

Abstract
Meperidine is an opioid analgesic that undergoes N-demethylation to form the neurotoxic metabolite normeperidine. Previous studies indicate that meperidine N-demethylation is catalyzed by cytochrome P450 2B6 (CYP2B6), CYP3A4, and CYP2C19.
The purpose of this study was to examine the relative P450 contributions to meperidine N-demethylation and to evaluate the effect of CYP2C19 polymorphism on normeperidine generation. Experiments were performed using recombinant P450 enzymes, selective chemical inhibitors, enzyme kinetic assays, and correlation analysis with individual CYP2C19-genotyped human liver microsomes.
The catalytic efficiency (kcat/Km) for meperidine N-demethylation was similar between recombinant CYP2B6 and CYP2C19, but markedly lower by CYP3A4.
In CYP2C19-genotyped human liver microsomes, normeperidine formation was significantly correlated with CYP2C19 activity (S-mephenytoin 4′-hydroxylation).
CYP2C19 inhibitor (+)-N-3-benzylnirvanol and CYP3A inhibitor ketoconazole significantly reduced microsomal normeperidine generation by an individual donor with high CYP2C19 activity, whereas donors with lower CYP2C19 activity were sensitive to inhibition by ketoconazole but not benzylnirvanol.
These findings demonstrate that the relative CYP3A4, CYP2B6, and CYP2C19 involvement in meperidine N-demethylation depends on the enzyme activities in individual human liver microsomal samples. CYP2C19 is likely an important contributor to normeperidine generation in individuals with high CYP2C19 activity, but additional factors influence inter-individual metabolite accumulation.
Keywords: cytochrome P450, meperidine, neurotoxic metabolite, reaction phenotyping, genetic polymorphism
Introduction
Meperidine (Demerol™) is a synthetic opioid that binds to mu opioid receptors in the central nervous system to produce analgesia. It is indicated for moderate to severe pain and is typically administered as a single parenteral dose (Hospira, 2015). Meperidine was once one of the most commonly prescribed analgesics in the U.S., with approximately 60% of physicians prescribing the agent for acute pain (Eisendrath et al., 1987). Serious adverse events have led to recommendations restricting its use. The Institute for Safe Medication Practices and the American Pain Society currently recommend a maximum dose of 600 mg/day with a total treatment period of <48 hours (Friesen et al., 2015).
Meperidine’s adverse effect profile is related to its metabolism in the liver. Meperidine undergoes extensive hepatic metabolism; only 5% of the parent compound is excreted in urine (Burns et al., 1955; Yeh et al., 1981). The major metabolic pathways of meperidine are hydrolysis by human liver carboxylesterase (hCE-1) to form the inactive metabolite meperidinic acid (Zhang et al., 1999), and N-demethylation via hepatic cytochrome P450 (CYP) enzymes to form normeperidine, a centrally active metabolite lacking analgesic activity (Fig. 1). Normeperidine may be hydrolyzed to form normeperidinic acid or hydroxylated in liver microsomes to form N-hydroxynormeperidine prior to excretion in the urine (Latta et al., 2002). The half-life of meperidine is 2.5 – 4 hours in patients with normal liver function, and is increased in cirrhotic patients (Clark et al., 1995). The half-life of normeperidine is 14 – 21 hours at therapeutic doses and may exceed 34 hours in patients with renal insufficiency (Clark et al., 1995). When administered at high doses or over prolonged periods, normeperidine may accumulate to cause agitation, tremors, and seizures (Kaiko et al., 1983; Clark et al., 1995). The exact mechanism of toxicity is not well understood, though its symptoms combined with meperidine’s suspected inhibition of neurotransmitter reuptake suggest that it may be related to serotonin accumulation (Latta et al., 2002). The risk for normeperidine neurotoxicity is increased in elderly patients or those with reduced renal or hepatic function. Oral administration is often avoided since extensive first-pass metabolism results in increased normeperidine exposure and reduced plasma concentrations of the analgesic parent compound (Clark et al., 1995). Some studies have suggested that meperidine may exhibit less tolerance and dependence relative to other opioids due to its poor activity as a P-glycoprotein substrate (Dagenais et al., 2004; Hamabe et al., 2006; Hassan et al., 2009). Identification of patients at a decreased risk for normeperidine toxicity may therefore allow meperidine to be considered as an alternative to more widely abused agents contributing to the current opioid epidemic.
Figure 1.
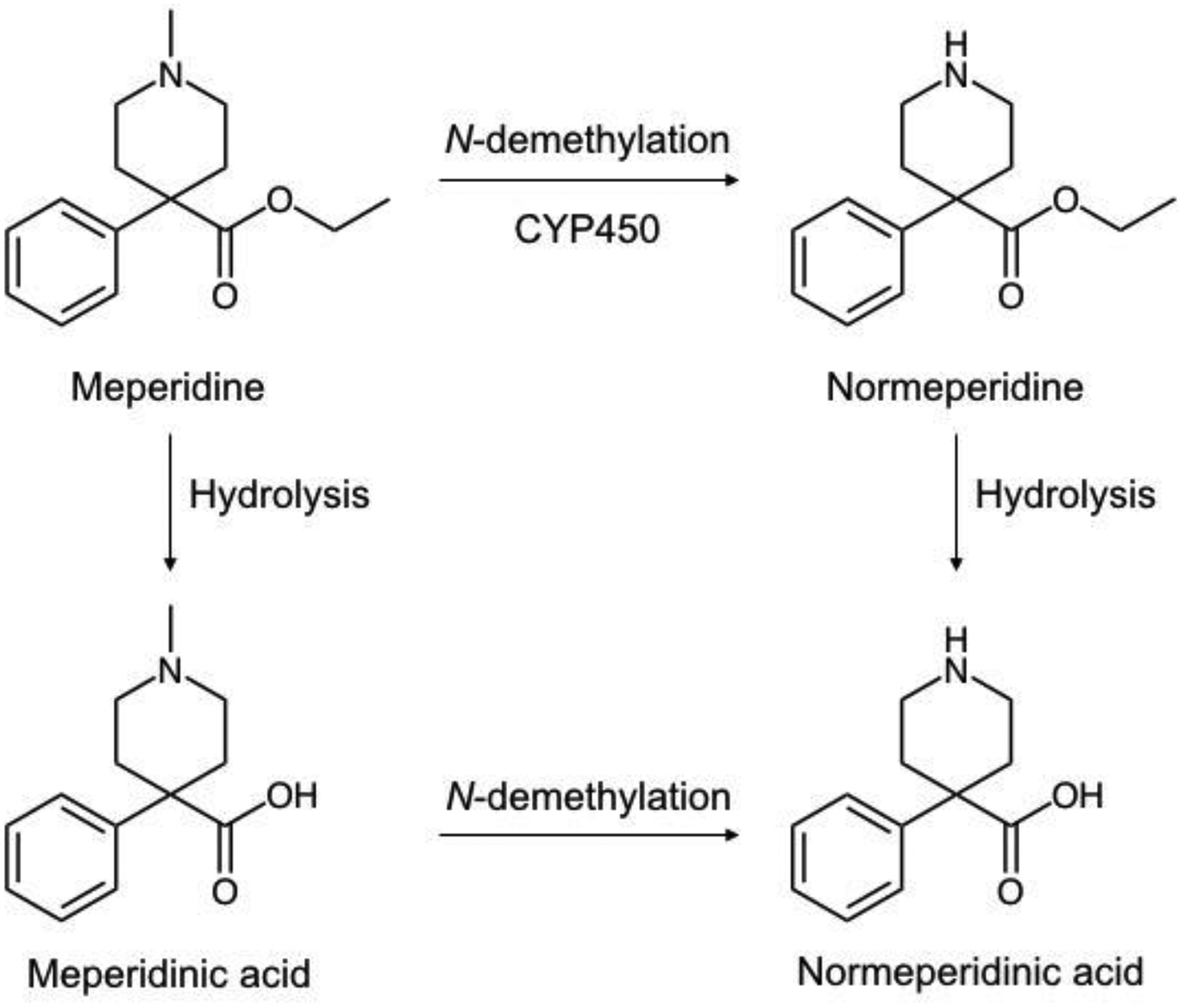
Structures of meperidine and meperidine metabolites.
A previous study reported that CYP2B6, CYP3A4, and CYP2C19 are the primary P450 enzymes responsible for normeperidine generation (Ramírez et al., 2004). Among these enzymes, CYP2B6 and CYP2C19 are highly polymorphic. Single nucleotide polymorphisms are known to alter P450 expression and activity among individuals, leading to notable differences in drug metabolism and potential variability in drug response (Zanger and Schwab, 2013). It is possible that P450 genetic variations contribute to differences in normeperidine generation among diverse patient populations, which may impact the risk of developing neurotoxicity with meperidine use. Whether CYP2B6 and CYP2C19 polymorphisms influence meperidine N-demethylation is not known.
The effects of CYP2C19 genetic variation on metabolism phenotype have been well-established for some CYP2C19 substrates, including clopidogrel, voriconazole, citalopram, and escitalopram (Scott et al., 2013; Moriyama et al., 2016; Hicks et al. 2015). The CYP2C19*1 wild-type allele leads to expression of functional protein, whereas the CYP2C19*2 variant allele results in truncated, nonfunctional protein due to altered mRNA splicing (de Morais et al., 1994). Inheritance of the CYP2C19*17 variant allele increases enzyme expression due to alterations in the promoter region, which contribute to increased transcription (Sim et al., 2006). Individuals may be classified as normal metabolizers (CYP2C19*1/*1), intermediate metabolizers (e.g. CYP2C19*1/*2), poor metabolizers (e.g. CYP2C19*2/*2), rapid metabolizers (CYP2C19*1/*17), or ultrarapid metabolizers (CYP2C19*17/*17) based on CYP2C19 genotype (Caudle et al., 2017; Scott et al., 2012). Single nucleotide polymorphisms in CYP2B6 have also been shown to affect CYP2B6 expression and activity; however, the influence of CYP2B6 genotype on CYP2B6 metabolism status is more complex, and conflicting results have been reported (reviewed by Zanger and Klein, 2013). Moreover, CYP2B6-genotyped human liver microsomes are not currently commercially available for in vitro investigations.
The purpose of this study was to examine the relative P450 enzyme contributions to meperidine N-demethylation and determine the impact CYP2C19 genetic polymorphisms on normeperidine generation. An integrated reaction phenotyping approach was performed using recombinant P450 enzymes, pooled human liver microsomal preparations with P450-selective chemical inhibitors, and correlation analysis with individual human liver microsomes. We examined normeperidine formation in human liver microsomal fractions from CYP2C19-genotyped donors to determine whether CYP2C19 genotype and activity influence meperidine metabolism. We provide evidence that CYP2C19 is a major contributor to normeperidine generation in individual human liver microsomal samples with high CYP2C19 activity.
Materials & Methods
Materials
Meperidine hydrochloride was purchased from Sigma Aldrich (St. Louis, MO). Normeperidine, deuterium-labeled standards of meperidine ([2H4]meperidine, meperidine-d4) and normeperidine ([2H4]normeperidine, normeperidine-d4), mephenytoin, 4′-hydroxymephenytoin, and [2H3]-4′-hydroxymephenytoin (4′-hydroxymephenytoin-d3) were purchased from Cerilliant (Round Rock, TX). Chemical inhibitors ketoconazole, ticlopidine, α-naphthoflavone, sulfaphenazole, quinidine, and 4-methylpyrazole were purchased from Sigma Aldrich. Furafylline, 2-phenyl-2-(1-piperidinyl)propane (PPP), and (+)-N-3-benzylnirvanol were purchased from Toronto Research Chemicals (Toronto, ON). An NADPH-regenerating system (solution A: 26 mM NADP+, 66 mM glucose 6-phosphate, 66 mM MgCl2; solution B: 40 U/ml glucose 6-phosphate dehydrogenase in 5 mM sodium citrate) was purchased from Corning (Woburn, MA). All other solvents and reagents were of analytical grade and purchased from standard commercial sources. Working solutions of meperidine and chemical inhibitors were prepared from stocks dissolved in DMSO and diluted into acetonitrile to make a solution in 1:9 DMSO/acetonitrile (v/v), unless otherwise specified.
Individual recombinant P450 enzymes, or Supersomes™, derived from baculovirus-infected insect cells containing cDNA-expressed CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9*1, CYP2C19, CYP2D6*1, CYP2E1, CYP3A4, or CYP3A5, co-expressed with P450 reductase and cytochrome b5, except CYP1A2 and CYP2D6*1 (expressed without cytochrome b5), were purchased from Corning. Pooled human liver microsomes (UltraPool™ HLM 150-donor samples of mixed gender, lot numbers 38291 and 38292) were purchased from Corning, similar to methods described by Towles et al. (2016). Human liver microsomes from individual donors genotyped as CYP2C19*1/*1, CYP2C19*1/*2, or CYP2C19*2/*2 were purchased from XenoTech, LLC (Lenexa, KS) and Corning Life Sciences. Human liver microsomes from individual donors genotyped as CYP2C19*1/*1, CYP2C19*1/*17, or CYP2C19*17/*17 were purchased from BioreclamationIVT (Baltimore, MD). Donor lots are listed as follows, with BioreclamationIVT lots indicated with three letters, XenoTech lots listed as six numbers, and Corning lots listed beginning with HH. Donor gender is marked with an M (male) or F (female): CYP2C19*1/*1 donors were 710444 (M), LLO (M), ONY (M), OED (M), IFF (M), and QLC (M); CYP2C19*1/*2 donors were 710456 (M) and 710415 (F); CYP2C19*2/*2 donors were 810010 (M), HH689 (M), HH863 (F), and HH40 (M); CYP2C19*1/*17 donors were GWN (M), RYQ (M), and TKM (M); CYP2C19*17/*17 donors were YEJ (M), ZKI (M), and ZGH (M).
Reaction phenotyping with recombinant P450 enzyme panel
Meperidine (10 μM) was incubated with recombinant P450 enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9*1, CYP2C19, CYP2D6*1, CYP2E1, CYP3A4, and CYP3A5) (20 nM) in 100 mM potassium phosphate buffer (pH 7.4) with NADPH-regenerating system. Final reaction mixtures contained organic solvent concentrations of 0.05% DMSO and 0.45% acetonitrile (v/v). Incubations were pre-warmed for 5 minutes at 37 °C in a shaking water bath prior to addition of NADPH-regenerating system to make a final reaction volume of 0.2 ml. Reactions were quenched after 10 minutes with twice the reaction volume (0.4 ml) of ice-cold methanol containing 0.4 μM meperidine-d4 (internal standard). Samples were then mixed with a vortex device for 10 seconds and centrifuged for 20 minutes (3740 × g at 4°C). Supernatants were collected and stored at −20 °C prior to LC-MS/MS analysis. Each incubation was performed in triplicate (n = 3). Control incubations did not contain NADPH-regenerating system. Three independent experiments were performed in triplicate each with the recombinant P450 enzyme panel. Normeperidine was detected by liquid chromatography – tandem mass spectrometry (LC-MS/MS). Relative metabolite levels were determined as a ratio of normeperidine to internal standard (meperidine-d4) peak areas.
Kinetic assays for meperidine N-demethylation by pooled human liver microsomes
To determine reaction conditions for kinetic assays, the linearity of normeperidine formation was determined with respect to protein concentration and time using pooled human liver microsomes. Meperidine (10 μM) was incubated with pooled human liver microsomes (lot 38291 at final concentrations of 0.05, 0.1, and 0.25 mg/ml) for 0, 2.5, 5, 10, 15, and 20 minutes. Incubations were pre-warmed for 5 minutes in a 37 °C shaking water bath prior to addition of NADPH-regenerating system to make a final reaction volume of 1.0 ml (final reaction mixtures contained 0.05% DMSO and 0.45% acetonitrile (v/v). At each time point, 100 μl of the incubation mixture was removed and added to 200 μL of ice-cold methanol containing meperidine-d4 (0.4 μM) to quench the reaction. Samples were then vortexed for 10 seconds and centrifuged for 20 minutes (3740 ° g, 4°C). Supernatants were collected and stored at −20 °C prior to LC-MS/MS analysis. Each incubation was performed in triplicate (n = 3) with control incubations that did not contain NADPH-regenerating system. This experiment was repeated a total of three times.
To determine the kinetics of normeperidine formation in human liver microsomes, meperidine (1 – 1000 μM) was incubated for 10 minutes with human liver microsomes (0.1 mg protein/ml). Reactions were initiated via addition of an NADPH-regenerating system following a 5-minute pre-warming period at 37 °C. Reactions were quenched by adding 0.4 ml of ice-cold methanol containing 0.4 μM meperidine-d4. Samples were then mixed with a vortex device for 10 seconds and centrifuged at 3740 × g, 4 °C, for 20 minutes. Supernatants were collected and stored at −20 °C prior to LC-MS/MS analysis. Experiments were performed in triplicate, and repeated three times.
Kinetic assays for meperidine N-demethylation by recombinant P450 enzymes
The protein-time linearity of normeperidine formation was examined with recombinant CYP2B6 in a manner similar to experiments with pooled human liver microsomes. Meperidine (10 μM) was incubated with recombinant CYP2B6 (Corning lots 3301984 and 5239002) at a range of enzyme concentrations (5, 10, and 20 nM) and incubation times (0, 2.5, 5, 10, 15, and 20 minutes) following the same procedure described above. Each experiment was performed in triplicate (n = 3) and repeated a total of three times. Control incubations did not contain NADPH-regenerating system.
The kinetics of normeperidine formation via P450 enzymes were determined by incubating meperidine (1 – 1000 μM) with 10 nM of recombinant CYP2B6, CYP2C19, or CYP3A4 for 10 minutes using the procedure described above. Experiments were performed in triplicate and repeated three times.
Normeperidine was quantified for kinetic assays by LC-MS/MS analysis using a standard curve with known concentrations of chemically synthesized normeperidine. When comparing normeperidine formation for enzyme kinetic assays, experimental samples were run on the same LC-MS/MS system (Shimadzu LCMS 8030 triple quadrupole mass spectrometer system interfaced with two HPLC LC-20AD XR pumps and a SIL-20AC XR autosampler) as described below. Samples were injected from a temperature-controlled autosampler maintained at 4°C. Each standard was prepared in a similar matrix as the experimental samples with inactive P450 enzyme or pooled human liver microsomes, 100 mM potassium phosphate buffer (pH 7.4), containing 0.1% DMSO, and 0.9% acetonitrile (all v/v). Quality control samples were included in analytical runs with calibration standards during method development and validation to assess accuracy, precision, and reproducibility. Quality control samples were prepared with recombinant CYP2B6, and the following normeperidine concentrations were tested: 0.02 μM (limit of quantitation), 0.04, 0.43, and 1.72 μM normeperidine. The limit of quantitation for normeperidine in human liver microsomes was also 0.02 μM. Five measurements of the same four normeperidine concentrations - 0.02, 0.04, 0.43, and 1.72 μM - were performed on the same day to test intra-assay variability. The intra-day percent % coefficient of variation and accuracy over this range of normeperidine concentrations were as follows: 2.4 and 115% at 0.02 μM; 2.3 and 109% at 0.04 μM; 0.92 and 108% at 0.43 μM; and 1.6 and 86% at 1.72 μM normeperidine, respectively. There was less than 0.03% carry-over from standards to blanks.
Calibration standards were prepared in the same matrix as experimental samples (without NADPH-regenerating system). For standard curves containing pooled human liver microsomes or inactive recombinant CYP3A4, an eight-point standard curve was prepared in the range of 0.02 – 4.29 μM normeperidine. For standard curves containing inactive recombinant CYP2B6 or CYP2C19, an 11-point standard curve was developed in the range of 0.02 – 21.6 μM normeperidine. Matrix-matched blanks were also included in analytical runs. Normeperidine standards were prepared and stored at −20 °C prior to addition to standard curves. Ice-cold methanol containing 0.4 μM meperidined4 (0.4 ml) was added to each standard prior to centrifugation and subsequent LC-MS/MS analysis. Calibration standards were run with experimental samples each day of the LC-MS/MS analysis for quantitation. Inter-assay variability was determined by comparing the same standard concentrations run in duplicate on three separate days. The inter-day percent % coefficient of variation and accuracy over a range of normeperidine concentrations were 11 and 111% at 0.02 μM (limit of quantitation); 4.9 and 98.3% at 0.04 μM; 0.26 and 97.9% at 0.43 μM; 1.2 and 98.4% at 2.15 μM; 0.03 and 100% at 4.29 μM normeperidine, respectively.
Reaction phenotyping with P450-selective chemical inhibitors
Meperidine (50 μM) was incubated with pooled human liver microsomes (0.1 mg protein/ml) in the presence of 5 μM ticlopidine, a CYP2C19 and CYP2B6 mechanism-based inhibitor, or 1 μM ketoconazole, a CYP3A inhibitor, or a co-incubation with both 5 μM ticlopidine and 1 μM ketoconazole following a 5-minute pre-warming period at 37 °C. Reactions were initiated with the addition of an NADPH-regenerating system and quenched after 10 minutes by adding 0.4 ml of ice-cold methanol containing internal standard (meperidine-d4, 0.4 μM). Normeperidine formation was detected by LC-MS/MS and expressed as a percentage compared to vehicle control incubations with the solvent (1:9 DMSO:acetonitrile, v/v) without inhibitor.
Meperidine (10 μM) was subsequently incubated with 150-donor pooled human liver microsomes (0.1 mg protein/ml) in the presence of various chemical inhibitors to estimate relative P450 contributions to meperidine N-demethylation. Reaction mixtures were co-incubated with P450-selective chemical inhibitors for the following enzymes: 25 μM furafylline (CYP1A2), 1 μM α-naphthoflavone (CYP1A2), 15 μM PPP (CYP2B6), 5 μM sulfaphenazole (CYP2C9), 5 μM (+)-N-3-benzylnirvanol (CYP2C19), 5 μM ticlopidine (CYP2C19 and CYP2B6), 2 μM quinidine (CYP2D6), 100 μM 4-methylpyrazole (CYP2E1), and 1 μM ketoconazole (CYP3A4 and CYP3A5). Following a 5-minute pre-warming period at 37 °C, reactions were initiated by addition of NADPH-regenerating system and incubated for 10 minutes. Final solvent concentration was 0.1% DMSO, 0.9% acetonitrile (v/v). Reactions were quenched with ice-cold methanol (0.4 ml) containing internal standard (meperidine-d4, 0.4 μM). Inhibitor concentrations were chosen based on previously published enzymatic studies (Rodrigues, 1999; Suzuki et al., 2002; Walsky and Obach, 2007; Khojasteh et al., 2011). Normeperidine formation was compared to vehicle controls containing 1:9 DMSO:acetonitrile (v/v) without inhibitors.
Meperidine N-demethylation with CYP2C19-genotyped human liver microsomes
To evaluate possible differences in normeperidine generation among allelic variants of CYP2C19, meperidine (50 μM) was incubated with individual CYP2C19-genotyped human liver microsomes (0.2 mg protein/ml) prepared from eighteen donors and 150-donor pooled human liver microsomes (0.2 mg protein/ml). Reaction mixtures (0.2 ml total) were pre-warmed for 5 minutes at 37 °C in a shaking water bath prior to addition of an NADPH-regenerating system. Following a 10-minute incubation period, reactions were quenched using twice the reaction volume (0.4 ml) of ice-cold methanol containing normeperidine-d4 (internal standard, 0.4 μM). Samples were then mixed with a vortex device for 10 seconds and centrifuged for 20 minutes (3740 × g at 4 °C). The supernatant was then collected and stored at −20 °C prior to LC-MS/MS analysis.
(S)-Mephenytoin 4′-hydroxylation with CYP2C19-genotyped human liver microsomes
In order to compare rates of normeperidine formation with a validated marker reaction for assessment of CYP2C19 enzyme activity in individual donors, rates of S-mephenytoin 4′-hydroxylation were determined in pooled and CYP2C19-genotyped human liver microsomes using previously published methods (Walsky and Obach, 2004). (S)-Mephenytoin (60 μM) was incubated with pooled human liver microsomes and eighteen individual CYP2C19-genotyped human liver microsomes (0.2 mg protein/ml) for 40 minutes. Reactions were quenched with the addition of 0.4 ml of ice-cold methanol containing 2 μM 4′-hydroxymephenytoin-d3 and prepared via centrifugation (3740 × g at 4 °C) for 20 minutes prior to LC-MS/MS analysis. Formation of 4′-hydroxymephenytoin was quantified by comparison to an eight-point standard curve containing commercial synthetic 4′-hydroxymephenytoin in the range of 50–10,000 nM. Standard curves were prepared each day following experiments using previously made 4′-hydroxymephenytoin stock solutions stored at −20 °C.
Effect of P450-selective chemical inhibitors on individual human liver microsomes
Additional experiments were conducted with a sub-set of individual human liver microsomal samples using chemical inhibitors of CYP2C19, CYP2B6, and CYP3A to evaluate the effects on meperidine N-demethylation. Individual donors that formed relatively high levels of normeperidine were selected: QLC (CYP2C19*1/*1), HH863 (CYP2C19*2/*2), and ZKI (CYP2C19*17/*17). Single-donor human liver microsomes (0.1 mg protein/ml) were incubated with meperidine (10 μM) in the presence of 5 μM (+)-N-3-benzylnirvanol (CYP2C19 inhibitor) and 1 μM ketoconazole (CYP3A inhibitor) for 10 minutes at 37 °C in a shaking water-bath. For the time-dependent inhibitors, microsomal samples were pre-incubated with 5 μM ticlopidine (CYP2B6/2C19 inhibitor), 30 μM PPP (CYP2B6 inhibitor), or vehicle for 15 minutes, and then meperidine (10 μM) was added for a 10-minute incubation. Following the incubation, normeperidine formation was measured by LC-MS/MS analysis as described above, and metabolite formation was compared with incubations without inhibitor (vehicle control). Meperidine-d4 (0.4 μM) was used as the internal standard for experiments with benzylnirvanol and ketoconazole, and normeperidine-d4 (0.4 μM) was used as the internal standard for experiments with ticlopidine and PPP.
LC-MS/MS analysis
Meperidine and normeperidine were analyzed by LC-MS/MS using two systems. LC-MS/MS system 1 consisted of a Thermo TSQ Quantum triple quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA) interfaced with an HPLC system and a Thermo PAL autoinjection system. LC-MS/MS system 2 consisted of a Shimadzu LCMS 8030 triple quadrupole mass spectrometer interfaced with two HPLC LC-20AD XR pumps and a SIL-20AC XR autosampler (Shimadzu, Kyoto, Japan) at the Lipscomb University College of Pharmacy. Pharmaceutical Sciences Bioanalytical Core laboratory. Compounds were detected from 20-μl sample injections onto a Kinetex C18 or EVO C18 column (2.6 μm, 50 mm × 2.1 mm, 100 Å) (Phenomenex, Torrance, CA) analytical grade HPLC column using a gradient elution scheme. For analysis of experiments containing meperidine, solvent A was Optima LC/MS grade water (Fisher Scientific, Hampton, NH) containing 0.1% formic acid (v/v), and solvent B was Optima LC/MS grade acetonitrile (Fisher Scientific, Hampton, NH) containing 0.1% formic acid (v/v). Samples were analyzed using a 0.3 ml/min solvent flow rate and a column temperature of 40 °C. The following gradient elution scheme was used: 95% A from 0–1 minutes, linear gradient from 95% to 5% A from 1–2 minutes, 5% A from 2–2.5 minutes, linear gradient from 5% to 95% A from 2.5–3 minutes, and 95% A from 3–5 minutes (v/v). LC-MS analysis was performed using electrospray ionization. Spectral data were analyzed using Thermo Xcalibur 2.0 (Thermo Fisher Scientific, Waltham, MA) and LabSolutions 5.85 (Shimadzu, Kyoto, Japan) software.
Meperidine, meperidine-d4, normeperidine-d4, and normeperidine were detected in the positive ion mode using multiple reaction monitoring (MRM) with the following precursor-to-product ion transitions: m/z 248.2 → 220.2 for meperidine, m/z 252.2 → 224.2 for meperidine-d4, m/z 238.2 → 164.2 for normeperidine-d4, and m/z 234.2 → 160.2 for normeperidine. Transitions were determined based on product ion fragmentation data collected using a collision energy of 20–21 V and previously published spectral data (Zhang et al., 2013).
Levels of 4′-hydroxymephenytoin and 4′-hydroxymephenytoin-d3 were detected using MRM with the following mass transitions: m/z 232.7 → 190.0 in the negative mode and m/z 235.0 → 149.9 in the positive mode for 4′-hydroxymephenytoin, and m/z 235.8 → 193.0 in the negative mode and m/z 238.0 → 150.0 in the positive mode for 4′-hydroxymephenytoin-d3. Transitions were selected based on published analytical methods (Stewart et al., 2011; Wang et al., 2014; Kus et al., 2015). For analysis of reactions containing (S)-mephenytoin, solvent A was Optima LC/MS grade water (Fisher Scientific, Hampton, NH) containing 0.05% formic acid (v/v), and solvent B was Optima LC/MS grade acetonitrile (Fisher Scientific) containing 0.05% formic acid (v/v) using the same gradient scheme described above.
Data analysis
All experiments were performed in triplicate and repeated three times unless otherwise stated. Data are represented as average ± standard deviation (SD) unless otherwise stated. Standard curves were assessed using r2 values with 1/C curve fitting in Shimadzu LabSolutions software. For reaction phenotyping experiments using chemical inhibitors, levels of normeperidine formation were compared to control incubations. Kinetic data were fit to the Michaelis-Menten equation, and estimates of Km and Vmax (kcat) were determined using non-linear regression analysis with GraphPad Prism 7 (GraphPad Software Inc., San Diego, CA, USA). The kcat/Km values from recombinant P450 enzymes were normalized to reported P450 abundances in human liver to calculate the adjusted estimated intrinsic clearance (Clint,expressed P450) for each enzyme using eq. 1 below (Kazui et al., 2010):
| (1) |
| (2) |
Linear regression r2 and Pearson r correlation coefficient was used to analyze the correlation between normeperidine formation and P450 activities in human liver microsomes, assuming a Gaussian distribution. Outlier analysis was performed using Grubbs’ test (α = 0.05). All statistical calculations were performed using GraphPad Prism 7 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was set at p < 0.05.
Results
Reaction phenotyping with recombinant CYP450 enzyme panel
Meperidine was incubated with a panel of recombinant P450 enzymes to assess the relative contributions of individual P450 enzymes to normeperidine generation. The results demonstrate that not only are CYP2B6 and CYP2C19 involved in meperidine N-demethylation (Fig. 2), as reported previously (Ramírez et al., 2004), but CYP2D6 and CYP1A2 are also capable of normeperidine formation. Recombinant CYP2B6 and CYP2C19 were found to generate relatively higher levels of normeperidine compared to other P450 enzymes in the following order (units shown are the peak area ratios of normeperidine to internal standard, mean ± SD): CYP2B6 (0.31 ± 0.15) ≈ CYP2C19 (0.19 ± 0.04) > CYP2D6 (0.09 ± 0.04) > CYP1A2 (0.04 ± 0.02) > CYP3A4 (0.01 ± 0.004). Lower normeperidine levels were detected in incubations with recombinant CYP2C8 (0.007 ± 0.003), CYP3A5 (0.005 ± 0.004), CYP2C9 (0.004 ± 0.002), CYP2A6 (0.003 ± 0.001), and CYP2E1 (0.002 ± 0.001).
Figure 2.
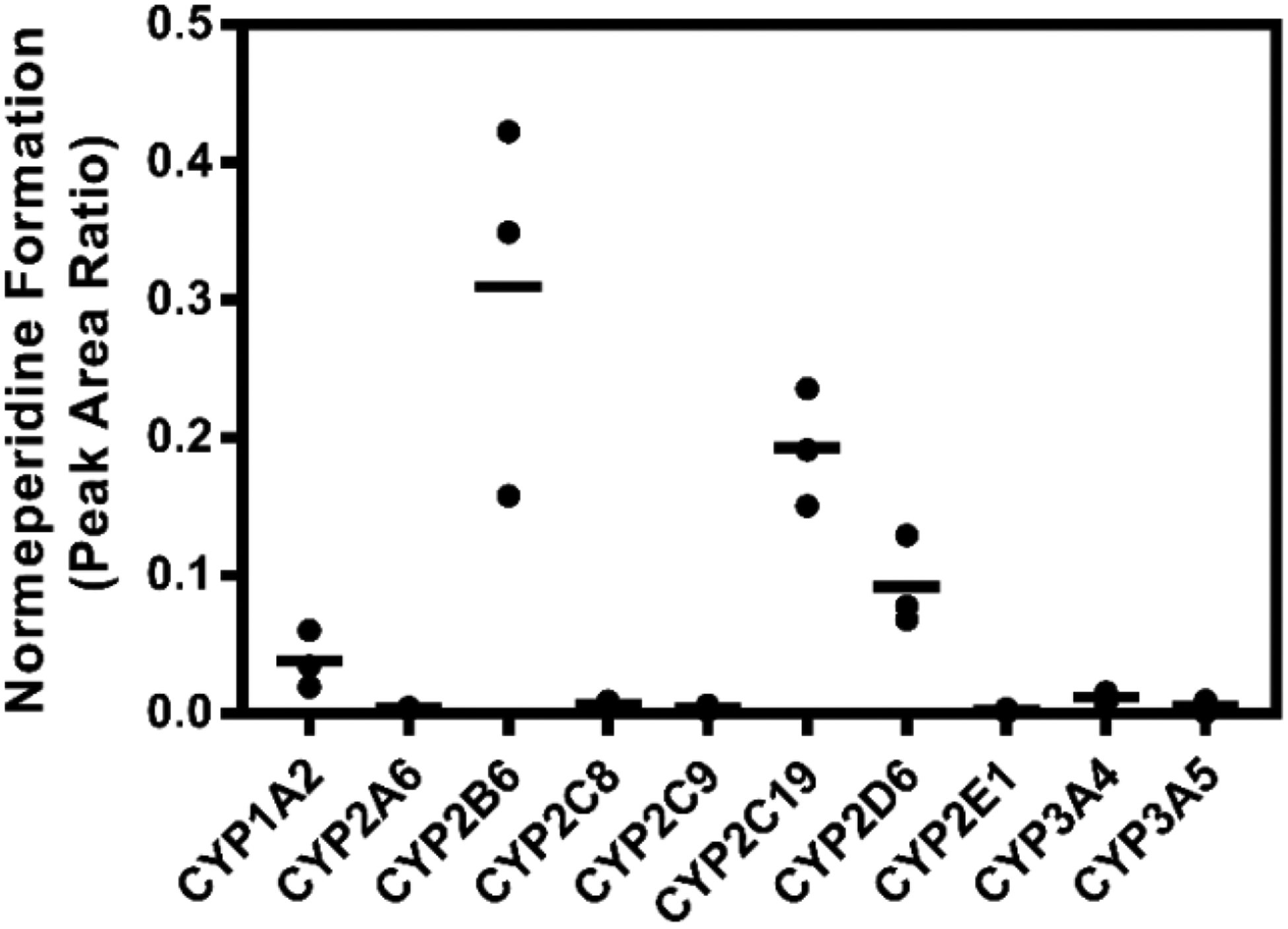
Normeperidine formation by recombinant P450 enzymes. Meperidine (10 μM) was incubated with recombinant P450 enzymes (20 nM) for 10 minutes. Normeperidine was detected by LC-MS/MS. Relative levels are represented as the peak area ratio of normeperidine (m/z 234.2→160.2) to internal standard (meperidine-d4, m/z 252.2→224.2). Each point represents the average of a single experiment performed in triplicate; bars are the mean values of normeperidine formation from three independent experiments (n = 3) performed in triplicate each.
Kinetic analysis of meperidine N-demethylation with pooled human liver microsomes
The kinetic parameters Km and Vmax of meperidine N-demethylation were also assessed using pooled human liver microsomes (Fig. 3A). The mean Vmax and apparent Km values were 1216 ± 152 pmol min−1 mg−1 protein and 622 ± 63 μM, respectively (mean ± SD). The catalytic efficiency (Vmax/Km) for this reaction was 2.0 ± 0.1 pmol/min/mg protein/μM. Hydrolysis of meperidine to meperidinic acid was also observed from microsomal incubations with meperidine in a concentration-dependent manner (Supplemental Figures S1 and S2). A detailed description of the LC-MS/MS detection and relative quantitation of meperidinic acid from human liver microsomal incubations is provided in the Supplementation Information. The estimated apparent Km for meperidinic acid formation in pooled human liver microsomes was 1.6 mM, which is similar to the Km (1.9 mM) described by Zhang et al. (1999) for human liver carboxylesterase hCE-1.
Figure 3.
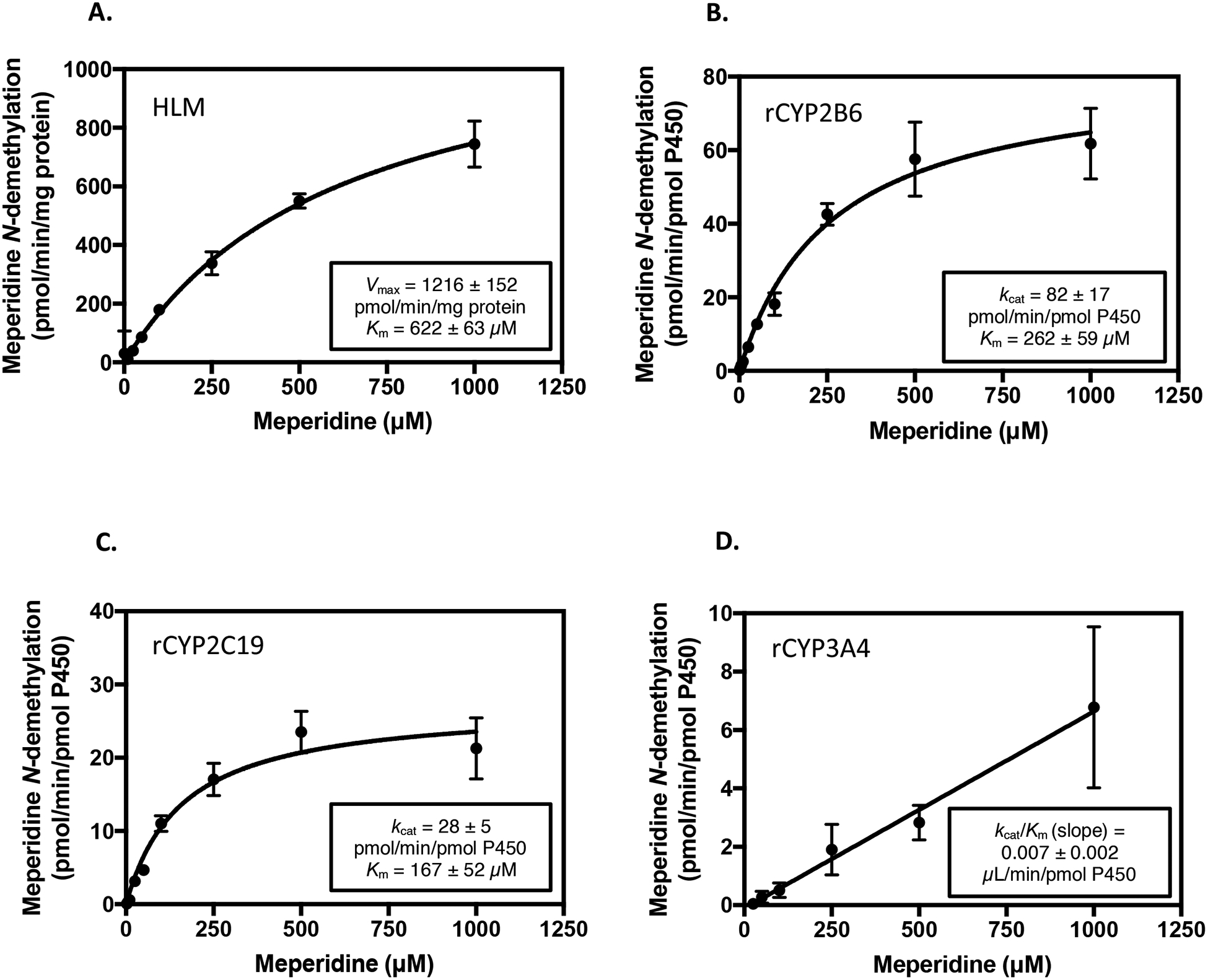
Kinetic analysis of normeperidine formation by pooled human liver microsomes and recombinant P450 enzymes. Meperidine (1–1000 μM) was incubated with (A) pooled human liver microsomes, HLM (0.1 mg protein/ml), (B) recombinant (r) CYP2B6, (C) rCYP2C19, and (D) rCYP3A4 (10 nM) for 10 minutes. Metabolite formation was detected by LC-MS/MS analysis and is expressed as pmol normeperidine formed/min/mg protein for incubations with pooled human liver microsomes, or pmol normeperidine formed/min/pmol P450 enzyme for recombinant P450s. Three independent experiments were conducted in triplicate each to assess reproducibility. Shown are the results (mean ± SD) from a representative experiment with each enzyme system performed in triplicate. Error bars are the standard deviation of the mean from three replicates in the same experiment. Kinetic data were fit to the Michaelis-Menten equation, and the apparent Km and Vmax (kcat for recombinant enzymes) were determined by nonlinear regression analysis using GraphPad Prism 7 Software. Values for apparent Km,Vmax, and kcat are the mean ± SD of triplicate determinations.
Kinetic analysis of meperidine N-demethylation with individual recombinant CYP450 enzymes
Kinetic parameters were similarly determined in individual recombinant P450 enzymes to obtain a quantitative measure of CYP2B6, CYP2C19, and CYP3A4 enzyme activity towards meperidine N-demethylation (Fig. 3B–D). The mean kcat and Km values (mean ± SD) were 82 ± 17 pmol/min/pmol P450 and 262 ± 59 μM for CYP2B6, and 28 ± 5 pmol/min/pmol P450 and 167 ± 52 μM for CYP2C19, respectively. The overall catalytic efficiency (kcat/Km) for meperidine N-demethylation by CYP2B6 and CYP2C19 was: CYP2B6 kcat/Km = 0.31 ± 0.02 min−1 μM−1; CYP2C19 kcat/Km = 0.17 ± 0.03 min−1 μM−1 (mean ± SD). In incubations with recombinant CYP3A4, meperidine N-demethylation did not reach saturation at substrate concentrations up to 1000 μM meperidine. However, the estimated catalytic efficiency of CYP3A4, as determined by the linear slope (kcat/Km) of the velocity vs. substrate concentration plot was 0.007 ± 0.002 min−1 μM−1 (mean ± SD).
Kinetic data (kcat/Km) from recombinant P450 enzymes were normalized to the specific P450 abundances in human liver reported from three studies (Rodrigues, 1999; Rowland Yeo et al., 2004; Achour et al., 2014) to estimate intrinsic clearance values and percent contribution of each P450 (Ramirez et al., 2004; Kazui et al., 2010). It should be noted that the P450 abundances in human liver reported differed for each study (Rodrigues, 1999; Rowland Yeo et al., 2004; Achour et al., 2014). For example, the reported mean abundance values for CYP2B6 were 11, 16, and 39 pmol P450/mg protein (Rowland Yeo et al. 2004; Achour et al. 2014; Rodrigues 1999). The mean abundance values reported for CYP2C19 were 14, 11, and 19 pmol P450/mg protein, and the reported average abundance values for CYP3A4 were 155, 93, and 108 pmol P450/mg protein (Rowland Yeo et al. 2004; Achour et al. 2014; Rodrigues 1999). The kcat/Km values determined in the present study for recombinant CYP2B6, CYP2C19, and CYP3A4 were multiplied by the enzyme abundances (pmol P450/mg protein) to calculate the adjusted predicted intrinsic clearance, Clint, P450 (μl/min/mg protein) for each enzyme. Based on the variation in the mean P450 abundances reported, the estimated percent (%) P450 contributions (fm, P450) for meperidine N-demethylation were CYP2B6 (49–75%), CYP2C19 (20–35%), and CYP3A4 (5–16%). These data are shown Table 1.
Table 1.
Kinetic parameters of meperidine N-demethylation by recombinant P450s and estimated enzyme contributions.
| Approach 1 | ||||
|---|---|---|---|---|
| Recombinant P450 |
kcat/Kma (min−1 μM−1) |
Enzyme Abundanceb (pmol P450/mg protein) | Adjusted Clint (μl/min/mg protein) | Percent Contribution (%) |
| CYP2B6 | 0.31 ± 0.02 | 11 | 3.4 | 49 |
| CYP2C19 | 0.17 ± 0.03 | 14 | 2.4 | 35 |
| CYP3A4 | 0.007 ± 0.002 | 155 | 1.1 | 16 |
| Approach 2 | ||||
| Recombinant P450 |
kcat/Km (min−1 μM−1) |
Enzyme Abundancec (pmol P450/mg protein) | Adjusted Clint (μl/min/mg protein) | Percent Contribution (%) |
| CYP2B6 | 0.31 ± 0.02 | 16 | 5.0 | 66 |
| CYP2C19 | 0.17 ± 0.03 | 11 | 1.9 | 25 |
| CYP3A4 | 0.007 ± 0.002 | 93 | 0.7 | 9 |
| Approach 3 | ||||
| Recombinant P450 |
kcat/Km (min−1 μM−1) |
Enzyme Abundanced (pmol P450/mg protein) | Adjusted Clint (μl/min/mg protein) | Percent Contribution (%) |
| CYP2B6 | 0.31 ± 0.02 | 39 | 12 | 75 |
| CYP2C19 | 0.17 ± 0.03 | 19 | 3.2 | 20 |
| CYP3A4 | 0.007 ± 0.002 | 108 | 0.8 | 5 |
Mean ± SD kcat/Km values are shown from a single experiment performed in triplicate. Kinetic parameters for recombinant CYP2B6 and CYP2C19 were estimated using non-linear regression analysis by fitting kinetic data to the Michaelis-Menten equation using GraphPad Prism 7 Software. The estimated kcat/Km value for CYP3A4 was based on the linear slope of the velocity vs. substrate concentration plot determined using linear regression analysis with GraphPad Prism 7 Software. Adjusted intrinsic clearance (Clint) was calculated by multiplying kcat/Km values by the reported P450 abundances.
Mean P450 abundances were obtained from data reported in Rowland-Yeo et al. (2004).
Mean P450 abundances were obtained from data reported in Achour et al. (2014).
Mean P450 abundances were obtained from data reported in Rodrigues (1999).
Reaction phenotyping with P450-selective chemical inhibitors
Preliminary inhibition experiments were performed with meperidine (50 μM) co-incubated in pooled human liver microsomes with ticlopidine (CYP2B6/CYP2C19 inhibitor) and ketoconazole (CYP3A inhibitor) to confirm the contributions of CYP2B6, CYP2C19, and CYP3A4 reported in the literature (Ramírez et al., 2004). Ticlopidine (5 μM) decreased normeperidine formation by 37 ± 8% compared to control, and ketoconazole (1 μM) reduced normeperidine generation by 23 ± 10% (mean ± SD) (Fig. 4). Co-incubation with both ticlopidine and ketoconazole reduced normeperidine formation by 50 ± 3%, compared to control incubations without inhibitor.
Figure 4.

Effect of chemical inhibition of CYP2B6, CYP2C19, and CYP3A on normeperidine formation by pooled human liver microsomes. Meperidine (50 μM) was incubated with pooled human liver microsomes (0.1 mg protein/ml) in the presence of 5 μM ticlopidine, a CYP2C19 and CYP2B6 mechanism-based inhibitor, and/or 1 μM ketoconazole, a CYP3A inhibitor, for 10 minutes. Normeperidine formation was detected by LC-MS/MS. Average levels of normeperidine formation from control incubations without inhibitor were: 0.05 ± 0.004 (expressed as peak area ratio, mean ± SD). Percent metabolite formation was determined by comparison to vehicle control incubations without inhibitor. The results are the mean ± SD of a single experiment performed in triplicate.
A full panel of P450-selective chemical inhibitors was added to pooled human liver microsomes to further evaluate the contributions of individual P450 enzymes to normeperidine formation (Fig. 5). In these experiments, inhibition of CYP3A by ketoconazole reduced normeperidine formation by 39 ± 10% (mean ± SD) compared to vehicle control. Ticlopidine (CYP2B6/CYP2C19 inhibitor) reduced normeperidine formation by 30 ± 5%. PPP, a selective CYP2B6 inhibitor, reduced normeperidine generation by 25 ± 13%. Selective inhibition of CYP2C19 by benzylnirvanol decreased meperidine N-demethylation by 21 ± 10%. Interestingly, the CYP1A2 inhibitors α-naphthoflavone and furafylline also reduced meperidine N-demethylation compared to control, although modestly (20 ± 12% inhibition by furafylline; 13 ± 8% inhibition by α-naphthoflavone).
Figure 5.
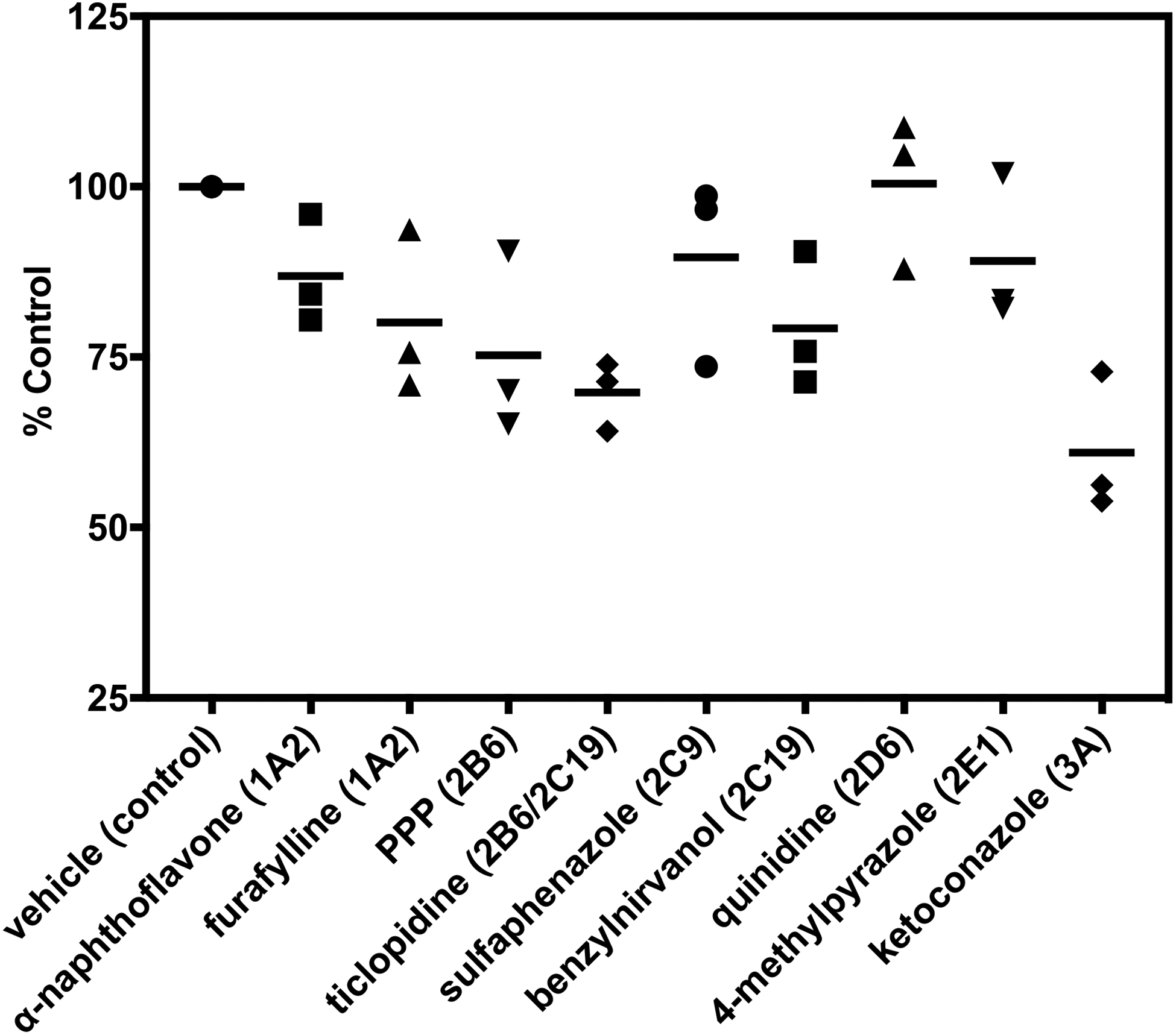
Effect of P450-selective chemical inhibitors on normeperidine formation by pooled human liver microsomes. Meperidine (10 μM) was incubated with pooled human liver microsomes (0.1 mg protein/ml) in the presence of P450-selective chemical inhibitors for 10 minutes (See “Materials and Methods” for inhibitor concentrations). Normeperidine formation was detected by LC-MS/MS. Average levels of normeperidine formation from control incubations without inhibitor were: 0.04 ± 0.02 (expressed as peak area ratio, mean ± SD). Percent metabolite formation was determined by comparison to vehicle control without inhibitor. Each point represents the average of a single experiment performed in triplicate, and bars are the mean of three independent experiments (n = 3) performed in triplicate each.
Meperidine N-demethylation by CYP2C19-genotyped human liver microsomes
The results from reaction phenotyping experiments using recombinant P450 enzymes and pooled human liver microsomal fractions demonstrated that multiple P450s are involved, and CYP2C19 may play a significant role in generating normeperidine in some individuals. CYP2C19 is highly polymorphic, with several well-known allelic variants, which impact the pharmacokinetics of commonly prescribed therapeutic agents (Scott et al., 2011; Sanford et al., 2013). Thus, we investigated whether CYP2C19 genetic variation impacts normeperidine formation. Meperidine N-demethylation was assessed in human liver microsomal fractions from individual CYP2C19-genotyped donors: CYP2C19*1/*1 (n = 6), CYP2C19*1/*2 (n = 2), CYP2C19*2/*2 (n = 4), CYP2C19*1/*17 (n = 3), and CYP2C19*17/*17 (n = 3). The CYP2C19 allelic variants were chosen to include both the wild-type and the most commonly expressed no function (*2) and increased function (*17) alleles. P450 enzyme activities were provided by the suppliers (Corning, XenoTech, and BioreclamationIVT). Single-donor microsomal lot HH863, a CYP2C19*2/*2 donor, was identified as an outlier, with respect to CYP3A4 activity (testosterone 6β-hydroxylation) among all human microsomal samples evaluated, based on Grubbs’ outlier analysis (α = 0.05). HH863 had the highest CYP3A4 activity (testosterone 6β-hydroxylation) compared to other CYP2C19*2/*2 lots (HH40 and HH689), according to the activities provided by the supplier (Corning). The testosterone 6β-hydroxylation activity for HH863 was 8700 pmol/min/mg protein, compared to HH40 at 2400 pmol/min/mg protein and HH689 at 2300 pmol/min/mg protein.
The results from incubation of meperidine (50 μM) with individual human liver microsomes are shown in Fig. 6. Donor lot HH863 was also identified as an outlier with respect to normeperidine formation; the rate of normeperidine formation was 6.8-fold higher by this donor (265 pmol/min/mg protein), compared to the mean rates of normeperidine generation (39 ± 4.4 pmol/min/mg protein) by other donors with the same genotype CYP2C19*2/*2 (lots HH40 and HH689) (Fig. 6A). Moreover, this donor had higher total P450 content, and higher CYP2B6, CYP2C19, and CYP3A4 activity compared to other microsomal donors with the same genotype (as determined by the supplier, Corning Life Sciences).
Figure 6.
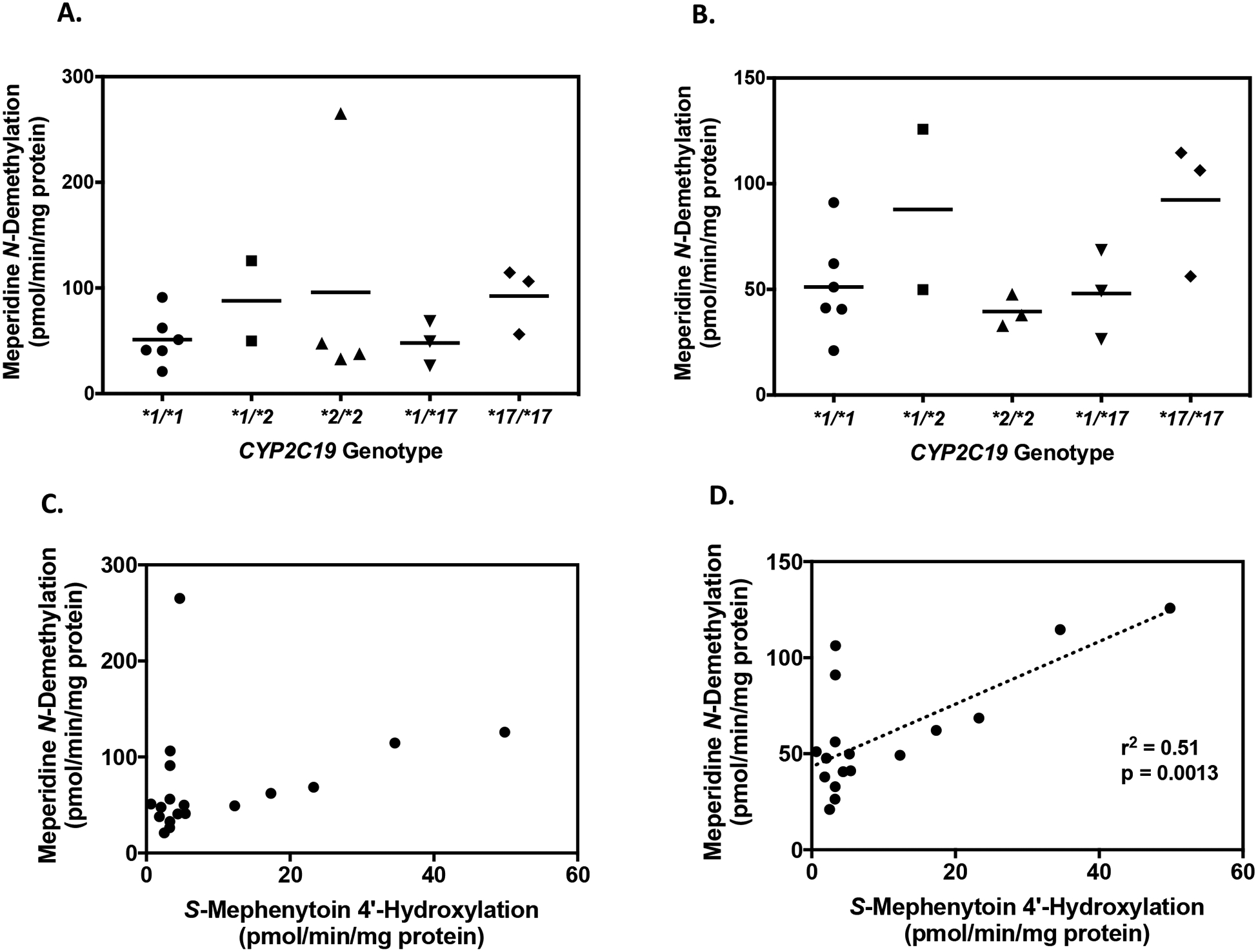
Normeperidine formation by CYP2C19-genotyped human liver microsomes. Meperidine (50 μM) was incubated with human liver microsomes (0.1 mg protein/ml) from 18 individual donors for 10 minutes to assess normeperidine formation. Genotype groups included the following: CYP2C19*1/*1 (n = 6), CYP2C19*1/*2 (n = 2), CYP2C19*2/*2 (n = 4), CYP2C19*1/*17 (n = 3), and CYP2C19*17/*17 (n = 3). (A) Comparison of normeperidine formation by CYP2C19 genotype. Bars represent the average metabolite formation for each CYP2C19 genotype. Each point represents the average metabolite formation for a single donor determined from three independent experiments, each performed in triplicate. Donor HH863 (CYP2C19*2/*2) was identified as an outlier with respect to normeperidine formation, based on Grubb’s outlier analysis (GraphPad Prism 7 Software). (B) Comparison of normeperidine formation by CYP2C19 genotype, excluding outlier HH863. (C) Correlation of meperidine N-demethylation with S-mephenytoin 4′-hydroxylation, a measure of CYP2C19 activity. S-Mephenytoin (60 μM) was incubated with CYP2C19-genotyped human liver microsomes (0.2 mg protein/ml) for 40 minutes. Formation of 4′-hydroxymephenytoin by individual human liver microsomes was measured from a single experiment performed in triplicate. Each point represents the average metabolite formation by individual donors (n = 18). (D) Correlation of meperidine N-demethylation with S-mephenytoin 4′-hydroxylation, excluding outlier HH863 (n = 17). Linear regression analysis was used to analyze the correlation between normeperidine formation and CYP2C19 activity (r2 = 0.51, p = 0.0013, n = 17). Statistical analysis was performed by Pearson r correlation using GraphPad Prism 7 Software.
Among the remaining 17 individual human liver microsomal samples tested, the mean normeperidine formation for each CYP2C19 genotype (expressed as means ± SD) were as follows: 51 ± 24 pmol/min/mg protein for CYP2C19*1/*1 donors (n = 6), 88 ± 54 pmol/min/mg protein for CYP2C19*1/*2 donors (n = 2), 39 ± 7.6 pmol/min/mg protein for CYP2C19*2/*2 donors (n = 3) (excluding outlier HH863), 48 ± 21 pmol/min/mg protein for CYP2C19*1/*17 donors (n = 3), and 92 ± 32 pmol/min/mg protein for CYP2C19*17/*17 donors (n = 3) (Fig. 6B).
Additional experiments were performed to test whether normeperidine formation was correlated with CYP2C19 activity in the same set of individual human liver microsomes. S-mephenytoin 4′-hydroxylation, which is a validated marker of CYP2C19 activity, was measured by our lab similar to the methods described previously (Walsky and Obach, 2004). Notably, CYP2C19 activity (formation of 4′-hydroxymephenytoin) did not display the expected gene-dose relationship across CYP2C19 genotypes in this set of individual genotyped human liver microsomes (Supplemental Figure S3). SMephenytoin 4′-hydroxylation varied widely within genotype groups and across different CYP2C19 genotypes (range among all donors: 0.65 to 50 pmol/min/mg protein). Based on our analysis, individual donor lot 710456 (genotyped as CYP2C19*1/*2) had the highest formation of 4′-hydroxymephenytoin (50 pmol/min/mg protein) and relatively high formation of normeperidine (126 pmol/min/mg protein) among the tissues tested. When outlier HH863 was excluded from the analysis, normeperidine formation was significantly correlated with CYP2C19 activity (r2 = 0.51, p = 0.0013, n = 17), (Fig. 6D). However, when HH863 was included in the analysis with other individual donors, the correlation was not present (r2 = 0.09, p = 0.24, n = 18) (Fig. 6C).
In a subset analysis of 11 individual genotyped human liver microsomes purchased from the same supplier (BioreclamationIVT), the correlation of normeperidine formation was also examined between CYP2B6, CYP3A4, and CYP2C19 activities, based on P450 activities provided by BioreclamationIVT using probe substrates (Supplemental Figure S4). In this analysis, normeperidine formation was significantly correlated with CYP2C19 activity, as measured by S-mephenytoin 4′-hydroxylation (r2 = 0.47, p = 0.02, n = 11). Normeperidine formation was not significantly correlated with CYP2B6 activity, bupropion hydroxylation (r2 = 0.03, p = 0.60, n = 11) or CYP3A4 activity, testosterone 6β-hydroxylation, (r2 = 0.30, p = 0.08, n = 11) in these microsomal samples.
Further studies were conducted using selective chemical inhibitors to evaluate the contributions of CYP2C19, CYP2B6, and CYP3A4 to normeperidine formation in human liver microsomal samples from different individual donors with variable P450 activities. Donors were selected based on having high relative normeperidine formation but different CYP2C19 genotypes: QLC (CYP2C19*1/*1), HH863 (CYP2C19*2/*2), and ZKI (CYP2C19*17/*17). The CYP2C19 activity (measured by S-mephenytoin 4′-hydroxylation) in donors QLC, HH863, and ZKI was: 3.3, 4.6, and 35 pmol/min/mg protein, respectively. Individual liver microsomal samples were incubated in the presence of the selective chemical inhibitors benzylnirvanol (CYP2C19), ketoconazole (CYP3A), ticlopidine (CYP2B6/2C19, 15-minute pre-incubation), and PPP (CYP2B6, 15-minute pre-incubation).
In human liver microsomes from donor ZKI (CYP2C19*17/*17), which had relatively high CYP2C19 activity, benzylnirvanol and ticlopidine reduced normeperidine levels by 43 ± 4.6% and 52 ± 7.5% (mean ± SD), respectively, compared to control incubations without inhibitor (Fig. 7). Ketoconazole decreased normeperidine formation by 48 ± 1.0%, compared to control. The CYP2B6 inhibitor PPP had minimal effect (<20% inhibition) on normeperidine levels, compared to control. These results indicate that CYP2C19 and CYP3A were the major contributors to microsomal normeperidine generation by donor ZKI (CYP2C19*17/*17).
Figure 7.
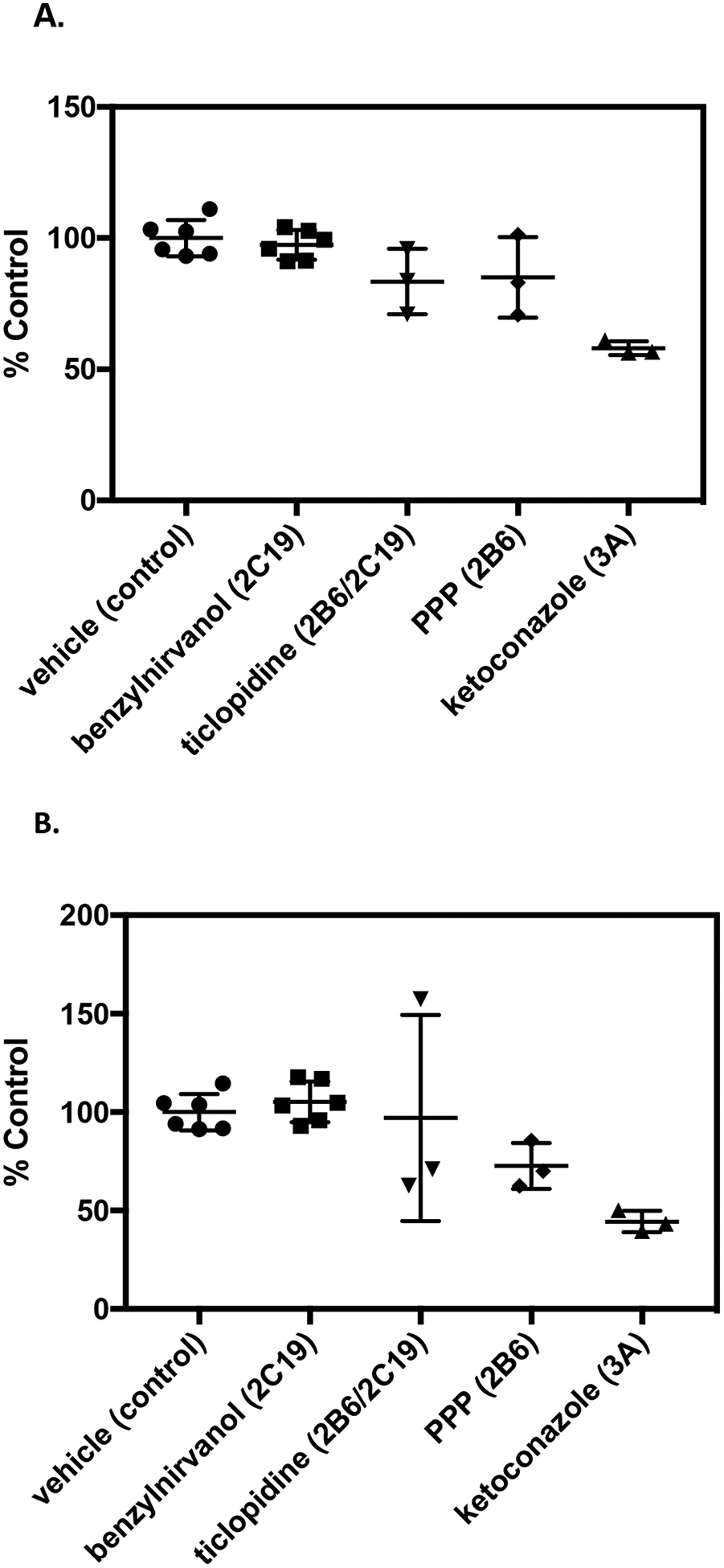
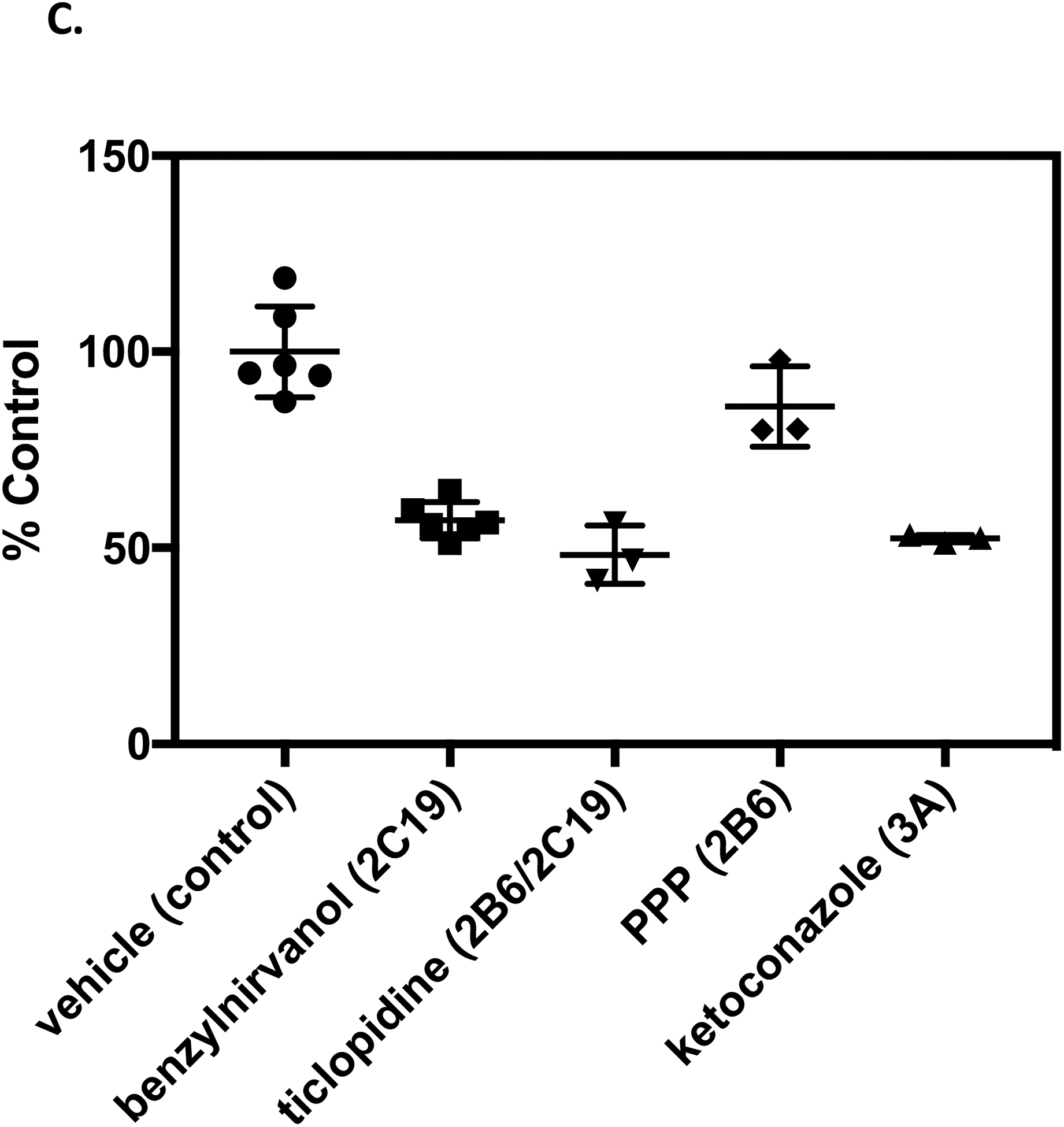
Effect of P450-selective chemical inhibitors on normeperidine formation by human liver microsomes from individual donors. Individual human liver microsomal samples: (A) QLC (CYP2C19*1/*1), (B) HH863 (CYP2C19*2/*2), and (C) ZKI (CYP2C19*17/*17), were selected based on donors that formed relatively high levels of normeperidine in previous studies (see Fig. 5). Meperidine (10 μM) was incubated with single-donor human liver microsomes (0.1 mg protein/ml) in the presence of 5 μM (+)-N-3-benzylnirvanol (CYP2C19 inhibitor) and 1 μM ketoconazole (CYP3A inhibitor) for 10 minutes. For time-dependent inhibitors, microsomal samples were pre-incubated with 5 μM ticlopidine (CYP2B6/2C19 inhibitor), 30 μM PPP (CYP2B6 inhibitor), or vehicle for 15 minutes, then meperidine (10 μM) was added for a 10-minute incubation. Normeperidine formation was detected by LC-MS/MS analysis. Average levels of normeperidine formation from control incubations without inhibitor were: (A) QLC (0.01 ± 0.002), (B) HH863 (0.02 ± 0.002), and (C) ZKI (0.02 ± 0.002) (expressed as peak area ratio, mean ± SD). Percent control metabolite formation was determined by comparison to vehicle control without inhibitor. Shown are the means ± SD (range) from two independent experiments performed in triplicate each for incubations with vehicle and benzylnirvanol, or a single experiment performed in triplicate for incubations with vehicle, ketoconazole, ticlopidine, and PPP. Each point is a replicate determination, and error bars are the standard deviation of the mean. Experiments with individual donor samples were performed on the same day under the same experimental conditions for comparison.
In incubations with human liver microsomes from QLC (CYP2C19*1/*1) and HH863 (CYP2C19*2/*2), which had relatively low CYP2C19 activity, benzylnirvanol and ticlopidine had little to no effect (<20% inhibition) on normeperidine formation, compared to vehicle control. Ketoconazole decreased normeperidine levels by 42 ± 3% and 56 ± 5% in incubations with QLC and HH863, respectively, compared to control. PPP had little effect (<20%) on normeperidine formation by donor QLC and showed a 27 ± 12% reduction with donor HH863, compared to control. These findings demonstrate that CYP3A played a dominant role in normeperidine generation by liver microsomal samples QLC and HH863; in addition to CYP3A, CYPB6 may have contributed to normeperidine generation by donor HH863 (Fig. 7).
Discussion
Understanding the contributions of P450 enzymes to formation of the neurotoxic metabolite normeperidine is important in determining the potential risk factors of meperidine toxicity. Meperidine is known to cause agitation, tremors, and seizures when administered at high doses or over prolonged periods (Latta et al., 2002). Meperidine neurotoxicity is believed to occur from accumulation of the N-demethylated metabolite normeperidine. Although normeperidine is implicated in meperidine neurotoxicity, very few studies have characterized the enzymology of normeperidine formation. Ramírez et al. (2004) reported that CYP2B6 and CYP3A4 are the primary enzymes responsible for generating normeperidine in human liver microsomes; CYP2C19 was suggested to have a minor role in normeperidine formation. The results of the present study suggest that CYP2C19 may play a significant role in meperidine N-demethylation in individuals with high CYP2C19 activity. This is relevant because CYP2C19 is highly polymorphic. Pharmacogenetic associations between P450 polymorphism and normeperidine formation have not been previously established. To our knowledge, this is the first study to investigate the effects of CYP2C19 genetic variation on meperidine N-demethylation.
An integrated reaction phenotyping approach was used to characterize normeperidine formation by P450 enzymes. Reaction phenotyping experiments with a panel of recombinant P450s revealed that CYP2B6 and CYP2C19 generated the highest levels of normeperidine compared to other recombinant enzymes in the following order: CYP2B6 ≈ CYP2C19 > CYP2D6 > CYP1A2 > CYP3A4 (Fig 2). Ramirez et al. (2004) reported similar observations for normeperidine formation by recombinant CYP2B6 > CYP2C19 > CYP2C18 > CYP3A4 > CYP2D6. Kinetic experiments demonstrated that recombinant CYP2B6 and CYP2C19 had catalytic efficiencies (kcat/Km) for meperidine N-demethylation (CYP2B6 kcat/Km = 0.31 min−1 μM−1; CYP2C19 kcat/Km = 0.17 min−1 μM−1) (Fig. 3) similar to the values reported by Ramirez et al. (2004) for meperidine N-demethylation (CYP2B6 Vmax/Km = 0.39 min−1 μM−1 and CYP2C19 Vmax/Km = 0.21 min−1 μM−1). The catalytic efficiency of recombinant CYP3A4 for meperidine N-demethylation was markedly lower (0.007 min−1 μM−1) in this study compared to that reported previously (0.07 min−1 μM−1) by Ramirez et al. (2004).
Kinetic data were normalized to the P450 abundances reported in human liver microsomes from multiple studies to estimate the relative enzyme contributions to normeperidine formation. When the kcat/Km values in the present study were normalized to the specific P450 abundances in human liver microsomes reported from three different studies (Rowland-Yeo et al. 2004; Achour et al. 2014; Rodrigues 1999), the estimated percent P450 contributions to meperidine N-demethylation were CYP2B6 (49–75%), CYP2C19 (20–35%), and CYP3A4 (5–16%). The relative P450 contributions for normeperidine formation reported by Ramírez et al., (2004) were CYP2B6 (57%), CYP2C19 (15%), and CYP3A4 (28%). The difference in the present study compared to the previous study may be due to differences in recombinant enzyme activity and the hepatic P450 abundance values used to calculate the adjusted Clint for each enzyme. For example, the nominal CYP2B6 abundance value used in the study by Ramírez et al. (2004) was 39 pmol P450/mg protein (Rodrigues 1999), which is greater than 2-fold higher than that reported in later studies: 11 and 16 pmol P450/mg protein (Rowland Yeo et al. 2004; Achour et al. 2014). This observation emphasizes the inter-individual variability in P450 expression, and it highlights the intersystem variations in mean P450 abundance values used for scaling in vitro data to make in vivo predictions (Achour et al. 2014).
The findings from recombinant CYP2B6 and CYP2C19 are supported by P450 inhibition experiments conducted with pooled human liver microsomal fractions. Initial experiments showed that ticlopidine, a CYP2B6 and CYP2C19 inhibitor, decreased normeperidine formation by 37%, and CYP3A inhibition by ketoconazole reduced normeperidine formation by 23% (Fig. 4). Because ticlopidine is a mechanism-based inactivator of CYP2B6 and CYP2C19 (Ritchie et al., 2004), the observed inhibition by ticlopidine may be due to a combination of competitive inhibition and progressive mechanism-based inactivation during the 10-minute incubation period in human liver microsomes. Further experiments with a panel of P450-selective chemical inhibitors demonstrated that ticlopidine reduced meperidine N-demethylation by 30%; whereas, the CYP2B6-selective inhibitor PPP and the CYP2C19-selective inhibitor benzylnirvanol decreased normeperidine generation by 25% and 21%, respectively (Fig. 5). In the later experiments, ketoconazole reduced meperidine N-demethylation by 39%. This finding suggests a significant contribution of CYP3A to normeperidine formation in addition to CYP2B6 and CYP2C19.
We investigated meperidine N-demethylation in CYP2C19-genotyped human liver microsomes because these samples are commercially available. CYP2B6genotyped human liver microsomes are not readily available; thus, the effect of CYP2B6 genotype on microsomal normeperidine generation was not analyzed in this study. Experiments with CYP2C19-genotyped human liver microsomes revealed high inter-individual variability (~13-fold) in normeperidine formation (range: 21 to 265 pmol/min/mg protein, n = 18). Liver microsomal donor HH863 (genotype CYP2C19*2/*2) was identified as an outlier among donors in this study with respect to normeperidine formation and CYP3A activity (according to testosterone 6β-hydroxylation measured by supplier). Microsomal incubations with HH863 were found to produce significantly higher rates of normeperidine (265 pmol/min/mg protein) compared to all other donors in this study, and among donors of the same genotype (CYP2C19*2/*2) (Fig. 6A). The markedly higher enzyme activity in this donor compared to other donors is a possible explanation for the increase in normeperidine formation; however, the reason for this large difference is unknown and warrants further investigation.
While meperidine N-demethylation was not significantly associated with CYP2C19 genotype across the microsomal samples tested, human liver microsomes from CYP2C19*17/*17 donors produced 80% higher rates of normeperidine formation (92 ± 32 pmol/min/mg protein, n = 3) compared to CYP2C19*1/*1 donors (51 ± 24 pmol/min/mg protein, n = 6) (Fig. 6B). A limitation to this study was the small sample size in each CYP2C19 genotype group (n = 2–6). Further studies with a larger number of genotyped donors should provide a more robust assessment of normeperidine generation among donors with various CYP2C19 genotypes.
Normeperidine formation was significantly correlated with CYP2C19 activity, as measured by S-mephenytoin 4′-hydroxylation (r2 = 0.51, p = 0.0013, n = 17), (Fig. 6D), when outlier HH863 was excluded from the analysis. Following assessment of meperidine N-demethylation compared to CYP2C19 activity in 17 donors, a correlation analysis was performed comparing normeperidine formation and P450 activities in a subset of 11 individual human liver microsomal samples obtained from the same supplier, BioreclamationIVT (BioIVT) (Supplemental Figure S4). CYP2B6, CYP2C19, and CYP3A4 activities based on probe substrate marker reactions were provided by the supplier. A significant correlation was observed between meperidine N-demethylation and CYP2C19 activity assessed by S-mephenytoin 4′-hydroxylation (r2 = 0.47, p = 0.02, n = 11) (Fig. S2); however, meperidine N-demethylation was not significantly correlated with CYP2B6 and CYP3A activities in these samples. In contrast to these findings, previous studies found significant correlations between normeperidine formation and both CYP2B6 activity (S-mephenytoin N-demethylation) and CYP3A activity (midazolam 1′-hydroxylation), whereas correlations with CYP2C19 activity were found to be insignificant (Ramírez et al., 2004). These present findings suggest that variation in CYP2C19 activity may impact meperidine N-demethylation; however, other factors also contribute to differences in normeperidine formation.
Studies using selective chemical inhibitors with single-donor human liver microsomes revealed that P450 contributions to meperidine N-demethylation may vary depending on the enzyme activities in different individual livers. CYP2C19 was shown to be a significant contributor to normeperidine formation in a human liver microsomal sample with high CYP2C19 activity (ZKI CYP2C19*17/*17). CYP2C19 inhibitor (+)-N-3-benzylnirvanol and CYP2B6/2C19 inhibitor ticlopidine reduced normeperidine formation by 43 and 52%, respectively, in incubations with ZKI (Fig. 7). The estimated percent % CYP2C19 contribution for this donor was in line with the predicted % contribution calculated from kinetic parameters with recombinant P450s in the present study, but higher than the estimated % CYP2C19 contribution reported previously (Ramirez et al., 2004). The difference in the results from pooled human liver microsomes and previous studies compared to the findings from individual donor ZKI could be explained in part by the high CYP2C19 activity in this sample, which may be masked in pooled liver microsomal samples with lower CYP2C19 content. The CYP2C19*17 allele is associated with increased enzyme activity compared to the wild-type *1 allele due to enhanced gene transcription (Sim et al., 2006). CYP2C19 protein may account for a substantial proportion of the total P450 content in donor ZKI due to increased expression. CYP2C19-mediated meperidine metabolism was minimal (<20%) in microsomal samples with relatively low CYP2C19 activity (QLC CYP2C19*1/*1 and HH863 CYP2C19*2/*2), as evidenced by the lack of an effect with benzylnirvanol and ticlopidine (Fig. 7). The CYP3A inhibitor ketoconazole reduced normeperidine formation by 42–56% in incubations with each individual donor, indicating that CYP3A plays a major role in catalyzing liver microsomal meperidine N-demethylation. The estimated percent % CYP3A contribution is consistent with predictions from pooled human liver microsomes, but higher than the % contribution predicted from kinetic parameters with recombinant P450s.
Previous studies support the contention that for some substrates, P450 contributions differ depending on the respective P450 content in liver microsomal samples from different individual donors. This may be especially the case for highly polymorphic P450s. Yamazaki et al. (1997) reported that the relative contributions of CYP2C19 and CYP3A in 5-hydroxylation of the proton pump inhibitor omeprazole were dependent upon the ratio of CYP2C19 and CYP3A content in human liver microsomes from different donors. Fluvoxamine was used previously to examine CYP2C19 involvement in normeperidine formation (Ramirez et al., 2004); however, fluvoxamine is also an inhibitor of CYP1A2 (Brøsen et al., 1993). In this study, we used the potent and selective CYP2C19 inhibitor (+)-N-3-benzylnirvanol (Suzuki et al., 2002) in CYP2C19-genotyped human liver microsomes with varying P450 activities. As noted above, the donor ZKI with high microsomal CYP2C19 activity was sensitive to inhibition by benzylnirvanol and ketoconazole, whereas donors QLC and HH863 with lower CYP2C19 activity were sensitive to inhibition by ketoconazole but not benzylnirvanol.
The observation that CYP2C19 genotype and activity alone are not predictive of individual microsomal normeperidine formation is consistent with the fact that multiple enzymes are involved in meperidine N-demethylation, including CYP3A4 and CYP2B6. In addition to CYP2C19, individual variations in CYP3A and CYP2B6 activity likely play a role in the extent of normeperidine formation. Additional investigations are needed to examine the relationship between CYP2B6 polymorphism and normeperidine formation. The most common CYP2B6 variant allele CYP2B6*6 is associated with decreased CYP2B6 activity toward some substrates and increased enzyme activity for other substrates (Hofmann et al., 2008; Zanger and Klein, 2013). One previous study explored the impact of CYP2B6 genotype on clinical response to meperidine in a small group of pediatric dental patients (Hua, 2009). However, the effects of CYP2B6 genotype on the pharmacokinetics of meperidine have not been reported, and this warrants further investigation. Large inter-individual variability in CYP2B6 expression and activity has been attributed to genetic and non-genetic factors (Pearce et al., 2016; reviewed by Hedrich et al., 2016). Thus, commercial sources of human liver microsomes and enzyme variants characterized for CYP2B6 genotype and phenotype are needed for future studies to determine the impact of CYP2B6 variation on the metabolism of meperidine and other CYP2B6 substrates.
Other factors have been shown to impact overall normeperidine exposure in patients administered meperidine. Based on findings by Plotnikoff et al. (1956), meperidine N-demethylation can be a significant clearance pathway for meperidine. Plotnikoff et al. (1956) demonstrated that N-demethylated products of meperidine (i.e. normeperidine, normeperidinic acid, and normeperidinic acid conjugates) can account for approximately 30–57% of the meperidine dose. Factors, such as age, renal- and hepatic impairment are known to influence normeperidine accumulation (Pond et al., 1981). Moreover, variations in the clearance pathways for normeperidine (i.e. hydrolysis and conjugative pathways) may also contribute to individual differences in normeperidine exposure. Indeed, hydrolysis of meperidine to meperidinic acid was observed in human liver microsomal incubations. The estimated apparent Km (1.6 mM) for meperidinic acid formation in pooled human liver microsomes was similar to the Km (1.9 mM) reported for human liver carboxylesterase hCE-1 (Zhang et al., 1999). It is possible that meperidine hydrolysis could be a competing pathway for meperidine metabolism in liver microsomal incubations; however, meperidine N-demethylation appeared to be the predominate metabolic pathway in pooled human liver microsomes in the present study.
Collectively, the results of this study confirm that multiple P450s (CYP2B6, CYP2C19, and CYP3A4) contribute to meperidine N-demethylation. The relative P450 involvement in normeperidine generation depends on the enzyme activities in human liver microsomal samples, which differ across individuals. We have shown for the first time that, in addition to CYP3A, CYP2C19 is a quantitatively important contributor to liver microsomal normeperidine formation in individuals with high CYP2C19 activity. Further research is necessary to examine the impact of P450 polymorphisms and other individual factors on normeperidine generation and neurotoxicity in vivo.
Supplementary Material
Acknowledgements
The authors thank Dr. F. Peter Guengerich and his group (Vanderbilt University) and Dr. Joseph E. Deweese (Lipscomb University) for valuable scientific discussions and editing during the manuscript preparation. The authors also thank Dr. Matthew Vergne for assistance with LC-MS/MS instrumentation and analysis in the Lipscomb University College of Pharmacy Bioanalytical Core Laboratory.
Funding
This research was supported by the American Foundation for Pharmaceutical Education Gateway to Research Scholarship, by the Lipscomb University College of Pharmacy Dean’s Grant, and by the National Cancer Institute of the National Institutes of Health under grant number K01CA190711. Research reported in this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- CYP
cytochrome P450
- DMSO
dimethyl sulfoxide
- HLM
human liver microsomes
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- PPP
2-phenyl-2-(1-piperidinyl)propane
Footnotes
Disclosure of Interest
The authors report no conflict of interest.
References
- Achour B, Barber J, Rostami-Hodjegan A (2014) Expression of hepatic drug-metabolizing cytochrome P450 enzymes and their intercorrelations: A meta-analysis. Drug Metab Dispos 42:1349–1356. [DOI] [PubMed] [Google Scholar]
- Brøsen K, Skjelbo E, Rasmussen BB, Poulsen HE, and Loft S (1993) Fluvoxamine is a potent inhibitor of cytochrome P4501A2. Biochem Pharmacol 45:1211–1214. [DOI] [PubMed] [Google Scholar]
- Burns JJ, Berger BL, Lief PA, Wollack A, Papper EM, and Brodie BB (1955) Physiological disposition and fate of meperidine (Demerol) in man and a method for its estimation in plasma. J Pharmacol Exp Ther 114:289–298. [PubMed] [Google Scholar]
- Caudle KE, Dunnenberger HM, Freimuth RR, Peterson JF, Burlison JD, Whirl-Carrillo M, Scott SA, Rehm HL, Williams MS, Klein TE, Relling MV, and Hoffman JM (2017) Standardizing terms for clinical pharmacogenetic test results: consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet Med 19:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RF, Wei EM, and Anderson PO (1995) Meperidine: therapeutic use and toxicity. J Emerg Med 13:797–802. [DOI] [PubMed] [Google Scholar]
- Dagenais C, Graff CL, and Pollack GM (2004) Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem Pharmacol 67:269–276. [DOI] [PubMed] [Google Scholar]
- De Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, and Goldstein JA (1994) The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 269:15419–15422. [PubMed] [Google Scholar]
- Eisendrath S, Goldman B, Douglas J, Dimatteo L, and Van Dyke C (1987) Meperidine-induced delirium. Am J Psychiatry 144:1062–1065. [DOI] [PubMed] [Google Scholar]
- Erstad BL, Meeks ML, Chow HH, Rappaport WD, and Levinson ML (1997) Site-specific pharmacokinetics and pharmacodynamics of intramuscular meperidine in elderly postoperative patients. Ann Pharmacother 31:23–28. [DOI] [PubMed] [Google Scholar]
- Friesen KJ, Falk J, and Bugden S (2015) Voluntary warnings and the limits of good prescribing behavior: the case for de-adoption of meperidine. J Pain Res 8:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamabe W, Maeda T, Fukazawa Y, Kumamoto K, Shang LQ, Yamamoto A, Yamamoto C, Tokuyama S, and Kishioka S (2006) P-glycoprotein ATPase activating effect of opioid analgesics and their P-glycoprotein-dependent antinociception in mice. Pharmacol Biochem Behav 85:629–636. [DOI] [PubMed] [Google Scholar]
- Hassan HE, Mercer SL, Cunningham CW, Coop A, and Eddington ND (2009) Evaluation of the P-glycoprotein (Abcb1) affinity status of a series of morphine analogs: comparative study with meperidine analogs to identify opioids with minimal P-glycoprotein interactions. Int J Pharm 375:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich WD, Hassan HE, and Wang H (2016) Insights into CYP2B6-mediated drug-drug interactions. Acta Pharm Sin B 6:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman RJ, McAllister CB, Branch RA, and Wilkinson GR (1985) Effects of age on meperidine disposition. Clin Pharmacol Ther (St Louis) 37:19–24. [DOI] [PubMed] [Google Scholar]
- Hofmann MH, Blievernicht JK, Klein K, Saussele T, Schaeffeler E, Schwab M, and Zanger UM (2008) Aberrant splicing caused by single nucleotide polymorphism c.516>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J Pharmacol Exp Ther 325:284–292. [DOI] [PubMed] [Google Scholar]
- Hospira (2015) Meperidine (Demerol™) Prescribing Information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021171s024s025lbl.pdf. [Last accessed 11 February 2019]
- Hicks JK, Bishop JR, Sangkuhl K, Muller DJ, Ji Y, Leckband SG, Leeder JS, Graham RL, Chiulli DL, LLerena A, Skaar TC, Scott SA, Stingl JC, Klein TE, Caudle KE, and Gaedigk A (2015) Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther 98:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S (2009) Genetic variations of CYP2B6 enzyme and the response to meperidine sedation. (Master’s thesis) Virginia Commonwealth University School of Dentistry; Available from scholarscompass.vcu.edu/etd/1709/. [Google Scholar]
- Kaiko RF, Foley KM, Grabinski PY, Heidrich G, Rogers AG, Inturrisi CE, and Reidenberg MM (1983) Central nervous system excitatory effects of meperidine in cancer patients. Ann Neurol 13:180–185. [DOI] [PubMed] [Google Scholar]
- Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O, Ikeda T, and Kurihara A (2010) Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos 38:92–99. [DOI] [PubMed] [Google Scholar]
- Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, and Lu AY (2011) Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re-evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 36:1–16. [DOI] [PubMed] [Google Scholar]
- Kus K, Zakrzewska A, Szafarz M, Walczak M, Gonciarz A, Kij A, Suraj J, and Szymura-Oleksiak J (2015) Validation of LC/MS/MS method for assessment of the in vitro activity of selected rat cytochrome P450 isoenzymes - Application to early drug metabolism screening. Acta Pol Pharm 72:1089–1099. [Google Scholar]
- Latta KS, Ginsberg B, and Barkin RL (2002) Meperidine: a critical review. Am J Ther 9:53–68. [DOI] [PubMed] [Google Scholar]
- Moriyama B, Obeng AO, Barbarino J, Penzak SR, Henning SA, Scott SA, Agúndez J, Wingard JR, McLeod HL, Klein TE, Cross SJ, Caudle KE, and Walsh TJ (2016) Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP2C19 and voriconazole therapy. Clin Pharmacol Ther 102:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy KV, Ma JD, Best BM, and Atayee RS (2014) Factors impacting variability of the urinary normeperidine-to-meperidine metabolic ratio in patients with chronic pain. J Anal Toxicol 38:1–7. [DOI] [PubMed] [Google Scholar]
- Pearce RE, Gaedigk R, Twist GP, Dai H, Riffel AK, Leeder JS, and Gaedigk A (2016) Developmental Expression of CYP2B6: A comprehensive analysis of mRNA expression, protein content and bupropion hydroxylase activity and the impact of genetic variation. Drug Metab Dispos 44:948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikoff NP, Way EL, and Elliott HW (1956) Biotransformation products of meperidine excreted in the urine of man. J Pharmacol Exp Ther 117:414–419. [PubMed] [Google Scholar]
- Plummer JL, Gourlay GK, and Cherry DA (2001) Norpethidine toxicity. Pain Rev 8:159–170. [Google Scholar]
- Pond SM, Tong T, Benowitz NL, Jacob P, and Rigod J (1981) Presystemic metabolism of meperidine to normeperidine in normal and cirrhotic subjects. Clin Pharmacol Ther 30:183–188. [DOI] [PubMed] [Google Scholar]
- Ramírez J, Innocenti F, Schuetz EG, Flockhart DA, Relling MV, Santucci R, and Ratain MJ (2004) CYP2B6, CYP3A4, and CYP2C19 are responsible for the in vitro N-demethlyation of meperidine in human liver microsomes. Drug Metab Dispos 32:930–936. [PubMed] [Google Scholar]
- Ritchie T, Mürdter TE, Heinkele G, Pleiss J, Tatzel S, Schwab M, Eichelbaum M, and Zanger UM (2004) Potent mechanism-based inhibition of human CYP2B6 by clopidogrel and ticlopidine. J Pharmacol Exp Ther 308:189–197. [DOI] [PubMed] [Google Scholar]
- Rodrigues AD (1999) Integrated cytochrome P450 reaction phenotyping: attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem Pharmacol 57:465–480. [DOI] [PubMed] [Google Scholar]
- Rowland-Yeo K, Rostami-Hodjegan A, and Tucker GT (2004) Abundance of cytochrome P450 in human liver: meta-analysis. Br J Clin Pharmacol 57:687:688. [Google Scholar]
- Sanford JC, Guo Y, Sadee W, and Wang D (2013) Regulatory polymorphisms in CYP2C19 affecting hepatic expression. Drug Metabol Drug Interact 28:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Sangkuhl K, Gardner EE, Stein CM, Hulot JS, Johnson JA, Roden DM, Klein TE, and Shuldiner AR (2011) Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450–2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther 90:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Sangkuhl K, Shuldiner AR, Hulot J-S, Thorn CF, Altman RB, and Klein TE (2012) PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 19. Pharmacogenet Genomics 22:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79:103–113. [DOI] [PubMed] [Google Scholar]
- Stewart NA, Buch SC, Conrads TP, and Branch RA (2011) A UPLC-MS/MS assay of the “Pittsburgh cocktail”: six CYP probe-drug/metabolites from human plasma and urine using stable isotope dilution. Analyst 136:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kneller MB, Haining RL, Trager WF, and Rettie AE (2002) (+)-N-3-Benzylnirvanol and (−)-N-3-benzyl-phenobarbital: new potent and selective in vitro inhibitors of CYP2C19. Drug Metab Dispos 30:235–239. [DOI] [PubMed] [Google Scholar]
- Towles JK, Clark RN, Wahlin MD, Uttamsingh V, Rettie AE, and Jackson KD (2016) Cytochrome P450 3A4 and CYP3A5-catalyzed bioactivation of lapatinib. Drug Metab Dispos 44:1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken JH, van der Vegt MH, van Kan HJ, and Kruis MR (2005) Plasma concentrations of meperidine and normeperidine following continuous intrathecal meperidine in patients with neuropathic cancer pain. Acta Anaesthesiol Scand 49:665–670. [DOI] [PubMed] [Google Scholar]
- Walsky RL and Obach RS (2004) Validated assays for human cytochrome P450 activities. Drug Metab Dispos 32:647–660. [DOI] [PubMed] [Google Scholar]
- Walsky RL and Obach RS (2007) A Comparison of 2-Phenyl-2-(1-piperidinyl)propane (PPP), 1,1′,1″-phosphinothioylidynetrisaziridine (ThioTEPA), Clopidogrel, and ticlopidine as selective inactivators of human cytochrome P450 2B6. Drug Metab Dispos 35:2053–2059. [DOI] [PubMed] [Google Scholar]
- Wang J-J, Guo J-J, Zhan J, Bu H-Z, and Lin JH (2014) An in-vitro cocktail assay for assessing compound-mediated inhibition of six major cytochrome P450 enzymes. J Pharm Anal 4:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Inoue K, Shaw PM, Checovich WJ, Guengerich FP, and Shimada T. (1997) Different contributions of cytochrome P450 2C19 and 3A4 in the oxidation of omeprazole by human liver microsomes: Effects of contents of these two forms in individual human samples. J Pharmacol Exp Ther 283:434–442. [PubMed] [Google Scholar]
- Yeh SY, Krebs HA, and Changchit A (1981) Urinary excretion of meperidine and its metabolites. J Pharm Sci 70:867–870. [DOI] [PubMed] [Google Scholar]
- Zanger UM and Klein K (2013) Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): Advances on polymorphisms, mechanisms, and clinical relevance. Front Genet 4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger UM and Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics 138:103–141. [DOI] [PubMed] [Google Scholar]
- Zhang C, Li X, Xu Y, Wu T, Ren X, and Liu D (2013) Simultaneous determination of pethidine and norpethidine in mouse plasma by liquid chromatography-electrospray ionization source-mass spectrometry. J Anal Toxicol 37:351–356. [DOI] [PubMed] [Google Scholar]
- Zhang C, Yu Z, Li X, Xu Y, and Liu D (2014) Chronopharmacodynamics and chronopharmacokinetics of pethidine in mice. PLoS ONE 9:e102054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Burnell JC, Dumaual N, and Bosron WF (1999) Binding and hydrolysis of meperidine by human liver carboxylesterase hCE-1. J Pharmacol Exp Ther 290:314–318. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.