

Abstract
Antibody‐drug conjugates are important molecular entities in the treatment of cancer, with 8 antibody‐drug conjugates approved by the US Food and Drug Administration since 2000 and many more in early‐ and late‐stage clinical development. These conjugates combine the target specificity of monoclonal antibodies with the potent anticancer activity of small‐molecule therapeutics. The complex structure of antibody‐drug conjugates poses unique challenges to pharmacokinetic (PK) and pharmacodynamic (PD) characterization because it requires a quantitative understanding of the PK and PD properties of multiple different molecular species (eg, conjugate, total antibody, and unconjugated payload) in different tissues. Quantitative clinical pharmacology using mathematical modeling and simulation provides an excellent approach to overcome these challenges, as it can simultaneously integrate the disposition, PK, and PD of antibody‐drug conjugates and their components in a quantitative manner. In this review, we highlight diverse quantitative clinical pharmacology approaches, ranging from system models (eg, physiologically based pharmacokinetic [PBPK] modeling) to mechanistic and empirical models (eg, population PK/PD modeling for single or multiple analytes, exposure‐response modeling, platform modeling by pooling data across multiple antibody‐drug conjugates). The impact of these PBPK and PK/PD models to provide insights into clinical dosing justification and inform drug development decisions is also highlighted.
Keywords: antibody‐drug conjugates, drug discovery and development, exposure‐response, PBPK, PK/PD, population PK
Antibody‐drug conjugates are an important class of anticancer therapeutic agents that combine the antigen‐targeting specificity and favorable pharmacokinetic properties of monoclonal antibodies (mAbs) with the cytotoxic potential of small‐molecule chemotherapeutics. 1 An antibody‐drug conjugate typically consists of 3 components, namely, a mAb to determine which cells are to be targeted, a cytotoxic drug to determine the mechanism of action by which cells are killed, and a chemical linker that attaches these 2 components together to determine how the drug is released. The mAb component of the antibody‐drug conjugate specifically targets cell surface antigens overexpressed in tumor cells. Upon binding, the antibody‐drug conjugate is internalized by the tumor cell, in which it undergoes lysosomal degradation, leading to the release of the cytotoxic drug and thus cell death.
Antibody‐drug conjugates have complex molecular structures, combining the molecular characteristics of small‐molecule drugs and those of large‐molecule biotherapeutics. The conjugation of the cytotoxic drug to the mAb often results in a heterogeneous mixture of antibody‐drug conjugate species, which differ not only in the number of cytotoxic drugs attached to the antibody (ie, drug‐to‐antibody ratio species), but also in the different attachment locations on the antibody. 2 For example, T‐DM1, which is conjugated at lysine residues, and brentuximab veodtin which is conjugated via cysteines derived from reduced interchain disulfides, both have a drug‐to‐antibody ratio ranging from 0 to 8, with an average drug‐to‐antibody ratio of approximately 3.5. With recent advances in antibody engineering and conjugation chemistry, the implementation of site‐specific conjugation, in which conjugation occurs only at engineered cysteine residues or unnatural amino acids, for example, has resulted in more homogeneous antibody‐drug conjugate production. 2 , 3 Even for more homogeneous antibody‐drug conjugates, it is still expected that biotransformation (eg, antibody‐drug conjugate catabolism and deconjugation) in vivo changes the concentration and relative fractions of individual drug‐to‐antibody ratio species with time, by converting high drug‐to‐antibody ratio species to low drug‐to‐antibody ratio species, resulting in a gradual decrease in average drug‐to‐antibody ratio over time. Considering the heterogeneity and complex changes in antibody‐drug conjugate concentration and composition after antibody‐drug conjugate administration, 3 different analytes, namely, conjugate (measured as conjugated payload or conjugated antibody), total antibody (fully conjugated, partially conjugated, and unconjugated antibody), and unconjugated payload, are typically measured in preclinical and clinical studies to characterize the pharmacokinetic (PK) properties of an antibody‐drug conjugate. 4 , 5
To date, 8 antibody‐drug conjugates have received US Food and Drug Administration (FDA) approval in various oncology indications. 1 In addition, several antibody‐drug conjugates showed promising clinical activities and have received a breakthrough therapy designate, with FDA approvals expected in the next 1 or 2 years. These antibody‐drug conjugates prove that the therapeutic index of otherwise untenable cytotoxic drugs can be improved to a therapeutically beneficial level by conjugating it to an antibody. Despite the great success of antibody‐drug conjugates, it is worth noting that the therapeutic window for antibody‐drug conjugates remains relatively narrow, with the maximal tolerated dose often reached before antibody‐drug conjugates achieve the maximal efficacious dose. In addition, the toxicities associated with the antibody‐drug conjugates might dictate the number of treatment cycles that the patients can tolerate and often result in dose delay, dose reduction, or study discontinuation. 6 In an attempt to optimize the benefit/risk profile of antibody‐drug conjugates, a great amount of effort has been made to understand the absorption, distribution, metabolism, and elimination (ADME), PK, and pharmacodynamics (PD) of antibody‐drug conjugates through development of quantitative physiologically based pharmacokinetic (PBPK) and/or PK/PD models using various quantitative approaches. This review summarized diverse PBPK and PK/PD modeling approaches for antibody‐drug conjugates and highlighted their impacts through the life cycle of drug development (Figure 1). Figure 2 illustrates our overall strategy, which comprehensively uses quantitative clinical pharmacology to support antibody‐drug conjugate discovery and development by integrating preclinical and clinical PK, biomarker, safety, and efficacy data.
Figure 1.
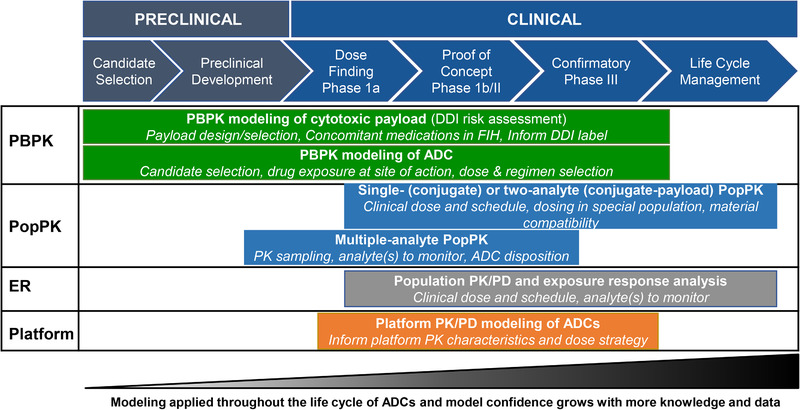
Impact of PBPK and PK/PD modeling on the preclinical and clinical development of antibody‐drug conjugates. DDI, drug‐drug interaction; ER, exposure‐response analysis; FIH, first in humans; PBPK, physiologically based pharmacokinetics; PopPK, population pharmacokinetics.
Figure 2.
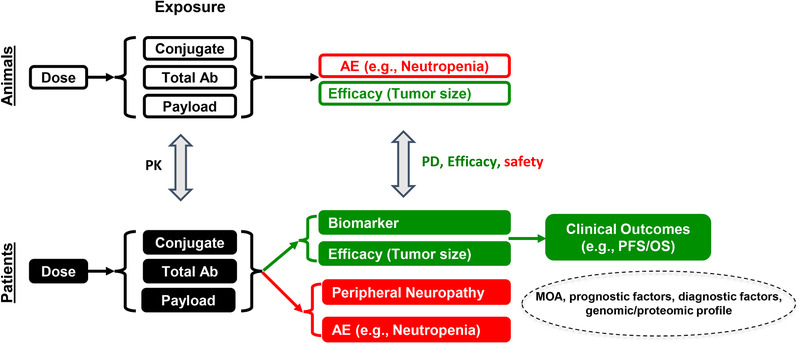
Quantitative clinical pharmacology strategy for antibody‐drug conjugate platform. AE, adverse event; MOA, mechanism of action; Total Ab, total antibody.
PBPK Modeling of Antibody‐Drug Conjugates
The PBPK model has become an important tool in drug development to understand and mechanistically characterize the exposure of the drug in different tissues. Typically, PBPK models are primarily composed of 2 types of parameters 7 : (1) system parameters based on the information related to the conserved anatomical and physiological structure of the body (eg, organ volume, blood flow, surface area, and expression level), which are usually obtained from the literature or determined a priori; (2) drug parameters that are specific for drugs under evaluation, including information related to physicochemical properties (eg, molecular weight, lipophilicity, and solubility) that are fully independent of physiology of the organism and drug‐biological properties (eg, fraction of unbound drug and tissue‐plasma partition coefficient) that are dependent on the interaction between drug and the physiology of the organism.
Antibody‐drug conjugates are designed to specifically target cancer cells by conjugating the highly potent small‐molecule chemotherapeutic agent (ie, payload) to a targeting mAb, for which efficacy is driven by the targeted delivery of a payload to the site of action in tumor, whereas toxicity is driven by unwanted tissue exposure to the cytotoxic payload. Therefore, understanding the tissue PK of an antibody‐drug conjugate and its components in different tissues becomes critical in understanding exposure‐response relationships and predicting the potential of drug‐drug interaction (DDI). Depending on the objectives of the PBPK modeling, 2 distinct PBPK models for antibody‐drug conjugates have been developed: PBPK modeling of the cytotoxic payload to inform DDI potential of the payload released from an antibody‐drug conjugate and PBPK modeling of antibody‐drug conjugates to provide enhanced mechanistic insight into the determinants of antibody‐drug conjugate and unconjugated payload disposition and to understand tissue exposure of antibody‐drug conjugates and released payloads.
PBPK Modeling of Cytotoxic Payloads
Upon formation via proteolytic degradation and/or deconjugation, the unconjugated cytotoxic payloads are expected to undergo enzyme and/or transporter‐mediated clearance mechanisms consistent with small molecules. Therefore, DDI may occur through modulation of these clearance pathways. Hence, evaluation of DDI potential associated with unconjugated cytotoxic payload is an important aspect of risk assessment to support clinical development of antibody‐drug conjugates.
Monomethyl auristatin E (MMAE) is one of the most commonly used cytotoxic payloads for auristatins antibody‐drug conjugates (∼50% of the antibody‐drug conjugates in clinical development). 8 The majority of the auristatin antibody‐drug conjugates use a dipeptide (valine‐citrulline [vc]) linker conjugated to cytotoxic payload MMAE via solvent‐accessible thiols present in mAb cysteines (vc‐MMAE antibody‐drug conjugates). 9 For most vc‐MMAE antibody‐drug conjugates, conjugate (measured as conjugated antibody or antibody conjugated MMAE [acMMAE]) and unconjugated MMAE in systemic circulation are usually monitored in the clinic, and MMAE was found to be the major circulating catabolite in humans. In vitro data suggest that MMAE is a substrate of cytochrome P450 (CYP) 3A and P‐glycoprotein, and also an inhibitor of CYP3A. A clinical DDI study showed that brentuximab vedotin, a vc‐MMAE antibody‐drug conjugate, did not affect the PK of midazolam, a sensitive CYP3A substrate. 10 Concomitant administration of rifampin (a strong CYP3A4 inducer) and ketoconazole (a strong CYP3A inhibitor) did not alter the PK of the antibody‐drug conjugate measured as conjugated antibody. However, exposure of unconjugated MMAE was reduced ∼46% by rifampin and increased ∼34% by ketoconazole coadministration. 10 As most of the vc‐MMAE antibody‐drug conjugates have similar PK characteristics for both conjugated and unconjugated MMAE, 11 , 12 it is conceivable that we could leverage the information learned from the brentuximab vedotin DDI study to inform DDI risk assessment for other vc‐MMAE antibody‐drug conjugates in clinical development.
For the first time, Chen et al proposed a novel approach using mechanistic PBPK modeling to enable the extrapolation of clinical observations with brentuximab vedotin to inform the prediction of the magnitude of DDIs for other vc‐MMAE antibody‐drug conjugates. 13 A PBPK model linking acMMAE as a parent drug to the catabolite unconjugated MMAE (cytotoxic payload) was developed using in vitro parameters and preclinical and clinical data (anti‐CD22‐vc‐MMAE antibody‐drug conjugate), a mixed bottom‐up and top‐down approach (Figure 3). Subsequently, model performance was verified by comparing predicted MMAE PK profiles with observed data from another vc‐MMAE antibody‐drug conjugate, that is, brentuximab vedotin. Finally, the validated model successfully predicted the clinically observed DDIs between brentuximab vedotin and midazolam, ketoconazole, and rifampin (Figure 3).
Figure 3.
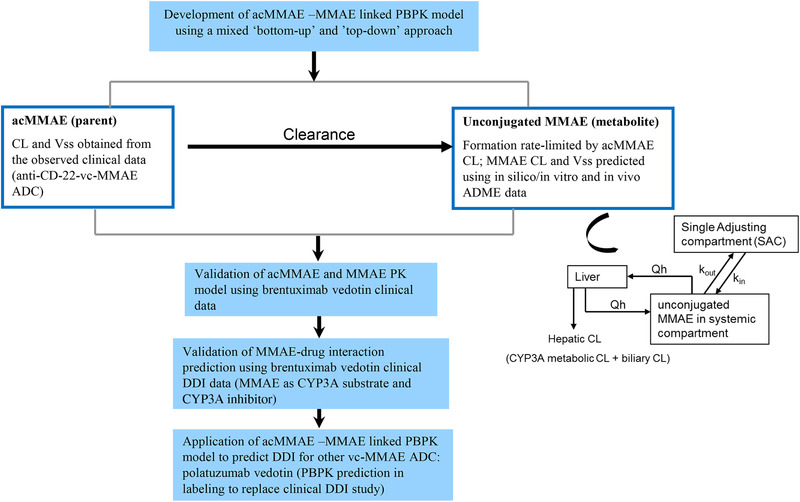
PBPK modeling approach to predict the payload‐mediated DDI risk for an antibody‐drug conjugate.
The main challenge in leveraging PBPK modeling to inform DDI risk assessment is the prediction of the PK profile of MMAE (the main driver of DDIs). This prediction relies on good mechanistic understanding of the relevant in vivo data about the formation and deposition of MMAE from an antibody‐drug conjugate in humans, which can be limited. A mixed bottom‐up and top‐down approach allows using both preclinical in vitro and in vivo and clinical data in the PBPK model development. For acMMAE, accurate prediction the of acMMAE PK profile is critical because it determines the level of MMAE formation. The model built (top down) using acMMAE clinical PK parameters (CL, Vss) in combination with PBPK distribution model best simulated the PK profile of acMMAE. The PK of MMAE treated as a metabolite of acMMAE in the model was determined simultaneously by its formation, clearance, and volume of distribution. The formation kinetic of MMAE was determined by acMMAE CL, which was calculated based on clinical PK data. The parameters of CL and Vss for MMAE were predicted based on in silico, in vitro data and preclinical in vivo information.
Prediction of MMAE clearance in humans is another critical component in the PBPK modeling of antibody‐drug conjugates. A mechanistic approach using all available data from in vitro hepatocyte incubation and preclinical and clinical mass‐balance studies was applied. By adding metabolic CL scaled using in vitro to in vivo extrapolation and nonmetabolic CL (biliary and urine, which are assumed to account for ∼50% of MMAE excretion based on mass‐balance study), the MMAE CL (∼8 L/h) was predicted, and the model was able to describe very well the PK of MMAE from dosing anti‐CD‐22‐vc‐MMAE antibody‐drug conjugate and brentuximab vedotin. In an effort to understand the disposition of cytotoxic payload (eg, MMAE) after release from an antibody‐drug conjugate in humans, preclinical PK studies with MMAE were conducted in cynomolgus monkeys. Using single‐species allometric scaling, MMAE CL, and Vss were predicted, which were 2‐ to 4‐fold higher than the predicted values using mechanistic approaches in the PBPK modeling. Using the CL and Vss values predicted from cynomolgus monkey, the simulated PK underpredicted the observed data, indicating that the disposition of MMAE in humans is different from that obtained from preclinical PK studies, especially when MMAE is administered as preformed catabolite instead of its conjugated form (antibody‐drug conjugate). Although the PBPK model was built using mechanistic approaches based on current knowledge of antibody‐drug conjugates, acMMAE was eventually catabolized to the unconjugated form of MMAE before being metabolized and/or excreted.
Nevertheless, the best utilization of existing clinical data for both model building and validation increased our confidence in using the model to evaluate the DDI risk for other vc‐MMAE antibody‐drug conjugates. Recently, this PBPK model‐based approach was applied to predict the CYP3A‐mediated DDI potential for polatuzumab vedotin, a vc‐MMAE antibody‐drug conjugate similar to brentuximab vedotin, with the ultimate goal of using PBPK simulations to support drug labeling without conducting any clinical DDI trials. Overall, the PBPK model‐based assessment showed that, polatuzumab vedotin has a limited drug interaction potential with strong CYP3A inhibitors (eg, predicted unconjugated MMAE AUC and Cmax ratios of 1.48 and 1.18, respectively, with/without ketoconazole) and strong inducers (eg, predicted unconjugated MMAE AUC and Cmax ratios of 0.49 and 0.69, respectively, with/without rifampin). The administered polatuzumab vedotin and the resulting unconjugated MMAE levels neither inhibit nor induce CYP3A. As a result, a dedicated clinical CYP3A‐based DDI study for polatuzumab vedotin is not warranted. Importantly, DDI results predicted by the PBPK model were included in polatuzumab vedotin US prescribing information (USPI). The health authority's acceptance of using PBPK modeling to replace dedicated clinical trials for antibody‐drug conjugate DDI assessment enabled accelerated drug approval without postmarket commitment. To our knowledge, this is the second biologic‐related PBPK submission to the FDA and the first PBPK application for an antibody‐drug conjugate.
Impact on Decision‐Making in Drug Development
Here we used a case study to illustrate how PBPK modeling impacts the late stage of the clinical development of an antibody‐drug conjugate. Instead of conducting a dedicated clinical DDI study, simulation results from the validated PBPK model can be used to inform the antibody‐drug conjugate prescribing information (ie, USPI or SmPC for European Medicine Agency). Given that many antibody‐drug conjugates may be against different targets and tumor indications but otherwise share the same payload and linker, it is conceivable that a PBPK model could be developed and validated for the entire platform and used to predict the magnitude of a DDI once clinical DDI data are available for one of the antibody‐drug conjugates. The PBPK model is extremely valuable, especially when the PK profiles of the unconjugated payload are different across the platform antibody‐drug conjugates because of different antibody‐drug conjugate doses and schedules and/or conjugation chemistry. For a novel payload, however, PBPK modeling might not be robust enough to inform drug labeling, given that there is no clinical DDI data to validate the model, but DDI risk assessment using the PBPK models with the novel payload could be used to support key discovery and development decision such as payload selection and concomitant medications permitted or prohibited in the clinical studies. By integrating the in silico, in vitro, and in vivo ADME and PK data and knowledge, PBPK models will inform overall risk‐based DDI strategy for an antibody‐drug conjugate, including the decision about whether a dedicated clinical DDI study is warranted.
PBPK Modeling of Antibody‐Drug Conjugates
Characterizing the ADME of an antibody‐drug conjugate is not a straightforward process, as both small‐molecule and mAb components have unique attributes to the antibody‐drug conjugate construct (eg, hydrophobicity of the small molecule, FcRn, and target binding of mAb). Furthermore, the conjugation of the payload to mAb as well as the selection of payload, linker, and site of conjugation all introduces new physicochemical properties and drug‐biological properties that need to be taken into account when characterizing the ADME of an antibody‐drug conjugate. Once released from the antibody‐drug conjugate, the small‐molecule chemotherapeutic agent may be further eliminated via enzyme‐ and/or transporter‐mediated clearance. Given the complex molecular structure and ADME properties of an antibody‐drug conjugate, PBPK modeling provides a great opportunity and platform to incorporate the in vitro, in vivo, and clinical results to understand the whole‐body disposition of an antibody‐drug conjugate and its implications for efficacy/safety.
Several efforts have been made to develop a PBPK model for an antibody‐drug conjugate. Zhao et al first developed the PBPK models to characterize plasma, tissue, and tumor PK of a dual radiolabeled SGN‐75, an anti‐CD70 maleimidocaproyl‐MMAF antibody‐drug conjugate (3H on the antibody backbone and 14C on MMAF) in tumor‐bearing mice. 14 Although the study provided the initial framework for PBPK modeling, many parameters associated with system and drug properties were estimated (eg, vascular and lymph reflection coefficients, clearance of an antibody‐drug conjugate in tissue, target densities in tumor [CD70 and tubulin], Kd between payload and tubulin) and thus limited its application. Later, Khot et al developed a PBPK model for T‐DM1, with the platform PBPK model for the mAb linked to the small‐molecule PBPK model for the payload through the proteolytic degradation and deconjugation processes of antibody‐drug conjugates. 15 The degradation and distribution of the antibody‐drug conjugate were assumed to be similar to the backbone mAb (ie, trastuzumab), and the deconjugation of DM1 from T‐DM1 over time was estimated using plasma PK of total trastuzumab and T‐DM1. The DM1 released from T‐DM1, through either proteolytic degradation or deconjugation, was then served as input of the small‐molecule PBPK model, which was established using biodistribution data of radiolabeled small‐molecule [3H]DM1 in rats. Several assumptions were made in this model: (1) antibody‐drug conjugate and backbone mAb share similar tissue distribution, FcRn binding, and intracellular degradation rate; and (2) the deconjugation rate is the same in both plasma and tissue. Overall, the model was able to describe the biodistribution of radiolabeled T‐[3H]DM1 in rats reasonably well. Beyond characterizing the plasma and tissue PK in rats, a translational effort was made to scale up the model for humans by simply replacing with the platform PBPK model for human mAbs as well as allometric scaling or using clinical reported values for parameters associated with the payload and deconjugation. Although the translated human PBPK model was able to capture the serum PK of total trastuzumab and conjugated antibody (ie, T‐DM1) reasonably well, the PK of DM1 was not captured well, indicating the potential to further improve the model.
Recently, Li et al developed a whole‐body PBPK model for vc‐MMAE antibody‐drug conjugates in rats. 16 Similar to the PBPK model for T‐DM1, the model was built on the platform PBPK model for mAb and linked to the small‐molecule PBPK model developed by Chen et al 13 through proteolytic degradation and deconjugation processes of an antibody‐drug conjugate (Figure 4). Antibody‐drug conjugate and backbone mAb were also assumed to have the same tissue distribution properties, FcRn binding, and intracellular degradation rate. Despite the similarity of overall model framework, Li et al used a variety of in vitro and in vivo data, and took a stepwise approach to characterize the model parameters: (1) rat IgG plasma and tissue profiles in rats were used to calibrate model parameters for rat IgG kinetics; (2) PK data of human IgG in rats were used to fine‐tune the parameters associated with rat FcRn binding and pinocytosis of human IgG; (3) the drug‐to‐antibody ratio‐dependent deconjugation rates in plasma were derived from in vitro rat plasma stability studies with antibody‐drug conjugates, and the deconjugation rates in tissue interstitial spaces were scaled down by taking 20% of plasma deconjugation rates given less tissue proteins (ie, albumin); and (4) drug‐to‐antibody ratio‐dependent plasma clearance of conjugated mAbs were derived using plasma profiles for the conjugated MMAE. 16 The model was validated using radiolabeled anti‐CD79b vc‐MMAE[3H] antibody‐drug conjugate and was capable of predicting both conjugated and unconjugated MMAE in both plasma and tissue in rats. 16
Figure 4.
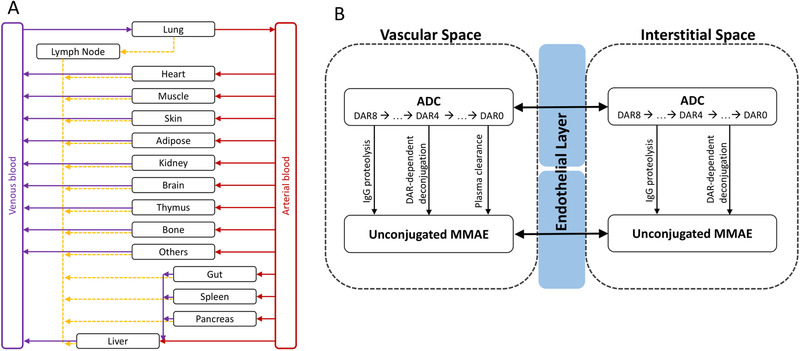
PBPK model of a vc‐MMAE antibody‐drug conjugate. (A) Structure of PBPK model at whole‐body level. Organs are represented by black rectangle and connected by blood flow (red and purple lines) and lymphatic flow (orange dashed line). (B) Structure of PBPK model at tissue level. Each tissue consists of vascular space, endothelial layer, and interstitial space. The distribution of antibody‐drug conjugate and unconjugated MMAE between vascular and interstitial space through convection, diffusion, or transcytosis is simplified and represented by the black double arrow. The formation of unconjugated MMAE from an antibody‐drug conjugate is linked by IgG proteolysis, drug‐to‐antibody ratio‐dependent deconjugation, and drug‐to‐antibody ratio‐dependent plasma clearance (vascular space only).
Both PBPK models for T‐DM1 and vc‐MMAE antibody‐drug conjugates, developed by Khot et al 15 and Li et al, 16 respectively, provided a “workflow” to derive parameter values associated with proteolytic degradation and deconjugation processes based on in vitro or in vivo data. Although this allows extrapolation of the models to other antibody‐drug conjugate molecules, it is important to bear in mind the strength and limitation of each derivation. For the proteolytic degradation process, it has been shown that higher drug‐to‐antibody ratio species exhibited faster clearance, which is closely related to the hydrophobicity of the payload and the amount of the payload conjugated to the backbone mAb. 17 In the PBPK model for T‐DM1, proteolytic degradation of T‐DM1 was assumed to be the same as backbone mAb, and there is no additional antibody‐drug conjugate‐specific or drug‐to‐antibody ratio‐dependent clearance included. Despite a lack of antibody‐drug conjugate‐specific clearance, the model was able to capture the PK profiles of T‐DM1 in both blood and tissue reasonably well. It is likely that the deconjugation parameter of the model is a composite parameter not only representing the deconjugation process but also accounting for additional clearance because of the change of hydrophobicity of the whole antibody‐drug conjugate molecule. In the PBPK model for vc‐MMAE antibody‐drug conjugates, additional drug‐to‐antibody ratio‐dependent plasma clearance in a linear relationship was introduced in the model, which allows for flexibility of the model to dissect the contribution of linker and payload on antibody‐drug conjugate clearance. For the deconjugation process, the PBPK model for T‐DM1 used the in vivo PK profile of total trastuzumab and conjugated antibody (ie, T‐DM1) to characterize the parameter. However, because of the format of the bioanalytical assay for conjugated antibody, both high and low drug‐to‐antibody ratio species were detected, and thus the deconjugation from high drug‐to‐antibody ratio species (eg, drug‐to‐antibody ratio of 4 → drug‐to‐antibody ratio of 3) cannot be captured for the parameter derivation. Therefore, the parameter only reflects the complete deconjugation of payload (ie, drug‐to‐antibody ratio of 1 → drug‐to‐antibody ratio of 0) and likely the additional clearance of antibody‐drug conjugate as mentioned above. In the PBPK model for vc‐MMAE antibody‐drug conjugates, the deconjugation process was characterized using in vitro plasma stability data with the deconjugation rate of each drug‐to‐antibody ratio species derived. Although this approach allows for distinguishing the deconjugation process from the antibody‐drug conjugate‐specific clearance in the body, the data being used were not able to discriminate loss of payload from different drug‐to‐antibody ratio species (eg, drug‐to‐antibody ratio of 8 → drug‐to‐antibody ratio of 7 vs drug‐to‐antibody ratio of 7 → drug‐to‐antibody ratio of 6). Indeed, several drug‐to‐antibody ratio‐dependent deconjugation relationships were described previously, 18 , 19 and the choice of the relationship would most likely depend on the properties of individual antibody‐drug conjugate (eg, linker and site of conjugation) and the observed data.
Impact on Decision‐Making in Drug Development
PBPK models could be a very valuable tool for antibody‐drug conjugate drug development. First, antibody‐drug conjugates are designed to maximize the exposure in tumor tissues while minimizing the exposure in normal tissues. Target expression has been shown to correlate with tumor uptake and the efficacy of an antibody‐drug conjugate in preclinical imaging studies. 20 Although characterizing the exposure to various tissues is desired, direct measurement of tissue concentration in clinical studies is not feasible, and imaging approaches such as immunoPET are often used to provide semiquantitative information of whole‐body distribution of an antibody‐drug conjugate. 21 To ensure high probability of success in clinical development of an antibody‐drug conjugate, a quantitative tool such as PBPK modeling represents a useful approach to accurately project human tissue exposure. Combined with a preclinical and clinical imaging study, the PBPK model could facilitate the selection of a dose regimen that optimizes the exposure difference between tumor and normal tissues. In addition, alternatives to conventional antibody‐drug conjugate dosing strategies, such as predose of unconjugated antibody to block undesirable uptake in nonmalignant tissues and potentially improve safety profile, can also be quantitatively evaluated. 22 Second, an antibody‐drug conjugate consists of various active moieties (eg, antibody‐drug conjugate with different drug‐to‐antibody ratios, payload, and sometimes the naked mAb) with the metabolism, elimination, and potency of each moiety differing significantly from the others. However, the bioanalytical methods commonly used only provide the quantitative information that represents a mixture of them, which sometimes limits the ability to characterize exposure‐response relationships for the key active moiety (eg, individual drug‐to‐antibody ratio species). By leveraging in vitro and in vivo data, the PBPK model provides a complementary tool to dissect and provide the full spectrum of dynamic change of each active moiety. The 3 publications discussed above illustrate the initial effort and different approaches to develop a PBPK model for an antibody‐drug conjugate. Learning from their modeling process, the drug developer of antibody‐drug conjugates will have an opportunity to collect the critical data package and develop the PBPK modeling strategy suited for individual antibody‐drug conjugate. It is also noted that the Simcyp Simulator has recently adapted the antibody‐drug conjugate module based on the model structure of vc‐MMAE antibody‐drug conjugates described above. Therefore, it is foreseeable that more applications of PBPK models will be seen in the near future.
Population PK/PD and Exposure‐Response Modeling for Antibody‐Drug Conjugates
Population PK Modeling
It is unique for an antibody‐drug conjugate that multiple analytes are quantified using various validated analytical methods after antibody‐drug conjugate dosing in both preclinical and clinical settings. 5 These analytes include conjugate, total antibody and unconjugated payload. Various modeling approaches, including a 1‐analyte model (eg, conjugate alone), 2‐analyte linked model (eg, conjugate‐unconjugated payload, total antibody‐conjugate), and multiple‐analyte modeling (eg, total antibody‐conjugate‐unconjugated payload or PK modeling including individual drug‐to‐antibody ratio species) have been performed using preclinical or clinical PK data. The strategies of PK modeling for antibody‐drug conjugates are in general dependent on the objectives and questions to be addressed at different stages of the antibody‐drug conjugate development.
Single‐analyte population PK models of antibody‐drug conjugates published up‐to‐date are all focused on the conjugate (measured as conjugated antibody or conjugated payload), 23 , 24 which is considered one of the important analytes to drive both efficacy and safety, based on the mechanism of actions (MOA) of the antibody‐drug conjugates. It was found that in general the conjugate PK is mainly driven by its antibody component instead of the payload. This single‐analyte population PK approach was often used to support regulatory interactions and filing of antibody‐drug conjugates, such as Kadcyla. 23 , 24
There are various types of 2‐analyte integrated population PK models. As summarized in Figure 5, 4 types of 2‐analyte integrated population PK models were reported in the literature, including integrated models of total antibody‐conjugated payload 25 (Figure 5A), total antibody‐conjugated antibody 26 (Figure 5B), conjugated payload‐unconjugated payload 27 (Figure 5C), and conjugated antibody‐unconjugated payload 28 (Figure 5D). The first 2 types are integrated models for the large‐molecule component‐related analytes after antibody‐drug conjugate dosing (eg, total antibody and conjugate). These models are often used to quantify the correlations among large‐molecule component‐related analytes (eg, correlation between total antibody and conjugated antibody or conjugated payload), infer the deconjugation rate, and inform the reduction of PK sampling for one of the large‐molecule component‐related analytes in late‐stage clinical trials. For example, in a total antibody‐conjugated payload integrated population PK model for 2 MMAE‐containing antibody‐drug conjugates (pinatuzumab vedotin and polatuzumab vedotin), 25 the deconjugation rate was estimated based on phase 1 intensive PK sampling of total antibody and conjugated MMAE. Based on the established model, removing the majority or all total antibody serum PK samples in phase 2, but keeping the original plasma PK sampling schedule of the conjugated payload, would not compromise the ability of the model to precisely estimate total antibody PK exposure parameters. The remaining 2 types are modeling the conjugate and unconjugated payload simultaneously. Unconjugated payload is considered a catabolite of its parent antibody‐drug conjugate and usually follows formation‐rate‐limited kinetics and may correlate with some safety events. This type of integrated model can not only characterize the conjugate PK and the impact of clinically relevant covariates but also the formation and elimination of unconjugated payload and the impact from the covariates. For polatuzumab vedotin, the conjugated payload‐unconjugated payload integrated model 27 was applied to support global regulatory filings: (1) to assess the impact of intrinsic factors (eg, body weight, sex, age, hepatic and renal impairment, ethnicity, line of therapy) and extrinsic factors (eg, drugs in combination, material manufacturing process) on PK; (2) to provide exposure metrics for subsequent exposure‐efficacy and exposure‐safety analyses for both conjugated and unconjugated payload; and (3) to justify label dose in special populations without conducting dedicated studies. 29
Figure 5.
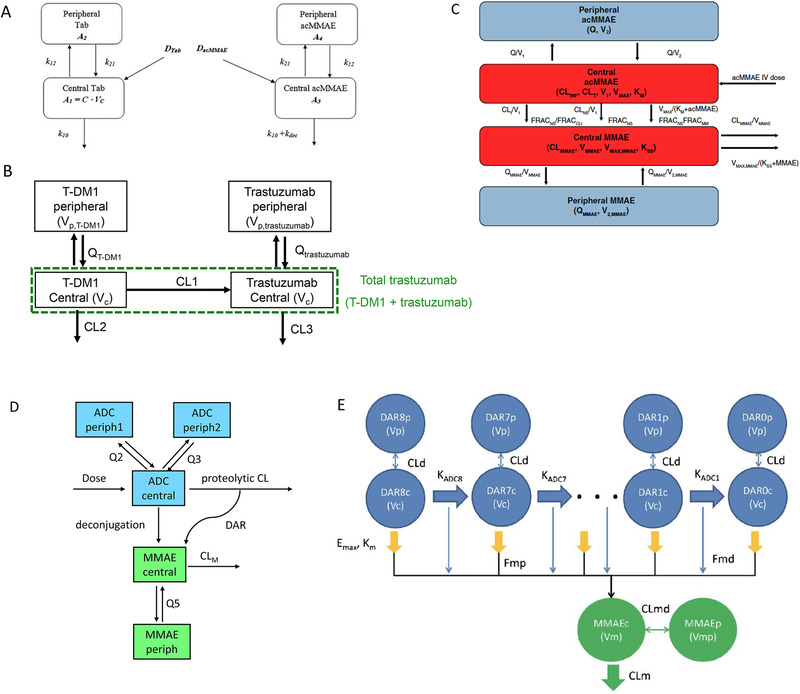
Population PK model schemes for antibody drug conjugates. (A) Example of total antibody‐acMMAE population PK model (after dosing of polatuzumab vedotin or pinatuzumab vedotin). 25 (B) Example of total antibody‐conjugated antibody population PK model after dosing of T‐DM1 (redrawn based on reference 26). (C) Example of conjugated payload‐unconjugated payload population PK model (after dosing of polatuzumab vedotin). 27 (D) Example of conjugated antibody‐unconjugated payload popPK model after dosing of brentuximab vedotin (redrawn based on reference 28). (E) Example of total antibody‐conjugated payload‐unconjugated payload popPK model (Tab‐acMMAE‐MMAE after dosing of an anti‐CD79b MMAE‐containing antibody‐drug conjugate to cynomolgus monkeys). 18
In addition, semimechanistic multiple‐analyte integrated PK models were also applied to understand the quantitative relationship of multiple analytes and to infer the mechanism and kinetic parameters related to proteolytic degradation, deconjugation, and unconjugated payload formation. These models are often built based on preclinical data, may include all 3 analytes (total antibody‐conjugate‐unconjugated payload), and may explicitly represent various drug‐to‐antibody ratio species. 18 , 19 , 30 The model structure may include drug‐to‐antibody ratio‐dependent sequential deconjugation of the drug, resulting in the conversion of higher drug‐to‐antibody ratio to lower drug‐to‐antibody ratio species and proteolytic degradation of the antibody component. Mechanistic insights were generated from these models regarding antibody‐drug conjugate disposition. For example, in a semimechanistic integrated 3‐analyte PK model for an MMAE containing antibody‐drug conjugate based on cynomolgus monkey data 18 (Figure 5E), it was found that conjugate is lost via both proteolytic degradation and deconjugation, whereas unconjugated MMAE in systemic circulation appears to be mainly released via proteolytic degradation instead of deconjugation.
Impact on Decision‐Making in Drug Development
The considerations of which population PK approach to use and which analyte(s) to model depend on the objectives of the modeling, the questions to answer by the model applications, the clinical relevance of the analytes quantified (ie, which analyte is the key driver of efficacy and safety), and the bioanalytical assay format that determines what are measured. If the population PK estimated exposures are used for exposure‐response analysis, the decision of the selection of analyte(s) for exposure‐response analysis, which are related to the clinical relevance of these analytes, is one of the key considerations to decide which analyte(s) to be included for the population PK analysis. The population PK‐derived exposure metrics of each analyte after antibody‐drug conjugate dosing are considered better for exposure‐response analysis than the observed exposure metrics that are often impacted by PK sampling schedule differences across trials and the variability of actual PK sampling time across individual patients.
Among the several types of population PK models discussed above, 1 analyte (conjugate alone) or 2‐analyte population PK models that link the parent antibody‐drug conjugate with the unconjugated payload (eg, conjugate‐unconjugated payload), which allow comprehensive covariate assessment and generate exposure metrics of key analyte(s) for exposure‐efficacy and exposure‐safety analyses, are favored in supporting the selection of phase 2/3 doses and regulatory submissions to support the dose selection in the pivotal study and label dose justification and inform the dosing strategy in a subgroup of patients based on intrinsic and extrinsic factors. At the labeling stage, the key regulatory questions may include justification of body weight‐based dosing and whether a dose capping should be applied for patients with body weight above a certain threshold, ethnic sensitivity analysis, dosing strategy in a specific population (geriatric population, patients with hepatic or renal function impairment), DDI risk assessment to inform the dosing strategy when the drug is given in combination, and the impact of material manufacturing process on PK.
For other types of multiple‐analyte PK models discussed above, such as the 2‐large‐molecule analyte (eg, total antibody‐conjugate) integrated model, and the multiple‐analyte integrated models are considered valuable in the context of answering specific questions and are often favored in the stages of preclinical development, preclinical to clinical translation, and early‐stage clinical development. By using the total antibody‐conjugate integrated model established from early‐stage clinical development data, the total antibody PK can be predicted from the conjugate, and thus PK sampling of total antibody in late‐stage clinical development can be reduced. The semimechanistic multiple‐analyte PK models provide valuable tools for the exploration of mechanisms governing disposition of antibody‐drug conjugates, the dynamic changes of high drug‐to‐antibody ratio species to low drug‐to‐antibody ratio species, and the formation of an unconjugated payload. These types of models are often built based on preclinical data; especially in some cases, the individual drug‐to‐antibody ratio species measurements were available to enable translational prediction of clinical disposition of antibody‐drug conjugates.
Exposure‐Response and PK/PD Modeling
As multiple analytes are quantified in antibody‐drug conjugate clinical studies, PK/PD and exposure‐response analysis are generally performed for the most relevant analyte(s) that are related to efficacy and safety based on the MOA of the antibody‐drug conjugates. The efficacy for antibody‐drug conjugates is driven by targeted delivery of payload by antibody to the site of action in tumor, whereas the toxicity is driven by unwanted tissue exposure to cytotoxic payload. In general, conjugate is considered the key analyte correlating with both efficacy and safety outcomes, for which exposure is often highly correlated with the antibody‐drug conjugate dose. Because there is a high correlation between conjugate exposure and total antibody exposure, 12 it is of limited value to perform exposure‐response analysis using both total antibody and conjugate. Thus, the key analyte for exposure‐response analysis is usually the conjugate. 31 , 32
A recent publication showed that for multiple vc‐MMAE antibody‐drug conjugates, efficacy (objective response rate [ORR]), and safety (grade 2+ peripheral neuropathy [PN]) end points appeared to correlate well with conjugate (acMMAE) exposure, but not with unconjugated MMAE over the doses tested in the phase 1 studies. 12 Similar exposure‐response findings were also observed for brentuximab vedotin. 33 Theoretically, the payload in the tissues can come from circulating antibody‐drug conjugate and/or payloads, but the exposure‐response results suggest that the circulating antibody‐drug conjugate appears to play a more important role in delivering payloads to the tissues compared with circulating payloads, and thus its exposure correlated better with the efficacy and safety outcomes. 12 Given the high potency of the payloads, unconjugated payload exposures may correlate with some safety end points in some cases and may be included in exposure‐safety analysis.
The efficacy end points often include ORR, progression‐free survival (PFS), and overall survival (OS), as all antibody‐drug conjugates approved up‐to‐date are for anticancer treatments. The safety end points of clinical relevance vary depending on different payloads. For example, for polatuzumab vedotin and brentuximab vedotin, the major safety end points include incidence of certain hematological adverse events (neutropenia) and PN. 29 , 33 For T‐DM1, the major safety end points include incidence of thrombocytopenia and alanine aminotransferase (AST)/alanine aminotransferase (ALT) elevation. In addition to the selection of which analyte(s), the methodologies, concepts, and interpretations of the exposure‐response results of antibody‐drug conjugates often follow a strategy similar to that of other mAbs. The approaches for the empirical exposure‐response models depend on the type of end points. For binary end points, such as the specific adverse events of special interest, grade ≥ 3 treatment‐emergent adverse events, and dose modification because of adverse events (AEs) for safety and the response rate for efficacy, logistic regression is used to assess the correlations between the exposure metrics (eg, AUC or Cmax) and the end point and the impact of covariates. For time‐to‐event end points such as OS, PFS, and time to the occurrence of delayed AEs (eg, grade ≥ 2 PN for polatuzumab vedotin), multivariate Cox proportional‐hazards model, and/or the time‐to‐event analysis could be used to assess the exposure‐response relationship. 29 , 31 , 32 , 34 It is worth noting that logistic analysis for the exposure‐safety assessment typically does not take the time component (eg, treatment duration) into account, which could be important in some cases if the AE of interest has a cumulative effect. In the case of polatuzumab vedotin, time‐to‐event PN modeling was conducted to assess the impact of treatment duration on the PN risk. 34 Although not routinely used, the longitudinal PK/PD analysis can be used to provide supporting evidence to justify dosing and schedule (eg, once every 3 weeks vs once weekly), or to evaluate the dosing regimens by the simulations with dose modification rules incorporated. 35 , 36 , 37
The key strategies and findings of exposure‐response analysis for T‐DM1 (a DM1‐containing antibody‐drug conjugate) and polatuzumab vedotin (an MMAE‐containing antibody‐drug conjugate) were summarized below as examples. For T‐DM1, the exposure‐response analysis was based on the conjugated antibody exposures. Although an apparent positive exposure‐efficacy response relationship was observed for the conjugated antibody within the exposure range of 3.6 mg/kg T‐DM1 dose, the patients at the lowest exposure quartile had numerically similar or better OS and PFS compared with the control group after adjustment of confounding covariates. 32 The analysis supported the approved T‐DM1 dose (3.6 mg/kg once every 3 weeks), which demonstrated a positive benefit/risk ratio versus control, even for the patients in the lowest exposure quartile. It is worth noting that the exposure‐efficacy relationship for antibody‐drug conjugates might be confounded (especially conducted at a single dose level) by prognostic factors similar to other mAbs, for which the conjugate exposure tends to be lower in patients with poor prognostic factors who generally also have poor efficacy because of their sicker disease status. 38 Although it is not considered a key analysis to support T‐DM1 dose, exposure‐response analysis with the unconjugated payload (DM1) exposures showed that no significant relationship was observed between these observed exposure metrics and efficacy or safety end points at the 3.6 mg/kg T‐DM1 dose. 32 Semimechanistic longitudinal PK/PD analysis of linking the conjugated antibody PK profile with the time course of PD end points (eg, platelet count decrease 35 or ALT/AST elevation 36 , 37 ) provided additional insight to support 3.6 mg/kg every 3 weeks as a well‐tolerated dose with minimal dose delays or reductions for thrombocytopenia, AST, and ALT, based on the rigorous simulations with the dose modification rules incorporated for T‐DM1.
The exposure‐response analysis for polatuzumab vedotin provided a strategy for how to leverage the exposure‐response outcome from the early studies (with the wider dose range and longer treatment duration, but different drug combinations from the pivotal study) and the observed risk‐benefit profile in the pivotal study (with 1 dose level) to justify the recommended dosing regimen for the pivotal study of 1.8 mg/kg up to 6 cycles given in combination with rituximab and bendamustine and supported the approved label dose. 39 In addition, the time‐to‐event analysis of delayed grade ≥ 2 peripheral neuropathy provided the rationale of capping the treatment duration to 6 to 8 cycles, as the capped duration is associated with an acceptable incidence of grade ≥ 2 peripheral neuropathy. 34
Impact on Decision‐Making in Drug Development
Given a relatively narrow therapeutic window of the antibody‐drug conjugates 6 compared with mAbs, exposure‐response analysis often plays a critical role for supporting phase 2/3 dose selection, label dose justification, and guidance of dose adjustment for antibody‐drug conjugates. The empirical models are routinely used, but the longitudinal PK/PD model might bring additional insight for the dose and regimen selection through mechanistic understanding and scenario simulation with adaptive dose modifications. Selecting the right analyte(s) as the driving force of responses (based on the MOA and bioanalytical strategy) for exposure‐efficacy and exposure‐safety analyses is often the most critical strategic consideration for antibody‐drug conjugates to inform its dosing strategy. The exposure‐response analysis conducted over a range of doses in the early clinical trials is critical to support the dose selection in the pivotal study, which is often conducted at a single dose. The outcomes of exposure‐response analysis are often important to infer the clinical relevance of PK exposure differences in a subgroup of patients based on intrinsic and extrinsic factors derived from the population PK analysis to support the justification of the dosing strategy in these patients.
Platform PK/PD Modeling of Antibody‐Drug Conjugates
Antibody‐drug conjugates may have different targets but otherwise share an identical construct (mAb, linker, and cytotoxic payload). The pharmacokinetics of these agents sharing the same construct is expected to be similar unless they undergo target‐mediated disposition to a significant extent.
vc‐MMAE antibody‐drug conjugates constitute the largest group of antibody‐drug conjugates that have been clinically tested. 9 To assess differences and similarities in the PK between different vc‐MMAE antibody‐drug conjugates with different targets, a platform population PK model was developed. 11 The increased sample size with a pooled data set also enabled a more robust assessment of covariate effects. The PK of all vc‐MMAE antibody‐drug conjugates was described by a 2‐compartment model with time‐dependent clearance. The results suggest that the PK was remarkably similar among the vc‐MMAE antibody‐drug conjugates, with a variability between compounds of 15% for clearance and 5% for central volume of distribution. It also showed that clearance and volume of distribution increased with body weight and that volume was higher for males. 11 This platform model can be applied to analyze and predict PK properties of a novel vc‐MMAE antibody‐drug conjugate under development.
Efficacy will be molecule specific because the vc‐MMAE antibody‐drug conjugates have different targets and are aiming to treat different cancer types. However, some safety events may be independent of target and hence shared across these antibody‐drug conjugates with the same cytotoxic payload. PN is a common nonhematological adverse event associated with a number of effective chemotherapeutic agents and is also the most frequent adverse event resulting in dose modifications and/or discontinuation of treatment for vc‐MMAE antibody‐drug conjugates. 40 To assess risk factors for developing PN and to evaluate if drug target plays a role, a pooled time‐to‐event analysis across 8 vc‐MMAE antibody‐drug conjugates (∼700 patients) was performed to evaluate the relationship between the antibody‐drug conjugate exposure and the risk for developing a clinically significant (grade ≥ 2) PN (Figure 6). 41 The analysis suggested that the risk for developing PN was largely independent of target and cancer type but increased with antibody‐drug conjugate exposure and treatment duration. In addition, large body weight and previously reported PN increased the risk for developing PN (Figure 6). Importantly, the effect of body weight did not appear to primarily be related to PK but rather to an inherent increased risk in heavy patients, likely because of larger nerve surface area in heavy patients. The derived relationship between treatment duration, dose, body weight, and prior PN, illustrated in Figure 6, can be used to optimize dosing regimen for new antibody‐drug conjugates, such as capping the treatment duration or adjusting dose based on body weight in drug development.
Figure 6.
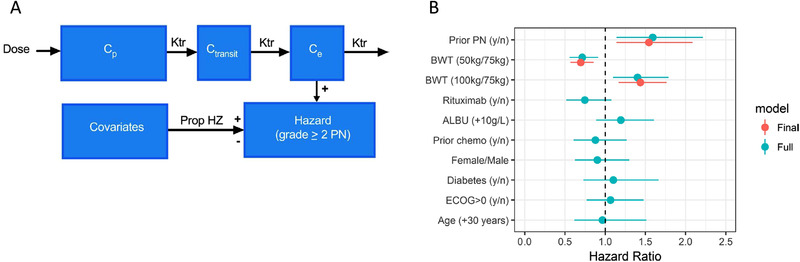
Time‐to‐event modeling of peripheral neuropathy—platform modeling. 41 (A) Model structure. The hazard in the time‐to‐event model is driven by individually predicted antibody‐drug conjugate plasma concentrations (Cp). Transit and effect compartments were included to account for the slow initial event rate. The hazard was linearly related to the concentration in the effect compartment (Ce). In addition, a Weibull function on top of the drug effect on hazard was added for slight improvement over time. Covariates in the full and final models were included assuming proportional hazards (Prop HZ). (B) Estimated hazard ratios (95%CI) for covariate effects based on full and final model. ALBU, albumin; BWT, body weight; CI, confidence interval; Ctransit, concentration in the transit compartment; ECOG, Eastern Cooperative Oncology Group; Ktr, first‐order transit rate; PN, peripheral neuropathy.
Impact on Decision‐Making in Drug Development
Platform modeling integrated data across multiple antibody‐drug conjugate molecules that shared the same linker and cytotoxic payload. This approach significantly increased sample size and enabled a more robust population PK and PK/PD evaluation. This knowledge integration is especially impactful in the early phase of clinical development, when a small sample size from an individual phase 1 study may significantly limit capability for covariate evaluation and exposure‐response assessment. The similarity in PK between vc‐MMAE antibody‐drug conjugates suggests that the developed platform PK model can be applied to predict PK properties of a novel vc‐MMAE antibody‐drug conjugate under development, estimate individual exposure for the subsequent PK/PD analysis, and project optimal dosing regimens and PK sampling times. Given that the risk for PN appears to be independent of target and cancer type, the platform model provides a means to predict the peripheral neuropathy risk at an early stage of clinical development of a new vc‐MMAE antibody‐drug conjugate. The platform modeling combined with exposure‐efficacy analysis for individual antibody‐drug conjugate can provide valuable information for the dose and regimen selection.
Conclusions
In this review, we have summarized diverse PBPK and PK/PD modeling for antibody‐drug conjugates including highlights of their impact through the drug development life cycle (Figure 1). The complex structure of antibody‐drug conjugates poses unique challenges to PK and PD characterization because it requires quantitative understanding about the PK and PD properties of multiple active molecular species in systemic circulation and/or tissues of interest (eg, tumors). Integration of diverse quantitative clinical pharmacology approaches, ranging from systems models (eg, PBPK modeling) to mechanistic and/or empirical models (eg, population PK modeling for single or multiple analytes, exposure‐response modeling) can provide insights into the PK, PD, and ADME properties of an antibody‐drug conjugate and inform drug development decisions and clinical dose selection. PBPK modeling of cytotoxic payloads can be used to predict DDI potential and ultimately inform drug labels if the model is validated with the clinical DDI data for the same payload. Conversely, full PBPK modeling of an antibody‐drug conjugate that integrates the in silico, in vitro and in vivo ADME data/knowledge could help to predict the tissue exposure to the antibody‐drug conjugate and payload and thus inform the potential safety risk for the tissues of interest. Clinical population PK/PD and exposure‐response models play a crucial role in optimizing the dose and schedule to maximize the therapeutic window of an antibody‐drug conjugate in the target population. Platform modeling by pooling the clinical data across antibody‐drug conjugates with the same payload‐linker offers unique value that maximized learning and quantitative knowledge integration across multiple molecules in the portfolio to best inform decision‐making for individual antibody‐drug conjugate molecules given clinical data were rather limited for each antibody‐drug conjugate in the early stage of clinical development. Although the present review focuses on the clinical application of PBPK and PK/PD modeling for antibody‐drug conjugates, it is worth emphasizing that PBPK and PK/PD modeling could start as early as the preclinical discovery stage. The models could be gradually improved by integrating more data and knowledge to inform decision‐making throughout development cycles of antibody‐drug conjugates. With more than 80 antibody‐drug conjugates tested in clinical development in nearly 600 clinical trials, 6 the PBPK and PK/PD learning and impact we summarized in this review could be leveraged to shed light on further optimization of M&S strategy and future development strategy for the next‐generation antibody‐drug conjugates.
Conflicts of Interest
All authors are employees of Genentech, Inc. and stockholders of the Roche group.
Funding
Third‐party assistance on reference compilation was provided by Anshin BioSolutions Corp and funded by F. Hoffmann La Roche.
[Correction added on 16 December 2020, after first online publication: the name of the author is changed from “Matts Kaagedal” to “Matts Kågedal”.]
References
- 1. Boni V, Sharma MR, Patnaik A. The resurgence of antibody drug conjugates in cancer therapeutics: novel targets and payloads. Am Soc Clin Oncol Educ Book. 2020;40:1‐17. [DOI] [PubMed] [Google Scholar]
- 2. Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site‐specific antibody drug conjugates for cancer therapy. MAbs. 2014;6(1):34‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody‐drug conjugates. Nat Rev Drug Discov. 2017;16(5):315‐337. [DOI] [PubMed] [Google Scholar]
- 4. Gorovits B, Alley SC, Bilic S, et al. Bioanalysis of antibody‐drug conjugates: American Association of Pharmaceutical Scientists Antibody‐Drug Conjugate Working Group position paper. Bioanalysis. 2013;5(9):997‐1006. [DOI] [PubMed] [Google Scholar]
- 5. Kaur S, Xu K, Saad OM, Dere RC, Carrasco‐Triguero M. Bioanalytical assay strategies for the development of antibody‐drug conjugate biotherapeutics. Bioanalysis. 2013;5(2):201‐226. [DOI] [PubMed] [Google Scholar]
- 6. Coats S, Williams M, Kebble B, et al. Antibody‐drug conjugates: future directions in clinical and translational strategies to improve the therapeutic index. Clin Cancer Res. 2019;25(18):5441‐5448. [DOI] [PubMed] [Google Scholar]
- 7. Kuepfer L, Niederalt C, Wendl T, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol. 2016;5(10):516‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sassoon I, Blanc V. Antibody‐drug conjugate (ADC) clinical pipeline: a review. Methods Mol Biol. 2013;1045:1‐27. [DOI] [PubMed] [Google Scholar]
- 9. Trail PA. Antibody drug conjugates as cancer therapeutics. Antibodies. 2013;2(1):113‐129. [Google Scholar]
- 10. Han TH, Gopal AK, Ramchandren R, et al. CYP3A‐mediated drug‐drug interaction potential and excretion of brentuximab vedotin, an antibody‐drug conjugate, in patients with CD30‐positive hematologic malignancies. J Clin Pharmacol. 2013;53(8):866‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kagedal M, Gibiansky L, Xu J, et al. Platform model describing pharmacokinetic properties of vc‐MMAE antibody‐drug conjugates. J Pharmacokinet Pharmacodyn. 2017;44(6):537‐548. [DOI] [PubMed] [Google Scholar]
- 12. Li C, Zhang C, Li Z, et al. Clinical pharmacology of vc‐MMAE antibody‐drug conjugates in cancer patients: learning from eight first‐in‐human Phase 1 studies. MAbs. 2020;12(1):1699768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y, Samineni D, Mukadam S, et al. Physiologically based pharmacokinetic modeling as a tool to predict drug interactions for antibody‐drug conjugates. Clin Pharmacokinet. 2015;54(1):81‐93. [DOI] [PubMed] [Google Scholar]
- 14. Zhao B, Zheng S, Alley SC. Physiologically‐based pharmacokinetic modeling of an anti‐CD70 auristatin antibody‐drug conjugate in tumor bearing mice. Conference of Pharmacometrics (ACoP). San Diego, California; 2011. [Google Scholar]
- 15. Khot A, Tibbitts J, Rock D, Shah DK. Development of a translational physiologically based pharmacokinetic model for antibody‐drug conjugates: a case study with T‐DM1. AAPS J. 2017;19(6):1715‐1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li L, Chen SC, Stader F, et al. Whole Body Physiologically Based Pharmacokinetic Model for Antibody Drug Conjugates —‐ Model Development and Verification in Rats. Presented at: Population Approach Group in Europe (PAGE); June 6‐9, 2017; Budapest, Hungary. [Google Scholar]
- 17. Lyon RP, Bovee TD, Doronina SO, et al. Reducing hydrophobicity of homogeneous antibody‐drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015;33(7):733‐735. [DOI] [PubMed] [Google Scholar]
- 18. Lu D, Jin JY, Girish S, et al. Semi‐mechanistic multiple‐analyte pharmacokinetic model for an antibody‐drug‐conjugate in cynomolgus monkeys. Pharm Res. 2015;32(6):1907‐1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sukumaran S, Zhang C, Leipold DD, et al. Development and translational application of an integrated, mechanistic model of antibody‐drug conjugate pharmacokinetics. AAPS J. 2017;19(1):130‐140. [DOI] [PubMed] [Google Scholar]
- 20. Williams SP, Ogasawara A, Tinianow JN, et al. ImmunoPET helps predicting the efficacy of antibody‐drug conjugates targeting TENB2 and STEAP1. Oncotarget. 2016;7(18):25103‐25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamberts LE, Menke‐van der Houven van Oordt CW, ter Weele EJ, et al. ImmunoPET with anti‐mesothelin antibody in patients with pancreatic and ovarian cancer before anti‐mesothelin antibody‐drug conjugate treatment. Clin Cancer Res. 2016;22(7):1642‐1652. [DOI] [PubMed] [Google Scholar]
- 22. Boswell CA, Yadav DB, Mundo EE, et al. Biodistribution and efficacy of an anti‐TENB2 antibody‐drug conjugate in a patient‐derived model of prostate cancer. Oncotarget. 2019;10(58):6234‐6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu D, Girish S, Gao Y, et al. Population pharmacokinetics of trastuzumab emtansine (T‐DM1), a HER2‐targeted antibody‐drug conjugate, in patients with HER2‐positive metastatic breast cancer: clinical implications of the effect of covariates. Cancer Chemother Pharmacol. 2014;74(2):399‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu D, Li C, Riggs M, et al. Pharmacokinetics of trastuzumab emtansine (T‐DM1) as a single agent or in combination with pertuzumab in HER2‐positive breast cancer patients with recurrent or locally advanced metastatic breast cancer. Cancer Chemother Pharmacol. 2019;84(1):175‐185. [DOI] [PubMed] [Google Scholar]
- 25. Lu D, Gibiansky L, Agarwal P, et al. Integrated two‐analyte population pharmacokinetic model for antibody‐drug conjugates in patients: implications for reducing pharmacokinetic sampling. CPT Pharmacometrics Syst Pharmacol. 2016;5(12):665‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu D, Joshi A, Wang B, et al. An integrated multiple‐analyte pharmacokinetic model to characterize trastuzumab emtansine (T‐DM1) clearance pathways and to evaluate reduced pharmacokinetic sampling in patients with HER2‐positive metastatic breast cancer. Clin Pharmacokinet. 2013;52(8):657‐672. [DOI] [PubMed] [Google Scholar]
- 27. Lu D, Lu T, Gibiansky L, et al. Integrated two‐analyte population pharmacokinetic model of polatuzumab vedotin in patients with non‐Hodgkin lymphoma. CPT Pharmacometrics Syst Pharmacol. 2020;9(1):48‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H, Han TH, Hunder NN, Jang G, Zhao B. Population pharmacokinetics of brentuximab vedotin in patients with CD30‐expressing hematologic malignancies. J Clin Pharmacol. 2017;57(9):1148‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. CDR. Center for Drug Evaluation and Research . Clinical Pharmacological Reviews. Original BLA (Priority Review). Polatuzumab vedotin. APPLICATION NUMBER:761121Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761121Orig1s000ClinPharmR.pdf.
- 30. Bender B, Leipold DD, Xu K, Shen BQ, Tibbitts J, Friberg LE. A mechanistic pharmacokinetic model elucidating the disposition of trastuzumab emtansine (T‐DM1), an antibody‐drug conjugate (ADC) for treatment of metastatic breast cancer. AAPS J. 2014;16(5):994‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen SC, Quartino A, Polhamus D, et al. Population pharmacokinetics and exposure‐response of trastuzumab emtansine in advanced breast cancer previously treated with ≥2 HER2‐targeted regimens. Br J Clin Pharmacol. 2017;83(12):2767‐2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li C, Wang B, Chen SC, et al. Exposure‐response analyses of trastuzumab emtansine in patients with HER2‐positive advanced breast cancer previously treated with trastuzumab and a taxane. Cancer Chemother Pharmacol. 2017;80(6):1079‐1090. [DOI] [PubMed] [Google Scholar]
- 33. Center for Drug Evaluation and Research . Clinical Pharmacology and Biopharmaceutics Review for Adcetris; Silver Spring, MD; 2011. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/125388Orig1s000ClinPharmR.pdf. Accessed December 2, 2019.
- 34. Lu D, Gillespie WR, Girish S, et al. Time‐to‐event analysis of polatuzumab vedotin‐induced peripheral neuropathy to assist in the comparison of clinical dosing regimens. CPT Pharmacometrics Syst Pharmacol. 2017;6(6):401‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bender BC, Schaedeli‐Stark F, Koch R, et al. A population pharmacokinetic/pharmacodynamic model of thrombocytopenia characterizing the effect of trastuzumab emtansine (T‐DM1) on platelet counts in patients with HER2‐positive metastatic breast cancer. Cancer Chemother Pharmacol. 2012;70(4):591‐601. [DOI] [PubMed] [Google Scholar]
- 36. Bender B. Pharmacometric models for antibody drug conjugates and taxanes in HER2+ and HER2‐breast cancer. Digital Comprehensive Summaries of Uppsala Dissertations. Sweden: Faculty of Pharmacy 217, Uppsala Universitet; 2016.
- 37. Bender BC, Quartino A, Li C, et al. An integrated pharmacokinetic‐pharmacodynamic modeling analysis of T‐DM1‐induced thrombocytopenia and hepatotoxicity in patients with HER2‐positive metastatic breast cancer. Presented at: Population Approach Group Europe (PAGE) Meeting June 7‐10, 2016; Lisbon, Portugal. [Google Scholar]
- 38. Dai H, Vugmeyster Y, Manga N. Characterizing exposure‐response relationship for therapeutic monoclonal antibodies in immuno‐oncology and beyond: challenges, perspectives and prospects [published online head of print 2020]. Clin Pharmacol Ther. 10.1002/cpt.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu T, Gibiansky L, Li X, et al. Exposure‐safety and exposure‐efficacy analyses of polatuzumab vedotin in patients with relapsed or refractory diffuse large B‐cell lymphoma [published online head of print 2020]. Leuk Lymphoma. 10.1080/10428194.2020.1795154. [DOI] [PubMed] [Google Scholar]
- 40. Stagg NJ, Shen BQ, Brunstein F, et al. Peripheral neuropathy with microtubule inhibitor containing antibody drug conjugates: challenges and perspectives in translatability from nonclinical toxicology studies to the clinic. Regul Toxicol Pharmacol. 2016;82:1‐13. [DOI] [PubMed] [Google Scholar]
- 41. Kagedal M, Samineni D, Gillespie WR, et al. Time‐to‐event modeling of peripheral neuropathy: platform analysis of eight valine‐citrulline‐monomethylauristatin E antibody‐drug conjugates. CPT Pharmacometrics Syst Pharmacol. 2019;8(8):606‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]