

Abstract
Mg2+ is an important cation in our body. It is an essential cofactor for many enzymes. Despite many works, nothing is known about the protective effects of MgSO4 against hypoxia-induced lethality and oxidative damage in brain mitochondria. In this study, antihypoxic and antioxidative activities of MgSO4 were evaluated by three experimental models of induced hypoxia (asphyctic, haemic, and circulatory) in mice. Mitochondria protective effects of MgSO4 were evaluated in mouse brain after induction of different models of hypoxia. Antihypoxic activity was especially pronounced in asphyctic hypoxia, where MgSO4 at dose 600 mg/kg showed the same activity as phenytoin, which used as a positive control (P < 0.001). In the haemic model, MgSO4 at all used doses significantly prolonged latency of death. In circulatory hypoxia, MgSO4 (600 mg/kg) doubles the survival time. MgSO4 significantly decreased lipid peroxidation and protein carbonyl and improved mitochondrial function and glutathione content in brain mitochondria compared to the control groups. The results obtained in this study showed that MgSO4 administration has protective effects against lethality induced by different models of hypoxia and improves brain mitochondria oxidative damage.
1. Introduction
The cation Mg2+ has an important role in the intracellular process regulation. It is necessary for ATP activity containing calcium current across and within cell membranes for tissues. Moreover, Mg2+ exists in more than 300 enzymatic systems through antioxidative properties and is the fourth most abundant cation in our body [1, 2]. Mg2+ is an essential cofactor for the activity of kinases and has effects on oxidative phosphorylation, DNA synthesis, lipid hydrolysis, and rates of glycolysis [3, 4]. Subsequently, Mg2+ is compulsory in all relating ATP reactions, and variations in this ion may regulate oxidative phosphorylation and substrate in the myocardium [5]. Cardiovascular disease has been associated with diminished Mg2+ levels, and a pivotal relationship may be present between mortality from nonocclusive ischemic heart disease and decreased cardiac Mg2+ [5, 6]. Myocardial damage consequence of oxidative stress in the heart cells can occur in several conduction problems such as hypoxia. There is evidence that hypoxia reduces cellular ATP, depletes mitochondrial energy, elevates ADP/ATP ratio mainly in the heart muscle cells, and induces oxidative stress [7]. The increased generation of ROS can cause a decrease in the rate of ATP synthesis in mitochondria because of the loss of cytochrome oxidase (Cox) activity. Cox is a chain in the mitochondrial respiratory process, which generates ATP by oxidative phosphorylation. The inhibition of Cox appears to be the main reason for the ATP depletion. A reduced capacity of Cox will cause an increase in ROS production and decrease ATP synthesis [5, 6, 8].
The imbalance between the supply of oxygen and its demand determines organ hypoxia. It occurs mainly in ischemia and heart diseases and leads to numerous deleterious effects and finally resulting in death [9]. Hypoxia causes oxidative stress involving the production of reactive oxygen species [6, 10]. It has been confirmed that the compounds with antioxidant activity can scavenge ROS and are able to show the antihypoxic property [11].
MgSO4 is a well-known cation that is vital for cell viability. To the best of our knowledge, there is no report about the protective effects of this ion against lethality and mitochondria oxidative brain damage induced by hypoxia. The present work is aimed at determining the antihypoxic activities and neuroprotective effects of MgSO4 to understand a possible mechanism of its action in hypoxia-induced lethality and brain mitochondria oxidative damage protection induced by different models of hypoxia.
2. Material and Methods
2.1. Chemicals
All chemical agents were prepared from Sigma-Aldrich Company and were used in this study.
2.2. Animals
Male Swiss albino mice (21 ± 3 g) were housed in polypropylene cages at 25 ± 1°C and 45-55% relative humidity, with a 12 h light : 12 h dark cycle (lights on at 7 a.m.). The animals had free access to standard pellet and water ad libitum. Experiments were conducted between 8:00 and 15:00 h. All the experimental procedures were performed following the NIH guidelines of the Laboratory Animal Care and Use. The Institutional Animal Ethical Committee of Mazandaran University of Medical Sciences also approved the experimental protocol (with ethics number IR.MAZUMS.REC.93.1001).
2.3. Animal Treatment
All of the 208 male mice were randomly divided into 26 groups (eight in each group). Thirteen groups were used for hypoxia-induced methods, and 13 groups were used for the evaluation of brain mitochondria oxidative stress biomarkers after induction of different hypoxia methods.
2.4. Asphyctic Hypoxia
The animals were subjected to hypoxia by putting them individually in a tightly closed 300 ml glass container, which was placed underwater in an aquarium of 25°C. The animals had convulsions and died from hypoxia. The latencies for death were recorded. The animals died approximately 1.5-2 min following convulsions. Mice received single i.p. injections of 300, 400, and 500 mg/kg doses of MgSO4 or phenytoin (50 mg/kg) as 30 min before they were subjected to hypoxia. The control group was treated with normal saline [12].
2.5. Haemic Hypoxia
Forty mice were divided into five groups, each containing eight mice. Mice were injected with sodium nitrite (NaNO2) in dose 360 mg/kg i.p. thirty minutes after the i.p. administration of 400, 500, or 600 mg/kg doses of MgSO4. The survival time (in min) for each animal was defined as time, measured from the hypoxia induction, caused by the introduction of hypoxia until death. The control group was treated with normal saline.
2.6. Circulatory Hypoxia
Forty mice were divided into five groups, each containing eight mice. The groups were treated with normal saline. Thirty minutes after i.p. administration of 400, 500, and 600 mg/kg doses of MgSO4, NaF (150 mg/kg) was applied i.p. to mice, and antihypoxic activity was estimated in minutes as the latent time of evidence of hypoxia [13].
2.7. Preparation of the Brain Mitochondria
All animals were sacrificed by cervical decapitation after seven minutes (in haemic and circulatory models) and 25 minutes in the asphyctic model based on our above results. Then, brain tissues were homogenized, and mitochondria were extracted from mouse brains using differential centrifugation. Tris buffer (0.05 M Tris-HCl, 20 mm KCl, 0.25 M sucrose, 2.0 mM MgCl2, and 1.0 mM Na2HPO4, pH = 7.4) was used for suspending of the final mitochondrial pellets. For ROS production assessment, mitochondria were suspended in respiration buffer (0.32 mM sucrose, 20 mM Mops, 10 mM Tris, 0.5 mM MgCl2, 50 μM EGTA, 0.1 mM KH2PO4, and 5 mM sodium succinate). All procedures were done at 4°C [14].
2.8. Protein Concentration
The Coomassie blue protein binding method [15] was used for the measurement of mitochondria protein content in the samples.
2.9. Lipid Peroxidation (LPO) Measurement
MDA malondialdehyde content was measured by the Zhang et al. method [16]. Briefly, phosphoric acid (0.25 ml, 0.05 M) was added to mitochondrial fractions (0.2 ml, 0.5 mg protein/ml), and then, thiobarbituric acid (TBA) (0.3 ml, 0.2%) was added to them. Afterward, the microtubes were located (30 min) in a boiling water bath. In the end, the samples were placed in an ice bath, and n-butanol (0.4 ml) was added to each sample. Finally, samples were centrifuged at 3500 × g for 10 min, and the MDA formed in the supernatant was determined at 532 nm with an ELISA reader (Tecan, Rainbow Thermo, Austria). Tetramethoxypropane as standard was used in this test.
2.10. Protein Carbonyl (PC) Determination
Protein carbonyl content in brain mitochondria was assessed by the spectrophotometric method. Briefly, 500 μl of 20% (w/v) trichloroacetic acid (TCA) was added to the samples (0.5 mg mitochondrial protein/ml). Afterward, the samples were stored at 4°C for 15 min. The precipitates of the samples were treated with 500 μl of 0.2% DNPH (2,4-dinitrophenylhydrazine) and incubated for 1 hour at room temperature. 55 μl of 100% TCA was used for precipitation of the proteins. Samples were centrifuged and washed with 1000 μl of the ethanol-ethyl acetate (1 : 1, v/v) mixture. Guanidine hydrochloride (200 μl, 6 M) was added to the samples, and the content of the carbonyl created was calculated by reading the absorbance at 365 nm wavelength [14].
2.11. Determination of GSH Content
The content of the GSH in the isolated mitochondria was measured using DTNB by the spectrophotometer method. The developed yellow color by DTNB was recorded at 412 nm. GSH content in each sample was expressed as μM [17].
2.12. Assessment of Mitochondrial Toxicity
Mitochondrial toxicity was assessed as an evaluation of mitochondria function in samples by measuring the reduction of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) with minor modification of Ghazi-Khansari et al. [18].
2.13. Statistical Analysis
Data were presented as mean ± SD. Analysis of variance (ANOVA) was performed. Newman-Keuls multiple comparison tests were used to determine the differences in means. All P values less than 0.05 were regarded as significant.
3. Results
The results of the asphyctic hypoxia are shown in Figure 1. The effect was dose dependent. MgSO4 at doses of 500 and 600 mg/kg significantly (P < 0.05 and P < 0.001) prolonged the latency for death concerning the control group. At dose 600 mg/kg, MgSO4 showed the same activity as phenytoin, which was used as a positive control (P > 0.001). Even at the lowest tested dose, 400 mg/kg, it has prolonged survival time (30.20 ± 5.27 min), but this prolongation was not statistically significant from the control (P > 0.05).
Figure 1.
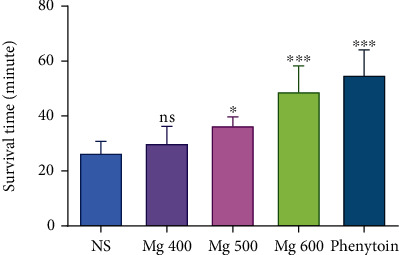
Antihypoxic activities of different doses of MgSO4 in the asphyctic hypoxia method in mice. Data are expressed as mean ± SD (n = 8).
MgSO4 showed good activity in the haemic model (Figure 2). The control group died by induced haemic hypoxia at 7.40 ± 0.46 min. MgSO4, at all tested doses, showed statistically significant activity concerning the control. This effect was dose dependent. At the highest measured dose, 600 mg/kg, it has prolonged latency for death to 13.72 ± 2.88 min, which was statistically (P < 0.01) significant from the control.
Figure 2.
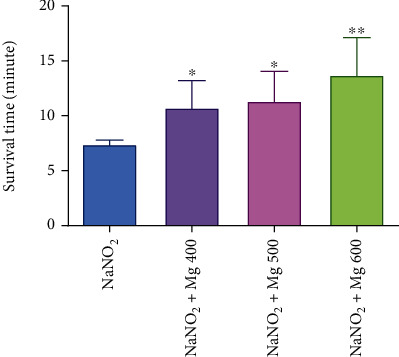
Antihypoxic activities of MgSO4 at different doses in induced haemic hypoxia in mice. Data are expressed as mean ± SD (n = 8); ∗P < 0.05 and ∗∗P < 0.01 compared to control (NaNO2).
The results of the circulatory hypoxia are shown in Figure 3. MgSO4 at doses of 500 and 600 mg/kg showed the most potent effect. It significantly prolonged the latency for death concerning the control group (>13.17 ± 2.06vs. 6.3 ± 0.74 min, P < 0.001). At these doses, it showed doubled survival time, and the effects were dose dependent. At the lowest tested dose (400 mg/kg), it also kept mice alive for 7.28 ± 1.40 min, but this effect was not statistically significant compared to the control (P > 0.05).
Figure 3.
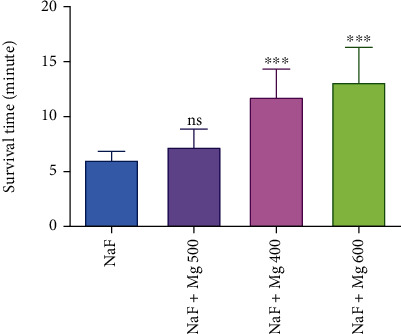
Antihypoxic activities of different doses of MgSO4 in circulatory-induced hypoxia in mice. Data are expressed as mean ± SD (n = 8); ns: not significant; ∗∗∗P < 0.001 compared to control (NaF).
MDA as a marker of lipid peroxidation was assayed in the brain mitochondria of mice following different methods of induced hypoxia. As shown in Table 1, the amount of MDA formation in mitochondria was decreased significantly (P < 0.05) by MgSO4 (500 and 600 mg/kg) in haemic induced hypoxia and by MgSO4 (600 mg/kg) in hypoxia induced by asphyctic and circulatory methods when compared with the control group of each.
Table 1.
Evaluation effects of different doses of MgSO4 on lipid peroxidation, protein carbonyl formation, and glutathione content against induced hypoxia by different methods.
| Parameter ⟶ | Group | LPO (μg/mg protein) | PC (μg/mg protein) | GSH (μg/mg protein) |
|---|---|---|---|---|
| Hypoxia type ↓ | ||||
| Asphyctic | Con (NS) | 0.8770 ± 0.036 | 1.320 ± 0.097 | 0.270 ± 0.021 |
| Phenytoin | 0.7560 ± 0.033 | 0.8527 ± 0.035∗ | 0.1187 ± 0.010∗ | |
| Mg 400 | 0.3000 ± 0.096 | 0.7573 ± 0.035∗ | 0.1813 ± 0.045 | |
| Mg 500 | 0.6270 ± 0.027 | 0.7883 ± 0.028∗ | 0.2123 ± 0.042∗ | |
| Mg 600 | 0.5020 ± 0.210∗ | 0.6873 ± 0.188∗∗ | 0.1233 ± 0.007 | |
|
| ||||
| Circulatory (NaF) | Con (NS+NaF) | 0.3468 ± 0.038 | 1.532 ± 0.426 | 0.1174 ± 0.011 |
| NaF+Mg 400 | 0.2307 ± 0.026 | 0.4417 ± 0.032∗ | 0.1077 ± 0.005 | |
| NaF+Mg 500 | 0.2470 ± 0.062 | 0.5247 ± 0.074∗ | 0.1143 ± 0.014 | |
| NaF+Mg 600 | 0.1470 ± 0.010∗ | 0.7007 ± 0.060 | 0.1163 ± 0.004 | |
|
| ||||
| Haemic (NaNO2) | Con (NS+NaNO2) | 0.6373 ± 0.039 | 1.532 ± 0.426 | 0.148 ± 0.022 |
| NaNO2+Mg 400 | 0.4913 ± 0.044 | 0.4343 ± 0.118∗ | 0.0950 ± 0.006 | |
| NaNO2+Mg 500 | 0.2920 ± 0.051∗ | 0.8483 ± 0.192 | 0.0846 ± 0.004∗ | |
| NaNO2+Mg 600 | 0.2860 ± 0.112∗ | 1.081 ± 0.152 | 0.0993 ± 0.005 | |
Data are presented as mean ± SD of 6 animals in each group. ∗P < 0.05 and ∗∗P < 0.01: significantly compared with each control group.
Due to previous findings on the beneficial effects of magnesium in mitochondrial function, we decided to assess the probable benefit role of this metal on the mitochondrial antioxidant status. Mitochondrial GSH was measured in isolated brain mitochondria after induced hypoxia with different methods. Levels of glutathione contents were significantly (P < 0.05) increased in the phenytoin and MgSO4 (500 mg/kg) groups compared to control in asphyctic condition, and it was increased in the MgSO4 (500 mg/kg) group in haemic (NaNO2) hypoxia condition compared to control (NS+NaNO2) group (Table 1). Protein carbonyl as a marker of protein oxidation in mitochondria was measured in this study. Compared to each control group (NS, NS+NaF, and NS+NaNO2), PC significantly increased by treatment with phenytoin, MgSO4 (400, 500, and 600 mg/kg) in asphyctic, MgSO4 (400 and 500 mg/kg) in circulatory, and MgSO4 (400 mg/kg) in haemic hypoxia groups (Table 1).
As shown in Figures 4 and 5, MTT reduction to Formosan, as a mitochondrial toxicity assessment, significantly (P < 0.05) increases by MgSO4 (600 mg/kg) in the hypoxia induced via asphyctic and haemic methods (44% and 66%, respectively). Mitochondria function was increased significantly (P < 0.05) by all doses of MgSO4 (23%, 33%, and 67%) in circulatory hypoxia when compared to the control group (Figure 6).
Figure 4.
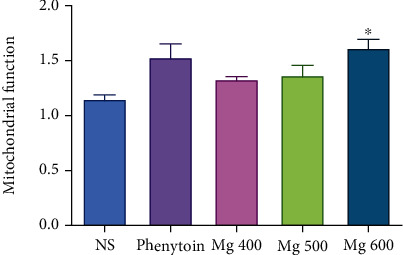
Effect of different doses of MgSO4 and phenytoin on mitochondrial function after induced hypoxia by the asphyctic method. Values represented as mean ± SD (n = 8). ∗P < 0.05 compared with the control group (NS).
Figure 5.
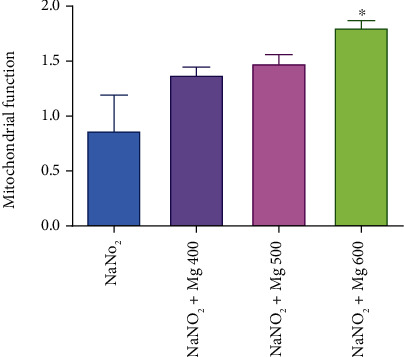
Effect of different doses of MgSO4 on mitochondrial function after induced hypoxia by the haemic (NaNO2) method. Data are presented as mean ± SD (n = 8). ∗P < 0.05 compared with the control group (NaNO2).
Figure 6.
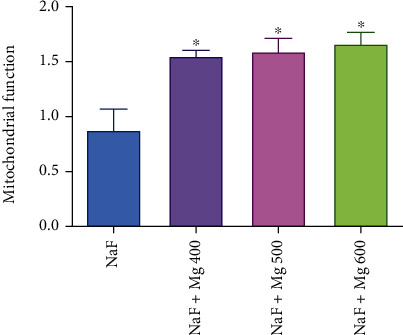
Effect of different doses of MgSO4 on mitochondrial function after induced hypoxia by the circulatory (NaF) method. Data are presented as mean ± SD (n = 8). ∗P < 0.05 compared with the control group (NaF).
4. Discussion
This study was undertaken to evaluate how probably the administration of MgSO4 would reduce oxidative damage in hypoxia induced with different methods. The results illustrated that MgSO4 improved oxidative stress induced by different hypoxia in a proper manner.
Hypoxia produces intense physiologic stress and induces a wide range of lethal effects at the cellular level. The brain with consumption of a large amount of oxygen is very vulnerable to the low levels of oxygen [11] because it has a high content of polyunsaturated fatty acids, which easily undergo oxidation [19]. The animals' survival time in a sealed container reflects the antihypoxic activity directly.
The lack of oxygen in the brain leads to inevitable changes in the structure and function of cerebral tissue. Thus, any medication that allows the brain to fight the consequences of ischemia or hypoxia would be a great therapeutic agent [19]. Throughout the past decades, a variety of different experimental models have been developed that could be used for testing antihypoxic and anti-ischemic drug effects in vivo.
Free radicals make the role of signaling species in different standard physiological procedures, but excessive production of such radicals leads to biological material damage. The elevated levels of ROS in hypoxia are the result of the accumulation of reducing equivalents in the mitochondrial electron transport system [20]. The ROS effects can be mainly evident in specific tissues such as the brain because it uses about one-fifth of the basal oxygen [21]. Given the efforts in developing treatments to reduce the oxidative stress effects, evidence indicates the protecting role of antioxidants in a variety of diseases [22].
In this study, statistically significant antihypoxic activities were established in some doses of MgSO4 in all experimental models of induced hypoxia in mice. The effects were dose dependent. Nearly at all tested doses, MgSO4 statistically showed significant activity with respect to the controls. A relationship between oxidative metabolism and cholinergic function has been reported during the investigations of NaNO2 on brain metabolism [23].
Chemical hypoxia induced by the injection of NaNO2 will reduce the blood oxygen capacity by changing hemoglobin to methemoglobin. The deadly dose is injected 30 min after the MgSO4 therapy. Immediately after injection, the animals are positioned in small cages, and the time between the NaNO2 injection and respiration cessation is recorded. Magnesium sulfate indicated an excellent activity in the haemic model (Figure 2).
There are literature data that the administration of NaF, which induces circulatory hypoxia, increases the blood histamine content and decreases the oxygen-carrying capacity. MgSO4 at 600 mg/kg showed the highest activity. The mechanism of induced protective action might be due to the MgSO4 antioxidant activity. Because there is no standard drug for haemic and circulatory hypoxic models, the results of this study were compared to those of control groups.
Mg is the most abundant cation with antioxidative properties that exist in more than 300 enzymatic systems. Mg is necessary for ATP activity and is vital in different processes, such as current the calcium across and within the cell membranes in tissues [1, 2].
Tissue hypoxia can induce cell death by inhibition of the activity of complexes I to IV in the mitochondria respiratory chain. Moreover, the reduction of mitochondrial transmembrane potential and depletion of intracellular ATP contents can occur, which finally elevates the ADP/ATP ratio in the cell [24, 25]. The consequence of these processes lactate dehydrogenase releases and damaging the cell membrane integrity can occur. It was demonstrated that hypoxia induces acidosis and depletion of mitochondrial energy in cells. Induced oxidative damage leads to the release of proteolytic enzymes and DNA fragmentations, processes which lead to cell death [26, 27]. It was reported that Mg has a beneficial role in the prevention of cell death by inhibiting calcium accumulation and improving cell metabolism [5]. The present results indicated significant improvement in oxidative biomarkers when MgSO4 was applied to induced hypoxic mice. The mechanism of Mg as an antioxidant defence system is not so precise and is still a matter of debate, but the beneficial function of Mg in oxidative stress and damage to molecules and cells has been proven by several studies [28, 29]. Hypoxia can induce ROS production that causes oxidative damage to molecules, cell organelles, and tissues. Many authors speculate that an imbalance between pro- and antioxidants results in an increased level of oxidative degradation of biomolecule products, such as lipid peroxidation products [30]. In our study, different models of hypoxia have been used to evaluate the cellular events associated with hypoxic injury. We induced hypoxia via various methods such as asphyctic, haemic (NaNO2), and circulatory (NaF) in mice.
In the haemic model, NaNO2 induces methemoglobinemia, and in the circulatory model, NaF increases the blood histamine content and decreases the oxygen-carrying capacity and finally inhibits mitochondrial respiration, which demonstrated that more than 90% of Mg2+ in the cell is bounded to adenine nucleotides, especially ATP [5]. Chemical hypoxia is distinguished by a reduction of electron carriers in mitochondria and depletion of cell ATP leading to cell ROS formation and, eventually, disruption of the plasma membrane permeability barrier with failure of cell viability [31]. It has been reported that inhibition of ATP-dependent ion transporters after ATP depletion associated with hypoxic conditions results in variation in the levels of cytosolic ions. After the hypoxia onset, a rise in cytosolic free Ca2+ occurs, which activates Ca-dependent degradative enzymes that lead to cell death. Also, chemical hypoxia can cause a significant increase in free Na+ and H+ in the cell [32–34]. It was stated that during hypoxia, oxygen-free radicals are produced, and peroxidation of proteins and membrane lipids contributes to cell damage. Administration of MgSO4 to the induced hypoxic mice showed significant attenuation of oxidative stress biomarkers compared to the control groups. Magnesium sulfate is a noncompetitive and voltage-dependent antagonist of the N-methyl-D-aspartate (NMDA) receptor-ion channel that may block all pathways and has been revealed to be neuroprotective in patients suffering from acute stroke and/or in various animal models of brain injury. Moreover, it was demonstrated that pretreatment with MgSO4 attenuates ROS generation and oxidative damage in the brain cells following induced hypoxia [35]. Our results are compatible with other studies that have shown MgSO4 decreases oxidative damage in the brain. The observations of oxidative damage to the membrane simultaneous with mitochondrial dysfunction lend credence to the assumption that an alteration of the mitochondrial membrane is a critical step in hypoxia-induced cell death. It seems that the administration of MgSO4 prevents the fall of Mg content in the brain and attenuates the hypoxia-induced neuronal mitochondria membrane damage. These results may appear to be a conflict with previous studies indicating no effect of MgSO4 in induced hypoxia on cerebral injury in animals [36, 37].
In the present study, we demonstrated that pretreatment of mice with MgSO4 before hypoxia induction not only attenuates the increased oxidative damage in the mouse brain but also improves mitochondria function by different doses of MgSO4 which is an essential distinction of our investigation with the previous studies. It was stated that irreversible brain damage in the acute phase of hypoxia could be due to primarily necrotic changes. In regard to hypoxic tissue, diminished cytosolic Mg2+ is associated with increased damage and formation of ROS while increases are related to the reverse [35]. The mechanism that Mg exerts its antioxidative effect is unclear, but it may be associated with inhibition of iron-driven lipid peroxidation [38]. Another feasibility is that Mg2+ blocks the excessive influx of Ca2+ by binding to the NMDA receptor ion channel, known as a trigger of oxygen free radical producing pathways, such as phospholipases, cyclooxygenase, and lipoxygenase [39]. It has been reported that Mg2+ alters the neuron sensitivity to an oxidative insult [40].
Our results are in agreement with prior studies that have indicated MgSO4 has a beneficial role in the decrease of oxidative stress biomarkers [6, 25, 35, 41]. It has been demonstrated that blockade of the ionic conductance and preventing of calcium ion influx during the NMDA receptor are the mechanism of MgSO4 prophylactic action in hypoxia [42]. Administration of MgSO4 before or after the exposure to hypoxia prevents the reduction in ATP levels in cerebral tissue [43]. It has been shown that acidosis can be created after hypoxia in cell tissues and consequence of that depletion of mitochondrial energy, induction of proteolytic enzymes, and oxidative damage can occur that finally result in cell death [44]. Therefore, magnesium improves myocardiocyte metabolism and energy, inhibits the accumulation of calcium, and ultimately prevents myocardial cell death, which enhances the delivery of oxygen to the hypoxic tissues, especially the brain, and prevents the cells from hypoxic damage. Moreover, regarding different beneficial results reported for Mg and magnetic Mg (25mg) via increase of the creatine kinase activity (2- to 4-fold) and ATP level, it is obvious that Mg by enhancement of both substrate and oxidative phosphorylation pathways can increase the ATP synthesis and prevent the oxidative damage and cell death [41, 45].
It was previously confirmed that following hypoxia, ROS generation, protein, and lipid peroxidation of the neuronal cell membrane and cell membrane dysfunction were increased in the animal's brain [35]. The present changes in LPO, GSH, PC, and mitochondrial function in our study support oxidative damage potential of induced hypoxia by different methods (Table 1 and Figures 4–6). Previous studies have shown that the generation of various free radicals such as nitric oxide, as a consequence of hypoxia, can increase in the cell [46]. Therefore, complex processes such as ATP depletion, the release of excitatory amino acids, the formation of ROS, and alkoxyl radicals are the series of intracellular events due to hypoxic cell injury. Moreover, previous studies have demonstrated that Mg is involved in ATP-dependent pump activity such as Na/K-ATPase and Ca-ATPase, which are sensitive to membrane lipid peroxidation [47, 48]. Therefore, Mg is useful in balancing the ATP and reduction of oxidative stress and has positive effects on cellular hypoxia. Practically, a dose of 600 mg/kg of MgSO4 showed the effective potential of antioxidants. Our findings showed that MgSO4 at dose 600 mg/kg improves hypoxia-induced oxidative stress status in mitochondria much better than other doses which may be due to more improvement of intracellular Mg levels and mitochondrial ATP.
Finally, this study demonstrated that during hypoxia, neuronal mitochondria oxidative damage can occur associated with a decrease of survival time in mice. Pretreatment with MgSO4 attenuated protein and lipid peroxidation and increased mitochondrial function in mice afflicted by different methods of hypoxia. The results of this study support the conclusion that Mg may be of worth in increasing survival time and preventing the mortality associated with asphyxiation.
5. Conclusion
MgSO4 showed an excellent protective effect against hypoxia in all tested models. Notably, it produced significant and dose-dependent beneficial effects on survival time and oxidative damage in different models of induced hypoxia.
Acknowledgments
The authors express their appreciation to the Vice-Chancellor for Research at Mazandaran University of Medical Sciences for the financial support of the current study.
Data Availability
The current study is the result of a dissertation code 437, School of Pharmacy, Mazandaran University of Medical Sciences, Sari, Iran. The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Disclosure
It is remarkable that the manuscript has been published as a preprint in bioRxiv database (DOI: 10.1101/2020.05.22.111435).
Conflicts of Interest
The authors declared no conflicts of interest.
References
- 1.Fox C., Ramsoomair D., Carter C. Magnesium: its proven and potential clinical significance. Southern medical journal. 2001;94(12):1195–1201. doi: 10.1097/00007611-200194120-00014. [DOI] [PubMed] [Google Scholar]
- 2.Garcia L. A., DeJong S. C., Martin S. M., Smith R. S., Buettner G. R., Kerber R. E. Magnesium reduces free radicals in an in vivo coronary occlusion-reperfusion model. Journal of the American College of Cardiology. 1998;32(2):536–539. doi: 10.1016/S0735-1097(98)00231-9. [DOI] [PubMed] [Google Scholar]
- 3.Pasternak K., Kocot J., Horecka A. Biochemistry of magnesium. Journal of Elementology. 2010;15:601–616. [Google Scholar]
- 4.Pilchova I., Klacanova K., Tatarkova Z., Kaplan P., Racay P. The involvement of Mg2+ in regulation of cellular and mitochondrial functions. Oxidative Medicine and Cellular Longevity. 2017;2017:8. doi: 10.1155/2017/6797460.6797460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fawcett W., Haxby E., Male D. Magnesium: physiology and pharmacology. British journal of anaesthesia. 1999;83(2):302–320. doi: 10.1093/bja/83.2.302. [DOI] [PubMed] [Google Scholar]
- 6.Spandou E., Soubasi V., Papoutsopoulou S., et al. Neuroprotective effect of long-term MgSO4 administration after cerebral hypoxia-ischemia in newborn rats is related to the severity of brain damage. Reproductive Sciences. 2007;14:667–677. doi: 10.1177/1933719107305864. [DOI] [PubMed] [Google Scholar]
- 7.Shechter M. Magnesium and cardiovascular system. Magnesium research. 2010;23(2):60–72. doi: 10.1684/mrh.2010.0202. [DOI] [PubMed] [Google Scholar]
- 8.Solaini G., Baracca A., Lenaz G., Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2010;1797(6-7):1171–1177. doi: 10.1016/j.bbabio.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 9.Kiang J. G., Tsen K. Biology of hypoxia. Chinese Journal of Physiology. 2006;49:p. 223. [PubMed] [Google Scholar]
- 10.Maiti P., Singh S. B., Sharma A. K., Muthuraju S., Banerjee P. K., Ilavazhagan G. Hypobaric hypoxia induces oxidative stress in rat brain. Neurochemistry International. 2006;49(8):709–716. doi: 10.1016/j.neuint.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Warner D. S., Sheng H., Batinić-Haberle I. Oxidants, antioxidants and the ischemic brain. Journal of experimental biology. 2004;207(18):3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- 12.Eslami B., Nabavi S., Nabavi S., Ebrahimzadeh M. A., Mahmoudi M. Pharmacological activities of Hypericum scabrum L. European Review for Medical and Pharmacological Sciences. 2011;15(5):532–537. [PubMed] [Google Scholar]
- 13.Ebrahimzadeh M. A., Nabavi S. F., Nabavi S. M., Eslami B. Antihypoxic and antioxidant activity of Hibiscus esculentus seeds. Grasas y Aceites. 2010;61(1):30–36. doi: 10.3989/gya.053809. [DOI] [Google Scholar]
- 14.Fathi H., Ebrahimzadeh M. A., Ziar A., Mohammadi H. Oxidative damage induced by retching; antiemetic and neuroprotective role of Sambucus ebulus L. Cell biology and Toxicology. 2015;31(4-5):231–239. doi: 10.1007/s10565-015-9307-8. [DOI] [PubMed] [Google Scholar]
- 15.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72(1-2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 16.Zhang F., Xu Z., Gao J., Xu B., Deng Y. In vitro effect of manganese chloride exposure on energy metabolism and oxidative damage of mitochondria isolated from rat brain. Environmental Toxicology and Pharmacology. 2008;26(2):232–236. doi: 10.1016/j.etap.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Sadegh C., Schreck R. P. The spectroscopic determination of aqueous sulfite using Ellman’s reagent. MURJ. 2003;8:39–43. [Google Scholar]
- 18.Ghazi-Khansari M., Mohammadi-Bardbori A., HOSSEINI M. J. Using janus green B to study paraquat toxicity in rat liver mitochondria: role of ACE inhibitors (thiol and nonthiol ACEi) Annals of the New York Academy of Sciences. 2006;1090(1):98–107. doi: 10.1196/annals.1378.010. [DOI] [PubMed] [Google Scholar]
- 19.Peruche B., Ahlemeyer B., Brungs H., Krieglstein J. Cultured neurons for testing antihypoxic drug effects. Journal of pharmacological methods. 1990;23(1):63–77. doi: 10.1016/0160-5402(90)90009-A. [DOI] [PubMed] [Google Scholar]
- 20.Bakonyi T., Radak Z. High altitude and free radicals. Journal of sports science & medicine. 2004;3:p. 64. [PMC free article] [PubMed] [Google Scholar]
- 21.Agranoff B. W., Siegel G. J. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. Raven Press; 1994. [Google Scholar]
- 22.Spencer J. P. Beyond antioxidants: the cellular and molecular interactions of flavonoids and how these underpin their actions on the brain. Proceedings of the Nutrition Society. 2010;69(2):244–260. doi: 10.1017/S0029665110000054. [DOI] [PubMed] [Google Scholar]
- 23.Gibson G. E., Shumada M., Blass J. P. Alterations in acetylcholine synthesis and cyclic nucleotides in mild cerebral hypoxia. Journal of Neurochemistry. 1978;31(4):757–760. doi: 10.1111/j.1471-4159.1978.tb00107.x. [DOI] [PubMed] [Google Scholar]
- 24.Fuhrmann D. C., Brüne B. Mitochondrial composition and function under the control of hypoxia. Redox biology. 2017;12:208–215. doi: 10.1016/j.redox.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maulik D., Zanelli S., Numagami Y., Ohnishi S. T., Mishra O. P., Delivoria-Papadopoulos M. Oxygen free radical generation during in-utero hypoxia in the fetal guinea pig brain: the effects of maturity and of magnesium sulfate administration. Brain research. 1999;817(1-2):117–122. doi: 10.1016/S0006-8993(98)01235-9. [DOI] [PubMed] [Google Scholar]
- 26.Regan R. F., Guo Y. Magnesium deprivation decreases cellular reduced glutathione and causes oxidative neuronal death in murine cortical cultures. Brain research. 2001;890(1):177–183. doi: 10.1016/S0006-8993(00)03156-5. [DOI] [PubMed] [Google Scholar]
- 27.Decking U. K., Schlieper G., Kroll K., Schrader J. ü. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circulation research. 1997;81(2):154–164. doi: 10.1161/01.RES.81.2.154. [DOI] [PubMed] [Google Scholar]
- 28.Hans C. P., Chaudhary D. P., Bansal D. D. Magnesium deficiency increases oxidative stress in rats. Indian Journal Experimental Biology. 2002;40(11):1275–1279. [PubMed] [Google Scholar]
- 29.Zheltova A. A., Kharitonova M. V., Iezhitsa I. N., Spasov A. A. Magnesium deficiency and oxidative stress: an update. Biomedicine. 2016;6(4):p. 20. doi: 10.7603/s40681-016-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacNee W., Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. 1999;160(supplement_1):S58–S65. doi: 10.1164/ajrccm.160.supplement_1.15. [DOI] [PubMed] [Google Scholar]
- 31.Kalogeris T., Baines C. P., Krenz M., Korthuis R. J. Cell biology of ischemia/reperfusion injury. International review of cell and molecular biology. 2012;298:p. 229. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kribben A., Wieder E. D., Wetzels J., et al. Evidence for role of cytosolic free calcium in hypoxia-induced proximal tubule injury. Journal of Clinical Investigation. 1994;93(5):1922–1929. doi: 10.1172/JCI117183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steenbergen C., Murphy E., Watts J. A., London R. E. Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischemic injury in perfused rat heart. Circulation research. 1990;66(1):135–146. doi: 10.1161/01.RES.66.1.135. [DOI] [PubMed] [Google Scholar]
- 34.Harman A. W., Nieminen A.-L., Lemasters J. J., Herman B. Cytosolic free magnesium, ATP, and blebbing during chemical hypoxia in cultured rat hepatocytes. Biochemical and biophysical research communications. 1990;170(2):477–483. doi: 10.1016/0006-291X(90)92116-H. [DOI] [PubMed] [Google Scholar]
- 35.Maulik D., Qayyum I., Powell S. R., Karantza M., Prakash Mishra O., Delivoria-Papadopoulos M. Post-hypoxic magnesium decreases nuclear oxidative damage in the fetal guinea pig brain. Brain research. 2001;890(1):130–136. doi: 10.1016/S0006-8993(00)03153-X. [DOI] [PubMed] [Google Scholar]
- 36.Penrice J., Amess P., Punwani S., et al. Magnesium sulfate after transient hypoxia-ischemia fails to prevent delayed cerebral energy failure in the newborn piglet. Pediatric Research. 1997;41(3):443–447. doi: 10.1203/00006450-199703000-00024. [DOI] [PubMed] [Google Scholar]
- 37.Sameshima H., Ota A., Ikenoue T. Pretreatment with magnesium sulfate protects against hypoxic-ischemic brain injury but postasphyxial treatment worsens brain damage in seven-day-old rats. American journal of obstetrics and gynecology. 1999;180(3):725–730. doi: 10.1016/S0002-9378(99)70279-6. [DOI] [PubMed] [Google Scholar]
- 38.Günther T., Vormann J., Höllriegl V. Effects of magnesium and iron on lipid peroxidation in cultured hepatocytes. Molecular and Cellular Biochemistry. 1995;144(2):141–145. doi: 10.1007/BF00944393. [DOI] [PubMed] [Google Scholar]
- 39.Lipton S. A., Nicotera P. Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium. 1998;23(2-3):165–171. doi: 10.1016/S0143-4160(98)90115-4. [DOI] [PubMed] [Google Scholar]
- 40.Regan R. F., Jasper E., Guo Y., Panter S. S. The effect of magnesium on oxidative neuronal injury in vitro. Journal of Neurochemistry. 1998;70(1):77–85. doi: 10.1046/j.1471-4159.1998.70010077.x. [DOI] [PubMed] [Google Scholar]
- 41.Mohammadi H., Karimi G., Mahdi Rezayat S., et al. Benefit of nanocarrier of magnetic magnesium in rat malathion-induced toxicity and cardiac failure using non-invasive monitoring of electrocardiogram and blood pressure. Toxicology and industrial health. 2011;27:417–429. doi: 10.1177/0748233710387634. [DOI] [PubMed] [Google Scholar]
- 42.Maulik D., Mishra O. P., Delivoria-Papadopoulos M. Effect of post-hypoxic MgSO4 administration in utero on Ca2+-influx and Ca2+/calmodulin kinase IV activity in cortical neuronal nuclei. Neuroscience Letters. 2005;386(2):127–132. doi: 10.1016/j.neulet.2005.05.064. [DOI] [PubMed] [Google Scholar]
- 43.Mami A. G., Ballesteros J., Mishra O. P., Delivoria-Papadopoulos M. Effects of magnesium sulfate administration during hypoxia on Ca2+ influx and IP 3 receptor modification in cerebral cortical neuronal nuclei of newborn piglets. Neurochemical Research. 2006;31(1):63–70. doi: 10.1007/s11064-005-9076-5. [DOI] [PubMed] [Google Scholar]
- 44.Chan J. Y., Chan S. H., Dai K., Cheng H. L., Chou J. L. J., Chang A. Y. W. Cholinergic receptor-independent dysfunction of mitochondrial respiratory chain enzymes, reduced mitochondrial transmembrane potential and ATP depletion underlie necrotic cell death induced by the organophosphate poison mevinphos. Neuropharmacology. 2006;51(7-8):1109–1119. doi: 10.1016/j.neuropharm.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 45.Shafiee H., Mohammadi H., Rezayat S. M., et al. Prevention of malathion-induced depletion of cardiac cells mitochondrial energy and free radical damage by a magnetic magnesium-carrying nanoparticle. Toxicology Mechanisms and Methods. 2010;20(9):538–543. doi: 10.3109/15376516.2010.518173. [DOI] [PubMed] [Google Scholar]
- 46.Mishra O., Zubrow A., Ashraf Q. Nitric oxide-mediated activation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) during hypoxia in cerebral cortical nuclei of newborn piglets. Neuroscience. 2004;123(1):179–186. doi: 10.1016/j.neuroscience.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Abad C., Teppa-Garrán A., Proverbio T., Piñero S., Proverbio F., Marín R. Effect of magnesium sulfate on the calcium-stimulated adenosine triphosphatase activity and lipid peroxidation of red blood cell membranes from preeclamptic women. Biochemical Pharmacology. 2005;70(11):1634–1641. doi: 10.1016/j.bcp.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 48.Chiarello D. I., Marín R., Proverbio F., et al. Effect of hypoxia on the calcium and magnesium content, lipid peroxidation level, and Ca2+-ATPase activity of syncytiotrophoblast plasma membranes from placental explants. BioMed Research International. 2014;2014:9. doi: 10.1155/2014/597357.597357 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The current study is the result of a dissertation code 437, School of Pharmacy, Mazandaran University of Medical Sciences, Sari, Iran. The data that support the findings of this study are available from the corresponding author, upon reasonable request.