

Abstract
Traumatic brain injury (TBI) is associated with increased blood content of fibrinogen (Fg), called hyperfibrinogenemia (HFg), which results in enhanced cerebrovascular permeability and leads to short-term memory (STM) reduction. Previously, we showed that extravasated Fg was deposited in the vasculo-astrocyte interface and was co-localized with cellular prion protein (PrPC) during mild-to-moderate TBI in mice. These effects were accompanied by neurodegeneration and STM reduction. However, there was no evidence presented that the described effects were the direct result of the HFg during TBI. We now present data indicating that inhibition of Fg synthesis can ameliorate TBI-induced cerebrovascular permeability and STM reduction. Cortical contusion injury (CCI) was induced in C57BL/6J mice. Then mice were treated with either Fg antisense oligonucleotide (Fg-ASO) or with control-ASO for two weeks. Cerebrovascular permeability to fluorescently conjugated bovine serum albumin was assessed in cortical venules following evaluation of STM with a Y-maze test. Separately, brain samples were collected in order to define the expression of PrPC via Western blotting while deposition and co-localization of Fg and PrPC, as well as gene expression of inflammatory marker activating transcription factor 3 (ATF3), were characterized with real-time PCR. Results showed that inhibition of Fg synthesis with Fg-ASO reduced overexpression of AFT3, ameliorated enhanced cerebrovascular permeability, decreased expression of PrPC and Fg deposition, decreased formation of Fg-PrPC complexes in brain, and improved STM. These data provide direct evidence that a CCI-induced inflammation-mediated HFg could be a triggering mechanism involved in vascular cognitive impairment seen previously in our studies during mild-to-moderate TBI.
Keywords: Cortical contusion injury, Fg-ASO, Fg-PrPC complex, short-term memory
1. Introduction
Mild traumatic brain injury (TBI) is a common neurological disorder and increasing health problem worldwide. Among the many examples of devastating clinical afflictions that results from mild TBI in patients are a decline in cognitive function and loss of memory (Cernich et al., 2010; Till et al., 2008). In particular, short term memory (STM) reduction is one of the problems associated with head injury in patients (Chuah et al., 2004). Transient long-term memory and longer lasting STM impairments are found during mild TBI (Gorman et al., 1993). It has been shown that STM deficits persist for a long time in patients with mild to moderate TBI (Chuah et al., 2004). Precise mechanisms of this STM reduction during mild head trauma are not clear. Lately, great attention has been focused on studies related to the vascular contributions to cognitive impairment (Corriveau et al., 2016; Gorelick Philip et al., 2011; Muradashvili et al., 2012a; Muradashvili and Lominadze, 2013; Muradashvili et al., 2016; Murphy et al., 2016).
It has been shown that in a mouse model of mild-to-moderate TBI, there are no ruptured microvessels nine days after head injury (Pleasant et al., 2011). Despite the intact microvessels, we found that systemic inflammation induced by TBI results in blood proteins traversing vascular walls (Muradashvili et al., 2015; Muradashvili et al., 2017). Most notably, we found that fibrinogen (Fg), a protein that is exclusively synthesized in hepatocytes (Fuller and Zhang, 2001; Vasse et al., 1996), extravasates and deposits in vasculo-astrocyte endfeet interface 14 days after cortical contusion injury (CCI) (Muradashvili et al., 2017). Fg/fibrin-containing plaque formations are the hallmark of neurodegenerative diseases associated with memory impairment such as Alzheimer’s disease (AD) (Ahn et al., 2010), multiple sclerosis (MS) (Vos et al., 2005), and TBI (Hay et al., 2015). Fibrin deposits were found many years after head injury in humans (Hay et al., 2015). Thus, our findings of TBI-induced Fg extravasation could be a causative explanation of the Fg/fibrin deposits present in the brains of trauma patents indicated above.
The high level of blood plasma protein Fg, called hyperfibrinogenemia (HFg), is both a marker of inflammation (Ross, 1999) and a cause of inflammatory responses (Kerlin et al., 2004; Muradashvili et al., 2011; Muradashvili et al., 2012a; Patibandla et al., 2009; Tyagi et al., 2008). It is known that the level of Fg in blood increases after local vascular injury (del Zoppo et al., 2009) and particularly during TBI (Muradashvili et al., 2015; Pahatouridis et al., 2010; Sun et al., 2011). It has been shown that the blood content of Fg remains elevated for more than 20 days after inflammatory insult (Gabay and Kushner, 1999). Since Fg synthesis exclusively occurs in hepatocytes with plasma half-life of 3–4 days (Martinez et al., 1974), its appearance in extravascular spaces can only occur due to increased vascular wall permeability. We found that increased cerebrovascular permeability during TBI results in enhanced deposition of Fg in extravascular space of the brain (Muradashvili and Lominadze, 2013; Muradashvili et al., 2017). Moreover, at an elevated level, Fg itself is the main cause of increased cerebrovascular permeability (Muradashvili et al., 2012a; Muradashvili et al., 2012b; Muradashvili and Lominadze, 2013; Muradashvili et al., 2014b; Muradashvili et al., 2016; Muradashvili et al., 2018).
Cellular Prion Protein PrPC is a cell surface, glycosylphosphatidylinositol-anchored glycoprotein, abundantly expressed in the nervous system including neurons (Westergard et al., 2007), glial (Westergard et al., 2007) and endothelial (Starke et al., 2002) cells. PrPC is involved in several distinct cellular phenomena, such as neurogenesis and neural differentiation (Westergard et al., 2007), cell signaling, changes in neuronal plasticity (Kanaani et al., 2005; Lauren et al., 2009), memory consolidation (Coitinho et al., 2007), as well as protection against both oxidative stress and apoptosis (Hetz et al.; Westergard et al., 2007). PrPC exhibits dual effects in the brain. In its native state, it provides neuroprotective effects, while when ligated, it results in neurotoxicity (Onodera, 2017). It has been shown that ligated PrPC induces production of reactive oxygen species, which results in neuronal and other brain cell toxicity and oxidative stress (Schneider et al., 2003). Thus it is not surprising that alterations in this protein should affect cognitive processes and result in memory reduction (Chung et al., 2010; Coitinho et al., 2007; Fischer et al., 2000; Zhou and Liu, 2013). It is known that the amyloid beta (Aβ) peptide is linked to AD severity. However, some studies indicate that the content of Aβ has limited effects on memory and point to a greater role of PrPC (Chung et al., 2010; Gimbel et al., 2010). It has been shown that PrPC is involved in TBI-associated memory reduction (Rubenstein et al., 2017).
Our previous study has shown that TBI-induced deposition of PrPC in brain tissue was enhanced by and was co-localized with extravasated Fg (Muradashvili et al., 2015) that was immobilized in the vasculo-astrocyte interface after CCI (Muradashvili et al., 2017). Lately, we showed a specific association of Fg and PrPC protein on astrocyte cell surface suggesting ligation of PrPC (Charkviani et al., 2020). All these effects were associated with the memory reduction following head injury in mice (Muradashvili et al., 2015; Muradashvili et al., 2017). Combined these results indicate neurotoxic effects of the ligated PrPC during TBI.
In this study, we aimed to define if a reduction of the blood content of Fg would decrease cerebrovascular permeability and ameliorate memory reduction during TBI in mice. To decrease Fg levels in blood we used Fg antisense oligonucleotide (Fg-ASO) that specifically decreases synthesis of Fg in mice (Yuasa et al., 2015).
2. Results
2.1. Fg-Aso reduced TBI-induced inflammation in brain.
Expression of ATF3, a marker of inflammation of brain cells, was increased in mice treated with control-ASO after CCI (Figure 1). In mice treated with Fg-ASO after CCI, expression of ATF3 was significantly reduced compared to that in mice treated with control-ASO after CCI (Figure 1). However, ATF3 expression was still greater in animals with CCI treated with Fg-ASO than that in sham-operated mice (Figure 1). Mann-Whitney statistical test was used to define the difference between the groups in this study.
Figure 1. Gene expression of Activating Transcription Factor 3 (ATF3) in brain of mice treated with fibrinogen antisense oligonucleotide (Fg-ASO).
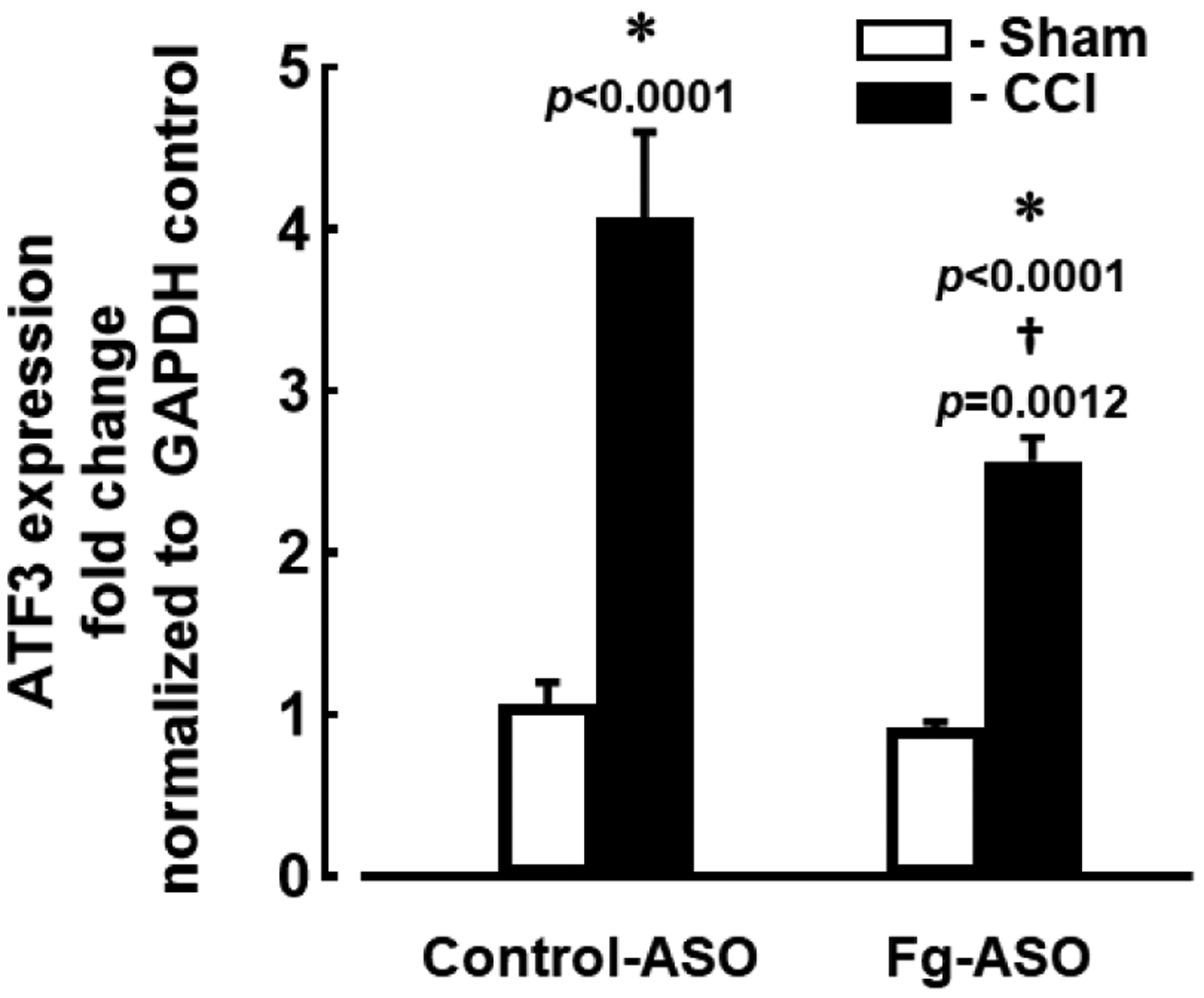
Quantitative PCR assessment of messenger RNA (mRNA) transcripts encoding ATF3 from brain samples of mice with CCI and sham-operation groups treated with control-ASO or Fg-ASO. Levels of ATF3 mRNA were assessed 14 days after CCI and presented as the fold change of the gene level over the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
P < 0.05 in all; * - vs. Sham, † - vs. CCI control-ASO; n=8/group. Mann-Whitney statistical test was used to define the difference between the groups.
2.2. Fg-ASO decreased cerebrovascular protein permeability.
14 days after CCI, pial venular permeability to Alexa Fluor 488-labelled BSA was significantly lower in mice treated with Fg ASO compared to that in mice given control-ASO (Figure 2). As expected, TBI resulted in greater cerebrovascular permeability in mice treated with control-ASO (Figure 2). Mann-Whitney statistical test was used to define the difference between the groups in this study.
Figure 2. Cerebrovascular permeability in mice treated with fibrinogen antisense oligonucleotide (Fg-ASO) after CCI.
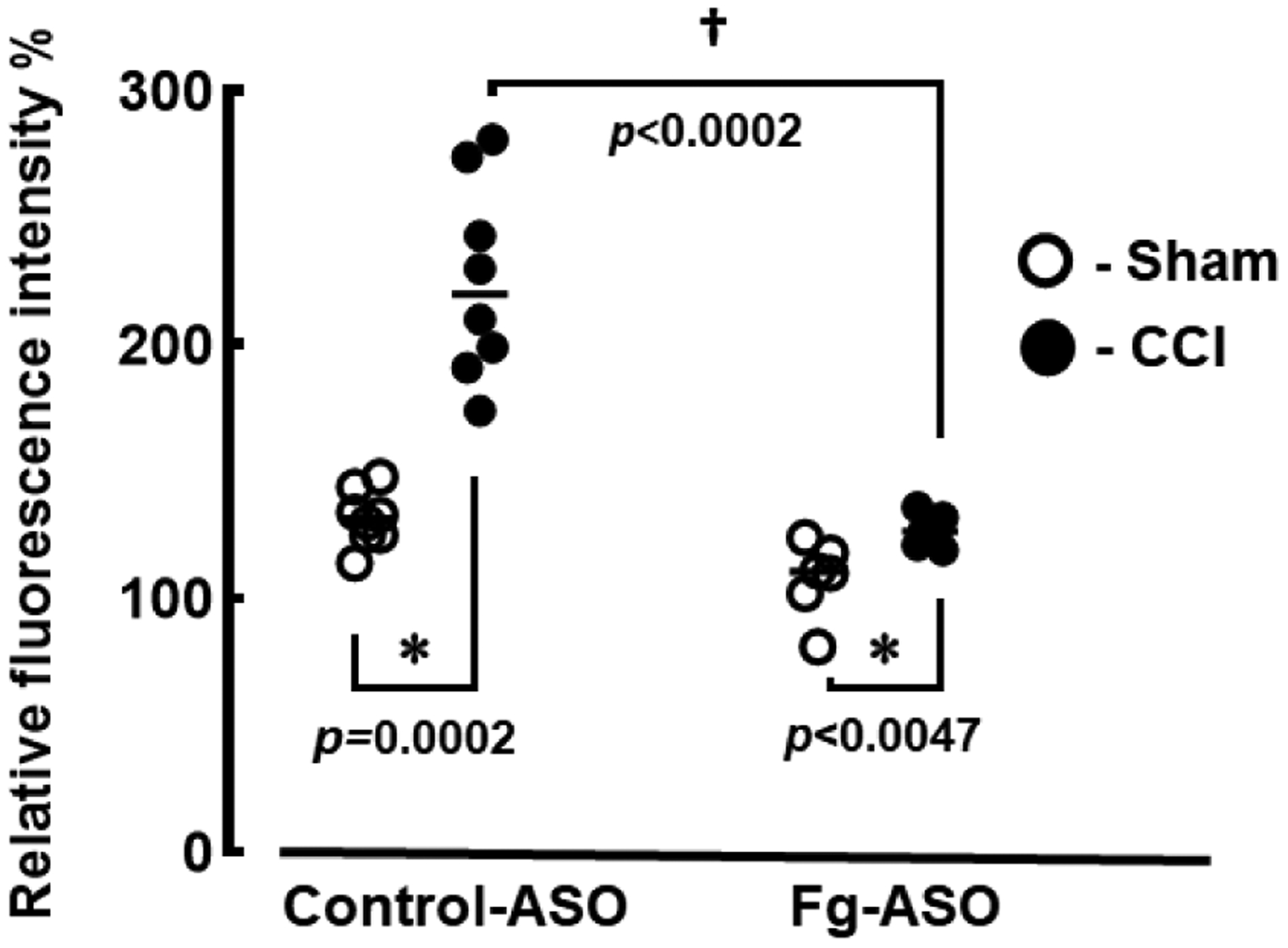
Pial venular permeability to bovine serum albumin (BSA) Alexa Fluor-488 fourteen days after CCI or sham-operation was assessed by comparison of ratios of fluorescence intensities of the dye measured along the line profile probe placed on a selected place outside of the vessel to that measured along the line profile placed inside of the vessel. Results are presented as a percent to baseline.
P<0.05 in all; * - vs. Sham, † - vs. CCI control-ASO; n=6/group. Mann-Whitney statistical test was used to define the difference between the groups.
2.3. Expression of PrPC in brains of Fg-ASO-treated mice after CCI.
Western blot analysis of mouse brain samples showed greater expression of brain PrPC after CCI when compared to that in the sham group of mice treated with control-ASO (Figure 3). Treatment with Fg-ASO significantly reduced PrPC expression in brains of both sham-operated mice and mice with TBI compared to their respective controls (Figure 3). No difference was found in expression of PrPC between sham-operated mice and mice with CCI treated with Fg-ASO. (Figure 3b). Kruskal-Wallis statistical test followed by Dunn’s multiple comparison post-hoc correction test were used to define the difference between the groups in this study.
Figure 3. Expression of cellular prion protein (PrPC) in mouse brain treated with fibrinogen antisense oligonucleotide (Fg-ASO) after CCI.
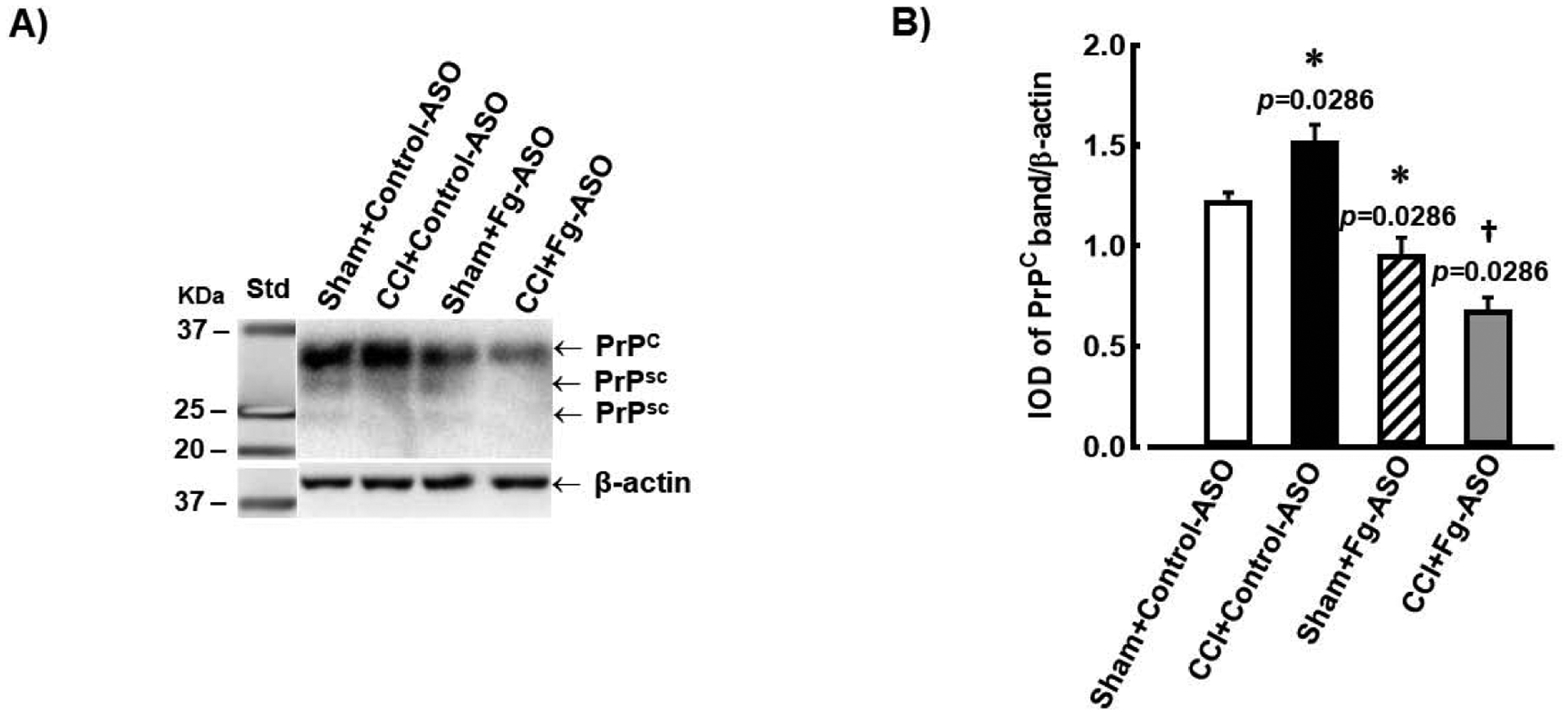
A) Examples of Western blot analyses images of PrPc expression in brain samples from mice treated with control-ASO or Fg-ASO undergone sham-operation or CCI. Samples were collected 14 days after CCI or sham-operation. An equal amount of protein was placed in each well of the SDS gel. Molecular weight standard (Std) is presented in the left lane. Twenty-seven and 22 kD scrape prion proteins (PrPSC), which reflect changes of the PrPC, are also visible
B) Data analysis of PrPC expression is shown. Relative protein expression in samples is presented as a ratio of integrated optical density (IOD) of each PrPC band to the IOD of the β-actin band in respective lane.
P<0.05 in all; * - vs. Sham Control-ASO, † - vs. CCI Control-ASO, n=4/group. Kruskal-Wallis statistical test followed by Dunn’s multiple comparison post-hoc correction test were used to define the difference between the groups.
2.4. Formation of Fg-PrPC complexes.
There was increased deposition of Fg and increased expression of PrPC in the pericontusional area of brain after CCI in mice treated with control-ASO compared to those of mice with sham-operation that were treated with control-ASO (Figure 4). Similarly, the number of spots of co-localized Fg and PrPC and thus, possible formation Fg-PrPC complexes along the Lycopersicon esculentum lectin-marked vessels were also greater in brains of mice with CCI treated with control-ASO (Figure 4). Treatment of mice with Fg-ASO significantly reduced deposition of Fg, expression of PrPC, and number of spots with co-localized Fg and PrPC in mice with CCI when compared to those in CCI mice treated with control-ASO (Figure 4). Mann-Whitney statistical test was used to define the difference between the groups in this study.
Figure 4. Deposition of Fg and expression of PrPC, and their co-localization in brain of mice treated with fibrinogen antisense oligonucleotide (Fg-ASO) after CCI.
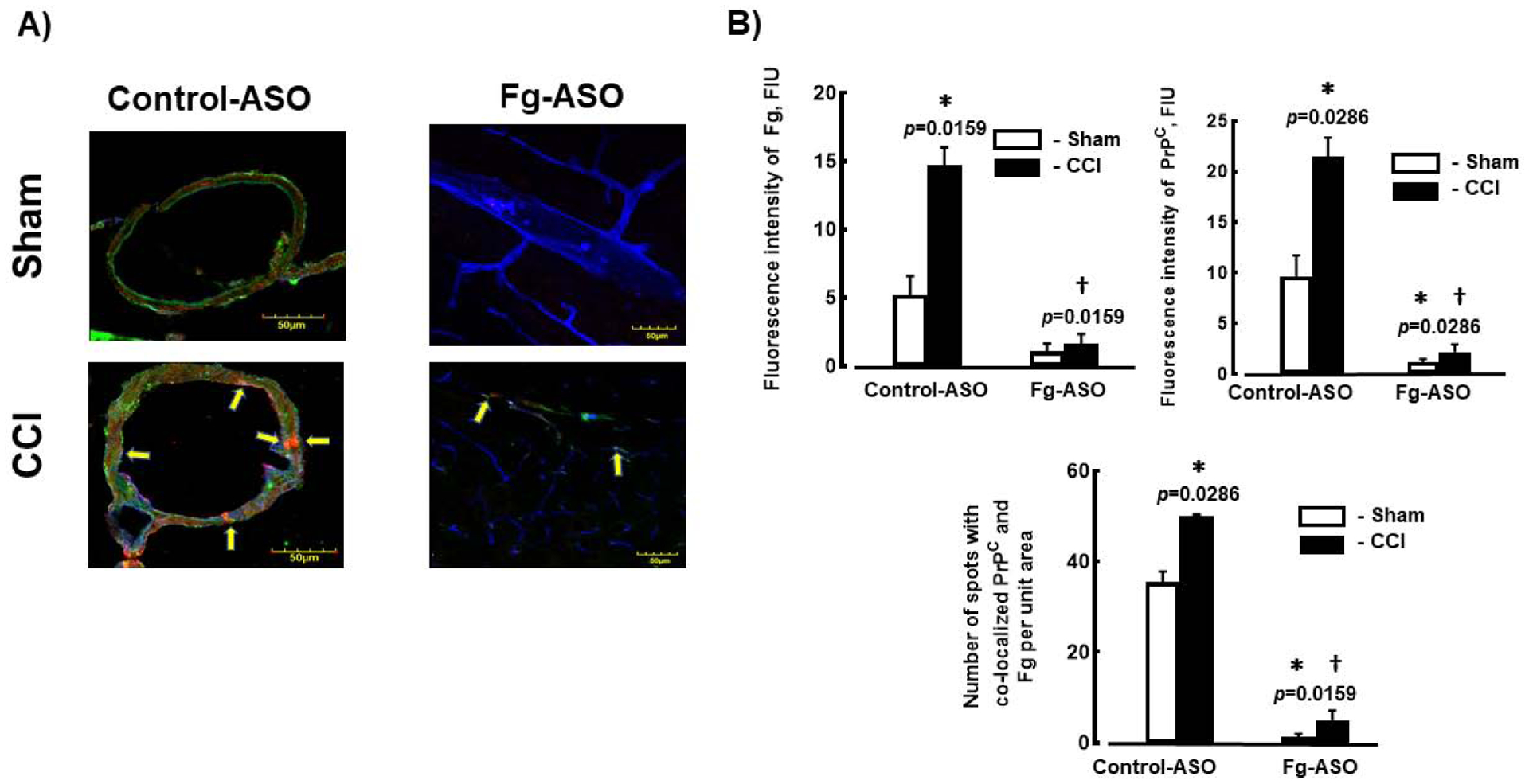
A) Examples of images showing expression of PrPC (green) and deposition of Fg (red), and their co-localization in mouse cortical vessels. Lycopersicon esculentum lectin, a marker of endothelium is shown in blue. Samples were collected 14 days after CCI or sham-operation.
B) Data analysis of fluorescence intensities of Fg and PrPC (presented as fluorescence intensity) are shown in the upper two plots. Co-localization of Fg and PrPC was assessed as a number of spots of co-localized respective fluorophores.
P<0.05 in all; * - vs. Sham Control-ASO, † - vs. CCI Control-ASO n=4/group. Mann-Whitney statistical test was used to define the difference between the groups.
2.5. Fg-ASO improved short-term memory (STM) deficit after CCI.
Both NORT and SAT (Y-maze test) showed significant improvement of STM in mice with CCI after treatment with Fg-ASO (Figure 5). Mann-Whitney statistical test was used define the difference between the groups in this study.
Figure 5. Short-term memory (STM) changes in mice treated with control-ASO or Fg-ASO after CCI.
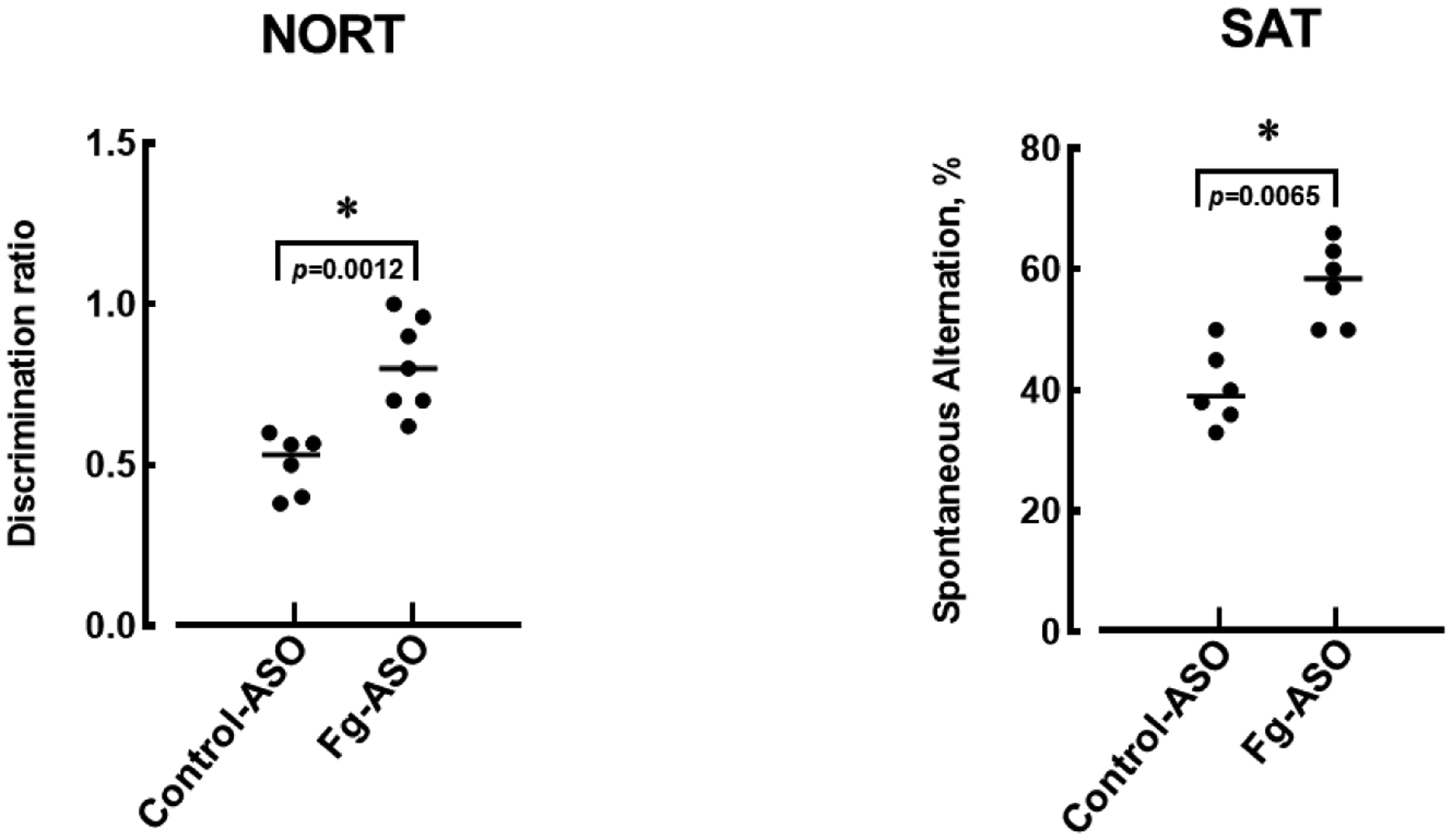
Changes in STM were assessed in mice 14 days after CCI or sham-operation using a novel object recognition test (NORT) and a Y-maze spontaneous alternation test (SAT). For STM assessment by NORT, discrimination ratio was calculated as a ratio of the time spent at the novel object to the time spent at both objects. The SAT was calculated as a ratio of actual alternations to possible alternations (the total number of arm entries minus two) and presented as a percentage.
P < 0.05 vs. Control-ASO; n=6/group. Mann-Whitney statistical test was used to define the difference between the groups.
3. Discussion
In several studies, blood levels of Fg was reduced with ancrod, a defibrinogenating agent derived from the venom of the Malayan pit viper, which did have some valuable results (del Zoppo et al., 2009; Paul et al., 2007; Schachtrup et al., 2010). However, it is known that ancrod decreases levels of most high molecular weight proteins such as fibronectin, von Willebrand factor, globulins, and various others. Therefore, ancrod cannot be considered as an agent specifically targeting blood level of Fg. Use of fenofibrate (brand name Tricor) in rodents have been shown to suppress Fg gene expression in liver and decrease the Fg level in blood (Kockx et al., 1999). However, it is mainly used to reduce low density lipoproteins and triglycerides (Genest et al., 2000) and thus, cannot be considered as a specific inhibitor of Fg synthesis. The majority of studies concerning Fg’s effects used models of reduced blood levels of Fg, such as Fg gene knock out (Fg−/−) mice (Ni et al., 2000; Wilberding et al., 2001). The results show that other proteins, e.g. fibronectin, can “assume” role of Fg in coagulation and/or thrombogenesis (Ni et al., 2000). Thus, all these manipulations with blood content of Fg mask its effects in circulation. On the other hand, Fg-ASO specifically and effectively decreases synthesis of Fg, and thus, blood level of Fg in mice (Yuasa et al., 2015).
It is well known that TBI is associated with HFg and the development of inflammation (Jenkins et al., 2018; Muradashvili and Lominadze, 2013; Sun et al., 2011). Our result showed that CCI induced expression of ATF3, a marker of inflammation. ATF3 is a member of the ATF/cAMP responsive element binding protein family that is typically greatly increased when cells are exposed to stress signals (Hai et al., 1999). Increase in ATF3 gene expression after CCI suggests that developed of TBI is accompanied with inflammation, which results in HFg and lead to enhanced Fg transcytosis. Our finding that treatment of mice with Fg-ASO after CCI caused lesser expression of ATF3 in brain, suggests that a decrease in Fg synthesis ameliorates inflammation. However, expression of ATF3 in mice with CCI was still greater than in sham-operated mice even after treatment with Fg-ASO. The difference in ATF3 gene expression levels in mice with CCI treated with control-ASO and Fg-ASO would indicate a level of Fg involvement in reduction of overall inflammation caused by TBI. In other words, the difference in ATF3 gene expressions between sham-operated animals and mice with CCI treated with Fg-ASO would point to the level of inflammation due to other factors than blood content of Fg.
We have previously shown that TBI-induced elevated blood levels of Fg caused a significant increase in pial venular permeability in WT mice (Muradashvili et al., 2012a). The changes in cerebrovascular permeability seen in our present study are perfect examples of inflammatory responses in this TBI model. It is well accepted that an increase in vascular permeability is a direct indication of inflammation (Mehta and Malik, 2006). In the same study, we also showed that the pial venular permeability 14 days after CCI was significantly lowered with Fg-ASO treatment. Thus, these data suggest a strong mechanistic role of Fg in cerebrovascular permeability and in inflammation. This confirms our hypothesis that CCI induced macromolecular traversing of vascular wall could be reduced by Fg-ASO resulting in reduction of blood content of Fg. The effect of reduced venular permeability potentially decreases the deposition of Fg in the extravascular space. Our previous data indicated that at a high level, Fg was involved in overexpression of PrPC and formation Fg-PrPC complexes (Muradashvili et al., 2015). Recently, we confirmed a strong association of Fg and astrocyte PrPC, indicating that PrPC can be ligated on the surface of activated astrocytes resulting in neurotoxicity (Charkviani et al., 2020). However, it has never been shown that the reduction of blood content of Fg would affect expression of PrPC and Fg-PrPC complex formations. Results reported here confirmed our previous findings that there is a greater deposition of Fg in the vascular and astrocyte endfeet interface in mice with TBI (Muradashvili et al., 2017) and greater deposition of PrPC that was associated with HFg (Muradashvili et al., 2016). In the current study, we found that the increased deposition of Fg, expression of PrPC, and formation of Fg-PrPC complexes observed during CCI (thus, during HFg) were significantly reduced with the use of Fg-ASO indicating a significant involvement of Fg in impairment of cerebrovascular function. Western blot results were confirmed with IHC experiments where a greater expression of PrPC protein was seen in mouse brains after CCI compared to that in sham-operated mice. These effects were significantly reduced in mice receiving Fg-ASO.
Results of the present study indicate that a decrease in Fg synthesis reduced not only expression of PrPC and formation of Fg-PrPC complexes, but also improved TBI-induced changes in STM. PrPC is known to be involved in memory reduction (Chung et al., 2010; Gimbel et al., 2010). It has been found that PrPC affects brain cell-redox homeostasis through reactive oxygen species (ROS) production (Schneider et al., 2003). In addition, toxic effects of ligated PrPC have been widely recognized (Onodera, 2017). We have previously shown that the increased deposition of Fg and expression of PrPC leading to possible a Fg-PrPC complex formation have been associated with reduced STM during HFg (Muradashvili et al., 2016) and TBI (Muradashvili et al., 2015). All these effects were correlated with neurodegeneration and memory reduction during TBI (Muradashvili et al., 2017). Therefore, if during the course of TBI there was a reduced level of Fg, it would result in lesser vascular permeability and lesser extravasation and deposition of Fg in extravascular space. These effects would diminish expression of PrPC and the resultant formation of Fg-PrPC complexes, which can result in lesser detriments in STM. Thus, our data indicate that Fg has a significant role in development of inflammation and the resultant inflammatory responses, such as increased cerebrovascular permeability, formation of Fg-PrPC complexes, generation of ROS, and reduction of STM during TBI. These results suggest that Fg could be considered as a potential target for therapy during TBI to mitigate or prevent vascular cognitive impairment.
4. Methods and Materials
4.1. Antibodies and reagents.
Human Fg depleted of plasminogen, von-Willebrand factor, and fibronectin was purchased from Enzyme Research Laboratories (South Bend, IN). Polyclonal rabbit antibody against human Fg (which is known to cross react with mouse Fg) was purchased from Dako Cytomation (Carpentaria, CA). Mouse monoclonal anti-prion protein antibody was obtained from Sigma-Aldrich (St. Louis, MO). Secondary antibodies conjugated with Alexa-Fluor 488 or Alexa-Fluor 594 were purchased from Invitrogen (Carlsbad, CA). Bovine serum albumin (BSA) was from Sigma and 10% normal donkey serum - from Jackson Immuno Research (West Grove, PA).
4.2. Animals.
In accordance with National Institute of Health Guidelines for animal research, all animal procedures for these experiments were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Louisville and University of South Florida. Twelve-week-old male wild-type (WT) C57BL/6J mice obtained from the Jackson Laboratory (Bar Harbor, ME) were used in the study.
4.3. Fg-ASO.
Fg-ASO (GCTTTGATCAGTTCTTTGGC), a gift from Ionis pharmaceuticals (Carlsbad, CA), was used to transiently decrease synthesis of Fg and thus, reduce its plasma level in the mice (Yuasa et al., 2015). Control-ASO (CCTTCCCTGAAGGTTCCTCC) which has no homologue in the mouse genome was used as a control compound. Animals were given Fg-ASO and control-ASO at the dose of 40 mg/kg (intraperitoneally) post-surgery. Then identical treatments (at the same dose) were given every third day for the duration of the experiment.
4.4. Cortical contusion injury (CCI).
Mice (26–30 g) were anesthetized with 2.5% isoflurane delivered by a nose cone (Kopf, Tujunga, CA). Heads were shaved and placed in a stereotaxic frame (Kopf). Briefly, a 4 mm diameter cranial window was created in the left parietal bone, centered at 2.5 mm caudal of bregma and 2.75 mm lateral to the midline according to the method described in detail previously (Muradashvili et al., 2015; Muradashvili et al., 2017). The impactor device (TBI 0310, Precision Systems & Instrumentation, Fairfax Station, VA) with 2 mm diameter flat tip was set to deliver an impact (0.5 mm impact depth, 3.5 m/s velocity, 500 ms simulation duration) to the cortical surface which is known to cause a mild-to-moderate injury (Muradashvili et al., 2015; Muradashvili et al., 2017; Pleasant et al., 2011). In control (sham-injured) group, all procedures but the cortical impact was performed (Muradashvili et al., 2015; Muradashvili et al., 2017). After injury, the brain was briefly superfused with artificial cerebrospinal fluid (Harvard Apparatus, Holliston, MA) and the skin was sutured. Then mice were placed on a heating pad to maintain normal body temperature until they were fully awake and returned to their home cages.
4.5. Cerebrovascular permeability assessment.
Pial venular permeability to BSA conjugated with Alexa-488 dye was performed as described previously (Muradashvili et al., 2014b; Muradashvili et al., 2015; Muradashvili et al., 2016). Briefly, after the mice were surgically prepared, 1 hour of equilibration period was given before microvascular permeability observation was performed. Initial autofluorescence of the observed area was recorded over a standard range of camera gain. The brain vasculature was observed using an epi-illumination system of an electron-multiplying charge-coupled device camera (Olympus BX61W1, Tokyo, Japan) and image acquisition system (Olympus cellSens Dimension Desktop 2.3, Tokyo, Japan). A mixture of 100μl of Fluor (300μg/ml) and 20 μl of BSA-Alexa Fluor-488 (3.3 mg/ml) in phosphate-buffered saline (PBS) was infused through the cannulated carotid artery with a syringe pump (Harvard apparatus) at 30μl/min. The solution was infused to circulate for 10 minutes and the pial circulation was observed to ensure there was no spontaneous leakage of the fluorophore-conjugated BSA. Pial venules were identified by the topology of the circulation and blood flow direction using the microscope. Selected third order venular segments were imaged and recorded. Readings were recorded at baseline, 10, 20, 40, 60 and 90 minutes. The area of interest was exposed to blue light (488 nm) with a power density of 3.5μW/cm2 to observe intravascular and extravascular BSA-Alexa Fluor-488. The camera output was standardized with a 50ng/nl fluorescein diacetate standard (Estman Kodak, Rochester, NY, USA). During the experiment, the lamp power and camera gain settings were kept constant and the camera response was verified for its linearity over the range used for these acquisitions. Cerebrovascular segment was observed with Olympus x20/0.40 (UPlanSApo) objective. Image analysis was performed with commercial analysis software (Image-Pro Plus 7.0, Media Cybernetics, Bethesda, MD, USA). A 30μm length line profile probe was positioned in the middle of the selected segment of the venule and outside of the venular wall, parallel to the vessel. Mean fluorescence intensities along the probes were measured. Extravasation of the BSA-Alexa-488 was assessed by changes in the ratio of fluorescence intensity in the interstitium to that inside the venule and calculated as a percent of baseline. Results were averaged for each experimental group.
4.6. Collection and preparation of mouse brain samples.
Fourteen days after CCI animals were exsanguinated and brain samples were collected for Western blot analysis as described previously (Muradashvili et al., 2014a; Muradashvili et al., 2015; Muradashvili et al., 2016). Briefly, whole brain samples were digested in radioimmunoprecipitation assay (RIPA) buffer (1 g of tissue/2 ml of RIPA) in the presence of protease inhibitor cocktail (Thermo Fisher Scientific). The samples were homogenized using Zirconium Beads and Beadmaster-24 homogenizer (Worldwide Life Science Division; Bristol, PA). After homogenization, samples were centrifuged at 7000g for 10 min at 4°C. The supernatant was collected, and total protein content was determined by the Bradford method.
4.7. Activating transcription factor 3 (ATF3) gene assessment.
To define the role of Fg in development of CCI-induced inflammation, we assessed gene expression of the inflammation marker ATF3 (Greer et al., 2011; Hai et al., 1999) in brain samples from mice with CCI or sham-operation treated with control-ASO or Fg-ASO. Total RNA was extracted from mice whole brain samples using a Trizol method according to the manufacturer’s instruction (Invitrogen Life Technologies, Carlsbad, CA, USA). Then, RNA quality was determined, by NanoDrop ND-1000, and RNA with high purity (260/280~2.00 and 260/230~2.00) was used for q-PCR analysis. RT-PCR was performed by as described previously using ImProm-II™ Reverse Transcription kit (Promega Corporation, Madison, WI). Complimentary DNA (cDNA) was prepared using 2 μg of the RNA. A reverse transcription program on the DNA Engine Peltier Thermal Cycler (Bio-Rad Laboratories, Hercules, CA) was used to synthesize cDNA. The program consisted of a denaturing cycle at 70°C for 6 min and then a reverse transcription cycle at 25°C for 2 min, 42°C for 50 min, and 75°C for 5 min. For qPCR, SYBR green-based kit was used to measure the relative expression of each mRNA specific primers (Förstner et al., 2018). A three-step cycling protocol was performed using 20 ng of cDNA template in a 20 μL reaction volume under the following conditions: denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 55°C for 1min, and 72°C for 34 s, in which fluorescence was acquired and detected by Roche LightCycler® 96 Real Time PCR System (Roche Diagnostics, IN). Following RT-qPCR, analysis of the melt curve was-performed to validate the specific generation of the expected PCR product. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an endogenous control (Quanta Biosciences, Beverly, MA). Although others have shown GAPDH expression changes dependent upon post-injury time and severity of the TBI (Zamani et al., 2020), in our study all the experimental animals received head trauma of a similar level of severity and were observed at the same time point after the injury. Therefore, in our study, expression of GAPDH would not be altered between the groups and thus, can be used as a reliable housekeeping protein. Moreover, the stability of GAPDH as a reference gene to efficiently normalize qRT-PCR gene expression in mouse model of TBI has been validated in work by others (Rhinn et al., 2008). The CT values were determined after baseline and threshold adjustment and the results are expressed in fold expression. The sequence of primers for real time PCR were for ATF3 Fwd-5’-GCT GGA GTC AGT TAC CGT CAA-3’; Rev-5’-CGC CTC CTT TTC CTC TCA T-3’ and for GAPDH Fwd-5′-TGGCAAAGTGGAGATTGTTGCC-3′ and Rev-5′-AAGATGGTGATGGGCTTCCCG-3′.
4.8. Immunohistochemistry (IHC).
Mouse brain tissue IHC was performed as previously described (Muradashvili et al., 2012a; Muradashvili et al., 2014c; Muradashvili et al., 2015; Muradashvili et al., 2016; Muradashvili et al., 2017). The investigator was blinded to the tissue sample sources. Briefly, deeply anesthetized mice were infused with fluorophore-conjugated Lycopersicon esculentum lectin via the carotid artery cannulation. Lycopersicon esculentum lectin was used to label the intravascular endothelial surface (Muradashvili et al., 2012a). Mice were exsanguinated by infusion of PBS followed by 4% paraformaldehyde (PFA) through the left ventricle. The brain samples were harvested and preserved in 4% PFA solution overnight prior to being kept in 30% sucrose for 3 days. Brain samples were mounted in protective matrix (Polyscience, Inc, Warrington, PA) and cryosectioned through coronal plane at 15μm thickness, placed on microscope slide, and stored at −80°C. Thawed slides were heated at 37°C for 20 minute and the mounting medium was removed. Ice cold 100% methanol was used for fixation for 10 minutes before washing with Tris-buffered saline and blocking with 0.1% TritonX-100TBS (TBS-T), 0.5% BSA and 10% normal donkey serum. Tissue samples were incubated with appropriate primary antibodies overnight at 4 °C followed with washing and incubation with fluorescently labeled secondary antibodies for 2 h at room temperature. The samples were cover slipped and sealed with ProLong™ Diamond Antifade mounting media (Thermo Fisher Scientific, Waltham, MA).
4.9. Confocal Microscopy.
A laser-scanning confocal microscope (Olympus FV1200 Spectral Inverted Laser Scanning Confocal Microscope, with objective x60) was used for the detection of Fg deposition, the expression of PrPC, and their co-localization in order to define the possible formation of Fg-PrPC complexes. The general area of interest was defined as the area of pial vasculature located at least 200μm away from the injury and no deeper than 200 μm from the brain surface. PrPC (the antibody was conjugated with Alexa-488 dye) was visualized using a multiline argon-ion laser (458/488/515 nm) to excite the dye, while light emission was observed above 519 nm. Fg (the antibody was conjugated with Alexa-594 dye) was visualized using a HeNe-Green laser (543 nm) to excite the dye, while light emission was observed above 620 nm.
4.10. Image Analysis
Expressions of Fg and PrPC were assessed for each experimental group using offline image analysis software, Image-Pro Plus (Media Cybernetics, Silver Spring, USA) as previously described (Muradashvili et al., 2012a; Muradashvili et al., 2014c; Muradashvili et al., 2015; Muradashvili et al., 2017). Fluorescence intensities of Fg or PrPC were measured in at least 4 randomly placed areas of interest of constant size along the vessel wall and normalized per length of the respective vascular segment. The results were averaged for each experimental group and presented as fluorescence intensity units. To identify co-localization of Fg with PrPC, the images were deconvoluted and “masks” were formed which demonstrated overlap of different colors as previously described (Muradashvili et al., 2014a; Muradashvili et al., 2017). The numbers of objects co-localized with fluorophores were counted.
4.11. Western Blot Analysis.
After assessing the total protein level in brain samples, equal volume (30 μl) of protein from each sample was loaded onto 10% SDS-PAGE gels, electrophoresed under reducing conditions, and then transferred onto polyvinylidene difluoride (PVDF) membranes. Non-specific sites on membranes were blocked with 5% nonfat dry milk in TBS-T and membranes were incubated with antibodies against target proteins overnight at 4°C. After probing with appropriate secondary antibodies for 2 hours at room temperature, the blots were visualized using a BioRad Molecular Imager (ChemiDoc XRS+, Hercules, CA). Then, blots were stripped with blot stripping buffer (RPI, Mt. Prospect, IL) and probed for the content of β-actin that was used as a loading control. Blot were visualized again. Obtained blot images were analyzed with Image Pro Plus. Contents of target proteins were assessed by measuring an integrated optical density (IOD) of their bands in each sample lane profile and presented relative to the IOD of the respective β-actin bands.
4.12. Memory assessment.
To investigate the effect of Fg content on memory-related behavior of the mice, a novel object recognition test (NORT) and a Y-maze spontaneous alternation test (SAT) were performed as we have described previously (Muradashvili et al., 2015; Muradashvili et al., 2016; Muradashvili et al., 2017). Behavioral tests were done by a person who was blinded to the animal conditions.
4.12.1. NORT was used to assess visual STM.
The mice were acclimatized and trained for two days prior the test for 10 minutes, twice a day. On the day of the test, each mouse was placed in the box at the mid-point of the wall opposite to two similar objects and allowed to investigate the objects for 5 minutes. After 1 hour, one of the objects was replaced with a new, different-shaped object and the animal was returned to the box for 3 min. TopScan behavioral analyzing system (Version 3.00; Clever Sys Inc., Reston, VA, USA) was used for the recording and analysis of mice behavior. A discrimination ratio was calculated as a ratio of the time spent at the novel object to the time spent at both objects.
4.12.2. SAT was used to assess spatial working STM.
This test is based on the concept of determining the willingness of rodents to explore new environment and thus their inquisitive nature to investigate a new arm of the maze. Age-matched mice were placed in the middle of Y-maze with all three arms opened and all arm entries were sequentially recorded for 8 minutes. The SAT was calculated as a ratio of actual alternations to possible alternations (the total number of arm entries minus two) and presented as a percent (Jadavji et al., 2015).
4.13. Data Analysis.
Data in Figures 1, 3 and 5 were presented as mean ± standard error of the mean while data in Figures 2 and 5 were presented as a scatter dot plot with a median line. The experimental groups were compared by Mann-Whitney or Kruskal-Wallis statistical test followed by Dunn’s multiple comparison post-hoc correction test, unless otherwise indicated. Differences were considered statistically significant at p < 0.05. Analyses were performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA. We used a blind study approach where it was possible. In vitro samples were coded. Experimental animals were also coded for behavioral tests. The decoding of data was done at the end the statistical analysis. An investigator was blinded while analyzing images for cerebral permeability.
Highlights.
Decrease of Fg synthesis reduced TBI-induced inflammation in brain
Decrease of Fg synthesis reduced expression of PrPC and formation of Fg-PrPC complex
Decrease of Fg synthesis improved TBI-induced changes in STM
Acknowledgements
This work was supported in part by funding from the National Institutes of Health (NS-084823 and HL-146832) and the American Heart Association (17SDG33670372).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Jeff Crosby is an employee of Ionis Pharmaceutical.
References
- Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S, 2010. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci USA. 107, 21812–21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernich A, Kurtz S, Mordecai K, Ryan P, 2010. Cognitive rehabilitation in traumatic brain injury. Current Treatment Options in Neurology. 12, 412–423. [DOI] [PubMed] [Google Scholar]
- Charkviani M, Muradashvili N, Sulimai NH, Lominadze D, 2020. Fibrinogen - Cellular Prion Protein Complex Formation on Astrocytes. Journal of Neurophysiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuah YML, Maybery MT, Fox AM, 2004. The long-term effects of mild head injury on short-term memory for visual form, spatial location, and their conjunction in well-functioning university students. Brain and Cognition. 56, 304–312. [DOI] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak R, Kascsak R, Mehta P, Strittmatter S, Wisniewski T, 2010. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 11, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coitinho AS, Lopes MH, Hajj GNM, Rossato JI, Freitas AR, Castro CC, Cammarota M, Brentani RR, Izquierdo I, Martins VR, 2007. Short-term memory formation and long-term memory consolidation are enhanced by cellular prion association to stress-inducible protein 1. Neurobiology of Disease. 26, 282–290. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Bosetti F, Emr M, Gladman JT, Koenig JI, Moy CS, Pahigiannis K, Waddy SP, Koroshetz W, 2016. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A Framework for Advancing Research Priorities in the Cerebrovascular Biology of Cognitive Decline. Cellular and Molecular Neurobiology. 36, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Levy DE, Wasiewski WW, Pancioli AM, Demchuk AM, Trammel J, Demaerschalk BM, Kaste M, Albers GW, Ringelstein EB, 2009. Hyperfibrinogenemia and functional outcome from acute ischemic stroke. Stroke. 40, 1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MB, Roeckl C, Parizek P, Schwarz HP, Aguzzi A, 2000. Binding of disease-associated prion protein to plasminogen. Nature. 408, 479–483. [DOI] [PubMed] [Google Scholar]
- Förstner P, Rehman R, Anastasiadou S, Haffner-Luntzer M, Sinske D, Ignatius A, Roselli F, Knöll B, 2018. Neuroinflammation after traumatic brain injury is enhanced in activating transcription factor 3 mutant mice. Journal of Neurotrauma. XX, 1–13. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Fuller GM, Zhang Z, 2001. Transcriptional control mechanism of fibrinogen gene expression. Ann N Y Acad Sci. 936, 469–79. [DOI] [PubMed] [Google Scholar]
- Gabay C, Kushner I, 1999. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 340, 448–454. [DOI] [PubMed] [Google Scholar]
- Genest JJ, Nguyen N-H, Theroux P, Davignon J, Cohn JS, 2000. Effect of Micronized Fenofibrate on Plasma Lipoprotein Levels and Hemostatic Parameters of Hypertriglyceridemic Patients with Low Levels of High-Density Lipoprotein Cholesterol in the Fed and Fasted State. Journal of Cardiovascular Pharmacology. 35, 164–172. [DOI] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM, 2010. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. The Journal of Neuroscience. 30, 6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick Philip B, Scuteri A, Black S, Decarli C, Greenberg S, Iadecola C, Launer L, Laurent S, Lopez O, Nyenhuis D, Petersen R, Schneider J, Tzourio C, Arnett D, Bennett D, Chui H, Higashida R, Lindquist R, Nilsson P, Roman G, Sellke F, Seshadri S, 2011. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 42, 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman L, Shook B, Becker D, 1993. Traumatic brain injury produces impairments in long-term and recent memory. Brain research. 614, 29–36. [DOI] [PubMed] [Google Scholar]
- Greer JE, McGinn MJ, Povlishock JT, 2011. Diffuse Traumatic Axonal Injury in the Mouse Induces Atrophy, c-Jun Activation, and Axonal Outgrowth in the Axotomized Neuronal Population. The Journal of Neuroscience. 31, 5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U, 1999. ATF3 and stress responses. Gene Expression. 7, 321–335. [PMC free article] [PubMed] [Google Scholar]
- Hay J,R, Johnson VE, Young AM, Smith DH, Stewart W, 2015. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. Journal of Neuropathology and Experimental Neurology. 74, 1147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Maundrell K, Soto C, 2003. Is loss of function of the prion protein the cause of prion disorders? Trends in Molecular Medicine. 9, 237–243. [DOI] [PubMed] [Google Scholar]
- Jadavji NM, Deng L, Malysheva O, Caudill MA, Rozen R, 2015. MTHFR deficiency or reduced intake of folate or choline in pregnant mice results in impaired short-term memory and increased apoptosis in the hippocampus of wild-type offspring. Neuroscience. 300, 1–9. [DOI] [PubMed] [Google Scholar]
- Jenkins DR, Craner MJ, Esiri MM, DeLuca GC, 2018. The contribution of fibrinogen to inflammation and neuronal density in human traumatic brain injury. Journal of Neurotrauma. 35, 2259–2271. [DOI] [PubMed] [Google Scholar]
- Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G, 2005. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. Journal of Neurochemistry. 95, 1373–1386. [DOI] [PubMed] [Google Scholar]
- Kerlin B, Cooley BC, Isermann BH, Hernandez I, Sood R, Zogg M, Hendrickson SB, Mosesson MW, Lord S, Weiler H, 2004. Cause-effect relation between hyperfibrinogenemia and vascular disease. Blood. 103, 1728–1734. [DOI] [PubMed] [Google Scholar]
- Kockx M, Gervois PP, Poulain P, Derudas B, Peters JM, Gonzalez FJ, Princen HMG, Kooistra T, Staels B, 1999. Fibrates suppress fibrinogen gene expression in rodents via activation of the peroxisome proliferator-activated receptor-α. Blood. 93, 2991–2998. [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM, 2009. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-[bgr] oligomers. Nature. 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Holburn RR, Shapiro SS, Erslev AJ, 1974. Fibrinogen Philadelphia: A hereditary hypodysfibrinogenemia characterized by fibrinogen hypercatabolism. J.Clin.Investigation 53, 600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Malik AB, 2006. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 86, 279–367. [DOI] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi N, Tyagi R, Munjal C, Lominadze D, 2011. Fibrinogen alters mouse brain endothelial cell layer integrity affecting vascular endothelial cadherin. Biochemical and Biophysical Research Communications 413, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Qipshidze N, Munjal C, Givvimani S, Benton RL, Roberts AM, Tyagi SC, Lominadze D, 2012a. Fibrinogen-induced increased pial venular permeability in mice. J Cereb Blood Flow Metab. 32, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi R, Lominadze D, 2012b. A dual-tracer method for differentiating transendothelial transport from paracellular leakage in vivo and in vitro. Frontiers in Physiology. 3, 166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Lominadze D, 2013. Role of fibrinogen in cerebrovascular dysfunction after traumatic brain injury. Brain Injury. 27, 1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Benton R, Tyagi R, Tyagi S, Lominadze D, 2014a. Elevated level of fibrinogen increases caveolae formation; Role of matrix metalloproteinase-9. Cell Biochem Biophys. 69, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Khundmiri SJ, Tyagi R, Gartung A, Dean WL, Lee M-J, Lominadze D, 2014b. Sphingolipids affect fibrinogen-induced caveolar transcytosis and cerebrovascular permeability. Am J Physiol Cell Physiol. 307, C169–C179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi R, Metreveli N, Tyagi SC, Lominadze D, 2014c. Ablation of MMP9 gene ameliorates paracellular permeability and fibrinogen-amyloid beta complex formation during hyperhomocysteinemia. J Cereb Blood Flow Metab. 34, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Benton RL, Saatman KE, Tyagi SC, Lominadze D, 2015. Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metabolic Brain Disease. 30, 411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi R, Tyagi N, Tyagi SC, Lominadze D, 2016. Cerebrovascular disorders caused by hyperfibrinogenemia. The Journal of Physiology. 594, 5941–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi SC, Lominadze D, 2017. Localization of fibrinogen in the vasculo-astrocyte interface after cortical contusion injury in mice. Brain Sciences. 7, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradashvili N, Tyagi SC, Lominadze D, 2018. Role of Fibrinogen in Vascular Cognitive Impairment in Traumatic Brain Injury In Traumatic Brain Injury. Vol., Gorbunov NV, Long JB, ed.êds. IntechOpen, pp. 105–119. [Google Scholar]
- Murphy M, Corriveau R, Wilcock D, 2016. Vascular contributions to cognitive impairment and dementia (VCID). Biochim. Biophys. Acta 1862, 857–859. [DOI] [PubMed] [Google Scholar]
- Ni H, Denis C, Subbarao S, Degen J, Sato T, Hynes R, Wagner D, 2000. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. Journal of Clinical Investigation. 106, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera T, 2017. Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proceedings of the Japan Academy. Series B, Physical and biological sciences. 93, 155–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahatouridis D, Alexiou G, Zigouris A, Mihos E, Drosos D, Voulgaris S, 2010. Coagulopathy in moderate head injury. The role of early administration of low molecular weight heparin. Brain Injury. 24, 1189–1192. [DOI] [PubMed] [Google Scholar]
- Patibandla PK, Tyagi N, Dean WL, Tyagi SC, Roberts AM, Lominadze D, 2009. Fibrinogen induces alterations of endothelial cell tight junction proteins. Journal of Cellular Physiology. 221, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul J, Strickland S, Melchor JP, 2007. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. The Journal of Experimental Medicine. 204, 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasant J, Carlson S, Mao H, Scheff S, Yang K, Saatman K, 2011. Rate of neurodegeneration in the mouse controlled cortical impact model is influenced by impactor tip shape: implications for mechanistic and therapeutic studies. Journal of Neurotrauma. 28, 2245–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn H, Marchand-Leroux C, Croci N, Plotkine M, Scherman D, Escriou V, 2008. Housekeeping while brain’s storming Validation of normalizing factors for gene expression studies in a murine model of traumatic brain injury. BMC molecular biology. 9, 62–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R, 1999. Atherosclerosis - an inflammatory disease. N Engl J Med. 340, 115–126. [DOI] [PubMed] [Google Scholar]
- Rubenstein R, Chang B, Grinkina N, Drummond E, Davies P, Ruditzky M, Sharma D, Wang K, Wisniewski T, 2017. Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein. Acta Neuropathologica Communications. 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K, 2010. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. The Journal of Neuroscience. 30, 5843–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider B, Mutel V, Mathéa Pietri M, Ermonval M, Mouillet-Richard S, Kellermann O, 2003. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells Proc Natl Acad Sci USA. 100, 13326–13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke R, Drummond O, MacGregor I, Biggerstaff J, Gale R, Camilleri R, Mackie I, Machin S, Harrison P, 2002. The expression of prion protein by endothelial cells: a source of the plasma form of prion protein? British Journal of Haematology. 119, 863–873. [DOI] [PubMed] [Google Scholar]
- Sun Y, Wang J, Wu X, Xi C, Gai Y, Liu H, Yuan Q, Wang E, Gao L, Hu J, Zhou L, 2011. Validating the incidence of coagulopathy and disseminated intravascular coagulation in patients with traumatic brain injury – analysis of 242 cases. British Journal of Neurosurgery 25, 363–368. [DOI] [PubMed] [Google Scholar]
- Till C, Colella B, Verwegen J, Green RE, 2008. Postrecovery Cognitive Decline in Adults With Traumatic Brain Injury. Archives of Physical Medicine and Rehabilitation. 89, S25–S34. [DOI] [PubMed] [Google Scholar]
- Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D, 2008. Fibrinogen induces endothelial cell permeability. Molecular & Cellular Biochemistry. 307, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasse M, Paysant J, Soria J, Collet JP, Vannier JP, Soria C, 1996. Regulation of fibrinogen biosynthesis by cytokines, consequences on the vascular risk. Haemostasis. 26, 331–339. [DOI] [PubMed] [Google Scholar]
- Vos CMP, Geurts JJG, Montagne L, van Haastert ES, Bö L, van der Valk P, Barkhof F, de Vries HE, 2005. Blood–brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiology of Disease. 20, 953–960. [DOI] [PubMed] [Google Scholar]
- Westergard L, Christensen HM, Harris DA, 2007. The Cellular Prion Protein (PrP(C)): Its Physiological Function and Role in Disease. Biochimica et biophysica acta. 1772, 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilberding JA, Ploplis VA, McLennan L, Liang Z, Cornelissen IVO, Feldman M, Deford ME, Rosen ED, Castellino FJ, 2001. Development of pulmonary fibrosis in fibrinogen-deficient mice. Annals of the New York Academy of Sciences. 936, 542–548. [DOI] [PubMed] [Google Scholar]
- Yuasa M, Mignemi NA, Nyman JS, Duvall CL, Schwartz HS, Okawa A, Yoshii T, Bhattacharjee G, Zhao C, Bible JE, Obremskey WT, Flick MJ, Degen JL, Barnett JV, Cates JMM, Schoenecker JG, 2015. Fibrinolysis is essential for fracture repair and prevention of heterotopic ossification. The Journal of Clinical Investigation. 125, 3117–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani A, Powell KL, May A, Semple BD, 2020. Validation of reference genes for gene expression analysis following experimental traumatic brain injury in a pediatric mouse model. Brain Research Bulletin. 156, 43–49. [DOI] [PubMed] [Google Scholar]
- Zhou J, Liu B, 2013. Alzheimer’s Disease and Prion Protein. Intractable & Rare Diseases Research. 2, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]