

Abstract
The liver is not the exclusive site of glucose production in humans in the postabsorptive state. Robust data support that the kidney is capable of gluconeogenesis and studies have demonstrated that renal glucose production can increase systemic glucose production. The kidney has a role in maintaining glucose body balance, not only as an organ for gluconeogenesis but by using glucose as a metabolic substrate. The kidneys reabsorb filtered glucose through the sodium‐glucose cotransporters sodium‐glucose cotransporter (SGLT) 1 and SGLT2, which are localized on the brush border membrane of the early proximal tubule with immune detection of their expression in the tubularized Bowman capsule. In patients with diabetes mellitus, the renal maximum glucose reabsorptive capacity, and the threshold for glucose passage into the urine, are higher and contribute to the hyperglycemic state. The administration of SGLT2 inhibitors to patients with diabetes mellitus enhances sodium and glucose excretion, leading to a reduction of the glycosuria threshold and tubular maximal transport of glucose. The net effects of SGLT2 inhibition are to drive a reduction in plasma glucose levels, improving insulin secretion and sensitivity. The benefit of SGLT2 inhibitors goes beyond glycemic control, since inhibition of renal glucose reabsorption affects blood pressure and improves the hemodynamic profile and the tubule glomerular feedback. This action acts to rebalance the dense macula response by restoring adenosine production and restraining renin‐angiotensin‐aldosterone activation. By improving renal and cardiovascular function, we explain the impressive reduction in adverse outcomes associated with heart failure supporting the current clinical perspective.
Keywords: cardiovascular disease, heart failure, sodium‐glucose cotransporter‐2 inhibitors, type 2 diabetes mellitus
Subject Categories: Cardiorenal Syndrome, Heart Failure
Nonstandard Abbreviations and Acronyms
- DAPA‐HF
Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure
- DM
diabetes mellitus
- EMPEROR‐Reduced
Empagliflozin Outcome Trial in Chronic Heart Failure With Reduced Ejection Fraction
- K+
potassium
- Na+
sodium
- NH2
aminyl radical
- RAAS
renin‐angiotensin‐aldosterone system
- SGLT
sodium‐glucose cotransporter
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- TmG
transport maximum glucose reabsorption
Heart failure (HF) is a complex syndrome with an incidence that is rapidly expanding in the population. It is estimated that there are >26 million people worldwide with HF. In Italy, ≈2% of the population is affected by either structural cardiac dysfunction or symptomatic HF. 1 The clinical deterioration linked to the progression of HF constitutes, after childbirth, the most frequent cause of hospitalization in all Western countries. Among patients with HF, concomitant diabetes mellitus (DM) is present in a percentage that varies from 30% to 40%. 2
Renal pathophysiology plays a pivotal role in contributing to the HF syndrome and driving its progression. HF occurs in a wide number of patients at risk for cardiovascular disease, reaching a 30% to 40% incidence rate among individuals with DM in Western countries. 2 The cause of the higher incidence of HF in individuals with DM is attributable to multiple factors including microvascular and macrovascular disease of the heart, metabolic damage to the myocardium, and chronic kidney disease. The sum of such condition is additive in patients with DM, particularly those with type 2 DM (T2DM) and HF, resulting in cardiovascular mortality >50% to 90% in comparison to the normoglycemic population and promoting the progression of HF until death. 3 , 4
Thus, in patients with DM, HF is a highly malignant disease and, even in patients without a previous myocardial infarction and/or peripheral arterial disease, 5 constitutes an important clinical comorbidity. Since the onset of HF after myocardial infarction is much more frequent in patients with DM, this contributes to the high prevalence of HF in this population. 6
Moreover, HF is prevalent in type 1 DM (T1DM), a much less frequent disease than T2DM, but which affects younger patients who are prematurely exposed to hyperglycemia and its harmful consequences, with a serious impact on quality of life and life expectancy. In fact, in patients with T1DM, the incidence of acute HF is directly related to the value of glycated hemoglobin (HbA1c). In T1DM, poor glycemic control combined with risk factors (such as smoking, hypertension, and dyslipidemia), has been associated with an increased risk of HF 6.37 times higher than patients who have DM with glycemic control and controlled risk factors. 7
Recently, studies have been conducted with 3 molecules of the gliflozin class, renal sodium‐glucose cotransporter (SGLT) type 2 inhibitors. Empagliflozin, canagliflozin, and dapagliflozin have unanimously been demonstrated to reduce the incidence of acute HF in patients with DM who have cardiovascular risk ranging from severe to mild. 8 The administration of these drugs improved glycemic status that was accompanied by a reduced incidence of HF. This reduction was of unexpected proportions and the results of the recent clinical studies appear to present a new treatment paradigm for HF, not only for patients with DM but also for patients without DM. In support of this, the administration of dapagliflozin has shown similar efficacy in reducing cardiovascular events, linked to the progression of HF, in both patients with and those without DM who have HF with reduced ejection fraction. 9
Kidney as a Glucometabolic Organ
Contrary to common belief, the liver is not the only gluconeogenic organ, although it does produce 80% of the endogenously derived glucose. The remaining 20% is produced by the kidney, which, like the liver, contains the necessary gluconeogenic enzymes. 10 Insulin acts as a potent inhibitor of gluconeogenesis in both the liver and the kidney 10 (Figure 1). While insulin acts on both the liver and the kidney, adrenaline stimulates the production of glucose in the kidneys, but not in the liver, and glucagon stimulates the production of glucose only in the liver and not in the kidney. 10 , 11 , 12 Normally, renal gluconeogenesis produces glucose at a rate that is consumed by the kidney. However, during the night, in the fasting phase, the renal parenchyma generates ≈2 mg of glucose per kg/min, which corresponds to the amount of glucose absorbed by the body tissues. 10 , 11 Remarkably, glutamine is the main gluconeogenic amino acid in the kidney, while alanine is the most represented gluconeogenic amino acid in the liver 11 (Figure 1).
Figure 1. The kidney's role in glucose metabolism.
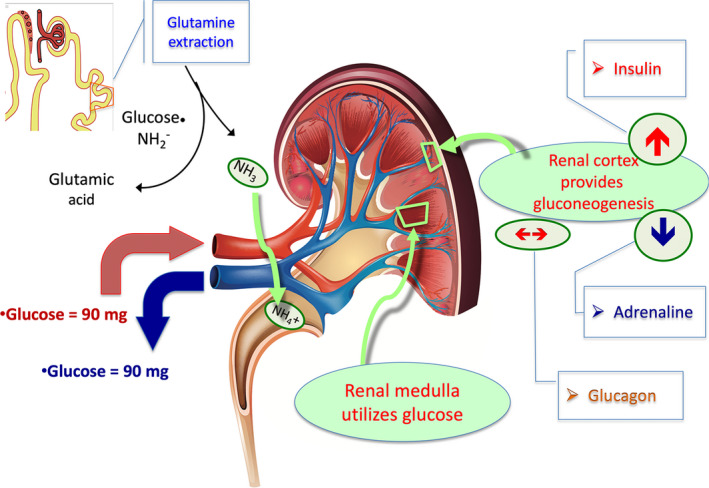
The kidney together with the liver is 1 of 2 body organs provided with gluconeogenesis capability and roughly contributes to 20% glycogen production in normal physiology. In the kidney, gluconeogenesis takes place in the cortical cells while the medullary cells metabolize glucose. It is relevant to observe that insulin inhibits while adrenaline stimulates glucose production in the kidney. Glucagon does not affect renal glucose production. Renal glucose metabolism supports glutamine extraction from tubular cells for the production of glutamic acid and ammonia (NH3). It is an energy‐based metabolic passage that plays a pivotal role in urine acid excretion (NH4 +). Ultimately, the kidney glucose metabolism generates a neutral balance between glucose amount entering with arterial blood and exiting with venous blood. NH2 indicates aminyl radical.
While kidney cells of the cortex generate glucose, this glucose is used in the cells of the renal medulla. As a result of the intrarenal metabolic distribution and the cortical‐medullary balance, the arteriovenous gradient of glucose in the kidney is neutral. 10 In patients with T2DM, even in the presence of high values of fasting glucose, the production of glucose in the kidney and liver increase together with the concentration of plasma insulin, indicative of the presence of insulin resistance in both organs. 13 It is important to underline that the gluconeogenesis generated by glutamine allows the production of ammonia with enhanced excretion of hydrogen ions in the form of ammonium, which allows the kidney to control metabolic acidosis 14 (Figure 1).
Structure, Mechanism of Action, and Regulation of Sodium and Glucose Reabsorption by SGLT in the Renal Tubule
In individuals without DM who have a normal estimated glomerular filtration rate (eGFR) at 180 L/d, the kidney filters ≈180 g of glucose over 24 hours (Figure 2). Thus, the filtered glucose is completely reabsorbed in the proximal tubule. For this reason, glucose is not present in the urine in people without DM. Most of the filtered glucose (80%–90%) is reabsorbed by the cotransporter enzyme SGLT2, which is located below the Bowman capsule, at the beginning of the S1 segment of the proximal tubule, while the remaining 10% to 20% is reabsorbed by the cotransporter SGLT1, which is located in the S2/S3 segment of the tubule in the most distal position (Figure 2). 15 The action of the SGLT2/SGLT1 cotransporters is to work in a complementary manner to avoid the loss of glucose in the urine. For this reason, they operate sequentially: SGLT2 works with high transport capacity, but with less affinity for the glucose molecule, while SGLT1 has less glucose transport capacity but higher binding affinity. Therefore, the SGLT1 is suitable for recovering glucose that the SGLT2 has not previously reabsorbed. SGLT1, under physiological conditions, operates within a limited functional regimen concerning the recovery potential of sugar and sodium. This explains how the glycosuria threshold is raised compared with the glucose present in the filtrate, even when hyperglycemia rises well beyond the threshold of 180 mg/dL. Glucose levels can increase to 220 mg/dL before glycosuria occurs. 13 , 15 , 16
Figure 2. Renal reabsorption of glucose.
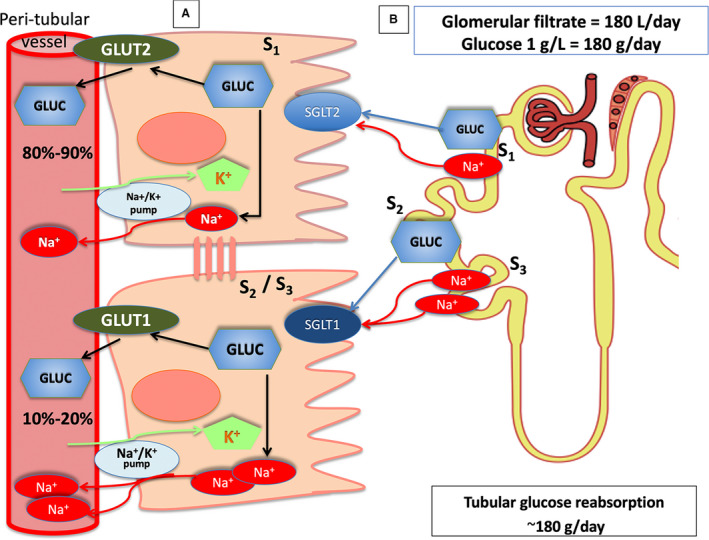
A, Reabsorption of glucose in the proximal tubule. Sodium‐glucose cotransporter (SGLT) type 2 is expressed on the apical membrane of epithelial cells in the S1 segment of the proximal tubule, while glucose transporter 2 is expressed on the basolateral membrane. Epithelial cells of the S2/S3 segment express SGLT1 on the apical membrane and glucose transporter (GLUT) 1 on the basolateral membrane. The Na+‐K+‐ATPase pump is also located in the basolateral membrane of the proximal tubule and provides the energy for sodium‐glucose transport (through both SGLT2 and SGLT1). The pump drives Na+ exchange for K+ taking sodium out of the cytoplasm. As the intracellular Na+ concentration declines, sodium moves passively with glucose from the tubular lumen to the intracellular domain via the SGLT transporters. Once glucose is close to the proximal tubular cell, the concentration gradient passively translocates the molecule down into the interstitial space via the GLUTs. SGLT2 and GLUT2 together with SGLT1 and GLUT1 represent a coupled transport mechanism. In normal physiology, the SGLT2 in the S1 segment of the proximal tubule reuptakes 80% to 90% of the filtered glucose, while the remaining 10% to 20% is reabsorbed by SGLT1 in the S2/S3 segment. B, The normal glomerular filtration rate is ≈180 L/d. With an average daily‐long plasma glucose concentration of 100 mg/dL, the kidney filters ≈180 g of glucose every day, without glucose present in the urine.
The SGLT2 transporter is mainly located in the kidney, but it is also found in α pancreatic cells and other tissues, such as the cerebellum. It is not found in the heart muscle. 16 At the level of the α pancreatic cell, the inhibition of SGLT2 stimulates the secretion of glucagon, which antagonizes the hypoglycemic effect of SGLT2 inhibitors by increasing liver gluconeogenesis. 17 This lessens the likelihood of hypoglycemia during the administration of the SGLT2 inhibitor drugs. This action is especially important since hypoglycemia remains a serious adverse event in patients with DM being treated with hypoglycemic drugs eliminated from the kidney, especially in the presence of chronic renal failure, which increases iatrogenic effects. SGLT1 has a wider distribution than SGLT2 and is found in the intestine wall and skeletal muscle, as well as in the heart and lungs. 18 The function of SGLT1 in organs other than the kidney remains unexplored. 18 , 19 In the intestine, SGLT1 is the primary cotransporter responsible for glucose absorption, and its inhibition is associated with glucose and galactose malabsorption. 19 , 20
Ability of the Kidney to Reabsorb Glucose
In healthy adult humans, the maximum renal glucose reabsorption capacity (transport maximum glucose reabsorption [TmG]) is ≈375 mg/min, slightly higher in men than in women. 13 , 15 , 16 In normal individuals, the rate at which glucose is filtered (ie, 180 g/d equal to 125 mg/min) is significantly lower than TmG. For this reason, the filtered glucose is completely reabsorbed and glycosuria is not present.
In contrast, in patients with DM, filtered glucose can exceed the threshold of tubular reabsorption and result in the appearance of glycosuria. For this reason, filtration and reabsorption can be in temporal disequilibrium in the patient with DM, with the nephron reabsorption capacity being lower than the filtering capacity of glucose. This functional misalignment can induce a modification of the glycosuria threshold. Therefore, the appearance of glycosuria can be attributable to different causes. It can depend both on the increase of the filtered glucose or on the relative substitution of the resorption capacity of the cells of the tubule or both factors.
The Na+/K+/ATPase‐dependent pump, present in the basal‐lateral membrane of the proximal tube, generates energy for the transport of sodium‐glucose through both SGLT2 and SGLT1. This pump allows sodium (Na+) to escape from the cell to be exchanged for potassium (K+). The decrease in intracellular Na+ concentration results in glucose moving from the tubular lumen to the intracellular environment via SGLT. Inside the cells of the renal proximal tubule, by a passive concentration gradient, the glucose transporters mobilize the glucose from the cell in the interstitial space. In the kidney tubule, SGLT2 with glucose transporter 2, as well as SGLT1 with glucose transporter 1, provide an integrated mechanism of transport of glucose 15 , 16 (Figure 2).
Reabsorption of Glucose During Hyperglycemia
In patients with DM, the TmG is increased compared with patients without DM, or compared with patients with DM who can maintain glycemic control. 21 In patients with T2DM, the renal threshold of glycosuria, as well as TmG, is higher. The TmG can correlate with the increase in HbA1c 13 , 16 and patients may appear to have good glycemic control. At the molecular level, increased TmG is associated by increased SGLT2 mRNA and protein in the proximal tubule. 13
The increase of the glycosuria threshold and TmG in the presence of hyperglycemia likely constitutes a phenomenon of an evolutionary adaptation of the kidney to avoid the loss of glucose as a necessary energy substrate in case of scarcity of food. Since the supply of sugars in the normal diet is abundant, T2DM has become a pandemic promoted by excessive food intake. As a result, the adaptive process of reabsorption of glucose in the tubule has turned into a process of maladaptation, which, by delaying the appearance of glycosuria, can delay the diagnosis of DM. When the concentration of Na+ decreases in the cytoplasm, Na+ reenters the cell down a chemical gradient, which is coupled with the transport of glucose. The ratio between the Na+ and glucose molecules is maintained 1:1 for SGLT2 and 2:1 for SGLT1 15 , 21 , 22 (Figure 2). Since Na+ and glucose travel together in the tubule by this mechanism, the increase in glucose reabsorption in patients with DM is associated with the increase in the Na+ content in the body, which constitutes the main cause of hypertension found in 70% of patients with T2DM. 23
Role of SGLT1 and SGLT2 in Glucose Homeostasis
The anatomical position of the 2 cotransporters determines that inhibition of SGLT2 alone increases urinary glucose excretion by only 80 g/d (≈50% of the filtered glucose) even through SGLT2 is responsible for 90% of filtered glucose (about 160 g in 24 hours in patients with normal glucose tolerance) 13 , 15 , 19 (Figure 3). Physiologically, SGLT1 is normally responsible for removing the remaining 10% to 20% of filtered glucose, a percentage that is far less than its maximum transport capacity. 19 Since SGLT2 inhibition decreases the reabsorption of large amounts of glucose, more glucose reabsorption takes place by SGLT1 because of its high glucose reabsorption capacity. This explains why only ≈50% of the filtered glucose is present in the urine of patients treated with SGLT2. 15 , 22
Figure 3. Sodium‐glucose cotransporter type 2 (SGLT2) inhibition and adenosine in healthy patients and those with diabetes mellitus.
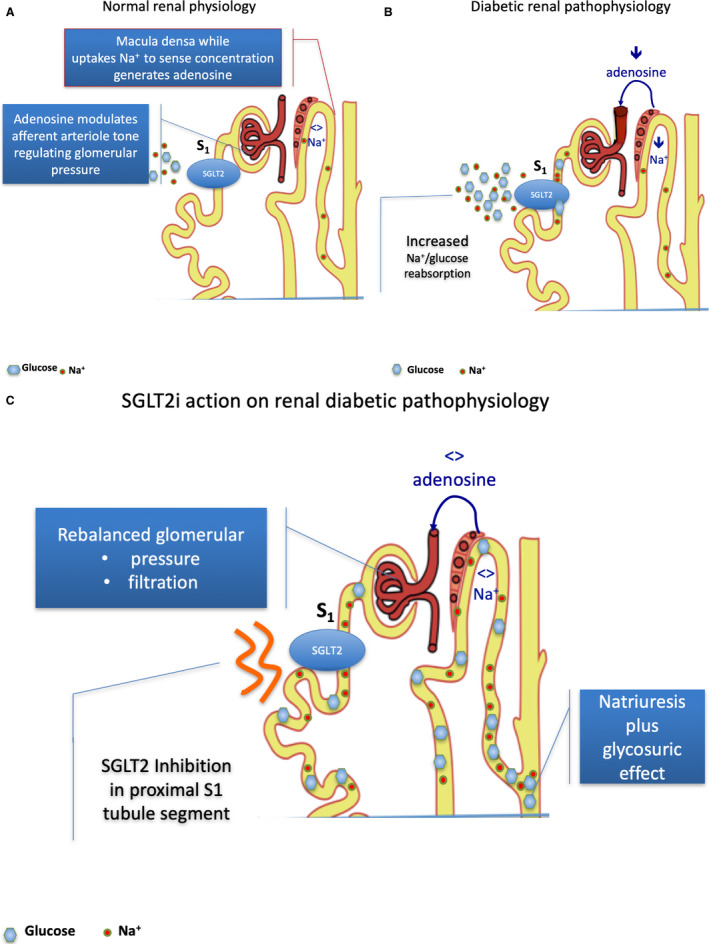
A, Normal renal physiology. The NaCl uptake by dense macula cells (juxtaglomerular apparatus) occurs largely via the Na/K/2Cl cotransporter. It is an energy‐requiring process leading to a breakdown of ATP to adenosine, which has a vasoconstrictive effect. In normal physiology, regular NaCl concentration in urine transit across dense macula (juxtaglomerular apparatus) cells balancing the amount of ATP release and the breakdown to adenosine. Adenosine acts modulating the afferent arteriole tone in relation to normal physiology occurrence. The mechanism is defined as tubular‐glomerular feedback (TGF). Afferent arteriole has a slightly larger diameter in comparison to efferent. The difference assures the intraglomerular pressure gradient to generate urine. Therefore, any vasodilating or vasoconstrictive action in the glomerular vasculature generates a disproportioned relative or absolute decrease in efferent vessel section with a corresponding increase in intraglomerular pressure. B, Diabetic renal pathophysiology. In diabetic renal pathophysiology, a lower urine NaCl concentration is driven by SGLT2‐increased Na+/glucose reabsorption, and therefore a decreased NaCl concentration transit across the dense macula (juxtaglomerular apparatus). The low NaCl concentration is sensed by the dense macula as a low volume, low perfusion state, leading to low adenosine production that induces vasodilating action prevailing on the afferent vessel and causing glomerular hypertension and glomerular hyperfiltration. 57 , 58 Decreased Na+ delivery to the dense macula entails decreased adenosine production with vasodilating effect on afferent glomerular arteriole, enhancing relative disproportion with efferent arteriole section and leading to intraglomerular hypertension and glomerular hyperfiltration. 59 , 60 C, SGLT2 inhibition and glomerular hyperphyltration correction. Selective SGLT2 inhibition by promoting glucose and NaCl. urination increases distal renal NaCl delivery, leading to adenosine production and afferent arteriole tone restoration, thus decreasing intraglomerular pressure and positively affecting hyperfiltration. Restoration of Na+ delivery to the dense macula normalizes adenosine production, reestablishing afferent glomerular arteriole tone and abolishing intraglomerular hypertension and glomerular hyperfiltration.
It would be natural to investigate what effects would result from the combined inhibition of the 2 cotransporters, SGLT2/SGLT1. Since glucose reabsorption by SGLT1 decreases glycosuria, the combined effect of inhibiting SGLT2 should significantly increase the excretion of urinary glucose and decrease the level of HbA1c. However, in the distal tract of the small intestine (ileum), the absorption of glucose and the secretion of glucagon‐like peptide 1 by L cells are closely linked, since these 2 actions are under the control of SGLT1 activity 24 and inhibition of glucagon‐like peptide 1. This combined effect worsens glycemic control by reducing insulin secretion. 24 This is supported by limited data where simultaneous inhibition of SGLT1 and SGLT2 was produced, which showed reduced gastrointestinal tolerability (abdominal pain and diarrhea). 18
It is worth noting that the presence of the SGLT1 protein has been documented through immune fixation studies in the capillaries of the myocardium, with its involvement in ischemia‐reperfusion injury. 25 Therefore, the combined SGLT2/SGLT1 inhibitor deserves appropriate scrutiny.
Mechanism of Action of the SGLT2 Inhibitors
Three SGLT2 inhibitors (dapagliflozin, canagliflozin, and empagliflozin) have been approved for clinical use in the United States and Europe, while many other molecules are currently under investigation. 26
In general, SGLT2 inhibitors can induce glycosuria by the following 3 mechanisms: (1) decreasing TmG, (2) lowering the threshold for glycosuria, and (3) decreasing the rate of reabsorption of filtered glucose. However, these mechanisms are insufficient to explain the induction of glycosuria in patients taking SGLT2 inhibitors who have normal fasting glucose, and this could be explained by methodological inaccuracy in measuring the threshold of glycosuria. In individuals without DM, the 3 SGLT2 inhibitors, dapagliflozin, empagliflozin, and canagliflozin, can increase the urinary glucose excretion by 60 to 80 g/d. 27 , 29
In a comparison study conducted in patients without DM, canagliflozin at a dose of 300 mg resulted in a daily excretion of glucose in the urine equal to 51.4 g, a significantly greater quantity than that obtained with the administration of dapagliflozin at a dose of 10 mg, which led to excretion of only 40.8 g of glucose. 30 The difference in urinary glucose excretion by these 2 molecules is the result of the longer half‐life of canagliflozin, which leads to increases in the plasma concentration of canagliflozin after 16 to 24 hours. The data are relevant in understanding how the different bioavailability of molecules that have similar pharmacological actions can lead to a different biologic efficacy. In T2DM, all 3 SGLT2 inhibitors produce a dose‐dependent increase in the elimination of glucose in the urine and a similar decrease in plasma fasting, postprandial glucose, and HbA1c levels. 29 , 30 , 31 , 32 However, no comparative study has yet to be performed to establish efficacy between different molecules in patients with DM. Controlled trials conducted with canagliflozin, empagliflozin, and dapagliflozin have shown that these SGLT2 inhibitors result in a reduction in HbA1c of 0.7% to 1.0% in a patient with T2DM without prior diabetic treatment, as well as in patients already treated with metformin, sulphonylureas, pioglitazone, metformin, and insulin, or in patients with HbA1c values of 7.8% to 8.2%. 32 , 33
If patients with T2DM who have normal renal function or moderately reduced renal function, all SGLT2 inhibitors are equally effective, both in the case of newly diagnosed T2DM and in the case of a T2DM diagnosis made many years before or in the presence of insulin resistance and reduced production of insulin. 27 Attributable to the SGLT2 inhibitor–induced glycosuric effect, patients with DM who have high HbA1c achieve greater reductions in HbA1c than in individuals with lower blood glucose values, regardless of the ongoing treatment for DM. 32 , 33 Logically, SGLT2 inhibitors can be effective in decreasing both glucose concentration and HbA1c more than other antidiabetic agents. Thus, some clinical studies prefigure their adoption as an alternative to treatment with high‐dose insulin. 34 This is attractive considering the documented effectiveness of these molecules in reducing body weight and blood pressure (BP). 8 , 16 , 35 These results could be explained by the fact that high plasma glucose concentrations induce high glucose concentration in the filtrate, which, in turn, represents the effective target for the action of the SGLT2 inhibitors. 27 , 28 , 29 These data have important clinical implications in patients with DM but also in patients without DM, patients with insulin resistance, and patients with clinical conditions of relative insulin resistance as in the course of HF. 36 , 37
Effect of Blood Glucose on the Response of the Macula Densa and the Consequence at the Level of Tubule‐Glomerular Feedback of SGLT2 Inhibitors
The inhibition of SGLT2 involves the combined reabsorption of Na+ and glucose in the proximal tubule. This is associated with a modestly negative balance of Na+ and water in the body, which generates a lasting reduction in the volume of water present in the vasculature as well in the extracellular space (third space). 38 , 39
The natriuretic effect of SGLT2 inhibition disappears within 48 to 72 hours and the balance of water and Na+ is restored; however, an ≈7% reduction in the volume of the plasma remains. 40
The modest, but not negligible, reduction of the extracellular volume involves a decline of 5 to 6 mm Hg of systolic BP and of 1 to 2 mm Hg of diastolic BP, coupled with body weight decline which is observed after 7 to 15 days from the beginning of treatment, both in patients with DM 41 and those without DM 9 , 42 already treated with renin‐angiotensin‐aldosterone system (RAAS) inhibitor drugs.
Importantly, after 6 to 12 months of SGLT2 inhibitor treatment, an increase in major hypoglycemic events and signs related to sympathetic activation such as reflex tachycardia were not observed.
In terms of activation of RAAS neurohormones, SGLT2 inhibition induces a modest rise in plasma RAAS hormones such as aldosterone and angiotensin II, but within the suppressed range that is typical of DM (the RAAS paradox) 43 and similarly increases levels of neurohormone markers in the urine (angiotensinogen, angiotensin‐converting enzyme, angiotensin‐converting enzyme 2), possibly as a consequence of plasma volume contraction. 40 , 44 The data provide clinical support for the synergistic adoption of SGLT2 inhibitors together with RAAS‐inhibiting drugs to achieve superior efficacy in BP control with a further decline of 3 to 4 mm Hg of systolic BP typically observed.
Whether this apparent synergy is based on plasma volume contraction leading to activation of RAAS, which is then pharmacologically tapered by drugs counteracting angiotensin II effect, allowing further BP lowering is unknown but opens up scrutiny on the way SGLT2 inhibitors can affect renal function and cardiovascular hemodynamics. 45 , 46
In animal models of hyperglycemia, glucose and Na+ reabsorption is increased in the proximal tubule. This is responsible for the decrease in Na+ content of the urine sensed at the juxtaglomerular apparatus. The resulting decreased urinary Na+ concentration is sensed as a decreased perfusion of the kidney. In response, the juxtaglomerular (or dense macula) apparatus activates the RAAS system with the formation of angiotensin II. 47 This hormone determines the constriction of the afferent and efferent arteriole, but with a different consequence on vessel conductance. The afferent arteriole has more represented smooth muscle, addressing the larger response to the vasoactive stimulus and is slightly larger in diameter than the efferent arteriole. In normal physiology, the diameter difference assures the intraglomerular pressure gradient to generate urine. 48 Therefore, any overimposed vasoconstrictive action in the glomerular vasculature generates a disproportioned decrease in efferent vessel section with a corresponding increase in intraglomerular pressure leading to renal hyperfiltration, a condition that drastically increases sodium and glucose urine content.
Renal hyperfiltration, in general, defined as a glomerular filtration rate ≥135 mL/min per 1.73 m2, is a marker of intraglomerular hypertension and is a risk factor for the initiation and progression of diabetic nephropathy 49 , 50 in animals as well as in humans, by the glomerular skein formation (often seen in elderly patients), which results in permanent glomerular damage. 51
Based on available data, the SGLT2 inhibitor class plays favorably on hyperfiltration pathophysiology with both important mechanistic and clinical shreds of evidence, because the underlying changes in intraglomerular pressure contribute to improvements in renal end points in all performed studies. 8 , 32
By considering the pattern of renal function change observed with SGLT2 inhibitors and the possible benefits, previous work has compared these salutary effects with what was achieved with RAAS‐inhibiting drugs. Inhibitors of the RAAS reduce intraglomerular pressure through efferent arteriolar vasodilatation, leading to reductions in intraglomerular hypertension and renal hyperfiltration and resulting in nephroprotection. 52
Analyzing patients with T2DM treated with RAAS blockade according to the change of eGFR over time, the best preservation of renal function occurred in patients who had the greatest tertile of eGFR decline within 3 months of treatment initiation. 53 Therefore, RAAS inhibition effects a delayed eGFR change characterized by a protective effect on glomerular function. 54
In contrast to the effects of RAAS blockers, the initial decline in eGFR observed with SGLT2 inhibition seems related to afferent arteriolar vasoconstriction through the tubule‐glomerular feedback. This is the autoadaptive mechanism regulating the glomerular filtration rate concerning NaCl concentration in the glomerular filtrate that reaches the dense macula. The proportion of NaCl reabsorption is positively related to the afferent arteriole tone and consequently to the glomerular filtration rate. This autoregulatory mechanism is intrinsic to the kidney and does not require neural or humoral mediators. 54 , 55
In an animal model, the response to an increase in NaCl delivery to the dense macula after blockade of proximal tubular NaCl reabsorption increases NaCl uptake by dense macula cells. It occurs prominently via the Na/K/2Cl cotransporter, which is an energy‐requiring process leading to the breakdown of ATP to adenosine. This pathway engages adenosine type 1 receptors located on the smooth muscle cells of the afferent arteriole leading to modulation of afferent vessel tone 56 (Figure 3A).
In patients without DM, SGLT2 is responsible for ≈5% of total renal NaCl reabsorption. However, in the context of hyperglycemia, SGLT2 and SGLT1 mRNA expression increase by 36% and 20%, respectively, suggesting their direct role in NaCl reabsorbtion. 57 , 58 , 59
As a consequence, SGLT1 and SGLT2 activity accounts for as much as 14% of total renal NaCl reabsorption in the setting of hyperglycemia in DM, thereby entailing a marked reduction in distal NaCl delivery to the dense macula, which, in turn, leads to maladaptive glomerular afferent arterial vasodilatation and increased intraglomerular pressure coupled with glomerular hyperfiltration 60 (Figure 3B). Again, the decline of NaCl delivery to the dense macula is sensed incorrectly as a reduction in effective circulating plasma volume by the juxtaglomerular apparatus, thereby by promoting avid Na+ uptake through RAAS activation.
In animals, nonspecific SGLT1/SGLT2 inhibitors 61 and selective SGLT2 inhibition increase distal renal NaCl delivery, leading to increased afferent tone and abolishing hyperfiltration. 62 This occurs in a paracrine fashion via adenosine type 1 receptors on afferent arteriolar vascular smooth muscle cells causing vasoconstriction independently by angiotensin II activation (Figure 3C).
The fast activation of the tubule‐glomerular feedback modulation by SGLT2 inhibitors has been elegantly documented in patients with T1DM. In this setting, the administration of empagliflozin, performed during hyperglycemic clamp technique, 63 in patients with DM who have normal blood glucose, was followed by a reduction of 33 mL/min per 1.73 m2 of filtration (glomerular filtration rate 172±23–139±25 mL/min per 1.73 m2, P<0.01). 44 The restoration of intraglomerular pressure has positive implications in the HF syndrome. 64 Improvements in renal function, driven by the use of SGLT2 inhibitors in addition to RAAS‐blocking agents, has been consistently linked to the fall of HF exacerbations in patients with DM as well those with DM who have HF. 42 The key role played by adenosine 63 , 65 , 66 , 67 has been strongly suggested by evidence generated with a mechanistic study performed in patients with T1DM. In the research, patients receiving empagliflozin under clamped hyperglycemic conditions 63 throughout an 8‐week investigation displayed increased urinary adenosine/creatinine ratio as measured by liquid chromatography‐tandem mass spectrometry. 68 Those data confirm that the tubule‐glomerular feedback modulation should become a matter of investigation in every clinical condition involving renal perfusion and cardiocirculatory balance. Moreover, it underscores that dysfunction of both the heart and the kidneys are intrinsic to the pathogenesis of HF. 69 , 70 , 71 , 72
Conclusions
The role of the kidney is critical to maintaining glucose homeostasis. This is closely linked to the Na+ homeostasis, which not only depends on the activation of the neuroendocrine system but, in a more direct fashion, the renal circulatory balance regulated by the tubule‐glomerular feedback. This mechanism is permanently active by the continuous sensing of NaCl in the urine content, allowing fine‐tuning of the filtration rate without external regulation. In the proximal segment of the tubular nephron, SGLT1 and SGLT2 provide highly efficient glucose reabsorption coupled with Na+ reuptake. The reabsorption mechanism influences urine sodium content that directly involves the tubule‐glomerular feedback with an impact on intraglomerular pressure. SGLT2 inhibition targets the exceedingly high glucose and sodium reabsorption occurring in DM as well as in other clinical conditions such as HF both with reduced and preserved ejection fraction. 72 , 73 , 74 , 75 The improved intrarenal regulation of nephron perfusion has been shown to reduce adverse events related to circulatory congestion, such as HF exacerbation. 73 Recent clinical data from the DAPA‐HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) and EMPEROR‐Reduced (Empagliflozin Outcome Trial in Chronic Heart Failure With Reduced Ejection Fraction) 75 trials showed impressive benefits afforded by the SGLT2 inhibitors in patients with and without DM who have HF. 42 , 75
Importantly, the liver and kidney are both sites of gluconeogenesis with purposes apparently disjointed. The glucose produced in the kidney is the substrate used to produce the energy consumed to produce ammonia, the urinary excretion of which is essential to regulate metabolic acidosis. The liver is, instead, the site of angiotensinogen production, the substrate of the renin enzyme produced by the kidney, from which angiotensin II is generated by the enzyme of conversion produced by the lung. The lung, through the ventilation of CO2, is the regulator of respiratory acidosis as well as of aerobic glycolysis. The linkage of glucose and Na+ metabolism and the neurohormonal response is a remarkable demonstration of how cardiovascular physiology is strongly and constantly provided by the feedback generated by the liver, kidney, and lung. These organs act as metabolic sensors and constantly regulate the vascular and cardiopulmonary pressure and chemoreceptor response.
Sources of Funding
None.
Disclosures
None.
Acknowledgments
We thank Dr Gary Lopaschuck (Canada) for his insightful suggestions and Giovanni Michele Napoli, Apple Developer (Sacro Cuore Institute, Naples, Italy), for his advice during figure assembling and preparation of the article.
(J Am Heart Assoc. 2020;9:e018889 DOI: 10.1161/JAHA.120.018889.)
For Sources of Funding and Disclosures, see page 10.
REFERENCES
- 1. Zucker IH, Xiao L, Haack KK. The central RAS and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond). 2014;126:695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE, Hansen TB, et al; ESC Scientific Document Group . 2019 ESC guidelines on diabetes, pre‐diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41:255–323. [DOI] [PubMed] [Google Scholar]
- 3. Cavender MA, Steg PG, Smith SC Jr, Eagle K, Ohman EM, Goto S, Kuder J, Im K, Wilson PW, Bhatt DL; REACH Registry Investigators . Impact of diabetes mellitus on hospitalization for heart failure, cardiovascular events, and death: outcomes at 4 years from the Reduction of Atherothrombosis for Continued Health (REACH) Registry. Circulation. 2015;132:923–931. [DOI] [PubMed] [Google Scholar]
- 4. MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, Solomon SD, Granger CB, Swedberg K, Yusuf S, et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur Heart J. 2008;29:1377–1385. [DOI] [PubMed] [Google Scholar]
- 5. Miceli M, Baldi D, Cavaliere C, Soricelli A, Salvatore M, Napoli C. Peripheral artery disease: the new frontiers of imaging techniques to evaluate the evolution of regenerative medicine. Expert Rev Cardiovasc Ther. 2019;17:511–532. [DOI] [PubMed] [Google Scholar]
- 6. Shah AD, Langenberg C, Rapsomaniki E, Denaxas S, Pujades‐Rodriguez M, Gale CP, Deanfield J, Smeeth L, Timmis A, Hemingway H. Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1.9 million people. Lancet Diabetes Endocrinol. 2015;3:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lind M, Bounias I, Olsson M, Gudbjornsdottir S, Svensson AM, Rosengren A. Glycaemic control and incidence of heart failure in 20 985 patients with type 1 diabetes: an observational study. Lancet. 2011;378:140–146. [DOI] [PubMed] [Google Scholar]
- 8. Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RH, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta‐analysis of cardiovascular outcome trials. Lancet. 2019;393:31–39. [DOI] [PubMed] [Google Scholar]
- 9. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, et al; DAPA‐HF Trial Committees and Investigators . Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. [DOI] [PubMed] [Google Scholar]
- 10. Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stumvoll M, Meyer C, Mitrakou A, Nadkarni V, Gerich JE. Renal glucose production and utilization: new aspects in humans. Diabetologia. 1997;40:749–757. [DOI] [PubMed] [Google Scholar]
- 12. Stumvoll M, Chintalapudi U, Perriello G, Welle S, Gutierrez O, Gerich J. Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest. 1995;96:2528–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeFronzo RA, Lecture B. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Veiner D, Verlander JW. Renal ammonia metabolism and transport. Compr Physiol. 2013;3:201–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu JJ, Defronzo RA. Why do SGLT2 inhibitors inhibit only 30–50% of renal glucose reabsorption in humans? Diabetes. 2012;61:2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeFronzo LA, Norton L, Abdul‐Ghani M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nature. 2017;13:11–26. [DOI] [PubMed] [Google Scholar]
- 17. Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, Radović N, Jadrijević S, Aleksic I, Walles T, et al. Localizations of Na‐D glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch. 2014;467:1881–1898. [DOI] [PubMed] [Google Scholar]
- 18. Bonner C, Kerr‐Conte J, Gmyr V, Queniat G, Moerman E, Thévenet J, Beaucamps C, Delalleau N, Popescu I, Malaisse WJ, et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med. 2015;21:512–517. [DOI] [PubMed] [Google Scholar]
- 19. Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;9:733–794. [DOI] [PubMed] [Google Scholar]
- 20. Tsimihodimos V, Filippas‐Ntekouan S, Elisaf M. SGLT1 inhibition: pros and cons. Eur J Pharmacol. 2018;838:153–156. [DOI] [PubMed] [Google Scholar]
- 21. Farber SJ, Berger EY, Earle DP. Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest. 1951;30:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abdul‐Ghani MA, DeFronzo RA, Norton L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes. 2013;62:3324–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Felicetta JV, Sowers J. Systemic hypertension in diabetes mellitus. Am J Cardiol. 1988;61:34H–40H. [DOI] [PubMed] [Google Scholar]
- 24. Powell DR, DaCosta CM, Smith M, Doree D, Harris A, Buhring L, Heydorn W, Nouraldeen A, Xiong W, Yalamanchili P, et al. Effect of LX4211 on glucose homeostasis and body composition in preclinical models. J Pharmacol Exp Ther. 2014;350:232–242. [DOI] [PubMed] [Google Scholar]
- 25. Li Z, Agrawal V, Ramratnam M, Sharma RK, D'Auria S, Sincoular A, Jakubiak M, Music ML, Kutschke WJ, Huang XN, et al. Cardiac sodium‐glucose co‐transporter 1 (SGLT1) is a novel mediator of ischemia/reperfusion injury. Cardiovasc Res. 2019;115:1646–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adeghate E, Mohsin S, Adi F, Ahmed F, Yahya A, Kalász H, Tekes K, Adeghate EA. An update of SGLT1 and SGLT2 inhibitors in early phase diabetes‐type 2 clinical trials. Expert Opin Investig Drugs. 2019;28:811–820. [DOI] [PubMed] [Google Scholar]
- 27. DeFronzo RA, Hompesch M, Kasichayanula S, Liu X, Hong Y, Pfister M, Morrow LA, Leslie BR, Boulton DW, Ching A, et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care. 2013;36:3169–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Al‐Jobori H, Daniele G, Cersosimo E, Triplitt C, Mehta R, Norton L, DeFronzo RA, Abdul‐Ghani M. Empagliflozin and kinetics of renal glucose transport in healthy individuals and individuals with type 2 diabetes. Diabetes. 2017;66:1999–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sha S, Devineni D, Ghosh A, Polidori D, Chien S, Wexler D, Shalayda K, Demarest K, Rothenberg P. Canagliflozin, a novel inhibitor of sodium glucose co‐transporter 2, dose dependently reduces calculated renal threshold for glucose excretion and increases urinary glucose excretion in healthy subjects. Diabetes Obes Metab. 2011;13:669–672. [DOI] [PubMed] [Google Scholar]
- 30. Sha S, Polidori D, Farrell K, Ghosh A, Natarajan J, Vaccaro N, Pinheiro J, Rothenberg P, Plum‐Mörschel L. Pharmacodynamic differences between canagliflozin and dapagliflozin: results of a randomized, double‐blind, crossover study. Diabetes Obes Metab. 2015;17:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharmacol Ther. 2009;85:513–519. [DOI] [PubMed] [Google Scholar]
- 32. DeFronzo RA, Stonehouse AH, Han J, Wintle ME. Relationship of baseline HbA1c and efficacy of current glucose‐lowering therapies: a meta‐analysis of randomized clinical trials. Diabet Med. 2010;27:309–317. [DOI] [PubMed] [Google Scholar]
- 33. Rosenstock J, Hansen L, Zee PL, Li Y, Cook W, Hirshberg B, Iqbal N. Dual add‐on therapy in type 2 diabetes poorly controlled with metformin monotherapy: a randomized double‐blind trial of saxagliptin plus dapagliflozin addition versus single addition of saxagliptin or dapagliflozin to metformin. Diabetes Care. 2015;38:376–383. [DOI] [PubMed] [Google Scholar]
- 34. Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double‐blind, placebo‐controlled, phase 3 trial. Diabetes Care. 2010;33:2217–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang L, Feng Y, List J, Kasichayanula S, Pfister M. Dapagliflozin treatment in patients with different stages of type 2 diabetes mellitus: effects on glycaemic control and body weight. Diabetes Obes Metab. 2010;12:510–516. [DOI] [PubMed] [Google Scholar]
- 36. Ingelsson E, Sundström J, Arnlöv J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294:334–341. [DOI] [PubMed] [Google Scholar]
- 37. Petrie MC, Verma S, Docherty KF, Inzucchi SE, Anand I, Belohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, et al. Effect of dapagliflozin on worsening heart failure and cardiovascular death in patients with heart failure with and without diabetes. JAMA. 2020;323:1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas MC, Cherney DZI. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia. 2018;61:2098–2107. [DOI] [PubMed] [Google Scholar]
- 39. Perkins BA, Udell JA, Cherney DZ. No need to sugarcoat the message: is cardiovascular risk reduction from SGLT2 inhibition related to natriuresis? Am J Kidney Dis. 2016;68:349–352. [DOI] [PubMed] [Google Scholar]
- 40. Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose‐regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab. 2013;15:853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weber MA, Mansfield TA, Cain VA, Iqbal N, Parikh S, Ptaszynska A. Blood pressure and glycaemic effects of dapagliflozin versus placebo in patients with type 2 diabetes on combination antihypertensive therapy: a randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Diabetes Endocrinol. 2016;4:211–220. [DOI] [PubMed] [Google Scholar]
- 42. Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, Brueckmann M, Ofstad AP, Pfarr E, Jamal W, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta‐analysis of the EMPEROR‐Reduced and DAPA‐HF trials. Lancet. 2020;396:819–829. [DOI] [PubMed] [Google Scholar]
- 43. Price DA, De'Oliveira JM, Fisher ND, Williams GH, Hollenberg NK. The state and responsiveness of the renin‐angiotensin‐aldosterone system in patients with type II diabetes mellitus. Am J Hypertens. 1999;12:348–355. [PubMed] [Google Scholar]
- 44. Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, Faga NM, Woerle HJ, Johansen OE, Broedl UC, et al. Renal hemodynamic effect of sodium‐glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129:587–597. [DOI] [PubMed] [Google Scholar]
- 45. Weber MA, Mansfield TA, Alessi F, Iqbal N, Parikh S, Ptaszynska A. Effects of dapagliflozin on blood pressure in hypertensive diabetic patients on renin‐angiotensin system blockade. Blood Press. 2016;25:93–103. [DOI] [PubMed] [Google Scholar]
- 46. Kojima N, Williams JM, Slaughter TN, Kato S, Takahashi T, Miyata N, Roman RJ. Renoprotective effects of combined SGLT2 and ACE inhibitor therapy in diabetic Dahl S rats. Physiol Rep. 2015;3:e12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hostetter TH, Troy JL, Brenner BM. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int. 1981;19:410–415. [DOI] [PubMed] [Google Scholar]
- 48. Neal CR, Arkill KP, Bell JS, Betteridge KB, Bates DO, Winlove CP, Salmon AH, Harper SJ. Novel hemodynamic structures in the human glomerulus. Am J Physiol Renal Physiol. 2018;315:F1370–F1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Magee GM, Bilous RW, Cardwell CR, Hunter SJ, Kee F, Fogarty DG. Is hyperfiltration associated with the future risk of developing diabetic nephropathy? A meta‐analysis. Diabetologia. 2009;52:691–697. [DOI] [PubMed] [Google Scholar]
- 50. Ruggenenti P, Porrini EL, Gaspari F, Motterlini N, Cannata A, Carrara F, Cella C, Ferrari S, Stucchi N, Parvanova A, et al; GFR Study Investigators . Glomerular hyperfiltration and renal disease progression in type 2 diabetes. Diabetes Care. 2012;35:2061–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hallow KM, Gebremichael Y, Helmlinger G, Vallon V. Primary proximal tubule hyper‐reabsorption and impaired tubular transport counterregulation determine glomerular hyperfiltration in diabetes: a modeling analysis. Am J Physiol Renal Physiol. 2017;312:F819–F835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zatz R, Dunn BR, Meyer TW, Anderson S, Rennke HG, Brenner BM. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest. 1986;77:1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Holtkamp FA, de Zeeuw D, Thomas MC, Cooper ME, de Graeff PA, Hillege HJ, Parving HH, Brenner BM, Shahinfar S, Lambers Heerspink HJ. An acute fall in estimated glomerular filtration rate during treatment with losartan predicts a slower decrease in long‐term renal function. Kidney Int. 2011;80:282–287. [DOI] [PubMed] [Google Scholar]
- 54. Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol. 2006;17:1703–1709. [DOI] [PubMed] [Google Scholar]
- 55. Arendshorst WJ, Bello‐Reuss E. Handbook of Cell Signaling. 2nd ed Academic Press, Elsevier; 2010:2707–2731. [Google Scholar]
- 56. Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–2576. [DOI] [PubMed] [Google Scholar]
- 57. Pollock CA, Lawrence JR, Field MJ. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am J Physiol. 1991;260:F946–F952. [DOI] [PubMed] [Google Scholar]
- 58. Vestri S, Okamoto MM, de Freitas HS, Aparecida Dos Santos R, Nunes MT, Morimatsu M, Heimann JC, Machado UF. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182:105–112. [DOI] [PubMed] [Google Scholar]
- 59. Bank N, Aynedjian HS. Progressive increases in luminal glucose stimulate proximal sodium absorption in normal and diabetic rats. J Clin Invest. 1990;86:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vallon V, Osswald H. Adenosine receptors and the kidney. Handb Exp Pharmacol Rev. 2009;86:443–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Malatiali S, Francis I, Barac‐Nieto M. Phlorizin prevents glomerular hyperfiltration but not hypertrophy in diabetic rats. Exp Diabetes Res. 2008;2008:305403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302:R75–R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. [DOI] [PubMed] [Google Scholar]
- 64. Vallon V, Miracle C, Thomson S. Adenosine and kidney function: potential implications in patients with heart failure. Eur J Heart Fail. 2008;10:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vallon V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia. 2018;61:2079–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kanbay M, Ertuglu LA, Afsar B, Ozdogan E, Kucuksumer ZS, Ortiz A, Covic A, Kuwabara M, Cherney DZI, van Raalte DH, et al. Renal hyperfiltration defined by high estimated glomerular filtration rate: a risk factor for cardiovascular disease and mortality. Diabetes Obes Metab. 2019;21:2368–2383. [DOI] [PubMed] [Google Scholar]
- 67. Packer M. Mitigation of the adverse consequences of nutrient excess on the kidney: a unified hypothesis to explain the renoprotective effects of sodium‐glucose cotransporter 2 inhibitors. Am J Nephrol. 2020;51:289–293. [DOI] [PubMed] [Google Scholar]
- 68. Rajasekeran H, Lytvyn Y, Bozovic A, Lovshin JA, Diamandis E, Cattran D, Husain M, Perkins BA, Advani A, Reich HN, et al. Urinary adenosine excretion in type 1 diabetes. Am J Physiol Renal Physiol. 2017;313:F184–F191. [DOI] [PubMed] [Google Scholar]
- 69. Napoli C, Casamassimi A, Crudele V, Infante T, Abbondanza C. Kidney and heart interactions during cardiorenal syndrome: a molecular and clinical pathogenic framework. Future Cardiol. 2011;7:485–497. [DOI] [PubMed] [Google Scholar]
- 70. Gronda E, Genovese S, Padeletti L, Cacciatore F, Vitale DF, Bragato R, Innocenti L, Schiano C, Sommese L, De Pascale MR, et al. Renal function impairment predicts mortality in patients with chronic heart failure treated with resynchronization therapy. Cardiol J. 2015;22:459–466. [DOI] [PubMed] [Google Scholar]
- 71. Gronda E, Vanoli E, Sacchi S, Grassi G, Ambrosio G, Napoli C. Risk of heart failure progression in patients with reduced ejection fraction: mechanisms and therapeutic options. Heart Fail Rev. 2020;25:295–303. [DOI] [PubMed] [Google Scholar]
- 72. Gronda E, Sacchi S, Benincasa G, Vanoli E, Napoli C. Unresolved issues in left ventricular postischemic remodeling and progression to heart failure. J Cardiovasc Med (Hagerstown). 2019;20:640–649. [DOI] [PubMed] [Google Scholar]
- 73. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co‐transporter 2 (SGLT2) inhibitors: a state‐of‐the‐art review. JACC Basic Transl Sci. 2020;5:632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart failure with reduced ejection fraction: a review. JAMA. 2020;324:488–504. [DOI] [PubMed] [Google Scholar]
- 75. Packer M. Molecular, cellular, and clinical evidence that Sodium‐Glucose Cotransporter 2 inhibitors act as neurohormonal antagonists when used for the treatment of chronic heart failure. J Am Heart Assoc. 2020;9:e016270 DOI: 10.1161/JAHA.120.016270 [DOI] [PMC free article] [PubMed] [Google Scholar]