

Abstract
Objective
Population‐based data on epilepsy syndromes and etiologies in early onset epilepsy are scarce. The use of next‐generation sequencing (NGS) has hitherto not been reported in this context. The aim of this study is to describe children with epilepsy onset before 2 years of age, and to explore to what degree whole exome and whole genome sequencing (WES/WGS) can help reveal a molecular genetic diagnosis.
Methods
Children presenting with a first unprovoked epileptic seizure before age 2 years and registered in the Stockholm Incidence Registry of Epilepsy (SIRE) between September 1, 2001 and December 31, 2006, were retrieved and their medical records up to age 7 years reviewed. Children who met the epilepsy criteria were included in the study cohort. WES/WGS was offered in cases of suspected genetic etiology regardless of whether a structural or metabolic diagnosis had been established.
Results
One hundred sixteen children were included, of which 88 had seizure onset during the first year of life and 28 during the second, corresponding to incidences of 139 and 42/100 000 person‐years, respectively. An epilepsy syndrome could be diagnosed in 54% of cases, corresponding to a birth prevalence of 1/1100. Structural etiology was revealed in 34% of cases, a genetic cause in 20%, and altogether etiology was known in 65% of children. The highest diagnostic yield was seen in magnetic resonance imaging (MRI) with 65% revealing an etiology. WES/WGS was performed in 26/116 cases (22%), with a diagnostic yield of 58%.
Significance
Epilepsy syndromes can be diagnosed and etiologies revealed in a majority of early onset cases. NGS can identify a molecular diagnosis in a substantial number of children, and should be included in the work‐up, especially in cases of epileptic encephalopathy, cerebral malformation, or metabolic disease without molecular diagnosis. A genetic diagnosis is essential to genetic counselling, prenatal diagnostics, and precision therapy.
Keywords: epilepsy syndrome, etiology, incidence, infantile epilepsy, next‐generation sequencing
Key Points.
An epilepsy syndrome can be diagnosed and etiology revealed in a majority of early onset epilepsy cases.
Whole exome/genome sequencing reveals a molecular diagnosis in cases both with and without a cerebral malformation or metabolic disease.
Next generation sequencing should be included in the work‐up of early onset epilepsy, especially in severe phenotypes.
A molecular diagnosis opens possibilities for the expanding field of precision medicine.
1. INTRODUCTION
A population‐based incidence approach is the best way to study clinical characteristics, etiology, and prognosis of a disease. 1 The comparison of available incidence studies of epilepsy is hampered by methodological differences, as case definitions and ascertainment methods have varied. 2 , 3 In addition, the number of cases is often small, especially when divided into subgroups.
Before the ongoing genetic revolution, the fraction of epilepsy cases with known etiology was unchanged for decades. Despite improved neuroimaging, the cause has been revealed in only a third of cases. 4 , 5 , 6 , 7 , 8 In epilepsy with infantile onset the proportion with known etiology is higher, 50%‐70% of cases. 9 , 10 , 11 In the last decade, new technologies like chromosomal microarrays and next‐generation sequencing (NGS) have made it possible to further explore the genetic basis for epilepsy, but hitherto mainly limited to laboratory‐ or hospital‐based cohorts. 12 , 13 , 14 , 15 , 16 , 17 , 18
Population‐based studies on epilepsy with onset early in life are scarce, and prospective incidence cohorts even more so. Incidence data on epilepsy syndromes and etiologies in this age group are uncertain or lacking. Until now, no population‐based studies have explored molecular genetic etiologies with the latest technologies. Only recently, a prospective nationwide Scottish study described epilepsy syndromes and genetic etiologies in children with onset of epileptic seizures before age 3 years, but excluded cases where an etiology, structural or other, that explained the seizures had been revealed. 19 The aim of the current study is to describe epilepsy syndromes and etiologies in children from a prospective incidence registry with a high ascertainment rate, and to investigate how NGS, in the form of whole exome and whole genome sequencing (WES/WGS), can help reveal a molecular genetic diagnosis.
2. METHODS
2.1. Study cohort
The study cohort was defined as all children living in the study area who had their first unprovoked epileptic seizure leading to medical attention before 2 years of age between September 1, 2001 and December 31, 2006, and met epilepsy criteria before age 7 years.
2.2. Case ascertainment through SIRE
From September 1, 2001, the prospective Stockholm Incidence Registry of Epilepsy (SIRE) aimed to register all cases in Northern Stockholm of a first unprovoked epileptic seizure leading to medical attention, referred to as the index seizure. The registry has been described in detail previously. 4 Pediatric cases were identified from four sources: All electroencephalography (EEG) referrals were checked; the medical records of all patients discharged from the Karolinska University Hospital Department of Pediatrics for the first time with an International Classification of Diseases (ICD) code of G40, G41, or R56.8 were reviewed; at the hospital´s neuropediatric outpatient clinic a standard form was used at every visit, with a checkbox for a newly diagnosed unprovoked epileptic seizure or epilepsy. Finally, the emergency room records of the two pediatric emergency departments in the study area were reviewed during two of the study years (2002‐2003), which added only one extra case (an older child not included in this study). Seizures during the first 4 weeks of life were considered provoked and not a basis for inclusion in the registry, except when seizures persisted after 4 weeks. Typical febrile seizures and seizures during the first week after acute brain injury/disease such as stroke, head trauma, or encephalitis were also considered provoked. 20 Febrile seizures clearly described as atypical were registered as index seizures in order to identify potential cases of Dravet syndrome and genetic epilepsy with febrile seizures plus (GEFS+). The Karolinska University Hospital has the only pediatric outpatient epilepsy unit in the area, the only inpatient child neurology ward, and the only unit for EEG interpretation. Thus all children with epilepsy in Northern Stockholm are expected to be treated at the Karolinska University Hospital.
2.3. Data collection
Children who had their index seizure during the study period and before 2 years of age were retrieved from SIRE. When all children had reached the age of 7 years or more, the hospital medical records were reviewed. In cases with incomplete medical records families were contacted by letter, followed up by a structured telephone interview, and asked for permission to obtain copies of medical records from relevant health care providers. Data on potential risk factors, work‐up, seizure types, epilepsy syndromes, and etiology were gathered from available sources up to age 7 years (Table S1). The work‐up of each case had been determined by the doctors caring for the patient. Results of neuroimaging are routinely reviewed by experienced pediatric neuroradiologists. All EEG studies were interpreted at the hospital EEG laboratory. Muscle biopsy for investigations of mitochondrial function and other metabolic work‐ups were done at the Centre for Inherited Metabolic Diseases (CMMS). Genetic tests that had been performed in‐house at the clinical genetics laboratory included chromosomal analysis, chromosomal microarrays and fluorescence in situ hybridization (FISH). Sanger sequencing of specific genes was performed mostly in‐house but in some cases at external laboratories. Genetic findings were included in the study also when acquired after 7 years of age.
2.4. Diagnosis and classification
In all cases the epilepsy diagnosis and the classification of seizure types, epilepsy syndromes, and etiology were evaluated by two of the authors (TS, PÅ) and discussed until consensus was reached.
2.4.1. Epilepsy diagnosis
Based on all information available up to age 7 years, all cases from SIRE were reassessed and classified as epilepsy, single unprovoked seizure, provoked seizures only, or nonepileptic seizures/events. As proposed by the International League Against Epilepsy (ILAE), epilepsy was defined as “an enduring predisposition to generate epileptic seizures,” 21 the epilepsy diagnosis being based on two or more unprovoked seizures, alternatively on a first seizure in cases with increased risk of further seizures due to certain etiologies or an epilepsy syndrome. In some cases, history revealed one or more unprovoked seizures before the index seizure, and the first seizure was then considered as onset seizure and onset of epilepsy. In cases of the febrile‐related epilepsies, Dravet syndrome, and GEFS+, index and onset seizures could be febrile.
2.4.2. Seizure classification
Based on documented seizure semiology and EEG findings, seizures from onset until 7 years of age were classified according to ILAEs revised classification of seizure types. 22 In unclear cases, other information up to age 7 years was considered, for example, neuroimaging findings, etiology, and epilepsy syndrome. We still found it difficult in many cases to classify, with reasonable certainty, specific seizure types. Therefore, seizure types were grouped into three broad categories based on onset: generalized, focal, and unknown. We added (infantile) spasms as a separate fourth category, because we wanted to distinguish spasms as a seizure type, but found it difficult in individual cases to decide whether they were generalized or focal.
2.4.3. Epilepsy syndrome classification
The classification of specific epilepsy syndromes was done according to ILAE recommendations. 23 , 24 Definitions were based on the syndrome descriptions in the ILAE online diagnostic manual EpilepsyDiagnosis.org. 25 Benign (self‐limited) familial infantile epilepsy (BFIE) required a first‐degree relative with a similar phenotype. GEFS+ required a confirmed genetic cause or a first‐degree relative with a similar phenotype.
2.4.4. Etiology
As proposed by the ILAE position paper on the classification of the epilepsies, 24 etiologies were grouped into six categories: structural, genetic, infectious, metabolic, immune, and unknown. Etiology was assessed based on all information available up to 7 years of age and on genetic findings at any age. If not clearly stated otherwise, genetic etiology is defined as a confirmed molecular diagnosis without a structural or metabolic disorder. Cases of BFIE and GEFS+ without a confirmed molecular diagnosis were considered presumed genetic.
2.5. Next‐generation sequencing
After review of the performed work‐up, WES or WGS was offered as part of the study in all cases without a genetic diagnosis and: (a) unknown etiology and drug‐resistant epilepsy at last follow‐up or significant neurological comorbidities like autism, intellectual disability, or cerebral palsy; (b) cerebral malformations; (c) suspected metabolic disease without diagnosis; and (d) West syndrome, even if seizure‐free and otherwise healthy. Mainly due to resource limitations, children with milder phenotypes like benign (self‐limited) infantile epilepsy (BIE), BFIE, and GEFS+, were not offered WES/WGS, but some cases already had a genetic diagnosis.
DNA from blood samples were extracted and converted to sequencing libraries using a polymerase chain reaction (PCR)–free paired‐end protocol (Illumina TruSeq DNA PCR‐free for >1000 ng input). Sequencing was done using the Hiseq X Ten or the Illumina NovaSeq 6000 platforms aiming at minimum 30× median coverage. The resulting sequences were analyzed using our in‐house Mutation Identification Pipeline as described previously. 26 All called variants are scored and ranked using the Mutation Identification Pipeline–weighted sum model, which uses multiple parameters but emphasizes Mendelian inheritance patterns and conserved, rare, and protein‐damaging variants. Data were filtered in silico for pre‐compiled clinically relevant gene lists relevant for, for example, epilepsy or inborn errors of metabolism depending on the clinical presentation of the patients. Panels are available at (https://www.karolinska.se/for‐vardgivare/karolinska‐universitetslaboratoriet/genomic‐medicine‐center‐karolinska/gmck‐rare‐diseases/). For some patients with specific symptoms or signs, for example, a cerebral malformation, customized panels were created using HPO terms. If a molecular finding was still not obtained and the suspicion of a rare genetic disease was high, the patients/families were offered a research‐setting analysis where the whole genome was considered.
2.6. Statistics
In analyzing data, descriptive statistics were applied and overall and syndrome‐ and etiology‐specific incidences were calculated. Differences between age groups were analyzed with the chi‐square test, using Yates correction when any number was below 10. 27 Population figures on December 31, 2001‐2006 were retrieved from Statistics Sweden. Northern Stockholm is an urban area with approximately one million inhabitants during the study period. Of these, 2.6% were below age 24 months and 1.3% were below 12 months. Approximately 13 000 children were born each year in the area. To calculate incidence, the number of index cases was divided by the number of person‐years at risk. The 95% confidence intervals (CI) were calculated by applying the exact method for single proportions.
2.7. Ethical approval
The study was approved by the Regional Ethical Review Board at the Karolinska Institutet, Stockholm, diary number 212/533‐31/1.
3. RESULTS
3.1. Cases and incidence
From SIRE, 163 cases registered with the index seizure before 2 years of age during the study period were retrieved. Six families were contacted by letter and telephone due to incomplete medical records after the families had moved from the study area. After reassessment of all available data up to age 7 years, 156 children were considered having had an epileptic seizure before 2 years of age, of which 116 cases (83% of all unprovoked seizure cases) met criteria for epilepsy (Figure 1), corresponding to an incidence of 89 (95% CI 72‐107)/100 000. All these children had recurrent seizures. Epilepsy criteria were considered fulfilled already after a first unprovoked seizure in two cases, based on revealed epileptogenic etiologies and epileptiform activity on the EEG studies. Incidence was higher before age 12 months, with 88 children having their onset during the first year of life (Figure 2), the incidence being 139 (95% CI 112‐171) compared to 42 (95% CI 29‐61)/100 000 for the second year of life.
FIGURE 1.
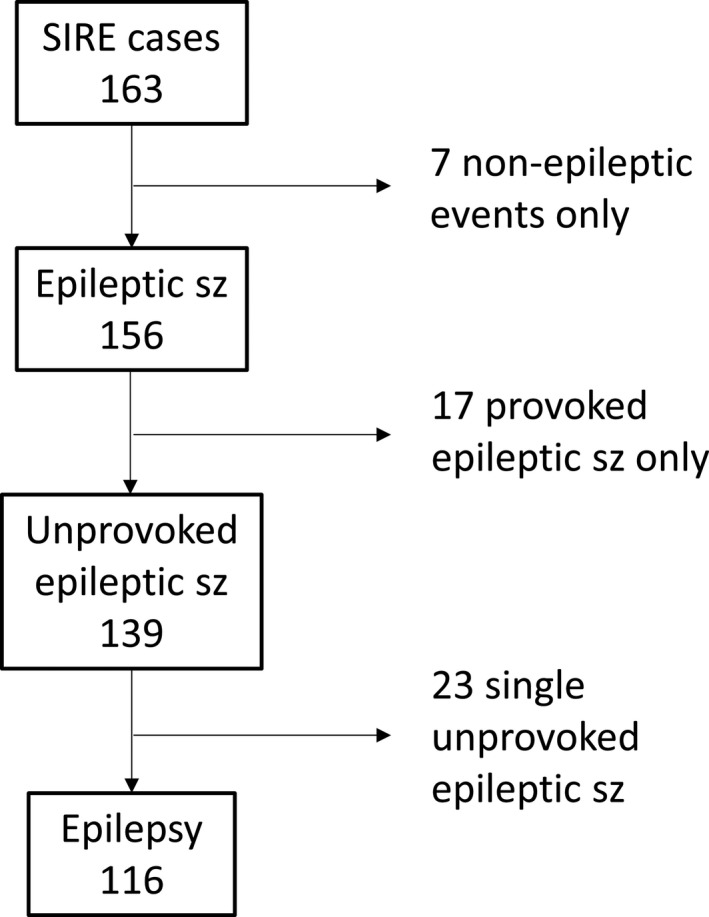
Classification of 163 cases retrieved from the Stockholm Incidence Registry of Epilepsy (SIRE), after reevaluation by two authors (TS and PÅ)
FIGURE 2.
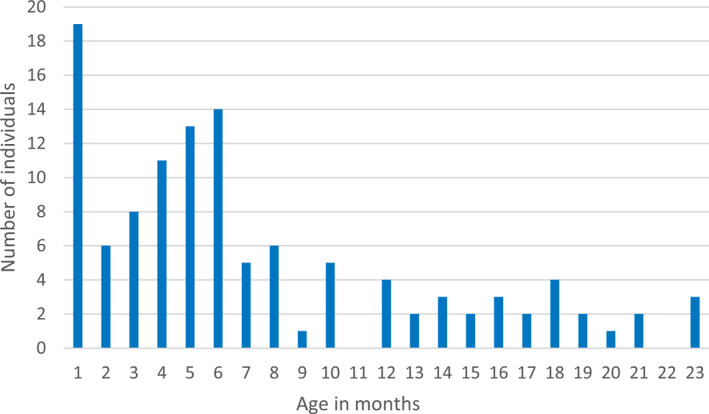
Age distribution of the first unprovoked epileptic seizure (onset seizure) in 116 cases of epilepsy
Background characteristics of the study cohort are summarized in Table 1. Male and female patients were equally represented. There were some differences in background characteristics between early (<12 months) and late (12‐23 months) onset. It was more common to have a first‐degree relative with febrile seizures in late onset (22% vs 2.4%, P = .005). In contrast, having a first‐degree relative with epilepsy tended to be more common in early onset (20% vs 3.7%, P = .08). If GEFS+ cases were excluded, significance was lost for febrile seizures (P = .92). Having had a seizure with fever before onset of afebrile seizures was more common in late compared to early onset (54% vs 9.7%, P < .001), and this remained when GEFS+ and Dravet syndrome were excluded. Developmental delay was present in 40% of children at epilepsy onset, and in 52% of West syndrome cases.
TABLE 1.
Background characteristics in 116 cases of epilepsy with onset before 2 years
|
Onset age <12 mo, n (%) |
Onset age >12 mo, n (%) |
Age difference, P‐value, significance at P < .05 |
All cases, onset age <24 mo, n (%) |
|
|---|---|---|---|---|
| All | 88/88 (100) | 28/28 (100) | 116/116 (100) | |
| Gender, female | 45/88 (51) | 12/28 (43) | Ns | 57/116 (49) |
| Epilepsy first‐degree relative | 18/88 (20) | 1/27 (3.7) | ns, P = .08 a | 19/115 (17) |
| Febrile seizures first‐degree relative | 2/83 (2.4) | 22 (5/23) | P = .005 b | 7/106 (6.6) |
| Type of birth: section | 24/86 (28) | 5/28 (18) | ns | 29/114 (25) |
| Gestation: <37 wk | 7/88 (8.0) | 2/28 (7.1) | ns | 9/116 (7.8) |
| Birth weight: <2.5 kg | 7/83 (8.4) | 2/24 (8.3) | ns | 9/107 (8.4) |
| Acute symptomatic seizures | 16/88 (18) | 4/28 (14) | ns | 20/116 (17) |
| Febrile before afebrile seizure | 8/88 (9.1) | 15/28 (54) | P < .001 c | 23/116 (20) |
| Developmental delay at onset | 34/88 (39) | 12/28 (43) | ns | 46/116 (40) |
Onset refers to the first unprovoked seizure. n, number of cases (% of all cases). Difference between age groups: chi‐square test with Yates correction. ns, non‐significant = P > .05.
P = .13 if GEFS+ cases are excluded.
P = .92 and not significant if GEFS+ excluded.
Still significant, P < .001, if GEFS+ and Dravet excluded.
Work‐up is summarized in Table S2. Structural neuroimaging was performed in 90% of cases, with computed tomography (CT; 78%) being more common than magnetic resonance imaging (MRI; 49%). Of all children, 47% had some kind of genetic investigation done including additional work‐up with NGS in 22% (7 WES, 19 WGS). Standard EEG studies, wake or sleep, were performed in all 116 children. The highest diagnostic yields were seen in MRI and WES/WGS with revealed etiology in 65% and 58%, respectively.
3.2. Seizures
In 102 cases the index seizure was the first unprovoked seizure (=onset seizure), but in 14 cases history revealed a previous first/onset seizure, on average 9 weeks earlier. The onset seizure occurred at a mean age of 7.4 months (median 6, span 1‐23), with the next unprovoked seizure occurring on average 0.5 months (median 0, range 0‐15) later. Of the onset seizures, 46% were focal, 6% generalized, 21% spasms, and 28% were unclassified. Up to 7 years of age 61% of children had focal seizures, 25% had generalized seizures, and 28% had spasms. In 59% of children only one seizure category occurred (31% focal, 0.9% generalized, 9% spasms, 18% unclassified). The rest had various combinations. In terms of seizure frequency 64% of cases had a period of at least 4 weeks with daily seizures. Status epilepticus occurred in 16 cases (14%), was the onset seizure in 7 children, and febrile in 6 of these 7 cases.
3.3. Epilepsy syndromes
Table 2 shows the diagnosed epilepsy syndromes. The approximate birth prevalences for the most common syndromes were 1/2100 for West syndrome, 1/5300 for BIE, 1/6900 for Lennox‐Gastaut syndrome (LGS), and 1/13 900 for BFIE. Of West syndrome cases, 31/33 had their onset seizure as well as their spasm onset before age 12 months, constituting 35% of first year onset epilepsy. Two children had spasms as their first seizure type at 12 and 13 months, respectively. In nine of the West syndrome cases, spasms were preceded by other seizure types, with focal being the most common. In 8/10 cases, LGS were preceded by West syndrome. Altogether 63 children (54%) developed an epilepsy syndrome before age 7 years, corresponding to a birth prevalence of 1/1100. For several of the syndromes, cases are few, which makes frequencies uncertain.
TABLE 2.
Epilepsy syndromes diagnosed before age 7 years
| Epilepsy syndrome |
Onset age <12 mo |
All cases, onset age <24 mo, n (%) |
Birth prevalence | |
|---|---|---|---|---|
| n (%) | Incidence (95% CI) | |||
| West syndrome | 31 (35) | 49 (34‐69) | 33 (28) | 1/2100 |
| Lennox‐Gastaut syndrome | 10 (11) | 16 (8‐29) | 10 (8.6) | 1/6900 a |
| EIMFS | 2 (2.3) | 2 (1.7) | ||
| Dravet | 1 (1.1) | 1 (0.9) | ||
| GEFS+ | 0 | 4 (3.4) | ||
| Myoclonic atonic epilepsy | 0 | 1 (0.9) | ||
| Benign familial infantile epilepsy | 5 (5.7) | 7.9 (3.3‐19) | 5 (4.3) | 1/13,900 |
| Benign infantile epilepsy | 13 (15) | 20 (12‐35) | 13 (11) | 1/5,300 |
| Benign neonatal epilepsy | 1 (1.1) | 1 (0.9) | ||
| Myoclonic epilepsy in infancy | 1 (1.1) | 1 (0.9) | ||
| Any epilepsy syndrome | 56 (64) | 88 (68‐115) | 63 (54) | 1/1,100 a |
| No syndrome | 32 (36) | 50 (36‐71) | 53 (46) | |
| Total | 88 (100) | 139 (112‐171) | 116 (100) | |
Onset refers to the first unprovoked seizure, not necessarily syndrome onset. CI, confidence interval; EIMFS, epilepsy of infancy with migrating focal seizures; GEFS+, genetic epilepsy with febrile seizures plus; n, number of cases (% of all cases). Incidence: cases/100 000 person‐years. Birth prevalence rounded to nearest hundred.
Potentially underestimations because Lennox‐Gastaut syndrome and some other syndromes may have seizure onset >24 mo of age.
3.4. Etiologies
Etiologies revealed are shown in Table 3. Overall 59% of cases had a known cause, which increased to 65% if familial, presumed genetic cases of BFIE and GEFS+ were included. The most common types of etiology were structural 34%, genetic 15% (20% if familial, presumed genetic cases were included), and metabolic 8.6%. Cerebral malformations, perinatal asphyxia, and stroke were the most common structural causes. The specific cerebral malformations and metabolic diseases are listed in Table S3 and S4, respectively. Monogenic etiology was verified in 28 cases (nine structural, seven metabolic, 12 without underlying condition) from 23 families (Table 4). The inheritance pattern was dominant in 11 families (de novo in nine), recessive in 8, and X‐linked in 4. The findings in four of the families were previously published. 28 , 29 , 30 , 31 The 27 pathogenic/likely pathogenic variants found were spread across 21 genes. Six children had chromosomal abnormalities. [Correction added on October 10, 2020, after first online publication: the sub‐heading '3.4 Etiologies' has been inserted for the above paragraph.]
TABLE 3.
Etiology in 116 cases of epilepsy with onset before 2 years
| Etiology |
Onset age <12 mo, n (%) |
Onset age >12 mo, n (%) |
All cases, onset age <24 mo, n (%) |
West syndrome n (%)[%] i |
|---|---|---|---|---|
| Structural a | 29 (33) | 10 (36) | 39 (34) | 14 (42)[36] |
| Cerebral malform b | 11 (13) | 5 (18) | 16 (14) | 4 (12)[25] |
| Tuberous sclerosis c | 5 (5.7) | 0 | 5 (4.3) | 3 (9.1)[60] |
| Asphyxia | 8 (9.1) | 2 (7.1) | 10 (8.6) | 4 (12)[40] |
| Stroke | 4 (4.5) | 1 (3.6) | 5 (4.3) | 2 (6.1)[40] |
| PVL/PVH preterm | 1 (1.1) | 0 | 1 (0.9) | 1 (3.0)[100] |
| Trauma | 0 | 2 (7.1) | 2 (1.7) | 0 |
| Metabolic disease d | 9 (10) | 1 (3.6) | 10 (8.6) | 3 (9.1)[30] |
| Infectious | 3 (3.4) | 0 | 3 (2.6) | 1 (3.0)[33] |
| Immune | 0 | 0 | 0 | 0 |
| Underlying condition e | 41 (47) | 11 (39) | 52 (45) | 18 (55)[35] |
| Genetic, confirmed and no underlying condition f | 14 (16) | 3 (11) | 17 (15) | 6 (18)[35] |
| Monogenic epilepsy | 9 (10) | 3 (11) | 12 (10) | 2 (6.1)[17] |
| Chromosomal syndr. | 5 (5.7) | 0 | 5 (4.3) | 4 (12)[80] |
| Known, total | 55 (62) | 14 (50) | 69 (59) | 24 (73)[35] |
| Unknown | 33 (38) | 14 (50) | 47 (41) | 9 (27)[19] |
| Total | 88 (100) | 28 (100) | 116 (100) | 33 (100)[28] |
| Genetic, confirmed with structural/metabolic | 16 (18) | 1 (3.6) | 17 (15) | 5 (15)[29] |
| Genetic, all confirmed | 30 (34) | 4 (14) | 34 (29) | 11 (33)[32] |
| Genetic, presumed g | 7 (8.0) | 5 (18) | 12 (10) | 2 (6.1)[17] |
| Structural/metabolic | 5 (5.7) | 1 (3.6) | 6 (5.2) | 2 (6.1)[33] |
| Familial syndrome | 2 (2.3) | 4 (14) | 6 (5.2) | 0 |
| Genetic, confirmed and presum., no underlying h | 16 (18) | 7 (25) | 23 (20) | 6 (18)[26] |
| Genetic, all confirmed and presumed | 32 (44) | 9 (32) | 46 (40) | 13 (39)[28] |
| Known total, including presumed genetic | 57 (65) | 18 (64) | 75 (65) | 24 (73)[32] |
[Correction added on October 10, 2020, after first online publication: the indent has been applied to "Monogenic epilepsy", "Chromosomal syndr.", "Structural/metabolic" and "Familial syndrome" since, they are subcategories.]
10/39 confirmed genetic cause.
8/16 confirmed genetic.
2/5 confirmed genetic.
7/10 confirmed genetic.
Structural + metabolic + infectious + immune.
No underlying structural or metabolic condition.
Cases of tuberous sclerosis (3), metabolic disease (3), BFIE (2) and GEFS+ (4), without revealed mutations.
Confirmed genetic without underlying structural or metabolic condition plus familial cases of BFIE and GEFS+, for comparison with other studies.
[%] = % of etiology.
TABLE 4.
Genetic diagnosis, phenotype and diagnostic method in 34 cases
| Case | Gene (Ref Seq) | Mutation | Inheritance | Epilepsy syndrome | Structural or metabolic phenotype | Age, ep onset, months | Previous negative genetic investigation | Diagnostic method |
|---|---|---|---|---|---|---|---|---|
| 1 |
AP4B1 |
c.1160_1161del; p.(Thr387Argfs*30) c.1540C > T; p.(Arg514*) |
AR, compound heterozygous | ‐ | Brain malformation | 23 | FRAX, FISH Prader‐Willi, array‐CGH, Sanger MECP2 and UBE3A | WGS trio |
| 2 | ASPA (NM_000049) | c.237‐1G > A; p.? | AR, homozygous | ‐ | Canavan disease | 5 | ‐ | Sanger |
| 3 |
ATP7A |
c.1081insT; p.(Ser361fs) | XL, hemizygous (maternally inherited) | ‐ | Menkes disease | 4 | ‐ | Sanger |
| 4a | BRAT1 (NM_152743) |
c.1771‐1G > C; p.? c.294dup; p.(Leu99Thrfs*92) |
AR compound heterozygous | ‐ | Brain malformation | 1 | Chrom. analysis, subtelom. analysis, array‐CGH, Sanger CTSD and MECP2 | WGS trio |
| 4b |
BRAT1 |
c.1771‐1G > C; p.? c.294dup; p.(Leu99Thrfs*92) |
AR compound heterozygous | ‐ | Brain malformation | 1 | ‐ | WGS trio |
| 4c | BRAT1 (NM_152743) |
c.1771‐1G > C; p.? c.294dup; p.(Leu99Thrfs*92) |
AR compound heterozygous | ‐ | Brain malformation | 1 | Chrom. analysis, subtelom. analysis | WGS trio |
| 5 | FGFR3 (NM_000142) | c.1620C > A; p.(Asn540Lys) | AD, de novo | West | Hypochondroplasia | 6 | ‐ | Sanger |
| 6 |
FLVCR2 31 |
c.1289C > T; p.(Thr430Met) | AR, homozygous | West, LGS | Brain malformation | 2 | Chrom. analysis, FISH Miller‐Dieker, array‐CGH, Sanger PAFAH1B1 | WES trio |
| 7 |
FOXRED1 |
c.874G > A; p.(Gly292Arg) | AR, homozygous | ‐ | Mitochondrial disease | 2 | Chrom. analysis, mtDNA sequenc. | WGS |
| 8 |
GABRB3 |
c.380A > G; p.(Lys127Arg) | AD, de novo | MAE | ‐ | 23 | Chrom. analysis, FRAX, array‐CGH | WES trio |
| 9 |
MID2 |
c.982C > T; p.(Arg328Trp) | XL, hemizygous (maternally inherited) | ‐ | ‐ | 5 | Chrom. analysis, FISH DiGeorge, array‐CGH | WES trio |
| 10 |
MMUT (MUT) (NM_0002553) |
c.655A > T; p.(Asn219Tyr) | AR, homozygous | West | Methylmal. aciduria | 2 | ‐ | Sanger |
| 11 |
OCRL |
c.689G > A; p.(Arg230Gln) |
XL (maternally inherited) |
‐ | Brain malformation | 7 | Chrom. analysis, subtelom. analysis, array‐CGH | WES trio |
| 12 |
POLG |
c.2243G > C; p.(Trp748Ser) c.2554C > T; p.(Arg852Cys) |
AR, compound heterozygous | ‐ | Mitochondrial disease | 6 | mtDNA sequenc. | Sanger |
| 13a |
PRRT2 29 |
c.649_650insC; p.(Arg217Profs*8) | AD (paternally inherited) | BFIE | ‐ | 4 | ‐ | Sanger |
| 13b |
PRRT2 29 |
c.649_650insC; p.(Arg217Profs*8) | AD (paternally inherited) | BFIE | ‐ | 4 | ‐ | Sanger |
| 14 |
SCN1A |
c.4003‐?_4284+?del; Deletion of exon 21 |
AD, de novo | Dravet | ‐ | 6 | Sanger SCN1A | MLPA |
| 15 |
SCN2A 28 |
c.1288G > C; p.(Glu430Gln) | AD (paternally inherited) | BFIE | ‐ | 5 | ‐ | Sanger |
| 16a |
SERAC1 |
c.1228T > C; p.(Trp410Arg) | AR, homozygous | West | Mitochondrial disease | 10 | mtDNA sequenc. Sanger POLG and SMN1 | Sanger |
| 16b |
SERAC1 |
c.1228T > C; p.(Trp410Arg) | AR, homozygous | ‐ | Mitochondrial disease | 10 | ‐ | Sanger |
| 17a |
SLC12A5 30 |
c.1277T > C; p.(Leu426Pro) c.1652G > A; p.(Gly551Asp) | AR, compound heterozygous | EIMFS | ‐ | 3 | Chrom. analysis, FISH Angelman, array‐CGH, Sanger UBE3A and SCN1A | WES trio |
| 17b |
SLC12A5 30 |
c.1277T > C; p.(Leu426Pro) c.1652G > A; p.(Gly551Asp) | AR, compound heterozygous | EIMFS | ‐ | 3 | ‐ | WES trio |
| 18 |
STXBP1 |
c.326‐2A > G; p.? | AD, de novo | ‐ | ‐ | 21 | Chrom. analysis, array‐CGH | WGS trio |
| 19 |
STXBP1 |
c.614_615del; p.(Ile205fs) | AD, de novo | West | ‐ | 1 | Chrom. analysis, FRAX, subtelomeric analys | WES trio |
| 20 |
TSC2 |
c.3779C > A; p.(Ser1254*) | AD, de novo | West | Tuberous sclerosis | 6 | ‐ | Sanger |
| 21 |
TSC2 |
c.1137 + 1G>C; p.? | AD, de novo | West | Tuberous sclerosis | 1 | ‐ | Sanger |
| 22 |
WDR45 |
c.38G > C; p.(Arg13Pro) | XL (de novo) | ‐ | ‐ | 18 | array‐CGH, mtDNA sequencing | WGS trio |
| 23 | ZEB2 (NM_014795) |
c.808‐2A > G; p.? |
AD, de novo | ‐ | Mowat‐Wilson syndrome and brain malformation | 16 | Chrom. analysis, array‐CGH, sequenc. POMGNT1, PPT1, CLN3 and CLN5 | WGS trio |
| Chromosomal abnormality | ||||||||
| 24,25,26,27 | Trisomy 21 | West in all 4 cases | Down syndrome | 3,5,5,6 | ‐ | Chromos. analysis | ||
| 28 | Del22q11.2 | ‐ | DiGeorge syndrome | 5 | ‐ | FISH | ||
| 29 | Partial tetrasomy 14q | ‐ | ‐ | 2 | ‐ | Array‐CGH | ||
AD, autosomal dominant; AR, autosomal recessive; BFIE, benign familial infantile epilepsy; EIFMS, epilepsy in infancy with migrating focal seizures; LGS, Lennox‐Gastaut syndrome; MAE, myoclonic atonic epilepsy; MLPA, multiplex ligation‐dependent probe amplification; Sanger, Sanger sequencing of the target gene only; Trio, proband + parents; WES, whole exome sequencing; WGS, whole genome sequencing; XLR, X‐linked recessive
For West syndrome, the cause was revealed in 73% of cases, structural etiology being the most common (42%) followed by genetic (18%) (Table 3). Genetic etiology increased to 33% if verified genetic cases with associated cerebral malformations, tuberous sclerosis, and metabolic disease were included and to 39% if two cases of presumed genetic cause (tuberous sclerosis and mitochondrial disease) were added. One third of children with structural, metabolic, and genetic etiology developed West syndrome, as did 4/4 Down syndrome cases and 3/5 children with tuberous sclerosis.
4. DISCUSSION
Our study confirms that the incidence of epilepsy is high during the first year of life and peaks during the first 6 months. We show that incidence quickly drops to less than a third from first to second year of life. Estimated incidences for the whole age group, and for the most common infantile epilepsy syndromes, are higher than those reported in comparable studies. 7 , 9 , 10 , 11 , 32 , 34 An infantile epilepsy syndrome could be diagnosed in two thirds of first year onset cases. Structural and genetic etiologies are common in early onset epilepsy, and the diagnostic yields for MRI and genetics were high. NGS in the form of whole exome/genome sequencing confirmed a molecular diagnosis in cases both with and without underlying structural or metabolic conditions, increasing the overall diagnostic yield substantially. Altogether 29% of all cases had a confirmed molecular genetic diagnosis, increasing to 40% if presumed genetic cases were included.
4.1. Strengths and weaknesses
The population‐based prospective case identification is a strength of our study. With the methods used we consider the ascertainment rate to be high and capture‐recapture calculations to adjust incidences for under‐ascertainment were not performed. All cases of childhood epilepsy in the study area are investigated, diagnosed, and managed at the Karolinska University Hospital. This enables not only a high ascertainment rate, but also a homogenous standard of care and easy access to all clinical information through electronic medical records. The cohort is comparatively large, but for some subgroup comparisons sample sizes are small and differences uncertain. All clinical data were extracted from medical records and not prospectively registered in a structured study protocol, which is a weakness. The lack of a fixed protocol for work‐up is to a certain degree compensated for by the structured written guidelines for work‐up of epilepsy in infants at the hospital. When we reviewed the work‐up previously performed, we did not find clinical indications to perform additional etiological investigations other than NGS. We did not perform WES/WGS in cases with seizure remission and normal neurodevelopment, thus underestimating genetic etiology. The study reflects the present clinical situation with NGS still being quite expensive, and epileptic encephalopathies and similar phenotypes being reasonable indications for NGS. Nine children who fulfilled our criteria for WES/WGS were not investigated (four had moved, one was deceased, two declined, and two were not reached).
4.2. Incidence
The incidence in infancy ranges from 82 to 124/100 000 in seven identified studies including >35 infants with epilepsy onset before age 12 months. 7 , 9 , 10 , 11 , 32 , 33 , 34 Of the seven studies only one collected cases prospectively, 9 three present seizure types at onset, 7 , 11 , 32 three report epilepsy syndromes, 9 , 10 , 11 and three describe etiology. 7 , 10 , 11 The reason that our first‐year incidence is higher than in previous studies most probably relates to a high ascertainment rate. Our definition that onset age is at the first unprovoked seizure, and not when epilepsy criteria are fulfilled, and our long follow‐up time until age 7 years for epilepsy to be diagnosed, could both potentially increase the calculated incidence compared to other studies. 9 , 10 , 33 However, this was not the case, since the time between first and second seizures were short, and 87/88 children with first‐year onset also fulfilled epilepsy criteria before age 12 months. Likewise, 112/116 children with onset during the first 2 years of life were assessed to have epilepsy before 24 months. For second‐year onset, our incidence was in line with 42‐58/100 000 in previous studies. 9 , 32 , 33
4.3. Background characteristics
There were some differences between early (<12 months) and late (12‐23 months) onset, in terms of epilepsy or febrile seizures in family members and whether the first seizure occurred in the context of fever or not, to which we found no comparable data in previous studies. The implications of these differences are not obvious.
4.4. Work‐up
MRI was performed in only half of children, which is low relative to our present guidelines recommending MRI in all small children with epilepsy. This was due to a shortage during the study period of the general anesthesia needed for MRI in small children, which led to the use of short CT scans as a suboptimal alternative. The use of MRI was lower but the yield higher at 65% (possibly due to selection bias), compared to three studies with MRI in 80%‐89% of children, and with yields 39%‐51%. 9 , 10 , 11 About half of all cases had some genetic investigation done, with a 67% diagnostic yield in early and 45% in late onset. WES/WGS was diagnostic in 15/26 tested cases, giving a higher yield than in previous research‐based or clinical studies on NGS. 12 , 13 , 14 , 15 , 16 This could be explained by differences in criteria for testing and methods. We sequenced trios (patient and both parents), which facilitates identification of pathogenic de novo variants, and in 19 cases we performed WGS, which enables detection of more variants, including intronic and structural, compared to other platforms. Today at our center, we perform WGS in a higher proportion of patients than the 22% investigated by NGS in this study. In comparable previous population‐based studies WES/WGS have not been reported. 9 , 10 , 11 A recent national study in Scotland performed a 104‐gene panel testing in 333 children with onset of epileptic seizures before age 3 years of age. 19 Children with revealed structural, metabolic, infectious, or immune etiology were excluded, and 80 cases who did not fulfill epilepsy criteria at follow‐up were included. The diagnostic yield of the gene panel in this cohort, with different inclusion criteria than our study, was 24%.
4.5. Seizures
The distribution of onset seizure types in our study was comparable to previous reports, with focal seizures predominating. 11 , 32 , 33 We found it difficult in many cases to specify seizure type with certainty.
4.6. Epilepsy syndromes
Four previous population‐based studies have reported epilepsy syndromes in the first years of life, 9 , 10 , 11 , 19 according to the latest ILAE syndrome classification. 23 , 24 Incidences vary, which can be due to small numbers/coincidence, different follow‐up times, and difficulties in distinguishing certain syndromes, for example, BIE. Our incidences are generally high, in our opinion mostly reflecting a high ascertainment rate, and show most resemblance to the Finnish study, which reported an infantile syndrome in 58% of all cases, to be compared to 64% of our first‐ year onset cases, and incidences corresponding to birth prevalences of 1/2400 for West syndrome, 1/6200 for BIE, and 1/14 300 for BFIE, 10 which are lower than our figures. The exception is the low Dravet syndrome occurrence in our cohort, for which we have no other explanation than coincidence.
4.7. Etiologies
If we add infectious etiology in our cohort (three cases) to a structural/metabolic group and define genetic as molecularly confirmed or familial (presumed genetic), without an underlying structural or metabolic condition, we can compare our results with three previous reports. 9 , 10 , 11 Results were similar but with some differences. Our results show a high proportion of overall known etiology and genetic etiology, which partly is explained by our use of next‐generation sequencing. WES/WGS raised the genetic etiology group from 14% to 20% of cases with onset before 24 months. If confirmed and presumed genetic etiologies with structural/metabolic abnormalities were included, the increase was from 27% to 40%. There were no significant differences between first‐ and second‐year onset for known, structural/metabolic, or genetic etiologies. This could be due partly to small numbers. No previous studies compared first‐ to second‐year onset, but in one report structural/metabolic etiology was significantly more common in onset months 2‐4 relative to months 5‐12 (52% vs 26%, P = .001) and had a worse prognosis. 10 Analyzing our data this way, we also found significantly more structural/metabolic etiologies for months 2‐4 (61% vs 38%, P = .04). Long‐term outcome in our cohort has not yet been analyzed and will be described in a coming report, but developmental delay at seizure onset was significantly more common in structural/metabolic etiology (65% vs 19%, P < .001).
For West syndrome, approximately three quarters of cases had a known etiology, which is higher than in comparable studies, mostly due to a higher rate of revealed genetic causes. 9 , 10 Of the 18 structural/metabolic West syndrome cases in our study, 5 also had a confirmed genetic diagnosis. In the UK infantile spasm study (UKISS), 35 although not population‐based, 61% of 207 children with infantile spasms had a revealed etiology. Several equally large, hospital‐based studies of West syndrome, report similar rates. 36 , 37 , 38 , 39
In accordance with the Scottish population‐based study 19 and several hospital‐based studies, 12 , 13 , 15 an autosomal dominant de novo inheritance pattern predominates among monogenic cases in our study. In our experience, this makes trio analysis (patient and parents) valuable in increasing both sensitivity and specificity. Our findings confirm the genetic heterogeneity of epilepsy with 27 variants in 21 genes. Only two genes, STXBP1 and TSC2, were disease‐causative in two families. Due to different inclusion criteria in terms of phenotypes and age at seizure onset, we cannot directly compare our gene findings with the Scottish study, where the three most frequent disease genes were PRRT2, SCN1A, and KCNQ2. We potentially underestimated genetic etiology by excluding cases with seizures exclusively in the neonatal period and not later (KCNQ2) and with febrile seizures only (SCN1A) and by not offering NGS in benign cases of GEFS+ (SCN1A) and BIE/BFIE (PRRT2).
Of the 15 patients who received a genetic diagnosis by NGS, 13 had gone through extensive investigations and multiple genetic tests (Table 4), which could have been avoided by earlier NGS. Five children were already deceased at the time of diagnosis. In the remaining 10 cases treatments were not significantly influenced.
5. CONCLUSIONS
Epilepsy syndromes can be diagnosed and etiologies revealed in a majority of early onset epilepsy cases, which is important for prognosis and treatment. NGS should be included in the work‐up of early onset epilepsy, especially in cases of epileptic encephalopathy, cerebral malformation, or metabolic disease without molecular diagnosis. At our hospital, whole exome and more recently whole genome sequencing of trios provide clinical diagnostics with short turn‐around time and a high diagnostic yield. The detection of pathogenic variants is based on a stepwise strategy: pre‐compiled clinically relevant gene lists that can be rapidly interrogated, gene lists created by using Human Phenotype Ontology (HPO) terms, reanalysis of sequence data with regularly updated gene lists, and finally extended analysis on a research basis in unsolved cases. In the present cohort, we discovered one novel disease gene, SLC12A5, this way. 30 A genetic diagnosis is essential to genetic counseling, prenatal diagnostics, and precision therapy. For some genetic and metabolic epilepsies, mechanism‐specific treatments are available. 40 , 41 This development is still in its beginning, but gives hope for the future to the one third of epilepsy patients, still being drug‐resistant. 42
CONFLICTS OF INTEREST
T Tomson received speaker's honoraria to his institution from Eisai, Sanofi, Sun Pharma, UCB, and Sandoz, and received research support from GSK, UCB, Eisai, and Bial. S Carlsson received grants from Novo Nordisk Foundation. The remaining authors have no conflicts of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the Clinical Genomics Stockholm facility at the Science for Life Laboratory for providing assistance in massive parallel sequencing and for the use of computer infrastructure resources.
Stödberg T, Tomson T, Barbaro M, et al. Epilepsy syndromes, etiologies, and the use of next‐generation sequencing in epilepsy presenting in the first 2 years of life: A population‐based study. Epilepsia. 2020;61:2486–2499. 10.1111/epi.16701
REFERENCES
- 1. Jager KJ, Zoccali C, Kramar R, Dekker FW. Measuring disease occurrence. Kidney Int. 2007;72:412–5. [DOI] [PubMed] [Google Scholar]
- 2. Sander JW, Hart YM, Johnson AL, Shorvon SD. National general practice study of epilepsy: newly diagnosed epileptic seizures in a general population. Lancet (London, England). 1990;336:1267–71. [DOI] [PubMed] [Google Scholar]
- 3. ILAE . Guidelines for epidemiologic studies on epilepsy. Commission on Epidemiology and Prognosis, International League Against Epilepsy. Epilepsia. 1993;34:592–6. [DOI] [PubMed] [Google Scholar]
- 4. Adelöw C, Åndell E, Åmark P, Andersson T, Hellebro E, Ahlbom A, et al. Newly diagnosed single unprovoked seizures and epilepsy in Stockholm, Sweden: first report from the Stockholm Incidence Registry of Epilepsy (SIRE). Epilepsia. 2009;50:1094–101. [DOI] [PubMed] [Google Scholar]
- 5. Granieri E, Rosati G, Tola R, Pavoni M, Paolino E, Pinna L, et al. A descriptive study of epilepsy in the district of Copparo, Italy, 1964–1978. Epilepsia. 1983;24:502–14. [DOI] [PubMed] [Google Scholar]
- 6. Gudmundsson G. Epilepsy in Iceland. A clinical and epidemiological investigation. Acta Neurol Scand. 1966;43(Suppl 25):21–124. [PubMed] [Google Scholar]
- 7. Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993;34:453–68. [DOI] [PubMed] [Google Scholar]
- 8. Thomas RH, Berkovic SF. The hidden genetics of epilepsy‐a clinically important new paradigm. Nat Rev Neurol. 2014;10:283–92. [DOI] [PubMed] [Google Scholar]
- 9. Eltze CM, Chong WK, Cox T, Whitney A, Cortina‐Borja M, Chin RFM, et al. A population‐based study of newly diagnosed epilepsy in infants. Epilepsia. 2013;54:437–45. [DOI] [PubMed] [Google Scholar]
- 10. Gaily E, Lommi M, Lapatto R, Lehesjoki AE. Incidence and outcome of epilepsy syndromes with onset in the first year of life: a retrospective population‐based study. Epilepsia. 2016;57:1594–601. [DOI] [PubMed] [Google Scholar]
- 11. Wirrell EC, Grossardt BR, Wong‐Kisiel LC, Nickels KC. Incidence and classification of new‐onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population‐based study. Epilepsy Res. 2011;95:110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18:898–905. [DOI] [PubMed] [Google Scholar]
- 13. Lindy AS, Stosser MB, Butler E, Downtain‐Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59:1062–71. [DOI] [PubMed] [Google Scholar]
- 14. Berg AT, Coryell J, Saneto RP, Grinspan ZM, Alexander JJ, Kekis M, et al. Early‐life epilepsies and the emerging role of genetic testing. JAMA Pediatr. 2017;171:863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early‐onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53:310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53:1387–98. [DOI] [PubMed] [Google Scholar]
- 17. Mefford HC, Yendle SC, Hsu C, Cook J, Geraghty E, McMahon JM, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol. 2011;70:974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Symonds JD, Zuberi SM, Stewart K, McLellan A, O‘Regan M, MacLeod S, et al Incidence and phenotypes of childhood‐onset genetic epilepsies: a prospective population‐based national cohort. Brain. 2019;142:2303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beghi E, Carpio A, Forsgren L, Hesdorffer DC, Malmgren K, Sander JW, et al. Recommendation for a definition of acute symptomatic seizure. Epilepsia. 2010;51:671–5. [DOI] [PubMed] [Google Scholar]
- 21. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475–82. [DOI] [PubMed] [Google Scholar]
- 22. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):522–30. [DOI] [PubMed] [Google Scholar]
- 23. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–85. [DOI] [PubMed] [Google Scholar]
- 24. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Epilepsy . ILAE. EpilepsyDiagnosis.org. Available at: https://www.epilepsydiagnosis.org/. Accessed April 1, 2020.
- 26. Stranneheim H, Engvall M, Naess K, Lesko N, Larsson P, Dahlberg M, et al. Rapid pulsed whole genome sequencing for comprehensive acute diagnostics of inborn errors of metabolism. BMC Genom. 2014;15:1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agresti A. Categorical data analysis, 2nd edn Hoboken, New Jersey: John Wiley and sons; 2002. [Google Scholar]
- 28. Herlenius E, Heron SE, Grinton BE, Keay D, Scheffer IE, Mulley JC, et al. SCN2A mutations and benign familial neonatal‐infantile seizures: the phenotypic spectrum. Epilepsia. 2007;48:1138–42. [DOI] [PubMed] [Google Scholar]
- 29. Heron S, Grinton B, Kivity S, Afawi Z, Zuberi S, Hughes J, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90:152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stödberg T, McTague A, Ruiz AJ, Hirata H, Zhen J, Long P, et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat Commun. 2015;6:8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kvarnung M, Taylan F, Nilsson D, Albåge M, Nordenskjöld M, Anderlid BM, et al. Mutations in FLVCR2 associated with Fowler syndrome and survival beyond infancy. Clin Genet. 2016;89:99–103. [DOI] [PubMed] [Google Scholar]
- 32. Camfield CS, Camfield PR, Gordon K, Wirrell E, Dooley JM. Incidence of epilepsy in childhood and adolescence: a population‐based study in Nova Scotia from 1977 to 1985. Epilepsia. 1996;37:19–23. [DOI] [PubMed] [Google Scholar]
- 33. Rantala H, Ingalsuo H. Occurrence and outcome of epilepsy in children younger than 2 years. J Pediatr. 1999;135:761–4. [DOI] [PubMed] [Google Scholar]
- 34. Saarinen MM, Sillanpaa M, Schmidt D, Virta LJ. Long‐term changes in the incidence of childhood epilepsy. A population study from Finland. Epilepsy Behav. 2016;58:81–5. [DOI] [PubMed] [Google Scholar]
- 35. Osborne JP, Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia. 2010;51:2168–74. [DOI] [PubMed] [Google Scholar]
- 36. Djuric M, Kravljanac R, Tadic B, Mrljes‐Popovic N, Appleton RE. Long‐term outcome in children with infantile spasms treated with vigabatrin: a cohort of 180 patients. Epilepsia. 2014;55:1918–25. [DOI] [PubMed] [Google Scholar]
- 37. Knupp KG, Coryell J, Nickels KC, Ryan N, Leister E, Loddenkemper T, et al. Response to treatment in a prospective national infantile spasms cohort. Ann Neurol. 2016;79:475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yilmaz S, Tekgul H, Serdaroglu G, Akcay A, Gokben S. Evaluation of ten prognostic factors affecting the outcome of West syndrome. Acta Neurol Belg. 2016;116:519–27. [DOI] [PubMed] [Google Scholar]
- 39. Riikonen R, Donner M. Incidence and aetiology of infantile spasms from 1960 to 1976: a population study in Finland. Dev Med Child Neurol. 1979;21:333–43. [DOI] [PubMed] [Google Scholar]
- 40. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–16. [DOI] [PubMed] [Google Scholar]
- 41. Truty R, Patil N, Sankar R, Sullivan J, Millichap J, Carvill G, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia open. 2019;4:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Z, Brodie MJ, Liew D, Kwan P. Treatment Outcomes In Patients With Newly Diagnosed Epilepsy Treated With Established And New Antiepileptic Drugs: A 30‐Year Longitudinal Cohort Study. JAMA Neurol. 2018;75:279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material