

Abstract
The 48‐FeIII‐containing 96‐tungsto‐16‐phosphate, [FeIII 48(OH)76(H2O)16(HP2W12O48)8]36− (Fe48), has been synthesized and structurally characterized. This polyanion comprises eight equivalent {FeIII 6P2W12} units that are linked in an end‐on fashion forming a macrocyclic assembly that contains more iron centers than any other polyoxometalate (POM) known to date. The novel Fe48 was synthesized by a simple one‐pot reaction of an {Fe22} coordination complex with the hexalacunary {P2W12} POM precursor in water. The title polyanion was characterized by single‐crystal XRD, FTIR, TGA, magnetic and electrochemical studies.
Keywords: electrochemistry, iron, macrocycle, magnetism, polyoxometalates
A very large, discrete 96‐tungsto‐16‐phosphate containing 48 FeIII centers, [FeIII 48(OH)76(H2O)16(HP2W12O48)8]36− (Fe48), has been synthesized and structurally characterized. Polyanion Fe48 comprises eight equivalent {Fe6P2W12} units which are linked end‐on to form a macrocyclic assembly containing more iron centers than any other polyoxometalate known to date (see figure).
Polyoxometalates (POMs) are discrete, anionic metal oxides of early d‐block elements in high oxidation states with edge‐ or corner‐shared MO6 octahedra (M = WVI, MoVI, VV, NbV and TaV) as building blocks. [1] Classical POMs such as the Keggin (e.g. [PW12O40]3−) or Well‐Dawson (e.g. [P2W18O62]6−) ions allow to remove one or more MO6 octahedra by controlled base hydrolysis, resulting in vacant (lacunary) derivatives, which can be considered as inorganic polydentate ligands. Such lacunary POMs can incorporate one or more transition‐metal, lanthanide, and actinide ions, resulting in a large variety of metal‐oxo clusters of various shape, size and composition, leading to a multitude of interesting physicochemical properties, which are of interest in catalysis, magnetism, biomedicine and material science.[ 2 , 3 ]
The number of 3d metal‐containing POMs is large and includes some FeIII‐containing species. [4] The multilacunary Well‐Dawson ion [H2P2W12O48]12− ({P2W12}) is a promising POM ligand for the incorporation of several metal ions, due to the six vacant sites in the polyanion. In 2005 Gouzerh's group reported that {P2W12} can incorporate 9 Fe3+ ions ([H4P2W12Fe9O56(OAc)7]6−) and the same group also made a tetrameric derivative containing 28 Fe3+ ions ([H56P8W48Fe28O248]28−. [5] In 2010 our group reported the cyclic, mixed‐valent 8‐vanadium‐substituted [Rb3⊂{VVVIV 3O7(H2O)6}2{H6P6W39O147(H2O)3}]15−. [6a] In 2011 Kögerler's group reported the 40‐manganese(III)‐containing polyanion [{(MnIII 4P2W14O60)(MnIII 3P2W15O58)2}4(P8W48O184)]144−, using the well‐known mixed‐valence ‘Mn12‐acetate’ [MnIV 4MnIII 8O12(O2CMe)16(H2O)4] as precursor. [6b] In 2014 our group reported the 4‐oxalato‐6‐titanium‐containing [Ti6(C2O4)4P4W32O124]20−. [6c] In 2016 Mizuno's group reported the 24‐manganese(III)‐containing species [{P2W12O48MnIII 4(C5H7O2)2(CH3CO2)}6]42− (using MnIII(acac)3 as precursor). [6d]
All these compounds are based on {P2W12} as POM precursor, which confirms that multilacunary POM ligands can stabilize high‐nuclearity 3d metal‐oxo clusters. However, the solution chemistry of {P2W12} is complicated as it tends to transform easily to Wells‐Dawson derivatives with fewer lacunarity sites. This is probably the main reason why the total number of known 3d transition metal derivatives containing the {P2W12} fragment is rather small.
Most iron‐containing polyoxotungstates are based on high‐spin FeIII ions (d 5, s=5/2) with five unpaired electrons and if two or more of these ions are present being linked by two bonds then antiferromagnetic exchange interactions are usually dominant. [7] On the other hand, if polynuclear iron‐oxo clusters inside POMs do have a large spin ground state (S) and uniaxial magnetic anisotropy (D) then they may behave as single molecule magnets (SMM). Mialane's group demonstrated SMM behavior for [Fe4(H2O)2(FeW9O34)2]10−, [8a] which had originally been prepared by Krebs’ group. [8b]
As part of our ongoing research on transition metal‐containing POMs, we decided to study further the interaction of the hexalacunary POM precursor {P2W12} with preformed multinuclear and water‐soluble coordination complexes, with the goal to replace the organic ligands by inorganic POM units. Following such strategy, we report herein on the synthesis of the gigantic, macrocyclic 48‐FeIII‐containing 96‐tungsto‐16‐phosphate, [Fe48(OH)76(H2O)16(HP2W12O48)8]36− (Fe48), which was isolated as the hydrated potassium salt K36[Fe48(OH)76(H2O)16(HP2W12O48)8]⋅400H2O (K‐Fe48). Reaction of the formally hexavacant 12‐tungsto‐2‐phosphate POM precursor [H2P2W12O48]12− ({P2W12}) with the 22‐iron(III)‐containing coordination complex [Fe22O14(OH)3(O2CMe)21(mda)6]⋅(ClO4)2 (mdaH2=N‐methyldiethanolamine) [9] ({Fe22}, see also Figure S1) in water (pH 5.4) resulted in the formation of Fe48, which was characterized in the solid state by single‐crystal XRD, IR, TGA, elemental analysis, as well as both solid‐ and solution‐state electrochemical studies. Single‐crystal X‐ray analysis revealed that K‐Fe48 crystallizes in the triclinic system with space group P−1. The crystallographic parameters are shown in the Supporting Information (Table S1).
Polyanion Fe48 consists of eight equivalent {Fe6P2W12} Dawson‐type subunits which are linked to each other via Fe−O−Fe/W bonds, forming a cyclic assembly with an inner dimension of ca. 24 Å × 13 Å (Figure 1, upper). The ring is not flat with the Dawson units pointing alternatingly up and down, resembling a chair conformation of cyclooctane with an idealized D2 point group symmetry, where both enantiomers are present in the crystal lattice due to the centrosymmetric space group. The inter‐Dawson linkage modes at the two caps of each {Fe6P2W12} unit are entirely different. At one end there are two equivalent Fe‐μ3O‐Fe/W bonds, whereas at the other end the linkage is accomplished by three Fe−OH−Fe bonds, resulting in the formation of a Fe3(OH)9 trigonal prism (see Figure S2). This alternating connectivity mode is preserved throughout the entire macrocycle of Fe48. The oxidation state of +3 for iron and monoprotonation of the bridging Fe−OH−Fe oxygens were confirmed by bond valence sum (BVS) calculations (see Tables S2–S3). [10] Interestingly, the Fe3(OH)9 trigonal‐prismatic motif has been observed before in Keggin dimers of the type [Fe6(OH)3(GeW9O34(OH)3)2]11−. [11]
Figure 1.
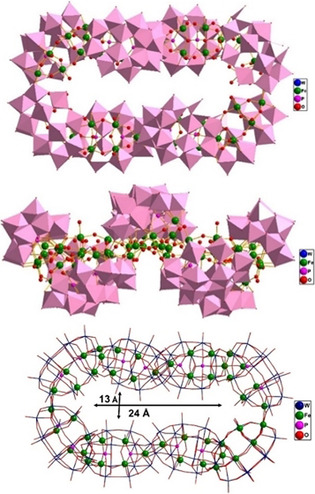
Polyhedral/ball‐and‐stick representation of Fe48, showing a top view (upper), a side‐view (middle) and a combined wireframe/ball‐and‐stick representation highlighting the 48 FeIII centers (lower). The pink octahedra represent WO6.
The Fe48 core is of interest for its magnetic properties, as this structure presents an intermediate between an infinite, 2‐dimensional, spin‐frustrated, triangular lattice and a molecular complex with a well‐defined spin ground state. Spin‐frustrated triangular lattices show a variety of phase transitions and magnetic structures.[ 2e , 12 , 13 ] Detailed magnetic susceptibility measurements over a range of 1.8–300 K and magnetization measurements in an applied field of 0–7 T were performed. Figure 1 (lower) shows the spin topology of the Fe48 core. The 48 exchange‐coupled FeIII ions (S=5/2) form a two‐dimensional ellipsoidal lattice comprising eight spin triangles and four trigonal‐prismatic lattice units with 30 slightly different exchange interactions. The magnetism of the Fe48 core unit is approximated by competing exchange interactions of localized magnetic moments in a quasi‐continuum limit. [2e] Figure 2 shows the temperature dependence of the magnetic susceptibility. At 300 K, the χmT product is 85.79 emu Kmol−1. The contribution of temperature‐independent paramagnetism (TIP) is considered and discussed below. The χmT value shows a gradual decrease to a value of 7.59 emu Kmol−1 at 1.8 K. This large decrease in χmT with decreasing temperature is indicative of the presence of antiferromagnetic exchange interactions. Because of the exchange coupling between the Fe centers, one can treat the Fe48 polyanion in the high temperature range (>50 K) as a system of exchange coupled FeIII ions with an averagely small coupling constant. It is shown in Figure 2 that the χmT above 50 K can be well fitted by the following equation [Eq. 1]:
| (1) |
Figure 2.
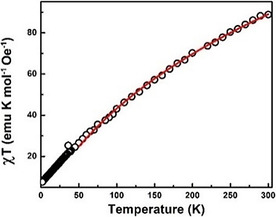
Plot of for a polycrystalline sample of K‐Fe48 measured in an applied magnetic field of 0.01 T. The solid line is a fit to Eqn. (2).
in which θ is the antiferromagnetic coupling parameter and χ TIP is the temperature independent susceptibility. Taking g=2 and S=5/2, the fitting gives a θ of −232 K and a χ TIP of 0.071 emu mol−1. As the 48 FeIII centers share a total of 60 exchange pathways (Figure 2), the averaged exchange coupling constant is estimated to be J av=−7.07 K considering [Eq. 2]:
| (2) |
in which z is the number of nearest neighbors of a given Fe center and is 60/48 on average for the Fe48 spin cluster.
The title polyanion Fe48 contains two types of redox‐active metals allowing for electron transfer, namely WVI/WV and FeIII/FeII, which renders electrochemical experiments attractive. The electrochemical diagnostic experiments described here include both (i) conventional voltammetry of K‐Fe48 dissolved in aqueous medium (see Supporting Information) and (ii) microelectrode based solid‐state‐type methodology,[ 14 , 15 , 16 ] to account for possible hydrolytic decomposition of the polyanion.
Due to a possible decomposition of Fe48 in aqueous medium we decided to also perform microelectrode‐based solid‐state electrochemistry on K‐Fe48. The diagnostic experiments were aimed at verifying the feasibility of fast and reversible electroreduction of WVI to WV, in addition to probing the redox transitions of the FeIII ions in Fe48.
Obviously, electron transfer between WVI and WV or FeIII and FeII could be operative in Fe48. It is apparent from the voltammetric characteristics shown in Figure 3 A that the reduction of the WVI ionic sites (to WV) occurs at potentials more negative than −0.2 V, whereas the FeIII/FeII redox transitions become operative at potentials higher than 0.2 V. The appearance of two peaks at potentials of ca. 0.3 and 0.7 V, respectively, may reflect the existence of two types of iron sites, possibly those bridged by oxo (Fe‐O‐W) or hydroxo (Fe−OH−Fe) ligands. It is noteworthy that, despite the scan rate being as low as 10 mV s−1, the voltammetric pattern of Figure 3 A does not show plateau currents typically observed when microelectrodes are used to study species dissolved in solution. Simply, under the present solid‐state‐type conditions, the effective diffusion (charge transport) coefficients are lower than for solution species; consequently, mixed linear/spherical, rather than typical spherical diffusional patterns are produced here. When the experiment of Figure 3 A was repeated at a faster scan rate such as 800 mV s−1, the overall charge propagation mechanism involved linear diffusion, and the voltammetric behavior of polyanion Fe48 started to develop voltammetric peaks both during reduction and oxidation (Figure 3 B). The fact that (at a scan rate as high as 800 mV s−1) the FeIII/FeII peaks are retained and both the cathodic and anodic peaks for WVI/WV redox transitions can be observed, implies fast electron transfers between tungsten ionic sites and the presence of electrochemically well‐behaved iron ionic sites, respectively.
Figure 3.
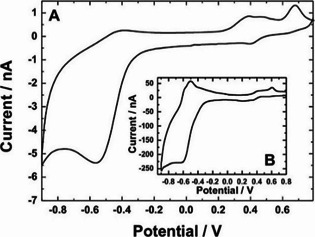
Microelectrode‐based cyclic voltammetry of Fe48 recorded at (A) 10 mVs−1 and (B) 800 mVs−1. The experiment was performed using a solid‐state cell with a planar three‐electrode configuration under nitrogen atmosphere. Microdisk diameter, 30 μm.
In summary, we have synthesized and structurally characterized the polyanion Fe48, which contains 48 FeIII centers and hence the largest number of iron centers incorporated in any POM known to date. The title polyanion Fe48 was prepared by a simple one‐pot reaction of the polynuclear coordination complex {Fe22} with the hexalacunary {P2W12} polyoxometalate (POM) precursor in water. The title compound was characterized by standard analytical techniques in the solid state and in solution. Magnetic studies indicate that the 48 FeIII centers in Fe48 share a total of 60 exchange pathways and the averaged exchange coupling constant is estimated to be J av=−7.07 K. No long‐range ferro or antiferromagnetic ordering is observed from 300 K down to 1.2 K. The title polyanion Fe48 is electrochemically well‐behaved, particularly during microelectrode‐based solid‐state‐type voltammetric experiments. The system exhibits redox transitions implying electroactivity of FeIII and WVI ionic sites. It is noteworthy that the WVI/WV redox transitions are fast and reversible even at fast scan rates (800 mV s−1). Under such conditions, the existence of two different structural types of electroactive iron sites can also be postulated.
Experimental Section
Synthesis of K36[Fe48(OH)76(H2O)16(HP2W12O48)8]⋅400 H2O (K‐Fe48)
The known coordination complex [Fe22O14(OH)3(O2CMe)21(mda)6]⋅ (ClO4)2 ({Fe22}) (mdaH2=N‐methyldiethanolamine) [9] (0.319 g, 0.076 mmol) was dissolved in H2O (40 mL) and then K12[H2P2W12O48]⋅24 H2O (0.200 g, 0.051 mmol) [17] was added. To this reaction mixture solid KCl (0.055 g 0.737 mmol) was added and the resulting turbid solution was stirred for 1 h at 70 °C and the pH was 5.4. Then the solution was allowed to stand at room temperature until the brown solid had settled at the bottom of the beaker, which was then removed by centrifugation and filtration. The solution (pH 5.4) was kept in an open vial at room temperature to allow for slow evaporation. After ca two months a yellow crystalline product started to appear. Evaporation was allowed to continue until about half the solvent had evaporated. The solid product was then collected by filtration and air dried. Yield 26 mg (11 %, based on W).
IR (2 % KBr pellet): ṽ=3415 (s), 1617 (s), 1462 (m), 1384 (m) 1086 (s), 1063 (s), 999 (w), 943 (s), 909 (s), 827 (sh), 777 (m), 705 (m), 616 (w), 556 (w), 485 (w), 418(w) cm−1. Elemental analysis (%) for K36[Fe48(OH)76(H2O)16(HP2W12O48)8]⋅400H2O (K‐Fe48), calcd: K 3.79, P 1.33, Fe 7.21, W 47.48; found K 3.48, P 1.44, Fe 7.76, W 46.84. Elemental analysis was performed at CREALINS (Villeurbanne, France).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
U.K. thanks the German Science Foundation (DFG, KO 2288/20‐1), the CMST COST Action CM1203 (PoCheMoN), and Jacobs University for research support. Figure 1 was generated using Diamond, Version 3.2 (copyright Crystal Impact GbR). I.A.R. and P.J.K. were supported by the National Science Center (NCN, Poland) under Opus Project 2018/29/B/ST5/02627. Open access funding enabled and organized by Projekt DEAL.
J. Goura, B. S. Bassil, J. K. Bindra, I. A. Rutkowska, P. J. Kulesza, N. S. Dalal, U. Kortz, Chem. Eur. J. 2020, 26, 15821.
Dedicated to Professor Imre Tóth on the occasion of his 70th birthday
Contributor Information
Dr. Joydeb Goura, http://ukortz.user.jacobs‐university.de/.
Prof. Ulrich Kortz, Email: u.kortz@jacobs-university.de.
References
- 1.
- 1a. Pope M. T., Heteropoly and Isopoly Oxometalates, Springer: Berlin, 1983; [Google Scholar]
- 1b. Izarova N. V., Pope M. T., Kortz U., Angew. Chem. Int. Ed. 2012, 51, 9492–9510; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9630–9649. [Google Scholar]
- 2.
- 2a. Mal S. S., Kortz U., Angew. Chem. Int. Ed. 2005, 44, 3777–3780; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3843–3846; [Google Scholar]
- 2b. Bassil B. S., Dickman M. H., Römer I., von der Kammer B., Kortz U., Angew. Chem. Int. Ed. 2007, 46, 6192–6195; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 6305–6308; [Google Scholar]
- 2c. Ritchie C., Ferguson A., Nojiri H., Miras H. N., Song Y.-F., Long D.-L., Burkholder E., Murrie M., Kögerler P., Brechin E. K., Cronin L., Angew. Chem. Int. Ed. 2008, 47, 5609–5612; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5691–5694; [Google Scholar]
- 2d. Ibrahim M., Lan Y., Bassil B. S., Xiang Y., Suchopar A., Powell A. K., Kortz U., Angew. Chem. Int. Ed. 2011, 50, 4708–4711; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4805–4808; [Google Scholar]
- 2e. Bassil B. S., Ibrahim M., Al-Oweini R., Asano M., Wang Z., van Tol J., Dalal N. S., Choi K.-Y., Biboum R. N., Keita B., Nadjo L., Kortz U., Angew. Chem. Int. Ed. 2011, 50, 5961–5964; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6083–6087; [Google Scholar]
- 2f. Huang L., Wang S.-S., Zhao J.-W., Cheng L., Yang G.-Y., J. Am. Chem. Soc. 2014, 136, 7637–7642; [DOI] [PubMed] [Google Scholar]
- 2g. Han X.-B., Li Y.-G., Zhang Z.-M., Tan H.-Q., Lu Y., Wang E.-B., J. Am. Chem. Soc. 2015, 137, 5486–5493; [DOI] [PubMed] [Google Scholar]
- 2h. Zhang C., Zhang M., Shi H., Zeng Q., Zhang D., Zhao Y., Wang Y., Ma P., Wang J., Niu J., Chem. Commun. 2018, 54, 5458–5461. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Pope M. T., Inorg. Chem. 1972, 11, 1973–1974; [Google Scholar]
- 3b. Sarafianos S. G., Kortz U., Pope M. T., Modak M. J., Biochem. J. 1996, 319, 619–626; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Coronado E., Day P., Chem. Rev. 2004, 104, 5419–5448; [DOI] [PubMed] [Google Scholar]
- 3d. Steitz T. A., Angew. Chem. Int. Ed. 2010, 49, 4381–4398; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4482–4500. [Google Scholar]
- 4.
- 4a. Ibrahim M., Haider A., Xiang Y., Bassil B. S., Carey A. M., Rullik L., Jameson G. B., Doungmene F., Mbomekallé I. M., de Oliveira P., Mereacre V., Kostakis G. E., Powell A. K., Kortz U., Inorg. Chem. 2015, 54, 6136–6146; [DOI] [PubMed] [Google Scholar]
- 4b. Winter R. S., Cameron J. M., Cronin L., J. Am. Chem. Soc. 2014, 136, 12753–12761; [DOI] [PubMed] [Google Scholar]
- 4c. Mal S. S., Dickman M. H., Kortz U., Todea A. M., Merca A., Bögge H., Glaser T., Müller A., Nellutla S., Kaur N., van Tol J., Dalal N. S., Keita B., Nadjo L., Chem. Eur. J. 2008, 14, 1186–1195; [DOI] [PubMed] [Google Scholar]
- 4d. Müller A., Sarkar S., Qaiser S., Shah N., Bögge H., Schmidtmann M., Sarkar S., Kögerler P., Hauptfleisch B., Trautwein A. X., Schünemann V., Angew. Chem. Int. Ed. 1999, 38, 3238–3241; [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 3435–3439. [Google Scholar]
- 5.
- 5a. Godin B., Chen Y.-G., Vaissermann J., Ruhlmann L., Verdaguer M., Gouzerh P., Angew. Chem. Int. Ed. 2005, 44, 3072–3075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3132–3135; [Google Scholar]
- 5b. Godin B., Vaissermann J., Herson P., Ruhlmann L., Verdaguer M., Gouzerh P., Chem. Commun. 2005, 5624–5626. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Assran A. S., Izarova N. V., Kortz U., CrystEngComm 2010, 12, 2684–2686; [Google Scholar]
- 6b. Fang X., Kögerler P., Furukawa Y., Speldrich M., Luban M., Angew. Chem. Int. Ed. 2011, 50, 5212–5216; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5318–5322; [Google Scholar]
- 6c. Al-Kadamany G., Bassil B. S., Raad F., Kortz U., J. Cluster Sci. J. Clust. Sci. 2014, 25, 867–878; [Google Scholar]
- 6d. Minato T., Suzuki K., Yamaguchi K., Mizuno N., Angew. Chem. Int. Ed. 2016, 55, 9630–9633; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9782–9785. [Google Scholar]
- 7.
- 7a. Christou G., Gatteschi D., Hendrickson D. N., Sessoli R., MRS Bull. 2000, 25, 66–71; [Google Scholar]
- 7b. Gatteschi D., Sessoli R., Angew. Chem. Int. Ed. 2003, 42, 268–297; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 278–309; [Google Scholar]
- 7c. Botar B., Geletii Y. V., Kögerler P., Musaev D. G., Morokuma K., Weinstock I. A., Hill C. L., J. Am. Chem. Soc. 2006, 128, 11268–11277; [DOI] [PubMed] [Google Scholar]
- 7d. Kortz U., Müller A., van Slageren J., Schnack J., Dalal N. S., Dressel M., Coord. Chem. Rev. 2009, 253, 2315–2327. [Google Scholar]
- 8.
- 8a. Compain J.-D., Mialane P., Dolbecq A., Mbomekallé I. M., Marrot J., Sécheresse F., Riviére E., Rogez G., Wernsdorfer W., Angew. Chem. Int. Ed. 2009, 48, 3077–3081; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3123–3127; [Google Scholar]
- 8b. Limanski E. M., Piepenbrink M., Droste E., Burgemeister K., Krebs B., J. Cluster Sci. 2002, 13, 369–379. [Google Scholar]
- 9. Foguet-Albiol D., Abboud K. A., Christou G., Chem. Commun. 2005, 4282–4284. [DOI] [PubMed] [Google Scholar]
- 10. Brown I. D., Altermatt D., Acta Crystallogr. Sect. B 1985, 41, 244–247. [Google Scholar]
- 11. Bi L.-H., Kortz U., Nellutla S., Stowe A. C., van Tol J., Dalal N. S., Keita B., Nadjo L., Inorg. Chem. 2005, 44, 896–903. [DOI] [PubMed] [Google Scholar]
- 12. Collins M. F., Petrenko O. A., Can. J. Phys. 1997, 75, 605. [Google Scholar]
- 13. Kawamura H., J. Phys.: Condens. Matter 1998, 10, 4707–4754. [Google Scholar]
- 14. Kulesza P. J., Malik M. A., Interfacial Electrochemistry: Theory Experiment and Applications Solid-State Voltammetry, Ed.: Wieckowski A., Marcel Dekker, New York, 1999, pp. 673. [Google Scholar]
- 15. Kulesza P. J., Cox J. A., Electroanalysis 1998, 10, 73–80. [Google Scholar]
- 16. Rutkowska I. A., Marszalek M., Orlowska J., Ozimek W., Zakeeruddin S. M., Kulesza P. J., Grätzel M., ChemSusChem 2015, 8, 2560. [DOI] [PubMed] [Google Scholar]
- 17. Contant R., Inorg. Synth. 1990, 27, 108–109. [Google Scholar]
- 18.Crystal Data for Fe48H916K36O876P16W96 (M=37172.79 g mol−1): triclinic, space group P−1, a=25.654(3) Å, b=38.137(4) Å, c=41.590(4) Å, α=113.739(3)°, β=90.052(3)°, γ=105.991(3)°, V=35516(6) Å3, Z=2, T=100 K, μ(MoKα)=16.818 mm−1, D calcd=3.476 g cm−3, 74012 reflections measured (6.79°≤2Θ≤41.63°), 51075 unique (Rint=0.172, Rsigma=0.1076) which were used in all calculations. The final R 1 was 0.1169 (I>2σ(I)) and wR 2 was 0.2851 (all data) (CSD-1895635).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary