

Abstract
Head and neck squamous cell carcinoma (HNSCC) encompasses a set of cancers arising from the epithelia of the upper aerodigestive tract, accounting for a significant burden of disease worldwide due to the disease’s mortality, morbidity, and predilection for recurrence. Prognosis of HNSCC in the recurrent and/or metastatic (R/M-HNSCC) setting is especially poor and effective treatment options increasingly rely on modulating T-cell antitumor responses. Still, immunotherapy response rates are generally low, prompting the exploration of novel strategies that incorporate other effector cells within the tumor microenvironment. Within the last decade, important advances have been made leveraging the powerful innate antitumor function of natural killer (NK) cells to treat solid tumors, including head and neck squamous cell carcinoma. NK cells are hybrid innate-adaptive effector cells capable of directly eliminating tumor cells in addition to initiating adaptive antitumor immune responses. In the setting of HNSCC, NK cells are important for tumor surveillance and control, and NK cell infiltration has repeatedly been associated with a favorable prognosis. Yet, HNSCC-infiltrating NK cells are susceptible to an array of immune evasion strategies employed by tumors that must be overcome to fully realize the antitumor potential of NK cells. We believe that a conceptual framework informed by the basic biological understanding of the mechanisms underlying NK cell activation can improve treatment of HNSCC, in part by selecting for patients most likely to respond to NK cell-based immunotherapy. Herein, we review the activity of NK cells in HNSCC, paying special attention to the role of environmental and genetic determinants of NK cell antitumor function. Moreover, we explore the evidence that NK cells are a crucial determinant of the efficacy of both established and emerging treatments for HNSCC.
Keywords: antigen presentation, costimulatory and inhibitory T-cell receptors, cytotoxicity, immunologic, killer cells, natural, head and neck neoplasms
Background
Head and neck squamous cell carcinoma (HNSCC) is an anatomically and etiologically diverse set of cancers arising in the upper aerodigestive tract, including the oral cavity, nasopharynx, oropharynx, hypopharynx, and larynx. The most commonly cited risk factors for HNSCC are environmental and include high-risk human papillomavirus (HPV) infection, tobacco smoke, and alcohol usage, among others.1 While the incidence of HPV-negative HNSCC has decreased in the USA over the last decades primarily due to decreased tobacco consumption, the incidence of HPV-positive HNSCC is rising rapidly.2 Although HPV-associated and HPV-unassociated HNSCC are anatomically related, their pathophysiology and clinical behaviors are remarkably distinct.3 Accordingly, HPV-associated and unassociated HNSCC tumors are now clinically regarded as separate entities, and where possible, we have treated them as such in this review. Immune checkpoint inhibitors that unleash T cells against recurrent HNSCC tumors have led to remarkable results for some patients, although in the majority of cases these agents will fail. As such, there is now interest in investigating other immune effector cells, including natural killer (NK) cells, as targets for immunotherapy.
NK cells are CD3-CD56+ innate lymphoid cells that develop in the bone marrow and secondary lymphoid tissue, including the spleen, tonsils, and lymph nodes.4 NK cells possess features of both innate and adaptive immune cells, in the sense that they do not undergo genetic recombination to assemble a diverse set of antigen-specific receptors, but are still capable of long-term ‘memory-like’ or ‘recall’ immune responses to antigens.5 6 NK cells are critical for the elimination of virally infected and transformed cells. Furthermore, NK-mediated cytolysis occurs more quickly than adaptive lymphocytes because NK cells are not reliant on in situ licensing.6 Taken together, these properties make NK cells attractive targets for emerging immunotherapies.
Regulation of NK cell activation
NK cell functions are mediated through a process of ‘education’ defined as the training of NK cells to distinguish diseased or allogeneic cells, which exhibit perturbed expression of human leukocyte antigen (HLA) class I.7 Broadly speaking, human NK cells are primarily educated via inhibitory signaling through either the NKG2A or KIR receptors, which bind HLA-E or HLA-A, HLA-B, HLA-C, HLA-F, and HLA-G, respectively7–9 (figure 1). The evidence suggests that the path through which NK cells are educated is dominantly determined by a single dimorphism at the −21 residue of the HLA-B leader sequence and the presence or absence of KIR ligands.8 HLA-B −21 methionine (−21 M) individuals (approximately 50 % allelic frequency7) encode a methionine at this residue in the signal sequence and are capable of producing HLA-B-derived leader peptides that provide stability to the HLA-E protein, whereas −21 threonine (−21 T) produces peptide that binds unstably to HLA-E and results in restricted cell-surface expression.8 10 The −21 M/T HLA-B dimorphism has significant implications for a range of clinically important immunological processes, such as HIV viral load control,11 response to interleukin (IL)-2 immunotherapy,12 and incidence of graft-versus-host disease after hematopoietic stem cell transplantation,13 although to date its potential role in solid tumor control has not been investigated.
Figure 1.
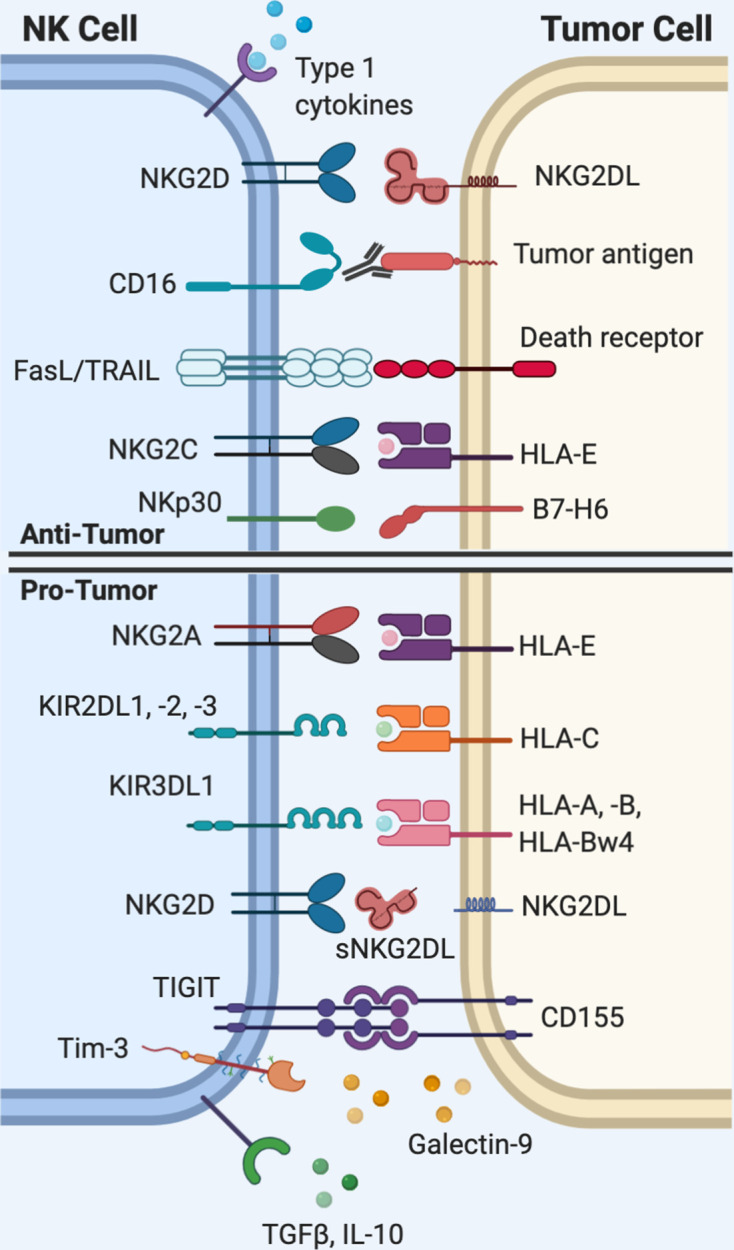
Natural killer (NK) cell interactions with tumor cells. Activation of NK cells requires that the net total of activating signals outweighs inhibitory signals. NK cells rely on a set of germline-encoded receptors to recognize ‘altered-self,’ or ‘missing-self’ patterns of protein expression on tumor cells. The diverse range of activating and inhibitory receptors expressed on NK cells are of interest as targets for immunotherapy. HLA, human leukocyte antigen; IL-10, interleukin-10; Tim-3, T cell immunoglobulin mucin-3; TRAIL, tumor necrosis factor-related apoptosis-inducingligand (TRAIL); TGFβ, transforming growth factor beta; TIGIT, T cell immunoreceptor with Ig and ITIM domains.
NK cells are directly activated via signaling through the CD16 receptor, which crosslinks after binding the Fc region of immunoglobulin (Ig)G bound to target cells and triggers antibody-dependent cellular cytoxicity (ADCC). Additionally, direct activating signals are generated through activating receptors expressed on the NK cell surface such as NKG2D and natural cytotoxicity receptors (NCRs) such as NKp30 and NKp46, which bind activating ligands that are upregulated on stressed, virally infected, and tumor cells.14 15 Common human NKG2D ligands include, but are not limited to, the major histocompatibility complex (MHC) class-1 chain-related protein A and B (MICA and MICB) and the ULBP protein family.15 Indirect activation, on the other hand, is accomplished through soluble factors, most notably interleukin (IL)-2, IL-12, IL-15, IL-18, IL-21, tumor necrosis factor-ɑ (TNF-ɑ), and type I interferon (IFN).16–18 Activation of NK cells requires that the net total of activating signals exceeds inhibitory signals.19
NK cells and cancer
There is extensive evidence that NK cells are critical components of tumor control. NK cells are a major source of IFN during early-phase immune responses, underscoring their role as rapid-acting immune effectors.20 IFNγ acts directly on tumor cells to enhance their immunogenicity and sculpts the immune response by differentiation of naive CD4+ T cells toward Th1 cells that promote cell-mediated antitumor responses (figure 2).20 IFNγ additionally strengthens crosstalk between myeloid cells (dendritic cells (DCs) and macrophages), effector memory CD4+ T cells, and naive effector CD8+ T cells.21 NK cells are also capable of producing the chemokines CCL5, XCL1, and Flt3L, which recruit conventional type I DCs (cDC1s) and naive effector CD8+ T cells that bolster antitumor immunity.22 23 In addition to their role as cytokine producers, NK cells are also able to directly kill tumor cells via cytotoxic granules or expression of death-receptor ligands, including but not limited to Fas-ligand and TNF-related apoptosis-inducing ligand.24 More generally, the TNF family of ligands and receptors plays a central role in tumor elimination.25
Figure 2.

Natural killer (NK) cells form dynamic interrelationships with immune, stromal and tumor cells with the tumor microenvironment (TME) of head and neck cancer. cDC1, conventional type 1 dendritic cells (DCs). (a) NK cells produce interferon (IFN)-gamma within the TME, which acts on both tumor and neighboring immune cells to enhance antitumor responses. NK cells both recruit cDC1 and provide differentiation and maturation signals for DCs with the TME. (b) Cell–cell interactions between NK cells and neighboring cells determine strength of activation. (c) Cetuximab, monalizumab, and ipilimumab represent some, but not all, monoclonal antibodies that at least in part rely on NK cell activation for full antitumor function. HLA, human leukocyte antigen.
Loss of inhibitory signals is a common pathway of NK activation in response to tumors. During the process of viral infection or oncogenic transformation, cells frequently lose their repertoire of surface MHC.26 While HLA-A, HLA-B, and HLA-C are necessary to promote tumor-specific activation of CD8 T cells, they may simultaneously inhibit NK cells through provision of inhibitory KIR ligands and by increasing expression of HLA-E. Therefore, loss of HLA permits evasion of adaptive immune responses that require antigen presentation via MHC, but it simultaneously disinhibits NK cells and allows for activation of their cytotoxic function. In other words, what is effective for triggering NK cell activation may prove suboptimal for CD8+ T cells and, conversely, what is optimal for CD8+ T cells may prove deleterious for NK cell activation. Elimination of tumors that have lost or altered their HLA expression pattern has been postulated as a mechanism of NK-mediated nascent tumor elimination, although measuring the true frequency of this phenomenon has not been established in vivo.6 The persistence of some tumors that lack HLA, however, implies that isolated HLA loss alone is not sufficient for NK-mediated tumor elimination in all cases.
NK cells in HNSCC
NK cell activity in the tumor microenvironment and the clinical prognosis of HNSCC
Pan-cancer transcriptomic analyses have revealed that both HPV+ and HPV- HNSCC are among the most highly infiltrated tumors, and that immune infiltration is higher among HPV+ than HPV- HNSCC.27 Furthermore, based on these transcriptional analyses, HNSCC exhibits the highest median CD56dim NK cell infiltration of any major tumor type.27 HPV+ HNSCC is more highly infiltrated with NK cells than HPV- HNSCC.28 Increased infiltration of NK cells, specifically CD56dim, is associated with improved disease-free and overall survival independent of HPV status.27–31 Peripheral NK cell activity also correlates with fewer pharyngeal cancer metastases.30 Compared with circulating NK cells, HNSCC-infiltrating NK cells more frequently express the inhibitory receptor NKG2A and less frequently express the inhibitory KIR (KIR3DL1 and KIR2DL1/2/3).32 33 Although the dominant NK cell subset in HNSCC tumors is mature CD56 dim NK cells, compared with other major tumor types, HNSCC is enriched in immature, cytokine-producing CD56bright, CD16dim/negative NK cells lacking expression of CD57.28 32 34 Basal ADCC activity in HNSCC tumors varies considerably.35
The presence or absence of activating and inhibitory NK ligands in the TME sculpts the NK response to HNSCC
NKG2D is an activating transmembrane receptor expressed on NK cells and on a subset of T cells.36 The ligands for NKG2D, including MICA/B, ULBP1/2/3, and Rae-1, are preferentially overexpressed on transformed cells and render them susceptible to NK-mediated tumor elimination.15 Tumors are capable of shedding soluble NKG2D ligands via proteolytic cleavage, which can paradoxically render NK cells less cytotoxic, possibly by non-functional occupation of the NKG2D receptor binding site.37 Levels of soluble NKG2D ligands (MICA/B and ULBP1/2/3) in the plasma of patients with HNSCC are significantly elevated compared with healthy controls, presumably due to shedding from the primary tumor after proteolytic cleavage.34 Primary healthy human NK cells incubated with plasma derived from patients with HNSCC were significantly less efficient at killing target cells in vitro; this effect was reversed by depletion of soluble NKG2D ligands prior to incubation, which suggests that NKG2D ligand shedding may be an important mechanism of HNSCC immune evasion.34
Defects in HLA expression in HNSCC are a common event.38 HLA class I expression is lower in HPV+ than HPV- oropharyngeal HNSCC.39 Interestingly, low HLA class I expression is associated with favorable prognosis in HPV+ HNSCC, but a poor prognosis in HPV- HNSCC.39 40 This may reflect the observation that HPV- HNSCC are relatively less infiltrated with NK cells compared with HPV+ HNSCC,28 41 and therefore may be protected from the consequences of HLA loss.
Distinguishing virally associated head and neck cancers
HPV+ and HPV- HNSCC primary tumors share a similar mutational and copy-number alteration burden; however, their mutational profiles, mechanism of malignant transformation, and clinical behavior are markedly distinct.42 43 HPV-associated tumorigenesis occurs via expression of the viral proteins E6 and E7, which inactivate p53 and retinoblastoma protein (RB), respectively. High-risk HPV infection also leads to upregulation of the cyclin-dependent kinase inhibitor p16, which is widely used as a surrogate marker for HPV positivity in diagnostic testing.44 HPV+ HNSCC most commonly arises in the oropharyngeal tonsillar tissue and is associated with a more favorable prognosis than HPV- HNSCC, even when controlling for tumor stage, smoking status, and other potential confounders.3 Recent large-scale tumor sequencing initiatives such as The Cancer Genome Atlas and MSK-IMPACT revealed that HPV- and HPV+ HNSCC harbor distinct mutational profiles.42 43 Overall, HPV- HNSCC mimicked the mutational landscape of lung and esophageal squamous cell carcinoma, which share tobacco smoke as a common risk factor.42 The tumor suppressor gene TP53 is mutated in the majority of HPV- HNSCC primary, locoregionally advanced, and recurrent/metastatic tumors, and it is independently associated with a poor prognosis.42 43 45 TP53 is rarely mutated in HPV+ HNSCC.45 Even when controlling for HPV and smoking status, TP53 mutations in HNSCC correlated with reduced NK cell infiltration and markers of NK cytolytic activity.46 The authors observed HLA downregulation in specimens with TP53 mutations, which may explain why these tumors were more immunosuppressed, even though they bore a higher mutational—and therefore neoantigen—burden.46
The Epstein-Barr virus belongs to the herpesvirus family and is an etiologic agent of nasopharyngeal carcinoma (NPC). The immune landscape of NPC and associated NK cell characterization is not as well-defined as non-nasopharyngeal HNSCC. NPC are characteristically well-infiltrated, ‘immune-hot’ tumors. Like oropharyngeal carcinoma, NK cell infiltration is correlated with longer overall survival and progression-free survival.31 Pretreatment blood sampling demonstrated NPC tended to have lower expression of the NCRs NKp30 and NKp46 compared with healthy donors.47 It is still unknown, however, how highly NKG2A, HLA-E, and other important ligands or receptors are expressed in NPC.
Over 50% of the US population is latently infected with cytomegalovirus (CMV), and some countries approach near 100% CMV seroprevalence.48 Although infection is not a risk factor for HNSCC, CMV exposure profoundly shapes the repertoire of NK cells (and T cells) within an individual. CMV infection serves as a useful case study of how environmental exposures imprint on NK and T cell diversity, with important consequences for immunotherapy. For instance, CMV infection may lead to stable increases in numbers of adaptive or ‘memory-like’ NK cells that express the activating isoform of NKG2A: NKG2C.49–51 Like NKG2A, NKG2C similarly recognizes HLA-E, but preferentially binds.51 Expansion of NKG2C+ NK cells in response to CMV infection is dependent on sequence variation in the viral UL40-derived peptides, which form a complex with HLA-E.50 This effect can be highly significant—as much as 60% of CD56dim NK cells in some CMV-seropositive individuals are composed of clonally expanded, educated NKG2C+ NK cells.51 NKG2A expression on NK cells mutually exclusive of NKG2C expression. CMV-expanded NKG2C+ NK cell populations have subsequently been found to bear epigenetic changes that alter inhibitory receptor expression, cytokine production and responsiveness, proliferative capacity, and signaling protein expression.52 53
Immunotherapy: moving beyond T cells
Although immunotherapy has heralded a new era in the treatment of HNSCC, rates of response to treatment have remained low. As a result, research has begun to focus on how combination regimens may synergize to improve responses by unlocking the activity of other effector cells. NK cells are at the nexus of these investigations. Given our understanding of the complex interrelationships between NK cells and other critically important antitumor effector cells, it is worth considering how NK cell function and dysfunction may contribute to the success or failure of immunotherapy in head and neck cancer.
Anti-epidermal growth-factor receptor
Before the approval of pembrolizumab and nivolumab, cetuximab was the only monoclonal antibody approved for the treatment of HNSCC.54–56 Cetuximab targets and prevents signaling through the transmembrane protein epidermal growth-factor receptor (EGFR), which is overexpressed on over 80% of head and neck cancers and is associated with increased risk of recurrence and worse overall survival.57 Cetuximab monotherapy benefits only 10% – 15% of patients with R/M-HNSCC; it is therefore important to understand what distinguishes responders from non-responders.57 The efficacy of cetuximab has been hypothesized to be due at least in part to its ability to activate NK cells and trigger ADCC and cytokine secretion.58 Evidence for this claim comes from the observation that HNSCC patients with higher baseline levels of ADCC and EGFR expression had greater overall survival after cetuximab treatment; additionally, other anti-EGFR agents that do not trigger NK-mediated ADCC fail to provide benefit for R/M-HNSCC.35 58 59 Cetuximab-activated NK cells promote DC maturation through IFN-gamma secretion, which leads to increased tumor antigen cross-presentation to CD8+ T cells and subsequent expansion of EGFR-specific T cells.60 NK:DC crosstalk is bidirectional—IFN-gamma produced by cetuximab-activated NK cells leads to increased DC expression of the NKG2D ligand MICA, which reciprocally bolsters NK cell activation (figure 2).61
FcR polymorphisms have been shown to predict cetuximab response rates in colorectal cancer, although a similar analysis in cetuximab-treated head and neck cancer patients did not reveal any association between FcRIIIa genotype and disease-specific survival.60 62 63 None of these studies, however, stratified patients according to HLA-B −21 M/T genotype, which has been shown experimentally to influence capacity for ADCC.8 Various agents are now being investigated, including IL-12, lenalidomide, monalizumab (anti-NKG2A), and the CD137/4-1BB agonist urelumab, in combination with cetuximab, in order to enhance NK activation and ADCC in head and neck cancer.33 64–66
Cetuximab is also associated with the induction of several immunosuppressive axes which may limit its overall effectiveness. Patients with locoregionally advanced HNSCC treated with cetuximab had expansion of CD39+ Foxp 3+ regulatory T cells (Treg) intratumorally, the extent of which was associated with a worse overall survival.67 HNSCC-derived Tregs suppress cetuximab-mediated NK cell ADCC in vitro. 67 Cetuximab treatment is also associated with increased expression of CTLA-4, TIM-3, and TGF-beta on intratumoral Tregs, likely involving NK-mediated DC maturation.67 CTLA-4 signaling in Treg cells leads to production of TGF-beta, which directly inhibits NK cell effector functions (figure 2).68 69 Because of these findings, the anti-CTLA4 mAb ipilimumab is now being studied in phase I trials in combination with cetuximab for HNSCC (NCT01935921).
Anti-PD-(L)1
Most previously published studies have provided evidence suggesting Programmed cell death protein 1 (PD-1) is not induced on human NK cells. However, several groups have shown PD-1 expression on NK cells in certain clinical settings,70 71 including head and neck cancer.72 To date, the data are inconclusive, and PD-1 expression on NK cells is outside the scope of this review. This topic has been discussed extensively by Judge et al.73 Regardless, the observation that depletion of NK cells eliminated the efficacy of PD-1/PD-L1 blockade in tumor-bearing mice suggests that PD-1-based immunotherapy for HNSCC may benefit from proper NK cell function.74 This is possibly by preventing immunosuppressive interactions between NK cells and other PD-1-expressing cells within the tumor microenvironment (TME), such as myeloid-derived suppressor cells (MDSCs).75 However, there are no data suggesting direct interactions between NK cells and PD-1-expressing MDSCs. Strikingly, high levels of pretreatment NK cell gene expression signatures in tumors predicted longer progression-free survival for patients with solid tumors—including HNSCC—after anti-PD-1 therapy, more than any other immune signature, including PD-1 expression and CD8+ T cell signatures.76 It is worth noting, however, that the NK cell gene signature in Prat et al was minimally defined by the expression of three genes (SPN, XCL2, NCR1). Further analyses are required, as these genes are not unique to NK cells. Tumors are capable of responding to IFN-gamma produced by NK cells, which leads to increased PD-L1 expression.77 Intratumoral NK cells are also indirectly responsible for PD-1 responsiveness by producing FLT3L, CCL5, and XCL1, which collectively enrich cDC1 in the TME that themselves closely correlate with PD-1 inhibitor responsiveness.22 23
Anti-NKG2A
NKG2A is an immunoreceptor tyrosine-based inhibition motif (ITIM)-bearing receptor that forms a heterodimer with CD94. On binding of NKG2A with its ligand HLA-E, the tyrosine phosphatase SHP-1 is recruited, which ultimately leads to downstream inhibition of NK-mediated cytotoxicity, including ADCC.78 NKG2A is stably expressed on approximately half of circulating and tumor-filtrating NK cells, and it was enriched on HNSCC-infiltrating CD8+ T cells.33 HLA-E expression protects tumor cells from NK-mediated killing.79 80 HLA-E is enriched on a broad range of carcinomas, including HNSCC, and is associated with poor prognosis.81–83 Expression of soluble HLA-E by tumors has been observed in ex vivo studies of human melanoma cells and in cell lines derived from various tumor types, but has not yet been directly described in HNSCC.84
In 2018, André et al reported the results of an ongoing trial investigating a humanized IgG4 anti-NKG2A monoclonal antibody, monalizumab, for R/M-HNSCC. In combination with cetuximab, monalizumab treatment led to an objective response rate (ORR) of 27.5% in patients with HNSCC, per Response Evaluation Criteria in Solid Tumors (RECIST) criteria.33 Although of course direct comparisons cannot be made without a randomized controlled trial, monalizumab-cetuximab compared favorably to single-agent cetuximab, which has a historical ORR of 13%.85 In vitro studies revealed that monalizumab enhanced NK cell and CD8+ T cell effector functions, and this effect synergized with both durvalumab and cetuximab separately.33 Monalizumab monotherapy is ineffective at promoting ADCC in vitro but synergizes with cetuximab to enhance ADCC. This suggests that monalizumab provides the additional benefit of potentiating cetuximab-induced NK-mediated ADCC. To date, no results have been published on monalizumab in combination with PD-(L)1 axis blockade in HNSCC, but this combination is actively being studied in microsatellite-stable colorectal cancer (NCT02671435), and will be included as an expansion cohort in NCT02643550. It remains to be seen how HLA genetics may stratify responders and non-responders, and whether HLA-E expression levels during NK cell education at steady-state predict the magnitude of response to monalizumab. Furthermore, stable expansion of NKG2C+ cells, such as occurs after CMV infection, may modulate the response to monalizumab, as NKG2C:HLA-E binding leads to NK cell activation rather than inhibition.83 Thus, we advocate that future studies of anti-NKG2A treatment substratify patients according to CMV-seropositivity, HLA-E expression and HLA-B genotype.
New directions
KIR blockade
The observation that allogeneic transfer of NK cells lacking inhibitory KIR for recipient HLA prevents relapse in patients with some hematological malignancies has generated interest in KIR blockade for cancer treatment.86–88 To that end, the anti-KIR2D monoclonal antibody lirilumab (IPH-2101) was developed to limit inhibitory KIR2DL1, KIR2DL2, and KIR2DL3 signaling and improve NK antitumor response.89 Lirilumab is capable of binding to and blocking KIR2DL1, KIR2DL2, and KIR2DL3 expressed on approximately half of peripheral NK cells, blocking their inhibitory interaction with HLA-C, in addition to some off target blockade of KIR2DS1 and KIR2DL2.89 Lirilumab showed considerable efficacy in preclinical mouse studies, where it was capable of clearing leukemic blasts after just a single injection.89 Lirilumab was studied in combination with nivolumab for patients with platinum-refractory R/M-HNSCC (NCT01714739). Early results of this phase I/II study showed an encouraging improvement in objective response rate, although the trial was abandoned in November 2017 due to lack of evidence for benefit provided by the combination regimen. Lirilumab is now being studied in the neoadjuvant setting alongside nivolumab for HNSCC (NCT03341936).
Why have the promising preclinical results of KIR blockade not yet been realized in the clinic? Separate analyses in patients with smoldering multiple myeloma have shown that NK cells derived from patients receiving lirilumab unexpectedly showed NK cell hyporesponsiveness.90 Lirilumab treatment induced significant contraction of KIR2DL-positive NK cells by triggering trogocytosis of lirilumab-bound KIR protein.90 This observation raises special concern for KIR2D+ NK cells that express no other self-reactive inhibitory KIR, as loss of KIR2D signaling may compromise the education of this subset of NK cells and induce functional hyporesponsiveness.
TIGIT blockade
The poliovirus receptor (PVR), or CD155, is the ligand for TIGIT, an inhibitory receptor expressed on NK cells and subsets of T and Treg cells.91 TIGIT signaling prevents NK-mediated cellular lysis and is associated with depressed capacity for cytokine production and degranulation capacity.92 TIGIT competes with the DNAM-1 receptor (CD226), which similarly binds CD155 but with lower affinity than TIGIT and provides an activating rather than inhibitory signal to NK cells93; DNAM-1 is enhanced on educated NK cells.94 Thus, TIGIT and DNAM-1 provide opposing regulatory signals to NK cells, and this opposition has important functional implications for tumor control. In melanoma, NK cells rely on DNAM-1:CD155 signaling to prevent metastasis.95 Antibody blockade of TIGIT in vitro and in mice reduces NK cell exhaustion, inhibits tumor growth, and enhances NK-mediated proinflammatory cytokine production.96 97 TIGIT blockade monotherapy was insufficient to induce tumor regression, but TIGIT/PD-1 co-blockade produced durable antitumor responses and prolonged survival compared with anti-PD-1 monotherapy in tumor-bearing mice.98 Based on these promising preclinical results, several anti-TIGIT mAbs have been developed and now are being investigated in phase I/II trials either as monotherapy or in combination with other checkpoint inhibitors for patients with advanced malignancies (table 1).99 The results of the ongoing phase III SKYSCRAPER trials in the setting of advanced lung cancer may be the first indication of the true clinical efficacy of anti-TIGIT agents (NCT04294810, NCT04256421).
Table 1.
Relevant clinical trials of NK-based immunotherapy for head and neck squamous cell carcinoma
| Agent | Target | Antibody type | Clinical trial | Type of trial | Condition | Treatment | Patient number | Notes | Reference |
| Monalizumab | NKG2A | Humanized IgG4 | NCT02643550 | Phase Ib/II | R/M-HNSCC±prior systemic therapy | Monalizumab with cetuximab±anti-PD(L)1 | 140 | 33 83 | |
| Ipilimumab | CLTA-4 | Fully human IgG1 | NCT01935921 | Phase Ib | LA-HNSCC | Cetuximab and IMRT with ipilimumab | 19 | ||
| Urelumab | CD137 (4-1BB) | Fully human IgG1 | NCT02110082 | Phase Ib | LA/R/M - HNSCC | Cetuximab with urelumab | 66 | Completed | 66 |
| IL-12 | N/A | N/A | NCT01468896 | Phase I/II | R/M-HNSCC | Cetuximab with IL-12 | 23 | Completed | 64 |
| Lenalidomide | N/A | N/A | NCT01254617P | hase I | Advanced solid tumors | Cetuximab with lenalidomide | 24 | Completed | 65 |
| Lirilumab | KIR2D | Fully human IgG4 | NCT03347123P | Phase I/II | Advanced solid tumors | Epacodostat with lirilumab with nivolumab | 11 | ||
| Lirilumab | KIR2D | Fully human IgG4 | NCT03341936 | Phase IIR | Recurrent HNSCC undergoing salvage surgery | Neoadjuvant lirilumab with nivolumab, followed by adjuvant lirilumab with nivolumab | 58 | ||
| Lirilumab | KIR2D | Fully human IgG4 | NCT01714739 | Phase I/II | Platinum-refractory R/M-HNSCC | Lirilumab with nivolumab | 337 | Completed | |
| AB154 | TIGIT | Humanized IgG1 | NCT03628677 | Phase I | Advanced solid tumors | AB154 with zimberelimab (αPD-1) | 66 | ||
| BMS-986207 | TIGIT | IgG1, FcγR null | NCT02913313 | Phase I/II | Advanced solid tumors | BMS-986207 with nivolumab | 170 | ||
| MBG453 | TIM-3 | Humanized IgG4 | NCT02608268 | Phase I-Ib/II | Advanced solid tumors | MBG453±PDR 001 (αPD-1) | 267 | ||
| BGB-A425 | TIM-3 | Humanized IgG1 | NCT03744468 | Phase I/II | Advanced solid tumors | BGB-A425±tislelizumab (αPD-1) | 162 | ||
| Relatlimab | LAG3 | Fully human IgG4 | NCT01968109 | Phase I/IIA | Advanced solid tumors | Relatlimab±nivolumab | 1500 | ||
| Sym022 | LAG3 | Fully human, Fc-inert | NCT03311412 | Phase I | Advanced solid tumors | Sym021 (αPD-1)±Sym 022 | 102 | ||
| FATE-NK100 | N/A | N/A | NCT03319459 | Phase I | Advanced solid tumors | FATE-NK100±cetuximab or trastuzumab | 100 | Ex vivo activated donor NK cells | |
| CIML NK cells | N/A | N/A | NCT04290546 | Phase I | Advanced head and neck cancer | CIML NK+IL-15±ipilimumab | 12 | Cytokine-induced memory-like NK cells | 114 |
IL-2, interleukin-2; LAG3, lymphocyte activating gene 3; NK, natural killer; R/M HNSCC, recurrent and/or metastatic head and neck squamous cell carcinoma; TIM-3, T cell immunoglobulin mucin-3.
The importance of TIGIT in head and neck cancer is poorly understood. CD155 is highly expressed on HNSCC tumor cells and intratumoral myeloid cells compared with healthy mucosa, and high expression independently associated with worse overall survival.100 Bulk expression analysis of HNSCC tumors shows that, compared with HPV- tumors, HPV+ tumors exhibit significantly higher expression of TIGIT, although it could not be assessed whether increased TIGIT expression was selective to NK cells, T cells, or both.41 Notably, in HPV+ HNSCC, high bulk TIGIT expression correlated with improved survival, whereas in HPV- HNSCC, TIGIT expression did not significantly correlate with either improved or worsened survival.41 TIGIT blockade delayed tumor growth and prevented T cell exhaustion in a mouse model of HNSCC.100 Taken together, these findings warrant further investigation into NK cell PVR-receptor expression dynamics and TIGIT blockade for HNSCC.
Anti-TIM3
Several early-phase clinical trials are now underway investigating the use of T cell immunoglobulin mucin-3 (TIM3) checkpoint inhibition for the treatment of advanced solid malignancies, including HNSCC (NCT02608268, NCT03744468). Although the clinical efficacy of these agents has yet to be determined, there is promising evidence that TIM3 plays a role in the progression of head and neck cancer. TIM3 is an inhibitory receptor that is upregulated on the NK cell surface after activation via the Fc receptor.101 102 The major ligand for TIM3 is Galectin-9. Although the precise functional status of TIM3+ NK cells remains controversial,102 previous studies in melanoma have shown that TIM3 inhibition can reverse NK cell exhaustion.103 Bulk expression analyses have shown that TIM3 is upregulated in HNSCC tumors compared with healthy or dysplastic mucosa and correlated with increased likelihood of a lymph node metastasis and disease recurrence.72 104 Further, anti-TIM3 suppressed tumor growth in a mouse model of HNSCC.104
Anti-LAG-3
Similarly, several large trials have now begun using anti-lymphocyte activating gene 3 (LAG3) monoclonal antibodies for the treatment of advanced solid tumors (NCT01968109, NCT03311412). LAG-3 is an inhibitory receptor expressed on the surface on some NK cells and binds to the major histocompatibility complex II (MHCII). LAG-3 is highly expressed on tumor-infiltrating lymphocytes (TILs) in HNSCC—especially in recurrent or metastatic disease—and correlates with worse overall survival.105 LAG-3 blockade in a mouse model of HNSCC restricted tumor growth.105 Still, the extent to which the beneficial effects of LAG-3 blockade can be attributed to NK cells is unknown, as LAG-3 expression is also observed on adaptive lymphocytes.
Other modalities
Several new modalities have emerged that do not rely on immune checkpoint inhibition, but instead unleash NK cell function through other various mechanisms. WEE1 is a nuclear kinase that activates the G2/M checkpoint, allowing for DNA repair prior to mitotic division. Conversely, granzyme B deactivates the G2/M checkpoint, which prevents cells from performing DNA repair and leads to mitotic catastrophe among cells with high DNA damage burden, such as irradiated cells.106 Aberrant tumor WEE1 expression is capable of preventing granzyme B-induced tumor cell apoptosis.107 Therefore, inhibition of WEE1 was hypothesized to resensitize tumor cells to granzyme B and render them vulnerable to NK and T-cell mediated cytotoxicity. Furthermore, because normal cells have a functioning G1 checkpoint, whereas TP53-mutated cancers rely solely on G2 for DNA repair, WEE1 inhibitors are potentially tumor-specific.
In vitro WEE1 inhibition using the small molecule inhibitor AZD1775 (adavosertib) effectively radiosensitized tumor cells, regardless of TP53 mutation status.108 The results of this study prompted a single-arm dose-escalation trial (NCT02037230) studying adavosertib in combination with gemcitabine for patients with unresectable locally advanced pancreatic cancer receiving chemoradiation. Compared with historical data on gemcitabine monotherapy, addition of adavosertib prolonged overall survival with tolerable side effects, and it is now heading to phase II studies.109 Although adavosertib sensitizes HNSCC tumor cells in vitro to NK-mediated killing, including via ADCC,110 no trials have investigated adavosertib for HNSCC. Given the available evidence, adavosertib is a promising combination agent to improve NK-driven treatments, such as cetuximab, for HNSCC.
As mentioned previously, HNSCC tumors tend to shed NKG2D ligands that lead to inhibition of NK cells.34 Several strategies have been tested in preclinical studies to counteract the effect of NKG2D ligand shedding in order to improve NK cell activity, including apheresis of ligands from peripheral blood,34 as well as antibody-mediated inhibition of MIC cleavage.111 Rhesus macaques injected with soluble MICA and subsequently apheresed had near-complete clearance of soluble MICA compared with non-apheresed controls.34 Antibody-mediated blockade of soluble MICA/MICB synergized with anti-PD-L1 to enhance the antitumor potential of intratumoral NK cells, CD8+ T cells, and DCs in a MIC+ mouse tumor model.112
Several clinical trials are underway investigating adoptive NK cell transfer for the treatment of solid tumors, such as HNSCC (NCT03319459). Adoptive NK cell transfer provides several advantages over T cell transfer, including the possibility of a universal ‘off-the-shelf’ product and a more favorable side-effect profile. The sources of NK cells for adoptive transfer are numerous and include autologous expansion, allogeneic transfer from KIR-ligand mismatched donors, and infusion of irradiated NK cells derived from transformed NK cell lines.113 Likewise, several strategies have emerged to maximize the antitumor activity of NK cells prior to infusion. It has been previously shown that preactivating NK cells with IL-12, IL-15, and IL-18 induces a memory-like NK cell phenotype114 capable of producing remission in patients with acute myeloid leukemia.115 This strategy is now being investigated in R/M-HNSCC in combination with CTLA-4 checkpoint inhibition (NCT04290546). Genetic engineering of NK cells using transduction of chimeric antigen receptors (CAR-NK) has shown promise in leukemia116 and is now being investigated in solid tumors, including HNSCC (NCT03415100).
Conclusion
The deluge of ongoing clinical trials investigating combination regimens shows the urgency of improving immunotherapy response rates for patients with HNSCC, while minimizing the toxicity of current therapy. Given their natural antitumor functionality, NK cells are attractive targets for future research. Determinants such as HLA-B −21 M/T genotype and CMV exposure demonstrate the remarkable importance of genetic and environmental factors in shaping an individual’s NK cell repertoires. Centering future development around rational considerations of the basic biological principles governing NK cell activation is imperative to maximizing the clinical success of NK-driven immunotherapy for head and neck cancer.
Footnotes
Twitter: @AmirHorowitz
Contributors: AJC, TE, RB, JS, BM, NB, and AH wrote the manuscript; all authors were involved in amendments and improvements in the text. AJC created the figures (using Biorender) and table. All authors read and approved the final manuscript.
Funding: This study was funded by the National Institute of Allergy and Infectious Diseases.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Argiris A, Karamouzis MV, Raben D, et al. . Head and neck cancer. The Lancet 2008;371:1695–709. 10.1016/S0140-6736(08)60728-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vokes EE, Agrawal N, Seiwert TY. HPV-Associated head and neck cancer. J Natl Cancer Inst 2015;107:djv344. 10.1093/jnci/djv344 [DOI] [PubMed] [Google Scholar]
- 3.Fakhry C, Westra WH, Li S, et al. . Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst 2008;100:261–9. 10.1093/jnci/djn011 [DOI] [PubMed] [Google Scholar]
- 4.Björkström NK, Ljunggren H-G, Michaëlsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol 2016;16:310–20. 10.1038/nri.2016.34 [DOI] [PubMed] [Google Scholar]
- 5.Fehniger TA, Cooper MA. Harnessing NK cell memory for cancer immunotherapy. Trends Immunol 2016;37:877–88. 10.1016/j.it.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 2016;16:7–19. 10.1038/nrc.2015.5 [DOI] [PubMed] [Google Scholar]
- 7.Parham P, Guethlein LA. Genetics of natural killer cells in human health, disease, and survival. Annu Rev Immunol 2018;36:519–48. 10.1146/annurev-immunol-042617-053149 [DOI] [PubMed] [Google Scholar]
- 8.Horowitz A, Djaoud Z, Nemat-Gorgani N, et al. . Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci Immunol 2016;1:eaag1672 10.1126/sciimmunol.aag1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anfossi N, André P, Guia S, et al. . Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006;25:331–42. 10.1016/j.immuni.2006.06.013 [DOI] [PubMed] [Google Scholar]
- 10.Yunis EJ, Romero V, Diaz-Giffero F, et al. . Natural killer cell receptor NKG2A/HLA-E interaction dependent differential thymopoiesis of hematopoietic progenitor cells influences the outcome of HIV infection. J Stem Cells 2007;2:237–48. [PMC free article] [PubMed] [Google Scholar]
- 11.Ramsuran V, Naranbhai V, Horowitz A, et al. . Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science 2018;359:86–90. 10.1126/science.aam8825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hallner A, Bernson E, Hussein BA, et al. . The HLA-B −21 dimorphism impacts on NK cell education and clinical outcome of immunotherapy in acute myeloid leukemia. Blood 2019;133:1479–88. 10.1182/blood-2018-09-874990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersdorf EW, Carrington M, O'hUigin C, et al. . Role of HLA-B exon 1 in graft-versus-host disease after unrelated haemopoietic cell transplantation: a retrospective cohort study. The Lancet Haematology 2020;7:e50–60. 10.1016/S2352-3026(19)30208-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanier LL. NK cell recognition. Annu Rev Immunol 2005;23:225–74. 10.1146/annurev.immunol.23.021704.115526 [DOI] [PubMed] [Google Scholar]
- 15.Raulet DH, Gasser S, Gowen BG, et al. . Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol 2013;31:413–41. 10.1146/annurev-immunol-032712-095951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol 2017;8:930. 10.3389/fimmu.2017.00930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1717.Smyth MJ, Kelly JM, Baxter AG, et al. . An essential role for tumor necrosis factor in natural killer cell-mediated tumor rejection in the peritoneum. J Exp Med 1998;188:1611–9. 10.1084/jem.188.9.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vujanovic L, Szymkowski DE, Alber S, et al. . Virally infected and matured human dendritic cells activate natural killer cells via cooperative activity of plasma membrane-bound TNF and IL-15. Blood 2010;116:575–83. 10.1182/blood-2009-08-240325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vivier E, Raulet DH, Moretta A, et al. . Innate or adaptive immunity? The example of natural killer cells. Science 2011;331:44–9. 10.1126/science.1198687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castro F, Cardoso AP, Gonçalves RM, et al. . Interferon-Gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol 2018;9:847 10.3389/fimmu.2018.00847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat Rev Immunol 2016;16:102–11. 10.1038/nri.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Böttcher JP, Bonavita E, Chakravarty P, et al. . NK cells stimulate recruitment of cdc1 into the tumor microenvironment promoting cancer immune control. Cell 2018;172:1022–37. 10.1016/j.cell.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barry KC, Hsu J, Broz ML, et al. . A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med 2018;24:1178–91. 10.1038/s41591-018-0085-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2424.Takeda K, Smyth MJ, Cretney E, et al. . Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med 2002;195:161–9. 10.1084/jem.20011171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kashii Y, Giorda R, Herberman RB, et al. . Constitutive expression and role of the TNF family ligands in apoptotic killing of tumor cells by human NK cells. J Immunol 1999;163:5358–66. [PubMed] [Google Scholar]
- 26.Garrido F, Aptsiauri N, Doorduijn EM, et al. . The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 2016;39:44–51. 10.1016/j.coi.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandal R, Şenbabaoğlu Y, Desrichard A, et al. . The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016;1:e89829 10.1172/jci.insight.89829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner S, Wittekindt C, Reuschenbach M, et al. . CD56-positive lymphocyte infiltration in relation to human papillomavirus association and prognostic significance in oropharyngeal squamous cell carcinoma. Int. J. Cancer 2016;138:2263–73. 10.1002/ijc.29962 [DOI] [PubMed] [Google Scholar]
- 29.Schantz SP, Savage HE, Racz T, et al. . Natural killer cells and metastases from pharyngeal carcinoma. Am J Surg 1989;158:361–6. 10.1016/0002-9610(89)90134-7 [DOI] [PubMed] [Google Scholar]
- 30.Schantz SP, Ordonez NG. Quantitation of natural killer cell function and risk of metastatic poorly differentiated head and neck cancer. Nat Immun Cell Growth Regul 1991;10:278–88. [PubMed] [Google Scholar]
- 31.Lu J, Chen X-M, Huang H-R, et al. . Detailed analysis of inflammatory cell infiltration and the prognostic impact on nasopharyngeal carcinoma. Head Neck 2018;40:1245–53. 10.1002/hed.25104 [DOI] [PubMed] [Google Scholar]
- 32.Habif G, Crinier A, André P, et al. . Targeting natural killer cells in solid tumors. Cell Mol Immunol 2019;16:415–22. 10.1038/s41423-019-0224-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.André P, Denis C, Soulas C, et al. . Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018;175:1731–43. 10.1016/j.cell.2018.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weil S, Memmer S, Lechner A, et al. . Natural killer group 2D ligand depletion reconstitutes natural killer cell immunosurveillance of head and neck squamous cell carcinoma. Front Immunol 2017;8:387. 10.3389/fimmu.2017.00387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lattanzio L, Denaro N, Vivenza D, et al. . Elevated basal antibody-dependent cell-mediated cytotoxicity (ADCC) and high epidermal growth factor receptor (EGFR) expression predict favourable outcome in patients with locally advanced head and neck cancer treated with cetuximab and radiotherapy. Cancer Immunol Immunother 2017;66:573–9. 10.1007/s00262-017-1960-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauer S, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999;285:727–9. 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 37.Kaiser BK, Yim D, Chow I-T, et al. . Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007;447:482–6. 10.1038/nature05768 [DOI] [PubMed] [Google Scholar]
- 38.Ferris RL, Hunt JL, Ferrone S. Human leukocyte antigen (HLA) class I defects in head and neck cancer: molecular mechanisms and clinical significance. Immunol Res 2005;33:113–34. 10.1385/IR:33:2:113 [DOI] [PubMed] [Google Scholar]
- 39.Näsman A, Andersson E, Marklund L, et al. . HLA class I and II expression in oropharyngeal squamous cell carcinoma in relation to tumor HPV status and clinical outcome. PLoS One 2013;8:e77025 10.1371/journal.pone.0077025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Näsman A, Andersson E, Nordfors C, et al. . MHC class I expression in HPV positive and negative tonsillar squamous cell carcinoma in correlation to clinical outcome. Int. J. Cancer 2013;132:72–81. 10.1002/ijc.27635 [DOI] [PubMed] [Google Scholar]
- 41.Gameiro SF, Ghasemi F, Barrett JW, et al. . Treatment-naïve HPV+ head and neck cancers display a T-cell-inflamed phenotype distinct from their HPV- counterparts that has implications for immunotherapy. Oncoimmunology 2018;7:e1498439 10.1080/2162402X.2018.1498439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seiwert TY, Zuo Z, Keck MK, et al. . Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res 2015;21:632–41. 10.1158/1078-0432.CCR-13-3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris LGT, Chandramohan R, West L, et al. . The molecular landscape of recurrent and metastatic head and neck cancers: insights from a precision oncology sequencing platform. JAMA Oncol 2017;3:244–55. 10.1001/jamaoncol.2016.1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albers AE, Qian X, Kaufmann AM, et al. . Meta analysis: HPV and p16 pattern determines survival in patients with HNSCC and identifies potential new biologic subtype. Sci Rep 2017;7:16715. 10.1038/s41598-017-16918-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawrence MS, Sougnez C, Lichtenstein L, et al. . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82. 10.1038/nature14129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyu H, Li M, Jiang Z, et al. . Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput Struct Biotechnol J 2019;17:1020–30. 10.1016/j.csbj.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Y, Zhou R, Huang C, et al. . Analysis of the expression of surface receptors on NK cells and NKG2D on immunocytes in peripheral blood of patients with nasopharyngeal carcinoma. Asian Pac J Cancer Prev 2018;19:661–5. 10.22034/APJCP.2018.19.3.661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 2010;20:202–13. 10.1002/rmv.655 [DOI] [PubMed] [Google Scholar]
- 49.Gumá Mónica, Angulo A, Vilches C, et al. . Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004;104:3664–71. 10.1182/blood-2004-05-2058 [DOI] [PubMed] [Google Scholar]
- 50.Hammer Q, Rückert T, Borst EM, et al. . Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol 2018;19:453–63. 10.1038/s41590-018-0082-6 [DOI] [PubMed] [Google Scholar]
- 51.Béziat V, Liu LL, Malmberg J-A, et al. . NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 2013;121:2678–88. 10.1182/blood-2012-10-459545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlums H, Cichocki F, Tesi B, et al. . Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015;42:443–56. 10.1016/j.immuni.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darboe A, Danso E, Clarke E, et al. . Enhancement of cytokine-driven NK cell IFN-γ production after vaccination of HCMV infected Africans. Eur J Immunol 2017;47:1040–50. 10.1002/eji.201746974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burtness B, Harrington KJ, Greil R, et al. . Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. The Lancet 2019;394:1915–28. 10.1016/S0140-6736(19)32591-7 [DOI] [PubMed] [Google Scholar]
- 55.Vermorken JB, Mesia R, Rivera F, et al. . Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008;359:1116–27. 10.1056/NEJMoa0802656 [DOI] [PubMed] [Google Scholar]
- 56.Ferris RL, Blumenschein G, Fayette J, et al. . Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 2016;375:1856–67. 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim S, Grandis JR, Rinaldo A, et al. . Emerging perspectives in epidermal growth factor receptor targeting in head and neck cancer. Head Neck 2008;30:667–74. 10.1002/hed.20859 [DOI] [PubMed] [Google Scholar]
- 58.García-Foncillas J, Sunakawa Y, Aderka D, et al. . Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front Oncol 2019;9:849. 10.3389/fonc.2019.00849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trivedi S, Srivastava RM, Concha-Benavente F, et al. . Anti-EGFR targeted monoclonal antibody isotype influences antitumor cellular immunity in head and neck cancer patients. Clin Cancer Res 2016;22:5229–37. 10.1158/1078-0432.CCR-15-2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Srivastava RM, Lee SC. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen–specific T-cell immunity in head and neck cancer patients. Clin Cancer Res 2013. https://clincancerres.aacrjournals.org/content/19/7/1858.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Srivastava RM, Lee SC, Andrade Filho PA, et al. . Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res 2013;19:1858–72. 10.1158/1078-0432.CCR-12-2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodríguez J, Zarate R, Bandres E, et al. . Fc gamma receptor polymorphisms as predictive markers of cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur J Cancer 2012;48:1774–80. 10.1016/j.ejca.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 63.Mellor JD, Brown MP, Irving HR, et al. . A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol 2013;6:1 10.1186/1756-8722-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMichael EL, Benner B, Atwal LS, et al. . A phase I/II trial of cetuximab in combination with interleukin-12 administered to patients with unresectable primary or recurrent head and neck squamous cell carcinoma. Clin Cancer Res 2019;25:4955–65. 10.1158/1078-0432.CCR-18-2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bertino EM, McMichael EL, Mo X, et al. . A phase I trial to evaluate antibody-dependent cellular cytotoxicity of cetuximab and lenalidomide in advanced colorectal and head and neck cancer. Mol Cancer Ther 2016;15:2244–50. 10.1158/1535-7163.MCT-15-0879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Srivastava RM, Trivedi S, Concha-Benavente F, et al. . CD137 stimulation enhances Cetuximab-Induced natural killer: dendritic cell priming of antitumor T-cell immunity in patients with head and neck cancer. Clin Cancer Res 2017;23:707–16. 10.1158/1078-0432.CCR-16-0879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jie H-B, Schuler PJ, Lee SC, et al. . CTLA-4 + Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res 2015;75:2200–10. 10.1158/0008-5472.CAN-14-2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oida T, Xu L, Weiner HL, et al. . TGF-Beta-Mediated suppression by CD4+CD25+ T cells is facilitated by CTLA-4 signaling. J Immunol 2006;177:2331–9. 10.4049/jimmunol.177.4.2331 [DOI] [PubMed] [Google Scholar]
- 69.Walker LSK, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol 2011;11:852–63. 10.1038/nri3108 [DOI] [PubMed] [Google Scholar]
- 70.Pesce S, Greppi M, Tabellini G, et al. . Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol 2017;139:335–46. 10.1016/j.jaci.2016.04.025 [DOI] [PubMed] [Google Scholar]
- 71.Della Chiesa M, Pesce S, Muccio L, et al. . Features of memory-like and PD-1+ human NK cell subsets. Front Immunol 2016;7:351 10.3389/fimmu.2016.00351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Concha-Benavente F, Kansy B, Moskovitz J, et al. . PD-L1 Mediates Dysfunction in Activated PD-1 + NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res 2018;6:1548–60. 10.1158/2326-6066.CIR-18-0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Judge SJ, Murphy WJ, Canter RJ. Characterizing the dysfunctional NK cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front Cell Infect Microbiol 2020;10:49. 10.3389/fcimb.2020.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hsu J, Hodgins JJ, Marathe M, et al. . Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest 2018;128:4654–68. 10.1172/JCI99317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strauss L, Mahmoud MAA, Weaver JD, et al. . Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020;5:eaay1863 10.1126/sciimmunol.aay1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prat A, Navarro A, Paré L, et al. . Immune-related gene expression profiling after PD-1 blockade in non–small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res 2017;77:3540–50. 10.1158/0008-5472.CAN-16-3556 [DOI] [PubMed] [Google Scholar]
- 77.Bellucci R, Martin A, Bommarito D, et al. . Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015;4:e1008824 10.1080/2162402X.2015.1008824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Braud VM, Allan DSJ, O'Callaghan CA, et al. . HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998;391:795–9. 10.1038/35869 [DOI] [PubMed] [Google Scholar]
- 79.Monaco EL, Tremante E, Cerboni C, et al. . Human leukocyte antigen E contributes to protect tumor cells from lysis by natural killer cells. Neoplasia 2011;13:822–IN14. 10.1593/neo.101684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Borrego F, Ulbrecht M, Weiss EH, et al. . Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med 1998;187:813–8. 10.1084/jem.187.5.813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Levy EM, Bianchini M, Von Euw EM, et al. . Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol 2008;32:633–41. 10.3892/ijo.32.3.633 [DOI] [PubMed] [Google Scholar]
- 82.de Kruijf EM, Sajet A, van Nes JGH, et al. . HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol 2010;185:7452–9. 10.4049/jimmunol.1002629 [DOI] [PubMed] [Google Scholar]
- 83.van Hall T, André P, Horowitz A, et al. . Monalizumab: inhibiting the novel immune checkpoint NKG2A. J. Immunotherapy Cancer 2019;7:263 10.1186/s40425-019-0761-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Derré L, Corvaisier M, Charreau B, et al. . Expression and release of HLA-E by melanoma cells and melanocytes: potential impact on the response of cytotoxic effector cells. J Immunol 2006;177:3100–7. 10.4049/jimmunol.177.5.3100 [DOI] [PubMed] [Google Scholar]
- 85.Vermorken JB, Trigo J, Hitt R, et al. . Open-Label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. JCO 2007;25:2171–7. 10.1200/JCO.2006.06.7447 [DOI] [PubMed] [Google Scholar]
- 86.Shi J, Tricot G, Szmania S, et al. . Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol 2008;143:641–53. 10.1111/j.1365-2141.2008.07340.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruggeri L, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002;295:2097–100. 10.1126/science.1068440 [DOI] [PubMed] [Google Scholar]
- 88.Thielens A, Vivier E, Romagné F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol 2012;24:239–45. 10.1016/j.coi.2012.01.001 [DOI] [PubMed] [Google Scholar]
- 89.Romagné F, André P, Spee P, et al. . Preclinical characterization of 1-7F9, a novel human anti–KIR receptor therapeutic antibody that augments natural killer–mediated killing of tumor cells. Blood 2009;114:2667–77. 10.1182/blood-2009-02-206532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carlsten M, Korde N, Kotecha R, et al. . Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clinical Cancer Research 2016;22:5211–22. 10.1158/1078-0432.CCR-16-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016;44:989–1004. 10.1016/j.immuni.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stanietsky N, Simic H, Arapovic J, et al. . The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A 2009;106:17858–63. 10.1073/pnas.0903474106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bottino C, Castriconi R, Pende D, et al. . Identification of PVR (CD155) and nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med 2003;198:557–67. 10.1084/jem.20030788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guillamón CF, Martínez-Sánchez MV, Gimeno L, et al. . NK cell education in tumor immune surveillance: DNAM-1/KIR receptor ratios as predictive biomarkers for solid tumor outcome. Cancer Immunol Res 2018;6:1537–47. 10.1158/2326-6066.CIR-18-0022 [DOI] [PubMed] [Google Scholar]
- 95.Chan CJ, Andrews DM, McLaughlin NM, et al. . DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J.i. 2010;184:902–11. 10.4049/jimmunol.0903225 [DOI] [PubMed] [Google Scholar]
- 96.Xu F, Sunderland A, Zhou Y, et al. . Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother 2017;66:1367–75. 10.1007/s00262-017-2031-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Q, Bi J, Zheng X, et al. . Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 2018;19:723–32. 10.1038/s41590-018-0132-0 [DOI] [PubMed] [Google Scholar]
- 98.Dixon KO, Schorer M, Nevin J, et al. . Functional Anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J.i. 2018;200:3000–7. 10.4049/jimmunol.1700407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol 2020;200:108–19. 10.1111/cei.13407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu L, Mao L, Liu J-F, et al. . Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res 2019;7:1700–13. 10.1158/2326-6066.CIR-18-0725 [DOI] [PubMed] [Google Scholar]
- 101.Ndhlovu LC, Lopez-Vergès S, Barbour JD, et al. . Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012;119:3734–43. 10.1182/blood-2011-11-392951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.So EC, Khaladj-Ghom A, Ji Y, et al. . NK cell expression of Tim-3: first impressions matter. Immunobiology 2019;224:362–70. 10.1016/j.imbio.2019.03.001 [DOI] [PubMed] [Google Scholar]
- 103.da Silva IP, Gallois A, Jimenez-Baranda S, et al. . Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res 2014;2:410–22. 10.1158/2326-6066.CIR-13-0171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu J-F, Ma S-R, Mao L, et al. . T-cell immunoglobulin mucin 3 blockade drives an antitumor immune response in head and neck cancer. Mol Oncol 2017;11:235–47. 10.1002/1878-0261.12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deng W-W, Mao L, Yu G-T, et al. . LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology 2016;5:e1239005. 10.1080/2162402X.2016.1239005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun L, Moore E, Berman R, et al. . Wee1 kinase inhibition reverses G2/M cell cycle checkpoint activation to sensitize cancer cells to immunotherapy. Oncoimmunology 2018;7:e1488359. 10.1080/2162402X.2018.1488359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen G, Shi L, Litchfield DW, et al. . Rescue from granzyme B-induced apoptosis by Wee1 kinase. J Exp Med 1995;181:2295–300. 10.1084/jem.181.6.2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cuneo KC, Morgan MA, Davis MA, et al. . Wee1 kinase inhibitor AZD1775 radiosensitizes hepatocellular carcinoma regardless of TP53 mutational status through induction of replication stress. Int J Radiat Oncol Biol Phys 2016;95:782–90. 10.1016/j.ijrobp.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cuneo KC, Morgan MA, Sahai V, et al. . Dose escalation trial of the Wee1 inhibitor Adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol 2019;37:2643–50. 10.1200/JCO.19.00730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Friedman J, Morisada M, Sun L, et al. . Inhibition of Wee1 kinase and cell cycle checkpoint activation sensitizes head and neck cancers to natural killer cell therapies. J Immunother Cancer 2018;6:59. 10.1186/s40425-018-0374-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ferrari de Andrade L, Tay RE, Pan D, et al. . Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018;359:1537–42. 10.1126/science.aao0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang J, Larrocha PS-L, Zhang B, et al. . Antibody targeting tumor-derived soluble NKG2D ligand sMIC provides dual co-stimulation of CD8 T cells and enables sMIC+ tumors respond to PD1/PD-L1 blockade therapy. J Immunother Cancer 2019;7:223. 10.1186/s40425-019-0693-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol 2016;17:1025–36. 10.1038/ni.3518 [DOI] [PubMed] [Google Scholar]
- 114.Romee R, Schneider SE, Leong JW, et al. . Cytokine activation induces human memory-like NK cells. Blood 2012;120:4751–60. 10.1182/blood-2012-04-419283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Romee R, Rosario M, Berrien-Elliott MM, et al. . Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 2016;8:ra123. 10.1126/scitranslmed.aaf2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu E, Marin D, Banerjee P, et al. . Use of CAR-Transduced natural killer cells in CD19-Positive lymphoid tumors. N Engl J Med 2020;382:545–53. 10.1056/NEJMoa1910607 [DOI] [PMC free article] [PubMed] [Google Scholar]