

ABSTRACT
Neutrophils are the most abundant effector cells of the innate immune system and represent the first line of defense against infection. However, in many common pathologies, including autoimmune diseases, excessive recruitment and activation of neutrophils can drive a chronic inflammatory response leading to unwanted tissue destruction. Several strategies have been investigated to tackle pathologic neutrophil biology, and thus provide a novel therapy for chronic inflammatory diseases. The chemokine receptor CXCR2 plays a crucial role in regulating neutrophil homeostasis and is a promising pharmaceutical target. In this study, we report the discovery and validation of a humanized anti-human CXCR2 monoclonal antibody. To enable in vivo studies, we developed a surrogate anti-mouse CXCR2 antibody, as well as a human knock-in CXCR2 mouse. When administered in models of atopic dermatitis (AD) and rheumatoid arthritis (RA), the antibodies rapidly clear inflammation. Our findings support further developments of anti-CXCR2 mAb approaches not only for RA and AD, but also for other neutrophil-mediated inflammatory conditions where neutrophils are pathogenic and medical needs are unmet.
KEYWORDS: CXCR2, neutrophil, atopic Dermatitis, arthritis, therapeutic antibody, inflammation
Introduction
Chemokines are low molecular weight proteins produced by a number of different cell types in response to infections or other inflammatory stimuli.1 Chemokine receptors are G protein-coupled receptors (GPCRs) present on the surface of various cell types. By binding to chemokine ligands, GPCRs trigger the movement and the positioning of immune cells,2 thus allowing the host’s immune system to fight an “aggression.” However, an excessive influx of immune cells in a specific tissue can also contribute to a wide variety of immune-mediated inflammatory pathologies, such as autoimmune diseases and chronic inflammatory disorders. Consequently, deleting and/or blocking these rogue cells through their chemokine receptor signatures constitute an attractive therapeutic approach. For instance, the accumulation of CXCR1 and CXCR2+ cells, interferon-γ producing T helper type 1 (Th1), and interleukin (IL)-17-producing T cells (Th17) have been strongly implicated in the pathogenesis of various autoimmune diseases.3–5 Therefore, specific depletion and/or inhibition of pathogenic immune cell subsets, without affecting other leukocyte functions required for protective immunity is a promising therapeutic approach to treat chronic inflammation and autoimmune diseases.6 This strategy has been successfully used in several studies, using a monoclonal antibody (mAb) targeting CXCR3, which is expressed by activated T cells and natural killer (NK) cells,7 lymphotoxin-α receptor expressed by Th1 and Th17 cells,8 and mAb against CCR6 expressed by IL-17 producing Th17 cells.9
The CXCR2 chemokine receptor is expressed on a range of leukocytes but prominently on neutrophils, and plays a critical role in regulating neutrophil homeostasis.10 Under a variety of inflammatory conditions, this receptor facilitates neutrophil trafficking to inflammatory sites.11–13 Neutrophils are the first cells rapidly recruited to inflamed sites and contribute to the cytokine and chemokine cascades. They also regulate inflammatory responses via cell–cell interactions, such as the recruitment and activation of antigen-presenting cells or can directly interact with T cells. In rheumatoid arthritis (RA), neutrophils are activated by immune complexes and pro-inflammatory molecules, which generate reactive oxygen species (ROS) and release the enzymes responsible for cartilage destruction. Activated neutrophils also produce chemoattractants such as IL-8 and leukotriene B4, creating a positive feedback loop that promotes further recruitment of neutrophils and enhances the acute inflammatory response.14,15 In inflamed joints, delayed neutrophil apoptosis results in persistent inflammation and tissue damage due to the continued release of ROS, granule enzymes, and cytokines.16,17 Several inflammatory disease model studies showed that blocking or eliminating CXCR2+ cells is highly effective in reducing excessive leukocyte recruitment to the sites of inflammation.18,19 The role of this receptor in regulating neutrophil recruitment is now better established using CXCR2 knockout strategies.10 Indeed, CXCR2-deficient mice show impaired neutrophil recruitment into the peritoneal cavity,20 lung,21 and the skin.22,23
Atopic dermatitis (AD) is a common chronic and relapsing inflammatory skin disease characterized by intense pruritic and eczematous lesions. Mechanisms thought to underlie AD pathogenesis include excessive type 2 inflammatory responses and impairment of barrier function.24,25 AD is also Th2/Th22-polarized with an attenuated Th17 axis and low levels of antimicrobial peptides, and AD patients show increased susceptibility to skin infections with fungus.26 While psoriasis is established as a disease in which neutrophils contribute to cutaneous inflammation, their role in AD is poorly understood. However, a recent study demonstrated that genes encoding for neutrophil chemoattractants were comparably elevated in both diseases.27
In this study, we isolated TAHX2, a mAb specific for human CXCR2 (hCXCR2). The humanized version of TAHX2 (HAHX2) showed similar binding and antagonistic properties as TAHX2. Furthermore, due to the lack of cross-reactivity of HAHX2 with mCXCR2, we developed hCXCR2 knock-in mice (hCXCR2-KI) for pre-clinical studies and a surrogate anti-mCXCR2 antibody (SAMX2). In chronic inflammatory mouse models of AD and arthritis, HAHX2 and SAMX2 showed significant anti-inflammatory therapeutic potential.
Materials and methods
Mice
C57BL/6 WT and human CXCR2 knock-in (hCXCR2-KI) female mice between 6 and 9 weeks of age were obtained from the Monash Animal Research Platform, Monash University, Victoria, Australia. Mice were maintained on a 12/12-h light/dark cycle and free access to food and water. All experimental procedures involving mice were carried out according to protocols approved by the Animal Ethics Committees of Monash University.
Generating hCXCR2/mCXCR2-/- KI mice
The CXCR2 humanized mice were generated by the MAGEC laboratory (WEHI) as previously described28 on a C57BL/6 background. To generate CXCR2 humanized mice, 20 ng/μl of Cas9 mRNA, 10 ng/μl of sgRNAs (TGTGGTTGTAATATACGTCC and ATTGGCCAGAAATTTCGCCA) and 5 ng/μl of targeting vector were injected into the pronucleus of fertilized one-cell stage embryos. Twenty-four hours later, two-cell stage embryos were transferred into the oviducts of pseudo-pregnant female mice. Viable offspring were genotyped by PCR.
Generation of anti-human and anti-mouse CXCR2 monoclonal antibodies
mAbs reactive with hCXCR2 were generated by immunizing C57BL/6 mice with 107 L1.2 human CXCR2 transfected cells, intraperitoneally (i.p.), five to six times at 2-week intervals. The final immunization was injected intravenously (i.v.). 4 days later, the spleen was removed and cells were fused with the SP2/0 cell line as described. The mouse anti-human CXCR2 antibodies were generated by immunizing human CXCR2 knock-in mice with L1.2 mouse CXCR2 transfected cells using the same protocol as described above.
Epitope mapping
Antibodies’ epitope was first determined by flow cytometry using chimeric human/mouse CXCR2 L1.2 cells. A panel of three overlapping synthetic peptides (Mimotopes) corresponding to the N-terminal region of hCXCR2 were next used in a standard ELISA for a finer epitope mapping. Peptides were coated directly at 1 μg/ml onto a 96-well plate (Nunc). Briefly, the plates were washed 3 times in phosphate-buffered saline (PBS) containing 0.05% Tween20 (T-PBS) and blocked for 1 h with Tween-PBS (T-PBS) plus 2% milk powder. After washing 3 times with T-PBS, the plate was incubated for 1 h with a 0.1 μg of purified mAbs in blocking buffer. The plate was then washed and incubated for 1 h at room temperature with a 1:5,000 dilution of goat anti-mouse IgG peroxidase conjugate (Jackson ImmunoResearch Laboratories; 115–035-008). After three final washes, bound IgG was detected with TMB substrate, and the absorbance was read at 450 nm.
Antibody engineering
Total cellular RNA was extracted from 5 × 106 hybridoma cells using TRIzol reagent (Invitrogen) and poly-A+ RNA was isolated from total cellular RNA using the Oligotex mRNA mini kit (Qiagen, Vic., Australia). Single-stranded (ss) cDNA was produced by reverse-transcription of RNA using the Omniscript reverse transcriptase kit (Qiagen). Reactions contained 500 ng of poly-A+ RNA and 10 pmol of isotype-specific anti-sense oligonucleotide primer (CH1-γ and CL-κ cDNA). A poly-G tail was appended to the 3ʹ-end of ss-cDNA using terminal transferase (NEB, MA, USA). Double-stranded cDNA was generated by PCR using Vent polymerase (NEB) with the poly-C anchor and CH-γ or CL-κ specific primers. The VH and VL genes were sequenced and analyzed by the IMGT® databases (the international ImMunoGeneTics information system®, http://www.imgt.org).
TAHX2 humanization was performed by the CDR-grafting method as previously described.29 Briefly, IMGT/V-QUEST30 and IMGT/Junctions31 analysis tools were used to identify human germline genes in which sequences from the variable regions of both the heavy and light chains were closely aligned with those of TAHX2. Framework sequences of these selected human germline genes were used as acceptor sequences for the CDRs. However, murine residues were retained in the critical “Vernier” zone. The humanized VH and VL genes were synthesized by GENEART AG to generated humanized TAHX2 (HAHX2). To produce the non-depleting (Fc-KO-L234F/L235E/P331S) humanized anti-hCXCR2 IgG1 (HAHX2 Fc-KO), standard mutagenesis techniques were used. The sequences were confirmed on both strands by DNA sequencing.
Similarly, the non-depleting surrogate antibody (SAMX2-FcKO) was generated by substituting the leucine residues at position 234 and 235 with alanine (LALA) in our mouse IgG2a expression vector.
Expression and purification of recombinant antibodies in CHO cells
The variable genes were amplified by PCR and cloned into mammalian expression vector containing either the human kappa and human heavy chain IgG1 FcKO constant regions or the mouse kappa and mouse IgG2a LALA constant region. The insertion was verified by restriction digestion. Recombinant DNA were transfected into CHO-DG44 cells with the AMAXA nucleofactor device (Lonza) according to manufacturer protocol. The transfected CHO cells were grown and maintained in serum-free medium (Invitrogen) for antibody production. Both
the humanized IgG1-FcKO and mIgG2a_LALA antibodies were purified from culture supernatant by affinity chromatography on a ProsepvA protein A column (Millipore). The bound mAbs were eluted with 0.2 M glycine/1 M NaCl, pH 3.0 into a neutralizing solution of 1 M Tris, pH 8.0, then buffer exchanged against PBS. Purified antibodies were endotoxin-free as determined by a chromogenic LAL assay (GenScript).
Chemotaxis assay
Human neutrophils were isolated from the peripheral venous blood of healthy volunteers after they provided informed consent. Blood samples collected into ethylenediamine tetraacetic acid (EDTA)–coated vacutainers were centrifuged at 400 g for 15 min and then the plasma and buffy coats were removed. After 1% dextran sedimentation for 30 min, the white blood cells were pelleted by centrifugation at 300 g for 5 min and washed with PBS. The cells were then centrifuged at 500 g for 30 min on a cushion of 65% Percoll (density 1.093 g/ml, Amersham Bioscience). After centrifugation, the neutrophils were resuspended in PBS. For the assay, the cells were resuspended at 106 cells ml–1 in assay buffer (RPMI 1640, 1% endotoxin-free bovine serum albumin, 100 U ml–1 penicillin, and 100 μg ml–1 streptomycin). Antibodies were preincubated with 100 μl of cells (1 × 105 cells) for 30 min and placed into the upper chamber of a transwell plate with 5-μm pores (Corning Life Sciences). In the lower chambers, the chemokines (30 min, 37°C) were placed and the assays were incubated at 37°C for 4 h (5% CO2). Live cells migrating to the lower chamber were counted using a BD LSRII flow cytometer.
MC903-induced Atopic dermatitis
Mice were anesthetized using 5% isoflurane. MC903 (calcipotriol; Cayman Chemicals, Ann Arbor, MI) was dissolved in 100% EtOH and topically applied on mouse ears (1 nmol in 20 µl, 10 µl per side of ear) on days 0, 2, 5, 7, 9, 12, 14. Same amount of EtOH was applied as vehicle control. Ear thickness was measured on each treatment day just before topical application of MC903 with a digital thickness gauge, and the change in percentage was calculated compared to the thickness on day 0. For the therapeutic regimen, mice ears were treated with MC903 until a 30% increase in ear thickness was reached. A loading dose of SAMX2-FcKO, HAHX2-FcKO, or isotype control mAb was then injected i.p. at 20 mg/kg of body weight (on day 5 for WT, and day 9 for hCXCR2-KI mice), followed by 5 mg/kg every other day for 1 week (See Figure 3(a), Figure 4(a) for treatment regimen). This antibody regimen was chosen based on unpublished experiments performed in our lab using an anti-C5aR antibody.
Figure 3.
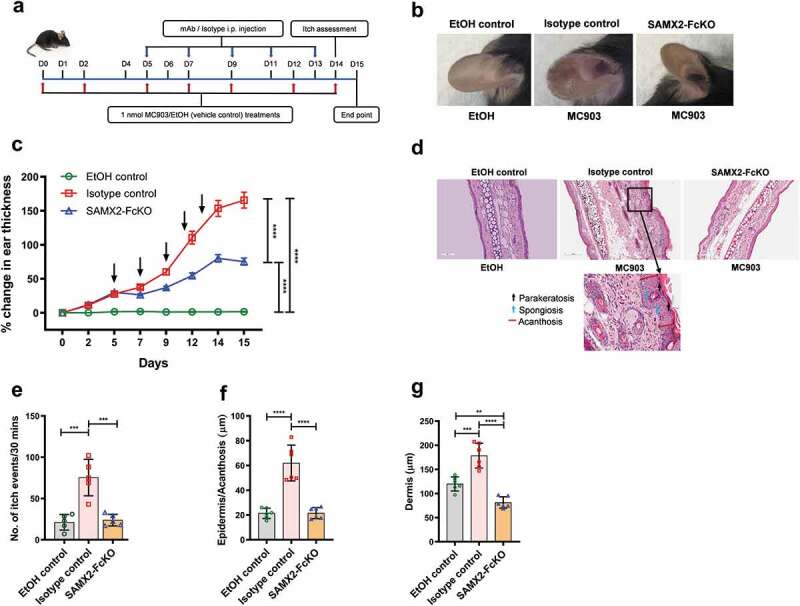
Surrogate anti-mCXCR2 mAb attenuates MC903-induced skin inflammation. C57BL/6 WT mice ears were topically administered with 1 nmol MC903 or EtOH (as vehicle control) for 14 days. On day 5, when ears exhibited symptoms of AD-like skin inflammation, a loading dose of SAMX2-FcKO or isotype control mAb at 20 mg/kg of body weight was injected i.p., followed by 5 mg/kg every other day for 1 week. (a) Schematic of the treatment regimen (b) Representative appearance of EtOH, MC903, and SAMX2-FcKO mAb-treated ears at the endpoint. (c) Ear thickness was measured at each treatment day. (d) Representative ear sections stained with H&E. Original magnification = × 20, scale bar = 200 μm. (e) Ear itch was assessed at day 14 (24 hours prior the endpoint). (f) Epidermis thickness (Acanthosis) and (g) Dermal thickening were measured in tissue sections and quantified by ImageJ software (National Institutes of Health). Each data point represents an individual ear or mouse, and n = 6 mice in each group. All data represented as mean ± standard error of the mean. Each graph is representative of three independent experiments. Ear thickening was analyzed with the two-way analysis of variance with Tukey posttest. Itch assessment and histological measurement was analyzed with the one-way analysis of variance with Tukey posttest. *P < .05, **P < .01, ***P < .005, ****P < .0001
Figure 4.
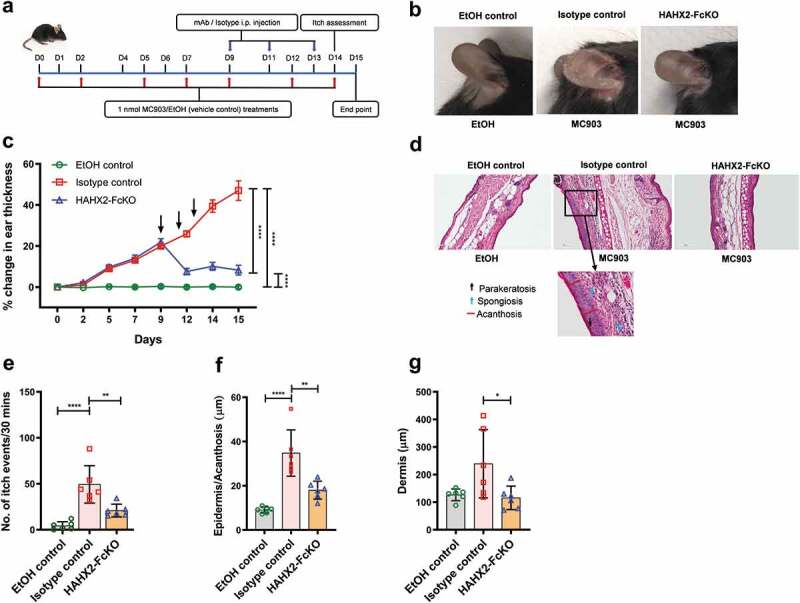
Humanized anti-hCXCR2 mAb suppress MC903-induced skin inflammation in human CXCR2 Knock-in mice. hCXCR2-KI mice ears were topically administered with 1 nmol MC903 or EtOH (as vehicle control) for 14 days. On day 9, when ears exhibited symptoms of AD-like skin inflammation, a loading dose of HAHX2-FcKO or isotype control mAb at 20 mg/kg of body weight was injected i.p., followed by 5 mg/kg every other day for 1 week. (a) Schematic of the treatment regimen (b) Representative appearance of EtOH, MC903, and HAHX2-FcKO mAb-treated ears at the endpoint. (c) Ear thickness was measured at each treatment day. (d) Representative ear sections stained with H&E. Original magnification = × 20, scale bar = 200 μm. (e) Ear itch was assessed at day 14 (24 hours prior the endpoint). (f) Epidermis thickness (Acanthosis) and (g) Dermal thickening were measured in tissue sections and quantified by ImageJ software (National Institutes of Health). Each data point represents an individual ear or mouse, and n = 6 mice in each group. All data represented as mean ± standard error of the mean. Each graph is representative of three independent experiments. Ear thickening was analyzed with the two-way analysis of variance with Tukey posttest. Itch assessment and histological measurement was analyzed with the one-way analysis of variance with Tukey posttest. *P < .05, **P < .01, ***P < .005, ****P < .0001
Itching frequency
Mice were recorded via time-lapse videography, and itch events were determined and quantified. This pathophysiological measurement was obtained from video observation of the relevant treatment groups 24 hours before the experimental endpoint.
K/BxN serum transfer-induced Arthritis
Experimental arthritis was induced in recipient C57BL/6 WT and hCXCR2-KI mice by injecting 200 µl of K/BxN serum i.p. on day 0 and 1. Disease progression was monitored, and ankle thickness and arthritis clinical scores were determined daily. The clinical score was calculated as described previously32 for each mouse by summing the scores for the four paws: 0, normal joint; 1, mild-to-moderate swelling of the ankle and/or one swollen digit; 2, swollen ankle or swelling in two or more digits; 3, severe swelling along all aspects of paw or all five digits swollen. On day 4 of K/BxN serum transfer, mice exhibited swelling in ankle joints and front paws with an average cumulative clinical score of 4. A loading dose (20 mg/kg of body weight) of SAMX2-FcKO or HAHX2-FcKO or isotype control mAb were then injected i.p. on that day, followed by 5 mg/kg every other day for 1 week (See Figure 7(a), Figure 8(a) for treatment regimen). On day 10, mice were killed, ankle joints and pLNs were collected for analyzing the cellular infiltrations and histology.
Figure 7.
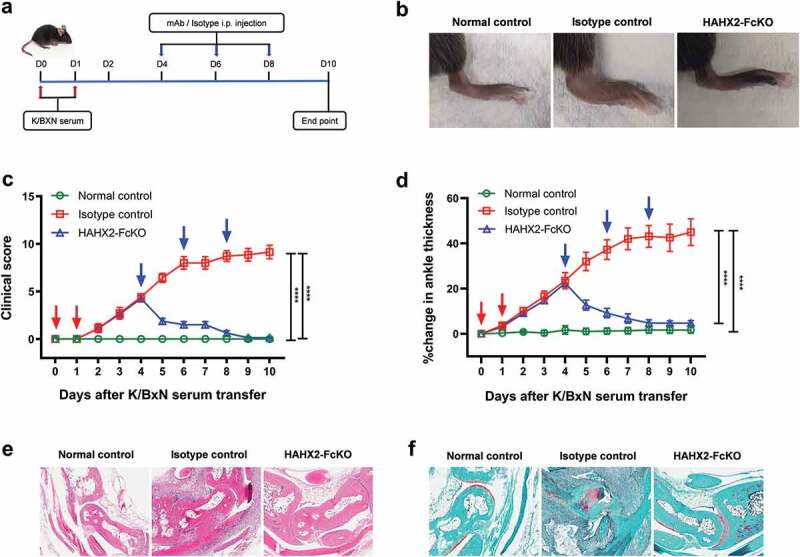
Humanized anti-hCXCR2 mAb reverse arthritis in human CXCR2 Knock-in mice. hCXCR2-KI mice were i.p. injected 200uL of K/BXN serum on day 0 and 1. The development of arthritis was assessed by measuring the ankle thickness and clinical index score every day until the experimental endpoint. When mice exhibiting symptoms of arthritis and cumulative clinical score reached at 4 (on day 4), mice split into two groups, injected i.p., HAHX2-FcKO or isotype control mAb antibody at 20 mg/kg of body weight, followed by 5 mg/kg every other day for 1 week (a) Scheme of the treatment regimen. (b) Representative images of ankle of mice at the endpoint. (c) Clinical score. (d) Ankle thickness. (e) and (f) Histological examination of the representative ankle joint stained with Hematoxylin and eosin (e), and Safranin O (f). Data are representative of three independent experiments with 6 animals per group. Mice that did not receive K/BXN serum or antibody treatment were include as a control group (normal control). All data represented as mean ± standard error of the mean. Statistics were calculated using two-way analysis of variance with Tukey posttest. ***p < .005, ****p < .0001
Figure 8.

Anti-CXCR2 mAbs treatment reduces leukocytes infiltration to the inflamed ankle and popliteal draining lymph nodes. C57BL/6 WT or hCXCR2 -KI mice were injected i.p. 200uL of K/BXN serum on day 0 and 1. When mice were exhibiting symptoms of arthritis and the cumulative clinical score reached 4 (on day 4), mice split into two groups and injected i.p. SAMX2-FcKO, HAHX2-FcKO, or isotype control mAb at a concentration of 20 mg/kg body weight, followed by 5 mg/kg every other day for 1 week. Flow cytometric analysis demonstrates the effect of SAMX2-FcKO and HAHX2-FcKO mAb treatment on the innate effector cells compare to a control group that did not receive K/BXN serum or antibody treatment (normal control): (a) C57BL/6 WT mice ankle and (b) hCXCR2-KI mice ankle, (c) C57BL/6 WT mice draining lymph nodes and (d) hCXCR2-KI mice draining lymph nodes, at the endpoint. Each data point represents an individual mouse, and n = 6 in each group. All data represented as mean ± SEM. Each graph is representative of three independent experiments. Statistics were calculated using one-way analysis of variance with Tukey posttest. *P < .05, **P < .01, ***P < .005, ****P < .0001
Histological evaluation and quantification
Mouse ankles and ear tissue was fixed in 10% neutral-buffered formalin for 24 hours, processed, followed by paraffin-embedded and sectioned at 4 µm. Ear sections were stained with hematoxylin and eosin (H&E) and evaluated at 20x magnification to quantify the acanthosis (epidermal thickening) and dermal thickening by using ImageJ software (National Institutes of Health). Mouse ankles sections were stained with H&E and Safranin O to examine the inflammatory cells infiltration and cartilage destruction.
Mouse tissue processing and cell preparation
For single-cell suspension from mouse ear skins, ear tissue was split into dorsal and ventral layers, was minced in RPMI-1640 medium (Gibco by Life Technologies) with 10% of fetal calf serum (FCS). The tissue was then digested with 2 mg mL−1 of Collagenase Type IV (Gibco by Life Technologies) and 120 µg mL−1 of DNase I (Roche Diagnostics) for 20 minutes at 37°C under agitation. After the digestion, the remaining tissue was passed through a 70 μm cell strainer and washed with cold RPMI-1640 containing 5 mM of EDTA.
To obtain a cell suspension from mouse ankle joints, legs cut at 0.7 cm above the ankle joints, and discard the finger portion. Bone marrow cells were flush out using a 27-gauge needle and 1 ml syringe filled with RPMI 1640. Ankles were then chopped up with scissors to 3–4 mm sized chunks and digested with 2.4 mg ml−1 of Hyaluronidase (Sigma-Aldrich), 1 mg ml−1 of Collagenase (Sigma-Aldrich) containing RPMI 1640 and 10% FCS for 1 h at 37°C under agitation, as previously described.33 The cells were filtered through a 70 μm cell strainer and washed with RPMI 1640 plus 10% FCS. For mouse popliteal and ear draining lymph nodes, lymph nodes were mechanically disrupted and passed through a 70 μm cell strainer to obtain single-cell suspension.
Flow cytometry
To assess the reactivity of mAbs against transfected cells, we used indirect immunofluorescence staining and flow cytometry. Cells were washed once with PBS and resuspended in 100 μl of fluorescence-activated cell sorting (FACS) buffer (PBS containing 2% FCS and 4 mM EDTA) and purified antibody. After 30 min at 4°C, cells were washed twice with FACS buffer and resuspended in 50 μl phycoerythrin (PE)-conjugated anti-human IgG (Jackson ImmunoResearch Laboratories; 109–115-098) diluted 1:500 in FACS buffer for the detection of humanized mAbs or 50 μl PE-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories; 115–116-071) for the detection of mouse mAbs. After incubating for 20 min at 4°C, cells were washed twice with FACS buffer and analyzed on BD LSRII flow cytometer (BD Biosciences). Dead cells were excluded using 7-aminoactinomycin D (7-AAD; BD Pharmingen).
To assess the reactivity of mAbs against human leukocytes, fresh human neutrophils were isolated from heparinized blood (obtained from healthy individuals after they provided informed consent) by centrifugation on a Percoll gradient. The purified neutrophils were then incubated with a biotinylated TAHX2 followed by streptavidin-PE and anti-hCD11b-APC (clone VIM12; ThermoFisher Scientific). The cells were analyzed on BD LSRII flow cytometer (BD Biosciences). 7-AAD (BD Pharmingen) staining was used to exclude dead cells.
To assess the infiltration of immune cell subsets into mice ear skin and ear draining lymph nodes, ankle joint, and popliteal lymph nodes, cells were resuspended with FACS buffer and treated with mouse FcR blocking reagent (Miltenyi Biotec) to block nonspecific antibody binding. For myeloid cells analysis, cells stained with anti-mouse antibodies for following analysis in two different panels: anti-CD45 (clone 30-F11, BD Pharmingen); anti-CD11b (clone M1/70, BD Pharmingen); anti-Ly6G (clone 1A8, BD Pharmingen); anti-Siglec-F (clone E50-2440, BD Pharmingen), anti-F4/80 (clone BM8, Biolegend); anti-CD11c (clone HL3, BD Pharmingen), anti-CD64 (clone X54-5/7.1, BD Pharmingen); anti-CD3 (clone 145–2C11, BD Pharmingen), anti-NK-1.1 (clone PK136, BD Horizon); anti-CD49b (clone DX5, eBioscience), anti-FcεRIα (clone MAR-1, Biolegend), anti-CD117 (clone 2B8, BD Horizon) and anti-I-A/I-E (clone M5/114.15.2, BD Horizon). For T cells analysis, cells stained with anti-mouse antibodies for following analysis panel: anti-CD45 (clone 30-F11, BD Pharmingen), anti-TCRβ (clone H57-597, BD Horizon), anti-TCRγδ (clone GL3, BioLegend), anti-CD4 (clone RM4-5, BD Horizon) and anti-CD8α (clone 53–6.7, BioLegend). Dead cells were excluded using 7-AAD (BD Pharmingen). Cell counts were determined using CountBright plus counting beads (Life Technologies) following manufacturer instructions. Cells were analyzed on BD LSRFortessa X-20 flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (Tree Star).
Statistics
Data analysis and all graphical representations of data were done using Prism 8.2.1 (GraphPad Software). Quantitative data were presented as mean ± standard error of the mean. Comparison between multiple groups was analyzed using one-way analysis of variance with Tukey’s multiple comparisons test, and multiple groups of different treatments using two-way analysis of variance with Tukey’s multiple comparisons test. P values <.05 were considered significant.
Results
Development of a human CXCR2 knock-in mouse using CRISPR/Cas9
In our previous studies, we have shown that transgenic mice expressing human receptors are a powerful tool to evaluate the therapeutic potential of human antigen-specific antibodies.9,32 In this study, we aimed to establish a human CXCR2 knock-in mouse using CRISPR/Cas9 technology. Importantly, previous studies have demonstrated that the mouse CXCR2 ligands were functional on human CXCR2.34 As shown in Figure 1(a), the Cas9 nuclease is directed by two guide RNAs (sgRNAs) to introduce double-stranded breaks within the exon 2 of the mouse CXCR2 locus. A targeting vector carrying the human exon 2 with mouse flanking regions (homology arms) was used as a template for Homology Directed Repair (HDR), thus allowing the integration of human CXCR2 exon 2 with concomitant replacement of the mouse Cxcr2 exon 2. F0 founders were obtained following transfer of microinjected zygotes in pseudopregnant mice. One positive founder was selected based on PCR screening, and expression of human CXCR2 was confirmed by flow cytometry on blood neutrophils. We further crossed the positive CXCR2 KI founder with C57BL/6 mice to confirm germline transmission. Three F1 mice displayed expression of human CXCR2 by PCR and flow cytometry (data not shown) and were used to establish a homozygous hCXCR2 KI colony.
Figure 1.
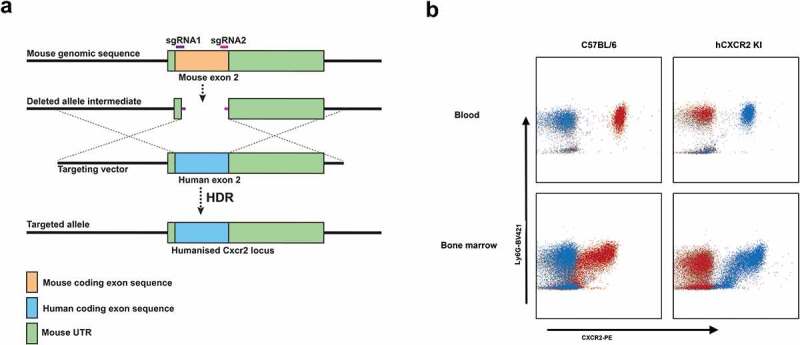
hCXCR2-KI mice design and characterization
(a) Strategy for the insertion of the human CXCR2 exon 2 gene in place of the mouse cxcr2 exon 2. (b) The expression of mCXCR2 (clone-SA044G4, red dots) or hCXCR2 (clone#5E8, blue dots) on peripheral blood neutrophils (top panels) or bone marrow neutrophils (bottom panels) was compared between C57BL/6 WT (left panels) and hCXCR2 (right panels) KI mice (right panels). The gray dots represent the isotype controls. Neutrophils were gated using anti-CD11b and anti-Ly6G antibodies.
Flow cytometry analysis on neutrophils isolated from blood or bone marrow mice revealed that the expression was similar between the C57BL/6 and hCXCR2 homozygote KI mice both in peripheral blood and in bone marrow (Figure 1(b)).
Generation and engineering of anti-human and anti-mouse CXCR2 mAbs
We sought to generate mAbs to human CXCR2 (hCXCR2) to inhibit the function of this receptor and to evaluate the effect of antagonizing hCXCR2 in certain inflammatory reactions, in vivo. mAbs to human CXCR2 were generated by immunizing C57BL/6 mice with the murine pre-B cell lymphoma line, L1.2, which expresses high levels of transfected human CXCR2. Amongst a dozen clones specific for hCXCR2, the clone TAHX2 showed the best reactivity. TAHX2 reacted specifically with hCXCR2-transfected L1.2 cells, but not with L1.2 cells expressing other chemokine receptors (Figure 2(a)). Moreover, TAHX2 showed reactivity against human blood neutrophils (Figure 2(b)).
Figure 2.
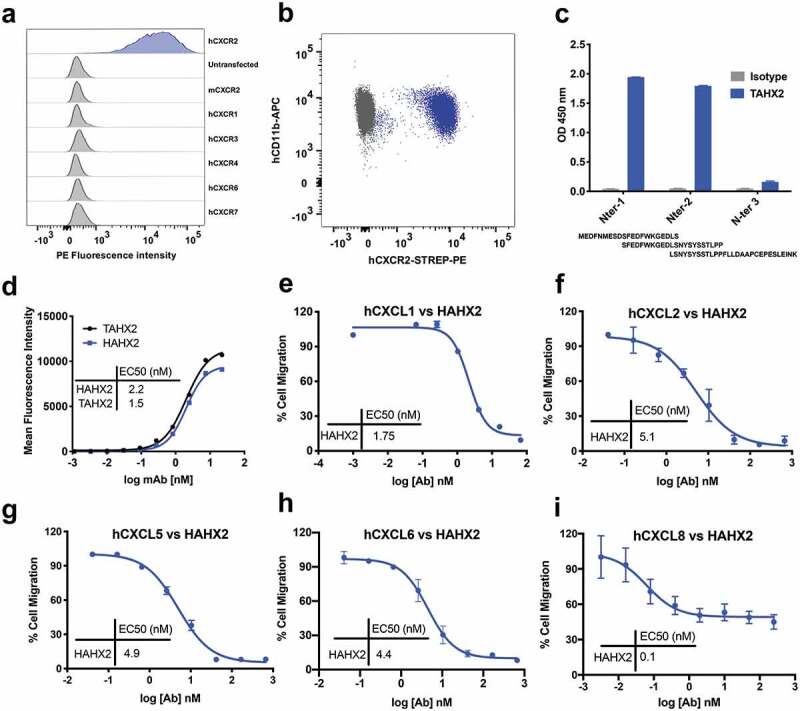
Anti-hCXCR2 mAb characterization
(a) TAHX2 staining of various L1.2 transfectants. Stable L1.2 transfectants expressing hCXCR2; mCXCR2; hCXCR1; hCXCR3; hCXCR4; hCXCR6 and hCXCR7 were stained with TAHX2 to assess the antibody specificity. (b) TAHX2 binding on CD11b neutrophils isolated from human peripheral blood. The gray dots represent the binding of an isotype control compare to TAHX2 (ble dots). (c) Epitope mapping of TAHX2 on the hCXCR2 N-Ter region using peptide ELISA. (d) Comparison of HAHX2 versus TAHX2 by flow cytometry. EC50 of HAHX2 and TAHX2 was measured by flow cytometry on hCXCR2 L1.2 cells using increasing concentrations of purified mAbs. (e;f;g;h;i) Inhibition of human neutrophils chemotaxis by HAHX2. Human neutrophils chemotaxis to hCXCR2’s ligands were carried out in the presence of increasing concentrations of purified HAHX2 mAbs. The percentage of total chemotaxis was calculated using the number of cells migrated in the absence of mAb as 100%.
To determine the epitope recognized by TAHX2, we first screened the mAb by flow cytometry using mouse/human CXCR2 chimeric transfectants. Only transfectants displaying the human CXCR2 N-Terminal region were recognized by TAHX2 (data not shown). We next used overlapping peptides covering the entire N-terminal region to define the core epitope. As shown in Figure 2(c), TAHX2’s epitope contains the AA residues SFEDFWKGEDL similar to other anti-CXCR2 antibodies previously described.35
TAHX2 was next humanized by complementarity-determining region (CDR)-grafting onto human germline sequences as previously described.29 The humanized TAHX2 mAb (HAHX2) genes were further cloned in a human IgG1 expression vector, and HAHX2 was purified from CHO-DG44 cell supernatant by affinity chromatography. The HAHX2 affinity was compared to the parental TAHX2 mAb using flow cytometry on hCXCR2 L1.2 cells. Both antibodies elicited a significant dose-dependent increase in the mean fluorescence intensity (MFI) with a half-maximal effective concentration (EC50) of 1.5 nM for TAHX2 and 2.2 nM for HAHX2. The EC50 values confirmed that minimal affinity toward hCXCR2 was lost during the humanization process (Figure 2(d)).
Since CXCR2 is expressed on neutrophils, and to avoid undesired neutropenia, we next introduced mutations in the hIgG1 Fc region9 to produce a humanized antibody devoid of effector functions (HAHX2-FcKO).
Generation and characterization of a surrogate anti-mCXCR2 mAb (SAMX2) with potent antagonistic activity
In addition to the anti-human mAb generation campaign described above, we sought to generate a surrogate anti-mouse CXCR2 mAb. For this, we used our hCXCR2-KI mice in order to break the immune tolerance to mouse CXCR2. Using this strategy, we isolated a surrogate antibody specific for mCXCR2 (SAMX2) that displayed antagonistic activity toward all mouse CXCL2 ligands (Figure S1). Next, we sequenced and cloned the VH and VK genes in a mouse IgG2a_LALA expression vector, allowing us to produce an Fc-KO surrogate antibody (SAMX2-FcKO) for in vivo use.
Chemotaxis inhibition assay
Chemotaxis assays were used to assess the potential of HAHX2 to antagonize hCXCR2 ligands-mediated cell migration. Purified human neutrophils were allowed to migrate in transwell plates toward hCXCL1; hCXCL2; hCXCL5; hCXCL6 and hCXCL8. HAHX2 was able to completely inhibit neutrophil migration toward hCXCL1; 2; 5 and 6, but partial inhibition was observed with CXCL8. This result may be explained by the presence of CXCR1, a high-affinity CXCL8 chemokine receptor that could still trigger human neutrophil chemotaxis.
Those data also confirm previous reports highlighting the importance of CXCR2 N-terminal region in ligand binding.35,36
Anti-CXCR2 mAb attenuates skin inflammation in the MC903-induced AD model
Topical application of the vitamin D3 analog MC903 can induce allergic skin inflammation characterized by pruritic eczematous skin lesions with elevated skin thickness in mice, similar to human AD.37–39 To determine whether inhibition of CXCR2 receptor could alter the clinical severity of the disease, we injected SAMX2-FcKO mAb to MC903-treated C57BL/6 wildtype (WT) mice already exhibiting symptoms of the disease (~30% increase in ear thickness on day 5; Figure 3 (a,c)). Ear thickening, a marker for ear skin inflammation, was significantly lower in SAMX2-FcKO mAb-treated mice compared to the isotype control-treated animals (Figure 3(c)). Histopathological analysis demonstrated that mice ear skin treated with MC903 for 14 days also exhibited other specific histologic markers of human AD, including acanthosis (thickening of the epidermis), parakeratosis (presence of nuclei in the corneocytes), presence of spongiosis (intercellular edema), and dermal thickening, compared to the vehicle control (ethanol (EtOH))-treated-animals (Figure 3(d)). In contrast, the injection of our mAb markedly reduced these disease parameters and significantly decreased the epidermal and dermal thickness compared with the isotype control group (Figure 3(f,g)).
Itching is a hallmark of AD, and neutrophils are the key initiators of itch in this disease.40 In accordance with altering the clinical severity of the disease shown above, SAMX2-FcKO mAb treatment significantly attenuated itch-evoked scratching compared to isotype control-treated mice (Figure 3(e)).
Bolstered by the results obtained with our surrogate mAb, we evaluated the therapeutic potential of HAHX2-FcKO, a blocking humanized anti-hCXCR2 mAb, in hCXCR2-KI mice. These mice developed ear skin inflammation similar to that observed in WT mice upon the topical application of MC903 to the ear. Using the same therapeutic protocol described above, we injected HAHX2-FcKO mAb when mice were exhibiting symptoms of AD (Figure 4(a)). Consistent with the results of WT mice cohorts, we found that HAHX2-FcKO mAb significantly reduced the disease severity, including ear thickening and itching frequency, compared with isotype control-treated mice (Figure 4(b,c,e)). Likewise, histological assessment of ear skin showed that mice in the isotype control group displayed signs of severe skin inflammation. In contrast, HAHX2-FcKO mAb-treated mice displayed less inflammation and with mostly healthy epidermal and dermal tissue as compared to isotype control-treated animals (Figure 4(d,f,g)).
Administration of anti-CXCR2 mAb reduces inflammatory cell infiltrations to inflamed skin and ear draining lymph nodes
In AD, a number of immune cell subsets infiltrate the skin at the early stage of the disease. Of these, neutrophils are the most abundant immune cell types and are the first one recruited to the skin.40 Therefore, we investigated the effect of blocking neutrophil migration using both our surrogate and humanized anti-CXCR2 mAbs on the long-term recruitment of selected myeloid and lymphoid cells subsets to the ear skin and ear draining lymph nodes (dLNs) after exposure to MC903 using flow cytometry. Using a rigorous gating strategy for myeloid cells (Figure S2), we noticed that MC903 treatment results in a drastic elevation of myeloid cell subsets, including neutrophils, macrophages, dendritic cells, basophils, and mast cells to inflamed ear skin and dLNs as compared to EtOH control both in WT and hCXCR2-KI mice (Figure 5(a,c,e,g)). These immune cells are known to contribute to the pathogenesis of AD.40–43 In contrast, our SAMX2-FcKO and HAHX2-FcKO mAbs treatment significantly reduced these cell numbers in ear skin and draining dLNs in comparison with isotype control-treated mice (Figure 5(a,c,e,g)). Eosinophils are another critical cell type for the development and maintenance of AD.39 Along with the reduction of other innate effector cells, our CXCR2 blocking strategy also significantly reduced the number of infiltrating eosinophils both in the ear skin and dLNs in WT and hCXCR2-KI mice as compared to isotype control-treated animals (Figure 5(a,c,e,g)).
Figure 5.
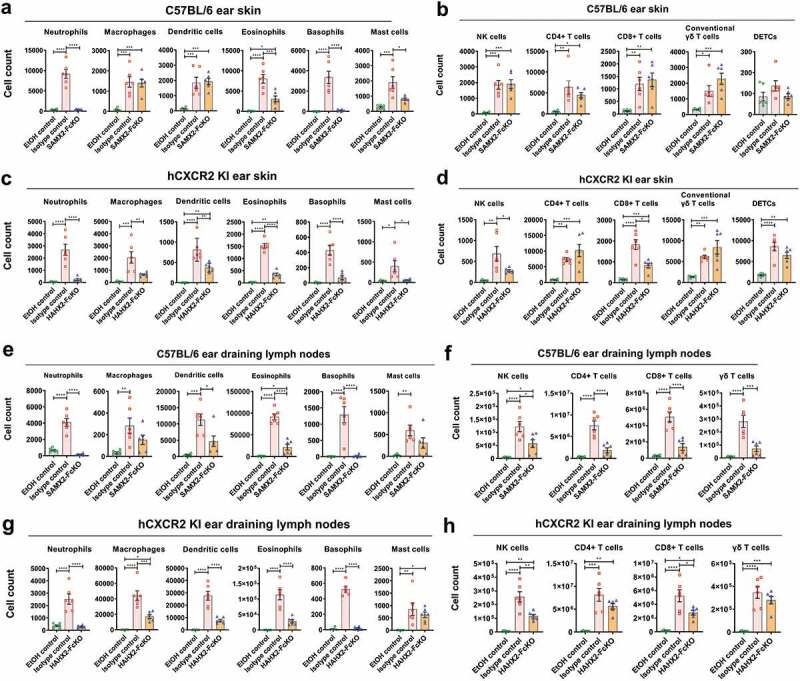
Anti-CXCR2 mAbs treatment reduces leukocytes infiltration in inflamed ear skin and ear draining lymph nodes. C57BL/6 WT and hCXCR2-KI mice were topically administered with 1 nmol MC903 or EtOH (vehicle control) for 14 days. When mice ear exhibiting symptoms of AD-like skin inflammation (on day 5 for WT, and day 9 for hCXCR2-KI mice), a loading dose of SAMX2-FcKO, HAHX2-FcKO, or isotype control mAb at 20 mg/kg of body weight was injected i.p., followed by 5 mg/kg every other day for 1 week. At the end of the experiment, single-cell preparation from ear tissues and ear draining lymph nodes (dLNs) were prepared and analyzed by flow cytometry. The effect of SAMX2-FcKO or HAHX2-FcKO mAb treatment on the infiltration of: (a) Myeloid cells in ear skin of C57BL/6 WT mice, (b) Lymphoid cells in ear skin of C57BL/6 WT mice, (c) Myeloid cells in ear skin of hCXCR2-KI mice, (d) Lymphoid cells in ear skin of hCXCR2-KI mice, (e) Myeloid cells in ear dLNs of C57BL/6 WT mice, (f) Lymphoid cells in ear dLNs of C57BL/6 WT mice, (g) Myeloid cells in ear dLNs of hCXCR2-KI mice, (h) Lymphoid cells in ear dLNs of hCXCR2-KI mice, at the endpoint. Each data point represents an individual mouse, and n = 6 in each group. All data represented as mean ± SEM. Each graph is representative of three independent experiments. Statistics were calculated using one-way analysis of variance with Tukey posttest. *P < .05, **P < .01, ***P < .005, ****P < .0001
Effector functions by T cells contribute to the pathogenesis of human AD.24 We found MC903 treatment results in a dramatic elevation of all T cell subsets in inflamed skin and dLNs in WT and hCXCR2-KI mice, including CD4 + T cells, CD8 + T cells, γδ high-expressing T cells (dendritic epidermal T cells, DETC), and γδ low-expressing T cells (conventional γδ T cells) (Figure 5(b,d,f,h)). Consistent with the results of attenuating the disease severity, our SAMX2-FcKO and HAHX2-FcKO mAb treatment reduced the number of these cells both in the ear skin and dLNs compared with isotype control-treated mice (Figure 5(b,d,f,h)). Surprisingly, our mAb treatment results in a modest increase of conventional γδ T cell infiltration in the ear skin (Figure 5(b,d)).
Our findings demonstrate that blocking CXCR2 using a specific mAb decreases cellular infiltration to cutaneous tissue and draining lymph nodes, which may explain the attenuation of AD-like skin inflammation in MC903-treated animals.
Administration of anti-CXCR2 mAb reverse Arthritis in the K/BXN serum-transfer model
Neutrophils are one of the primary effector cells that drives the joint-specific inflammatory reaction.15 There is a strong correlation between neutrophil recruitment to inflamed joints and the severity of disease. Indeed, the inhibition of neutrophil recruitment with anti-C5aR mAb has been showed to reverse arthritis symptoms.32 Another important receptor involved in neutrophil recruitment to the joints in RA is CXCR2.44,45 Therefore, we investigated the role of blocking CXCR2 in the development of arthritis after intraperitoneally injecting K/BXN serum to C57BL/6 WT and hCXCR2-KI mice. Following the injection of K/BXN serum, hCXCR2-KI mice developed joint inflammation in a similar fashion to that observed in WT mice. To determine whether inhibition of CXCR2 receptor could alter the clinical severity of the disease, we injected SAMX2-FcKO and HAHX2-FcKO mAbs to WT and hCXCR2-KI mice already exhibiting symptoms of arthritis (on day 4 of the serum transfer; Figure 6(a) and Figure 7(a)). Remarkably, the injection of our anti-CXCR2 mAb quickly reversed arthritic clinical symptoms such as ankle thickness, which serves as a surrogate marker of arthritis, in comparison with isotype control-treated mice (Figure 7(b,c,d) and Figure 8(b,c,d)).
Figure 6.
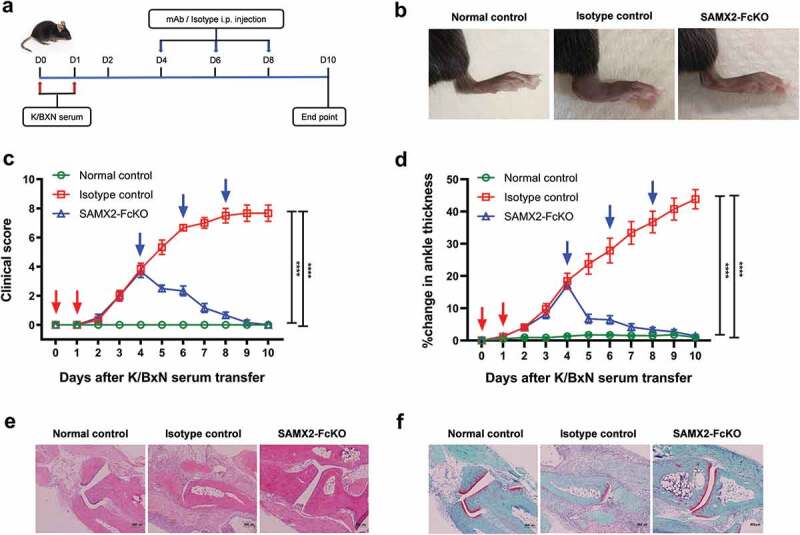
Surrogate anti-mCXCR2 mAb reverse the K/BXN-serum transfer arthritis. C57BL/6 WT mice were i.p. injected 200uL of K/BXN serum on day 0 and 1. The development of arthritis was assessed by measuring the ankle thickness and clinical index score every day until the experimental endpoint. When mice exhibiting symptoms of arthritis and cumulative clinical score reached at 4 (on day 4), mice split into two groups, injected i.p., SAMX2-FcKO or isotype control mAb at 20 mg/kg of body weight, followed by 5 mg/kg every other day for 1 week (a) Scheme of the treatment regimen. (b) Representative images of ankle of mice at the endpoint. (c) Clinical score. (d) Ankle thickness. (e) and (f) Histological examination of the representative ankle joint stained with H&E (e), and Safranin O (f). Mice that did not receive K/BXN serum or antibody treatment were include as a control group (normal control). Data are representative of three independent experiments with 6 animals per group. All data represented as mean ± standard error of the mean. Statistics were calculated using two-way analysis of variance with Tukey posttest. ***p < .005, ****p < .0001
Histological analysis of the ankle joint showed that substantial cellular infiltrates in isotype control-treated animals (Figure 7(e), Figure 8(e)). To examine the joints’ articular surface, sections were also stained with safranin O. As shown in Figure 7(f) and Figure 8(f), there was a marked loss of cartilage proteoglycan in isotype control-treated animals. Additionally, synovial infiltrate adhered and extended into the eroding cartilage within the ankle joint. In contrast, sections of joints from mAb-treated animals showed a marked decrease in the infiltration of leukocytes in the synovial space, minimal loss of cartilage and mostly normal ankle morphology, which were virtually indistinguishable from control animals (Figure 7(e,f) and Figure 8(e,f)).
Anti-CXCR2 mAbs reduces inflammatory cell infiltration to the ankle and draining popliteal lymph nodes
Since neutrophils play a crucial role in the initiation and progression of inflammation in RA,15 we investigated what would be the effect of blocking CXCR2 on the recruitment of various myeloid and lymphoid cell subsets involved in this model. The neutrophils,15 macrophages,46 dendritic cells,47,48 mast cells49 and NK cells50,51 population (gating strategies shown in Figure S3) were analyzed in inflamed joints and draining popliteal lymph nodes (pLNs) using flow cytometry. As expected, SAMX2-FcKO mAb treatment markedly reduced numbers of infiltrating neutrophils in the ankle and draining pLNs compared with isotype control-treated mice (Figure 8(a,c)). Similarly, this mAb treatment significantly reduced the recruitment of other innate effector cells, including dendritic cells, mast cells, and NK cells in the ankle joints and draining pLNs compared to the isotype control-treated mice (Figure 8(a,c)).
Macrophages play a pivotal role in the development of RA and are present in high numbers in the inflamed synovial membrane and at the cartilage-pannus junction.52,53 In accordance with previous studies, we observed a higher number of macrophages and Ly6C+ macrophages in inflamed joint and draining dLNs of isotype control-treated mice, while SAMX2-FcKO mAb treatment significantly reduced the infiltration of cell types (Figure 8(a,c)). We observed similar results when hCXCR2-KI mice were treated with the HAHX2-FcKO mAb (Figure 8(b,d)).
These findings indicate that the administration of anti-CXCR2 mAb decreases the infiltration of innate effector cells to the ankle joints and draining lymph nodes, which ultimately results in the reversal of joint inflammation in this model.
Discussion
The CXCR2 chemokine receptor and its cognate ligands are implicated in various inflammatory diseases, making it a promising anti-inflammatory therapeutics target. Apart from targeting inflammatory cytokines, blocking this receptor leads to inhibition of polymorphonuclear neutrophils homing to inflammatory sites, thus reducing inflammation. This therapeutic approach has been validated by genetic depletion or small molecule receptor antagonists, and has often been shown to be efficacious in treating inflammatory diseases in numerous animal models.10,45,54,55 Consequently, the blockade of the recruitment of inflammatory cells by targeting chemokine receptors has long been seen as an attractive proposition in human inflammatory diseases.
An important outcome of this study was the isolation of a therapeutic anti-hCXCR2 mAb that blocks neutrophil recruitment and inflammatory response. A humanization approach was designed to maximize similarity to the human germline sequence and, by engineering the Fc part of HAHX2, we were able to solely assess the significance of antagonizing CXCR2, rather than CXCR2+ cells depletion, which may cause neutropenia.
A crucial step in therapeutic mAb development is to adequately test the mAb in relevant animal models, but the lack of cross-reactivity of biological drugs with orthologous targets makes this difficult. The preferred routes to overcome this hurdle are: 1) to develop a surrogate mAb with functional characteristics similar to the lead candidate, or 2) knocking-in the human target gene in the desired animal model. Here, we applied both strategies. Using a CRISPR/Cas approach, we produced a human CXCR2 knock-in mouse that allowed us to translate in vivo the humanized antibody efficacy observed in vitro. Moreover, we used the hCXCR2 KI mouse to generate a unique surrogate anti-mCXCR2 blocking antibody. We next monitored the efficacies of both blocking mAbs in two inflammatory disease mouse models. We found that our therapeutic antibodies exhibited remarkable effectiveness and rapidly reduced inflammation by attenuating all disease parameters in a therapeutic setting in both models. Indeed, in the AD mouse model, both the surrogate and humanized anti-CXCR2 mAbs treatment attenuated AD phenotypes, including suppressing the thickness of ear skin, dermal, and epidermal thickness compared to isotype control-treated mice. More impressively, the treatments significantly reduced itch-evoked scratching (pruritus), which is the hallmark of this disease56 in comparison with isotype control-treated animals. These results were unexpected and provide proof that neutrophils are involved in the inflammatory reaction and play a key role inducing pruritus in this disease. Of studies carried out on the abnormalities of genes and immune cells, and their interactions with chronic itching,27,57–59 only a few of them reported that neutrophil induces itchiness and scratching during allergic skin inflammation.40,42 Consistent with the reduced AD phenotypes, our anti-CXCR2 mAb treatment also altered the infiltration of inflammatory cells in the ear skin and draining dLNs. Our results suggest the importance of CXCR2 for the migration of inflammatory cells and anti-CXCR2 mAb suppressed this disease by restricting the immune cell recruitment to the inflamed skin.
Similarly, in the K/BXN serum-induced arthritis model, therapeutic blockade of CXCR2 using mAb treatment had a striking effect in the reversal of ankle inflammation and other arthritic clinical symptoms, including massive inflammatory cell infiltration and loss of cartilage proteoglycan in the ankle joints.54 Consistent with the lessening of the disease severity, our antibody treatment markedly reduced the number of neutrophils, total macrophages, and Ly6C+ macrophages in the ankle joint and draining pLNs. Neutrophils are highly migratory and thought to play a pathogenic role in several inflammatory diseases. Recruitment of neutrophils to the inflamed joints is CXCR2 dependent,45 and blocking neutrophil trafficking to inflammation sites suppressed local production of proinflammatory cytokines, e.g., IL-1β, IL-6, and chemokines KC and MCP-1, which are associated with the driving tissue damage.54 Several previous inflammatory disease model studies showed that blocking or eliminating CXCR2+ cells is highly effective in reducing excessive leukocyte recruitment to inflammation sites.18,19 Remarkably, Ly6C+ macrophage numbers were higher in the inflamed joints. These cells are most likely recently accumulated to the joints in response to inflammation and are derived from circulating Ly6C+ monocytes.60 A recent study showed that infliximab treatment increased the apoptosis of Ly6C+ macrophages in the ankles and draining lymph nodes, thus reducing the expression of CCL2 and monocyte migration into the ankles, and attenuated arthritic disease symptoms.60
In summary, our findings indicate that targeted inhibition of the proinflammatory pathway of the CXCR2 receptor with anti-CXCR2 mAb is an excellent therapeutic approach and may be useful in the treatment of AD and arthritis. Our results support the development of anti-CXCR2 therapy for these and possibly other immune-mediated disorders where CXCR2+ cells play a pathogenic role.
Supplementary Material
Acknowledgments
The authors acknowledge FlowCore for assistance with flow cytometry analysis. The generation of the hCXCR2-KI mice used in this study was supported by the Australian Phenomics Network (APN) and the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program.
Funding Statement
This work was supported by the National Health and Medical Research Council.
Disclosure of interest
The authors have no conflicts of interest relevant to this article to disclose.
Abbreviations
| AD: | Atopic dermatitis |
| dLN: | Draining lymph node |
| EtOH: | Ethanol |
| GPCR: | G protein-coupled receptor |
| HAHX2: | Humanized version of human CXCR2 specific mAb |
| hCXCR2 KI: | Human CXCR2 knock-in mice |
| KO: | Knockout |
| mAb: | Monoclonal antibody |
| pLN: | Popliteal lymph node |
| RA: | Rheumatoid Arthritis |
| SAMX2: | Surrogate anti-mouse CXCR2 mAb |
| TAHX2: | Human CXCR2 specific mAb |
| Th1: | T helper type 1 |
| Th17: | T helper 17 cell |
| WT: | Wild Type |
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250:91–15. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- 2.Mackay CR. Chemokines: immunology’s high impact factors. Nat Immunol. 2001;2:95–101. doi: 10.1038/84298. [DOI] [PubMed] [Google Scholar]
- 3.Farooq SM, Stillie R, Svensson M, Svanborg C, Strieter RM, Stadnyk AW. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2009;329:123–29. doi: 10.1124/jpet.108.145862. [DOI] [PubMed] [Google Scholar]
- 4.Liston A, Kohler RE, Townley S, Haylock-Jacobs S, Comerford I, Caon AC, Webster J, Harrison JM, Swann J, Clark-Lewis I, et al. Inhibition of CCR6 function reduces the severity of experimental autoimmune encephalomyelitis via effects on the priming phase of the immune response. J Immunol. 2009;182:3121–30. doi: 10.4049/jimmunol.0713169. [DOI] [PubMed] [Google Scholar]
- 5.Min SH, Wang Y, Gonsiorek W, Anilkumar G, Kozlowski J, Lundell D, Fine JS, Grant EP. Pharmacological targeting reveals distinct roles for CXCR2/CXCR1 and CCR2 in a mouse model of arthritis. Biochem Biophys Res Commun. 2010;391:1080–86. doi: 10.1016/j.bbrc.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 6.Mackay CR. Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nat Immunol. 2008;9:988–98. doi: 10.1038/ni.f.210. [DOI] [PubMed] [Google Scholar]
- 7.Dai Z, Xing L, Cerise J, Wang EH, Jabbari A, de Jong A, Petukhova L, Christiano AM, Clynes R. CXCR3 blockade inhibits T cell migration into the skin and prevents development of Alopecia Areata. J Immunol. 2016;197:1089–99. doi: 10.4049/jimmunol.1501798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang EY, Kolumam GA, Yu X, Francesco M, Ivelja S, Peng I, Gribling P, Shu J, Lee WP, Refino CJ, et al. Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat Med. 2009;15:766–73. doi: 10.1038/nm.1984. [DOI] [PubMed] [Google Scholar]
- 9.Robert R, Ang C, Sun G, Juglair L, Lim EX, Mason LJ, Bernard CC, Mackay CR. Essential role for CCR6 in certain inflammatory diseases demonstrated using specific antagonist and knockin mice. JCI Insight. 2017;2(15):e94821. doi: 10.1172/jci.insight.94821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boppana NB, Devarajan A, Gopal K, Barathan M, Bakar SA, Shankar EM, Ebrahim AS, Farooq SM. Blockade of CXCR2 signalling: a potential therapeutic target for preventing neutrophil-mediated inflammatory diseases. Exp Biol Med (Maywood). 2014;239:509–18. doi: 10.1177/1535370213520110. [DOI] [PubMed] [Google Scholar]
- 11.Chapman RW, Minnicozzi M, Celly CS, Phillips JE, Kung TT, Hipkin RW, Fan X, Rindgen D, Deno G, Bond R, et al. A novel, orally active CXCR1/2 receptor antagonist, Sch527123, inhibits neutrophil recruitment, mucus production, and goblet cell hyperplasia in animal models of pulmonary inflammation. J Pharmacol Exp Ther. 2007;322(2):486–93. doi: 10.1124/jpet.106.119040. [DOI] [PubMed] [Google Scholar]
- 12.Holz O, Khalilieh S, Ludwig-Sengpiel A, Watz H, Stryszak P, Soni P, Tsai M, Sadeh J, Magnussen H. SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced neutrophilia in healthy subjects. Eur Respir J. 2010;35(3):564–70. doi: 10.1183/09031936.00048509. [DOI] [PubMed] [Google Scholar]
- 13.Matzer SP, Zombou J, Sarau HM, Rollinghoff M, Beuscher HU. A synthetic, non-peptide CXCR2 antagonist blocks MIP-2-induced neutrophil migration in mice. Immunobiology. 2004;209(3):225–33. doi: 10.1016/j.imbio.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203:837–42. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–08. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 16.Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371:531–39. doi: 10.1007/s00441-017-2785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilgus TA, Roy S, McDaniel JC. Neutrophils and wound repair: positive actions and negative reactions. Adv Wound Care (New Rochelle). 2013;2:379–88. doi: 10.1089/wound.2012.0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosoki K, Rajarathnam K, Sur S. Attenuation of murine allergic airway inflammation with a CXCR1/CXCR2 chemokine receptor inhibitor. Clin Exp Allergy. 2019;49:130–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerner CA, Lei W, Sundar IK, Rahman I. Genetic ablation of CXCR2 protects against cigarette smoke-induced lung inflammation and injury. Front Pharmacol. 2016;7:391. doi: 10.3389/fphar.2016.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood W, Moore M. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–84. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 21.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cattani F, Gallese A, Mosca M, Buanne P, Biordi L, Francavilla S, Coletti G, Pellegrini L, Melillo G, Bertini R. The role of CXCR2 activity in the contact hypersensitivity response in mice. Eur Cytokine Netw. 2006;17:42–48. [PubMed] [Google Scholar]
- 23.Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, Richmond A. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–44. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dainichi T, Kitoh A, Otsuka A, Nakajima S, Nomura T, Kaplan DH, Kabashima K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat Immunol. 2018;19:1286–98. doi: 10.1038/s41590-018-0256-2. [DOI] [PubMed] [Google Scholar]
- 25.Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387:1109–22. doi: 10.1016/S0140-6736(15)00149-X. [DOI] [PubMed] [Google Scholar]
- 26.Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis–part I: clinical and pathologic concepts. J Allergy Clin Immunol. 2011;127:1110–18. doi: 10.1016/j.jaci.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 27.Choy DF, Hsu DK, Seshasayee D, Fung MA, Modrusan Z, Martin F, Liu F-T, Arron JR. Comparative transcriptomic analyses of atopic dermatitis and psoriasis reveal shared neutrophilic inflammation. J Allergy Clin Immunol. 2012;130:1335–43 e5. doi: 10.1016/j.jaci.2012.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kueh AJ, Pal M, Tai L, Liao Y, Smyth GK, Shi W, Herold MJ. An update on using CRISPR/Cas9 in the one-cell stage mouse embryo for generating complex mutant alleles. Cell Death Differ. 2017;24:1821–22. doi: 10.1038/cdd.2017.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robert R, Streltsov VA, Newman J, Pearce LA, Wark KL, Dolezal O. Germline humanization of a murine Abeta antibody and crystal structure of the humanized recombinant Fab fragment. Protein Sci. 2010;19:299–308. doi: 10.1002/pro.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giudicelli V, Chaume D, Lefranc MP. IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic Acids Res. 2004;32:W435–40. doi: 10.1093/nar/gkh412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yousfi Monod M, Giudicelli V, Chaume D, Lefranc MP. IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J JUNCTIONs. Bioinformatics. 2004;20(Suppl 1):i379–85. doi: 10.1093/bioinformatics/bth945. [DOI] [PubMed] [Google Scholar]
- 32.Lee H, Zahra D, Vogelzang A, Newton R, Thatcher J, Quan A, So T, Zwirner J, Koentgen F, Padkjær SB, et al. Human C5aR knock-in mice facilitate the production and assessment of anti-inflammatory monoclonal antibodies. Nat Biotechnol. 2006;24:1279–84. doi: 10.1038/nbt1248. [DOI] [PubMed] [Google Scholar]
- 33.Akitsu A, Ishigame H, Kakuta S, Chung SH, Ikeda S, Shimizu K, Kubo S, Liu Y, Umemura M, Matsuzaki G, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vgamma6(+)gammadelta T cells. Nat Commun. 2015;6:7464. doi: 10.1038/ncomms8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mihara K, Smit MJ, Krajnc-Franken M, Gossen J, Rooseboom M, Dokter W. Human CXCR2 (hCXCR2) takes over functionalities of its murine homolog in hCXCR2 knockin mice. Eur J Immunol. 2005;35:2573–82. doi: 10.1002/eji.200526021. [DOI] [PubMed] [Google Scholar]
- 35.Wu L, Ruffing N, Shi X, Newman W, Soler D, Mackay CR, Qin S. Discrete steps in binding and signaling of interleukin-8 with its receptor. J Biol Chem. 1996;271:31202–09. doi: 10.1074/jbc.271.49.31202. [DOI] [PubMed] [Google Scholar]
- 36.Rossant CJ, Carroll D, Huang L, Elvin J, Neal F, Walker E, Benschop JJ, Kim EE, Barry ST, Vaughan TJ, et al. Phage display and hybridoma generation of antibodies to human CXCR2 yields antibodies with distinct mechanisms and epitopes. MAbs. 2014;6:1425–38. doi: 10.4161/mabs.34376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, Hepworth MR, Van Voorhees AS, Comeau MR, Artis D, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5:170ra16. doi: 10.1126/scitranslmed.3005374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M, Hener P, Zhang Z, Kato S, Metzger D, Chambon P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A. 2006;103:11736–41. doi: 10.1073/pnas.0604575103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naidoo K, Jagot F, van den Elsen L, Pellefigues C, Jones A, Luo H, Johnston K, Painter G, Roediger B, Lee J, et al. Eosinophils determine dermal thickening and water loss in an MC903 model of atopic dermatitis. J Invest Dermatol. 2018;138:2606–16. doi: 10.1016/j.jid.2018.06.168. [DOI] [PubMed] [Google Scholar]
- 40. Walsh CM, Hill RZ, Schwendinger-Schreck J, Deguine J, Brock EC, Kucirek N, Rifi Z ,Wei J, Gronert K, Brem RB, et al. Neutrophils promote CXCR3-dependent itch in the development of atopic dermatitis. Elife. 2019;8:e48448. doi: 10.7554/eLife.48448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu FT, Goodarzi H, Chen HY. IgE, mast cells, and eosinophils in atopic dermatitis. Clin Rev Allergy Immunol. 2011;41:298–310. doi: 10.1007/s12016-011-8252-4. [DOI] [PubMed] [Google Scholar]
- 42.Oyoshi MK, He R, Li Y, Mondal S, Yoon J, Afshar R, Chen M, Lee D, Luo H, Luster A, et al. Leukotriene B4-driven neutrophil recruitment to the skin is essential for allergic skin inflammation. Immunity. 2012;37:747–58. doi: 10.1016/j.immuni.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siracusa MC, Kim BS, Spergel JM, Artis D. Basophils and allergic inflammation. J Allergy Clin Immunol. 2013;132:789–801. quiz 788. doi: 10.1016/j.jaci.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, Haribabu B, Iwakura Y, Luster AD. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33:266–78. doi: 10.1016/j.immuni.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobs JP, Ortiz-Lopez A, Campbell JJ, Gerard CJ, Mathis D, Benoist C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 2010;62:1921–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solomon S, Rajasekaran N, Jeisy-Walder E, Snapper SB, Illges H. A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur J Immunol. 2005;35:3064–73. doi: 10.1002/eji.200526167. [DOI] [PubMed] [Google Scholar]
- 47.Lebre MC, Jongbloed SL, Tas SW, Smeets TJ, McInnes IB, Tak PP. Rheumatoid arthritis synovium contains two subsets of CD83-DC-LAMP- dendritic cells with distinct cytokine profiles. Am J Pathol. 2008;172:940–50. doi: 10.2353/ajpath.2008.070703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas R, MacDonald KP, Pettit AR, Cavanagh LL, Padmanabha J, Zehntner S. Dendritic cells and the pathogenesis of rheumatoid arthritis. J Leukoc Biol. 1999;66:286–92. doi: 10.1002/jlb.66.2.286. [DOI] [PubMed] [Google Scholar]
- 49.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–92. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 50.Conigliaro P, Scrivo R, Valesini G, Perricone R. Emerging role for NK cells in the pathogenesis of inflammatory arthropathies. Autoimmun Rev. 2011;10:577–81. doi: 10.1016/j.autrev.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 51.Yamin R, Berhani O, Peleg H, Aamar S, Stein N, Gamliel M, Hindi I, Scheiman-Elazary A, Gur C. High percentages and activity of synovial fluid NK cells present in patients with advanced stage active rheumatoid arthritis. Sci Rep. 2019;9:1351. doi: 10.1038/s41598-018-37448-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blom AB, van Lent PL, van Vuuren H, Holthuysen AE, Jacobs C, van de Putte LB, van de Winkel JG, van den Berg WB. Fc gamma R expression on macrophages is related to severity and chronicity of synovial inflammation and cartilage destruction during experimental immune-complex-mediated arthritis (ICA). Arthritis Res. 2000;2:489–503. doi: 10.1186/ar131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. 2000;2:189–202. doi: 10.1186/ar86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell IK, Leong D, Edwards KM, Rayzman V, Ng M, Goldberg GL, Wilson NJ, Scalzo-Inguanti K, Mackenzie-Kludas C, Lawlor KE, et al. Therapeutic targeting of the G-CSF receptor reduces neutrophil trafficking and joint inflammation in antibody-mediated inflammatory arthritis. J Immunol. 2016;197:4392–402. doi: 10.4049/jimmunol.1600121. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Guo R, Kambara H, Ma F, Luo HR. The role of CXCR2 in acute inflammatory responses and its antagonists as anti-inflammatory therapeutics. Curr Opin Hematol. 2019;26:28–33. doi: 10.1097/MOH.0000000000000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wahlgren CF. Itch and atopic dermatitis: an overview. J Dermatol. 1999;26:770–79. doi: 10.1111/j.1346-8138.1999.tb02090.x. [DOI] [PubMed] [Google Scholar]
- 57.Ewald DA, Noda S, Oliva M, Litman T, Nakajima S, Li X, Xu H, Workman CT, Scheipers P, Svitacheva N, et al. Major differences between human atopic dermatitis and murine models, as determined by using global transcriptomic profiling. J Allergy Clin Immunol. 2017;139:562–71. doi: 10.1016/j.jaci.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 58.Guttman-Yassky E, Suarez-Farinas M, Chiricozzi A, Nograles KE, Shemer A, Fuentes-Duculan J, Cardinale I, Lin P, Bergman R, Bowcock AM, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol. 2009;124:1235–44 e58. doi: 10.1016/j.jaci.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 59.Suarez-Farinas M, Dhingra N, Gittler J, Shemer A, Cardinale I, de Guzman Strong C, Krueger JG, Guttman-Yassky E. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013;132:361–70. doi: 10.1016/j.jaci.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang QQ, Birkett R, Doyle R, Shi B, Roberts EL, Mao Q, Pope RM. The role of macrophages in the response to TNF inhibition in experimental arthritis. J Immunol. 2018;200:130–38. doi: 10.4049/jimmunol.1700229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.