

Abstract
The antitumor activities of triazine dendrimers bearing paclitaxel, a well-known mitotic inhibitor, are evaluated in SCID mice bearing human prostate cancer xenografts. To increase the activity of a first generation prodrug 1 that contained twelve paclitaxel molecules tethered via an ester linkage, the new construct described here, prodrug 2, tethers paclitaxel with linkers containing both an ester and disulfide. While PEGylation is necessary for solubility, and may improve biocompatibility and increase plasma half-life, it increases the heterogeneity of the sample with an average of eight to nine PEG chains (2 kDa each) incorporated. The heterogeneous population of PEGylated materials was used without fractionation based on models obtained from molecular dynamics simulations. Three models were examined; hexaPEGylated, nonaPEGylated, and dodecaPEGylated constructs. Intravenous delivery of prodrug 2 was performed by single, double or triple dosing regimes with doses spaced by one week. The administrated doses varied from 50 mg paclitaxel/kg to 200 mg paclitaxel/kg. Tumor growth arrest and regression was observed over the 10-week treatment period without mortality for mice treated with the 50 mg paclitaxel/kg triple dosing regime.
Keywords: Paclitaxel, triazine, dendrimer, prostate cancer, xenografts, drug delivery, molecular dynamics, simulation
Graphical Abstract
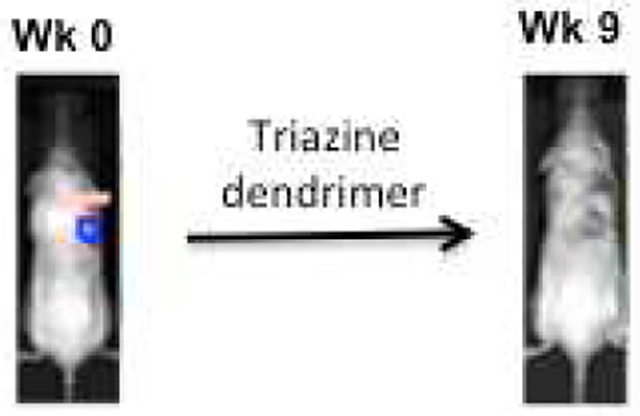
Efficacy is seen at 50 mg paclitaxel/kg mouse when administer once a week for three weeks.
Introduction
Paclitaxel (Taxol, PTX) is a well-known anti-mitosis agent that has been extensively studied since its discovery in the early 1960s due to its therapeutic efficacy against a wide range of cancers including those of the breast, ovaries, prostate, lung, bladder, esophagus, and head and neck.1–4 Initially, advances in clinical development were slowed by both the short supply and low solubility of paclitaxel. In the late 1980s, however, the development of a semisynthetic route5 to paclitaxel production as well as co-solvent systems6,7 to combat solubility challenges accelerated clinical studies with success. The co-solvents—Cremophor EL (CrEL) or polysorbate 80 formulations—introduced challenges with some patients reporting severe hypersensitivity reactions.7,8 Co-solvents also impact absorption upon oral administration, and affect pharmacokinetic and pharmacodynamic behaviors.9,10
The use of polymer vehicles11–18 represents one alternative to co-solvents wherein the drug can be associated with the polymer platform by covalent or non-covalent methods. For such applications, dendrimers19–22 stand among one of the most promising scaffolds for drug delivery because they are structurally and compositionally well-defined, multivalent, and often orthogonally functionalized. Dendrimer vehicles may be able to deliver high payloads of active drugs to target areas with reproducible therapeutic efficacy either with or without additional groups of interest (targeting or diagnostic). Ranging from 2–10 nanometers in diameter, these macromolecules may intrinsically accumulate in solid tumors due to the so-called enhanced permeability and retention (EPR) effect23,24 which derives from a leaky tumor vasculature and impaired lymphatic drainage system.
In comparison with other polymers, dendrimers have been less popular for delivery of paclitaxel presumably due to the difficulties arising in preparation of the drug constructs. Triazine dendrimers have advantages as drug vehicles since they are readily prepared, nearly monodisperse, stable during long-term storage, potentially functionalized with multiple ligands, and available at kilogram scale for lower generations.25–28 Previously, we reported synthesis of triazine dendrimers containing 12 paclitaxel groups.29 Paclitaxel was attached by ester linkage for prodrug 1, while prodrug 2 contains both ester and disulfide linkages. Prior to PEGylation, the composition was readily described. Indeed, prodrugs 1 and 2 appear to be single chemical entity materials before PEGylation. Upon PEGylation with 2 kDa PEGs using PEG-NHS esters, heterogeneity is introduced. Prodrugs 1 and 2 displayed an average of eight to nine PEG chains. Both prodrugs 1 and 2 carry a high drug payload (26–30 wt% drug).
Of the many factors to be considered in the design, synthesis and assessment of a nanomedicine, our current focus is on its biocompatibility, systemic toxicity, targeting, controlled release, and dosing plan to achieve an efficacious therapy. This multi-dimensional space can be reduced to a more manageable size by considering benchmarks set by the clinical standards of care. For paclitaxel, these standards are Taxol30–32 and Abraxane.11,33,34 Apart from basic research, the long term value of the triazine targets rests in competitive advantage in dose size, schedule and/or route of delivery when compared to these clinical standards.
We recently reported on studies of the efficacy and toxicity of prodrug 1.35 As summarized in Table 1, the drug was administered via i.v. injection through the tail vein of SCID mice bearing PC-3-h-luc xenografts with a single or double dosing (spaced by 4 days) of 100 mg of PTX/kg or 200 mg of PTX/kg. During the 10-week treatment period, mice dosed with 1 showed tumor regression or suppression at all doses when compared with the control group as monitored by noninvasive bioluminescence imaging (BLI). The double dosing of 200 mg/kg was most efficacious, but the tumor measured by BLI was still present at the end of treatment and the group suffered a mortal rate of 40%. Of note, these dosages are 3–6 times greater than those of paclitaxel formulated as Abraxane or Taxol in similar murine models.11 To achieve an efficacious therapy and in the meanwhile reduce the exposure to the nanomedicine scaffold, we sought to achieve a similar efficacy at 50mg/kg. We hypothesized that drug release is one of the important factors influencing efficacy.
Table 1.
Dosage Regimen of Prodrugs 1 and 2 in Mice Bearing Human Prostate Cancer Xenografts and Mortality during the Treatment. Prodrug 1 data was reported earlier,35 but is included for comparison.
| Drug | Dosage (mg PTX/kg) | Treatment (i.v. tail vein injection) | Name | Study duration | Mortality |
|---|---|---|---|---|---|
| Prodrug 1 | 100 | Single dose | 1–100s | 10 weeks | 0% |
| 100 | Double dose | 1–100d | 0% | ||
| 200 | Single dose | 1–200s | 20% | ||
| 200 | Double dose | 1–200d | 40% | ||
| Prodrug 2 | 50 | Double dose | 2–50d | 10 weeks | 0% |
| 50 | Triple dose | 2–50t | 0% | ||
| 100 | Single dose | 2–100s | 40% | ||
| 100 | Double dose | 2–100d | 40% | ||
| 200 | Single dose | 2–200s | 60% |
Here, we address this challenge using prodrug 2. This prodrug is expected to be more toxic than 1 because paclitaxel is tethered by two different bio-degradable linkages (ester and disulfide). We hypothesized that disulfide-mediated release would ultimately increase the amount of available free drug (Chart 1 and Table 1). Indeed, release mediated by disulfide cleavage with an endogenous reductant like glutathione, cysteine or homocysteine whose intracellular concentrations can exceed those of the vasculature by 103 is an increasingly attractive strategy in nanomedicines.36 Molecular dynamics (MD) simulations helped to reinforce this model—the linkages appear to be assessable to reductants over a range of PEGylation states. Discussions of these simulations and the therapeutic efficacy and toxicity profile of prodrug 2 follow.
Chart 1.
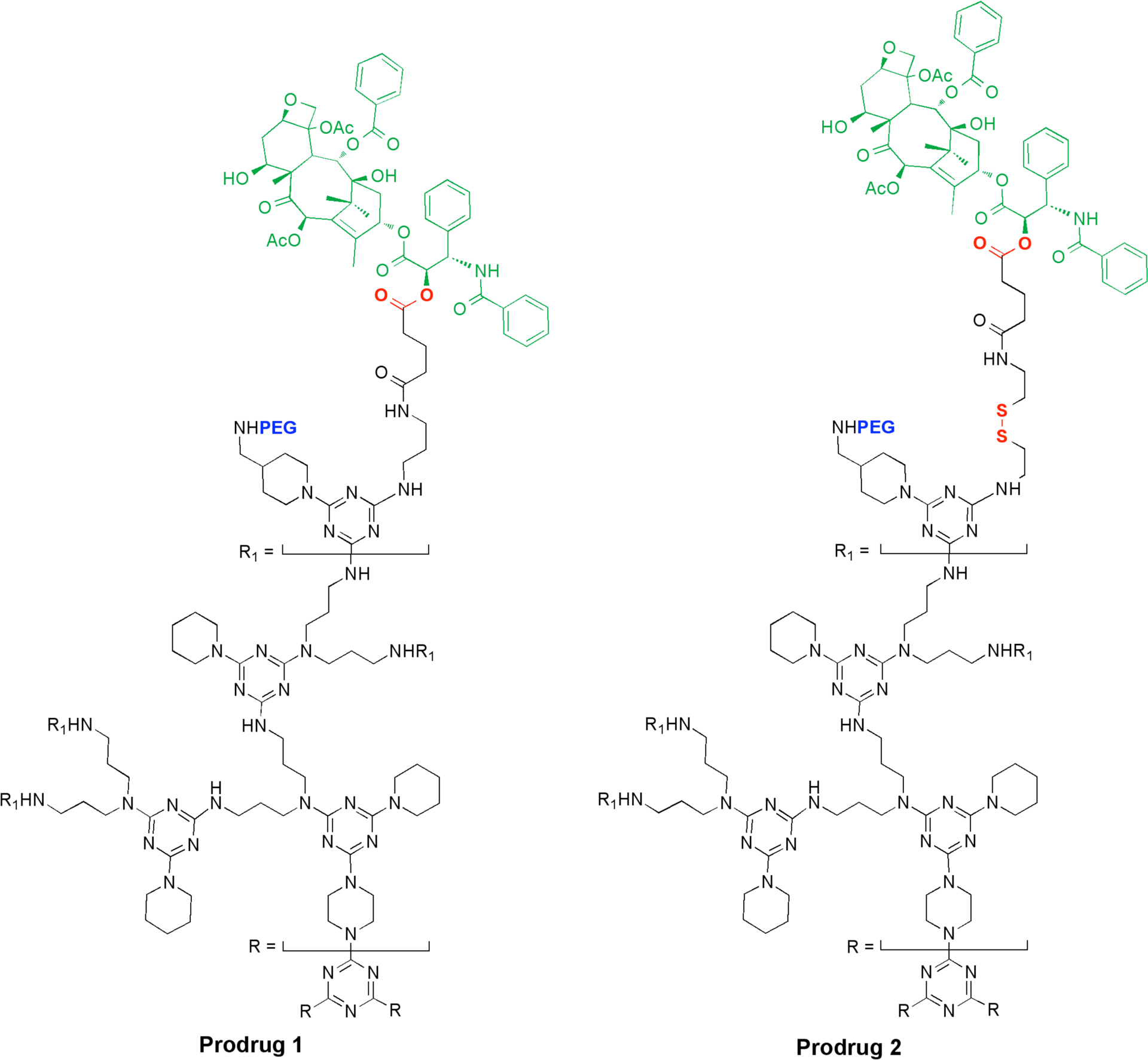
Chemical Structure of Prodrug 1 and 2: Paclitaxel is shown in green with biolabile esters and disulfides in red. The 2 kDa PEG chains are indicated in blue. The triazine dendrimer core is shown in black. Idealized structures contain 12 paclitaxel and PEG groups
Experimental Section
Synthesis of Prodrugs.
The prodrugs were prepared and characterized by published methods.29 Characterization rested on NMR spectroscopy, MALDI-TOF mass spectrometry, and gel permeation chromatography. Prodrug 2 has average 8–9 PEG chains and 12 paclitaxel groups (as determined by NMR and mass spectrometry) installed on a second generation dendrimer. Prodrug 2 is assigned as 28% wt drug.
Animals and Cell Lines.
All animals were handled according to the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of University of Texas Southwestern Medical Center at Dallas (UT Southwestern). These animals were kept under defined-flora pathogen-free condition at the animal facility of UT Southwestern. Human prostate cancer cell line, PC3-h-luc, which stably expressed firefly luciferase was maintained in T-medium with 10% fetal bovine serum, 100U/ml penicillin, and 100 μg/ml of streptomycin. Studies involving tumor-bearing animals were performed by rigorously following the UT Southwestern guidelines on tumor burden in rodents. Mice were sacrificed immediately when the tumor burden was measured more than 10% of total body weight.
Human Tumor Xenograft Model.
Twenty nine male SCID mice with 6–8 weeks of age were purchased from the animal breeding facility of UT Southwestern. The human prostate cancer xenograft model was established by subcutaneous injection of 2 ×106 PC3-h-luc cells in 100 μL into the back of each mouse. Tumors were allowed to grow for three weeks before starting the treatment when the size of tumors ranged from 1.5 – 125 mm3.
Therapeutic Efficacy.
The 29 tumor-bearing mice were randomized into 6 groups: five treatment groups and one control group (Table 1). Mice in the treatment groups were intravenously administered with prodrug 2 via the tail vein (2–50d: double doses of 50 mg of PTX/kg; 2–50t: triple doses of 50 mg of PTX/kg; 2–100s: single dose of 100 mg of PTX/kg; 2–100d: double doses of 100 mg of PTX/kg; and 2–200s: single dose of 200 mg of PTX/kg). The multiple doses were spaced one week. The control mice were injected with phosphate-buffered saline (PBS) only. During the course of treatment, the tumor volume was measured by caliber every other day and calculated with the equation: volume = (π/6) W2 × L, where “L” indicates the longest diameter of the tumor and “W” is the tumor width perpendicular to the “L”. Tumor volume change was analyzed by the tumor volume ratio (tumor volume / day 0 tumor volume) change in each animal. In addition, the animal whole body weight was monitored every other day for general toxicity evaluation. The tumor viability was evaluated using a Xenogen Bioluminescence Imaging system (Xenogen Corp., Alameda, CA).
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism. A p value less than 0.05 (unpaired two-tailed t test) was considered statistically significant. All results are presented as mean ± standard deviation. The survival rates of the treatment groups were compared using Mantel-Cox test, showing that the dosing of 200s significantly decreased the survival rate (p = 0.0494) while other dosing groups did not.
Methods and Instruments in Molecular Dynamics Simulation.
The molecular models for the hexaPEGylated, nonaPEGylated and dodecaPEGylated pacitaxel dendrimers were created using Material Studio 5.0 (Accelrys, San Diego, CA, USA). The dendritic structures were composed of different residues according to previous studies on similar dendrimers.37 Since the triazine dendrimers are symmetrical and grows around a three-branched core, the degree of PEGylation was assumed to be uniformly spread on the three branches (i.e. 2, 3 and 4 PEG chains decorating the surface of hexaPEGylated, nonaPEGylated and dodecaPEGylated dendrimers respectively). All of the nonstandard residues appearing in the dendrimers were obtained according to a well-validated procedure adopted in precedence by our group for the simulation of dendrimers38,39 and dendrons.40,41 At pH 7.4, each free surface amine of the dendrimers was considered to be protonated, for this reason, due to surface PEGylation (partial or complete), dendrimers decorated with 6, 9 and 12 PEG chains carried +6 e, +3 e and 0 e global charge respectively. All calculations were conducted using the AMBER 11 suite of programs.42 The three dendrimers were solvated in a TIP3P43 periodic water box with 14 Å of buffer, and a suitable number of Cl− and Na+ ions were added to the system with the leap module of AMBER 11 to guarantee overall neutrality and to reproduce the experimental salt concentration of 150 mM NaCl. The systems were first minimized and then equilibrated for 20 ns in NPT condition at the temperature of 300 K and pressure of 1 atm until all of the molecular systems converged to the equilibrium with good stability. Simulations were conducted using a time step of 2 fs, a Langevin thermostat, a 10 Å cut-off, the particle mesh Ewald44 (PME) approach for long-range electrostatic interactions and the SHAKE algorithm.45,46 The parm99 all-atom force field by Cornell et al.47 was used for all of the standard residues present in the systems.
The solvation energies ΔGsol for the three dendrimers were calculated over 200 snapshots taken from the equilibrated phase of the dynamic trajectories according to the MM-PBSA approach48 The total solvation energy ΔGsol was calculated as the sum of a polar component (ΔGPB), which was calculated using the Poisson-Boltzmann approach,49 and of a non-polar solvation term (ΔGNP). ΔGNP was calculated as ΔGNP = γ (SASA) + β, in which γ = 0.00542 kcal/Å2, β = 0.92 kcal/mol, where the solvent-accessible surface area (SASA) was obtained with the MSMS program.50 The radius of gyration (Rg) and the radial distribution functions (RDF) of the dendrimers were calculated by processing the equilibrated dynamic trajectories with the ptraj module of AMBER 11.
Results and Discussion
Design.
Before pursuing in vivo assessment of prodrug 2 in light of the behavior of prodrug 1, computational modeling of these platforms was performed to ensure that the biolabile linkers were indeed accessible to solvolysis and reductants. In the earlier report of 1,35 gas phase simulations anchored our intuition of size and structure. The simulations showed PEG chains wrapped around the globular dendrimer and concealed much of the drug and linker. If such a model accurately depicted reality, reducing the degree of PEGylation or presenting the drug on the end of a more hydrophilic linker could be valuable. While hydrophobic drug might still associate with the hydrophobic dendrimer, a hydrophilic linker might present the biolabile groups in solvent accessible surface loops. We turned to molecular dynamics simulations with explicit molecules of solvent to obtain a more reliable model of 2.
Molecular Dynamics Simulations.
Molecular dynamics (MD) simulations were performed in salt solution (150 mM) to emulate physiological conditions. As PEGylation is heterogeneous and incomplete, we chose to examine dendrimers with 6, 9 and 12 PEG chains. Consistent with expectation, fewer PEGs lead to a more open and accessible structure (Figure 2). Indeed, the model of 2 with twelve PEG chains has very compact and dense architecture similar to the gas phase simulations (Figure 2C).
Figure 2.
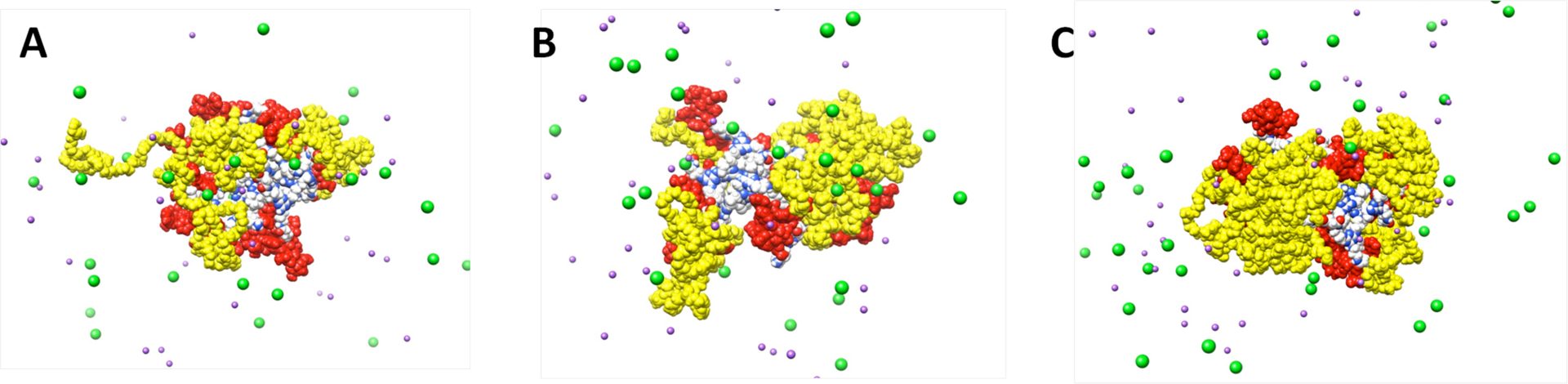
Equilibrated snapshots (A for the hexaPEGylated model, B for the nonaPEGylated model, and C for the dodecaPEGylated model) taken from MD simulations. PEG and linker are colored in yellow, paclitaxel in red, triazine dendrimer colored per atom, Na+ in purple, and Cl− in green. Explicit molecules of water are omitted for clarity.
These differences in structure may be more subtle than just steric bulk. PEG chains are installed on available surface amines. In the absence of a PEG chain, the dendrimer displays an amine group that is likely to be (and modelled as) cationic under physiological conditions. That is, these models vary in charge from 0 for the dodecaPEGylated model, +3 for the nonaPEGylated model, to +6 for hexaPEGylated model. As a result, ions (Na+ and Cl−) and solvation play an important role in opening the structures of cationic dendrimers, respectively. We would hypothesize that fewer PEG chains should lead to more facile release of drug. However, models that display fewer PEG chains also show additional hydrophobic surface area that could facilitate aggregation.
Radial distribution functions (RDFs) provide additional insight into the structural differences resulting from incomplete PEGylation (Figure 3). The RDFs of all the models are expected to be similar since the globular, triazine “rigid” part is constant. As RDF graphs are averaged over the dynamic steps of the simulation, a peak in this plot represents both “spatial density” (i.e. atoms in a given zone in the space) and also “temporal density” (i.e. atoms staying in the same zone in the space). A high peak is interpretted as not only high occupancy, but also high residency of atoms at a given distance from the “center” of dendrimer.
Figure 3.
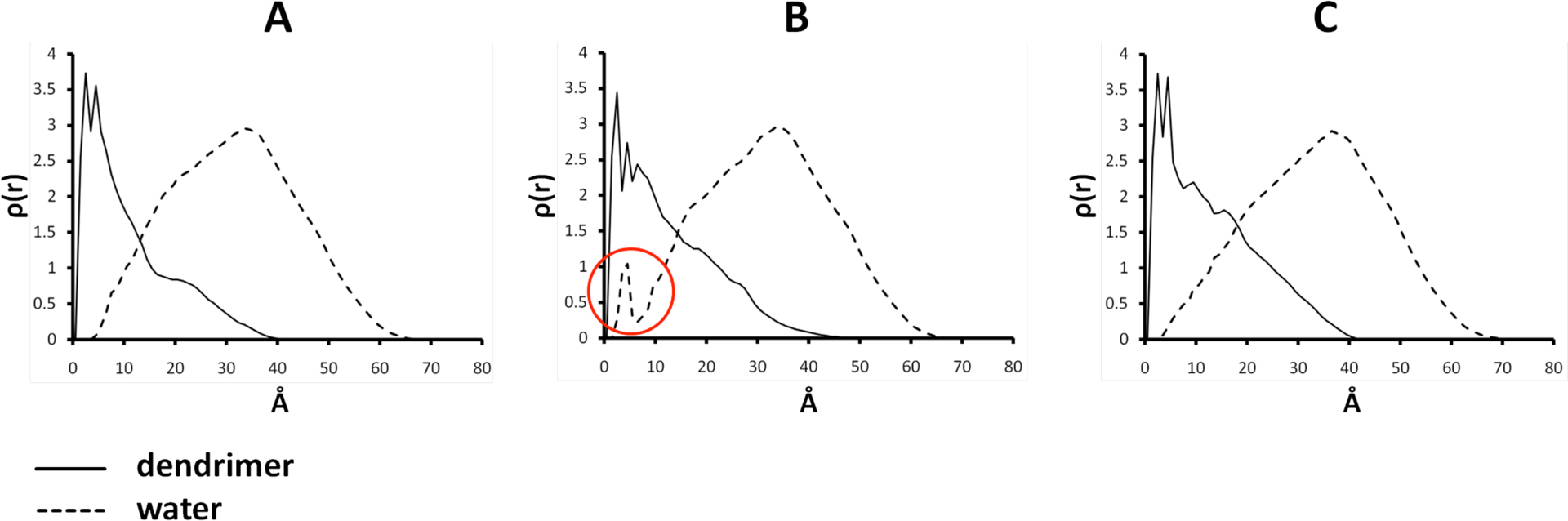
Radial distribution function plots (y-axis is ϱ(r) and x-axis represents the distance from the center of mass of the dendrimers measured in Å): A is the hexaPEGylated model, B is the nonaPEGylated model, and C is the dodecaPEGylated model.
The RDF plots reveal differences in density of atoms of 2 as well as atoms of water as a function of the degree of PEGylation. In general, the triazine portion of the models appears globular and tightly packed with a diameter of about 12 Å represented by the peak between 0 and 12 Å. At distances greater than 12 Å, the contributions of the PEG chains appear. The shape produced by these contributions is interesting. For the hexaPEGylated material, much of the PEG coats the dendrimer effectively (interpretted as a shoulder between 15–25 Å), but clearly extends outward into solvent to distances of 40 Å (a rapid fall in ϱ(r) identifies high surface flexibility and oscillation of PEG chains which are evidenced also in Figure 3A). If PEG chains behave similarly, this shoulder should simply increase as more PEG chains are added. In all cases the similar amounts of PEG extend out into solvent. However, these plots reveal that increasing PEGylation leads to a significant crowding near the surface of the triazine/PEG interface where the sites of the biolabile linkers that faciliate drug release are located. This distribution of biolabile ester groups can be quantified using a similar strategy. The data shows (Supporting Information) that these groups are similarly disposed across the hexaPEGylated, nonaPEGylated, and dodecaPEGylated models with most linkages at or near the dendrimer-solvent interface, and with only two paclitaxel groups back-folded into the dendrimer.
There are other features in these plots that suggest both voids and trapped molecules of water (peak in the nonaPEGylated material RDF at 5 Å shown in red circle in Figure 3B), but these are not yet sufficiently understood to merit additional comment at this time.
PEGylation is performed to insure solubility of these materials in water. Indeed, if 2 bears fewer than 6 PEG chains, it is insoluble. Solvation energies for these three models were calculated. The values of ΔGsol were −1848.5 ± 26.5 kcal/mol for the hexaPEGylated material, −1946.8 ± 126.2 for nonaPEGylated material, and −1620.1 ± 61.4 for dodecaPEGylated material. The differences may be rationalized in terms of a balance of polar (electrostatic) and non-polar (shape, accessible area, cavity, and van der waals) energetic terms, and the degree of water penetration as revealed in the RDF plots. Radii of gyration in 150 mM salt solution were also calculated as 23.3 Å for the hexaPEGylated model, 24.4 Å for the nonaPEGylated model, and 25.1 Å for the dodecaPEGylated model. Data related to the size of these dendrimers in solution are consistent with the results previously reported.35
In Vivo Toxicity.
The in vivo toxicities of Prodrug 2 were assessed by animal behavior, body weight, whole blood cell count, renal, and hepatic functions. All animals showed normal behavior in the entire experiment duration. However, the treatment groups exhibited different levels of body weight loss during the treatment course. While the control group showed a body weight loss at the late stages of the experiment as a result of tumor burden, the treatment groups of 2–50t, 2–100s, 2–100d, and 2–200s showed early body weight loss consistent with delivery of cytotoxin. After 20 days of treatment, the body weight of the treatment groups rebounded and returned to the initial values within the range of ± 10% at day 40. The 2–50d group stayed at the same body weight level during the entire study period (Figure 4A).
Figure 4.
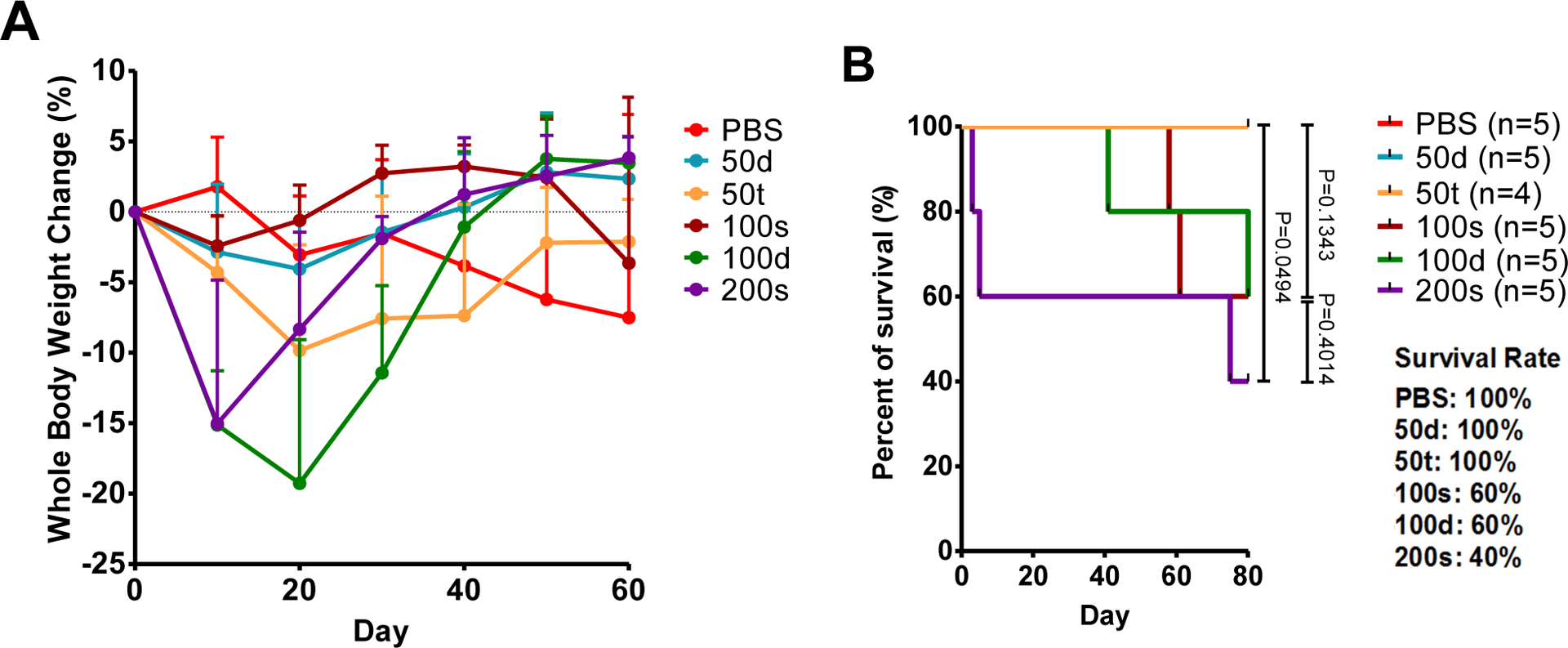
Whole body weight change (A) as a function of % weight change and survival curve (B) for mice administered with prodrug 2 or PBS control. The final survival rates for the 80 day experiment are tabulated and show a correlation between dose size and mortality. The difference im survival between the highest dose (200s) and other doses is statistically significant.
To evaluate the renal and hepatic toxicities, blood samples were collected in week 2, 5, and 8 from the mice groups administered with 2–50d and 2–50t (Table 2). In week 5 and 8, blood samples were collected for whole blood cell counts. The white blood cell (WBC), red blood cell (RBC), and platelet counts in all groups were in the normal range (Table 2), but persistant decreases in WBC counts could suggest myelosuppression. A trend toward a decrease in platelet count is also observed. For renal and hepatic toxicity evaluation, blood samples were collected in week 2, 5, and 8. Low hepatic toxicity was observed in the 2–50d and 2–50t groups as measured by the slightly higher alanine transaminase (ALT) level in week 2 and week 5 possibly suggestive of hepatic and kidney toxicity. However, in week 8, the ALT level of all groups returned to the normal range (Table 2). The renal function measured by the blood urea nitrogen (BUN) level stayed in normal range during the entire study period.
Table 2.
| PBS | 2–50d | 2–50t | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Week | 2 | 5 | 8 | 2 | 5 | 8 | 2 | 5 | 8 |
| ALT (IU/L) | 26 | 19 | 20 | 46 | 27.5 | 20.3 | 42 | 45 | 29 |
| BUN (mg/dL) | 20 | 20 | 18.7 | 26.3 | 21.5 | 20.7 | 27 | 25.7 | 20 |
| WBC (K/μL) | n.d. | 7.86 | 6.37 | nd | 3.47 | 3.47 | n.d. | 3.33 | 3.89 |
| RBC (M/μL) | n.d. | 9.54 | 6.34 | nd | 8.86 | 6.75 | n.d. | 8.06 | 6.60 |
| Platelet (K/μL) | n.d. | 1543 | 1112 | nd | 1163 | 972 | n.d. | 1064 | 906 |
ALT normal range: 10–35 IU/L; BUN normal range: 9–30 mg/dl; n.d.: not determined.
Data are presented as mean values from three mice of each group.
Therapeutic Efficacy Evaluation.
As shown in Figure 5, all five treatment groups (2–50d, 2–50t, 2–100s, 2–100d, and 2–200s) showed significant tumor growth suppression starting from day 20 (p < 0.0001) when compared with the control group (Figure 5B). Of the treatment groups, the single dose treatment of 200 mg PTX/kg (2–200s) completely suppressed the tumor growth, but three mice perished during the treatment. Encouragingly, the groups administered 50mg/kg showed the high efficacy and 100% survival. Compared to the 2–50d, the group given with 2–50t suppressed the tumor growth more effectively (Figure 5C).
Figure 5.
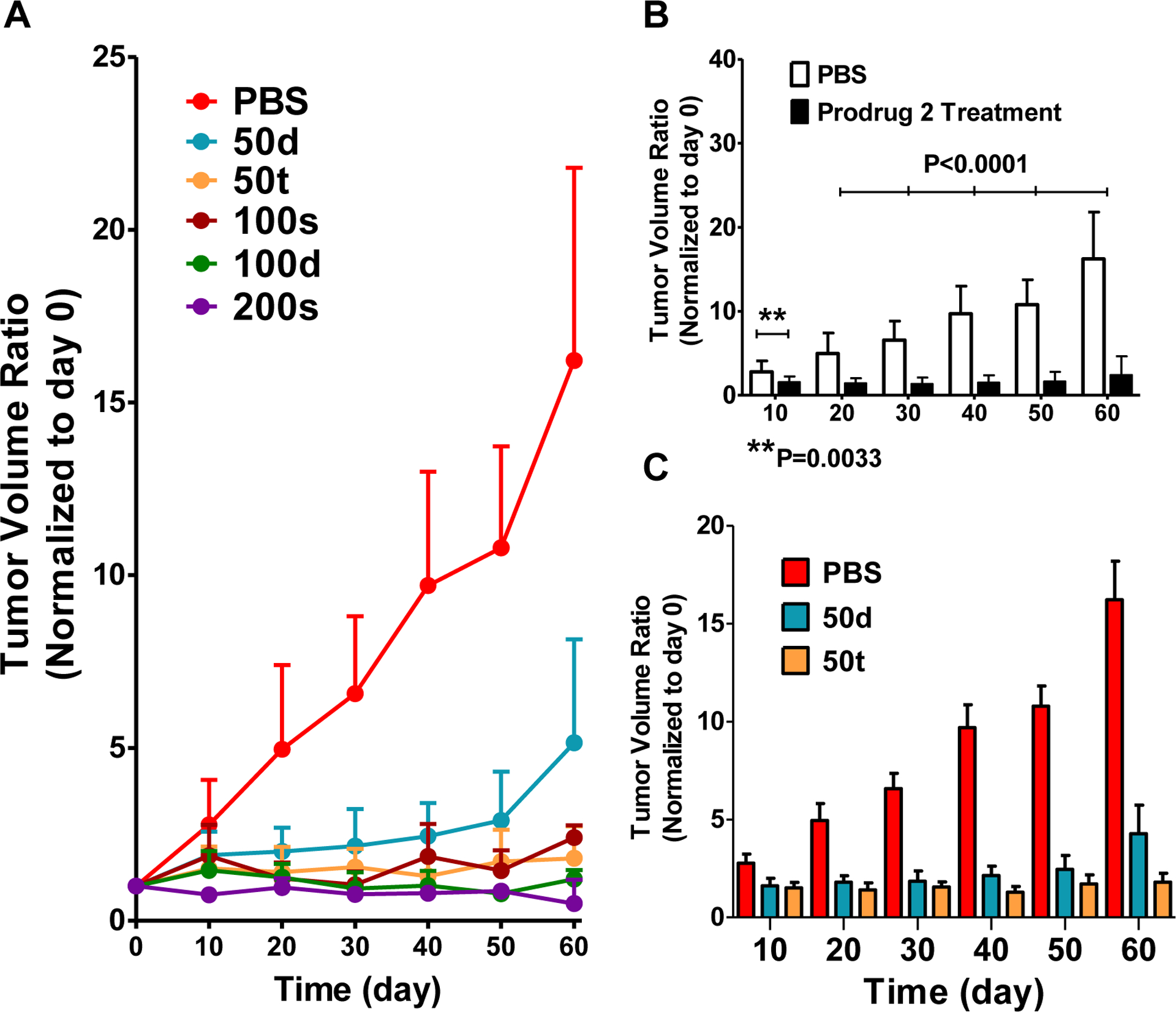
Therapeutic efficacy evaluation of produg 2 in SCID mice bearing PC3-h-luc xenografts. (A) Tumor volume changes measured by caliper during the 60-day treatment period (tumor volume ratio = tumor size at a given time / tumor size on day 0). (B) Tumor growth comparison between the treatment groups (data pooled) and the control group. The treatment with prodrug 2 significantly suppressed the tumor growth (p < 0.0001) from day 20 post treatment. (C) Tumor volume ratio observed with the treatment plans of 2–50d, 2–50t, and PBS.
The tumor viability was longitudinally monitored by BLI during the 10-week treatment experiment period. The BLI results in Figure 6 display tumor growth of all the groups treated with prodrug 2 was suppressed while the tumor burden of mice given with PBS control increased continuously throughout the study. As shown in Figure 6A–C, the treatment plan of 2–50t showed the most efficacious tumor growth suppression. Even more promising, the BLI signal—which reflects the metabolic activity of the tissue—in the 2–50t group was virtually gone in spite of the fact that the tumor was still physically palpable. The images also reveal that tumors rebound following treatment as in the 2–50d and 2–100s animals. The images present a single representative data point: Tumor heterogeneity clearly exists across all groups including the 2–50t and 2–50d groups shown.
Figure 6.
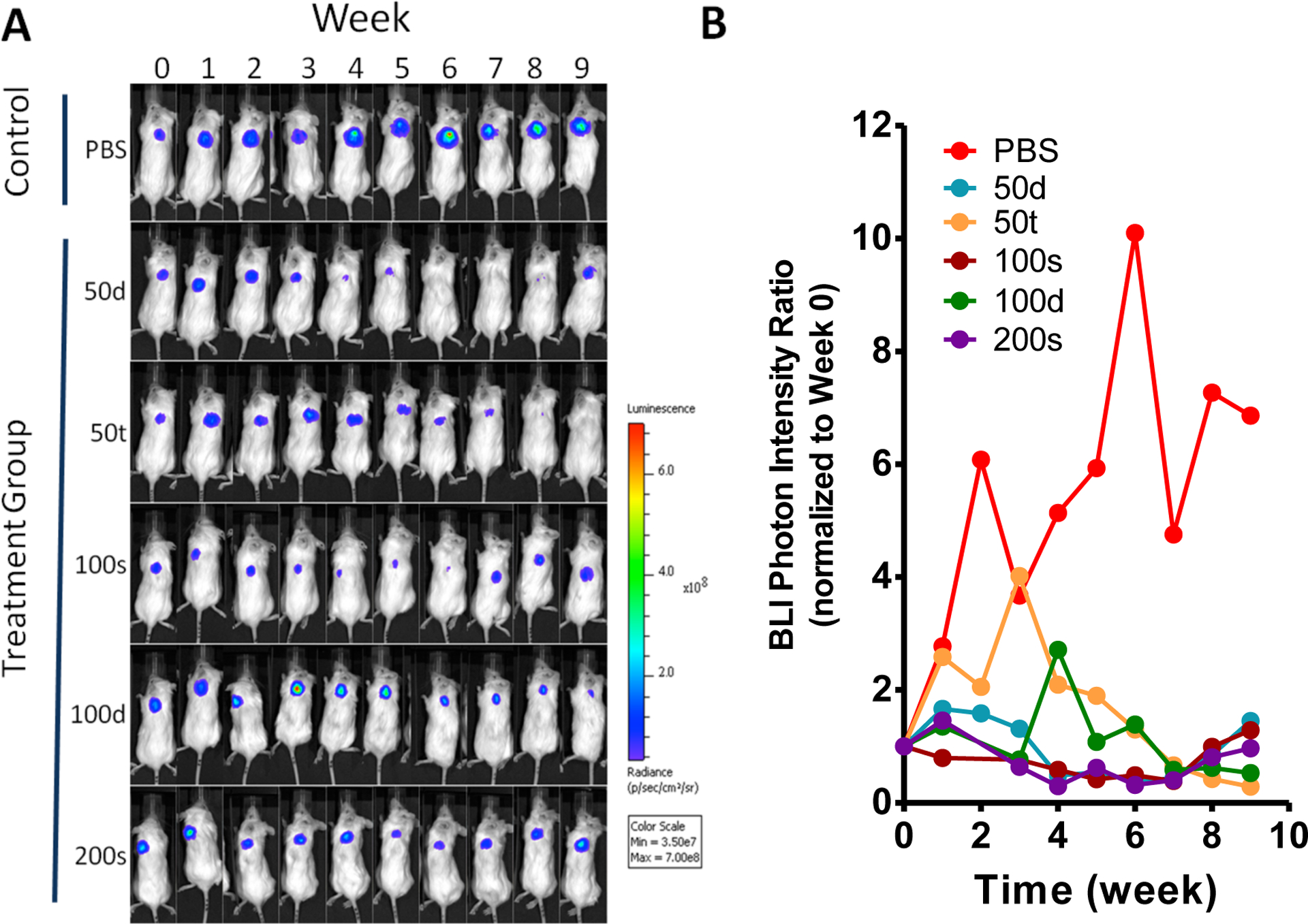
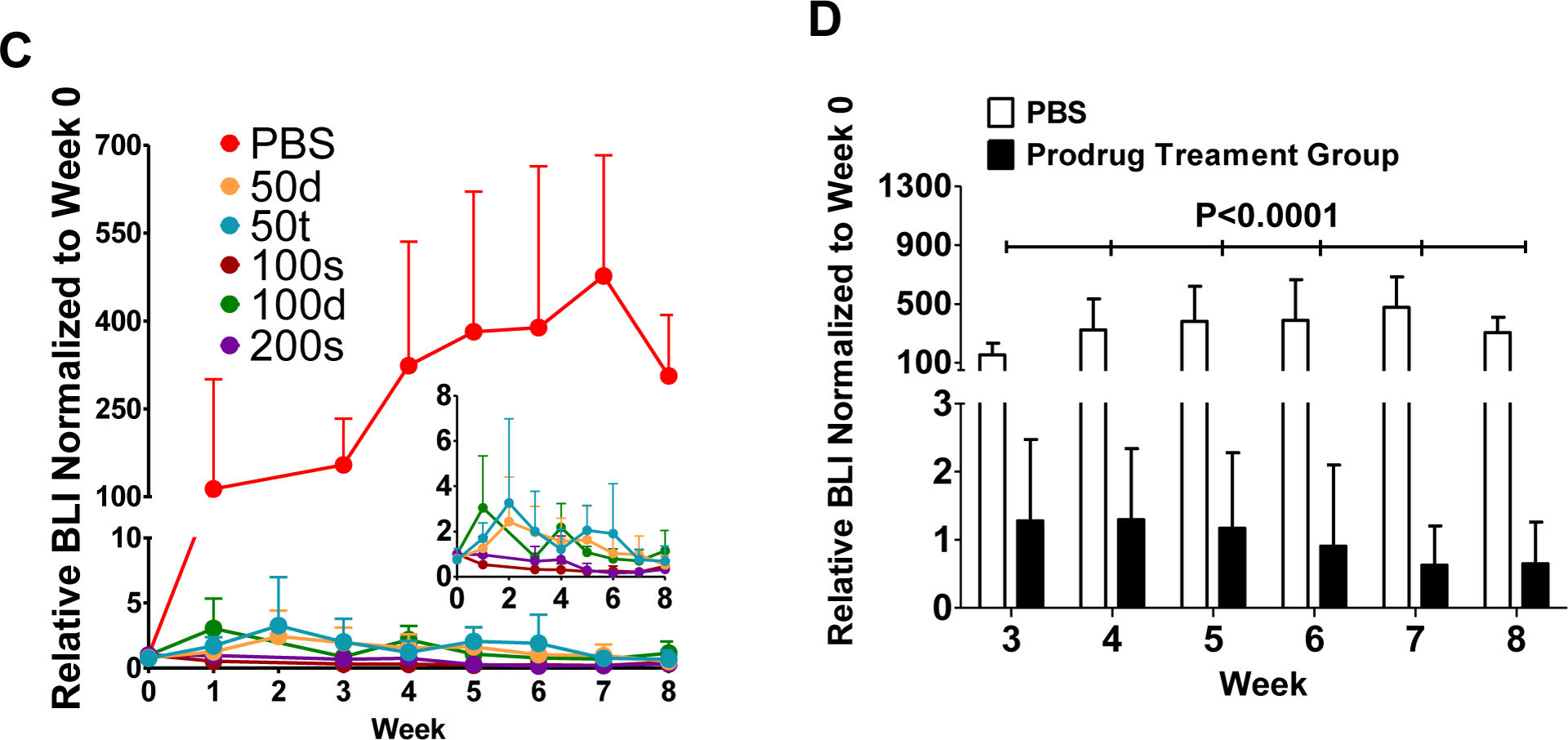
BLI evaluation of the therapeutic efficacy of prodrug 2 in SCID mice bearing PC3-h-luc xenografts. (A) Representative BLI images of five treatment groups and PBS control. The photon intensity of the BLI images is shown on the same scale. (B) Comparative presentation of tumor BLI photon intensities in all groups. The relative photon intensity at the given week were normalized to the data acquired on week 0 (before treatment) which set as 100% for all individual mice. (C) Comparative BLI data from the treatment groups (2–50d, 2–50t, 2–100s, 2–100d, and 2–200s) and PBS control group. The data was shown in relative photon intensity at the given week normalized to the data of week 0 for all individual mice. The figure insert is an expanded portion of the treatment groups. (D) Statistical comparison between the combined treatment groups and the PBS control.
Conclusions
The guiding hypothesis—that introduction of a labile disulfide into the linker domain of platform 1 to yield 2 will provide a more toxic prodrug—appears to be borne out. While our intuitive model posits that there is causality between the increased lability conveyed by a disulfide and increased efficacy, the lack of clear evidence to this end must be noted. Indeed, while these materials are also more toxic in cell culture with the addition of dithiothreitol, the role of the disulfide bond here and in vivo is as yet unestablished. At the equivalent doses (100s, 100d, and 200s), prodrug 2 displayed the higher mortality than prodrug 1. This increased toxicity provides opportunities for efficacy at lower doses. Indeed, at doses similar to the target doses for competitive efficacy with Taxol and Abraxane, 2–50d and 2–50t show great promise. On this basis, competitive efficacy studies may be warranted. Critical to such future studies are those aimed at ruling out an additional toxicity associated with the construct in comparison to clinically efficacious formulations of paclitaxel. However, competitive efficacy alone is not compelling argument for clinical relevance. More labile linkers may lead to increased toxicity and efficacy at lower doses of paclitaxel.
Molecular dynamics simulations lead us to consider additional hypotheses as we move forward. The heterogenous distribution of 2 with materials ranging from hexaPEGylated to dodecaPEGylated platforms is both a cause for concern and springboard for curiosity. Simulations reveal that water penetration and accessibility of linkers vary with the degree of PEGylation. The apparent size of the hydrophobic patch visible on the surface also varies. These patches could promote aggregation (a phenomena that has been observed but not rigorously quantified), and ultimately liver uptake. Prodrugs 1 and 2 show significant liver uptake, and accordingly the amount of nanomedicine available to the tumor is reduced.29 This data is reviewed in the supporting information. Efficacy is assumed to be mediated by the small fraction of the injected dose (~3%) that localizes in the tumor. Understanding the nature of tumor and liver uptake has merit. Specifically, the role that aggregation may be playing requires attention. Such studies start with examining the relationship between PEGylation and aggregation, and ultimately efficacy. Targeting using ligands to enhance localization over the passive strategies we currently rely on represents another strategy.
Supplementary Material
Acknowledgment.
We thank the NIH for support of these efforts (NIGMS R01 64560; EES). EES holds the Robert A. Welch Chair of Chemistry at TCU (0008). GMP was supported by the Swiss State Secretariat for Education and Research (SER).
Footnotes
Supporting Information Available: Additional in vivo toxicity and efficacy data. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).Rowinsky EK The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu. Rev. Med 1997, 48, 353–374. [DOI] [PubMed] [Google Scholar]
- (2).Scripture CD; Figg WD; Sparreboom A Paclitaxel chemotherapy: from empiricism to a mechanism-based formulation strategy. Ther. Clin. Risk Manag 2005, 1, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Crown J; O’Leary M; Ooi WS Docetaxel and paclitaxel in the treatment of breast cancer: a review of clinical experience. Oncologist 2004, 9, 24–32. [DOI] [PubMed] [Google Scholar]
- (4).Kingston DG; Newman DJ Taxoids: cancer-fighting compounds from nature. Curr. Opin. Drug Discov. Devel 2007, 10, 130–144. [PubMed] [Google Scholar]
- (5).Holton RA; Biediger RJ; Boatman PD Semisynthesis of taxol and taxotere In: Suffness M (Ed.) Taxol: science and applications. CRC press, Boca Raton, 1995. pp. 97–121. [Google Scholar]
- (6).Hennenfent KL; Govindan R Novel formulations of taxanes: a review. Old wine in a new bottle? Ann. Oncol 2006, 17, 735–749. [DOI] [PubMed] [Google Scholar]
- (7).Terwogt JM; Nuijen B; Huinink WW; Beijnen JH Alternative formulations of paclitaxel. Cancer Treat. Rev 1997, 23, 87–95. [DOI] [PubMed] [Google Scholar]
- (8).Liebmann J; Cook JA; Mitchell JB Cremophor EL, solvent for paclitaxel, and toxicity. Lancet 1993, 342, 1428. [DOI] [PubMed] [Google Scholar]
- (9).Malingré MM; Schellens JH; Van Tellingen O; Ouwehand M; Bardelmeijer HA; Rosing H; Koopman FJ; Schot ME; Ten Bokkel Huinink WW; Beijnen JH The co-solvent Cremophor EL limits absorption of orally administered paclitaxel in cancer patients. Br. J. Cancer 2001, 85, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sparreboom A; van Tellingen O; Nooijen WJ; Beijnen JH Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res. 1996, 56, 2112–2115. [PubMed] [Google Scholar]
- (11).Feng Z; Zhao G; Yu L; Gough D; Howell SB Preclinical efficacy studies of a novel nanoparticle-based formulation of paclitaxel that out-performs Abraxane. Cancer Chemother. Pharmacol 2010, 65, 923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Meerum Terwogt JM; ten Bokkel Huinink WW; Schellens JH; Schot M; Mandjes IA; Zurlo MG; Rocchetti M; Rosing H; Koopman FJ; Beijnen JH Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs 2001, 12, 315–323. [DOI] [PubMed] [Google Scholar]
- (13).Zou Y; Fu H; Ghosh S; Farquhar D; Klostergaard J Antitumor activity of hydrophilic paclitaxel copolymer prodrug using locoregional delivery in human orthotopic nonsmall cell lung cancer xenograft models. Clin. Cancer Res 2004, 10, 7382–7391. [DOI] [PubMed] [Google Scholar]
- (14).Etrych T; Sírová M; Starovoytova L; Ríhová B; Ulbrich K HPMA copolymer conjugates of paclitaxel and docetaxel with pH-controlled drug release. Mol. Pharmaceutics 2010. 7, 1015–1026. [DOI] [PubMed] [Google Scholar]
- (15).Sugahara S; Kajiki M; Kuriyama H; Kobayashi TR Complete regression of xenografted human carcinomas by a paclitaxel-carboxymethyl dextran conjugate (AZ10992). J. Control. Release 2007, 117, 40–50. [DOI] [PubMed] [Google Scholar]
- (16).Galic VL; Herzog TJ; Wright JD; Lewin SN Paclitaxel poliglumex for ovarian cancer. Expert Opin. Investig. Drugs 2011, 20, 813–821. [DOI] [PubMed] [Google Scholar]
- (17).Dhanikula AB; Singh DR; Panchagnula R In vivo pharmacokinetic and tissue distribution studies in mice of alternative formulations for local and systemic delivery of paclitaxel: gel, film, prodrug, liposomes and micelles. Curr. Drug Deliv 2005, 2, 35–44. [DOI] [PubMed] [Google Scholar]
- (18).Dosio F; Reddy LH; Ferrero A; Stella B; Cattel L; Couvreur P Novel nanoassemblies composed of squalenoyl-paclitaxel derivatives: synthesis, characterization, and biological evaluation. Bioconjug. Chem 2010, 21, 1349–1361. [DOI] [PubMed] [Google Scholar]
- (19).Boas U; Heegaard PMH Dendrimers in drug research. Chem. Soc. Rev 2004, 33, 43–63. [DOI] [PubMed] [Google Scholar]
- (20).Duncan R The dawning era of polymer therapeutics. Nat. Rev. Drug Discovery 2003, 2, 347–360. [DOI] [PubMed] [Google Scholar]
- (21).Majoros IJ; Myc A; Thomas T; Mehta CB; Baker JR PAMAM dendrimer-based multifunctional conjugate for cancer therapy: synthesis, characterization, and functionality. Biomacromolecules 2006, 7, 572–579. [DOI] [PubMed] [Google Scholar]
- (22).Lee CC; MacKay JA; Fréchet JM; Szoka FC Designing dendrimers for biological applications. Nat. Biotechnol 2005, 23, 1517–1526. [DOI] [PubMed] [Google Scholar]
- (23).Matsumura Y; Maeda H A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 6, 6387–6392. [PubMed] [Google Scholar]
- (24).Maeda H; Matsumura Y EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv Drug Deliv Rev. 2011, 63, 129–130. [DOI] [PubMed] [Google Scholar]
- (25).Steffensen MB; Simanek EE Chemoselective building blocks for dendrimers from relative reactivity data. Org. Lett 2003, 5, 2359–2361. [DOI] [PubMed] [Google Scholar]
- (26).Chouai A; Venditto VJ; Simanek EE Vanderplas BC; Ragan JA Large scale, green synthesis of a generation-1 melamine (triazine) dendrimer. Organic Synth. 2009, 86, 151. [PMC free article] [PubMed] [Google Scholar]
- (27).Lim J; Mintzer MA; Perez LM; Simanek EE Synthesis of odd generation triazine dendrimers using a divergent, macromonomer approach. Org Lett. 2010, 12, 1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Steffensen MB; Simanek EE Synthesis and manipulation of orthogonally protected dendrimers: building blocks for library synthesis. Angew. Chem. Int. Ed 2004, 43, 5178–5180. [DOI] [PubMed] [Google Scholar]
- (29).Lim J; Chouai A; Lo S-T; Liu W; Sun X; Simanek EE Design, synthesis, characterization, and biological evaluation of triazine dendrimers bearing paclitaxel using ester and ester/disulfide linkages. Biconjugate Chem. 2009, 20, 2154–2161. [DOI] [PubMed] [Google Scholar]
- (30).Holmes FA; Walters RS; Theriault RL; Forman AD; Newton LK; Raber MN; Buzdar AU; Frye DK; Hortobagyi GN Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. J. Natl. Cancer Inst 1991, 83, 1797–1805. [DOI] [PubMed] [Google Scholar]
- (31).Harper E; Dang W; Lapidus RG; Garver RI Jr. Enhanced efficacy of a novel controlled release paclitaxel formulation (PACLIMER delivery system) for local-regional therapy of lung cancer tumor nodules in mice. Clin. Cancer Res 1999, 5, 4242–4248. [PubMed] [Google Scholar]
- (32).Seidman AD; Tiersten A; Hudis C; Gollub M; Barrett S; Yao TJ; Lepore J; Gilewski T; Currie V; Crown J Phase II trial of paclitaxel by 3-hour infusion as initial and salvage chemotherapy for metastatic breast cancer. J. Clin. Oncol 1995, 13, 2575–2581. [DOI] [PubMed] [Google Scholar]
- (33).Gradishar WJ Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006, 7, 1041–1053. [DOI] [PubMed] [Google Scholar]
- (34).Desai N; Trieu V; Yao Z; Louie L; Ci S; Yang A; Tao C; De T; Beals B; Dykes D; Noker P; Yao R; Labao E; Hawkins M; Soon-Shiong P Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res 2006, 12, 1317–1324. [DOI] [PubMed] [Google Scholar]
- (35).Lo ST; Stern S; Clogston JD; Zheng J; Adiseshaiah PP; Dobrovolskaia M; Lim J; Patri AK; Sun X; Simanek EE Biological assessment of triazine dendrimer: toxicological profiles, solution behavior, biodistribution, drug release and efficacy in a PEGylated, paclitaxel construct. Mol. Pharmaceutics 2010, 7, 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cheng R; Feng F; Meng F; Deng C; Feijen J; Zhong Z Glutathione-responsive nanovehicles as a promising platform for targeted intracellular drug and gene delivery. J. Cont. Rel 2011, 152, 2–12. [DOI] [PubMed] [Google Scholar]
- (37).Pavan GM; Mintzer MA; Simanek EE; Merkel OM; Kissel T; Danani A Computational insights into the interactions between DNA and siRNA with “rigid” and “flexible” triazine dendrimers. Biomacromolecules 2010, 11, 721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shema-Mizrachi M; Pavan GM; Levin E; Danani A; Lemchoff NG Catalytic chameleon dendrimers. J. Am. Chem. Soc 2011, 133, 14359–14367. [DOI] [PubMed] [Google Scholar]
- (39).Merkel OM; Zeng M; Mintzer MA; Pavan GM; Librizzi D; Maly M; Höffken H; Danani A; Simanek EE; Kissel T Molecular modeling and in vivo imaging can identify successful flexible triazine dendrimer-based siRNA delivery systems. J. Contr. Release 2011, 153, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Doni D; Kostiainen MA; Danani A; Pavan GM Generation-dependent molecular recognition controls self-assembly in supramolecular dendron-virus complexes. Nano Lett. 2011, 11, 723–728. [DOI] [PubMed] [Google Scholar]
- (41).Pavan GM; Kostiainen MA; Danani A Computational approach for understanding the interactions of UV-degradable dendrons with DNA and siRNA. J. Phys. Chem. B, 2010, 114, 5686–5693. [DOI] [PubMed] [Google Scholar]
- (42).Case DA; Darden TA; Cheatham TE III; Simmerling CL; Wang J; Duke RE; Luo R; Walker RC; Zhang W; Merz KM; Robertson B; Wang B; Hayik S; Roitberg A; Seabra G; Kolossvary I; Wong KF; Paesani F; Vanicek J; Liu J; Wu X; Brozell S; Steinbrecher T; Gohlke H; Cai Q; Ye X; Wang J; Hsieh M-J; Cui G; Roe DR; Mathews DH; Seetin MG; Sangui C; Babin V; Luchko T; Gusarov S; Kovalenko A; Kollman PA, AMBER 11. In University of California, San Francisco, 2010. [Google Scholar]
- (43).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (44).Darden T; York D; Pedersen L Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (45).Ryckaert JP; Ciccotti G; Berendsen HJC Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (46).Krautler V; Van Gunsteren WF; Hunenberger PH A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem 2001, 22, 501–508. [Google Scholar]
- (47).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (48).Srinivasan J; Cheatham TE Cieplak P; Kollman PA; Case DA Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate–DNA helices. J. Am. Chem. Soc 1998, 120, 9401–9409. [Google Scholar]
- (49).Jayaram B; Sprous D; Beveridge DL Solvation free energy of biomacromolecules: Parameters for a modified generalized Born model consistent with the AMBER force field. J. Phys. Chem. B 1998, 102, 9571–9576. [Google Scholar]
- (50).Sitkoff D; Lockhart DJ; Sharp KA; Honig B Calculation of electrostatic effects at the amino terminus of an alpha helix. Biophys. J 1994, 67, 2251–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.