

Abstract
As a mitotic-specific target widely deregulated in various human cancers, polo-like kinase 1 (Plk1) has been extensively explored for anticancer activity and drug discovery. Although multiple catalytic domain inhibitors were tested in preclinical and clinical studies, their efficacies are limited by dose-limiting cytotoxicity, mainly from off-target cross reactivity. The C-terminal noncatalytic polo-box domain (PBD) of Plk1 has emerged as an attractive target for generating new protein–protein interaction inhibitors. Here, we identified a 1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one scaffold that efficiently inhibits Plk1 PBD but not its related Plk2 and Plk3 PBDs. Structure–activity relationship studies led to multiple inhibitors having ≥10-fold higher inhibitory activity than the previously characterized Plk1 PBD-specific phosphopeptide, PLHSpT (Kd ~ 450 nM). In addition, S-methyl prodrugs effectively inhibited mitotic progression and cell proliferation and their metabolic stability was determined. These data describe a novel class of small-molecule inhibitors that offer a promising avenue for future drug discovery against Plk1-addicted cancers.
Graphical Abstract
INTRODUCTION
Members of the polo subfamily of Ser/Thr protein kinases [collectively, polo-like kinases (Plks)] play a key role in regulating various aspects of the cell cycle and cell proliferation.1 Among them, Plk1 is critically required for proper mitotic progression, whereas other members play distinct roles during interphase progression and exhibit little functional overlap with other Plk family members.1,2 In accordance with its importance in promoting mitosis, Plk1 is largely upregulated in a broad range of human cancers and its level of overexpression appears to correlate with aggressiveness and poor prognosis for a wide spectrum of human cancers.3,4 Notably, various cancer cells, but not their isogenic normal cells, are addicted to high Plk1 levels and consequently require Plk1 overexpression for their viability.5–7 As the reversal of addicted protein functions in cancer cells has proven to be an attractive strategy to selectively kill cancer cells,8–10 Plk1 is considered a discriminating target for anticancer therapy.
Plk1 contains an N-terminal kinase domain (KD) for ATP-dependent catalysis and is characterized by the presence of the C-terminal noncatalytic, but functionally essential, polo-box domain (PBD).11,12 The PBD plays a key role in mediating Plk1 functions by targeting its N-terminal catalytic activity to distinct subcellular structures, such as centrosomes, kineto-chores, and midbody, through specific protein–protein interactions (PPIs).12,13 For more than a decade, extensive efforts were made to develop Plk1 inhibitors targeting the KD, resulting in several advanced Plk1 ATP-competitive inhibitors (Chart 1), such as volasertib/BI6727 1,14 BI2536 2,15 GSK461364 3,16 NMS-P937 4,17 and TAK-960 5,18 that have been examined against various cancers.2 However, they all showed limited efficacy with more-than-acceptable dose-limiting toxicity in various preclinical or clinical trials. As dose-limiting toxicity arises mainly from nonspecific activity of these inhibitors,19 improving the Plk1 specificity is likely the major concern that needs to be addressed to achieve better clinical outcomes. In fact, one of the common problems associated with the currently available Plk1 ATP-competitive inhibitors appears to be their low degree of selectivity against other kinases,20,21 including two closely related Plk2 and Plk3 with potential tumor suppressor roles.22,23
Chart 1.
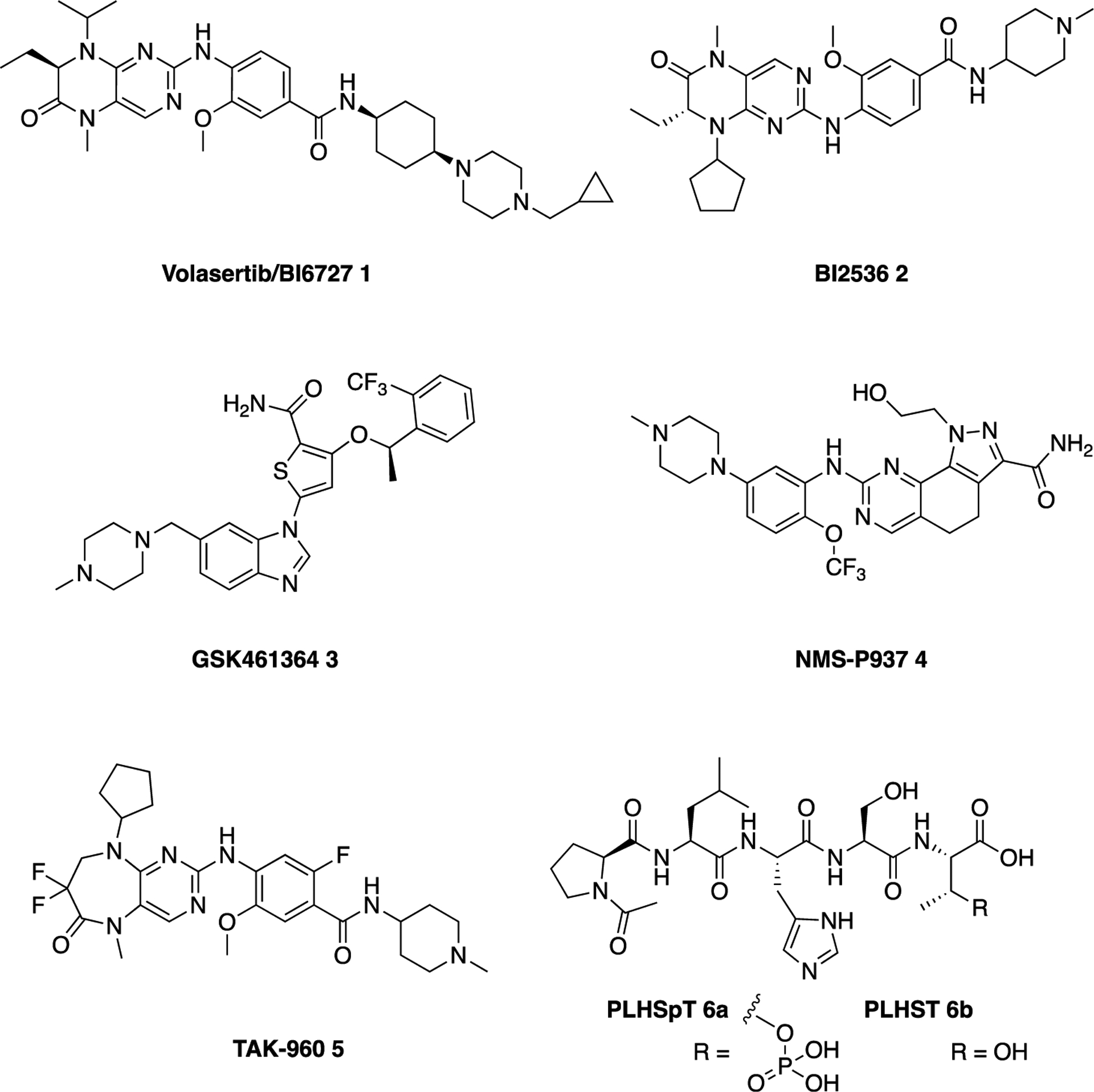
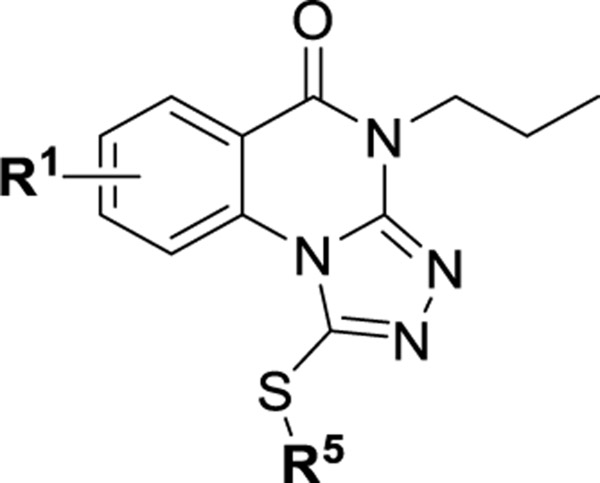
Structures of Inhibitors of the KD (1–5) and the PBD (6) of Plk1
Unlike the KD inhibitors aimed at annihilating all Plk1 catalytic activity-dependent processes, inhibiting the Plk1 PBD is expected to be less drastic because it could selectively mitigate a subset of essential Plk1 functions requiring PBD-dependent interactions.7,24 In addition, given a high affinity (low nM Kd values) interaction between Plk1 PBD and its binding target sequence,25 the effect of PBD inhibition is expected to be less severe than that of KD inhibition. As Plk1 is also required for the viability of normal cells, a moderate level of inhibition could serve as a fail-safe mechanism that helps prevent undesirable normal cell killing.
At the structural level, Plk1 PBD represents a well-characterized PPI domain that interacts with its short phosphopeptide targets with a high affinity.11 The unique nature of PBD, which requires strict structural elements for binding, holds the promise of generating specific inhibitors capable of overcoming nonspecific toxicities associated with the KD inhibitors.20,21 Intriguingly, the blockade of Plk1 PBD-dependent PPIs by short peptides or peptide mimetics is sufficient for effectively abrogating the proper mitotic progression, thus imposing potent mitotic arrest and apoptotic cell death in cancer cells but less in normal cells.7,25,26 These findings suggest that PBD is an attractive target amenable for in vitro PPI- and in vivo cell-based assays required to carry out optimization and preclinical studies for potential leads.
Over the years, various Plk1 PBD inhibitors were generated by either derivatizing the well-characterized minimal phosphopeptide, PLHSpT 6a,27 or isolating small molecules interfering with Plk1 PBD-dependent PPI.20,21 However, although peptide-derived inhibitors provide valuable insights into the binding interface of Plk1 PBD-dependent interactions, whether they can serve as a lead for developing anti-PBD agents is disputable, largely because of their poor permeability and limited bioavailability.21 In addition, although multiple small molecule inhibitors targeting the Plk1 PBD were reported, most exhibit a low level (IC50 of 50–1000 μM) of anti-Plk1 activity in cell-based assays,28–35 while others are not sufficiently characterized at the cellular level.36–41
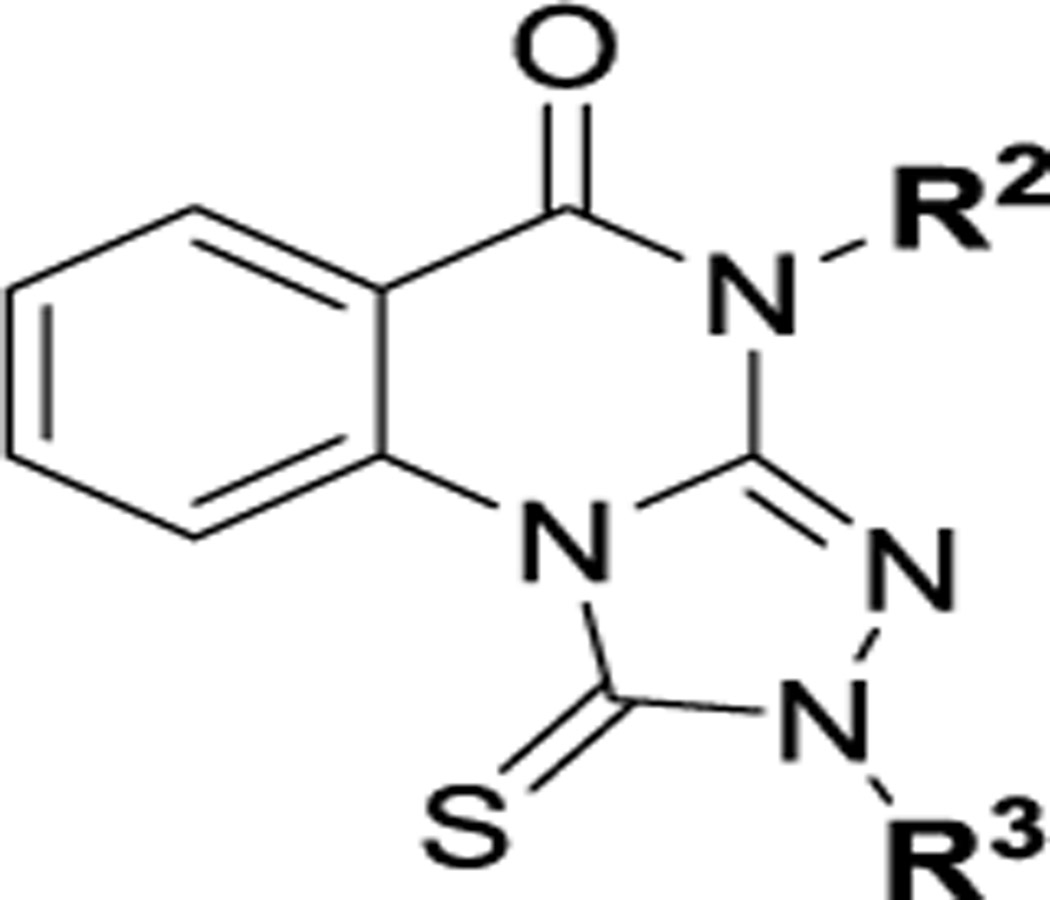
Here, we report the identification and exploration of triazoloquinazolinone-based small-molecule compounds that exhibit specific anti-Plk1 PBD activity in both in vitro biochemical and cell-based assays. These compounds are anticipated not to be chemically reactive as are many of the current probes of Plk1 PBD that contain electrophilic groups.42,43 Their low molecular weight (MW) (generally ~300–400 Da) along with moderate hydrophobicity and activity of uncharged analogues predict facile availability in intracellular compartments. Notably, the two heterocyclic moieties of the inhibitors (i.e., quinazoline and triazole) are frequently found in various FDA-approved drugs, suggesting the potential druggability of this inhibitor class. We propose the triazoloquinazolinone-based compounds described here as attractive compounds that may lead to the development of a new class of anti-PBD therapeutics against various Plk1-addicted cancers.
RESULTS
Screening Small Molecules to Target Plk1 PBD.
A small chemical library of ~400 drug-like molecules in the Molecular Recognition Section, National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (NIH), was screened for the ability to bind to the PBD of Plk1. The principle screening assay consisted of an ELISA-based Plk1 PBD inhibition assay that utilizes the specific interaction between the full-length human Plk1 and a specific phospho-Thr (pT)-containing peptide (Biotin-Ahx-C-ETFDPPLHSpTAI-NH2) derived from a kinetochore-localizing Plk1-binding protein, PBIP1.27,44 When necessary, secondary fluorescence polarization (FP) assays were carried out using a 5-carboxyfluorescein-labeled peptide (FITC-Ahx-DPPLHSpTAI-NH2)26 to confirm the anti-Plk1 PBD activity from the primary ELISA-based assay and eliminate false positives.
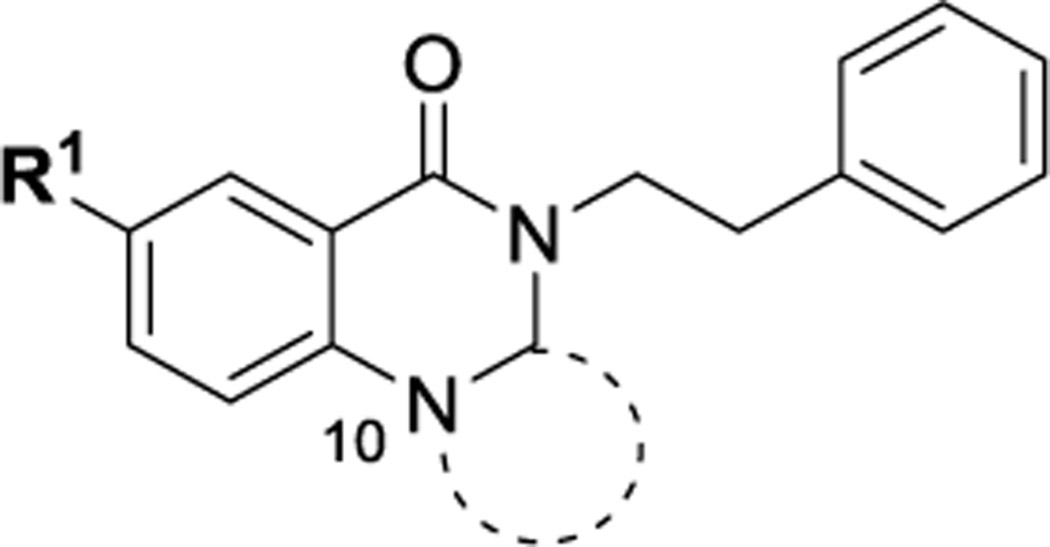
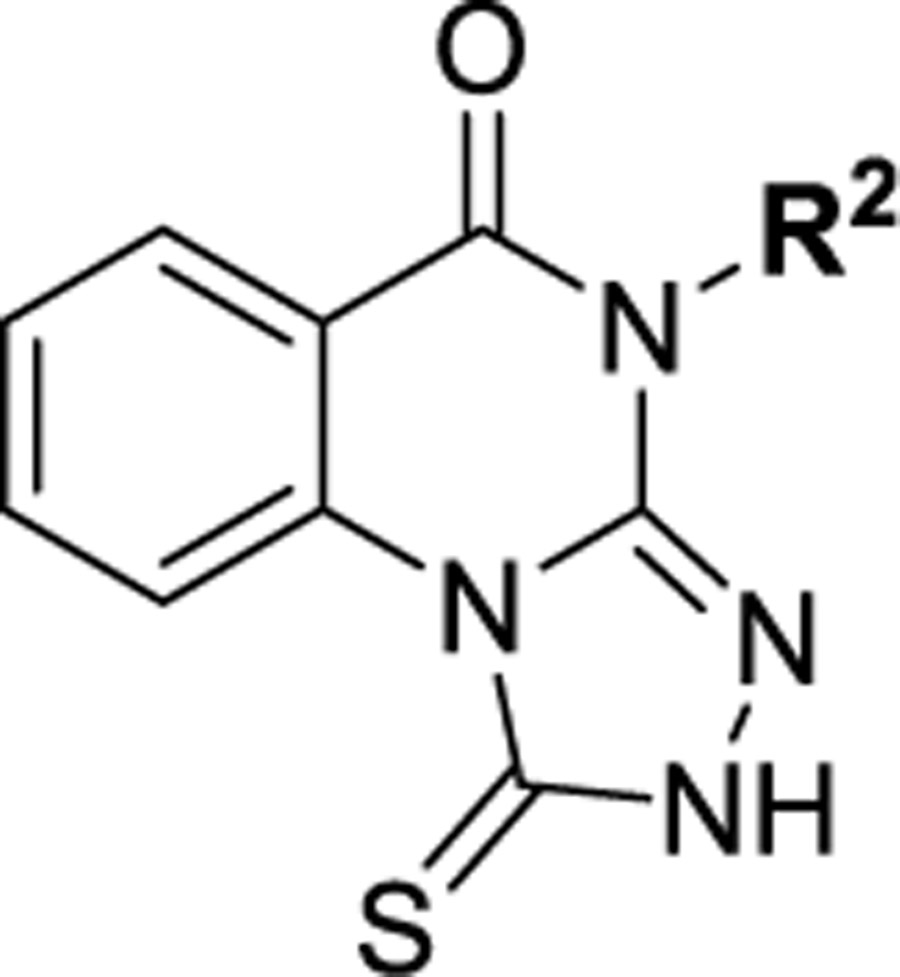
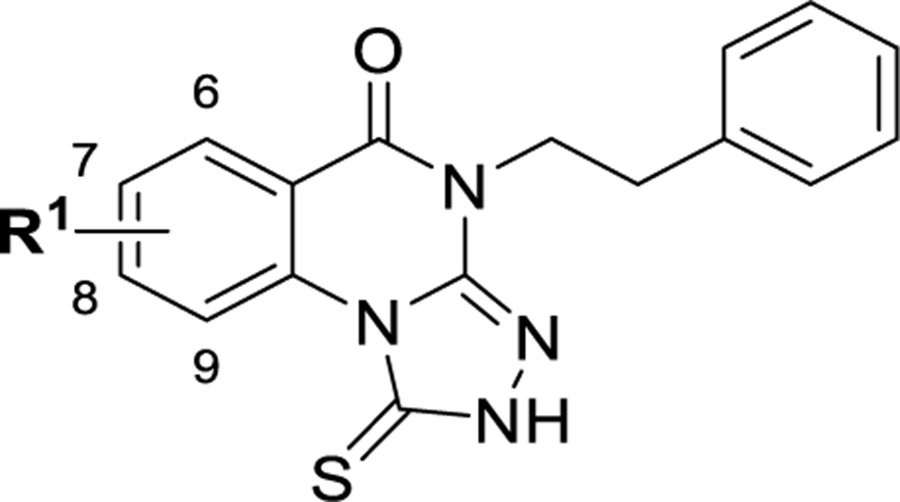
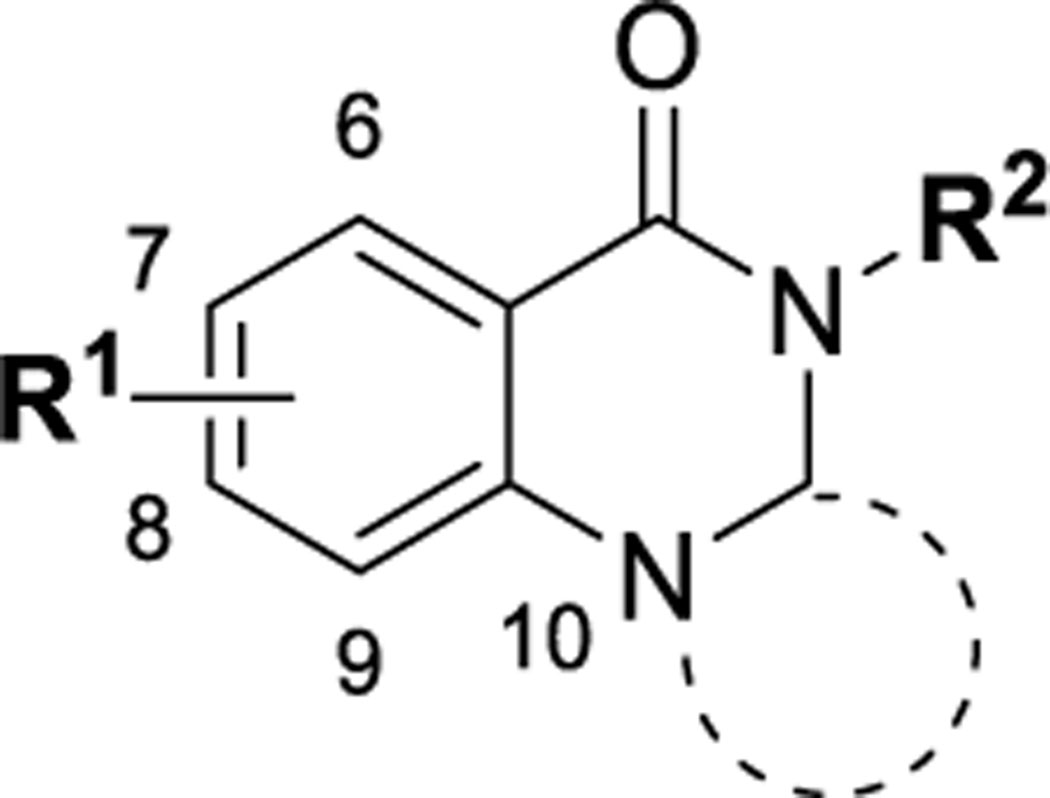
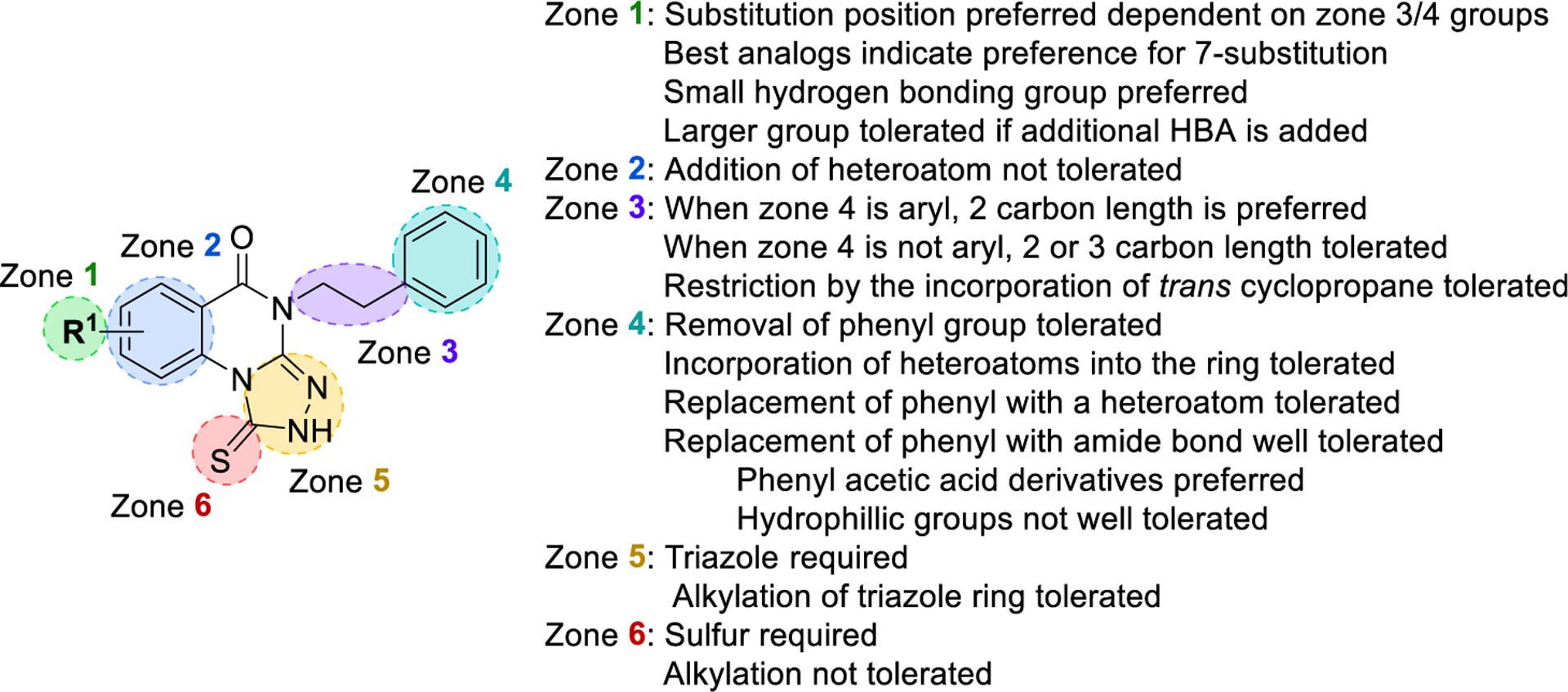
Among four hits identified was triazoloquinazolinone 7 (Chart 1), which inhibited PBD binding in the ELISA assay with an IC50 of 4.38 μM. When compared to the previously characterized phosphopeptide, PLHSpT 6a (IC50 of 14.74 μM), showing a Kd of ~450 nM,27 the affinity of the lead compound 7 is anticipated to be at least threefold higher than that of peptide 6a (Table 1). Compound 7 was previously reported as a weak hit in a structure-based in silico screen to identify ligands of the adenosine receptors.45 From this hit, a family of congeners was synthesized by substitutions introduced on all of the moieties of the lead compound 7 (Figure 1). Various substituents were introduced as R1 groups, and aza analogues of the phenyl ring were also introduced. Many alkyl, heteroalkyl, and arylalkyl modifications of the R2 group (phenylethyl in compound 7) were included. Some of the analogues were obtained from commercial sources and their identity confirmed by 1H NMR and mass spectrometry.
Table 1.
| Cmpd | Triazole | R1= | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1e-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|---|
| PLHSpT 6a |
-- | -- | 14.74 ± 0.48 (12) | ND | ND | ND |
| 7 | 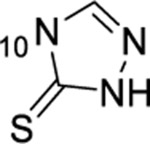 |
H | 4.38 ± 0.41 (6) | ND | ND | ND |
| 15 | 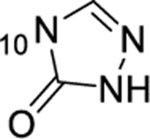 |
H | >50 | ND | ND | <1 |
| 16 | 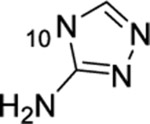 |
F | >50 | 29.4 | 885 | 2.2 |
| 17 | 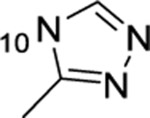 |
F | >50 | 10.7 | 1084 | 11.0 |
| 18 | 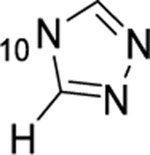 |
F | >50 | 11.8 | 924 | 14.8 |
| 19 | 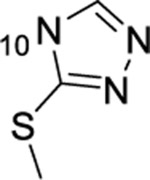 |
H | >50 | ND | ND | ND |
(IC50 values are n = 3 unless noted in parentheses) and microsomal half-life, PAMPA assays, and aqueous solubility.
ND, not determined.
Figure 1.
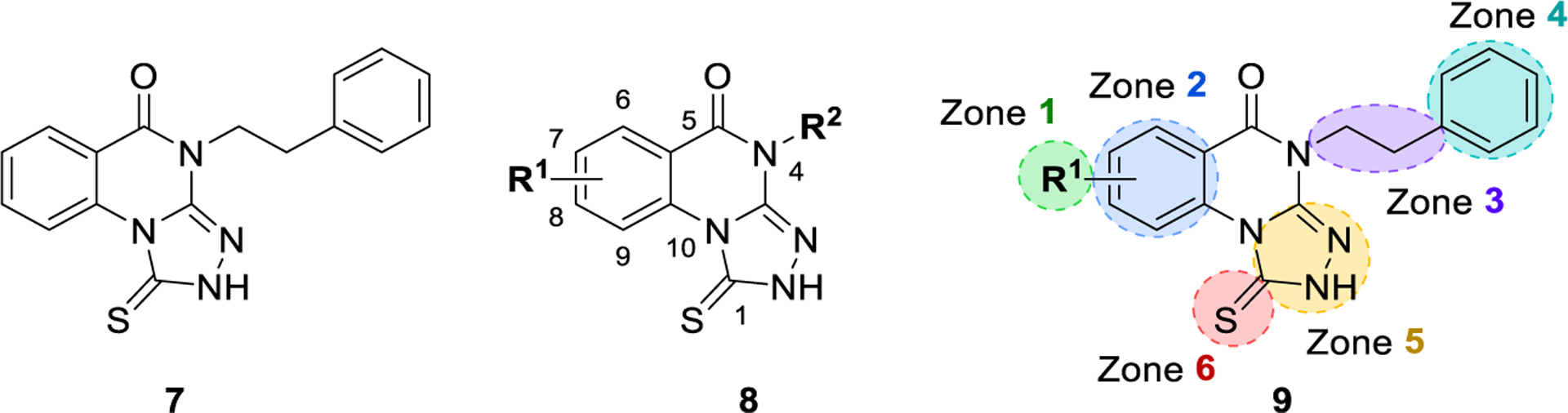
Delineation of zones of SAR focus in the triazoloquinazolinone series.
Chemical Synthesis.
The synthesis of analogues of 7 generally began with either the corresponding isatoic anhydride (Scheme 1) or anthranilic acid (Supporting Information). An isatoic anhydride (e.g., 10) was heated in the presence of the desired amine to give the corresponding intermediate 2-aminobenzamide (structure not shown). This intermediate was heated in the presence of carbon disulfide and potassium hydroxide to give the cyclized 2-thioxo-2,3-dihydroquinazolin-4(1H)-one derivative (e.g., 11), which was isolated by precipitation with treatment of water, followed by filtration. The corresponding 3-thione-1,2,4-triazole (e.g., 12) was then typically formed in a one-pot procedure by first stirring with hydrazine in refluxing ethanol to form the hydrazine intermediate. This intermediate, after cooling, was treated with pyridine and carbon disulfide. The desired triazoloquinazolinone 12 was formed after heating the reaction again to 80 °C. The 2-thioxo-2,3-dihydroquinazolin-4(1H)-one intermediate could also be formed by reacting the desired 2-aminobenzoic acid with an isothiocyanate. However, because of the limited availability of isothiocyanates, the preferred route for preparation of the triazoloquinazolinones is shown in Scheme 1. With the core triazoloquinazolinone formed, additional analogues could be generated with a terminal amine (e.g., 13, Scheme 2). Amides of this amino congener (e.g., 14) were selected as a primary focus, and (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholinocarbenium hexafluorophosphate (COMU) was found to be the best coupling agent and therefore used to synthesize all additional analogues.
Scheme 1.
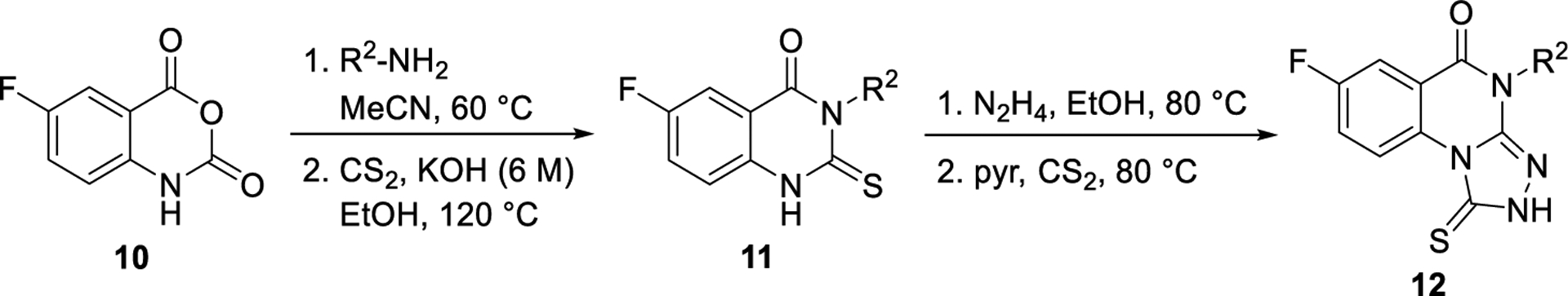
General Synthesis of 7-Fluoro Analogues
Scheme 2.
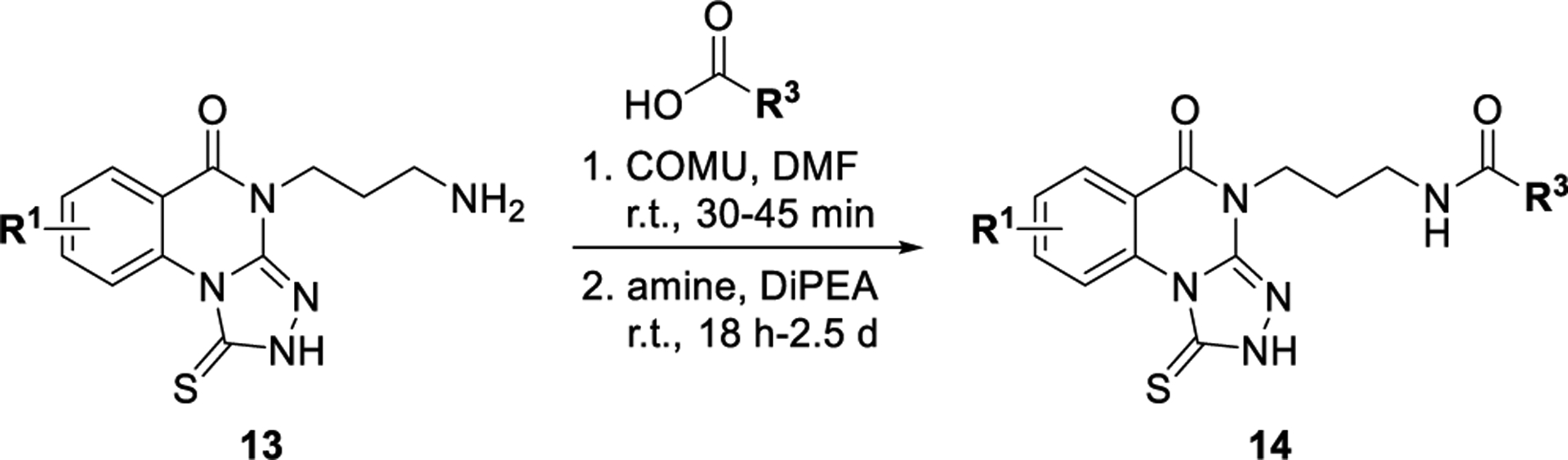
Synthesis of Zone 4 Amides
Structure–Activity Relationship.
The structure–activity relationship (SAR) of the 1-thioxo-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one scaffold as PBD inhibitors was explored (compounds 15–138, Tables 1–9). All compounds were evaluated for their efficacy against the full-length human Plk1 in an ELISA assay (representative data shown in Figure 2) and for their in vitro physiochemical properties (half-life, permeability, and solubility). Because of the lack of structural information for the binding of 7 to the PBD domain of Plk1, early SAR studies began by exploring the 2,4-dihydro-3H-1,2,4-triazole-3-thione moiety to determine empirically the requirements for binding. Despite the prevalence of sulfur atoms in small-molecule drug discovery, we began by replacing and modifying the thiourea portion of the molecule. First, we replaced the sulfur with a range of additional atoms including oxygen in 15. We found that any replacement of the sulfur resulted in complete loss of activity, indicating that a free 1-thioxo group is essential for activity (Table 1). To determine whether the thiocarbonyl was required, sulfur was methylated and product 19 tested, but this methyl thioether was also inactive. In addition, many alkyl groups were tried, and all were found to be inactive (see Table S1, Supporting Information). This showed that unmodified thiourea was required for activity.
Table 9.
Inhibitory Activity of Prodrug Triazoloquinazolinones and Their Corresponding Parent Drugs at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses)
| cmpd | R1= | R5= | IC50 (μM) |
|---|---|---|---|
| 21 | H | Ha | 1.03 ± 0.08 (9) |
| 64 | 7-F | Ha | 1.16 ± 0.06 (12) |
| 139 | 9-F | Ha | 2.06 ± 0.07 (4) |
| 140 | 9-C1 | Ha | 2.49 ± 0.02 |
| 141 | 9-OMe | Ha | 2.43 ± 0.01 |
| 142 | H | Ac | 1.33 ± 0.08 |
| 143 | H | Me | >50 |
| 144 | 7-F | Me | >50 |
| 145 | 9-F | Me | >50 |
| 146 | 9-C1 | Me | >50 |
| 147 | 9-OMe | Me | >50 |
Shown as the enol tautomers, while this series is shown in the thiocarbonyl form elsewhere. ND, not determined.
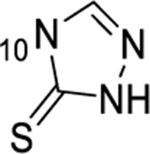
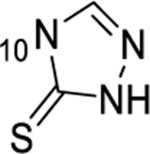
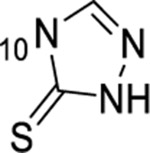
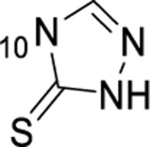
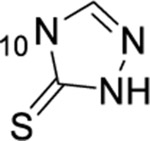
1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one scaffold for inhibition of Plk1 PBD.
Figure 2.
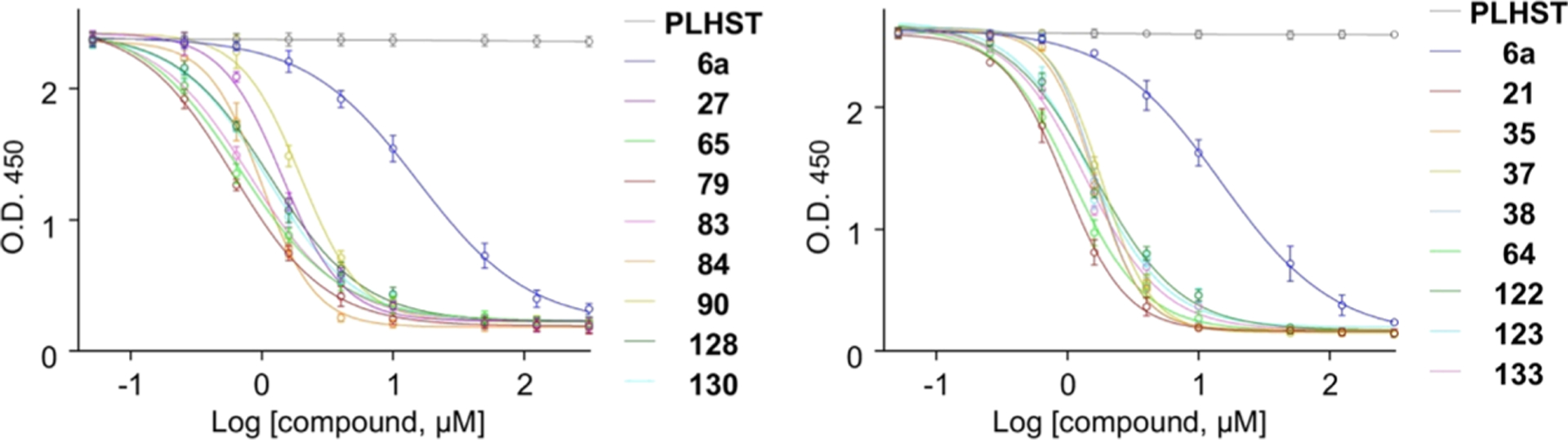
Representative inhibition curves in the ELISA assay. A previously characterized phosphopeptide, PLHSpT 6a (Kd of ~450 nM27), is included for comparison. The curve numbers correspond to compound numbers.
We also explored S-methyl and S-acetyl derivatives (compounds 139–144, Table 9) as potential prodrugs of the corresponding active 1-thioxo derivatives. By decreasing the polarity of the molecules, we expected to achieve better intracellular levels of the active species, with the expectation that the masking moiety on the S could be labile prior to reaching the site of action.
We then explored the SAR of the side chain, R2, by systematically modifying the length and composition of the linker as well as the end group of the side chain. First, we simplified the side chain by removing the terminal phenyl ring and maintaining the simpler alkyl chain of various lengths (Table 2, 20–34). All alkyl chain lengths tested, from the shorter ethyl group through the pentyl group, which maintain a linear length similar to that of 7, showed improved potency relative to the aryl-containing 7. This suggests a potential size restriction in the area of the pocket where the side chain binds. To further explore this, branching was introduced into the alkyl side chain with the 3-methylbutane of 25 and the isopropyl group of 26. However, these bulkier alkyl groups, relative to their linear counterparts 23 and 20, respectively, maintained potency in the range of 1–2 μM, suggesting that the improvement in potency was not strictly due to removal of the bulkier phenyl group.
Table 2.
Inhibitory Activity of Early Triazoloquinazolinones Modified in Zones 3 and 4 at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubility
| Cmpd | R2 = | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1e-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|
| 7 | 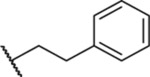 |
4.38 ± 0.41 (6) | ND | ND | ND |
| 20 | Et | 1.49 ± 0.22 (6) | >30 | 427 | >36 |
| 21 | Pr | 1.03 ± 0.08 (9) | >30 | 37.0 | 21.3 |
| 22 |  |
1.31 ± 0.03 | ND | ND | 19.4 |
| 23 | Bu | 1.03 ± 0.14 (5) | >30 | 77.5 | 8.5 |
| 24 | Pent | 1.97 ± 0.11 (6) | ND | ND | 4.2 |
| 25 | 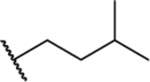 |
1.73 ± 0.08 (6) | ND | ND | 8.0 |
| 26 | i-Pr | 1.30 ± 0.06 | ND | ND | 19.5 |
| 27 | 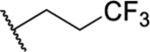 |
1.25 ± 0.11(5) | >30, | 82 | 23.3 |
| 28 | 1.75 ± 0.19 | ND | ND | 31.2 | |
| 29 | 1.68 ± 0.11 | ND | ND | 15.5 | |
| 30a |  |
3.23 ± 0.44 | ND | ND | ND |
| 31 |  |
3.90 ± 0.43 | ND | ND | ND |
| 32 | 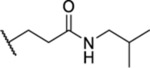 |
2.92 ± 0.56 | ND | ND | 30.8 |
| 33 | 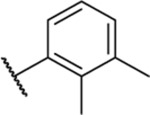 |
1.85 ± 0.12 | ND | ND | ND |
| 34 | 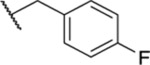 |
3.54 ± 0.57 | ND | ND | ND |
Racemic. ND, not determined.
Next, we tried to evaluate if incorporation of heteroatoms on the alkyl group (R2) was tolerated (Table 2, 27–29). The CF3 group of 27 also had the added benefit of blocking potential cytochrome P450 (CYP)-mediated metabolism of the side chain. Though there was no observable change in the metabolic stability of 27 relative to 21, 27 did maintain a potency relative to that of 21. Incorporation of oxygen to give ether side chains also improved the activity of 28 and 29 relative to 7 and maintained the activity relative to the hydrocarbon chains. We also incorporated amide groups into the side chain to see if there were additional H-bonding interactions that could be established with the PBD (Table 2, 30–32). All three amide-containing analogues showed a slight improvement over 7 (IC50 = 2.92–3.90 μM vs IC50 = 4.38 μM, respectively) but reduced activity relative to the alkyl chain-containing analogues (IC50 = 2.92–3.90 μM vs IC50 = 1.03–1.97 μM, respectively). This reduction in potency relative to the alkyl chain-containing analogues could be due to the branching of the fragment attached at the nitrogen, although branching appeared to be tolerated for analogues 25 and 26. In addition, the combination of the branching along with the carbonyl of the amide resulted in an apparently less favorable orientation of the side chain and reduced potency. The final side chains tested for these early SARs studied included shorter-chain aryl groups (Table 2, 33 and 34). For analogue 33, the 2,3-dimethylphenyl group was attached directly to the core and resulted in an improved activity relative to 7 (IC50 = 1.85 μM vs 4.38 μM, respectively). However, when the 4-fluorobenzyl group was present, it failed to show significant improvement over 7 (IC50 = 3.54 μM vs 4.38 μM, respectively). This indicates that, unless directly connected to the core, phenyl groups are not well tolerated when a linker is present.
Once an initial SAR was analyzed for the 2,4-dihydro-3H-1,2,4-triazole-3-thione and the side chain, we chose to revisit the triazole and determined whether alkylation of N-2 could be tolerated. Four analogues (35–38) included a tertiary amine with various small alkyl groups (Table 3). All four N-substituted triazole analogues showed good activity, particularly the three analogues where R2 was the longer propyl group (IC50 = 1.63–1.83 μM). The ketone-containing analogue 39 lacking a basic nitrogen was inactive (IC50 > 50 μM).
Table 3.
Inhibitory Activity of Zone 5-Modified Triazoloquinazolinone Derivatives at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubilitya
| Cmpd | R2 = | R3 = | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1e-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|---|
| 21 | Pr | H | 1.03 ± 0.08 (9) | >30 | 37 | 21.3 |
| 35 | Pr | 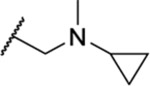 |
1.63 ± 0.05 | ND | ND | Insol. |
| 36 | CH3 | 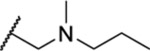 |
1.20 ± 0.03 | ND | ND | ND |
| 37 | Pr | 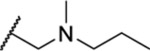 |
1.83 ± 0.04 | ND | ND | ND |
| 38 | Pr | 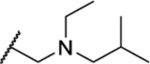 |
1.67 ± 0.06 | ND | ND | 8.7 |
| 39 | Pr | 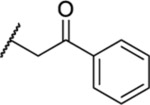 |
>50 | ND | ND | ND |
ND, not determined
We then explored the fused phenyl ring of the core to determine whether substitution would be tolerated in this region. First, we began with halogenation at the 7-position (Table 4, 40–42). As the size of the halogen increased, the affinity decreased (F, IC50 = 12.9 μM; Br, IC50 = 14.7 μM; I, IC50 = 30.7 μM). The larger, hydrophobic 7-methyl group (43) showed a potency similar to that of the 7-F analogue. However, these analogues were still less potent overall than the unsubstituted 7. We then wanted to determine if larger substituents would be tolerated if a heteroatom was introduced to make additional interactions. The 7-acetamide 44, 7-dimethylamine 45, and 7-(1-morpholino) 46 were prepared (Table 4). All three of these analogues with H-bonding capabilities were approximately twofold more potent than 7 (IC50 = 1.54–2.77 μM vs IC50 = 4.38 μM, respectively). Next, we explored the substitution pattern around the ring. To do so, the mono-fluoro analogues were prepared including 7-F (40), 8-F (41), and 9-F (42). They were shown to increase in potency as the fluorine moved around the ring from the 7- to 8- to 9-position (IC50 = 12.9, 8.29, and 2.58 μM, respectively). Finally, we explored the replacement of the phenyl ring with pyridine. The 6-aza (49), 8-aza (50), and 9-aza (51) analogues were prepared; however, these heteroaryl derivatives were found to be less potent than 7 (>2–6-fold).
Table 4.
Inhibitory Activity of Triazoloquinazolinones Modified in Zones 1 and 2 at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubilitya
| cmpd | R1= | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1 × 10−6 cm/s) | solub. (μg/mL) |
|---|---|---|---|---|---|
| 7 | H | 4.38 ± 0.41 (6) | ND | ND | ND |
| 40 | 7-F | 12.92 ± 1.8S | >30 | 77.0 | ND |
| 41 | 7-Br | 14.74 ± 0.81 | >30 | 162 | <1 |
| 42 | 7-I | 30.71 ± 1.31 | >30 | 1025 | <1 |
| 43 | 7-Me | 11.29 ± 0.92 | >30 | ND | <1 |
| 44 | 7-NHAc | 2.19 ± 0.10 | >30 | 253 | 2.2 |
| 45 | 7-N(CH3)2 | 2.77 ± 0.33 | 13.3 | 356 | <1 |
| 46 | 7-(1-morpholino) | 1.54 ± 0.21 | >30 | 1.2 | 3.4 |
| 47 | 8-F | 8.29 ± 0.59 | 22.7 | 91.0 | <1 |
| 48 | 9-F | 2.58 ± 0.12 | >30 | 141 | 3.4 |
| 49 | H (6-aza) | 6.16 ± 0.44 | >30 | 16.8 | 26.9 |
| 50 | H (8-aza) | 27.78 ± 4.07 | 15.8 | 202 | 17.3 |
| 51 | H (9-aza) | 7.55 ± 0.44 | >30 | 51.8 | 4.9 |
ND, not determined.
The next stage of our SAR exploration involved combining features from each of the regions explored so far (Table 5). First, we incorporated substitutions on the phenyl ring and modified the side chain. On the 7-fluoro core, we shortened (52) and extended (53), the alkyl phenyl side chain. Both the longer propyl and shorter methyl homologues showed similar activity that was slightly less potent than that of the parent 7 with the ethyl linker (propyl, IC50 = 6.63 μM; methyl, IC50 = 6.42 μM; ethyl, IC50 = 4.38 μM). We then revisited the alkyl chains with the 7-chloro core (Table 5, 54–57). Compound 54 with the propyl side chain showed a small decrease in potency from the unsubstituted 21 (IC50 = 1.77 μM and IC50 = 1.03 μM, respectively). For compound 55 with the 3,3,3-trifluoropropane side chain, the 7-chloro core resulted in a nearly twofold loss of potency compared to its unsubstituted counterpart, 27 (IC50 = 2.14 μM and IC50 = 1.25 μM, respectively). The ether-containing analogue 56 was equipotent to its most similar 29. The two amide-containing side chain analogues 57 and 58 with the 7-chloro and 7-methyl cores, respectively, were about twofold more potent than the unsubstituted 31 (IC50 = 1.49 μM, IC50 = 2.01 μM, and IC50 = 3.90 μM, respectively).
Table 5.
Inhibitory Activity of Triazoloquinazolinones Modified in Combined Zones 1, 2, 3, and 4 at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubilitya
| Cmpd | R1 = | R2 = | Triazole | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1e-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|---|---|
| 52 | 7-F | 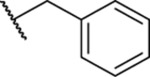 |
 |
6.42 ± 0.44 | >30 | 841 | 8.3 |
| 53 | 7-F |  |
 |
6.63 ± 0.39 | 8.8 | 1269 | <1 |
| 54 | 7-C1 |  |
 |
1.77 ± 0.15 (6) | ND | ND | ND |
| 55 | 7-C1 | 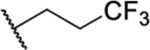 |
 |
2.14 ± 0.12 | >30 | 257 | 5.1 |
| 56 | 7-C1 | 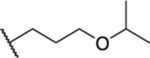 |
 |
1.62 ± 0.17 | ND | ND | 8.6 |
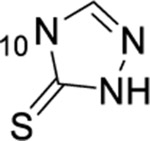
| 57 | 7-C1 | 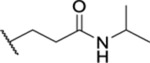 |
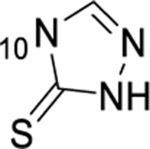 |
1.49 ± 0.09 | ND | ND | 8.1 |
| 58 | 7-CH3 | 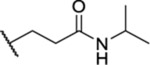 |
 |
2.01 ± 0.21 | ND | ND | ND |
| 59 |  |
 |
 |
3.01 ± 0.10 | ND | ND | ND |
| 60 | 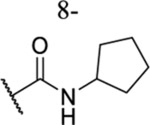 |
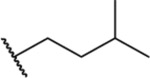 |
 |
11.11 ± 0.90 | ND | ND | ND |
| 61 | 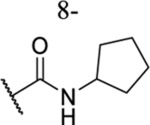 |
CH3 | 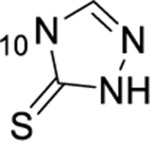 |
2.75 ± 0.17 | ND | ND | ND |
| 62 | 7-F |  |
 |
>50 | 2.2 | 161 | 4.5 |
ND, not determined.
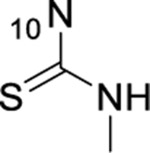
After the promising results shown in Table 4 for the 7-acetamide 44, three additional amides were prepared this time in the 8-position to determine the tolerability of a larger group in the 8-position. The 8-isopropyl amide with the propyl chain (59, IC50 = 3.01 μM) was shown to be less potent than the unsubstituted 21 (IC50 = 1.03 μM) but more potent than either the 8-fluoro (47, IC50 = 8.29 μM) or the 8-aza (50, IC50 = 27.8 μM). Compounds 60 and 61 have an 8-cyclopentyl amide substitution with alkyl side chains. However, when comparing 60 and 25, both of which have a 3-methylbutane side chain, the substitution in the 8-position was not beneficial as 60 was found to be sixfold less potent than 25. We had already discovered that modifying the thiourea portion of the 2,4-dihydro-3H-1,2,4-triazole-3-thione was not tolerated; however, we wanted to determine whether the triazole ring itself was required. To test this, we prepared the thiourea, 62, which maintained the thiocarbonyl as well as the NH of the 2,4-dihydro-3H-1,2,4-triazole-3-thione. Similar to most other modifications of the 2,4-dihydro-3H-1,2,4-triazole-3-thione, the thiourea of 62 was not tolerated and also resulted in a highly metabolically labile analogue (IC50 > 50 μM, t1/2 RLM = 2.2 min).
Based on the observed results thus far, it became clear that we could make the most significant positive modifications to the chemotype in the side chain. Therefore, we made a large number of analogues on the 7-fluoro core and varied the side chain to expand our SAR. We began with linear alkyl groups, which we had previously found to lead to low micromolar affinity. The shorter ethyl group (63) led to a small reduction in affinity compared to the unsubstituted 20 (IC50 = 1.98 μM and IC50 = 1.49 μM, respectively), while the propyl analogue (64) was found to be equipotent to the unsubstituted equivalent (21). Previously the 3,3,3-trifluoropropyl group led to one of the most potent analogues. Therefore, we combined this group with the 7-F and found that the 7-F analogue 65 was at least as potent as the corresponding unsubstituted 27. We then chose to introduce additional H-bonding groups that might increase interactions within the binding site. To this end, hydroxy (66), amino (67–76), and phosphorus (77, 78) derivatives were synthesized and tested. The 1-propanol group, which is similar in length to the butyl analogue (23), maintained good potency at 1.44 μM. For the primary amines, both ethyl and propyl chains were tested, and the three-carbon linker was found to be twice as potent as the two-carbon linker. Also, as the 9-fluoro substitution was previously shown to be the most potent of the mono-fluoro substituents, the 9-F analogue with a propylamine chain (69) was prepared but found to be less potent than the 7-F derivative 68. This is unexpected as it is opposite to the trend seen for the phenethyl series. When relatively simple dimethyl amine analogues 70 and 71 were prepared, 70 was shown to be slightly more potent than the equivalent primary amine 67, while the branched 1-dimethylaminoprop-2-yl analogue (71) was slightly less potent. In the case of amide (72–74) and sulfonamide (75 and 76) derivatives, regardless of the linker length or the type of derivative, all analogues essentially performed similarly (IC50 = ~2.0 μM) with the benzamide 74 showing the most potent affinity of the five. The phosphorus-containing analogues, phosphonic acid 77 and phosphonate diester 78, both showed significantly weaker affinity, with the phosphonic acid being essentially inactive. In addition to these heteroatom-containing analogues, several cyclopropane derivatives were prepared (79 and 80). Both 79 and 80, which differ in the location of the cyclopropane in the chain, showed good potency of approximately 1.0 μM, indicating that although heteroatoms are well tolerated in the side chain, they are not required.
Next, we explored the phenethyl side chain (Table 6), first, by substituting the phenyl moiety with chloro groups (81 and 82). While the 4-chloro analogue maintained an affinity similar to that of 7 (82, IC50 = 5.71 μM) and was over twofold more potent than 40, the 2-chloro analogue was less potent than either 7 or 40 (81, IC50 = 14.4 μM). This indicates that ortho substitution is not well tolerated, which supports the earlier conclusion that for the phenethyl derivatives the binding pocket around the phenyl group is restrictive, and substituents that make it bulkier led to negative interactions. Concluding that the space around the phenyl ring was sterically limited, we next chose to replace it with a pyridine to determine if H-bonding opportunities are available without increasing the steric bulk. All three pyridine analogues were synthesized, and all were found to be ≥12-fold more potent than the phenyl-containing 40. The trend for the potency (4-pyridine ≈ 2-pyridine > 3-pyridine, best to worst) differs from that observed for the chloro substitution, which clearly indicated that the 4- position is the most favorable for affinity. However, in the case of the pyridines, all three N positions were well tolerated. This could be due to the chloro group extending into the pocket, creating steric clashes, which were absent for the pyridines. Nonaromatic heterocycles were also tested to determine whether they would be more beneficial than the aromatic heterocycles. Azetidine 86, pyrrolidine 87, and piperidine 88 were all prepared to determine if there was a size preference for the ring, and a clear preference was observed for the six-membered piperidine (IC50 = 5.67 μM, IC50 = 5.67 μM, and IC50 = 1.51 μM, respectively). With this information, we then explored whether additional heteroatoms in the 4-position would be tolerated. The 4,4-difluoropiperidine 89 and the N-morpholino 90 were equipotent to 88, that is, all near IC50 = 1.5 μM. However, when a second nitrogen was incorporated into the ring in analogues 91 and 92, a discrepancy was observed in the SAR. Based on the chloro-substituted phenyl rings, both 91 and 92 were expected to be potent; however, only 92 showed an affinity similar to the other six-membered ring heterocycles. Because the methyl group is much smaller than the Boc group, the difference in affinity is not due to the size of the group. The more likely source of the difference is the basicity of the nitrogen in the 4-position, given that the Boc group significantly decreases the pKa of the nitrogen relative to 91. Finally, we also prepared the carbocyclic version of 89 to determine if the heteroatom in the ring was required at all. Compound 93 maintained the difluoro substitution, which could potentially generate H-bonding interactions. However, there were no other heteroatoms with which to form H-bonds or salt bridges, and as a result, the potency showed a small (approximately twofold) decrease, indicating that though the nitrogen is not required, it is beneficial.
Table 6.
Inhibitory Activity at the Plk1 PBD of Triazoloquinazolinones Modified in Zones 3 and 4 with a Preferred 7-Fluoro Substitution, Except for 69 with 9-Fluoro (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubility
| Cmpd | R2 = | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1e-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|
| 63 | Et | 1.98 ± 0.07 | >30 | 39.5 | >39 |
| 64 | Pr | 1.16 ± 0.06 (12) | >30 | 195 | >28 |
| 65 | 0.75 ± 0.06 (5) | >30 | 117 | 17.3 | |
| 66 | 1.44 ± 0.10 (9) | >30 | 222 | >43 | |
| 67 | 4.33 ± 0.18 | >30 | 8.2 | >41 | |
| 68 | 2.26 ± 0.07 (4) | >30c >30d |
<lc 16.8d |
>43c 27d |
|
| 69 | 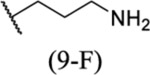 |
5.07 ± 0.16 | >30 | 4.2 | 15.9 |
| 70 | 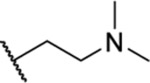 |
2.45 ± 0.25 | >30 | 177 | >45 |
| 71a | 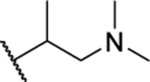 |
6.12 ± 0.17 | >30 | 186 | >47 |
| 72 | 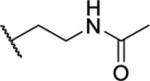 |
2.48 ± 0.20 | >30 | 12.9 | >47 |
| 73 | 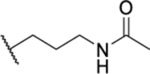 |
2.48 ± 0.08 (6) | >30 | 18.0 | >50 |
| 74 | 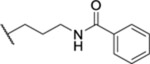 |
1.60 ± 0.19 (4) | >30 | 267 | 21.9 |
| 75 | 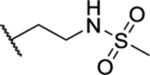 |
2.77 ± 0.31 | >30 | 8.9 | >53 |
| 76 | 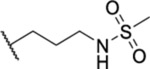 |
1.87 ± 0.06 | >30 | 10.8 | 34 |
| 77 | 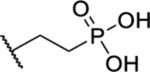 |
21.70 ± 1.73 | ND | 9.3 | >51 |
| 78 | 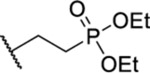 |
13.89 ± 0.59 | >30 | 16.7 | >59 |
| 79 | 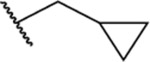 |
0.77 ± 0.08 (8) | >30 | 46.0 | 16.2 |
| 80b | 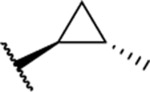 |
1.19 ± 0.05 (4) | >30 | 188 | 13.2 |
| 81 | 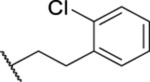 |
14.37 ± 0.33 | 22.8 | 56.3 | 3.0 |
| 82 | 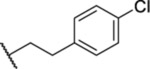 |
5.71 ± 0.44 | >30 | <1 | 1.2 |
| 83 |  |
0.87 ± 0.07 (4) | >30 | 355 | 13.0 |
| 84 |  |
1.07 ± 0.13 (9) | >30 | <1 | 25.1 |
| 85 | 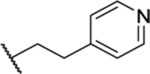 |
0.89 ± 0.05 (4) | >30 | 32.7 | 10.9 |
| 86 | 5.67 ± 0.30 | ND | 229 | 15 | |
| 87a | 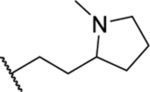 |
5.67 ± 0.42 | >30 | 53.8 | 17.8 |
| 88 | 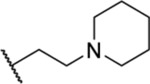 |
1.51 ± 0.19 (4) | >30 | 358 | 2.4 |
| 89 | 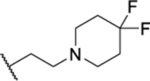 |
1.37 ± 0.08 (5) | >30 | 234 | 20.2 |
| 90 | 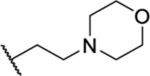 |
1.63 ± 0.22 (5) | >30 | 67.0 | >52 |
| 91 | 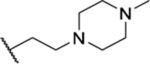 |
6.72 ± 0.17 | >30 | 12.7 | >54 |
| 92 | 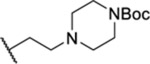 |
1.84 ± 0.01 | 26.3 | 33.1 | 20.5 |
| 93 | 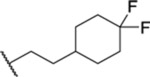 |
2.83 ± 0.36 | >30 | 201 | <1 |
Racemic.
Relative stereochemistry, trans.
TFA salt.
Free base. ND, not determined.
Expanding upon the SAR established above, we continued to explore the side chain region by focusing on the propyl amide motif, given that this serves as a good handle for further modification to probe the binding pocket (Table 7) and that compound 74 with the benzamide was one of the more potent analogues thus far. First, we prepared analogues 94 and 95 with polyethylene glycol (PEG) linkers to extend the primary amine further into the pocket. While both compounds maintained single-digit micromolar affinity, the Boc-protected 95 was more potent than the free amine 94 (IC50 = 3.22 μM and IC50 = 5.36 μM, respectively), which suggests a preference for a less basic group and that the size of the terminal group is unimportant. To determine whether this was related to a preference for uncharged species or unprotonated species, the l-Glu derivatives were prepared (96 and 97). While the fully deprotected 96 was shown to be >twofold less potent than 94, the t-butyl ester-protected 97 was <twofold less potent. This does support the conclusion that, in the region where these highly extended compounds reside, an uncharged species is preferred. We then chose to look at increasingly shorter chains to determine their tolerability in the binding pocket. The PEG3 chain analogues (98 and 99) showed the same preference for the less basic, Boc-protected amine that was observed for the PEG4 chain. However, when the chain was shorted further to the hexylamine, the primary amine and the corresponding Boc-protected amine were equipotent, indicating that the preference for uncharged species begins at greater distances from the main binding site for the chemotype. The Boc-d-Glu-OtBu (102) and Boc-l-Glu-OtBu (103) derivatives showed a preference for the natural amino acid derivative (>twofold more potent than 102). Compound 103 was then deprotected, giving either the Boc deprotected L-Glu-OtBu 104 or the fully deprotected 105. These analogues show that a negatively charged species is not well tolerated (105 IC50 = 12.7 μM). The succinic acid derivative, 106, still exhibited an intolerance for negatively charged species as well, indicating that the two charges likely present in 105 are not beneficial either. Moreover, the sulfonamide 107, which is thought to maintain many of the same potential interactions without being negatively charged, was shown to be the most potent of these extended linear analogues with an IC50 = 2.18 μM.
Table 7.
Inhibitory Activity of Amide-Containing Triazoloquinazolinones Modified in Zones 3 and 4 with Preferred 7-Fluorine at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubility
| Cmpd | R2 = | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (le-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|
| 94 | 5.36 ± 0.40 | ND | ND | ND | |
| 95 | 3.22 ± 0.12 | ND | ND | ND | |
| 96 | 13.13 ± 0.36 | >30 | 13.4 | >99 | |
| 97 | 9.40 ± 0.35 | >30 | 18.1 | >107 | |
| 98 | 5.02 ± 0.29 | >30 | 8.8 | 36.1 | |
| 99 | 2.88 ± 0.21 | 27.9 | 44.3 | >88 | |
| 100 | 3.04 ± 0.16 | >30 | 3.5 | 31.8 | |
| 101 | 3.23 ± 0.15 | >30 | 405 | >75 | |
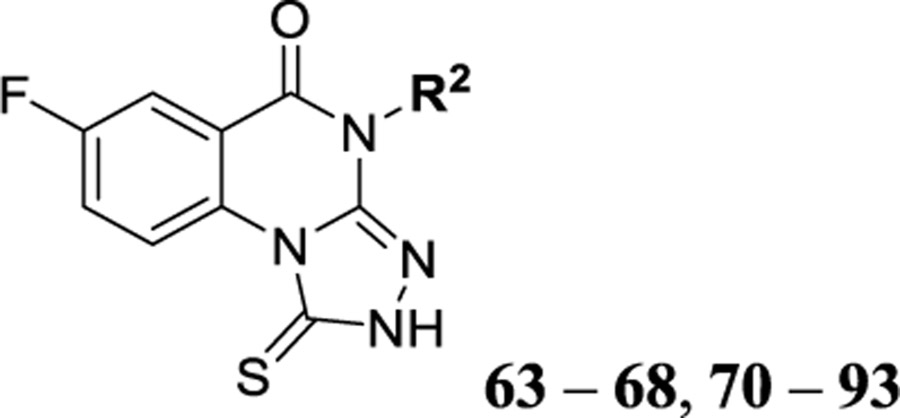
| 102 | 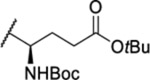 |
8.19 ± 0.26 | 25.1 | 1214 | 20.4 |
| 103 | 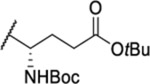 |
3.31 ± 0.14 | 3.8 | 744 | 41.6 |
| 104 | 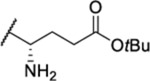 |
2.33 ± 0.13 | >30 | 8.7 | 37.5 |
| 105 | 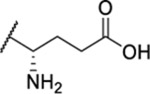 |
12.65 ± 0.44 | >30 | 20.1 | 32.0 |
| 106 | 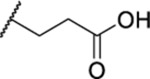 |
5.05 ± 0.22 | >30 | <1 | >58 |
| 107 | 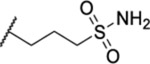 |
2.18 ± 0.19 | >30 | 3.0 | 42.4 |
| 108 | 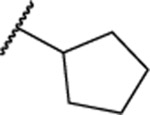 |
1.47 ± 0.18 | >30 | 155 | >58 |
| 109a | 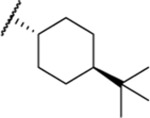 |
>50 | >30 | 66.9 | 1.1 |
| 110b | 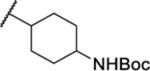 |
2.38 ± 0.74 | 24.8 | 330 | >77 |
| 111b | 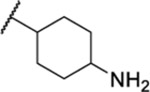 |
3.97 ± 0.26 | >30 | 1.6 | 38.8 |
| 112b | 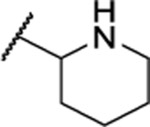 |
27.66 ± 1.25 | >30 | ND | >60 |
| 113b | 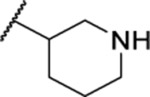 |
30.20 ± 1.09 | >30 | ND | 29.6 |
| 114 | 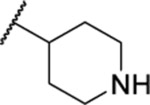 |
44.01 ± 2.32 | >30 | ND | >60 |
| 115 | 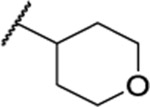 |
1.79 ± 0.11 (4) | ND | 8.0 | <1 |
| 116 | 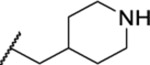 |
3.96 ± 0.37 | >30 | 4.4 | 37.5 |
| 117 | 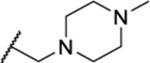 |
4.66 ± 0.23 | >30 | <1 | >64 |
| 118 | 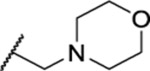 |
3.77 ± 0.19 | >30 | <1 | >62 |
| 119 | 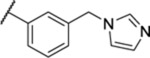 |
1.48 ± 0.12 (4) | >30 | 46.8 | 13.7 |
| 120 | 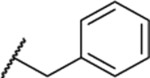 |
1.23 ± 0.09 (6) | 18.7 | 248 | 31.7 |
| 121 | 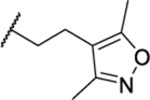 |
1.78 ± 0.13 (4) | 6.3 | 27.6 | 37.6 |
Relative stereochemistry, trans.
Racemic. ND, not determined.
After determining that the shorter chain molecules were preferred, we focused on smaller, cyclic analogues. Beginning with carbocyclic groups, the cyclopentyl amide 108 showed a relatively good potency (IC50 = 1.47 μM), but the sterically much bulkier trans-4-(t-butyl)cyclohexane amide 109 was inactive. Consistent with the finding that a highly bulky group was not tolerated, the 4-Boc-aminocyclohexane amide 110 was found to be moderately potent (IC50 = 2.38 μM). However, once again, when the Boc group was removed (111), the potency decreased slightly, suggesting the preference for uncharged species. A series of piperidine analogues (112–114) were prepared to further assess this. In agreement with the data above, all three piperidines, despite the location of the nitrogen in the ring, were significantly less active and nearly inactive (IC50 = 27.7–44.0 μM). Notably, 4-tetrahydropyran 115 was found to show good potency (IC50 = 1.79 μM), indicating that the protonation of the amine, which likely occurs at physiological pH, is the feature not tolerated rather than the presence of a heteroatom in the ring. Three additional analogues with a methylene linker between the amide carbonyl and the heterocycle were also prepared (116–118). All three analogues contained a basic nitrogen in the ring and all three showed moderate affinity, reinforcing the preference for neutral compounds in this side chain region. Finally, a series of aromatic analogues of differing lengths were prepared (119–121). All three analogues, despite the presence of heteroatoms or the overall length of the side chain, showed good affinity (IC50 = 1.23–1.78 μM).
Given that compound 120 showed the best affinity of these amide analogues, we decided to explore substitution around the phenylacetamide portion (Table 8). Beginning with simple halogen-containing analogues, compounds 122–125 were prepared. Though not all positions were tested for each halogen, a general trend did emerge. When comparing 122 and 123, the position of the halogen was found not to have a significant impact on the potency (meta IC50 = 1.42 μM and para IC50 = 1.35 μM). Also, the halogen size had no effect on the affinity of the analogue as seen by all four analogues showing essentially the same potency, indicating that the size of the group does not have a large impact on binding. Next, we looked at substituents that would modulate the electronic character of the ring (126–134). For electron-donating groups, methyl (126), hydroxy (127), and methoxy (128–130) analogues were prepared. Overall, electron-donating groups were well tolerated, maintaining a similar affinity relative to 120. Similar to the trend observed for the halogens, the location of these groups did not have a large impact on the potency of the compound either. The electron-withdrawing groups, trifluoromethyl (131–132) and cyano (133–134), showed similar results. They were also well tolerated in all positions, but the 3-CF3 derivative (132) was the least potent but only slightly (<twofold) less potent than all other substituted phenylacetamide analogues. The Boc-protected 4-amino analogue was also twofold less potent than the parent 120. Finally, in the case of a series of pyridine analogues prepared (136–138), all three analogues were less potent than the phenyl analogue 120 and 2-pyridine was the weakest at IC50 = 5.34 μM.
Table 8.
Inhibitory Activity of 4 Phenylacetic Acid Amide Triazoloquinazolinone Derivatives Modified in Zones 3 and 4 with Preferred 7-Fluorine at the Plk1 PBD (IC50 Values Are n = 3 Unless Noted in Parentheses) and Microsomal Half-Life, PAMPA Assays, and Aqueous Solubilitya
| Cmpd | R4 = | IC50 (μM, ELISA) | t1/2 (min, RLM) | PAMPA (1e-6 cm/s) | Solub. (μg/ml) |
|---|---|---|---|---|---|
| 122 | 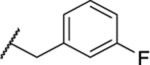 |
1.42 ± 0.18 (4) | 26.5 | 364 | >63 |
| 123 | 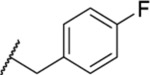 |
1.35 ± 0.17 (4) | 26.3 | 8.5 | 8.5 |
| 124 | 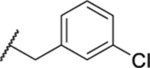 |
1.15 ± 0.03 (4) | 8.4 | 861 | >66 |
| 125 | 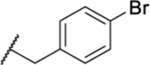 |
1.59 ± 0.12 | 20.6 | 767 | <1 |
| 126 | 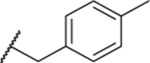 |
1.18 ± 0.01 | 23.9 | 621 | 12.9 |
| 127 | 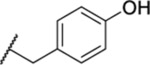 |
1.16 ± 0.06 | >30 | 3.7 | 31.0 |
| 128 | 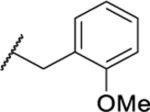 |
1.15 ± 0.11 (5) | 16.1 | 379 | >65 |
| 129 | 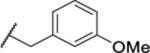 |
0.96 ± 0.08 (4) | 20.1 | 300 | >65 |
| 130 | 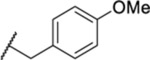 |
1.06 ± 0.08 (5) | >30 | 233 | 3.2 |
| 131 | 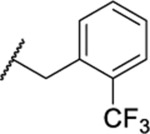 |
1.40 ± 0.07 (4) | 3.7 | 825 | 40.3 |
| 132 | 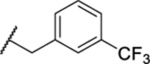 |
2.23 ± 0.33 | 17.3 | 1088 | 26.8 |
| 133 | 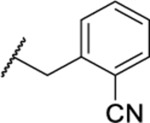 |
1.24 ± 0.14 (4) | >30 | 156 | 20.9 |
| 134 | 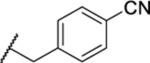 |
0.99 ± 0.14 | >30 | 172 | 2.0 |
| 135 | 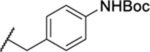 |
2.72 ± 0.63 | >30 | 910 | 17.4 |
| 136 | 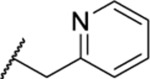 |
5.34 ± 0.64 | >30 | 68.0 | 41.4 |
| 137 | 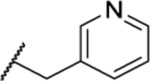 |
2.50 ± 0.28 | >30 | 21.0 | 31.8 |
| 138 | 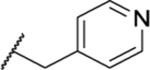 |
3.01 ± 0.26 | >30 | <1 | 9.6 |
ND, not determined.
Plk1 PBD Specificity.
To determine whether the above modified compounds show Plk1 PBD-binding specificity, we carried out FP-based inhibition assays using fluorescein isothiocyanate (FITC)-labeled peptides that specifically bind to each of the PBDs from Plk1, Plk2, and Plk3.26 Under these experimental settings, PLHSpT 6, but not its respective nonphosphorylated peptide, specifically inhibited Plk1 PBD with an IC50 of 22 μM (Figure 3). Under the same conditions, representative, potent compound 79 inhibited Plk1 PBD with an IC50 of 0.47 μM and failed to exhibit any detectable level of inhibition against the PBDs from Plk2 and Plk3. An approximately 40-fold increased potency for compound 79 over PLHSpT 6 is largely in good agreement with the data obtained with ELISA-based assays described above. Similar to this observation, expanded FP assays showed that several additional compounds tested showed approximately the same degree of Plk1 PBD specificity (Figure S3, Supporting Information). The all-or-none selectivity for Plk1 PBD is significant, given that the well-characterized Plk1 KD inhibitor BI2536 exhibits Plk1 selectivity of only around fourfold.49 These data collectively suggest that, while the exact binding mode of these compounds is yet to be determined, they may at least partially block the canonical S-pT/pS-P/X-binding region of Plk1 PBD described previously.11,27
Figure 3.
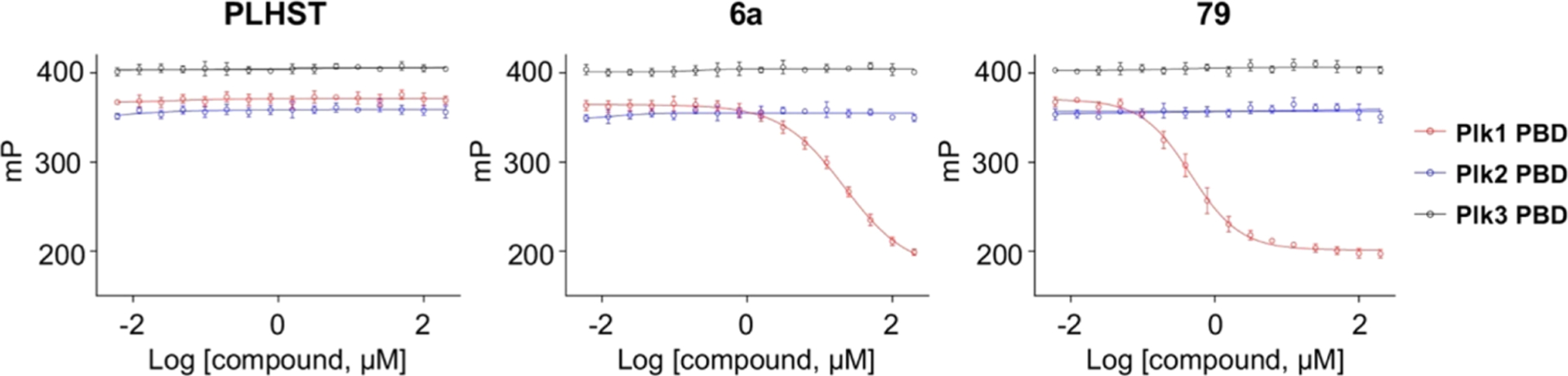
Comparative FP-based assays showing that compound 79 potently inhibits the PBD of Plk1 but not Plk2 and Plk3 with an IC50 of 0.47 μM. Under the same experimental conditions, PLHSpT 6a shows an IC50 of 22 μM, whereas its control nonphosphorylated form, PLHST 6b, exhibits no detectable activity. Bars, mean ± standard deviation (s.d.).
Prodrug and Cellular Efficacy.
Having confirmed that many of the compounds generated through SAR exhibited submicromolar IC50 values with a superb specificity for Plk1 PBD, we then examined their activities in whole-cell studies. However, likely due to limited intracellular bioavailability arising from low membrane permeability, we failed to observe mitotic arrest, the phenotype that can be expected by interfering with the function of Plk1 PBD.25,26 We reasoned that alkylating the thiocarbonyl could improve the cellular uptake of the compounds. In addition, it would directly block oxidative metabolism of the thiourea as well as prevent potential oxidation of the phenyl ring via steric factors, consequently reducing clearance and allowing for greater exposure of the drug at a site of action. Therefore, we selected a small alkyl group or small alkyl ester as a starting point as they should be relatively readily cleaved within the body and could allow us to gauge the level of stability necessary for the prodrug to be beneficial. Accordingly, compounds 142–147 were prepared as thioester or S-methyl prodrugs and examined in the ELISA assay (Table 9) and in metabolic studies (see below). The corresponding parent drugs were unsubstituted 21 for 142 and 143, 7-F analogue 64 for 144, 9-F analogue 139 for 145, 9-Cl analogue 140 for 146, and 9-OMe analogue 141 for 147. Not surprisingly, these prodrugs, except the S-acyl group-containing 142, were inactive with IC50 > 50 μM (Table 9). Compound 142 appeared to rapidly become active by releasing the active parental compound 21 because of the reactivity of the S-acyl group toward electron-rich moieties (see below and Figure S4, Supporting Information).
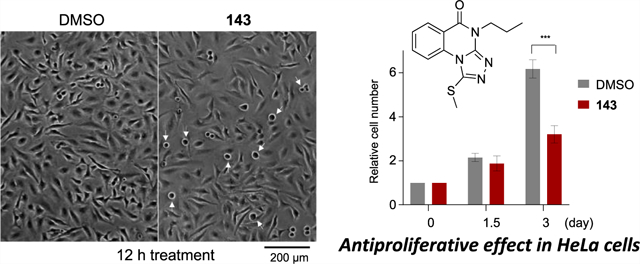
In HeLa cells, compounds 143, 144, and 145 were compared for their ability to induce mitotic arrest and an antiproliferation effect, which are characteristics of Plk1 PBD inhibition.25,27 Results showed that treatment of cells with 100 μM of either compound 143 or 145 effectively induced rounded cells with apoptotic or aberrant chromosome morphologies (Figure 4A,B), indicative of a sustained mitotic arrest. Consequently, both compounds inhibited cell proliferation, decreasing the total cell population nearly by 50% 3 days after treatment. Considering that blocking the function of Plk1 PBD-dependent PPI is destined to be less drastic than that of Plk1 KD20,21 and that several purported inhibitors reported to date show a low level (IC50 of 50–1000 μM) of cellular activities,28–35 the activity of 143 or 145 is a modest but clear improvement. Treatment with compound 144 also induced mitotic arrest but at a somewhat reduced level, whereas control DMSO failed to induce any detectable phenotype under the same conditions. As expected, if the arrest was caused by interfering with the PBD-dependent Plk1 function, compound 143 significantly inhibited Plk1 localization to centrosomes (approximately 20% inhibition) and kinetochores (approximately 50% inhibition), frequently yielding cytosolic aggregates of delocalized Plk1 as observed previously44 (Figure 4C). A weaker blocking of Plk1 localization to the centrosome is likely due to the presence of PBD-independent Plk1-binding targets at this structure. Consistent with impaired Plk1 localization, spindle bipolarity was significantly compromised in 143-treated cells (Figure 4D). Under the same conditions, the parent drugs, that is, 21, 64, and 139, lacked a detectable level of cellular response. These findings suggest that, although the prodrugs themselves failed to inhibit Plk1 PBD (Table 9) because of the absence of a free 1-thioxy group, the S-methyl thioethers had a more pronounced response, presumably by promoting cell membrane permeability while regenerating the parent drug intracellularly.
Figure 4.
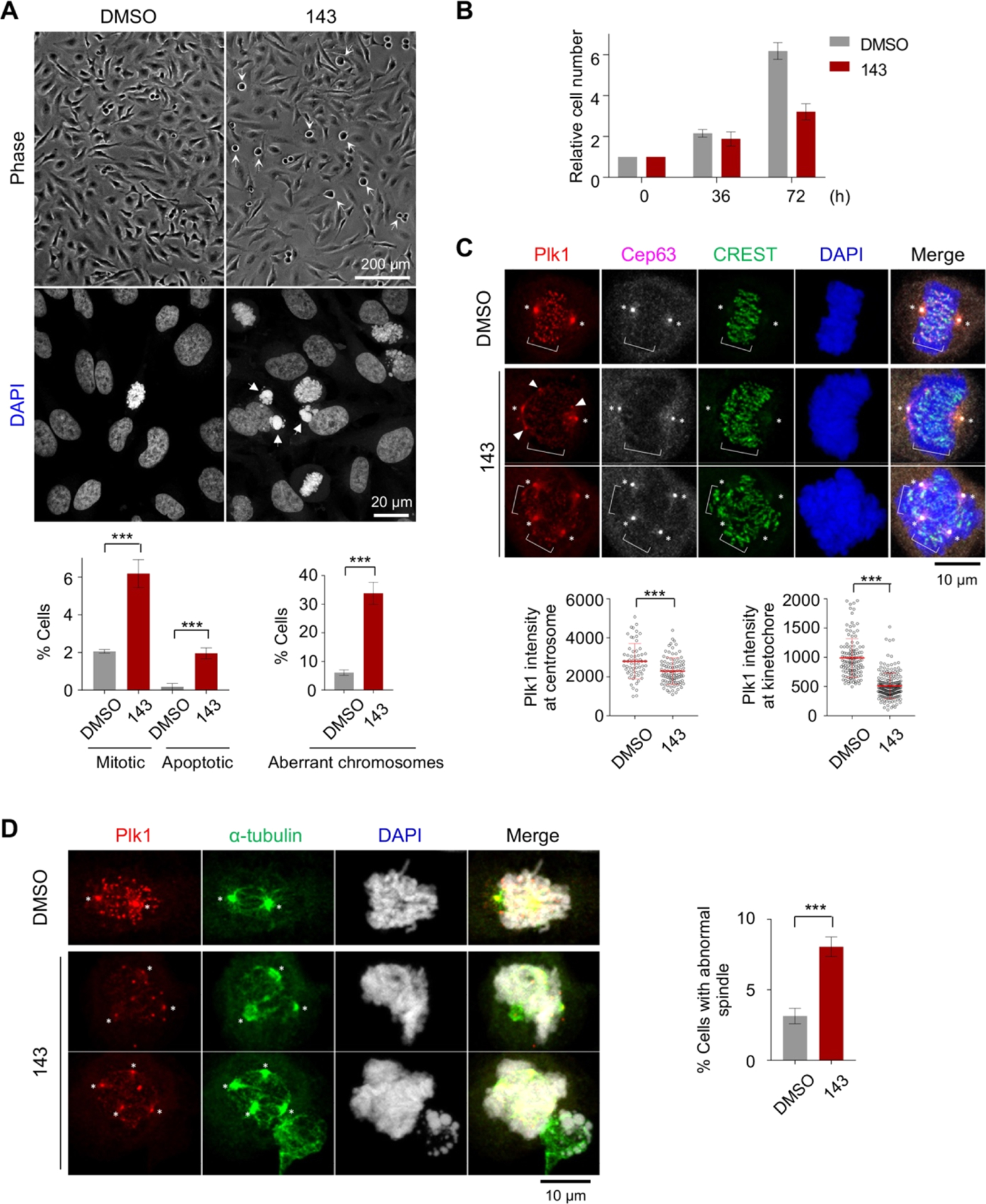
Antiproliferative effect of 143 in HeLa cells. (A) Asynchronously growing HeLa cells were treated with 100 μM of compound 143 for 12 h, subjected to phase-contrast micrography (cells with a rounded morphology are marked with barbed arrows) and stained with DAPI. Cells exhibiting mitotic and apoptotic DNA morphologies (arrows in DAPI) (bottom left) and abnormally misaligned or lagging chromosomes (bottom right) were quantified from three independent experiments (n > 515 cells/sample/experiment). Bars, mean ± s.d., ***, P < 0.001 (unpaired two-tailed t-test). (B) Cell numbers at the indicated time points were quantified from three independent experiments (n = an average of 2547 cells/experiment for DMSO and 1673 cells/experiment for 143). Bars, mean ± s.d. (C,D) Cells in (A) were immunostained with the indicated antibodies to reveal delocalized Plk1 signals from centrosomes (marker: Cep63) and kinetochores (marker: CREST) (C) and abnormal spindle morphologies (marker: α-tubulin) (D). Asterisks, centrosome-localized Plk1; brackets, kinetochore-associated Plk1. Arrowheads in (C), delocalized Plk1 aggregates in the cytosol. Quantification was carried out from three independent experiments [≥20 (DMSO) or 32 (143) centrosomes/sample/experiment and ≥42 (DMSO) or 64 (143) kinetochores (~4 kinetochores per cell)/sample/experiment for (C); ≥106 (DMSO) or 102 (143) cells/sample/experiment for (D)]. Bars, means ± s.d., ***, P < 0.001 (unpaired two-tailed t-test).
Analysis of Pharmacokinetics.
To obtain metabolic stability information, the half-life of the selected compounds was determined in rat liver microsomes (RLMs) containing the NADPH-generating system, which permits CYP-mediated metabolic reactions.46 Parallel artificial membrane permeability assay (PAMPA) permeability was measured using a high-throughput protocol, as reported.47 Aqueous solubility was determined by a published procedure.48 Most compounds tested showed a half-life of >30 min, indicating that they could be relatively stable in the CYP system. To further elucidate their metabolic pathway and to prioritize compounds for in vivo testing, several were selected as representative compounds from each group with different substitution patterns to characterize the metabolites that may be generated during incubation with CYP using mouse liver microsomes (MLMs). By inspection of the triazoloquinazolinone structures, they display a potential for CYP-mediated hydroxylation on the main quinazolinone aromatic ring as a preferred target. To our surprise, not only were the expected hydroxylated metabolites undetectable, but no other metabolites were produced in the CYP incubation system for most of the tested compounds. To further understand the metabolic pathways, similar MLM incubations of the selected compound set were conducted with uridine 5′-diphospho-glucuronic acid (UDPGA) as a cofactor to allow glucuronidation reactions. As shown in Figure S1, a new peak with 176 Da increase of the corresponding parent, which is a characteristic gain for a glucuronide, appeared for each of the tested compounds. It should be noted that for some of these compounds, two peaks with the MWs corresponding to one and two glucuronide additions were apparent, but for the remainder, only one peak appeared with the fragmentation pattern and MW representing a glucuronide. To characterize the glucuronidated site, the MS/MS analysis was performed to generate a fragment pattern for each of the glucuronides. Upon entering the source, however, the parent ions of the glucuronides fragmented into the daughter ions, which are generated from the parent ion, rather than giving a characteristic fragment to indicate the glucuronidation site (Figure S2). Examining the structure of these compounds, the thiocarbonyl S and the triazole ring secondary amine are two sites that could potentially be attacked by uridine 5′-diphospho-glucuronosyltransferase (UGT). Thus, it is highly likely that one of the two glucuronides was generated from the S-glucuronidation and the other from N-glucuronidation. Remarkably, a considerable amount of parent ion remained for the earlier-eluted glucuronides, while the parent ions of the later-eluted glucuronides almost completely fragmented. This is indicative of an N-glucuronide for the former and an S-glucuronide for the latter, given that the C–N bond linking the parent and the glucuronic acid in the former is much more stable than the corresponding C–S bond in the latter. In addition, only the S-glucuronide could be generated for the amide- or pyridine-containing compounds as the parent ions of their glucuronides were also completely fragmented. Thus, it is reasonable to hypothesize that the hydrogen-bonding capabilities of the amide- or pyridine-containing compounds may account for their altered glucuronidation profile where they interacted with UGT in a pattern from which N-glucuronidation was not favored. These results demonstrated that noncirculating metabolites other than the circulating products are expected to appear in vivo when these triazoloquinazolinones are administrated to mice.
Stability of Selected Compounds and Their Prodrugs in Mice.
Despite the in vitro data suggesting that most analogues were stable in RLMs up to 30 min (Tables 1–8), further assays were required to fully understand the stability in both in vitro and in vivo systems. Therefore, for selected compounds, additional microsome-based stability assays were performed to determine the major metabolites and class of enzymes responsible for the modifications. Several molecules including early hit-compounds (23, 46) and compounds with submicromolar affinity (i.e., 65, 79, 83, and 134) were subjected to in vitro MLM assay. LC–MS/MS analysis revealed that glucuronidation is the major metabolite. Note that, in vitro glucuronidation resulted in two products, S- and N-glucuronides in varying amounts, which resulted in two glucuronidation peaks in LC-traces for some compounds (Figures S1 and S2, Supporting Information). To mitigate this issue, the S-acetyl thioester prodrug 142 was made and found to be active but also toxic and rapidly cleaved by liver microsomes. The instability and toxicity of this compound was expected, given the reactivity of the S-acyl group toward electron-rich moieties. To confirm this hypothesis, a model reaction on 142 with ammonium acetate at pH 7.4 was performed, which resulted in efficient acyl transfer releasing the parent drug 21 (Figure S4, Supporting Information). Therefore, to enhance the stability of prodrugs, our attention was turned to S-methyl thioether analogues (143–145), which exhibited anti-Plk1 PBD activity in HeLa cells (Figure 4). Our analyses with two of these analogues (143 and 144) revealed that although they were stable in human, mouse, and rat cytosol with t1/2 values up to 120 min, unlike their parental 21 and 64, they exhibited much shorter t1/2 values when reacted with the microsomes from the corresponding three species (Table S2). These findings suggest that the membrane-bound enzymes in the microsomes are mainly responsible for the metabolism of these prodrugs rather than the soluble cytosolic enzymes.
The CYP-mediated reactions are the most likely metabolic pathways these prodrugs would undergo given the nature of the NADPH-containing microsome-based incubation system. To further identify the metabolites generated in the microsome reaction, 143 was incubated with MLMs for 1 h, and the resulting mixture processed and subjected to LC–MS/MS analysis (Figure S5A, Supporting Information). As expected, CYP catalysis generated a hydroxylated product, which was further shown by MS/MS fragmentography to be hydroxylated on the main aromatic ring of the quinazolinone (Figure S5B, Supporting Information). In addition, an S-demethylated metabolite, also mediated by CYP, was found, for which the structure was determined by comparing the fragmentation pattern with that of the parent drug, 21. As demonstrated by the in vitro results of MLM-treated nonprodrugs (Figures S1 and S2, Supporting Information), a glucuronidation could also be expected for the demethylated product. To detect a subsequent glucuronidation, 143 was incubated in a mixed system containing both NADPH and UDPGA, the cofactor for UDP-glucuronosyltransferase. After MS analysis, glucuronidation of the demethylated metabolite was evidenced by the appearance of a new peak having a 176 Da increase compared to the demethylated product (Figure S5A, top, Supporting Information).
Next, to achieve a comprehensive understanding of the metabolic stability of the prodrugs, we performed the in vivo experiment by injecting 143 in mice [20 mg/kg, intraperitoneal (IP) injection] under a protocol approved by the National Cancer Institute Animal Care and Use Committee. During the whole period of the experiment, no signs of toxicity was observed under these conditions. Serum was collected at 15, 30, 60, 120, and 240 min after injection. In close agreement with in vitro results in Figure S5, both the hydroxylated and demethylated products were detected in the serum ≥15 min postinjection (Figure S6, Supporting Information). While both metabolites decreased time-dependently, the demethylated product markedly increased in the serum ≥240 min postinjection (Figure S6, Supporting Information). A trace amount of the subsequent glucuronide of the demethylated metabolite was also seen in the serum as it could be promptly excreted from the liver to the bile, once generated, and can then undergo enterohepatic circulation through which the demethylated product is reabsorbed to the circulation. This could also serve as an explanation for the increased demethylated metabolite at the last time point. Compound 145 yielded similar metabolites with a somewhat improved t1/2 value of 51.26 min (Table S3 and Figure S6, Supporting Information). These results illustrated that the prodrugs could release the corresponding parent drugs enzymatically, and the released parent drugs levels could be longer lasting because of an enterohepatic circulation. As the glucuronides are considered pharmacologically inactive, whether the hydroxylated metabolite could also interact with Plk1 to exert an inhibitory effect requires further investigation.
DISCUSSION
Antimitotic drugs, such as taxanes and vinca alkaloids, that are directed at inhibiting the dynamic function of microtubules (MTs), have proven to be effective in the treatment of cancer.50 However, given the significant side effects of these conventional anti-MT agents, targeting mitosis-specific and cancer cell-addicted Plk1 has been considered an attractive strategy for generating a cancer cell-selective therapeutic agent.3,20 Indeed, a large body of studies show that several Plk1 ATP-competitive inhibitors developed over the years exhibit significant activities against hematological malignancies51–55 and several advanced solid tumors,56–60 although their less-than-acceptable specificities and dose-limiting toxicities have hampered further clinical applications.
PBD has emerged as an alternative target for developing a new class of Plk1 inhibitors that can potentially overcome the hurdles facing ATP-competitive inhibitors. As PBD inhibitors interfere only with the PBD-dependent Plk1 functions, they are anticipated to incur mitotic stress sufficient to induce cell death in cancer cells but not in normal cells.7 Completely disrupting Plk1 function would be detrimental even for normal cell proliferation. While a high level of target specificity is one of the inherent advantages that PPI inhibitors can achieve, the primary challenge has been developing small (<500 Da) molecules that still exhibit high affinity to the shallow and often nondescript interface of a target PPI.61,62 Because of this difficulty, most of the purported PBD inhibitors reported to date exhibit a poor (IC50 of 50–1000 μM) anti-Plk1 activity in cell-based assays.28–35 In addition, many of these compounds appear to exhibit cross-reacting alkylating activities42,43 that could impose dose-limiting nonspecific cytotoxicity.
SAR Summary.
The overall conclusions of our SAR studies are summarized in Figure 5. Our SAR studies identified six analogues which showed submicromolar affinity (65, 79, 83, 85, 129, and 134), which is at least an order of magnitude more potent than the previously characterized Plk1 PBD-specific phosphopeptide, PLHSpT 6a (Kd ~450 nM). All the six compounds appeared to share two distinct features: (1) 7-F substitution in zone 1 and (2) an unsubstituted triazole (zone 5). These are distinct because in our initial analogues exploring zone 1, there was no obvious preference for the 7-substitution. Then, when we combined early zone 1 and zone 3/4 modification unsubstituted triazoles, the fluorine was not obviously the preferred group. The inactivity of thiourea-containing analogue 62 and preference for an intact triazole indicated that a π-stacking interaction rather than H-bonding of the free NH is the important interaction. In addition, five of the six analogues contained zone 4 groups that could form H-bonding interactions, either CF3 (65), pyridine (83, 85), or amides and an additional H-bond acceptor (HBA) (129, 134). All five of these analogues contain HBA in positions that would all be in the same general region of the binding pocket. Compounds 129 and 134 have methoxy and cyano groups that might interact in adjacent regions of the pocket. The only submicromolar analogue that did not have additional H-bonding capabilities, that is, the methyl cyclopropane zone 3/4 analogue (79), might still establish stabilizing hydrophobic interactions.
Figure 5.
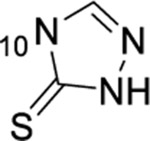
Summary of SAR studies on a 1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one scaffold for inhibition of Plk1 PBD.
Absorption distribution metabolism excretion (ADME) data were compared for the six most potent analogues (65, 79, 83, 85, 129, and 134). Five of the six analogues had half-lives of >30 min in RLMs, and the sixth (129) had a fairly good (20.1 min) half-life (Tables 6 and 8). Notably, the PAMPA permeability at pH 7.4 was favorable for six compounds (65, 83, 129, and 134) with moderate to good permeability (100 to >200 × 10−6 cm/s). However, only three analogues (65, 79, and 129) showed moderate to good solubility (10 to >60 μg/mL). Only compound 129 displayed both good permeability and solubility (P = 311 × 10−6 cm/s, S >65 μg/mL) (Table 8). Despite its relatively shorter half-life than other analogues, 129 may represent the best balance of PLK1 PBD affinity and ADME properties.
Some regions of the scaffold tolerated broad structural modification, while a few did not. In zone 1, interdependence on the groups in zones 3 and 4 defined the preferred substitution. This suggests that the core (zones 1, 2, and the quinazolinone) binding region might have been unfilled to allow for flexibility in the binding pose of molecules with various zone 1 substitutions. However, for the best analogues, the 7-position with a small H-bonding group is preferred; larger groups are tolerated but only with additional HBA groups. Zone 2 did not tolerate modification; the phenyl ring was optimal. Zones 3 and 4 proved to be the most generally amenable to modification. When zone 4 was cyclic, the linker length was best unmodified; however, when zone 4 was acyclic (an alkyl chain, a heteroatom, or another functional group), the entire chain could contain two or three carbons. This linker could also be sterically restricted with a trans cyclopropane, but this conclusion may not apply to all analogues differing in zone 4. In zone 4, removal and replacement of the phenyl group with unsubstituted heteroaryl groups was well tolerated, as was replacement with amides, especially for phenylacetyl derivatives. However, beyond the amide, hydrophilic or negatively charged moieties were disfavored. This extreme difference in affinity between phosphorus derivatives 77 and 78 and the other heteroatom-containing analogues could be due to the much larger size of these groups or the highly negatively charged phosphonic acid 77 at physiological pH.
All three nitrogen atoms of the triazole played an important role in binding, and alkylation of the triazole NH but not the S was accommodated. The tertiary amine substituents in 35–38 likely reside in a region that was formerly not accessed by the scaffold and therefore can establish new interactions, allowing for the improvement in affinity. The reason for the inactivity of N-substituted triazole analogue 39 is undetermined tertiary amines can form salt bridges, which ketones cannot form, and this could account for the potency differences. The difference could be due to either electrostatic or steric interactions (the acetophenone group in 39 is both larger and less flexible than the tertiary amines 35–38). Given that both tertiary amines and carbonyl oxygens can form H-bonding interactions, H-bonding may not account for the significant affinity difference.
The results provided here suggest that a 1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one scaffold offers promising structural and chemical features that can be exploited for anti-Plk1 PBD drug discovery. While the triazole moiety is one of the main components of antifungal drugs widely used for the prophylaxis and treatment of invasive fungal disease,63,64 the quinazoline moiety is frequently found in several FDA-approved EGFR or HER2 inhibitors, such as gefitinib, erlotinib, lapatinib, afatinib, and vandetanib.65 As these approved drugs have been extensively tested in humans for safety, available information from their prior clinical trials could facilitate further development of the above-described triazoloquinazolinone-based Plk1 PBD inhibitors. Drugs containing the conjoined triazoloquinazolinone backbone have not yet been reported in the drug depository (Drugbank; https://www.drugbank.ca/), indicating the uniqueness of the current triazoloquinazolinone leads. Remarkably, unlike previously reported several Plk1 PBD inhibitors with unacceptable alkylating activities,42,43 the identified triazoloquinazolinone chemotype does not bear any hallmark structural features that could be a reactive liability. In addition, triazoloquinazolinone-based compounds generally remain inert in multiple inhibitor screenings66–69 and specific modifications, such as a methyl group at N-3 of the quinazolinone lead, are required to induce biological efficacy.70,71 These chemical properties suggest a lower likelihood of off-target effects when dosed. In fact, potential off-target interactions for the initial hit, 7, and three other high-affinity compounds (21, 68, and 127) were not detected at 10 μM, when they were screened at 45 receptors and channels by the National Institute of Mental Health Psychoactive Drug Screening Program (https://pdsp.unc.edu/pdspweb/) using standard radioligand binding and functional assay methods.72 The only hits among these compounds at 10 μM causing >50% binding inhibition were for the primary amine 68 at M3 (100%), M4 (73%), and M5 (84%) muscarinic acetylcholine receptors. Thus, there was no pharmacological promiscuity observed for these four representative analogues.
The low MWs (300–450 Da) of several promising triazoloquinazolinone-based inhibitors described here offer considerable room for optimizations. Our extensive SAR analyses revealed distinct patterns (Figure 3) that could serve as a roadmap for further development. Notably, the ELISA-based IC50 values for several compounds (65, 79, 83, 85, 129, and 134) reached <1 μM. As the previously characterized Plk1 PBD-binding PLHSpT 6a (Kd of ~450 nM)27 shows an IC50 of 14.74 μM under the same conditions, these compounds are expected to have at least an order-of-magnitude-higher affinity than 6a. Moreover, the superb Plk1 PBD specificity of triazoloquinazolinone-derived inhibitors (Figure 4) along with the cellular efficacy of multiple prodrugs (143, 144, and 145) (Figure 5) provides clear proof-of-principle evidence that intervene in the Plk1 PBD-mediated PPI is feasible with small drug-like compounds amenable for potential clinical applications.
CONCLUSIONS AND PERSPECTIVE
In this study, we used a 1-thioxo-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one scaffold to synthesize S-methyl prodrugs that effectively inhibit PBD-dependent Plk1 function in cultured cells. Further development of these inhibitors may yield a new class of Plk1 PBD inhibitors that could offer superb specificity with versatile applicability either as a single agent or as a part of combination chemotherapy. Combination therapeutic regimens with various chemotherapeutic agents, including cisplatin, cytarabine, methotrexate, and doxorubicin, have already shown an improved antitumor activity with Plk1’s ATP-competitive inhibitors in numerous studies.52,54,73–77 In addition, as the most advanced ATP-competitive inhibitor 114 was doomed in phase III clinical trials because of its less-than-acceptable tolerability, combination treatment strategy with this inhibitor may allow to achieve the synergistic anti-Plk1 efficacy without causing the dose-limiting toxicity that narrowed their therapeutic windows. As a promising avenue for anti-Plk1 drug discovery, PBD inhibitors may prove to be effective in overcoming the hurdles facing current anti-Plk1 therapy and improving clinical outcomes.
EXPERIMENTAL SECTION
Chemical Synthesis.
General Methods for Chemistry.
All reactions were carried out under the nitrogen atmosphere using anhydrous solvents. All moisture-sensitive reactions were also performed with oven-dried glassware. Chemical reagents and anhydrous solvents were obtained from commercial sources and used as-is. Room temperature or rt refers to 25 ± 2 °C. Preparative purification was performed on a Waters semipreparative HPLC equipment. The column used was a Phenomenex Luna C18 (5 μm, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10–50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). Initial analytical analysis during compound synthesis was performed on an Agilent 1200 LC–MS system (Agilent Technologies) using a 3-min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8-min run time at a flow rate of 1 mL/min. The purity of compounds newly synthesized was demonstrated on an Agilent 1200 LC–MS system (Agilent Technologies) using a 7-min linear gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) followed by a 4.5 min run time at a flow rate of 1 mL/min and a Phenomenex Luna C18 column (3 μm, 3 × 75 mm) at 50 °C. The purity of the purchased compounds was determined using an Agilent ZORBAX Eclipse XDB C18 column (5 mm, 4.6 × 250 mm) with a linear gradient of 5–95% acetonitrile in water (containing 10 mM triethylammonium acetate) for 20 min at a flow rate of 1.0 mL/min. 1H and 13C NMR spectra were recorded on either a Varian 400 (100) MHz spectrometer or a Bruker 400 MHz spectrometer. Chemical shifts are given in ppm (δ), calibrated to the residual solvent signals and frequency calibrated internally by solvent for 19F NMR (BrukerTopspin/MestReNova 10.0.2 or 14.1.0). High-resolution mass spectrometry was recorded on either an Agilent 6210 time-of-flight LCMS system or a Waters Micromass spectrometer equipped with a standard electrospray ionization (ESI) and modular Lock-SprayTM interface. The purity of all the tested compounds (including both newly synthesized and purchased, active compounds) were demonstrated to be >95% at 254 nm, except commercially procured compound 58, which was 93.5% pure. The synthesis of compounds 7–64 is described in the Supporting information.
General Procedure A.
To a solution of appropriate 2-aminobenzoic acid (1 equiv) and isothiocyanate (1.2 equiv) in EtOH (0.37 M) was added triethylamine (1.2 equiv). The reaction was stirred at 80 °C for 1–2 h. The reaction was cooled to room temperature (RT) and diluted with water. The solid (intermediate A) was filtered and dried under vacuum and used without further purification.
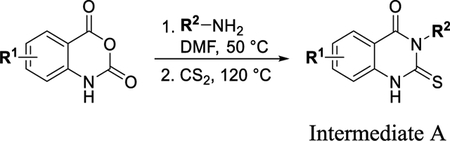
General Procedure B.
A solution of isatoic anhydride (1 equiv) in acetonitrile (0.67 M) was added to the amine (1.5 equiv). In the case of amine salts, triethylamine (1.5 equiv) was added to a solution of the amine in acetonitrile (0.67 M), the salt was removed by filtration, and the free based amine solution was added to the reaction. The reaction was heated at 50 °C for 30 min. The reaction was cooled to RT and carbon disulfide (7 equiv) was added cold. The reaction was then heated to 120 °C for 45 min. The reaction was diluted with Et2O, filtered, rinsed with Et2O, and dried. The solid (intermediate A) was dried under vacuum and used without further purification.
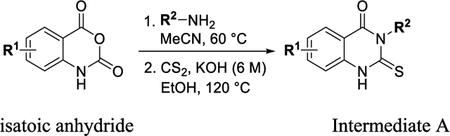
General Procedure C.
A solution of isatoic anhydride (1 equiv) in acetonitrile (0.67 M) was added to the amine (1.5 equiv). In the case of amine salts, triethylamine (1.5 equiv) was added to a solution of the amine in acetonitrile (0.67 M), the salt was removed by filtration, and the free based amine solution was added to the reaction. The reaction was heated at 60 °C for 3 h. The reaction was concentrated and the residue dissolved in EtOH (0.67 M), and aq KOH (1.2 equiv) was added, followed by carbon disulfide (2 equiv). The reaction was then heated to 120 °C for 2 h. The reaction was cooled to RT, diluted with water, and washed with Et2O. The solid (intermediate A) was dried under vacuum and used without further purification.
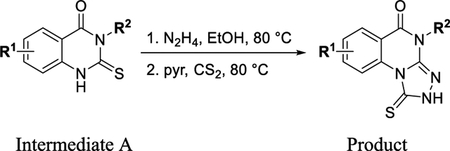
General Procedure D.
To a solution of intermediate A (1 equiv) in EtOH (0.25–0.35 M) was added hydrazine (7 equiv). The reaction was then heated to 80 °C for 4 h. The reaction was then cooled to RT and pyridine (10 equiv) and carbon disulfide (10 equiv) were added. The reaction was heated to 80 °C for 1–2 h. The reaction was poured into cold water, and the product was filtered and washed with water or purified by HPLC.
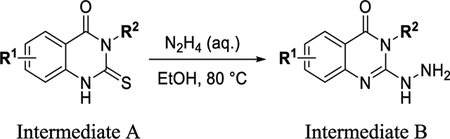
General Procedure E.
To a solution of intermediate A (1 equiv) in EtOH (0.35 M) was added water (2.8 equiv), followed by hydrazine (7 equiv). The reaction was then heated to 80 °C for 4 h. The reaction was then poured into cold water and concentrated. The crude material was purified on a Teledyne ISCO CombiFlash System by (dry-loading) (EtOAc/DCM: 0–3%) to give intermediate B.
General Procedure F.
To a solution of carboxylic acid (1.5 equiv) in DMF (0.2 M) was added COMU (1.5 equiv) and the reaction stirred at RT for 30–45 min. Then, the amine (1 equiv) was added, followed by DIPEA (2.2 equiv). The reaction was then stirred for 18 h or 2.5 d at RT. The crude material was purified by HPLC.
4-Phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (7).
Note: Compound 7 previously appeared as an in silico hit in a screen for A2A adenosine receptor antagonists {compound 30 in PMC2865168}.
2-Amino-N-phenethylbenzamide: A mixture of methylanthranilate (0.856 mL, 6.615 mmol) and phenethylamine (1.3 mL, 10 mmol) was stirred in a round-bottom flask at 190 °C for 5 h. The product was purified by silica-gel column chromatography (0.44 g, 28%). 1H NMR (400 MHz, CDCl3): δ 7.40–7.14 (m, 6H), 6.68 (dd, J = 1.1, 8.1 Hz, 1H), 6.62 (ddd, J = 1.2, 7.2, 8.1 Hz, 1H), 6.06 (s, 1H), 5.49 (s, 2H),3.70 (td, J = 5.8, 6.8 Hz, 2H), 2.94 (t, J = 6.9 Hz, 2H); HRMS: (M +H) for C15H16N2O calcd 241.1341; found, 241.1339.
3-Phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one: Route 1: To a solution of 2-amino-N-phenethylbenzamide (100 mg, 0.416 mmol) in anhydrous DMF (2 mL) at RT was added carbon disulfide (72 μL, 1.248 mmol) and DBU (136 μL, 0.916 mmol) sequentially. After stirring the reaction mixture for 18 h at RT, cold 1 N aq HCl was added with vigorous stirring. The precipitate was collected by filtration, washed with cold water and hexanes, and dried to afford a practically pure product (60 mg, 50%). Route 2: To a solution of 2-amino-N-phenethylbenzamide (100 mg, 0.416 mmol) in anhydrous ethanol (2 mL) at RT were added carbon disulfide (75 μL, 1.25 mmol) and solid KOH (32 mg, 0.916 mmol) sequentially. The reaction mixture was stirred at 80 °C for 18 h at RT, cooled, and was added to a cold 1 N aq HCl with vigorous stirring. The precipitate was collected by filtration, washed with cold water and hexanes, and dried to afford a practically pure product (70 mg, 60%). Note: The product was purified further by silica-gel column chromatography using 0–2% methanol in dichloromethane as an eluent. The purified product gave better yield in the next step. 1H NMR (400 MHz, CDCl3): δ 9.99 (s, 1H), 8.25–8.10 (m, 1H), 7.69 (ddd, J = 1.5, 7.3, 8.4 Hz, 1H), 7.45–7.40 (m, 2H), 7.38–7.32 (m, 2H), 7.27 (d, J = 5.6 Hz, 1H), 7.13 (dt, J = 0.7, 8.2 Hz, 1H), 4.82–4.65 (m, 2H), 3.19–3.07 (m, 2H); HRMS: (M + H) for C16H14N2OS calcd, 283.0905; found, 283.0909.
2-Hydrazineyl-3-phenethylquinazolin-4(3H)-one: To a suspension of 3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (200 mg, 0.708 mmol) in anhydrous ethanol (5 mL) was added anhydrous hydrazine (0.33 mL, 10.62 mmol) and heated at 85 °C for 18 h. The volatiles were evaporated under high vacuum, followed by coevaporation with toluene (2×), giving 2-hydrazineyl-3-phenethylquinazolin-4(3H)-one as a yellow solid and was used as such without further purification (200 mg, quantitative). 1H NMR (400 MHz, CDCl3): δ 8.16 (dd, J = 1.6, 7.9 Hz, 1H), 7.68–7.56 (m, 1H), 7.45–7.14 (m, 5H), 4.21 (dd, J = 6.7, 8.4 Hz, 2H), 3.09–2.99 (m, 1H); HRMS: (M + H) for C16H16N4O calcd, 281.1402; found, 281.1405.
4-Phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one: 2-Hydrazineyl-3-phenethylquinazolin-4(3H)-one (200 mg, 0.713 mmol) was dissolved in anhydrous ethanol (10 mL). To the solution was added carbon disulfide (0.13 mL, 2.14 mmol), followed by solid KOH (120 mg, 2.14 mmol), and it was heated to 80 °C for 18 h. The reaction mixture was cooled and poured into cold 1 N aq HCl solution. The precipitates were collected and purified by silica-gel column chromatography using 0–2% methanol in dichloromethane as an eluent to afford a pure product as a white solid (200 mg, 87%). 1H NMR (400 MHz, DMSO-d6): δ 14.10 (s, 1H), 10.23 (dd, J = 1.1, 8.6 Hz, 1H), 8.22 (dd, J = 1.6, 7.9 Hz, 1H), 7.91 (ddd, J = 1.7, 7.3, 8.7 Hz, 1H), 7.62 (td, J = 1.1, 7.6 Hz, 1H), 7.38–7.15 (m, 4H), 4.32–4.15 (m, 2H), 3.08–2.92 (m, 2H); HRMS: (M + H) for C17H14N4OS, calcd 323.0967; found, 323.0966.
4-Phenethyl-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazoline-1,5-dione (15).
2-Hydrazineyl-3-phenethylquinazolin-4(3H)-one (35 mg, 0.117 mmol) was suspended in anhydrous toluene (2 mL), and to it was added carbonyl diimidazole (22 mg, 0.135 mmol) and heated to reflux (115 °C) for 1.5 h. The volatiles evaporated under vacuum and the residue was treated with 1 N aq HCl and 5% isopropanol-dichloromethane. The organic layer was separated, dried over Na2SO4, evaporated, and the residue was purified by silica-gel column chromatography to afford 7b as a white solid (13 mg, 34%). 1H NMR (400 MHz, DMSO-d6): δ 12.07 (s, 1H), 8.65 (dd, J = 4.6, 9.1 Hz, 1H), 7.84 (dd, J = 3.0, 8.6 Hz, 1H), 7.75 (td, J = 3.1, 8.7 Hz, 1H),7.35–7.19 (m, 4H), 4.26–4.05 (m, 2H), 3.08–2.92 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.33 (td, J = 4.5, 8.1 Hz); HRMS: (M + H) for C17H13N4O2F calcd, 325.1101; found, 325.1107.
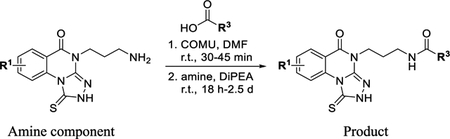
1-Amino-7-fluoro-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (16).
6-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. 6-Fluoro-2-hydrazineyl-3-phenethylquinazolin-4(3H)-one was synthesized according to General Procedure E. Then, to a solution of 6-fluoro-2-hydrazinyl-3-phenethylquinazolin-4(3H)-one (0.1 g, 0.335 mmol) and cyanogen bromide (0.036 g, 0.335 mmol) in ethanol (16.8 mL) was added sodium hydroxide (0.067 g, 1.675 mmol) and the reaction was stirred at RT for 3 h. The reaction was neutralized with sat. NaHCO3 and the solid filtered. The solid was redissolved and purified by HPLC to give 1-amino-7-fluoro-4-phenethyl-[1,2,4]-triazolo[4,3-a]quinazolin-5(4H)-one, 2 TFA: 1H NMR (400 MHz, DMSO-d6): δ 8.29 (dd, J = 9.2, 4.2 Hz, 1H), 7.92 (dd, J = 8.5, 3.1 Hz, 1H), 7.82 (td, J = 8.6, 3.1 Hz, 1H), 7.34–7.24 (m, 4H), 7.22 (d, J = 7.0 Hz, 1H), 6.90 (s, 2H), 4.29 (dd, J = 9.5, 6.5 Hz, 2H), 3.06–2.98 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −114.22; HRMS (ES-API) m/z: calcd for C17H15FN5O (M + H), 324.1255; found, 324.1264.
7-Fluoro-1-methyl-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (17).
6-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. 6-Fluoro-2-hydrazineyl-3-phenethylquinazolin-4(3H)-one was synthesized according to General Procedure E. Then, 6-fluoro-2-hydrazinyl-3-phenethylquinazolin-4(3H)-one (0.1 g, 0.335 mmol) was stirred in Ac2O (16.8 mL) at 100 °C for 5 h. The reaction was concentrated, filtered, and washed with water. The solid was dried and crystallized from EtOH to give 7-fluoro-1-methyl-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (0.0453 g, 42%): 1H NMR (400 MHz, DMSO-d6): δ 8.11 (dd, J = 9.2, 4.2 Hz, 1H), 7.95 (dd, J = 8.5, 3.1 Hz, 1H), 7.80 (ddd, J = 9.2, 8.0, 3.1 Hz, 1H), 7.29 (td, J = 9.0, 8.4, 6.1 Hz, 4H), 7.21 (td, J = 6.5, 2.2 Hz, 1H), 4.42–4.33 (m, 2H), 3.08–2.99 (m, 2H), 2.84 (s, 3H); 19F NMR (376 MHz, DMSO-d6): δ −114.29 (td, J = 8.2, 4.0 Hz).
7-Fluoro-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (18).
6-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. 6-Fluoro-2-hydrazineyl-3-phenethylquinazolin-4(3H)-one was synthesized according to General Procedure E. Then, 6-fluoro-2-hydrazinyl-3-phenethylquinazolin-4(3H)-one (0.1 g, 0.335 mmol) was stirred in formic acid (16.8 mL) at 100 °C for 5 h. The reaction was concentrated, redissolved, and purified by HPLC to give 7-fluoro-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one, TFA: 1H NMR (400 MHz, DMSO-d6): δ 9.50 (s, 1H), 8.27 (dd, J = 8.8, 4.3 Hz, 1H), 7.89 (ddd, J = 11.0, 7.1, 3.9 Hz, 2H), 7.34–7.25 (m, 4H),7.25–7.17 (m, 1H), 4.37 (dd, J = 9.2, 6.8 Hz, 2H), 3.09–3.01 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.78 (td, J = 8.3, 4.1 Hz); HRMS (ES-API) m/z: calcd for C17H14FN4O (M + H), 309.1146; found, 309.1155.
1-(Methylthio)-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (19).
3-Phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D. Then, 1-(methylthio)-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one was synthesized as follows: to a solution of 4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.092 g, 0.223 mmol) in DMF (1.113 mL) was added K2CO3 (0.037 g, 0.267 mmol) and MeI (0.017 mL, 0.267 mmol). The reaction was quenched with methanol and concentrated. The reaction was stirred for 3 h and quenched with methanol. The crude mixture was concentrated and purified by HPLC to give 1-(methylthio)-4-phenethyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one, TFA.
4-Ethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (20).
3-Ethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 4-ethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.20 (d, J = 8.5 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 7.87 (t, J = 8.0 Hz, 1H), 7.59 (t, J = 7.7 Hz, 1H), 4.05 (q, J = 7.1 Hz, 2H), 1.23 (t, J = 7.2 Hz, 3H); HRMS (ES-API) m/z: calcd for C11H10N4NaOS (M + Na), 269.0468; found, 269.0476.
4-Propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (21).
4-Propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-Allyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (22).
4-Allyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-Butyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (23).
4-Butyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-Pentyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (24).
4-Pentyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one was purchased Enamine (Monmouth Jct., NJ).
4-Isopentyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (25).
4-Isopentyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-Isopropyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (26).
4-Isopropyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was purchased from ChemDiv (San Diego, CA).
1-Thioxo-4-(3,3,3-trifluoropropyl)-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one (27).
2-Thioxo-3-(3,3,3-trifluoropropyl)-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 1-thioxo-4-(3,3,3-trifluoropropyl)-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.19 (d, J = 8.5 Hz, 1H), 8.21 (d, J = 7.8 Hz, 1H),7.90 (t, J = 8.1 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 4.25 (t, J = 7.3 Hz, 2H), 2.72 (dt, J = 11.5, 7.4 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −64.31 (t, J = 11.3 Hz).
4-(3-Methoxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (28).
4-(3-Methoxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-(3-Ethoxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (29).
4-(3-Ethoxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
N-(sec-Butyl)-3-(5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propenamide (30).
N-(sec-Butyl)-3-(5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propenamide was purchased from ChemDiv (San Diego, CA).
N-Isopropyl-3-(5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propenamide (31).
N-Isopropyl-3-(5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propenamide was purchased from ChemDiv (San Diego, CA).
N-Isobutyl-3-(5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propenamide (32).
N-Isobutyl-3-(5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propenamide was purchased from ChemDiv (San Diego, CA).
4-(2,3-Dimethylphenyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (33).
4-(2,3-Dimethylphenyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-(4-Fluorobenzyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (34).
4-(4-Fluorobenzyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from ChemDiv (San Diego, CA).
2-((Cyclopropyl(methyl)amino)methyl)-4-propyl-1-thioxo-2,4-di-hydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (35).
2-((Cyclopropyl(methyl)amino)methyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
4-Methyl-2-((methyl(propyl)amino)methyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (36).
4-Methyl-2-((methyl(propyl)amino)methyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
2-((Methyl(propyl)amino)methyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (37).
2-((Methyl-(propyl)amino)methyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
2-(((Cyclopropylmethyl) (ethyl)amino)methyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (38).
2-(((Cyclopropylmethyl) (ethyl)amino)methyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from Enamine (Monmouth Jct., NJ).
2-(2-Oxo-2-phenylethyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (39).
2-(2-Oxo-2-phenylethyl)-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from ChemDiv (San Diego, CA).
7-Fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (40).
2-Amino-5-fluoro-N-phenethylbenzamide: To a solution of 2-amino-5-fluorobenzoic acid (250 mg, 1.61 mmol), phenethylamine (0.243 mL, 1.93 mmol), dimethylaminopyridine (100 mg, 0.81 mmol), and triethylamine (0.45 mL, 3.22 mmol) in anhydrous dichloromethane (5 mL) was added EDC·HCl (370 mg, 1.93 mmol) and stirred at RT for 18 h. Saturated aq NaHCO3 solution and dichloromethane were added and stirred. The organic layer was separated, dried over Na2SO4, and evaporated under reduced pressure. The residue obtained was purified by silica-gel chromatography to afford the desired product as a white solid (200 mg, 48%). 1H NMR (400 MHz, CDCl3): δ 7.36 (t, J = 7.3 Hz, 2H),7.27 (q, J = 6.7, 7.6 Hz, 3H), 6.96 (td, J = 2.9, 8.4 Hz, 1H), 6.89 (dd, J = 2.8, 9.2 Hz, 1H), 6.64 (dd, J = 4.6, 9.0 Hz, 1H), 6.01 (s, 1H), 5.24 (s, 2H), 3.69 (q, J = 6.6 Hz, 2H), 2.94 (t, J = 6.9 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ −111.91; HRMS: (M + H) for C16H15N2OF calcd, 259.1247; found, 259.1247.
6-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one: To a solution of 2-amino-5-fluoro-N-phenethylbenzamide (100 mg, 0.387 mmol) in anhydrous DMF (2 mL) at RT was added carbon disulfide (70 μL, 1.16 mmol) and DBU (127 μL, 0.851 mmol) sequentially. After stirring the reaction mixture for 18 h at RT, cold 1 N aq HCl was added with vigorous stirring. The precipitate was collected by filtration, washed with cold water and hexanes, and dried to give a white solid (110 mg, 95%). 1H NMR (400 MHz, CDCl3): δ 9.94 (s, 1H), 7.82 (dd, J = 2.9, 8.1 Hz, 1H), 7.40 (d, J = 7.7 Hz, 3H),7.35 (t, J = 7.3 Hz, 1H), 7.27 (d, J = 4.9 Hz, 2H), 7.19–7.03 (m, 1H),4.74 (dd, J = 6.4, 10.2 Hz, 2H), 3.11 (t, J = 8.3 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ −114.59; HRMS: (M + H) for C16H13N2OSF calcd, 301.0811; found, 301.0813.
6-Fluoro-2-hydrazineyl-3-phenethylquinazolin-4(3H)-one: Following the procedure mentioned for the synthesis of 2-hydrazineyl-3-phenethylquinazolin-4(3H)-one, 6-fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (110 mg, 0.366 mmol) gave product 6-fluoro-2-hydrazineyl-3-phenethylquinazolin-4(3H)-one as a white solid (80 mg, 73%). 1H NMR (400 MHz, CDCl3): δ 7.81 (dt, J = 1.9, 8.5 Hz, 1H), 7.41–7.20 (m, 8H), 4.20 (t, J = 7.4 Hz, 2H), 3.04 (t, J = 7.4 Hz, 2H); 19F NMR (376 MHz, CDCl3): δ −117.99 (td, J = 4.8, 8.3 Hz); HRMS: (M + H) for C16H15N4OF calcd, 299.1308; found, 299.1307.
7-Fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one: Following the procedure described for the synthesis of 4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one, 6-fluoro-2-hydrazineyl-3-phenethylquinazolin-4(3H)-one (50 mg, 0.168 mmol) gave product 7-fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one as a white solid (20 mg, 35%). 1H NMR (400 MHz, DMSO-d6): δ 14.16 (s, 1H), 10.31 (dd, J = 4.7, 9.3 Hz, 1H), 7.93 (dd, J = 3.1, 8.6 Hz, 1H), 7.83 (td, J = 3.1, 8.6 Hz, 1H), 7.27 (ddt, J = 7.3, 14.2, 19.9 Hz, 4H), 4.34–4.15 (m, 2H), 3.09–2.93 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −112.99 (td, J = 4.7, 8.3 Hz); HRMS: (M + H) for C17H13N4OSF calcd, 341.0872; found, 341.0871.
7-Bromo-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (41).
6-Bromo-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 7-bromo-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.17 (d, J = 9.1 Hz, 1H), 8.25 (d, J = 2.4 Hz, 1H), 8.11 (dd, J = 9.0, 2.5 Hz, 1H), 7.34–7.17 (m, 5H), 4.25–4.17 (m, 2H), 3.02–2.94 (m, 2H); HRMS (ES-API) m/z: calcd for C17H14FN4OS (M + H), 403.0046; found, 403.0046.
7-Iodo-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (42).
6-Iodo-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 7-iodo-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.00 (d, J = 8.8 Hz, 1H), 8.41 (d, J = 2.1 Hz, 1H), 8.24 (dd, J = 8.9, 2.2 Hz, 1H), 7.32–7.18 (m, 5H), 4.24–4.16 (m, 2H), 3.02–2.93 (m, 2H).
7-Methyl-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (43).
6-Methyl-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 7-methyl-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.06 (d, J = 8.6 Hz, 1H), 8.02–7.97 (m, 1Hl), 7.71 (dd, J = 8.8, 2.2 Hz, 1H), 7.33–7.16 (m, 5H), 4.26–4.18 (m, 2H), 3.03–2.94 (m, 2H), 2.44 (s, 3H).
N-(5-Oxo-4-phenethyl-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]-triazolo[4,3-a]quinazolin-7-yl)acetamide (44).
N-(4-Oxo-3-phenethyl-2-thioxo-1,2,3,4-tetrahydroquinazolin-6-yl)acetamide was synthesized according to General Procedure A. Then, N-(5-oxo-4-phenethyl-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]triazolo[4,3-a]-quinazolin-7-yl)acetamide was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.37 (s, 1H),10.09 (d, J = 9.1 Hz, 1H), 8.51 (d, J = 2.6 Hz, 1H), 7.96 (dd, J = 9.1, 2.7 Hz, 1H), 7.26 (ddd, J = 19.6, 13.5, 7.3 Hz, 5H), 4.22 (dd, J = 9.4, 6.5 Hz, 2H), 2.99 (t, J = 7.9 Hz, 2H), 2.48 (t, J = 3.0 Hz, 2H), 2.08 (s, 3H); HRMS (ES-API) m/z: calcd for C19H18N5O2S (M + H), 380.1176; found, 380.1174.
7-(Dimethylamino)-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (45).
6-(Dimethylamino)-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 7-(dimethylamino)-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.92 (s, 1H), 9.98 (d, J = 9.3 Hz, 1H), 7.33 (d, J = 3.1 Hz, 1H), 7.32–7.16 (m, 6H), 4.26–4.18 (m, 2H), 2.99 (s, 6H), 3.00–2.94 (m, 2H); HRMS (ES-API) m/z: calcd for C19H20N5OS (M + H), 366.1383; found, 366.1388; purity (HPLC) 94.83%.
7-Morpholino-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one (46).
6-Morpholino-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 7-morpholino-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.97 (s, 1H), 10.02 (d, J = 9.3 Hz, 1H), 7.57 (d, J = 3.0 Hz, 1H), 7.53 (dd, J = 9.3, 3.0 Hz, 1H), 7.31–7.17 (m, 5H),4.26–4.18 (m, 2H), 3.76 (t, J = 4.8 Hz, 4H), 3.22 (dd, J = 6.1, 3.6 Hz, 4H), 3.02–2.94 (m, 2H); HRMS (ES-API) m/z: calcd for C21H22N5O2S (M + H), 408.1489; found, 408.1490.
8-Fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (47).
7-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 8-fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.11 (dd, J = 11.4, 2.5 Hz, 1H), 8.27 (dd, J = 8.8, 6.3 Hz, 1H), 7.48 (td, J = 8.4, 2.6 Hz, 1H), 7.26 (ddt, J = 13.0, 11.5, 7.1 Hz, 5H), 4.25–4.16 (m, 2H), 3.02–2.94 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −101.36 (dt, J = 11.9, 7.1 Hz); HRMS (ES-API) m/z: calcd for C17H14FN4OS (M + H), 341.0867; found, 341.0882.
9-Fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (48).
8-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 9-fluoro-4-phenethyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.88 (s, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.83–7.73 (m, 1H),7.62 (td, J = 8.0, 4.0 Hz, 1H), 7.25 (ddd, J = 18.6, 13.0, 7.3 Hz, 5H),4.16 (dd, J = 9.2, 6.7 Hz, 2H), 3.02–2.94 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −93.38 (dd, J = 11.6, 4.1 Hz); HRMS (ES-API) m/z: calcd for C17H14FN4OS (M + H), 341.0867; found, 341.0859.
4-Phenethyl-1-thioxo-2,4-dihydropyrido[2,3-e][1,2,4]triazolo-[4,3-a]pyrimidin-5(1H)-one (49).
3-Phenethyl-2-thioxo-2,3-dihydropyrido[3,2-d]pyrimidin-4(1H)-one was synthesized according to General Procedure A. Then, 4-phenethyl-1-thioxo-2,4-dihydropyrido[2,3-e][1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-one, TFA, was synthesized according to General Procedure D.
4-Phenethyl-1-thioxo-2,4-dihydropyrido[4,3-e][1,2,4]triazolo-[4,3-a]pyrimidin-5(1H)-one (50).
3-Phenethyl-2-thioxo-2,3-dihydropyrido[3,4-d]pyrimidin-4(1H)-one was synthesized according to General Procedure A. Then, 4-phenethyl-1-thioxo-2,4-dihydropyrido[4,3-e][1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 11.36 (s, 1H), 8.82 (d, J = 5.0 Hz, 1H),8.06 (d, J = 5.0 Hz, 1H), 7.33–7.22 (m, 4H), 7.20 (t, J = 7.1 Hz, 1H),4.24–4.17 (m, 2H), 2.97 (t, J = 8.0 Hz, 2H).
6-Phenethyl-9-thioxo-8,9-dihydropyrido[3,2-e][1,2,4]triazolo-[4,3-a]pyrimidin-5(6H)-one (51).
3-Phenethyl-2-thioxo-2,3-dihydropyrido[2,3-d]pyrimidin-4(1H)-one was synthesized according to General Procedure A. Then, 6-phenethyl-9-thioxo-8,9-dihydropyrido[3,2-e][1,2,4]triazolo[4,3-a]pyrimidin-5(6H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.84 (s, 1H), 8.83 (dd, J = 4.7, 1.9 Hz, 1H), 8.52 (dd, J = 7.8, 1.9 Hz, 1H), 7.63 (dd, J = 7.8, 4.8 Hz, 1H),7.24 (ddt, J = 20.7, 14.0, 7.3 Hz, 5H), 4.23–4.14 (m, 2H), 2.97 (dd, J = 9.4, 6.4 Hz, 2H).
4-Benzyl-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (52).
3-Benzyl-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 4-benzyl-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D and purified by pouring into ice water and drops of acetic acid were added. The solid was filtered and recrystallized from dioxane to give 4-benzyl-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (0.0226 g, 25%): 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.4, 4.7 Hz, 1H), 7.95 (dd, J = 8.6,3.1 Hz, 1H), 7.82 (ddd, J = 9.3, 7.9, 3.1 Hz, 1H), 7.43–7.36 (m, 2H),7.30 (dd, J = 8.1, 6.2 Hz, 2H), 7.29–7.21 (m, 1H), 5.22 (s, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.06 (td, J = 8.4, 4.9 Hz); HRMS (ES-API) m/z: calcd for C16H12FN4OS (M + H), 327.0710; found, 327.0716.
7-Fluoro-4-(3-phenylpropyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (53).
6-Fluoro-3-(3-phenylpropyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 7-fluoro-4-(3-phenylpropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.27 (dd, J = 9.3, 4.6 Hz, 1H), 7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.79 (td, J = 8.6, 3.1 Hz, 1H), 7.25–7.14 (m, 4H), 7.10 (t, J = 7.0 Hz, 1H), 4.07 (t, J = 7.2 Hz, 2H), 2.67 (t, J = 7.7 Hz, 2H), 2.02 (p, J = 7.4 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19 (td, J = 8.4, 4.9 Hz).
7-Chloro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (54).
7-Chloro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one one was purchased from ChemDiv (San Diego, CA).
7-Chloro-1-thioxo-4-(3,3,3-trifluoropropyl)-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (55).
6-Chloro-2-thioxo-3-(3,3,3-trifluoropropyl)-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-chloro-1-thioxo-4-(3,3,3-trifluoropropyl)-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.23 (d, J = 9.1 Hz, 1H), 8.14 (d, J = 2.5 Hz, 1H), 8.00 (dd, J = 9.1, 2.5 Hz, 1H), 4.25 (t, J = 7.3 Hz, 2H), 2.72 (dt, J = 17.7, 9.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −64.33 (t, J = 11.3 Hz); HRMS (ES-API) m/z: calcd for C12H9ClF3N4OS (M + H), 349.0132; found, 349.0141; purity (HPLC) 91.99%.
7-Chloro-4-(3-isopropoxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (56).
7-Chloro-4-(3-isopropoxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from ChemDiv (San Diego, CA).
3-(7-Chloro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)-N-isopropylpropanamide (57).
3-(7-Chloro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-N-isopropylpropanamide one was purchased from ChemDiv (San Diego, CA).
N-Isopropyl-3-(7-methyl-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propanamide (58).
N-Isopropyl-3-(7-methyl-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propanamide one was purchased from ChemDiv (San Diego, CA). Purity (HPLC) 93.49%.
N-Isopropyl-5-oxo-4-propyl-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]-triazolo[4,3-a]quinazoline-8-carboxamide (59).
N-Isopropyl-5-oxo-4-propyl-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]triazolo[4,3-a]-quinazoline-8-carboxamide one was purchased from ChemDiv (San Diego, CA).
N-Cyclopentyl-4-isopentyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]triazolo[4,3-a]quinazoline-8-carboxamide (60).
N-Cyclopentyl-4-isopentyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]triazolo[4,3-a]quinazoline-8-carboxamideone was purchased from ChemDiv (San Diego, CA).
N-Cyclopentyl-4-methyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]triazolo[4,3-a]quinazoline-8-carboxamide (61).
N-Cyclopentyl-4-methyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro-[1,2,4]triazolo[4,3-a]-quinazoline-8-carboxamide one was purchased from ChemDiv (San Diego, CA).
6-Fluoro-N-methyl-4-oxo-3-phenethyl-3,4-dihydroquinazoline-1(2H)-carbothioamide (62).
6-Fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 6-fluoro-3-phenethyl-2,3-dihydroquinazolin-4(1H)-one was synthesized as follows: to a solution of 6-fluoro-3-phenethyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (0.2 g, 0.666 mmol) and nickel(II)chloride (0.604 g, 4.66 mmol) in MeOH (5 mL) was added sodium borohydride (0.529 g, 13.98 mmol) carefully. The reaction was stirred at RT for 30 min. The reaction was then filtered through Celite and the solid washed with MeOH. The filtrate was taken up in EtOAc and washed with water. The organics were dried over MgSO4, filtered, and concentrated. The product was recrystallized from EtOH.
6-Fluoro-N-methyl-4-oxo-3-phenethyl-3,4-dihydroquinazoline-1(2H)-carbothioamide, TFA, was synthesized as follows: to a solution of 6-fluoro-3-phenethyl-2,3-dihydroquinazolin-4(1H)-one (0.05 g, 0.185 mmol) in DCM (0.462 mL) was added 1,1′-thiocarbonyldiimidazole (0.049 g, 0.277 mmol) and the reaction was stirred for 2 h at RT. Methylamine hydrochloride (0.125 g, 1.850 mmol) and triethylamine (0.284 mL, 2.035 mmol) were added, and the reaction was stirred for 24 h. The reaction was concentrated under a stream of N2 and purified by HPLC: 1H NMR (400 MHz, DMSO-d6): δ 8.42 (s, 1H), 7.51 (d, J = 8.8 Hz, 1H), 7.41 (d, J = 6.3 Hz, 2H), 7.19 (dq, J = 14.0, 7.2 Hz, 4H), 5.43 (s, 2H), 3.66 (t, J = 7.3 Hz, 2H), 2.93 (d, J = 4.2 Hz, 3H), 2.83 (t, J = 7.4 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −115.99; HRMS (ES-API) m/z: calcd for C18H18F3N3NaOS (M + Na), 366.1047; found, 366.1052.
4-Ethyl-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (63).
3-Ethyl-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 4-ethyl-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.28 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.79 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H); 19F NMR (376 MHz, DMSO-d6): δ −113.15 (td, J = 8.5, 4.8 Hz); HRMS (ES-API) m/z: calcd for C11H10FN4OS (M + H), 265.0554; found, 265.0557; purity (HPLC) 91.89%.
7-Fluoro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (64).
7-Fluoro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was purchased from ChemDiv (San Diego, CA).
7-Fluoro-1-thioxo-4-(3,3,3-trifluoropropyl)-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (65).
6-Fluoro-2-thioxo-3-(3,3,3-trifluoropropyl)-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-1-thioxo-4-(3,3,3-trifluoropropyl)-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.27 (d, J = 6.5 Hz, 1H), 7.93 (dd, J = 8.6, 3.0 Hz, 1H), 7.82 (td, J = 9.3, 8.7, 3.1 Hz, 1H), 4.26 (t, J = 7.2 Hz, 2H), 2.72 (dtd, J = 18.6, 11.3, 7.7 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −64.33 (t, J = 11.3 Hz), −112.81; HRMS (ES-API) m/z: calcd for C12H8F4N4NaOS (M + Na), 355.0247; found, 355.0245.
7-Fluoro-4-(3-hydroxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (66).
6-Fluoro-3-(3-hydroxypropyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(3-hydroxypropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.28 (dd, J = 9.4, 4.6 Hz, 1H), 7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.78 (td, J = 8.5, 3.0 Hz, 1H), 4.48 (t, J = 5.1 Hz, 1H), 4.08 (t, J = 7.4 Hz, 2H), 3.47 (q, J = 5.8 Hz, 2H), 1.89–1.77 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.18 (q, J = 7.4 Hz); HRMS (ES-API) m/z: calcd for C12H12FN4O2S (M + H), 294.0660; found, 294.0664.
4-(2-Aminoethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one (67).
tert-Butyl (2-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)carbamate was synthesized according to General Procedure C. Then, tert-butyl (2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)carbamate was synthesized according to General Procedure D. Then, 4-(2-aminoethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized by treating tert-butyl (2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)carbamate (0.112 g, 0.295 mmol) in DCM (1.230 mL) and MeOH (0.246 mL) with TFA(0.989 mL, 12.84 mmol). The reaction was stirred at RT for 3 h, then blown down, redissolved, and purified by HPLC: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.6 Hz, 1H), 7.95 (dd, J = 8.5, 3.1 Hz, 1H), 7.85 (ddd, J = 9.2, 7.6, 3.2 Hz, 3H), 4.32 (t, J = 5.8 Hz, 2H), 3.20 (t, J = 6.0 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −112.75 (td, J = 8.3, 4.7 Hz); HRMS (ES-API) m/z: calcd for C11H10FN5NaOS (M + Na), 302.0482; found, 302.0493.
4-(3-Aminopropyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one (68).
tert-Butyl (3-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)propyl)carbamate was synthesized according to General Procedure C. Then, tert-butyl (3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamate was synthesized according to General Procedure D. Then, 4-(3-aminopropyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized by treating a solution of tert-butyl (3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamate(0.056 g, 0.142 mmol) in DCM (0.593 mL), MeOH (0.119 mL) was added to TFA (0.477 mL, 6.19 mmol), and the reaction stirred at RT for 3 h. The reaction was then concentrated, and the product was purified by ISCO (DCM/MeOH +3.3% NH4OH, 0–100%): 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.4, 4.7 Hz, 1H), 7.92 (dd, J = 8.5, 3.1 Hz, 1H), 7.83 (ddd, J = 9.3, 7.9, 3.1 Hz, 1H), 7.65 (s, 3H), 4.10 (t, J = 6.8 Hz, 2H), 2.89 (q, J = 8.4, 7.3 Hz, 2H), 2.00 (p, J = 7.0 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.06 (td, J = 8.3, 4.8 Hz); HRMS (ES-API) m/z: calcd for C12H13FN5OS (M + H), 294.0819; found, 294.0825.
4-(3-Aminopropyl)-9-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-5(1H)-one (69).
tert-Butyl (3-(8-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)propyl)carbamate was synthesized according to General Procedure A. Then, tert-butyl (3-(9-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamate was synthesized according to General Procedure D. Then, 4-(3-aminopropyl)-9-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was synthesized as follows: tert-butyl (3-(9-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)carbamate (0.170 g, 0.432 mmol) was dissolved in HCl in dioxane (1.512 mL, 6.05 mmol) and the solution stirred at RT for 5 h. The reaction was quenched with triethylamine (0.843 mL, 6.05 mmol), filtered, and concentrated. The product was purified by ISCO (EtOAc/hexanes, 0–100%) to give 4-(3-amino-propyl)-9-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one, di-TFA salt: 19F NMR (376 MHz, DMSO-d6): δ −93.31 (dd, J = 11.6, 4.2 Hz).
4-(2-(Dimethylamino)ethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (70).
3-(2-(Dimethylamino)ethyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(2-(dimethylamino)ethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.6 Hz, 1H), 9.10 (s, 1H), 7.95 (dd, J = 8.5, 3.1 Hz, 1H), 7.86 (td, J = 8.5, 3.1 Hz, 1H), 4.40 (t, J = 5.6 Hz, 2H), 3.50–3.43 (m, 2H), 2.85 (s, 6H); 19F NMR (376 MHz, DMSO-d6): δ −112.62 (q, J = 7.5 Hz); HRMS (ES-API) m/z: calcd for C13H15FN5OS (M + H), 308.0976; found, 308.0989.
4-(1-(Dimethylamino)propan-2-yl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (71).
3-(1-(Dimethylamino)propan-2-yl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(1-(dimethylamino)propan-2-yl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.34 (dd, J = 9.3, 4.6 Hz, 1H), 9.24 (s, 1H),7.94 (dd, J = 8.6, 3.1 Hz, 1H), 7.85 (td, J = 9.2, 8.7, 3.1 Hz, 1H), 4.05 (t, J = 12.3 Hz, 1H), 3.35 (s, 2H), 2.80 (d, J = 17.7 Hz, 6H), 1.52 (d, J = 6.8 Hz, 3H); 19F NMR (376 MHz, DMSO-d6): δ −112.68 (q, J = 7.8 Hz); HRMS (ES-API) m/z: calcd for C14H17FN5OS (M + H), 322.1132; found, 322.1139.
N-(2-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)ethyl)acetamide (72).
N-(2-(6-Fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)acetamide was synthesized according to General Procedure B. Then, N-(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-ethyl)acetamide, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3,4.6 Hz, 1H), 7.92 (dd, J = 8.6, 3.0 Hz, 1H), 7.88–7.76 (m, 2H), 4.08 (t, J = 5.8 Hz, 2H), 3.39 (q, J = 6.1 Hz, 2H), 1.64 (s, 3H); 19F NMR (376 MHz, DMSO-d6): δ −113.00 (q, J = 7.6, 6.7 Hz); HRMS (ES-API) m/z: calcd for C13H13FN5O2S (M + H), 322.0769; found, 322.0780.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)acetamide (73).
tert-Butyl (3-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)propyl)carbamate was synthesized according to General Procedure C.
3-(3-Aminopropyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one: TFA (1.476 mL, 19.15 mmol) was added to a solution of crude tert-butyl (3-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)propyl)carbamate (0.1556 g, 0.440 mmol) in DCM (2.201 mL), and the reaction was stirred at RT for 2 h. The reaction was then blown down, the residue dissolved in MeOH, gravity filtered through a PL-HCO3 SPE cartridge, and concentrated. The crude product was used without further purification.
N-(3-(6-Fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)-propyl)acetamide: to a solution of 3-(3-aminopropyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (0.05 g, 0.197 mmol) in pyridine (0.6 mL) was added acetyl chloride (0.021 mL, 0.296 mmol) and the reaction stirred at 0 °C for 2 h. The reaction was then cooled to RT and poured into ice water and filtered, and the solid was washed with water and Et2O and dried. The crude product was used without further purification.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)acetamide, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.4, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H),7.86–7.75 (m, 2H), 4.02 (dd, J = 8.2, 6.3 Hz, 2H), 3.09 (q, J = 6.6 Hz, 2H), 1.82 (p, J = 7.2 Hz, 2H), 1.76 (s, 3H); 19F NMR (376 MHz, DMSO-d6): δ −113.17 (td, J = 8.2, 4.5 Hz); HRMS (ES-API) m/z: calcd for C17H18F3N4OS (M + H), 336.0925; found, 336.0929; purity (HPLC) 90.11%.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)benzamide (74).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propyl)benzamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.7 Hz, 1H), 8.46 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H),7.85–7.78 (m, 3H), 7.54–7.49 (m, 1H), 7.48–7.42 (m, 2H), 4.11 (dd, J = 8.1, 6.6 Hz, 2H), 3.36 (q, J = 6.6 Hz, 2H), 1.99 (p, J = 6.9 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.15 (td, J = 8.2, 4.6 Hz); HRMS (ES-API) m/z: calcd for C19H17FN5O2S (M + H), 398.1082; found, 398.1063.
N-(2-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)ethyl)methanesulfonamide (75).
N-(2-(6-Fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)-methanesulfonamide was synthesized according to General Procedure C. Then, N-(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-4(5H)-yl)ethyl)methanesulfonamide, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.7 Hz, 1H), 7.93 (dd, J = 8.6, 3.1 Hz, 1H), 7.81 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.17 (t, J = 6.4 Hz, 1H),4.18 (t, J = 6.2 Hz, 2H), 3.36 (q, J = 6.2 Hz, 2H), 2.88 (s, 3H); 19F NMR (376 MHz, DMSO-d6): δ −112.99 (td, J = 8.3, 4.7 Hz); HRMS (ES-API) m/z: calcd for C12H13FN5O3S2 (M + H), 358.0438; found, 358.0441.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)methanesulfonamide (76).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)methanesulfonamide was synthesized by treating 4-(3-aminopropyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (0.05 g, 0.170 mmol) in DCM (0.852 mL) at RT with methanesulfonyl chloride (0.015 mL, 0.188 mmol) and triethylamine (0.029 mL, 0.205 mmol). The reaction was stirred at RT for 2 h, concentrated, and purified by HPLC: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.92 (dd, J = 8.6, 3.1 Hz, 1H), 7.81 (ddd, J = 9.3, 8.0, 3.2 Hz, 1H), 7.00 (t, J = 5.9 Hz, 1H), 4.07 (t, J = 7.1 Hz, 2H), 3.02 (q, J = 6.7 Hz, 2H), 2.86 (s, 3H),1.89 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.18 (td, J = 8.3, 5.0 Hz); HRMS (ES-API) m/z: calcd for C13H15FN5O3S2 (M + H), 372.0595; found, 372.0601; purity (HPLC) 94.02%.
(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)ethyl)phosphonic Acid (77).
(2-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-ethyl)phosphonic acid was synthesized by treating a solution of diethyl (2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)phosphonate (0.111 g, 0.277 mmol) in DCM (4.62 mL) at 0 °C with bromotrimethylsilane (0.216 mL, 1.663 mmol) dropwise (fast). The reaction was then warmed to RT, and the solution was stirred for 8 h. The reaction was concentrated, redissolved, and purified by HPLC: 1H NMR (400 MHz, DMSO-d6): δ 10.27 (dd, J = 9.3, 4.6 Hz, 1H), 7.90 (dd, J = 8.7, 3.1 Hz, 1H),7.79 (td, J = 8.5, 3.1 Hz, 1H), 4.24–4.14 (m, 2H), 2.06–1.93 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.10 (q, J = 7.5 Hz); 31P NMR (162 MHz, DMSO-d6): δ 20.70.
Diethyl (2-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-4(5H)-yl)ethyl)phosphonate (78).
Diethyl (2-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)-phosphonate was synthesized according to General Procedure B. Then, diethyl (2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-4(5H)-yl)ethyl)phosphonate, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.32 (d, J = 7.3 Hz, 1H), 7.92 (dd, J = 8.5, 3.1 Hz, 1H), 7.80 (ddd, J = 9.1, 7.9, 3.1 Hz, 1H), 4.25–4.15 (m, 2H), 4.00 (dqd, J = 8.1, 7.0, 3.5 Hz, 4H), 3.26 (s, 1H), 2.28–2.15 (m, 2H), 1.22 (t, J = 7.0 Hz, 6H); 19F NMR (376 MHz, DMSO-d6): δ −113.07; 31P NMR (162 MHz, DMSO-d6): δ 26.69; HRMS (ES-API) m/z: calcd for C15H19FN4O4PS (M + H), 401.0843; found, 401.0856.
4-(Cyclopropylmethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (79).
3-(Cyclopropylmethyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(cyclopropylmethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.92 (dd, J = 8.6, 3.0 Hz, 1H), 7.80 (ddd, J = 9.3, 7.9, 3.1 Hz, 1H), 3.92 (d, J = 7.1 Hz, 2H), 1.27 (s, 1H), 0.49–0.37 (m, 4H); 19F NMR (376 MHz, DMSO-d6): δ −113.02 (td, J = 8.3, 4.7 Hz); HRMS (ES-API) m/z: calcd for C13H12FN4OS (M + H), 291.0710; found, 291.0720.
7-Fluoro-4-(trans-2-methylcyclopropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (80).
6-Fluoro-3-(trans-2-methylcyclopropyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(trans-2-methylcyclopropyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.99 (s, 1H), 10.26 (dd, J = 9.3, 4.6 Hz, 1H), 7.86 (dd, J = 8.8, 3.1 Hz, 1H),7.76 (ddd, J = 10.7, 8.2, 3.0 Hz, 1H), 2.56 (dt, J = 7.3, 3.5 Hz, 1H),1.27–1.19 (m, 1H), 1.15 (d, J = 6.0 Hz, 3H), 1.07 (dt, J = 9.6, 4.8 Hz, 1H), 0.90 (q, J = 6.6 Hz, 1H); 19F NMR (376 MHz, DMSO-d6): δ −113.28 (q, J = 7.3, 6.8 Hz); HRMS (ES-API) m/z: calcd for C13H12FN4OS (M + H), 291.0710; found, 291.0717.
4-(2-Chlorophenethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (81).
3-(2-Chlorophenethyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(2-chlorophenethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.28 (s, 1H), 7.88 (dd, J = 8.6, 2.9 Hz, 1H), 7.80 (t, J = 8.7 Hz, 1H), 7.39 (dd, J = 5.6, 3.7 Hz, 1H),7.36–7.29 (m, 1H), 7.23 (dd, J = 5.9, 3.5 Hz, 2H), 4.27 (t, J = 7.5 Hz, 2H), 3.13 (t, J = 7.3 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −112.98; HRMS (ES-API) m/z: calcd for C17H13ClFN4OS (M + H), 375.0477; found, 375.0481.
4-(4-Chlorophenethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (82).
3-(4-Chlorophenethyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(4-chlorophenethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.28 (dd, J = 9.4, 4.6 Hz, 1H), 7.89 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 7.9, 3.1 Hz, 1H), 7.32 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H), 4.21 (t, J = 7.7 Hz, 2H),2.98 (t, J = 7.6 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.00 (td, J = 8.4, 5.0 Hz); HRMS (ES-API) m/z: calcd for C17H13ClFN4OS (M + H), 375.0477; found, 375.0480.
7-Fluoro-4-(2-(pyridin-2-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (83).
6-Fluoro-3-(2-(pyridin-2-yl)ethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-(pyridin-2-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.4, 4.6 Hz, 1H), 8.46 (d, J = 5.0 Hz, 1H), 7.88 (dd, J = 8.6, 3.0 Hz, 1H), 7.80 (td, J = 8.6, 3.1 Hz, 1H), 7.76–7.73 (m, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.29–7.24 (m, 1H), 4.38 (t, J = 7.6 Hz, 2H), 3.20–3.11 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −112.97 to −113.04(m); HRMS (ES-API) m/z: calcd for C16H13FN5OS (M + H), 342.0819; found, 342.0818; purity (HPLC) 94.92%.
7-Fluoro-4-(2-(pyridin-3-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (84).
6-Fluoro-3-(2-(pyridin-3-yl)ethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-(pyridin-3-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.7 Hz, 1H), 8.56 (d, J = 2.2 Hz, 1H), 8.50 (dd, J = 4.9, 1.6 Hz, 1H), 7.92–7.85 (m, 2H), 7.82 (td, J = 8.6, 3.1 Hz, 1H), 7.46 (dd, J = 7.9, 5.0 Hz, 1H),4.29 (t, J = 7.2 Hz, 2H), 3.08 (t, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.00 (td, J = 8.2, 4.6 Hz); HRMS (ES-API) m/z: calcd for C16H13FN5OS (M + H), 342.0819; found, 342.0811.
7-Fluoro-4-(2-(pyridin-4-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (85).
6-Fluoro-3-(2-(pyridin-4-yl)ethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-(pyridin-4-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.7 Hz, 1H), 8.65–8.59 (m, 2H), 7.91–7.77 (m, 2H), 7.66 (d, J = 5.6 Hz, 2H), 4.34 (t, J = 7.1 Hz, 2H), 3.17 (t, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −112.98 (td, J = 8.2, 4.7 Hz); HRMS (ES-API) m/z: calcd for C16H13FN5OS (M + H), 342.0819; found, 342.0825.
tert-Butyl 3-(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)azetidine-1-carboxylate (86).
tert-Butyl 3-(2-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)azetidine-1-carboxylate was synthesized according to General Procedure B. Then, tert-butyl 3-(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)-azetidine-1-carboxylate, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.28 (dd, J = 9.3, 4.6 Hz, 1H), 7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.79 (ddd, J = 9.3, 7.9, 3.1 Hz, 1H), 3.98 (t, J = 6.8 Hz, 2H), 3.84 (s, 2H), 3.47 (s, 2H), 2.59–2.49 (m, 1H), 1.95 (q, J = 7.1 Hz, 2H), 1.33 (s, 8H); 19F NMR (376 MHz, DMSO-d6): δ −113.14 (q, J = 7.5 Hz); HRMS (ES-API) m/z: calcd for C19H22FN5NaO3S (M + Na), 442.1320; found, 442.1332.
7-Fluoro-4-(2-(1-methylpyrrolidin-2-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (87).
6-Fluoro-3-(2-(1-methylpyrrolidin-2-yl)ethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-(1-methylpyrrolidin-2-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.6 Hz, 1H), 9.43 (s, 1H), 7.92 (dd, J = 8.5, 3.1 Hz, 1H), 7.83 (td, J = 8.5, 3.1 Hz, 1H), 4.11 (dp, J = 28.3, 7.4 Hz, 2H), 3.51 (d, J = 11.9 Hz, 1H), 3.01 (d, J = 10.1 Hz, 1H),2.73 (s, 3H), 2.65 (d, J = 10.2 Hz, 1H), 2.43–2.25 (m, 1H), 2.04–1.79 (m, 3H), 1.72 (dq, J = 16.0, 8.5 Hz, 1H); 19F NMR (376 MHz, DMSO-d6): δ −112.96 (dt, J = 12.5, 6.1 Hz); HRMS (ES-API) m/z: calcd for C16H18FN5NaOS (M + Na), 370.1108; found, 370.1121.
7-Fluoro-4-(2-(piperidin-1-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (88).
6-Fluoro-3-(2-(piperidin-1-yl)ethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-(piperidin-1-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.6 Hz, 1H), 8.78 (s, 1H), 7.96 (dd, J = 8.5, 3.0 Hz, 1H), 7.86 (td, J = 8.6, 3.0 Hz, 1H), 4.41 (t, J = 6.1 Hz, 2H), 3.61 (s, 2H), 3.44 (s, 2H), 2.96 (s, 3H), 1.89–1.28 (m, 5H); 19F NMR (376 MHz, DMSO-d6): δ −112.56 (q, J = 7.4 Hz); HRMS (ES-API) m/z: calcd for C16H19FN5OS (M + H), 348.1289; found, 348.1305.
4-(2-(4,4-Difluoropiperidin-1-yl)ethyl)-7-fluoro-1-thioxo-2,4-di-hydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (89).
3-(2-(4,4-Difluoropiperidin-1-yl)ethyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(2-(4,4-difluoropiperidin-1-yl)ethyl)-7-fluoro-1-thioxo-2,4-di-hydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: 19F NMR (376 MHz, DMSO-d6): δ −95.47 (d, J = 213.5 Hz), −100.80 (d, J = 228.4 Hz), −112.68; HRMS (ES-API) m/z: calcd for C16H16F3N5NaOS (M + Na), 406.0920; found, 406.0937.
7-Fluoro-4-(2-morpholinoethyl)-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one (90).
6-Fluoro-3-(2-morpholinoethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-morpholinoethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.4, 4.5 Hz, 1H), 9.48 (s, 1H), 7.95 (dd, J = 8.6, 3.1 Hz, 1H), 7.86 (t, J = 8.9 Hz, 1H), 4.41 (s, 2H), 3.98 (s, 2H), 3.57 (dd, J = 25.5, 15.2 Hz, 2H), 3.35 (s, 4H), 3.18 (s, 2H); 19F NMR (376 MHz, DMSO-d6): δ −112.55; HRMS (ES-API) m/z: calcd for C15H17FN5O2S (M +H), 350.1082; found, 350.1091.
7-Fluoro-4-(2-(4-methylpiperazin-1-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (91).
6-Fluoro-3-(2-(4-methylpiperazin-1-yl)ethyl)-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 7-fluoro-4-(2-(4-methylpiperazin-1-yl)ethyl)-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, di-TFA salt, was synthesized according to General Procedure D: HRMS (ES-API) m/z: calcd for C17H18F3N4OS (M + H), 363.1398; found, 363.1384.
tert-Butyl 4-(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)piperazine-1-carboxylate (92).
tert-Butyl 4-(2-(6-fluoro-4-oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)piperazine-1-carboxylate was synthesized according to General Procedure B. Then, tert-butyl 4-(2-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)ethyl)-piperazine-1-carboxylate, di-TFA salt, was synthesized according to General Procedure D: HRMS (ES-API) m/z: calcd for C20H26FN6O3S (M + H), 449.1766; found, 449.1786.
4-(2-(4,4-Difluorocyclohexyl)ethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (93).
3-(2-(4,4-Difluorocyclohexyl)ethyl)-6-fluoro-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure B. Then, 4-(2-(4,4-difluorocyclohexyl)ethyl)-7-fluoro-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 10.28 (dd, J = 9.4, 4.6 Hz, 1H), 7.91 (dd, J = 8.7, 3.0 Hz, 1H), 7.84–7.74 (m, 1H), 4.05 (t, J = 7.4 Hz, 2H), 1.96 (d, J = 10.9 Hz, 2H), 1.88–1.58 (m, 6H), 1.46 (s, 1H), 1.16 (q, J = 12.3 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −89.54 (d, J = 231.6 Hz), −99.44 (d, J = 231.4 Hz), −113.14 (q, J = 7.1, 6.2 Hz); HRMS (ES-API) m/z: calcd for C17H18F3N4OS (M + H), 383.1148; found, 383.1166.
1-Amino-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-3,6,9,12-tetraoxapentadecan-15-amide (94).
To a solution of 95 (0.025 g, 0.039 mmol) in DCM (0.195 mL) was added TFA (0.066 mL, 0.858 mmol) and the reaction stirred at RT overnight. The reaction was then blown down and redissolved. The crude material was purified by RP-ISCO (H2O/MeCN + 0.2% NH4OH, 10–100%) to give 1-amino-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-3,6,9,12-tetraoxapentadecan-15-amide, di-TFA salt: 1H NMR (400 MHz, CD2Cl2): δ 13.38 (s, 1H), 10.25 (dd, J = 9.3, 4.6 Hz, 1H), 8.04 (s, 3H), 7.95 (dd, J = 8.3, 3.0 Hz, 1H), 7.45 (ddd, J = 9.4, 7.5, 3.1 Hz, 1H), 7.40 (s, 1H), 5.53 (s, 1H), 4.21 (t, J = 6.7 Hz, 2H), 3.89–3.55 (m, 16H), 3.27 (d, J = 22.8 Hz, 4H), 2.57 (s, 2H),2.01 (s, 2H); 19F NMR (376 MHz, CD2Cl2): δ −112.54 (td, J = 7.9,4.4 Hz).
tert-Butyl (19-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)-15-oxo-3,6,9,12-tetraoxa-16-azanonadecyl)carbamate (95).
tert-Butyl (19-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-15-oxo-3,6,9,12-tetraoxa-16-azanonadecyl)carbamate, TFA salt, was synthesized according to General Procedure F.
(S)-22-Amino-1-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)-5,21-dioxo-8,11,14,17-tetraoxa-4,20-diazapentacosan-25-oic Acid (96) and tert-Butyl (S)-22-Amino-1-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-5,21-dioxo-8,11,14,17-tetraoxa-4,20-diazapentacosan-25-oate (97).
tert-Butyl (S)-22-((tert-butoxycarbonyl)-amino)-1-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)-5,21-dioxo-8,11,14,17-tetraoxa-4,20-diazapentacosan-25-oate: To a solution of 95 (0.04 g, 0.074 mmol) in DMF (0.190 mL) was added a solution of (S)-5-(tert-butoxy)-2-((tertbutoxycarbonyl)amino)-5-oxopentanoic acid (Boc-L-Glu-γ-t-Bu-ester, 0.067 g, 0.222 mmol) and COMU (0.063 g, 0.148 mmol) followed by DIPEA (0.013 mL, 0.074 mmol). The reaction was then stirred overnight at RT. The reaction was quenched with water and concentrated. The product was purified by RP-ISCO (H2O/MeCN +3.3% NH4OH, 10–75%) to give the protected amino acid intermediate (0.0245 g, 40%): MS (ES-API) found (M + H), 826.2 and (M + Na), 848.1.
Compounds 96 and 97 were then prepared according to the following method. TFA (0.022 mL, 0.291 mmol) and triisopropylsilane (5.95 μL, 0.029 mmol) were added to a solution of the protected amino acid intermediate (0.012 g, 0.015 mmol) in DCM (0.145 mL). The reaction was stirred at RT for 2 h and concentrated. The mixture of products was purified by HPLC to give 96 (2 TFA) and 97 (2 TFA): 96: HRMS (ES-API) m/z: calcd for C28H41FN7O9S (M + H), 670.2665; found, 670.2672. 97: 1H NMR (400 MHz, dmso): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.47 (t, J = 5.7 Hz, 1H), 8.07 (s, 3H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.86 (s, 1H), 7.83–7.76 (m, 1H), 4.02 (t, J = 7.3 Hz, 2H), 3.72 (s, 1H), 3.56 (t, J = 6.5 Hz, 2H), 3.46–3.39 (m, 9H), 3.26–3.15 (m, 2H), 3.11 (q, J = 6.7 Hz, 2H), 2.27 (t, J = 7.2 Hz, 3H), 1.85 (dt, J = 23.5, 7.2 Hz, 3H), 1.37 (s, 9H); 19F NMR (376 MHz, dmso): δ −113.13 (dd, J = 8.2, 4.8 Hz); HRMS (ES-API) m/z: calcd for C32H49FN7O9S (M + H), 726.3291; found, 726.3284.
3-(2-(2-(2-Aminoethoxy)ethoxy)ethoxy)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propyl)propenamide (98).
To a solution of 99 (0.1496 g, 0.251 mmol) in HCl in dioxane (0.627 mL, 2.507 mmol) was added triisopropylsilane (0.079 g, 0.501 mmol), and the solution was stirred at RT for 2 h and then concentrated. The product was purified by HPLC to give 3-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)propenamide, di-TFA salt: HRMS (ES-API) m/z: calcd for C21H30FN6O5S (M + H), 497.1977; found, 497.1973.
tert-Butyl (16-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)-12-oxo-3,6,9-trioxa-13-azahexadecyl)carbamate (99).
tert-Butyl (16-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-12-oxo-3,6,9-trioxa-13-azahexadecyl)carbamate, TFA salt, was synthesized according to General Procedure F: HRMS (ES-API) m/z: calcd for C26H37FN6NaO7S (M + Na), 619.2321; found, 619.2330.
6-Amino-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)hexanamide (100).
Triisopropylsilane (0.031 g, 0.193 mmol) was added to a solution of 101 (0.049 g, 0.097 mmol) in HCl in dioxane (0.242 mL, 0.967 mmol), and the solution was stirred at RT for 2 h. The reaction was concentrated and purified by HPLC to give 6-amino-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)hexanamide, di-TFA salt: 1H NMR (400 MHz, dmso): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.81 (td, J = 7.9, 2.9 Hz, 2H), 7.61 (s, 3H), 4.02 (t, J = 7.4 Hz, 2H), 3.11 (q, J = 6.6 Hz, 2H), 2.75 (h, J = 6.0 Hz, 2H), 2.03 (t, J = 7.3 Hz, 2H),1.82 (p, J = 7.2 Hz, 2H), 1.55–1.41 (m, 4H), 1.31–1.19 (m, 2H); 19F NMR (376 MHz, dmso): δ −113.13 (td, J = 8.3, 4.8 Hz); HRMS (ES-API) m/z: calcd for C18H23FN6NaO2S (M + Na), 430.1506; found, 430.1512.
tert-Butyl (6-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-6-oxohexyl)-carbamate (101).
tert-Butyl (6-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-6-oxohexyl)carbamate was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.5, 3.1 Hz, 1H), 7.80 (ddd, J = 9.1, 7.8, 2.8 Hz, 2H),6.73 (t, J = 5.6 Hz, 1H), 4.02 (dd, J = 8.3, 6.3 Hz, 2H), 3.10 (q, J = 6.6 Hz, 2H), 2.86 (q, J = 6.6 Hz, 2H), 2.00 (t, J = 7.4 Hz, 2H), 1.82 (p, J = 7.0 Hz, 2H), 1.44 (p, J = 7.4 Hz, 2H), 1.37–1.27 (m, 11H),1.24–1.11 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19; HRMS (ES-API) m/z: calcd for C23H31FN6NaO4S (M + Na), 529.2004; found, 529.1994.
tert-Butyl (R)-4-((tert-butoxycarbonyl)amino)-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoate (102).
tert-Butyl (R)-4-((tertbutoxycarbonyl)amino)-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoate, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H),7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.85–7.76 (m, 2H), 6.82 (d, J = 8.2 Hz, 1H), 4.03 (t, J = 7.2 Hz, 2H), 3.90–3.80 (m, 1H), 3.12 (dp, J = 20.2, 6.6 Hz, 2H), 2.18 (t, J = 7.7 Hz, 2H), 1.83 (dt, J = 13.7, 6.5 Hz, 3H), 1.66 (dt, J = 14.3, 7.6 Hz, 1H), 1.36 (s, 9H), 1.35 (s, 9H); 19F NMR (376 MHz, DMSO-d6): δ −113.21 (t, J = 6.8 Hz); HRMS (ES-API) m/z: calcd for C26H35FN6NaO6S (M + Na), 601.2215; found, 601.2214.
tert-Butyl (S)-4-((tert-butoxycarbonyl)amino)-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoate (103).
tert-Butyl (S)-4-((tertbutoxycarbonyl)amino)-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoate, TFA salt, was synthesized according to General Procedure F: 19F NMR (376 MHz, DMSO-d6): δ −113.15 (d, J = 11.0 Hz); HRMS (ES-API) m/z: calcd for C26H35FN6NaO6S (M + Na), 601.2215; found, 601.2216.
tert-Butyl (S)-4-Amino-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoate (104) and (S)-4-Amino-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-amino)-5-oxopentanoic Acid (105).
TFA (0.041 mL, 0.532 mmol) and triisopropylsilane (10.90 μL, 0.053 mmol) were added to a solution of 103 (0.0154 g, 0.027 mmol) in DCM (0.266 mL). The reaction was stirred at RT for 2 h and concentrated. The products were purified by HPLC to give tert-butyl (S)-4-amino-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoate, di-TFA salt, and (S)-4-amino-5-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)amino)-5-oxopentanoic acid; 104: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.8 Hz, 1H),8.25 (s, 3H), 8.01 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H),7.82 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 4.07–3.99 (m, 2H), 3.92 (t, J = 6.3 Hz, 1H), 3.13 (q, J = 6.7 Hz, 2H), 2.22 (ddt, J = 22.8, 15.3, 7.4 Hz, 2H), 1.95 (q, J = 7.4 Hz, 2H), 1.85 (p, J = 7.1 Hz, 2H), 1.45 (s, 9H); 19F NMR (376 MHz, DMSO-d6): δ −113.12 (dt, J = 7.8, 4.1 Hz); HRMS (ES-API) m/z: calcd for C21H28FN6O4S (M + H), 479.1871; found, 479.1866. 105: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.7 Hz, 1H), 8.26 (s, 3H), 8.02 (t, J = 5.7 Hz, 1H),7.92 (dd, J = 8.5, 3.1 Hz, 1H), 7.83 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H),4.05 (dd, J = 8.1, 6.4 Hz, 2H), 3.93 (t, J = 6.6 Hz, 1H), 3.15 (q, J = 6.7 Hz, 2H), 2.54 (s, 0H), 2.24 (ddt, J = 23.0, 15.4, 7.5 Hz, 2H), 1.97 (q, J = 7.4 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H), 1.46 (s, 9H); 19F NMR (376 MHz, DMSO-d6): δ −113.10 (td, J = 8.2, 4.8 Hz); HRMS (ES-API) m/z: calcd for C17H20FN6O4S (M + H), 423.1245; found, 423.1232.
4-((3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-4-oxobutanoic Acid (106).
4-((3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)amino)-4-oxobutanoic acid, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.34 (s, 1H), 7.92 (dd, J = 8.6, 3.1 Hz, 1H),7.86 (t, J = 5.6 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 4.05 (t, J = 7.3 Hz, 2H), 3.12 (q, J = 6.7 Hz, 2H), 2.45–2.37 (m, 2H), 2.32–2.24 (m, 3H), 1.84 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.28; HRMS (ES-API) m/z: calcd for C16H17FN5O4S (M +H), 394.0980; found, 394.0979.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-4-sulfamoylbutanamide (107).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-4-sulfamoylbutanamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.7 Hz, 1H), 7.93 (dd, J = 8.6, 3.1 Hz, 1H), 7.87 (t, J = 5.6 Hz, 1H), 7.85–7.78 (m, 1H), 6.75 (s, 2H),4.05 (dd, J = 8.1, 6.3 Hz, 2H), 3.13 (q, J = 6.6 Hz, 2H), 3.00–2.92 (m, 2H), 2.20 (t, J = 7.4 Hz, 2H), 1.94–1.81 (m, 4H); 19F NMR (376 MHz, DMSO-d6): δ −113.14; HRMS (ES-API) m/z: calcd for C16H20FN6O4S2 (M + H), 443.0966; found, 443.0977.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)cyclopentanecarboxamide (108).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)cyclopentanecarboxamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.75 (t, J = 5.6 Hz, 1H),4.02 (dd, J = 8.2, 6.5 Hz, 2H), 3.10 (q, J = 6.6 Hz, 2H), 1.82 (p, J = 7.0 Hz, 2H), 1.73–1.63 (m, 2H), 1.63–1.51 (m, 4H), 1.50–1.40 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19 (td, J = 8.2, 4.6 Hz); HRMS (ES-API) m/z: calcd for C18H21FN5O2S (M + H), 390.1395; found, 390.1387.
trans-4-(tert-Butyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)cyclohexane-1-carboxamide (109).
trans-4-(tert-Butyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-cyclohexane-1-carboxamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.67 (t, J = 5.7 Hz, 1H), 4.01 (dd, J = 8.4, 6.4 Hz, 2H), 3.09 (q, J = 6.5 Hz, 2H), 1.93 (td, J = 12.2, 2.9 Hz, 1H), 1.81 (p, J = 6.9 Hz, 2H), 1.73 (d, J = 9.8 Hz, 4H), 1.26 (q, J = 12.2 Hz, 2H),0.97–0.87 (m, 3H), 0.80 (s, 9H); 19F NMR (376 MHz, DMSO-d6): δ −113.21 (td, J = 8.3, 4.6 Hz); HRMS (ES-API) m/z: calcd for C23H31FN5O2S (M + H), 460.2177; found, 460.2177.
tert-Butyl (4-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)cyclohexyl)-carbamate (110).
tert-Butyl (4-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-carbamoyl)cyclohexyl)carbamate, di-TFA salt was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H),7.80 (ddd, J = 9.4, 8.0, 3.2 Hz, 1H), 7.67 (t, J = 5.7 Hz, 1H), 6.65 (d, J = 6.5 Hz, 1H), 4.06–3.98 (m, 2H), 3.43 (s, 1H), 3.11 (q, J = 6.5 Hz, 2H), 2.10 (dq, J = 12.2, 3.8 Hz, 1H), 1.82 (p, J = 7.7, 7.3 Hz, 2H),1.77–1.68 (m, 1H), 1.64–1.53 (m, 2H), 1.47–1.37 (m, 5H), 1.36 (s, 9H); 19F NMR (376 MHz, DMSO-d6): δ −113.17 (td, J = 8.3, 4.8 Hz); HRMS (ES-API) m/z: calcd for C24H31FN6NaO4S (M + Na), 541.2004; found, 541.2011.
4-Amino-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)cyclohexane-1-carboxamide (111).
tert-Butyl (4-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)-cyclohexyl)carbamate (0.0282 g, 0.054 mmol) was dissolved in HCl in dioxane (0.136 mL, 0.544 mmol) and the solution stirred at RT for 2 h. The reaction was concentrated and purified by HPLC to give 4-amino-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)cyclohexane-1-carboxamide, di-TFA salt: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.7 Hz, 1H), 7.93 (dd, J = 8.5, 3.1 Hz, 1H), 7.88–7.76 (m, 2H), 7.71 (s, 3H),4.04 (dd, J = 8.3, 6.4 Hz, 2H), 3.14 (q, J = 6.6 Hz, 2H), 2.33–2.23 (m, 0H), 1.91–1.78 (m, 5H), 1.72–1.63 (m, 6H), 1.58–1.47 (m, 1H); 19F NMR (376 MHz, DMSO-d6): δ −113.09 (td, J = 8.4, 4.8 Hz); HRMS (ES-API) m/z: calcd for C19H24FN6O2S (M + H), 419.1660; found, 419.1642.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)piperidine-2-carboxamide (112).
tert-Butyl 2-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)piperidine-1-carboxylate was synthesized according to General Procedure F. Then, N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)piperidine-2-carboxamide was synthesized as follows: tert-butyl 2-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)piperidine-1-carboxylate (0.172 g, 0.341 mmol) was dissolved in HCl in dioxane (0.852 mL, 3.41 mmol) and the solution stirred at RT for 2 h. The reaction was concentrated and purified by HPLC (H2O + 0.1% NH4OH/MeCN, 10–50%, 8 min, Waters XBridge Prep C18 OBD, 5 μm, 30 × 75 mm) to give N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)piperidine-2-carboxamide: 1H NMR (400 MHz, dmso): δ 10.46 (dd, J = 9.3, 4.8 Hz, 1H), 8.28–8.22 (m, 1H), 7.87 (dd, J = 8.6, 3.2 Hz, 1H), 7.77 (td, J = 8.7, 3.1 Hz, 1H), 4.07 (t, J = 7.2 Hz, 2H), 3.49 (s, 1H), 3.22–3.06 (m, 3H), 2.79 (t, J = 11.8 Hz, 1H), 2.00–1.93 (m, 1H), 1.91–1.82 (m, 2H), 1.76–1.71 (m, 1H), 1.61 (d, J = 12.4 Hz, 1H), 1.49–1.37 (m, 4H); HRMS (ES-API) m/z: calcd for C18H22FN6O2S (M + H), 405.1503; found, 405.1517.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)piperidine-3-carboxamide (113).
tert-Butyl 2-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)piperidine-1-carboxylate was synthesized according to General Procedure F. Then, N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)piperidine-3-carboxamide was synthesized as follows: tert-butyl 3-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)piperidine-1-carboxylate (0.172 g, 0.341 mmol) was dissolved in HCl in dioxane (0.852 mL, 3.41 mmol) and the solution stirred at RT for 2 h. The reaction was concentrated and purified by HPLC (H2O + 0.1% NH4OH/MeCN, 10–50%, 8 min, Waters XBridge Prep C18 OBD, 5 μm, 30 × 75 mm) to give N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)piperidine-3-carboxamide: 1H NMR (400 MHz, dmso): δ 10.43 (s, 1H), 8.14 (t, J = 5.8 Hz, 1H), 7.87 (dd, J = 8.6, 3.2 Hz, 1H), 7.82–7.73 (m, 1H), 4.05 (t, J = 7.5 Hz, 2H), 3.11 (dq, J = 19.6, 7.8, 6.2 Hz, 5H), 2.96–2.90 (m, 1H), 2.89–2.80 (m, 2H), 1.84 (p, J = 7.1 Hz, 4H), 1.74 (app s, 1H), 1.56 (t, J = 9.7 Hz, 2H); HRMS (ES-API) m/z: calcd for C18H22FN6O2S (M + H), 405.1503; found, 405.1491.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)piperidine-4-carboxamide (114).
tert-Butyl 4-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)piperidine-1-carboxylate was synthesized according to General Procedure F. Then, N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)piperidine-4-carboxamide was synthesized as follows: tert-butyl 4-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)carbamoyl)piperidine-1-carboxylate (0.172 g, 0.341 mmol) was dissolved in HCl in dioxane (0.852 mL, 3.41 mmol) and the solution stirred at RT for 2 h. The reaction was concentrated and purified by HPLC (H2O + 0.1% NH4OH/MeCN, 10–50%, 8 min, Waters XBridge Prep C18 OBD, 5 μm, 30 × 75 mm) to give N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)piperidine-4-carboxamide: 1H NMR (400 MHz, dmso): δ 10.34 (s, 1H), 7.96 (t, J = 5.5 Hz, 1H), 7.91–7.87 (m, 1H), 7.83–7.76 (m, 1H), 4.02 (t, J = 7.4 Hz, 2H), 3.12 (dd, J = 12.4, 6.1 Hz, 2H), 2.87 (t, J = 12.2 Hz, 2H), 2.41–2.32 (m, 1H), 1.81 (d, J = 12.4 Hz, 5H), 1.69 (d, J = 13.1 Hz, 2H); HRMS (ES-API) m/z: calcd for C18H22FN6O2S (M + H), 405.1503; found, 405.1511; purity (HPLC) 93.09%.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)tetrahydro-2H-pyran-4-carboxamide (115).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)tetrahydro-2H-pyran-4-carboxamide, TFA salt, was synthesized according to General Procedure F: HRMS (ES-API) m/z: calcd for C18H21FN5O3S (M + H), 406.1344; found, 406.1355.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(piperidin-4-yl)acetamide (116).
tert-Butyl 4-(2-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-2-oxoethyl)piperidine-1-carboxylate was synthesized according to General Procedure F. Then, N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(piperidin-4-yl)acetamide was synthesized as follows: tert-butyl 4-(2-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-2-oxoethyl)piperidine-1-carboxylate (0.0209 g, 0.040 mmol) was dissolved in HCl in dioxane (0.101 mL, 0.403 mmol) and the solution stirred at RT for 2 h. The reaction was concentrated and purified by HPLC to give N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-2-(piperidin-4-yl)-acetamide, TFA: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.7 Hz, 1H), 8.43 (s, 1H), 8.12 (s, 1H), 7.96–7.89 (m, 2H), 7.83 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 4.08–4.00 (m, 2H), 3.24 (d, J = 13.1 Hz, 2H), 3.13 (q, J = 6.6 Hz, 2H), 2.86 (dd, J = 21.5, 10.8 Hz, 3H),2.02 (d, J = 7.0 Hz, 2H), 1.97–1.89 (m, 1H), 1.84 (p, J = 7.0 Hz, 2H), 1.77 (d, J = 14.0 Hz, 2H), 1.35–1.22 (m, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.06 to −113.17 (m).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-methylpiperazin-1-yl)acetamide (117).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-2-(4-methylpiperazin-1-yl)acetamide, di-TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.7 Hz, 1H), 9.49 (s, 1H), 7.97 (t, J = 5.5 Hz, 1H), 7.93 (dd, J = 8.5, 3.1 Hz, 1H), 7.82 (ddd, J = 9.3, 8.0, 3.2 Hz, 1H), 4.02 (t, J = 7.2 Hz, 2H), 3.40 (d, J = 10.2 Hz, 1H), 3.21–3.11 (m, 2H), 3.08 (s, 4H), 2.99 (d, J = 13.1 Hz, 3H), 2.78 (s, 3H), 1.84 (p, J = 6.8 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.07 (td, J = 8.2, 4.6 Hz); HRMS (ES-API) m/z: calcd for C19H25FN7O2S (M + H), 434.1769; found, 434.1763.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-morpholinoacetamide (118).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-morpholinoacetamide, di-TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 10.18 (s, 1H),8.55 (s, 1H), 7.92 (dd, J = 8.6, 3.1 Hz, 1H), 7.82 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 4.05 (t, J = 7.1 Hz, 2H), 3.97–3.67 (m, 8H), 3.22 (q, J = 6.6 Hz, 2H), 1.89 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.07 (td, J = 8.3, 4.6 Hz); HRMS (ES-API) m/z: calcd for C18H22FN6O3S (M + H), 421.1453; found, 421.1449.
4-((1H-Imidazole-1-yl)methyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-benzamide (119).
4-((1H-Imidazole-1-yl)methyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propyl)benzamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.7 Hz, 1H), 9.13 (t, J = 1.5 Hz, 1H), 8.52 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.5, 3.1 Hz, 1H), 7.86–7.77 (m, 3H), 7.75 (t, J = 1.7 Hz, 1H), 7.64 (t, J = 1.6 Hz, 1H), 7.57–7.45 (m, 2H), 5.46 (s, 2H), 4.11 (dd, J = 8.3, 6.3 Hz, 2H), 3.37–3.33 (m, 2H), 1.99 (p, J = 7.0 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.10 (td, J = 8.3, 4.7 Hz); HRMS (ES-API) m/z: calcd for C23H21FN7O2S (M + H), 478.1456; found, 478.1454.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-phenylacetamide (120).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-2-phenylacetamide was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3, 4.7 Hz, 1H), 8.05 (t, J = 5.7 Hz, 1H), 7.92 (dd, J = 8.6, 3.1 Hz, 1H), 7.81 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.33–7.15 (m, 5H), 4.04 (dd, J = 8.2, 6.4 Hz, 2H), 3.38 (s, 2H), 3.14 (q, J = 6.7 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.17 (td, J = 8.3, 4.8 Hz); HRMS (ES-API) m/z: calcd for C19H17FN5O2S (M + H), 398.1082; found, 398.1063.
3-(3,5-Dimethylisoxazol-4-yl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-propenamide (121).
3-(3,5-Dimethylisoxazol-4-yl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propyl)propenamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.31 (dd, J = 9.3,4.7 Hz, 1H), 7.92 (dd, J = 8.6, 3.0 Hz, 1H), 7.85–7.78 (m, 2H), 4.01 (t, J = 7.2 Hz, 2H), 3.32–3.23 (m, 2H), 3.10 (q, J = 6.7 Hz, 2H), 2.25 (s, 3H), 2.19 (dd, J = 7.9, 7.0 Hz, 2H), 2.13 (s, 3H), 1.81 (p, J = 7.0 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.14 (dd, J = 8.3,5.0 Hz); HRMS (ES-API) m/z: calcd for C20H22FN6O3S (M + H), 445.1453; found, 445.1475.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(3-fluorophenyl)acetamide (122).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(3-fluorophenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.08 (t, J = 5.6 Hz, 1H), 7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.31 (td, J = 8.1, 6.5 Hz, 1H), 7.08–6.99 (m, 3H), 4.03 (t, J = 7.3 Hz, 2H), 3.40 (s, 2H), 3.13 (q, J = 6.6 Hz, 2H), 1.84 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19 (q, J = 7.7, 7.1 Hz), −113.86 to −113.98 (m); HRMS (ES-API) m/z: calcd for C20H18F2N5O2S (M + H), 430.1144; found, 430.1153.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-fluorophenyl)acetamide (123).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-fluorophenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.4, 4.7 Hz, 1H), 8.05 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.30–7.22 (m, 2H), 7.14–7.05 (m, 2H), 4.02 (t, J = 7.3 Hz, 2H), 3.36 (s, 2H), 3.12 (q, J = 6.6 Hz, 2H), 1.84 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.18 (td, J = 8.3, 4.8 Hz), −116.95 (ddd, J = 14.6, 9.1, 5.5 Hz); HRMS (ES-API) m/z: calcd for C20H18F2N5O2S (M + H), 430.1144; found, 430.1141; purity (HPLC) 93.64%.
2-(3-Chlorophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide (124).
2-(3-Chlorophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.09 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.33–7.24 (m, 3H), 7.19 (dt, J = 7.5, 1.5 Hz, 1H), 4.03 (t, J = 7.2 Hz, 2H), 3.39 (s, 2H), 3.27 (s, 1H), 3.12 (q, J = 6.7 Hz, 2H),1.85 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19 (d, J = 10.0 Hz); HRMS (ES-API) m/z: calcd for C20H18ClFN5O2S (M + H), 446.0848; found, 446.0867; purity (HPLC) 94.66%.
2-(4-Bromophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide (125).
2-(4-Bromophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.6 Hz, 1H), 8.08 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.2, 7.9, 3.1 Hz, 1H), 7.50–7.42 (m, 2H), 7.22–7.15 (m, 2H), 4.02 (t, J = 7.3 Hz, 2H), 3.36 (s, 2H), 3.12 (q, J = 6.6 Hz, 2H), 1.84 (p, J = 7.0 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19; purity (HPLC) 88.26%.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(p-tolyl)acetamide (126).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-2-(p-tolyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.4, 4.6 Hz, 1H), 7.99 (t, J = 5.8 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 10.9, 8.0, 3.1 Hz, 1H), 7.09 (q, J = 8.0 Hz, 4H), 4.02 (t, J = 7.3 Hz, 2H), 3.11 (q, J = 6.6 Hz, 2H), 2.24 (s, 3H), 1.83 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.20; purity (HPLC) 89.66%.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-hydroxyphenyl)acetamide (127).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-hydroxyphenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 9.16 (s, 1H), 7.94–7.87 (m, 2H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.04–6.98 (m, 2H), 6.69–6.60 (m, 2H), 4.06–3.98 (m, 2H), 3.23 (s, 2H),3.10 (q, J = 6.6 Hz, 2H), 1.83 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19; purity (HPLC) 90.60%.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(2-methoxyphenyl)acetamide (128).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(2-methoxyphenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.2, 7.8, 3.0 Hz, 2H), 7.22–7.16 (m, 1H), 7.13 (dd, J = 7.5, 1.7 Hz, 1H), 6.92 (dd, J = 8.2, 1.1 Hz, 1H), 6.86 (td, J = 7.4, 1.1 Hz, 1H), 4.03 (t, J = 7.3 Hz, 2H), 3.74 (s, 3H), 3.34 (s, 2H), 3.13 (q, J = 6.6 Hz, 2H), 1.84 (p, J = 7.0 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.18 (td, J = 8.3, 4.7 Hz); HRMS (ES-API) m/z: calcd for C21H21FN5O3S (M + H), 442.1344; found, 442.1360.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(3-methoxyphenyl)acetamide (129).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(3-methoxyphenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.6 Hz, 1H), 8.01 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.22–7.13 (m, 1H), 6.82–6.73 (m, 3H), 4.03 (t, J = 7.2 Hz, 2H), 3.71 (s, 3H), 3.33 (s, 2H), 3.12 (q, J = 6.7 Hz, 2H), 1.84 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19.
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-methoxyphenyl)acetamide (130).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(4-methoxyphenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 7.96 (t, J = 5.8 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.17–7.09 (m, 2H), 6.87–6.79 (m, 2H), 4.02 (t, J = 7.3 Hz, 2H), 3.69 (s, 3H), 3.29 (s, 2H), 3.11 (q, J = 6.6 Hz, 2H), 1.83 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.19 (td, J = 8.2, 4.7 Hz).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(2-(trifluoromethyl)phenyl)-acetamide (131).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-2-(2-(trifluoromethyl)phenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.05 (t, J = 5.7 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.2, 7.9, 3.1 Hz, 1H), 7.65 (d, J = 7.7 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.44 (d, J = 7.8 Hz, 2H), 4.04 (t, J = 7.3 Hz, 2H), 3.61 (s, 2H), 3.15 (q, J = 6.7 Hz, 2H), 1.85 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −58.53, −113.20 (td, J = 8.3, 4.8 Hz).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(3-(trifluoromethyl)phenyl)-acetamide (132).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)-2-(3-(trifluoromethyl)phenyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.14 (t, J = 5.6 Hz, 1H), 7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.61–7.49 (m, 4H), 4.03 (dd, J = 8.1, 6.3 Hz, 2H), 3.50 (s, 2H), 3.13 (q, J = 6.8 Hz, 2H), 1.85 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −61.01, −113.20 (td, J = 8.2, 4.7 Hz); HRMS (ES-API) m/z: calcd for C21H18F4N5O2S (M + H), 480.1112; found, 480.1126.
2-(2-Cyanophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide (133).
2-(2-Cyanophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.20 (t, J = 5.6 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.85–7.73 (m, 2H), 7.63 (td, J = 7.7, 1.4 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.42 (td, J = 7.6, 1.2 Hz, 1H), 4.05 (t, J = 7.2 Hz, 2H), 3.65 (s, 2H), 3.16 (q, J = 6.7 Hz, 2H), 1.87 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.18 (td, J = 8.4, 4.8 Hz); HRMS (ES-API) m/z: calcd for C21H18FN6O2S (M + H) 437.1190; found, 437.1194.
2-(4-Cyanophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide (134).
2-(4-Cyanophenyl)-N-(3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)acetamide, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.16 (t, J = 5.6 Hz, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.77–7.73 (m, 2H), 7.47–7.40 (m, 2H), 4.02 (t, J = 7.3 Hz, 2H), 3.50 (s, 2H), 3.18–3.09 (m, 2H), 1.84 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.17.
tert-Butyl (4-(2-((3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-4(5H)-yl)propyl)amino)-2-oxoethyl)-phenyl)carbamate (135).
tert-Butyl (4-(2-((3-(7-fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-4(5H)-yl)-propyl)amino)-2-oxoethyl)phenyl)carbamate, TFA salt, was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.30 (dd, J = 9.3, 4.7 Hz, 1H), 9.23 (s, 1H), 7.98 (t, J = 5.7 Hz, 1H), 7.92 (dd, J = 8.6, 3.1 Hz, 1H), 7.81 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.34 (d, J = 8.5 Hz, 2H), 7.15–7.05 (m, 2H), 4.03 (dd, J = 8.1, 6.5 Hz, 2H), 3.30 (s, 2H), 3.12 (q, J = 6.7 Hz, 2H), 1.84 (p, J = 7.1 Hz, 2H), 1.46 (s, 9H); 19F NMR (376 MHz, DMSO-d6): δ −113.16 (td, J = 8.2, 4.8 Hz).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(pyridine-2-yl)acetamide (136).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(pyridine-2-yl)acetamide was synthesized according to General Procedure F: 1H NMR (400 MHz, dmso): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.48–8.42 (m, 1H), 8.13 (t, J = 5.8 Hz, 1H), 7.91 (dd, J = 8.6, 3.2 Hz, 1H), 7.80 (td, J = 8.6, 3.2 Hz, 1H),7.71 (td, J = 7.8, 1.8 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.22 (dd, J = 7.5, 5.0 Hz, 1H), 4.04 (t, J = 7.2 Hz, 2H), 3.57 (s, 2H), 3.14 (q, J = 6.7 Hz, 2H), 2.52 (s, 1H), 1.85 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, dmso): δ −113.14 to −113.23 (m).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(pyridine-3-yl)acetamide (137).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(pyridine-3-yl)acetamide was synthesized according to General Procedure F: 1H NMR (400 MHz, DMSO-d6): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.44–8.37 (m, 2H),8.14 (t, J = 5.7 Hz, 1H), 7.90 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (ddd, J = 9.3, 8.0, 3.1 Hz, 1H), 7.64 (dt, J = 7.8, 2.0 Hz, 1H), 7.31 (dd, J = 7.8, 4.8 Hz, 1H), 4.03 (dd, J = 8.3, 6.3 Hz, 2H), 3.42 (s, 2H), 3.13 (q, J = 6.5 Hz, 2H), 1.85 (p, J = 7.1 Hz, 2H); 19F NMR (376 MHz, DMSO-d6): δ −113.20 (d, J = 9.2 Hz).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(pyridine-4-yl)acetamide (138).
N-(3-(7-Fluoro-5-oxo-1-thioxo-1,2-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-4(5H)-yl)propyl)-2-(pyridine-4-yl)acetamide was synthesized according to General Procedure F: 1H NMR (400 MHz, dmso): δ 10.29 (dd, J = 9.3, 4.7 Hz, 1H), 8.51–8.44 (m, 2H), 8.23–8.16 (m, 1H), 7.91 (dd, J = 8.6, 3.1 Hz, 1H), 7.80 (td, J = 8.6, 3.2 Hz, 1H),7.28 (s, 2H), 4.03 (t, J = 7.3 Hz, 2H), 3.44 (d, J = 4.1 Hz, 2H), 3.14 (q, J = 6.6 Hz, 2H), 1.85 (p, J = 7.2 Hz, 2H); 19F NMR (376 MHz, dmso): δ −113.17 (td, J = 8.3, 4.7 Hz).
9-Fluoro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (139).
8-Fluoro-3-propyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one, TFA salt, was synthesized according to General Procedure A. Then, 9-fluoro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.83 (s, 1H), 8.02 (dd, J = 7.9, 1.5 Hz, 1H), 7.77 (ddd, J = 11.5, 8.3, 1.5 Hz, 1H), 7.61 (td, J = 8.0, 4.1 Hz, 1H), 3.97–3.89 (m, 2H), 1.70 (sext, J = 7.5 Hz, 2H), 0.89 (t, J = 7.5 Hz, 3H); 19F NMR (376 MHz, DMSO-d6): δ −93.30 (dd, J = 11.5, 4.2 Hz).
9-Chloro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (140).
8-Chloro-3-propyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one, TFA salt, was synthesized according to General Procedure A. Then, 9-chloro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, DMSO-d6): δ 13.78 (s, 1H), 8.10 (dd, J = 7.7, 1.5 Hz, 1H), 7.91 (dd, J = 8.0, 1.5 Hz, 1H), 7.60 (t, J = 7.9 Hz, 1H), 3.94–3.86 (m, 2H), 1.70 (sext, J = 7.5 Hz, 2H), 0.89 (t, J = 7.4 Hz, 3H).
9-Methoxy-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one (141).
8-Methoxy-3-propyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 9-methoxy-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]-triazolo[4,3-a]quinazolin-5(1H)-one was synthesized according to General Procedure D: 1H NMR (400 MHz, dmso): δ 13.67 (s, 1H),7.71 (dd, J = 7.3, 1.7 Hz, 1H), 7.62–7.50 (m, 2H), 3.95–3.87 (m, 5H), 1.68 (sext, J = 7.5 Hz, 2H), 0.87 (t, J = 7.5 Hz, 3H).
S-(5-Oxo-4-propyl-4,5-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-1-yl) Ethanethioate (142).
3-Propyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one was synthesized according to General Procedure A. Then, 4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one, TFA salt, was synthesized according to General Procedure D. Then, S-(5-oxo-4-propyl-4,5-dihydro-[1,2,4]triazolo-[4,3-a]quinazolin-1-yl) ethanethioate was synthesized by the following method. Triethylamine (0.054 mL, 0.384 mmol) and acetic anhydride (0.022 mL, 0.230 mmol) were added to a solution of 4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.05 g, 0.192 mmol) in acetonitrile (0.549 mL). The reaction was stirred for 2 h and quenched with water. The crude mixture was concentrated and purified by HPLC to give S-(5-oxo-4-propyl-4,5-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-1-yl) ethanethioate, TFA: 1H NMR (400 MHz, DMSO-d6): δ 10.32 (dd, J = 8.6, 1.0 Hz, 1H), 8.24 (dd, J = 7.9, 1.6 Hz, 1H), 7.90 (ddd, J = 8.8, 7.3, 1.7 Hz, 1H), 7.64 (td, J = 7.6, 1.1 Hz, 1H), 4.06–4.00 (m, 2H), 2.65 (s, 3H),1.77 (sext, J = 7.4 Hz, 2H), 0.93 (t, J = 7.5 Hz, 3H).
1-(Methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one (143).
4-Propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]-quinazolin-5(1H)-one was synthesized as described above. Then, 1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one was synthesized by the following method. K2CO3 (0.032 g, 0.230 mmol) and MeI (0.012 mL, 0.192 mmol) were added to a solution of 4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.05 g, 0.192 mmol) in DMF (0.549 mL). The reaction was stirred for 2 h and quenched with methanol. The crude mixture was concentrated and purified by HPLC (conditions) to give 1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one, TFA.
7-Fluoro-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]-quinazolin-5(4H)-one (144).
To a solution of 7-fluoro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.05 g, 0.180 mmol) in DMF (0.513 mL) was added K2CO3 (0.030 g, 0.216 mmol) and MeI (0.011 mL, 0.180 mmol). The reaction was stirred for 2 h and quenched with methanol. The crude mixture was concentrated and purified by HPLC (conditions) to give 7-fluoro-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one, TFA.
9-Fluoro-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]-quinazolin-5(4H)-one (145).
K2CO3 (0.027 g, 0.199 mmol) and MeI (10.36 μL, 0.166 mmol) were added to a solution of 9-fluoro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.0461 g, 0.166 mmol) in DMF (0.473 mL). The reaction was stirred for 3 h and quenched with methanol. The crude mixture was concentrated and purified by HPLC to give 9-fluoro-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one, TFA.
9-Chloro-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]-quinazolin-5(4H)-one (146).
K2CO3 (0.066 g, 0.480 mmol) and MeI (0.030 mL, 0.480 mmol) were added to a solution of 9-chloro-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.1179 g, 0.400 mmol) in DMF (2.000 mL). The reaction was stirred for 3 h and quenched with methanol. The crude mixture was concentrated and purified by ISCO (EtOAc/hexanes, 0–75%) to give 9-chloro-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one.
9-Methoxy-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]-quinazolin-5(4H)-one (147).
K2CO3 (0.059 g, 0.426 mmol) and MeI (0.027 mL, 0.426 mmol) were added to a solution of 9-methoxy-4-propyl-1-thioxo-2,4-dihydro-[1,2,4]triazolo[4,3-a]quinazolin-5(1H)-one (0.103 g, 0.355 mmol) in DMF (1.774 mL). The reaction was stirred for 3 h and quenched with methanol. The crude mixture was concentrated and purified by ISCO (EtOAc/hexanes, 0–90%) to give 9-methoxy-1-(methylthio)-4-propyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one.
Biochemical Assays.
ELISA-Based PBD-Binding Inhibition Assay.
The assay was performed essentially as described previously. Briefly, a biotinylated PBIP1 p-T78 peptide (i.e., Biotin-Ahx-CETFDPPLHSpTAI-NH2) was first diluted with 1× coating solution (SeraCare, Gaithersburg, MD) to the final concentration of 0.3 μM, and then 100 μL of the resulting solution was immobilized onto a 96-well streptavidin-coated plate (Nalgene Nunc, Rochester, NY). The wells were washed once with PBS + 0.05% Tween 20 (PBST) and incubated with 200 μL of PBS plus 1% BSA (blocking buffer) for 1 h to prevent nonspecific binding. HEK293A lysates expressing HAEGFP-Plk1 were prepared in a TBSN [20 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.5% NP-40, 5 mM EGTA, 2 mM MgCl2, 1.5 mM EDTA, and protease inhibitor cocktail (Roche)] + 20% DMSO buffer (~20 μg total lysates in 100 μL buffer), mixed with the indicated amount of the competitors (p-T78 peptide and its derivative compounds) for 30 min, provided onto the biotinylated peptide-coated ELISA wells, and then incubated with constant rocking for 1 h at 25 °C. Following the incubation, ELISA plates were washed five times with PBST. To detect bound HA-EGFP-Plk1, the plates were probed for 2 h with 100 μL/well of anti-HA antibody at a concentration of 0.5 μg/mL in the blocking buffer and then washed five times. The plates were further probed for 1 h with 100 μL/well of HRP-conjugated secondary antibody (GE Healthcare) at a 1:1000 dilution in the blocking buffer. The plates were washed five times with PBST and incubated with 100 μL/well of 3,3′,5,5′-tetramethylbenzidine solution (Sigma-Aldrich, St. Louis, MO) as a substrate until a desired absorbance was reached. The reactions were stopped by the addition of 100 μL/well of stop solution (Cell Signaling Technology, Danvers, MA). The optical density was measured at 450 nm by using a PerkinElmer Enspire Multimode Plate reader (PerkinElmer, Inc., Boston, MA). Data obtained from more than three independent experiments were analyzed by GraphPad (San Diego, CA) Prism software version 7.
PBD FP Binding Assays for Plk1 Specificity.
FP assays were carried out essentially as described previously.26 Briefly, an appropriate 5-carboxyfluorescein-labeled PBD-binding peptide for Plk1, Plk2, or Plk3 was incubated at a final concentration of 5 nM with various concentrations of the bacterially expressed and purified PBD of Plk1, Plk2 or Plk3, respectively, in a binding buffer [10 mM Tris (pH 8.0), 1 mM EDTA, 50 mM NaCl, and 0.01% Nonidet P-40]. Samples were analyzed 30 min after mixing all components in a 384-well format using the Molecular Devices (San Jose, CA). To eliminate the possibility that the apparent anti-Plk1 PBD activity is not the result of the compounds’ ability to inhibit the ligand peptide used for the Plk1 FP assay above (i.e., FITC-Ahx-DPPLHSpTAI-NH2), a second Plk1 PBD-binding peptide (FITC-Ahx-GPMQSpTPLNG-NH2)25 was used to confirm the activity (Figure S3B, Supporting Information). SpectraMax Paradigm multimode microplate detection platform. All experiments were performed in triplicate for each sample. The obtained data were plotted using GraphPad Prism software version 7.
ADME Measurements.
PAMPA permeability was measured using a high-throughput protocol, as previously reported.47 Aqueous solubility was determined by a published procedure.48 Microsomal metabolic stability assays were performed as reported previously.46
Cell-Based Assays.
Culture, Immunostaining, and Confocal Microscopy.
HeLa cells were cultured as recommended by the American Type Culture Collection (ATCC). Asynchronously growing cells were treated with control DMSO or the indicated concentration of 143. To reveal chromosome morphologies, cells were treated with DAPI, fixed, and subjected to confocal microscopy.
Immunostaining was carried out using anti-Plk1 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Cep63 (MilliporeSigma), anti-CREST (Antibodies Incorp, Davis, CA), and anti-α-tubulin (Sigma) antibodies. Confocal images were acquired using a Zeiss LSM780 equipped with a plan-apochromat 63× (N.A. 1.4) oil-immersion objective lens, 34-channel GaAsP spectral detector (Carl Zeiss Microscopy, LLC.), and 8-bit, 0.5 μm z-steps.
Image Quantification and Statistical Analysis.
To quantify fluorescence signal intensities, images were acquired under the same settings and the images obtained after the maximum-intensity projection of z-stacks were analyzed using the Zeiss ZEN v2.1 software (Carl Zeiss Microscopy, LLC). All the quantifications were performed with randomly chosen fields from at least three independent experiments. All values are given as mean ± s. d. P values were calculated by an unpaired t-test from the mean data of each group in GraphPad Prism (***P < 0.001). The statistical details including the definitions and value of n (e.g., number of experimental replicates, cells, etc.) and standard deviations are provided in the figure legends.
Metabolism Study.
Animals.
Male C57BL/6 mice from Charles River Laboratories (Wilmington, MA) were housed in the National Cancer Institute animal facility that is a pathogen-free environment controlled for temperature, light (25 °C, 12 h light/dark cycle), and humidity (45–65%) with free access to food and water. The National Cancer Institute Animal Care and Use Committee approved all animal experiments conducted in this study.
In Vivo Mouse Pharmacokinetics Study.
6–8-week-old male C57BL/6 mice were selected for metabolite profiling. In brief, 200 μL/20 g mice of WA (20 mg/kg dissolved in 5% DMSO and 5% Tween 80-contained saline) was administered intraperitoneally to mice. Control mice were treated with saline containing 5% DMSO and 5% Tween 80 alone. Each group consisted of three mice. Blood samples were collected by retro-orbital bleeding at 4 h postdose. After centrifugation for 10 min at 14,000g, serum was obtained and prepared by mixing 20 μL serum with 180 μL 50% aqueous acetonitrile. After centrifugation at 14,000g for 20 min, a 5 μL aliquot of the supernatants was injected into a Waters UPLC-ESI-QTOFMS system (Waters Corporation, Milford, MA).
In Vitro MLM Assay.
The incubation system (200 μL) contained 100 mM phosphate buffer solution (pH = 7.4), 1.0 mg·mL−1 MLMs, 5 mM MgCl2, and 20 μM test compound. The mixture was preincubated at 37 °C for 3 min and initiated with 2 mM NADPH or 2 mM UDPGA. The reaction was stopped by an aliquot of 200 μL of ice-cold acetonitrile after 60 min. After centrifugation at 14,000g for 10 min, a 5 μL aliquot of the supernatant was injected into a UHPLCESIQTOFMS.
UHPLC-ESI-QTOFMS and UHPLC-ESI-TQMS Analysis.
Metabolite profiling and identification were performed on an Acquity UPLC/Synapt HDMS Q-TOF MS system (Waters Corp., Milford, MA) with an ESI source. Separation was achieved on an Acquity C18 BEH UPLC column (1.7 mm, 2.1 × 50 mm; Waters Corp.). The mobile phase consisted of water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The initial condition of 2% B was held for 0.5 min, with the following linear gradient employed: 20% B at 4 min, 95% B at 8 min, to 99% B at 9 min, held for 1 min for column flushing, then returning to initial conditions for 2 min for column equilibration before the next injection. The flow rate of the mobile phase was set at 0.4 mL/min. Column temperature was maintained at 40 °C throughout the run. Data were collected in positive ion mode, which was operated in full-scan mode at 50–950 m/z. Nitrogen was used as both cone gas (50 L/h) and desolvation gas (950 L/h). Source temperature and desolvation temperature were set at 150 and 400 °C, respectively. The capillary voltage and cone voltage were 3000 and 30 V, respectively. The structures of metabolites of interest were elucidated by tandem MS fragmentography with collision energies ramping from 15 to 40 eV, employing the same chromatographic conditions described above.
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert O’Connor and Stewart R. Durell for structural assistance and Juan Marugan and Anton Simeonov for helpful advice. This research was supported by the Intramural Research Program of the National Institutes of Health NCI FLEX (K.S.L.), NIDDK ZIADK31117 (K.A.J.), and NCI ZIA BC005562 (F.J.G.).
ABBREVIATIONS
- ADME
absorption distribution metabolism excretion
- ATP
adenosine triphosphate
- Boc
tert-butyloxycarbonyl
- COMU
(1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholino-carbenium hexafluorophosphate
- CYP
cytochrome P450
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- DMSO
dime-thylsulfoxide
- EGFR
epidermal growth factor receptor
- ELISA
enzyme-linked immunosorbent assay
- FDA
Food and Drug Administration
- FITC
fluorescein-5-isothiocyanate
- FP
fluorescence polarization assay
- HBA
H-bond acceptor
- HER2
human epidermal growth factor receptor 2
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- IP
intraperitoneal
- KD
kinase domain
- LCMS
liquid chromatography–mass spectrometry
- MLM
mouse liver microsome
- MS/MS
tandem mass spectrometry
- MT
microtubule
- NAPDH
reduced nicotinamide adenine dinucleotide phosphate
- PAMPA
parallel artificial membrane permeability assay
- PBD
polo-box domain
- PBIP1
centromeric protein U
- PEG
polyethylene glycol
- PPI
protein–protein interaction
- pyr
pyridine
- RLM
rat liver microsome
- RT
room temperature
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
- UDPGA
uridine 5′-diphosphoglucuronic acid
- UGT
uridine diphosphate glucuronosyltransferase
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01669
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01669.
Extracted ion chromatograms of selected compounds and their glucuronides in MLM incubations with UDPGA after 60 min; MS/MS spectrum of selected compounds and their glucuronides; comparative FP-based assays showing the ability of additional triazoloquinazolinone-derived compounds (20, 27, 89, and 90) to specifically inhibit Plk1 PBD; acyl-transfer from 142; extracted ion chromatograms of 143 and its metabolites in MLM incubations with NADPH and UDPGA after 60 min, MS/MS spectrum of 143, its hydroxylated metabolite, and its demethylated metabolite, and proposed metabolic pathways of 143 in mice; extracted ion chromatograms of 143 and its metabolites from MS after 20 mg/kg IP injection of C57BL/6 mice analyzed 4 h post injection and extracted ion chromatograms of 145 and its metabolites from MS after 20 mg/kg IP injection of C57BL/6 mice analyzed 4 h post injection; inactive S-alkylated compounds; multispecies microsome and cytosol stability of selected compounds; and in vitro and in vivo stability of selected prodrugs (PDF)
Molecular formula strings (CSV)
The authors declare no competing financial interest.
Contributor Information
Celeste N. Alverez, Chemistry Section, Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States; Division of Preclinical Innovation, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Jung-Eun Park, Chemistry Section, Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States.
Kiran S. Toti, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Yangliu Xia, Chemistry Section, Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States.
Kristopher W. Krausz, Chemistry Section, Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States
Ganesha Rai, Division of Preclinical Innovation, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Jeong K. Bang, Division of Magnetic Resonance, Korea Basic Science Institute, Cheongju 28119, Republic of Korea.
Frank J. Gonzalez, Chemistry Section, Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Kyung S. Lee, Chemistry Section, Laboratory of Metabolism, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States.
REFERENCES
- (1).Zitouni S; Nabais C; Jana SC; Guerrero A; Bettencourt-Dias M Polo-like Kinases: Structural Variations Lead to Multiple Functions. Nat. Rev. Mol. Cell Biol 2014, 15, 433–452. [DOI] [PubMed] [Google Scholar]
- (2).Lee KS; Burke TR Jr.; Park J-E; Bang JK; Lee E Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol. Sci 2015, 36, 858–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Strebhardt K Multifaceted Polo-like Kinases: Drug Targets and Antitargets for Cancer Therapy. Nat. Rev. Drug Discovery 2010, 9, 643–660. [DOI] [PubMed] [Google Scholar]
- (4).de Cárcer G The Mitotic Cancer Target Polo-like Kinase 1: Oncogene or Tumor Suppressor? Genes 2019, 10, 208–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Luo J; Emanuele MJ; Li D; Creighton CJ; Schlabach MR; Westbrook TF; Wong K-K; Elledge SJ A Genome-Wide RNAi Screen Identifies Multiple Synthetic Lethal Interactions with the Ras Oncogene. Cell 2009, 137, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sur S; Pagliarini R; Bunz F; Rago C; Diaz LA; Kinzler KW; Vogelstein B; Papadopoulos N A Panel of Isogenic Human Cancer Cells Suggests a Therapeutic Approach for Cancers with Inactivated p53. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Park J-E; Kim T-S; Kim BY; Lee KS Selective Blockade of Cancer Cell Proliferation and Anchorage-Independent Growth by Plk1 Activity–Dependent Suicidal Inhibition of its Polo-Box Domain. Cell Cycle 2015, 14, 3624–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Weinstein IB Addiction to Oncogenes – The Achilles Heal of Cancer. Science 2002, 297, 63–64. [DOI] [PubMed] [Google Scholar]
- (9).McMurray HR; Sampson ER; Compitello G; Kinsey C; Newman L; Smith B; Chen S-R; Klebanov L; Salzman P; Yakovlev A; Land H Synergistic Response to Oncogenic Mutations Defines Gene Class Critical to Cancer Phenotype. Nature 2008, 453, 1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Luo J; Solimini NL; Elledge SJ Principles of Cancer Therapy: Oncogene and Non-Oncogene Addiction. Cell 2009, 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Elia AEH; Rellos P; Haire LF; Chao JW; Ivins FJ; Hoepker K; Mohammad D; Cantley LC; Smerdon SJ; Yaffe MB The Molecular Basis for Phosphodependent Substrate Targeting and Regulation of Plks by the Polo-Box Domain. Cell 2003, 115, 83–95. [DOI] [PubMed] [Google Scholar]
- (12).Lee KS; Grenfell TZ; Yarm FR; Erikson RL Mutation of the Polo-Box Disrupts Localization and Mitotic Functions of the Mammalian Polo Kinase Plk. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Seong Y-S; Kamijo K; Lee J-S; Fernandez E; Kuriyama R; Miki T; Lee KS A Spindle Checkpoint Arrest and a Cytokinesis Failure by the Dominant-Negative Polo-Box Domain of Plk1 in U-2 OS Cells. J. Biol. Chem 2002, 277, 32282–32293. [DOI] [PubMed] [Google Scholar]
- (14).Rudolph D; Steegmaier M; Hoffmann M; Grauert M; Baum A; Quant J; Haslinger C; Garin-Chesa P; Adolf GR BI 6727, a Polo-like Kinase Inhibitor with Improved Pharmacokinetic Profile and Broad Antitumor Activity. Clin. Cancer Res 2009, 15, 3094–3102. [DOI] [PubMed] [Google Scholar]
- (15).Steegmaier M; Hoffmann M; Baum A; Lénárt P; Petronczki M; Krššák M; Gürtler U; Garin-Chesa P; Lieb S; Quant J; Grauert M; Adolf GR; Kraut N; Peters J-M; Rettig WJ BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth in Vivo. Curr. Biol 2007, 17, 316–322. [DOI] [PubMed] [Google Scholar]
- (16).Russo MA; Kang KS; Di Cristofano A The PLK1 Inhibitor GSK461364A is Effective in Poorly Differentiated and Anaplastic Thyroid Carcinoma Cells, Independent of the Nature of Their Driver Mutations. Thyroid 2013, 23, 1284–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Beria I; Bossi RT; Brasca MG; Caruso M; Ceccarelli W; Fachin G; Fasolini M; Forte B; Fiorentini F; Pesenti E; Pezzetta D; Posteri H; Scolaro A; Depaolini SR; Valsasina B NMS-P937, a 4,5-dihydro-1H-Pyrazolo[4,3-h]Quinazoline Derivative as Potent and Selective Polo-like Kinase 1 Inhibitor. Bioorg. Med. Chem. Lett 2011, 21, 2969–2974. [DOI] [PubMed] [Google Scholar]
- (18).Nie Z; Feher V; Natala S; McBride C; Kiryanov A; Jones B; Lam B; Liu Y; Kaldor S; Stafford J; Hikami K; Uchiyama N; Kawamoto T; Hikichi Y; Matsumoto S.-i.; Amano N; Zhang L; Hosfield D; Skene R; Zou H; Cao X; Ichikawa T Discovery of TAK-960: An Orally Available Small Molecule Inhibitor of Polo-like Kinase 1 (PLK1). Bioorg. Med. Chem. Lett 2013, 23, 3662–3666. [DOI] [PubMed] [Google Scholar]
- (19).Karaman MW; Herrgard S; Treiber DK; Gallant P; Atteridge CE; Campbell BT; Chan KW; Ciceri P; Davis MI; Edeen PT; Faraoni R; Floyd M; Hunt JP; Lockhart DJ; Milanov ZV; Morrison MJ; Pallares G; Patel HK; Pritchard S; Wodicka LM; Zarrinkar PP A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- (20).Lee KS; Burke TR Jr.; Park J-E; Bang JK; Lee E Recent Advances and New Strategies in Targeting Plk1 for Anticancer Therapy. Trends Pharmacol. Sci 2015, 36, 858–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Park J-E; Hymel D; Burke TR Jr.; Lee KS Current Progress and Future Perspectives in the Development of Anti-Polo-like Kinase 1 Therapeutic Agents. F1000Research 2017, 6, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Syed N; Smith P; Sullivan A; Spender LC; Dyer M; Karran L; O’Nions J; Allday M; Hoffmann I; Crawford D; Griffin B; Farrell PJ; Crook T Transcriptional Silencing of Polo-like Kinase 2 (SNK/PLK2) is a Frequent Event in B-cell Malignancies. Blood 2006, 107, 250–256. [DOI] [PubMed] [Google Scholar]
- (23).Yang Y; Bai J; Shen R; Brown SAN; Komissarova E; Huang Y; Jiang N; Alberts GF; Costa M; Lu L; Winkles JA; Dai W Polo-like Kinase 3 Functions as a Tumor Suppressor and is a Negative Regulator of Hypoxia-Inducible Factor-1 Alpha Under Hypoxic Conditions. Cancer Res. 2008, 68, 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hanisch A; Wehner A; Nigg EA; Silljé HHW Different Plk1 Functions Show Distinct Dependencies on Polo-Box Domain-Mediated Targeting. Mol. Biol. Cell 2006, 17, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu F; Park J-E; Qian W-J; Lim D; Gräber M; Berg T; Yaffe MB; Lee KS; Burke TR Jr. Serendipitous Alkylation of a Plk1 Ligand Uncovers a New Binding Channel. Nat. Chem. Biol 2011, 7, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Qian W-J; Park J-E; Lim D; Lai CC; Kelley JA; Park S-Y; Lee KW; Yaffe MB; Lee KS; Burke TR Jr. Mono-anionic Phosphopeptides Produced by Unexpected Histidine Alkylation Exhibit High Plk1 Polo-Box Domain-Binding Affinities and Enhanced Antiproliferative Effects in HeLa Cells. Biopolymers 2014, 102, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yun S-M; Moulaei T; Lim D; Bang JK; Park J-E; Shenoy SR; Liu F; Kang YH; Liao C; Soung N-K; Lee S; Yoon D-Y; Lim Y; Lee D-H; Otaka A; Appella E; McMahon JB; Nicklaus MC; Burke TR Jr.; Yaffe MB; Wlodawer A; Lee KS Structural and Functional Analyses of Minimal Phosphopeptides Targeting the Polo-Box Domain of Polo-Like Kinase 1. Nat. Struct. Mol. Biol 2009, 16, 876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huggins DJ; Hardwick BS; Sharma P; Emery A; Laraia L; Zhang F; Narvaez AJ; Roberts-Thomson M; Crooks AT; Boyle RG; Boyce R; Walker DW; Mateu N; McKenzie GJ; Spring DR; Venkitaraman AR Development of a Novel Cell-Permeable Protein-Protein Interaction Inhibitor for the Polo-box Domain of Polo-like Kinase 1. ACS Omega 2020, 5, 822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sharma P; Mahen R; Rossmann M; Stokes JE; Hardwick B; Huggins DJ; Emery A; Kunciw DL; Hyvonen M; Spring DR; McKenzie GJ; Venkitaraman AR A Cryptic Hydrophobic Pocket in the Polo-Box Domain of the Polo-like Kinase PLK1 Regulates Substrate Recognition and Mitotic Chromosome Segregation. Sci. Rep 2019, 9, 15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rubner S; Scharow A; Schubert S; Berg T Selective Degradation of Polo-like Kinase 1 by a Hydrophobically Tagged Inhibitor of the Polo-Box Domain. Angew. Chem., Int. Ed 2018, 57, 17043–17047. [DOI] [PubMed] [Google Scholar]
- (31).Abdelfatah S; Berg A; Huang Q; Yang LJ; Hamdoun S; Klinger A; Greten HJ; Fleischer E; Berg T; Wong VKW; Efferth T MCC1019, a Selective Inhibitor of the Polo-Box Domain of Polo-like Kinase 1 as Novel, Potent Anticancer Candidate. Acta Pharm. Sin. B 2019, 9, 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Reindl W; Yuan J; Krämer A; Strebhardt K; Berg T A Pan-Specific Inhibitor of the Polo-Box Domains of Polo-like Kinases Arrests Cancer Cells in Mitosis. ChemBioChem 2009, 10, 1145–1148. [DOI] [PubMed] [Google Scholar]
- (33).Reindl W; Yuan J; Krämer A; Strebhardt K; Berg T Inhibition of Polo-Like Kinase 1 by Blocking Polo-Box Domain-Dependent Protein-Protein Interactions. Chem. Biol 2008, 15, 459–466. [DOI] [PubMed] [Google Scholar]
- (34).Watanabe N; Sekine T; Takagi M; Iwasaki J.-i.; Imamoto N; Kawasaki H; Osada H Deficiency in Chromosome Congression by the Inhibition of Plk1 Polo Box Domain-Dependent Recognition. J. Biol. Chem 2009, 284, 2344–2353. [DOI] [PubMed] [Google Scholar]
- (35).Normandin K; Lavallee JF; Futter M; Beautrait A; Duchaine J; Guiral S; Marinier A; Archambault V Identification of Polo-like Kinase 1 Interaction Inhibitors Using a Novel Cell-Based Assay. Sci. Rep 2016, 6, 37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Scharow A; Knappe D; Reindl W; Hoffmann R; Berg T Development of Bifunctional Inhibitors of Polo-like Kinase 1 with Low-Nanomolar Activities Against the Polo-Box Domain. ChemBioChem 2015, 17, 759–767. [DOI] [PubMed] [Google Scholar]
- (37).Chen Y; Zhang J; Li D; Jiang J; Wang Y; Si S Identification of a Novel Polo-like Kinase 1 Inhibitor that Specifically Blocks the Functions of Polo-Box Domain. Oncotarget 2017, 8, 1234–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kim TG; Lee JH; Lee MY; Kim K-U; Lee JH; Park CH; Lee BH; Oh K-S Development of a High-Throughput Assay for Inhibitors of the Polo-Box Domain of Polo-Like Kinase 1 Based on Time-Resolved Fluorescence Energy Transfer. Biol. Pharm. Bull 2017, 40, 1454–1462. [DOI] [PubMed] [Google Scholar]
- (39).Chen Y; Li Z; Liu Y; Lin T; Sun H; Yang D; Jiang C Identification of Novel and Selective Non-Peptide Inhibitors Targeting the Polo-Box Domain of Polo-Like Kinase 1. Bioorg. Chem 2018, 81, 278–288. [DOI] [PubMed] [Google Scholar]
- (40).Abdelfatah S; Berg A; Böckers M; Efferth T A Selective Inhibitor of the Polo-Box Domain of Polo-like Kinase 1 Identified by Virtual Screening. J. Adv. Res 2019, 16, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou Y; Yan F; Huo X; Niu M-M Discovery of a Potent PLK1-PBD Small-Molecule Inhibitor as an Anticancer Drug Candidate Through Structure-Based Design. Molecules 2019, 24, 4351–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lee KS; Idle JR Pinning Down the Polo-Box Domain. Chem. Biol 2008, 15, 415–416. [DOI] [PubMed] [Google Scholar]
- (43).Archambault V; Normandin K Several Inhibitors of the Plk1 Polo-Box Domain Turn Out to Be Non-Specific Protein Alkylators. Cell Cycle 2017, 16, 1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kang YH; Park J-E; Yu L-R; Soung N-K; Yun S-M; Bang JK; Seong Y-S; Yu H; Garfield S; Veenstra TD; Lee KS Self-regulated Plk1 Recruitment to Kinetochores by the Plk1-PBIP1 Interaction Is Critical for Proper Chromosome Segregation. Mol. Cell 2006, 24, 409–422. [DOI] [PubMed] [Google Scholar]
- (45).Carlsson J; Yoo L; Gao Z-G; Irwin JJ; Shoichet BK; Jacobson KA Structure-Based Discovery of A2A Adenosine Receptor Ligands. J. Med. Chem 2010, 53, 3748–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Urban DJ; Martinez NJ; Davis MI; Brimacombe KR; Cheff DM; Lee TD; Henderson MJ; Titus SA; Pragani R; Rohde JM; Liu L; Fang Y; Karavadhi S; Shah P; Lee OW; Wang A; McIver A; Zheng H; Wang X; Xu X; Jadhav A; Simeonov A; Shen M; Boxer MB; Hall MD Assessing Inhibitors of Mutant Isocitrate Dehydrogenase Using a Suite of Pre-Clinical Discovery Assays. Sci. Rep 2017, 7, 12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sun H; Nguyen K; Kerns E; Yan Z; Yu KR; Shah P; Jadhav A; Xu X Highly Predictive and Interpretable Models for PAMPA Permeability. Bioorg. Med. Chem 2017, 25, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sun H; Shah P; Nguyen K; Yu KR; Kerns E; Kabir M; Wang Y; Xu X Predictive Models of Aqueous Solubility of Organic Compounds Built on a Large Dataset of High Integrity. Bioorg. Med. Chem 2019, 27, 3110–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lénárt P; Petronczki M; Steegmaier M; Di Fiore B; Lipp JJ; Hoffmann M; Rettig WJ; Kraut N; Peters J-M The Small-Molecule Inhibitor BI 2536 Reveals Novel Insights Into Mitotic Roles of Polo-Like Kinase 1. Curr. Biol 2007, 17, 304–315. [DOI] [PubMed] [Google Scholar]
- (50).Jackson JR; Patrick DR; Dar MM; Huang PS Targeted Anti-Mitotic Therapies: Can We Improve on Tubulin Agents? Nat. Rev. Cancer 2007, 7, 107–117. [DOI] [PubMed] [Google Scholar]
- (51).Münch C; Dragoi D; Frey A-V; Thurig K; Lübbert M; Wäsch R; Bogatyreva L; Hauschke D; Lassmann S; Werner M; May AM Therapeutic Polo-Like Kinase 1 Inhibition Results in Mitotic Arrest and Subsequent Cell Death of Blasts in the Bone Marrow of AML Patients and Has Similar Effects in Non-Neoplastic Cell Lines. Leuk. Res 2015, 39, 462–470. [DOI] [PubMed] [Google Scholar]
- (52).Döhner H; Lübbert M; Fiedler W; Fouillard L; Haaland A; Brandwein JM; Lepretre S; Reman O; Turlure P; Ottmann OG; Müller-Tidow C; Krämer A; Raffoux E; Döhner K; Schlenk RF; Voss F; Taube T; Fritsch H; Maertens J Randomized, Phase 2 Trial of Low-Dose Cytarabine With or Without Volasertib in AML Patients Not Suitable for Induction Therapy. Blood 2014, 124, 1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Janning M; Fiedler W Volasertib for the Treatment of Acute Myeloid Leukemia: A Review of Preclinical and Clinical Development. Future Oncol. 2014, 10, 1157–1165. [DOI] [PubMed] [Google Scholar]
- (54).Awada A; Dumez H; Aftimos PG; Costermans J; Bartholomeus S; Forceville K; Berghmans T; Meeus M-A; Cescutti J; Munzert G; Pilz K; Liu D; Schöffski P Phase I Trial of Volasertib, a Polo-like Kinase Inhibitor, Plus Platinum Agents in Solid Tumors: Safety, Pharmacokinetics and Activity. Invest. New Drugs 2015, 33, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Willemze R; Suciu S; Meloni G; Labar B; Marie J-P; Halkes CJM; Muus P; Mistrik M; Amadori S; Specchia G; Fabbiano F; Nobile F; Sborgia M; Camera A; Selleslag DLD; Lefrére F; Magro D; Sica S; Cantore N; Beksac M; Berneman Z; Thomas X; Melillo L; Guimaraes JE; Leoni P; Luppi M; Mitra ME; Mitra D; Bron G; Marijt EWA; Venditti A; Hagemeijer A; Mancini M; Jansen J; Cilloni D; Meert L; Fazi P; Vignetti M; Trisolini SM; Mandelli F; de Witte T; de Witte T High-Dose Cytarabine in Induction Treatment Improves the Outcome of Adult Patients Younger Than Age 46 Years With Acute Myeloid Leukemia: Results of the EORTC-GIMEMA AML-12 Trial. J. Clin. Oncol 2014, 32, 219–228. [DOI] [PubMed] [Google Scholar]
- (56).Lin C-C; Su W-C; Yen C-J; Hsu C-H; Su W-P; Yeh K-H; Lu Y-S; Cheng A-L; Huang DC-L; Fritsch H; Voss F; Taube T; Yang JC-H A Phase I Study of Two Dosing Schedules of Volasertib (BI 6727), an Intravenous Polo-Like Kinase Inhibitor, in Patients With Advanced Solid Malignancies. Br. J. Cancer 2014, 110, 2434–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Machiels J-P; Peeters M; Herremans C; Surmont V; Specenier P; De Smet M; Pilz K; Strelkowa N; Liu D; Rottey S A Phase I Study of Volasertib Combined With Afatinib, in Advanced Solid Tumors. Cancer Chemother. Pharmacol 2015, 76, 843–851. [DOI] [PubMed] [Google Scholar]
- (58).de Braud F; Cascinu S; Spitaleri G; Pilz K; Clementi L; Liu D; Sikken P; De Pas T A Phase I, Dose-Escalation Study of Volasertib Combined With Nintedanib in Advanced Solid Tumors. Ann. Oncol 2015, 26, 2341–2346. [DOI] [PubMed] [Google Scholar]
- (59).Olmos D; Barker D; Sharma R; Brunetto AT; Yap TA; Taegtmeyer AB; Barriuso J; Medani H; Degenhardt YY; Allred AJ; Smith DA; Murray SC; Lampkin TA; Dar MM; Wilson R; de Bono JS; Blagden SP Phase I Study of GSK461364, a Specific and Competitive Polo-like Kinase 1 Inhibitor, in Patients With Advanced Solid Malignancies. Clin. Cancer Res 2011, 17, 3420–3430. [DOI] [PubMed] [Google Scholar]
- (60).Schöffski P; Awada A; Dumez H; Gil T; Bartholomeus S; Wolter P; Taton M; Fritsch H; Glomb P; Munzert G A Phase I, Dose-Escalation Study of the Novel Polo-like Kinase Inhibitor Volasertib (BI 6727) In Patients With Advanced Solid Tumours. Eur. J. Cancer 2012, 48, 179–186. [DOI] [PubMed] [Google Scholar]
- (61).Bojadzic D; Buchwald P Toward Small-Molecule Inhibition of Protein-Protein Interactions: General Aspects and Recent Progress in Targeting Costimulatory and Coinhibitory (Immune Checkpoint) Interactions. Curr. Top. Med. Chem 2018, 18, 674–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Rognan D Rational design of protein–protein interaction inhibitors. MedChemComm 2015, 6, 51–60. [Google Scholar]
- (63).Ceesay MM; Couchman L; Smith M; Wade J; Flanagan RJ; Pagliuca A Triazole Antifungals Used for Prophylaxis and Treatment of Invasive Fungal Disease in Adult Haematology Patients: Trough Serum Concentrations in Relation to Outcome. Med. Mycol 2016, 54, 691–698. [DOI] [PubMed] [Google Scholar]
- (64).Lass-Flörl C Triazole Antifungal Agents in Invasive Fungal Infections: A Comparative Review. Drugs 2011, 71, 2405–2419. [DOI] [PubMed] [Google Scholar]
- (65).Shagufta S; Ahmad I An Insight Into the Therapeutic Potential of Quinazoline Derivatives as Anticancer Agents. MedChemComm 2017, 8, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ritschel T; Hermans SMA; Schreurs M; van den Heuvel JJMW; Koenderink JB; Greupink R; Russel FGM In Silico Identification and in Vitro Validation of Potential Cholestatic Compounds Through 3D Ligand-Based Pharmacophore Modeling of BSEP Inhibitors. Chem. Res. Toxicol 2014, 27, 873–881. [DOI] [PubMed] [Google Scholar]
- (67).Zhang H-J; Jin P; Wang S-B; Li F-N; Guan L-P; Quan Z-S Synthesis and Anticonvulsant Activity Evaluation of 4-Phenyl-[1,2,4]triazolo[4,3-a]quinazolin-5(4H)-one and Its Derivatives. Arch. Pharm 2015, 348, 564–574. [DOI] [PubMed] [Google Scholar]
- (68).Zhang H-J; Wang S-B; Quan Z-S Synthesis and Antidepressant Activities of 4-(substituted-phenyl)tetrazolo[1,5-a]-quinazolin-5(4H)-ones and Their Derivatives. Mol. Divers 2015, 19, 817–828. [DOI] [PubMed] [Google Scholar]
- (69).Agarwal S; Nagpal N; Fong Y PAPD5 Inhibitors and Methods of Use Thereof. U.S. Patent WO 2019084271 A1, May 2, 2019.
- (70).Ram VJ; Srimal RC; Kushwaha DS; Mishra L Chemoterapeutic Agents. XIX. Synthesis of [1,2,4]-Triazoloquinazolinones and Related Compounds as Antihypertensive Agents. J. Prakt. Chem 1990, 332, 629–639. [Google Scholar]
- (71).Ram VJ; Goel A; Verma M; Kaul IB; Kapil A Latent Leishmanicidal Activity of Quinazolinones and 1,2,4-Triazoloquinazolinones. Bioorg. Med. Chem. Lett 1994, 4, 2087–2090. [Google Scholar]
- (72).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Brassesco MS; Pezuk JA; Morales AG; Carvalho de Oliveira J; Roberto GM; Nicioli da Silva G; Francisco de Oliveira H; Scrideli CA; Tone LG In Vitro Targeting of Polo-like Kinase 1 in Bladder Carcinoma: Comparative Effects of Four Potent Inhibitors. Canc. Biol. Ther 2013, 14, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Bogado RFE; Pezuk JA; de Oliveira HF; Tone LG; Brassesco MS 6727 and GSK461364 Suppress Growth and Radiosensitize Osteosarcoma Cells, but Show Limited Cytotoxic Effects When Combined With Conventional Treatments. Anticancer Drugs 2015, 26, 56–63. [DOI] [PubMed] [Google Scholar]
- (75).Rudolph D; Impagnatiello MA; Blaukopf C; Sommer C; Gerlich DW; Roth M; Tontsch-Grunt U; Wernitznig A; Savarese F; Hofmann MH; Albrecht C; Geiselmann L; Reschke M; Garin-Chesa P; Zuber J; Moll J; Adolf GR; Kraut N Efficacy and Mechanism of Action of Volasertib, a Potent and Selective Inhibitor of Polo-like Kinases, in Preclinical Models of Acute Myeloid Leukemia. J. Pharmacol. Exp. Ther 2015, 352, 579–589. [DOI] [PubMed] [Google Scholar]
- (76).Gjertsen BT; Schöffski P Discovery and Development of the Polo-like Kinase Inhibitor Volasertib in Cancer Therapy. Leukemia 2015, 29, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Erba HP Finding the Optimal Combination Therapy for the Treatment of Newly Diagnosed AML in Older Patients Unfit for Intensive Therapy. Leuk. Res 2015, 39, 183–191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.