hepatic infection might therefore start in hepatocytes, which express the required receptor binding proteins and are in direct contact with the portal circulation. Subsequently, SARS‐CoV‐2 virions exiting infected hepatocytes by transcytotic vesicular pathways could reach the bile, which has tested positive in some studies, 76 although this remains controversial. 49 As a result, cholangiocytes might also get in contact with and infected by SARS‐CoV‐2. Since the biliary tract provides a direct link between liver and gut, SARS‐CoV‐2 may thereby reach and infect the intestine via bile, causing in turn a second wave of infection.
Thus, the here proposed speculative mechanism could generate a vicious circle, which increases the chances of survival for the virus and might explain the worse overall outcome in patients manifesting hepatic and intestinal symptoms upon SARS‐CoV‐2 infection. On the other hand, COVID‐19 with fatal outcome seems to be associated with severe damage of lung tissue, whereas the intestines are only mildly altered, most commonly by focal ischaemic changes in the intestinal mucosa. 24 Whether biliary tropism and requirement of bile/bile acids for viral attachment and entry into cholangiocytes and enterocytes 171 , 172 also play a role for SARS‐CoV‐2 remains to be determined. Given the functional and physiological similarities between bile ducts and ducts of the exocrine pancreas, and the observations concerning a potential pancreatic involvement in COVID‐19 infection, 49 , 173 , 174 the research of a common mechanism allowing infection of the two tissues might help in uncovering further determinants of SARS‐CoV‐2 tropism.
9. CONCLUSIONS AND PERSPECTIVES
Over the last months, several studies have highlighted the potential role of liver involvement in COVID‐19 infection and pathology. In this review, we analysed the published experimental and clinical findings concerning SARS‐CoV‐2 and previous coronavirus pandemics and proposed mechanisms concerning a putative SARS‐CoV‐2 hepatic tropism and the interplay between cytopathic and systemic effects in hepatic COVID‐19 pathophysiology.
Elevated liver enzymes reflecting hepatic injury are common in COVID‐19 patients both with and without chronic liver diseases. 35 , 38 , 43 , 44 Interestingly, while early clinical studies identified significant raises exclusively in serum ALT and AST upon SARS‐CoV‐2 infection, which reflect hepatocellular damage, recent investigations and metanalyses also highlighted significant increases in ALP and γ‐GT and therefore cholangiocellular injury. 35 , 45 However, it is still not clear whether elevated serum liver biochemistries are causative for the worse outcome, or a consequence of the severe disease course.
In COVID‐19 patients without pre‐existing hepatic conditions who experienced liver damage, the injury is mostly mild. However, given the central role of the liver in endo‐ and xenobiotic/drug metabolism, coagulation, albumin and acute phase reactant production, hepatic dysfunction may impact on systemic disease pathophysiology of COVID‐19. Long‐term follow‐up studies are required to explore potential long‐term sequels of SARS‐CoV‐2 infection such as fibrosis.
Crucial questions remain open and need to be answered by future research: Which specific hepatic cells are infected by SARS‐CoV‐2? Which molecular processes are dysregulated by the infection? What is the real contribution of direct cytopathic effects, cytokine storm, DILI or hypoxia in hepatic dysfunction? By which means could liver injury promote respiratory failure and predispose to a severe course of COVID‐19?
The establishment of international registries collecting clinical reports of patients with liver diseases also tested positive for COVID‐19, such as the COVID‐Hep 175 and the SECURE‐Cirrhosis, 176 together with molecular and translational research will surely help us shed some light on these intriguing questions and to set up more effective hepatoprotective programs for future pandemics.
CONFLICT OF INTERESTS
The Medical Universities of Graz and Vienna have filed patents for the medical use of norUDCA and MT is listed as co‐inventor. MT has served as a speaker for Falk Foundation, Gilead, Intercept and MSD; he has advised for Albireo, BiomX, Boehringer Ingelheim, Falk Pharma GmbH, Genfit, Gilead, Intercept, Jannsen, MSD, Novartis, Phenex, Regulus and Shire. He further received travel grants from Abbvie, Falk, Gilead and Intercept and research grants from Albireo, CymaBay, Falk, Gilead, Intercept, MSD and Takeda. SL has received personal fees from Roche, AstraZeneca, Novartis and Biogena outside the submitted work, Authors not named here have disclosed no conflicts of interest.
AUTHORS’ CONTRIBUTION
MT and ADN planned the project, MT, AND and MS outlined the content and contributed to writing and editing of the manuscript. ADN, MB and EDM contributed to the research, discussion of content and writing of the molecular basic science sections of the manuscript. MT, MS and SFL contributed to the research, discussion of content and writing of the clinical sections of the manuscript. ADN, MB and MS generated the figures of the manuscript. All authors corrected and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
This work was supported by grant F7310‐B21 from the Austrian Science Foundation (to MT). We thank Jelena Remetic, Claudia D. Fuchs, Veronika Mlitz and Daniel Steinacher, for their valuable input and discussion. Figure 1 and Figure 2 have been created with BioRender.com.
Nardo AD, Schneeweiss‐Gleixner M, Bakail M, Dixon ED, Lax SF, Trauner M. Pathophysiological mechanisms of liver injury in COVID‐19. Liver Int. 2021;41:20–32. 10.1111/liv.14730
Handling editor: Luca Valenti
Alexander D. Nardo and Mathias Schneeweiss‐Gleixner Contributed equally, shared first authorship.
REFERENCES
- 1. John Hopkins University and Medicine . COVID‐19 Map ‐ Johns Hopkins Coronavirus Resource Center. John Hopkins Coronavirus Resource Center 1 https://coronavirus.jhu.edu/map.html (2020)
- 2. Xie M, Chen Q. Insight into 2019 novel coronavirus — An updated interim review and lessons from SARS‐CoV and MERS‐CoV. Int J Infect Dis. 2020;94:119‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang C, Shi L, Wang FS. Liver injury in COVID‐19: management and challenges. The Lancet Gastroenterol Hepatol. 2020;5:428‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu L, Liu J, Lu M, Yang D, Zheng X. Liver injury during highly pathogenic human coronavirus infections. Liver Int. 2020;40:liv.14435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun J, Aghemo A, Forner A, Valenti L. COVID‐19 and liver disease. Liver Int. 2020. 10.1111/liv.14470 [DOI] [PubMed] [Google Scholar]
- 6. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID‐19): A Review. JAMA ‐ J Am Med Assoc. 2020;324:782‐793. [DOI] [PubMed] [Google Scholar]
- 7. Li H, Liu L, Zhang D, et al. SARS‐CoV‐2 and viral sepsis: observations and hypotheses. The Lancet. 2020;395:1517‐1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marra MA, Jones SJ, Astell CR, et al. The genome sequence of the SARS‐associated coronavirus. Science (80‐. ). 2003;300:1399‐1404. [DOI] [PubMed] [Google Scholar]
- 9. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Nat Med. 2020;26:450‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: Recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14:523‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor Article SARS‐CoV‐2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glowacka I, Bertram S, Müller MA, et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J Virol. 2011;85:4122‐4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perlman S, Netland J. Coronaviruses post‐SARS: Update on replication and pathogenesis. Nat Rev Microbiol. 2009;7:439‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang H, Yang P, Liu K, et al. SARS coronavirus entry into host cells through a novel clathrin‐ and caveolae‐independent endocytic pathway. Cell Res. 2008;18:290‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fehr AR, Perlman S. Coronaviruses: An overview of their replication and pathogenesis. in Coronaviruses: Methods and Protocols, vol. 1282 1–23 (Springer, New York, 2015). [DOI] [PMC free article] [PubMed]
- 17. Sharma K, Surjit M, Satija N, et al. The 3a accessory protein of SARS coronavirus specifically interacts with the 5′UTR of its genomic RNA, using a unique 75 amino acid interaction domain. Biochemistry. 2007;46:6488‐6499. [DOI] [PubMed] [Google Scholar]
- 18. Tan YJ, Lim SG, Hong W. Understanding the accessory viral proteins unique to the severe acute respiratory syndrome (SARS) coronavirus. Antiviral Res. 2006;72:78‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Snijder EJ, Van Der Meer Y, Zevenhoven‐Dobbe J, et al. Ultrastructure and Origin of Membrane Vesicles Associated with the Severe Acute Respiratory Syndrome Coronavirus Replication Complex. J Virol. 2006;80:5927‐5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knoops K, Kikkert M, Van Den Worm SH, et al. SARS‐Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stertz S, Reichelt M, Spiegel M, et al. The intracellular sites of early replication and budding of SARS‐coronavirus. Virology. 2007;361:304‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guan W, Ni ZY, Hu Y, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382:1708‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skok K, Stelzl E, Trauner M, Kessler HH, Lax SF. Post‐mortem viral dynamics and tropism in COVID‐19 patients in correlation with organ damage. Virchows Arch. 2020. 10.1007/s00428-020-02903-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li R, Qiao S, Zhang G. Analysis of angiotensin‐converting enzyme 2 (ACE2) from different species sheds some light on cross‐species receptor usage of a novel coronavirus 2019‐nCoV. J Infect. 2020;80:469‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu H, Zhong L, Deng J, et al. High expression of ACE2 receptor of 2019‐nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020;12:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turner AJ, Hiscox JA, Hooper NM. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol Sci. 2004;25:291‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oyelade T, Alqahtani J, Canciani G. Prognosis of COVID‐19 in Patients with Liver and Kidney Diseases: An Early Systematic Review and Meta‐Analysis. Trop Med Infect Dis. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu C, Chen X, Cai Y, et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. 2020. 10.1001/jamainternmed.2020.0994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang C, Shi L, Wang F‐S. Comment Liver injury in COVID‐19: management and challenges. Lancet Gastroenterol Hepatol. 2020;5:428‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bangash MN, Patel J, Parekh D. COVID‐19 and the liver: little cause for concern. Lancet Gastroenterol Hepatol. 2020;5:529‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boeckmans J, Rodrigues RM, Demuyser T, et al. COVID‐19 and drug‐induced liver injury: a problem of plenty or a petty point? Arch Toxicol. 2020;94:1367‐1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garrido I, Liberal R, Macedo G. Review article: COVID‐19 and liver disease ‐ what we know on 1st May 2020. Aliment Pharmacol Ther. 2020. 10.1111/apt.15813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kulkarni AV, Kumar P, Tevethia HV, et al. Systematic review with meta‐analysis: liver manifestations and outcomes in COVID‐19. Aliment Pharmacol Ther. 2020;52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bertolini A, van de Peppel IP, Bodewes FA, et al. Abnormal liver function tests in COVID‐19 patients: relevance and potential pathogenesis. Hepatology. 2020. 10.1002/hep.31480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu J, Song S, Cao HC, Li LJ. Liver diseases in COVID‐19: Etiology, treatment and prognosis. World J Gastroenterol. 2020;26:2286‐2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yadav DK, Singh A, Zhang Q, et al. Involvement of liver in COVID‐19: systematic review and meta‐analysis. Gut. 2020. 10.1136/gutjnl-2020-322072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M. COVID‐19 and Liver. J. Hepatol. 2020. 10.1016/j.jhep.2020.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Y, Liu S, Liu H, et al. SARS‐CoV‐2 infection of the liver directly contributes to hepatic impairment in patients with COVID‐19. J Hepatol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Lu F, Zhao J. Reply to: Correspondence relating to “SARS‐CoV‐2 infection of the liver directly contributes to hepatic impairment in patients with COVID‐19”. J Hepatol. 2020. 10.1016/j.jhep.2020.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kucharski AJ,Russell TW, Diamond C, et al. Early dynamics of transmission and control of COVID‐19: a mathematical modelling study. Lancet Infect Dis. 2020;20:553‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar‐M P, Mishra S, Jha DK, et al. Coronavirus disease (COVID‐19) and the liver: a comprehensive systematic review and meta‐analysis. Hepatol Int. 2020. 10.1007/s12072-020-10071-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Paliogiannis P, Zinellu A. Bilirubin levels in patients with mild and severe Covid‐19: A pooled analysis. Liver International. 2020;40:1787‐1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parasa S, Desai M, Chandrasekar VT, et al. Prevalence of Gastrointestinal Symptoms and Fecal Viral Shedding in Patients With Coronavirus Disease 2019. JAMA Netw Open. 2020;3:e2011335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cai Q, Huang D, Yu H, et al. COVID‐19: Abnormal liver function tests. J Hepatol. 2020. 10.1016/j.jhep.2020.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bernal‐Monterde V, Casas‐Deza D, Letona‐Giménez L, et al. SARS‐CoV‐2 Infection Induces a Dual Response in Liver Function Tests: Association with Mortality during Hospitalization. Biomedicines. 2020;8:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID‐19. The Lancet. 2020;395:1417‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lax SF, Skok K, Zechner P, et al. Pulmonary arterial thrombosis in covid‐19 with fatal outcome: results from a prospective, single‐center, clinicopathologic case series. Ann Intern Med. 2020;M20–2566: 10.7326/M20-2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ji D, Qin E, Xu J, et al. Non‐alcoholic fatty liver diseases in patients with COVID‐19: A retrospective study. J Hepatol. 2020. 10.1016/j.jhep.2020.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sonzogni A, Previtali G, Seghezzi M, et al. Liver histopathology in severe COVID 19 respiratory failure is suggestive of vascular alterations. Liver Int. 2020;40:2110‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Puelles VG, Lütgehetmann M, Lindenmeyer MT, et al. Multiorgan and Renal Tropism of SARS‐CoV‐2. N Engl J Med. 2020. 10.1056/NEJMc2011400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chappell JD, Dermody TS. Biology of Viruses and Viral Diseases. In Bennet JE, Dolin R, Blase MJ. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases (vol. 2, 1681‐1693.e4). Elsevier Inc.; 2014. [Google Scholar]
- 54. Chai X, Hu L, Zhang Y, et al. Specific ACE2 Expression in Cholangiocytes May Cause Liver Damage After 2019‐nCoV Infection. bioRxiv. 2020. 10.1101/2020.02.03.931766 [DOI] [Google Scholar]
- 55. Pirola CJ, Sookoian S. SARS‐CoV‐2 virus and liver expression of host receptors: Putative mechanisms of liver involvement in COVID‐19. Liver Int. 2020;40:2038‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hamming I, Timens W, Bulthuis MLC, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid‐19. N Engl J Med. 2020. 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Paizis G, Tikellis C, Cooper ME, et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut. 2005;54:1790‐1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang Q, Xie Q, Shi CC, et al. Expression of angiotensin‐converting enzyme 2 in CCL4‐induced rat liver fibrosis. Int J Mol Med. 2009;23:717‐723. [DOI] [PubMed] [Google Scholar]
- 60. Suárez‐Fariñas M, Tokuyama M, Wei G, et al. Intestinal inflammation modulates the expression of ACE2 and TMPRSS2 and potentially overlaps with the pathogenesis of SARS‐CoV‐2 related disease. Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gkogkou E, Barnasas G, Vougas K, Trougakos IP. Expression profiling meta‐analysis of ACE2 and TMPRSS2, the putative anti‐inflammatory receptor and priming protease of SARS‐CoV‐2 in human cells, and identification of putative modulators. Redox Biol. 2020;36:101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Muhović D, Bojović J, Bulatović A, et al. First case of drug‐induced liver injury associated with the use of tocilizumab in a patient with COVID‐19. Liver Int. 2020;40:1901‐1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS‐CoV‐2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Koshikawa N, Hasegawa S, Nagashima Y, et al. Expression of trypsin by epithelial cells of various tissues, leukocytes, and neurons in human and mouse. Am J Pathol. 1998;153:937‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mohammed FF, Khokha R. Thinking outside the cell: Proteases regulate hepatocyte division. Trends Cell Biol. 2005;15:555‐563. [DOI] [PubMed] [Google Scholar]
- 66. Shulla A, Heald‐Sargent T, Subramanya G, et al. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85:873‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Coutard B, Valle C, de Lamballerie X, et al. The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fantini J, Di Scala C, Chahinian H, Yahi N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS‐CoV‐2 infection. Int J Antimicrob Agents. 2020. 10.1016/j.ijantimicag.2020.105960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nat Commun. 2020;11:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chu H, Chan JFW, Yuen TTT, et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS‐CoV‐2 and SARS‐CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID‐19: an observational study. The Lancet Microbe. 2020;1:e14‐e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harcourt J, Tamin A, Lu X, et al. Severe acute respiratory syndrome coronavirus 2 from patient with coronavirus disease, United States. Emerg Infect Dis. 2020;26:1266‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang H, Zhou P, Wei Y, et al. Histopathologic Changes and SARS‐CoV‐2 Immunostaining in the Lung of a Patient With COVID‐19. Ann Intern Med. 2020;172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang L, Han Y, Nilsson‐Payant BE, et al. A Human Pluripotent Stem Cell‐based Platform to Study SARS‐CoV‐2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell. 2020;27:125‐136.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Banales JM, Huebert RC, Karlsen T, et al. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol. 2019;16:269‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhao B, Ni C, Gao R, et al. Recapitulation of SARS‐CoV‐2 infection and cholangiocyte damage with human liver ductal organoids. Protein and Cell. 2020;1. 10.1007/s13238-020-00718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Han D, Fang Q, Wang X. SARS‐CoV‐2 was found in the bile juice from a patient with severe COVID‐19. J Med Virol. 2020. 10.1002/jmv.26169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Moon AM, Webb GJ, Aloman C, et al. High Mortality Rates for SARS‐CoV‐2 Infection in Patients with Pre‐existing Chronic Liver Disease and Cirrhosis: Preliminary Results from an International Registry. J Hepatol. 2020. 10.1016/j.jhep.2020.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shim KY, Eom YW, Kim MY, Kang SH, Baik SK. Role of the renin‐angiotensin system in hepatic fibrosis and portal hypertension. Korean J Intern Med. 2018;33:453‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lubel JS, Herath CB, Tchongue J, et al. Angiotensin‐(1–7), an alternative metabolite of the renin‐angiotensin system, is up‐regulated in human liver disease and has antifibrotic activity in the bile‐duct‐ligated rat. Clin Sci. 2009;117:375‐386. [DOI] [PubMed] [Google Scholar]
- 81. De Minicis S, Seki E, Uchinami H, et al. Gene Expression Profiles During Hepatic Stellate Cell Activation in Culture and In Vivo. Gastroenterology. 2007;132:1937‐1946. [DOI] [PubMed] [Google Scholar]
- 82. Marcher AB, Bendixen SM, Terkelsen MK, et al. Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci Rep. 2019;9:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Akil A, Endsley M, Shanmugam S, et al. Fibrogenic Gene Expression in Hepatic Stellate Cells Induced by HCV and HIV Replication in a Three Cell Co‐Culture Model System. Sci Rep. 2019;9:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Keidar S, Gamliel‐Lazarovich A, Kaplan M, et al. Mineralocorticoid receptor blocker increases angiotensin‐converting enzyme 2 activity in congestive heart failure patients. Circ Res. 2005;97:946‐953. [DOI] [PubMed] [Google Scholar]
- 85. Shieh WJ, Hsiao CH, Paddock CD, et al. Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS‐associated coronavirus in lung of a fatal case of severe acute respiratory syndrome in Taiwan. Hum Pathol. 2005;36:303‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schaefer I‐M, Padera RF, Solomon IH, et al. In situ detection of SARS‐CoV‐2 in lungs and airways of patients with COVID‐19. Mod Pathol. 2020. 10.1038/s41379-020-0595-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhu Y, Jiang M, Gao L, Huang X. Single Cell Analysis of ACE2 Expression Reveals the Potential Targets for 2019‐nCoV. Preprints.org. 2020;1‐14. 10.20944/preprints202002.0221.v1 [DOI] [Google Scholar]
- 88. Qi F, Qian S, Zhang S, Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun. 2020;526:135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Scott CL, Zheng F, De Baetselier P, et al. Bone marrow‐derived monocytes give rise to self‐renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blériot C, Dupuis T, Jouvion G, et al. Liver‐Resident Macrophage Necroptosis Orchestrates Type 1 Microbicidal Inflammation and Type‐2‐Mediated Tissue Repair during Bacterial Infection. Immunity. 2015;42:145‐158. [DOI] [PubMed] [Google Scholar]
- 91. David BA, Rezende RM, Antunes MM, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology. 2016;151:1176‐1191. [DOI] [PubMed] [Google Scholar]
- 92. Guillot A, Tacke F. Liver Macrophages: Old Dogmas and New Insights. Hepatol Commun. 2019;3:730‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Abassi Z, Knaney Y, Karram T, Heyman SN. The Lung Macrophage in SARS‐CoV‐2 Infection: A Friend or a Foe? Front Immunol. 2020;11:1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tandra S, Yeh MM, Brunt EM, et al. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J Hepatol. 2011;55:654‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miller B, Silverstein A, Flores M, et al. SARS‐CoV‐2 induces a unique mitochondrial transcriptome signature. Prepr. 2020. 10.21203/RS.3.RS-36568/V1. https://www.researchsquare.com/article/rs‐36568/v1 [DOI] [Google Scholar]
- 96. Koliaki C, Szendroedi J, Kaul K, et al. Adaptation of hepatic mitochondrial function in humans with non‐alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739‐746. [DOI] [PubMed] [Google Scholar]
- 97. Rutkowski DT, Wu J, Back SH, et al. UPR Pathways Combine to Prevent Hepatic Steatosis Caused by ER Stress‐Mediated Suppression of Transcriptional Master Regulators. Dev Cell. 2008;15:829‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chan C‐P, Siu KL, Chin KT, et al. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2006;80:9279‐9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Versteeg GA, van de Nes PS, Bredenbeek PJ, Spaan WJM. The Coronavirus Spike Protein Induces Endoplasmic Reticulum Stress and Upregulation of Intracellular Chemokine mRNA Concentrations. J Virol. 2007;81:10981‐10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Minakshi R, Padhan K, Rani M, et al. The SARS Coronavirus 3a Protein Causes Endoplasmic Reticulum Stress and Induces Ligand‐Independent Downregulation of the Type 1 Interferon Receptor. PLoS One. 2009;4:e8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Krähling V, Stein DA, Spiegel M, Weber F, Mühlberger E. Severe Acute Respiratory Syndrome Coronavirus Triggers Apoptosis via Protein Kinase R but Is Resistant to Its Antiviral Activity. J Virol. 2009;83:2298‐2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Peterson T, Sengupta S, Harris T, et al. MTOR complex 1 regulates lipin 1 localization to control the srebp pathway. Cell. 2011;146:408‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus Replication Complex Formation Utilizes Components of Cellular Autophagy. J Biol Chem. 2004;279:10136‐10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cottam EM, Maier HJ, Manifava M, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7:1335‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cottam EM, Whelband MC, Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. 2014;10:1426‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kindrachuk J, Ork B, Hart BJ, et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015;59:1088‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gassen NC, Papies J, Bajaj T, et al. Analysis of SARS‐CoV‐2‐controlled autophagy reveals spermidine, MK‐2206, and niclosamide as putative antiviral therapeutics. bioRxiv. 2020.04.15.997254 (2020). 10.1101/2020.04.15.997254 [DOI] [Google Scholar]
- 109. He M, Shi X, Yang M, et al. Mesenchymal stem cells‐derived IL‐6 activates AMPK/mTOR signaling to inhibit the proliferation of reactive astrocytes induced by hypoxic‐ischemic brain damage. Exp Neurol. 2019;311:15‐32. [DOI] [PubMed] [Google Scholar]
- 110. Brunn GJ, Hudson CC, Sekulić A, et al. Phosphorylation of the translational repressor PHAS‐I by the mammalian target of rapamycin. Science (80‐. ). 1997;277:99‐101. [DOI] [PubMed] [Google Scholar]
- 111. Hannan KM, Brandenburger Y, Jenkins A, et al. mTOR‐Dependent Regulation of Ribosomal Gene Transcription Requires S6K1 and Is Mediated by Phosphorylation of the Carboxy‐Terminal Activation Domain of the Nucleolar Transcription Factor UBF†. Mol Cell Biol. 2003;23:8862‐8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hosokawa N, Hara T, Kaizuka T, et al. Nutrient‐dependent mTORCl association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981‐1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kim YM, Jung CH, Seo M, et al. MTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015;57:207‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577‐590. [DOI] [PubMed] [Google Scholar]
- 116. Harrington LS, Findlay GM, Gray A, et al. The TSC1‐2 tumor suppressor controls insulin‐PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hsu PP, Kang SA, Rameseder J, et al. The mTOR‐regulated phosphoproteome reveals a mechanism of mTORC1‐mediated inhibition of growth factor signaling. Science (80‐. ). 2011;332:1317‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science (80‐. ). 2011;332:1322‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lagana SM, Kudose S, Iuga AC, et al. Hepatic pathology in patients dying of COVID‐19: a series of 40 cases including clinical, histologic, and virologic data. Mod Pathol. 2020;33(11):2147‐2155. 10.1038/s41379-020-00649-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dhainaut JF, Marin N, Mignon A, Vinsonneau C, Sprung C. Hepatic response to sepsis: Interaction between coagulation and inflammatory processes. Crit Care Med. 2001;29: (Lippincott Williams and Wilkins). [DOI] [PubMed] [Google Scholar]
- 121. Geier A, Fickert P, Trauner M. Mechanisms of disease: Mechanisms and clinical implications of cholestasis in sepsis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:574‐585. [DOI] [PubMed] [Google Scholar]
- 122. Horvatits T, Drolz A, Trauner M, Fuhrmann V. Liver Injury and Failure in Critical Illness. Hepatology. 2019;70:2204‐2215. [DOI] [PubMed] [Google Scholar]
- 123. Trauner M, Fickert P, Stauber RE. Inflammation‐induced cholestasis. J Gastroenterol Hepatol (Australia). 1999;14:946‐959. [DOI] [PubMed] [Google Scholar]
- 124. Moseley RH. Sepsis‐associated cholestasis. Gastroenterology. 1997;112:302‐306. [DOI] [PubMed] [Google Scholar]
- 125. Bauer M, Press AT, Trauner M. The liver in sepsis: Patterns of response and injury. Curr Opin Crit Care. 2013;19:123‐127. [DOI] [PubMed] [Google Scholar]
- 126. Recknagel P, Gonnert FA, Westermann M, et al. Liver dysfunction and phosphatidylinositol‐3‐kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS Med. 2012;9:e1001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Xu Z, Karlsson J, Huang Z. Modeling the Dynamics of Acute Phase Protein Expression in Human Hepatoma Cells Stimulated by IL‐6. Processes. 2015;3:50‐70. [Google Scholar]
- 128. Yokomuro S, Tsuji H, Lunz JG, et al. Growth control of human biliary epithelial cells by interleukin 6, hepatocyte growth factor, transforming growth factor β1, and activin A: Comparison of a cholangiocarcinoma cell line with primary cultures of non‐ neoplastic biliary epithelial cells. Hepatology. 2000;32:26‐35. [DOI] [PubMed] [Google Scholar]
- 129. O’Hara SP, Tabibian JH, Splinter PL, Larusso NF. The dynamic biliary epithelia: Molecules, pathways, and disease. J Hepatol. 2013;58:575‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gelbmann CM, Rümmele P, Wimmer M, et al. Ischemic‐like cholangiopathy with secondary sclerosing cholangitis in critically ill patients. Am J Gastroenterol. 2007;102:1221‐1229. [DOI] [PubMed] [Google Scholar]
- 131. Zilkens C, Friese J, Koeller M, Muhr G, Schinkel C. Hepatic Failure After Injury ‐ A Common Pathogenesis With Sclerosing Cholangitis? Eur J Med Res. 2008;13:309‐313. [PubMed] [Google Scholar]
- 132. Fiorotto R, Spirlì C, Fabris L, et al. Ursodeoxycholic Acid Stimulates Cholangiocyte Fluid Secretion in Mice via CFTR‐Dependent ATP Secretion. Gastroenterology. 2007;133:1603‐1613. [DOI] [PubMed] [Google Scholar]
- 133. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J Hepatol. 2015;62:S25‐S37. [DOI] [PubMed] [Google Scholar]
- 134. Fickert P, Hirschfield GM, Denk G, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. 2017;67:549‐558. [DOI] [PubMed] [Google Scholar]
- 135. Horvatits T, Trauner M, Fuhrmann V. Hypoxic liver injury and cholestasis in critically ill patients. Curr Opin Crit Care. 2013;19:128‐132. [DOI] [PubMed] [Google Scholar]
- 136. Waseem N, Chen P‐H. Hypoxic Hepatitis: A Review and Clinical Update. J Clin Transl Hepatol. 2016;4:263‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Henrion J, Descamps O, Luwaert R, et al. Hypoxic hepatitis in patients with cardiac failure: incidence in a coronary care unit and measurement of hepatic blood flow. J Hepatol. 1994;21:696‐703. [DOI] [PubMed] [Google Scholar]
- 138. Henrion J. Hypoxic hepatitis. Liver International. 2012;32:1039‐1052. [DOI] [PubMed] [Google Scholar]
- 139. Birrer R, Takuda Y, Takara T. Hypoxic hepatopathy: Pathophysiology and prognosis. Intern Med. 2007;46:1063‐1070. [DOI] [PubMed] [Google Scholar]
- 140. Seeto RK, Fenn B, Rockey DC. Ischemic hepatitis: Clinical presentation and pathogenesis. Am J Med. 2000;109:109‐113. [DOI] [PubMed] [Google Scholar]
- 141. Mallory FB. Necroses of the Liver. J Med Res. 1901;6:264‐280.7. [PMC free article] [PubMed] [Google Scholar]
- 142. Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS‐CoV‐2 pneumonia in Wuhan, China: a single‐centered, retrospective, observational study. Lancet Respir Med. 2020;8:475‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Qiu H, Tong Z, Ma P, et al. Intensive care during the coronavirus epidemic. Intensive Care Med. 2020;46:576‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Arabi YM, Fowler R, Hayden FG. Critical care management of adults with community‐acquired severe respiratory viral infection. Intensive Care Med. 2020;46:315‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Alhazzani W, Møller MH, Arabi YM, et al. Surviving Sepsis Campaign: guidelines on the management of critically ill adults with Coronavirus Disease 2019 (COVID‐19). Intensive Care Med. 2020;46:854‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Vieillard‐Baron A, Naeije R, Haddad F, et al. Diagnostic workup, etiologies and management of acute right ventricle failure: A state‐of‐the‐art paper. Intensive Care Med. 2018;44:774‐790. [DOI] [PubMed] [Google Scholar]
- 147. Fayssoil A, Mustafic H, Mansencal N. The Right Ventricle in COVID‐19 Patients. J Clean Prod. 2020;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Grillet F, Behr J, Calame P, Aubry S, Delabrousse E. Acute Pulmonary Embolism Associated with COVID‐19 Pneumonia Detected with Pulmonary CT Angiography. Radiology. 2020;296:E186‐E188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Bloom PP, Meyerowitz EA, Reinus Z, et al. Liver Biochemistries in Hospitalized Patients With COVID‐19. Hepatology. hep.31326 (2020): 10.1002/hep.31326 [DOI] [PubMed] [Google Scholar]
- 150. Hao SR, Zhang SY, Lian JS, et al. liver enzyme elevation in coronavirus disease 2019: a multicenter, retrospective, Cross‐Sectional Study. Am J Gastroenterol. 2020;115:1075‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hundt MA, Deng Y, Ciarleglio MM, Nathanson MH, Lim JK. Abnormal Liver Tests in COVID‐19: A Retrospective Observational Cohort Study of 1827 Patients in a Major U.S. Hospital Network. Hepatology. 2020. 10.1002/hep.31487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yip TCF, Lui GCY, Wong VWS, et al. Liver injury is independently associated with adverse clinical outcomes in patients with COVID‐19. Gut. 2020. 10.1136/gutjnl-2020-321726 [DOI] [PubMed] [Google Scholar]
- 153. Iavarone M, D’Ambrosio R, Soria A, et al. High rates of 30‐day mortality in patients with cirrhosis and COVID‐19. J Hepatol. 2020. 10.1016/j.jhep.2020.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Jiang S, Wang R, Li L, et al. Liver Injury in Critically Ill and Non‐critically Ill COVID‐19 Patients: A Multicenter, Retrospective, Observational Study. Front Med. 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Corticosteroids for COVID‐19. https://www.who.int/publications/i/item/WHO‐2019‐nCoV‐Corticosteroids‐2020.1
- 156. LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury [Internet]. LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury (National Institute of Diabetes and Digestive and Kidney Diseases, 2012). [PubMed]
- 157. Tocilizumab . LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury (National Institute of Diabetes and Digestive and Kidney Diseases, 2012). [PubMed]
- 158. Wong SH, Lui RNS, Sung JJY. Covid‐19 and the digestive system. J Gastroenterol Hepatol (Australia). 2020. 10.1111/jgh.15047 [DOI] [PubMed] [Google Scholar]
- 159. Wang W, Xu Y, Gao R, et al. Detection of SARS‐CoV‐2 in Different Types of Clinical Specimens. JAMA ‐ Journal of the American Medical Association. 2020. 10.1001/jama.2020.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Xiao F, Tang M, Zheng X, et al. Evidence for Gastrointestinal Infection of SARS‐CoV‐2. Gastroenterology. 2020;158:1831‐1833.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Zhang JC, Wang SB, Xue YD. Fecal specimen diagnosis 2019 novel coronavirus–infected pneumonia. J Med Virol. 2020;92:680‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Amirian ES. Potential Fecal Transmission of SARS‐CoV‐2: Current Evidence and Implications for Public Health. Int J Infect Dis. 2020;95:363‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Jin X, Lian JS, Hu JH, et al. Epidemiological, clinical and virological characteristics of 74 cases of coronavirus‐infected disease 2019 (COVID‐19) with gastrointestinal symptoms. Gut. 2020. 10.1136/gutjnl-2020-320926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Lin L, Jiang X, Zhang Z, et al. Gastrointestinal symptoms of 95 cases with SARS‐CoV‐2 infection. Gut. 2020. 10.1136/gutjnl-2020-321013 [DOI] [PubMed] [Google Scholar]
- 165. Pan L, Mu M, Yang P, et al. Clinical Characteristics of COVID‐19 Patients With Digestive Symptoms in Hubei, China. Am J Gastroenterol. 2020;1: 10.14309/ajg.0000000000000620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Feng Z, Wang Y, Qi W. The small intestine, an underestimated site of SARS‐CoV‐2 infection: from Red Queen effect to probiotics. Preprints doi:10.20944/preprints202003.0161.v1. 2020. 10.20944/preprints202003.0161.v1 [DOI]
- 167. Zhang H, Li HB, Lyu JR, et al. Specific ACE2 Expression in Small Intestinal Enterocytes may Cause Gastrointestinal Symptoms and Injury after 2019‐nCoV Infection. Int J Infect Dis. 2020;96:19‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhang H, Kang Z, Gong H, et al. Digestive system is a potential route of COVID‐19: An analysis of single‐cell coexpression pattern of key proteins in viral entry process. Gut. 2020;69:1010‐1018. [Google Scholar]
- 169. Lamers MM, et al. SARS‐CoV‐2 productively infects human gut enterocytes. Science (80‐. ). eabc1669 (2020). 10.1126/science.abc1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Hindson J. COVID‐19: faecal–oral transmission? Nat Rev Gastroenterol Hepatol. 2020;17:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Murakami K, Tenge VR, Karandikar UC, et al. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc Natl Acad Sci USA. 2020;117:1700‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Grau KR, Zhu S, Peterson ST, et al. The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid‐mediated priming of type III interferon. Nature Microbiology. 2020;5:84‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Liu F, Long X, Zhang B, et al. ACE2 Expression in Pancreas May Cause Pancreatic Damage After SARS‐CoV‐2 Infection. Clin Gastroenterol Hepatol. 2020. 10.1016/j.cgh.2020.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Pal R, Banerjee M. COVID‐19 and the endocrine system: exploring the unexplored. J Endocrinol Invest. 2020;1–5: 10.1007/s40618-020-01276-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. COVID‐Hep registry . https://www.covid‐hep.net/
- 176. SECURE Cirrhosis Registry . https://covidcirrhosis.web.unc.edu/