

Abstract
Potassium often has a negative connotation in Nephrology as patients with chronic kidney disease (CKD) are prone to develop hyperkalaemia. Approaches to the management of chronic hyperkalaemia include a low potassium diet or potassium binders. Yet, emerging data indicate that dietary potassium may be beneficial for patients with CKD. Epidemiological studies have shown that a higher urinary potassium excretion (as proxy for higher dietary potassium intake) is associated with lower blood pressure (BP) and lower cardiovascular risk, as well as better kidney outcomes. Considering that the composition of our current diet is characterized by a high sodium and low potassium content, increasing dietary potassium may be equally important as reducing sodium. Recent studies have revealed that dietary potassium modulates the activity of the thiazide-sensitive sodium-chloride cotransporter in the distal convoluted tubule (DCT). The DCT acts as a potassium sensor to control the delivery of sodium to the collecting duct, the potassium-secreting portion of the kidney. Physiologically, this allows immediate kaliuresis after a potassium load, and conservation of potassium during potassium deficiency. Clinically, it provides a novel explanation for the inverse relationship between dietary potassium and BP. Moreover, increasing dietary potassium intake can exert BP-independent effects on the kidney by relieving the deleterious effects of a low potassium diet (inflammation, oxidative stress and fibrosis). The aim of this comprehensive review is to link physiology with clinical medicine by proposing that the same mechanisms that allow us to excrete an acute potassium load also protect us from hypertension, cardiovascular disease and CKD.
Keywords: albuminuria, aldosterone, blood pressure, CKD, hyperkalaemia, hypertension, nutrition
INTRODUCTION
The diet of our hunter-gatherer ancestors consisted mainly of fruit, vegetables and game, and provided small amounts of sodium (Na+) and large amounts of potassium (K+). During the late Palaeolithic times, estimated dietary Na+ intake was ∼30 mmol/day (∼690 mg/day) and dietary K+ intake was ∼280 mmol/day (∼11 g/day) [1]. This is very different from our current diet, which contains large amounts of Na+ and small amounts of K+. Studies based on 24-h dietary recalls or urinary excretion estimate that the current average Na+ intake is between 148 mmol/day (3.4 g/day) [2] and 213 mmol/day (4.9 g/day) [3], and that the average K+ intake is between 54 mmol/day (2.1 g/day) [3] and 67 mmol/day (2.6 g/day) [2]. This dietary transition may explain, at least in part, the high prevalence of hypertension, cardiovascular disease and chronic kidney disease (CKD) [4–6]. Current evidence links dietary Na+ intake directly to blood pressure (BP). Accordingly, reducing dietary Na+ intake decreases BP [3, 7–10]. Based on the assumption that any approach to reduce BP will result in a lower incidence of cardiovascular disease, public health campaigns and research efforts have primarily focused on interventions to reduce dietary Na+ intake. For example, the World Health Organization recommends a reduction to <2 g/day Na+ (equivalent to 5 g sodium chloride per day) in adults [11]. On the other hand, strategies to raise dietary K+ intake have received less attention despite accumulating evidence that higher dietary K+ intake benefits health. First, several systematic reviews and meta-analyses have shown that increasing dietary K+ intake decreases BP []. Secondly, large population studies have shown that higher dietary K+ intake is associated with lower incidence of cardiovascular disease and stroke [3, 12–14]. Thirdly, several observational studies found an association between higher urinary K+ excretion (as proxy for dietary intake) and better kidney outcomes [15]. Finally, experimental studies have uncovered underlying mechanisms that may explain the health benefits of dietary K+. Our aim is to review the health effects of dietary Na+ and K+, novel physiological insights in Na+ and K+ handling by the kidney and how these mechanisms may explain the effects of dietary K+ on BP, the cardiovascular system and the kidney.
DIETARY SODIUM, POTASSIUM AND HUMAN HEALTH: A BRIEF HISTORY
Globally, hypertension is considered the leading risk factor for cardiovascular and kidney disease, including ischaemic heart disease, congestive heart failure, stroke and CKD [16]. The link between a high-salt (sodium chloride) diet and the incidence of hypertension emerged during the early 1900s [17, 18]. In the 1960s, Dahl [19] found that societies with higher-than-average salt intake also had a higher incidence of hypertension. However, each population also consisted of individuals who did not develop hypertension even though they consistently consumed a high-salt diet. Thus, people can be classified as salt-sensitive or salt-resistant. Dahl et al. suggested that hypertension results from genetic and environmental susceptibility. To demonstrate this, they separated strains of salt-sensitive and salt-resistant rats based on their BP response to high salt [20]. Their findings indicated that salt-sensitivity is a genetic trait, and that salt intake should be seen as an environmental factor. Subsequently, they studied whether dietary K+ influences salt-sensitive hypertension [21]. One of the reasons to do so was that early findings had shown that dietary Na+ and K+ have opposite effects on BP. For example, in 1928, Addison published a report of five patients with hypertension of various aetiology in whom administration of different K+ salts decreased BP [22]. In the 1950s, Meneely et al. [23] found that high dietary K+ intake had prominent anti-hypertensive effects on rats during high dietary Na+ intake, and increased their lifespan. Dahl et al. found that in rats on a high Na+ diet, BP was reduced in the groups for which the diet also consisted of high dietary K+. For example, in Dahl salt-sensitive rats on a high-salt diet for 12 months, rats with a dietary Na+/K+ ratio of 10 had a mean systolic BP (SBP) of 157 mmHg, whereas rats on the same NaCl diet with a Na+/K+ ratio of 1 had a mean SBP of 127 mmHg [21]. While the effects of K+ on BP were impressive, at this time, the mechanisms explaining the beneficial effects of dietary K+ were unknown. Over the following decades, many epidemiologic and interventional studies reported an inverse relationship between dietary K+ intake and BP. For example, in the 1980s, the INTERSALT investigators studied 24-h urine electrolyte excretion and BP in 10 079 people (aged from 20 to 59 years from 32 countries), and found that 24-h urinary K+ excretion was inversely associated with BP [7]. This association remained after adjustment for confounders that usually correlate with BP such as urine Na+ excretion, body mass index and alcohol consumption. The association between BP and the urinary Na+/K+ excretion ratio was stronger than the association between BP and either Na+ or K+ excretion alone [7]. The INTERSALT study also included a particularly interesting Brazilian Indian population called the Yanomamo and Xingu. Their diet is characterized by a low Na+ and high K+ content, similar to our ancestors’ diet in Palaeolithic times [1]. In the Yanomamo and Xingu, the urinary Na+/K+ ratio was extremely low (<0.01 in the Yanomamo and <0.08 in the Xingu), and there was virtually no BP rise with age [7, 24]. A large meta-analysis of 32 randomized controlled trials (including a total of 2609 participants) between 1981 and 1995 showed that K+ supplementation (median of 75 mmol/day) caused a significant reduction in SBP by 3.11 mmHg and diastolic BP (DBP) by 1.97 mmHg [25]. The BP-lowering effect was higher when only studies of individuals on a high Na+ diet were included. Two other smaller meta-analyses also found stronger BP-lowering effects of higher dietary K+ intake in subjects with hypertension [26, 27]. The Dietary Approaches to Stop Hypertension (DASH)-Sodium study enrolled 412 participants, and showed that the DASH diet, containing ∼120 mmol/day K+, reduced BP [8]. This BP-lowering effect was more prominent in subjects consuming a high Na+ diet. The fact that the BP-lowering effects of K+ are more pronounced in individuals that consume a high Na+ diet suggests that K+ influences salt-sensitivity. This was further explored by Krishna et al. [28], who performed a randomized placebo-controlled cross-over study in 10 healthy males who were placed on a constant high Na+ diet (120–200 mmol/day) and a controlled low K+ diet (10 mmol/day). Participants were randomly supplemented with either placebo or 80 mmol of K+/day (via potassium chloride tablets) for 9 days, maintaining their dietary K+ intake at 10 or 90 mmol/day. Each individual was studied on both the low and higher K+ diets in 4–8 weeks apart. During the low K+ diet, urinary Na+ excretion decreased, Na+ balance became positive (396 ± 63 mmol over the 9-day period) and BP increased (mean arterial pressure increased from 90.9 ± 2.2 to 95.0 ± 2.2 mmHg). During K+ supplementation, urinary Na+ excretion was higher and BP decreased (mean arterial pressure decreased from 91.1 ± 1.8 to 88.9 ± 2.3 mmHg). Natriuresis occurred as soon as the participants started with K+ supplementation. Similar results were observed in 12 patients with essential hypertension [29]. More recently, Aburto et al. [12] conducted a meta-analysis of 21 randomized controlled trials (including a total of 1892 subjects) and found that increasing dietary K+ intake to a level of 90 mmol/day reduced SBP and DBP by 3.49 and 1.96 mmHg, respectively, in the overall population. When including only the hypertensive individuals or those on a high Na+ diet (>4 g Na+/day), the effects were even stronger: SBP and DBP were reduced by 5.32 and 3.10 mmHg in the hypertensive groups and by 6.91 and 2.87 mmHg in the high Na+ groups, respectively. Furthermore, increasing dietary K+ intake to 90–120 mmol/day was associated with a lower risk of stroke in 11 cohort studies (including a total of 127 038 participants). On the basis of this meta-analysis, the World Health Organization recommended a dietary K+ intake of at least 90 mmol/day [11]. The Institute of Medicine recommends a dietary K+ intake of 120 mmol/day [30]. This is based mostly on the same studies, but also taking into account the amount of K+ in the DASH diet (120 mmol/day) [31]. Recently, the Prospective Urban Rural Epidemiology (PURE) study, a cohort study of 102 216 people throughout the world, estimated dietary K+ intake on the basis of spot urine K+ excretion, and found that daily K+ intake modified the effect of Na+ intake on BP [3]. The researchers divided their participants into three levels of urinary Na+ excretion (>5 g/day; 3–5 g/day; <3 g/day), and found that within each group, higher urinary K+ excretion was associated with lower BP. Again, the modifying effect of K+ on BP reduction was most pronounced in the individuals with hypertension, as well as in the elderly. Of note, the association between urinary K+ and BP extended to relevant clinical outcomes. PURE showed that a higher urinary K+ excretion was associated with a lower risk of death or cardiovascular events, and this association remained significant after correction for physical activity, dietary factors and BP [13]. These and other studies on the influence of K+ on cardiovascular and kidney outcomes will be discussed in more detail in this review. In the next section, we will first review current understanding of Na+ and K+ handling in the distal nephron and how this pertains to the BP-lowering effects of K+.
DISTAL NEPHRON SODIUM AND POTASSIUM HANDLING
Potassium secretion in the distal nephron
The three key factors that regulate K+ secretion in the aldosterone-sensitive distal nephron (ASDN) are [1] aldosterone, [2] Na+ delivery and [3] tubular fluid flow rate [32] (Figure 1). The ASDN comprises the late distal convoluted tubule (also called DCT2), the connecting tubule (CNT) and the cortical collecting duct (CCD). The major apical Na+ transporter for electrogenic Na+ reabsorption along the ASDN is the epithelial sodium channel (ENaC), which is expressed in principal cells of the DCT2, CNT and CCD, and is upregulated by aldosterone [33]. Electrogenic movement of Na+ through ENaC generates a lumen-negative transepithelial voltage that drives K+ secretion through the renal outer medullary K+ (ROMK) channel. In addition to ROMK channels, the ASDN also expresses big-K+ channels (BK, or maxi-K+ channels), which play an important role in flow-dependent K+ secretion [34]. Therefore, the ASDN is able to switch between electroneutral Na+ reabsorption [by the sodium-chloride cotransporter (NCC)] to electrogenic Na+ reabsorption (by ENaC). Accordingly, it is equipped to change urinary K+ and Na+ excretion in response to variations in dietary K+ and Na+ intake. The natriuresis upon K+ intake suggests a ‘potassium switch’ that regulates both K+ and Na+ handling by the kidney. Current insights indicate that this switch occurs in the DCT, which is upstream of the major K+-secreting segment of the nephron (Figure 1). The potassium switch regulates downstream delivery of Na+ to the ASDN.
FIGURE 1.
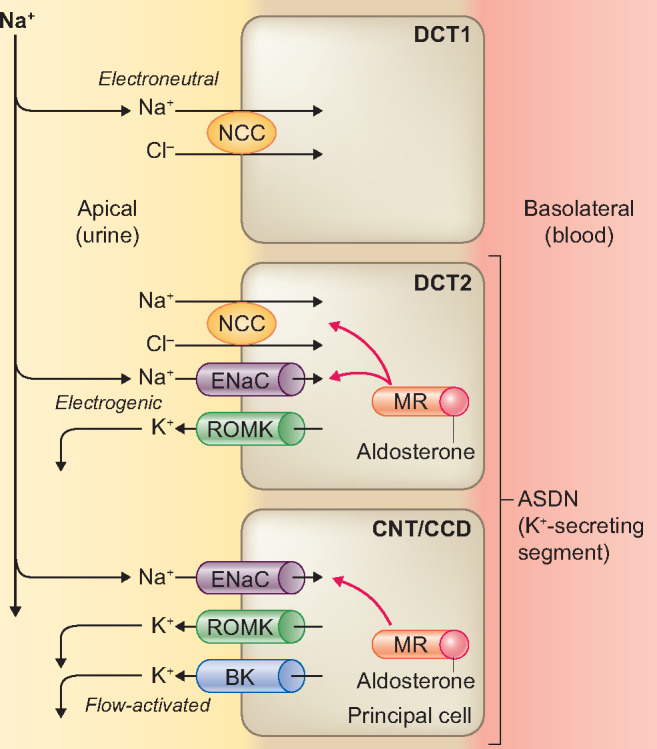
Overview of the main apical transport pathways contributing to Na+ reabsorption and K+ secretion in the ASDN. In the initial two-thirds of the DCT (DCT1), the apical NCC is the major Na+ transporter, which mediates electroneutral Na+ reabsorption. In the final one-third of the DCT (DCT2), NCC is co-expressed with the ENaC and ROMK. K+ secretion progressively increases in the principal cells of DCT2, CNT and CCD that express ENaC and ROMK. Na+ reabsorption by ENaC is electrogenic and is regulated by aldosterone. Na+ reabsorption through ENaC generates a lumen-negative transepithelial voltage that drives K+ secretion via ROMK. BK channels mediate flow-dependent K+ secretion in the collecting duct. The three major factors that promote K+ secretion are (i) Na+ delivery to CNT/CCD, (ii) tubular flow rate and (iii) aldosterone.
Role of the DCT
Recent insights suggest that the DCT, upstream of the ASDN, acts as a K+ sensor and affects downstream K+ handling by regulating Na+ delivery [35–37]. The electroneutral apical Na+ transporter in the DCT is the NCC (gene symbol SLC12A3) [38, 39], which plays an important role in fine-tuning Na+ delivery downstream to the ASDN. The functional relevance of the NCC for the handling of Na+ and K+ homoeostasis as well as BP is illustrated by two tubulopathies that affect NCC activity, including Gitelman syndrome and Gordon syndrome [40, 41]. Gitelman syndrome is caused by loss-of-function of NCC due to inactivating mutations in SLC12A3. Patients with Gitelman syndrome present with urinary Na+ wasting, hypokalaemia and low-to-normal BP. Conversely, Gordon syndrome, also called familial hyperkalaemic hypertension or pseudohypoaldosteronism Type II, is caused by gain-of-function of NCC due to mutations in genes encoding regulatory proteins of NCC [40, 42–44]. Gordon syndrome patients present with the mirror image of Gitelman syndrome, including salt-sensitive hypertension and hyperkalaemia. NCC is activated by phosphorylation and inactivated by dephosphorylation [45, 46]. Several experimental studies have demonstrated that a low K+ diet increases total and phosphorylated NCC (pNCC) [36, 47–52], resulting in decreased natriuresis, decreased kaliuresis and elevated BP. This can be reversed by administration of a thiazide diuretic, further supporting a role of NCC [36, 48]. Conversely, acute K+ loading rapidly dephosphorylates NCC, regardless of whether K+ is administered in a high K+ diet [53, 54], through oral gavage [55, 56] or by intravenous infusion [57, 58]. The K+ loading is followed by increased natriuresis and kaliuresis occurring as soon as 30–60 min. Chronic dietary K+ loading reduces phosphorylated, total and surface NCC abundance [47, 59–63]. Pooled data from different rat and mouse experiments showed that pNCC levels are inversely correlated with plasma K+ within the physiological range [54, 64]. These data indicate that NCC is regulated in response to variations in plasma K+. High plasma K+ increases Na+ delivery to the ASDN and low plasma K+ decreases Na+ delivery (Figure 2). The K+ sensor in the DCT and K+ secretion in ASDN work in concert in response to dietary K+. A high K+ diet dephosphorylates NCC, thereby reducing its activity and inhibiting electroneutral Na+ reabsorption. In other words, K+ loading has a thiazide-like effect. This increases Na+ delivery to the downstream ASDN for electrogenic Na+ reabsorption by ENaC, causing an electrochemical gradient that drives K+ secretion through ROMK. Increasing tubular flow also stimulates flow-dependent K+ secretion through BK (Figure 2). The physiological consequences are natriuresis and kaliuresis. Conversely, low dietary K+ induces NCC phosphorylation, resulting in electroneutral Na+ reabsorption through NCC. This will reduce distal Na+ delivery and prevent Na+ reabsorption through ENaC and K+ secretion through ROMK. The physiological consequences are Na+ retention and K+ conservation (Figure 2).
FIGURE 2.

Model of how the DCT and ASDN work in concert in response to a low or high K+ diet. (A) A low K+ diet leads to phosphorylation (activation) of the NCC in the DCT, resulting in increased electroneutral Na+ reabsorption through NCC. This will reduce Na+ delivery to the ASDN and inhibit electrogenic Na+ reabsorption through the ENaC and K+ secretion through ROMK. (B) A high K+ diet leads to dephosphorylation (inactivation) of NCC, thereby reducing electroneutral Na+ reabsorption. This increases Na+ delivery to the ASDN for electrogenic Na+ reabsorption through ENaC and drives K+ secretion through ROMK.
Prioritizing between sodium and potassium
In addition to dietary K+, dietary Na+ also regulates NCC phosphorylation. NCC phosphorylation is suppressed by a high Na+ diet and is promoted by a low Na+ diet [65]. A relevant question is what happens when there is a high Na+ diet and a low K+ diet, characteristic of our current diet. Terker et al. [36] showed that NCC is activated by a low K+ diet despite a high Na+ diet and that this results in Na+ retention and a rise in BP. Conversely, of interest is to analyse which signal will dominate in the setting of a low Na+ and high K+ diet (the ‘Paleolithic’ diet). Previously, we combined a low Na+ and high K+ diet and showed that there was still a decreased NCC abundance despite a low Na+ diet and maximal activation of the renin–angiotensin system [61]. The overall result was that potassium-induced natriuresis was maintained, even in the context of a low Na+ diet. This also illustrates that the effect of high K+ can override the NCC stimulating effects of angiotensin II and aldosterone [66]. In agreement, Jensen et al. found that acute K+ loading induced NCC dephosphorylation and natriuresis in mice on a high, normal or low Na+ diet [53]. Taken together, these studies support that K+ homoeostasis is prioritized over Na+ and volume regulation.
LOW POTASSIUM DIET ACTIVATES NCC
When consuming a high Na+ and low K+ diet, the kidneys prioritize conservation of K+ over excretion of Na+ and switch NCC ‘on’. During K+ deficiency, this mechanism serves well to inhibit K+ excretion. However, a long-term low K+ diet can cause chronic Na+ reabsorption by upregulating NCC with subsequent plasma volume expansion and hypertension. Therefore, low K+ diet-induced Na+ reabsorption through NCC may be a key driver in the pathogenesis of salt-sensitive hypertension. Recently, several studies have provided insights in the molecular pathways that increase NCC activity during low K+ diet (Figure 3). In the basolateral plasma membrane of DCT cells, potassium conductance is mediated by Kir4.1 (encoded by KCNJ10) and Kir5.1 (encoded by KCNJ16) [67–69]. These K+ channels form a heterotetramer (Kir 4.1/5.1) which is responsive to plasma K+ and initiates a signalling cascade that results in the phosphorylation of NCC [70–72]. A low extracellular K+ concentration stimulates efflux of K+ through Kir4.1/5.1, leading to membrane hyperpolarization. This leads to chloride efflux through a basolateral voltage-gated Cl− channel (ClC-Kb), resulting in a reduction in the intracellular Cl− concentration [73–75]. ClC-Kb requires the beta-subunit barttin for its function [76]. Disruption of any of these transporters blocks NCC phosphorylation. The importance of this pathway has been supported by in vitro and in vivo studies [36, 70, 73, 77–79]. For example, NCC phosphorylation by low K+ was blocked in KCNJ10 knockout mice [70, 78], in ClC-K2 (a murine ortholog of human ClC-Kb) knockout mice [73], and in barttin hypomorphic mice [79]. In humans, loss-of-function mutations in the KCNJ10 gene cause a syndrome with a Gitelman-like phenotype [80]. Intracellular chloride normally inhibits chloride-sensitive kinases called with-no-lysine (K) kinases (WNKs) by binding to their catalytic domain, and thereby blocking their autophosphorylation (Figure 3) [81]. When intracellular chloride is reduced, this inhibition is relieved and WNKs are able to autophosphorylate [82]. Phosphorylated WNKs activate the intermediary kinases Ste-20-related proline alanine-rich protein kinase (SPAK) and oxidative stress-responsive kinase 1 (OSR1), which activate NCC [36]. Animal studies from several groups have implicated and supported the importance of the intracellular chloride-WNK-SPAK/OSR1-pNCC pathway in the response to a low K+ diet by studying wild-type or genetically altered mice [36, 48–52, 83, 84]. Multiple WNKs exist, but WNK4 appears to be the dominant form in the regulation of NCC. For example, Cl− inhibits WNK4 kinase activity at lower concentrations than it inhibits other WNKs and these concentrations are analogous to those in the human DCT [64]. Furthermore, WNK4 knockout mice on a low K+ diet failed to increase pNCC [49, 52], whereas WNK1 or WNK3 siRNA knockdown mice still had increased phosphorylation of SPAK/OSR1 and NCC in response to a low K+ diet [36]. The role of WNK4 in Cl− sensing in vivo was further supported by knock-in mice carrying Cl−-insensitive mutant WNK4 (WNK4L319F/L321F), which failed to increase total and pNCC in response to dietary K+ restriction [84]. A study in Drosophila kidney tubule also found that this chloride-WNK4 sensing mechanism requires the kinase scaffolding protein Mo25 [85, 86]. Taken together, the effect of a low K+ diet on NCC activity is explained by the relationship between Kir4.1/5.1, membrane voltage, intracellular chloride and WNK4-SPAK/OSR1 activation (Figure 3). Furthermore, ubiquitin ligases are also involved in this pathway. For example, Kelch-like 3 (KLHL3) normally degrades WNK4. A low K+ diet increased phosphorylated (inactivated) KLHL3, and this abolished its activity to degrade WNK4, ultimately leading to increased WNK4 activity [87]. However, other signals or additional mechanisms may be present, because in SPAK knockout mice, a low K+ diet was still able to phosphorylate NCC [83], and SPAK/kidney-specific OSR1 double knockout mice on a low K+ diet had variable results with either blunted [36] or absent [51] phosphorylation of NCC.
FIGURE 3.
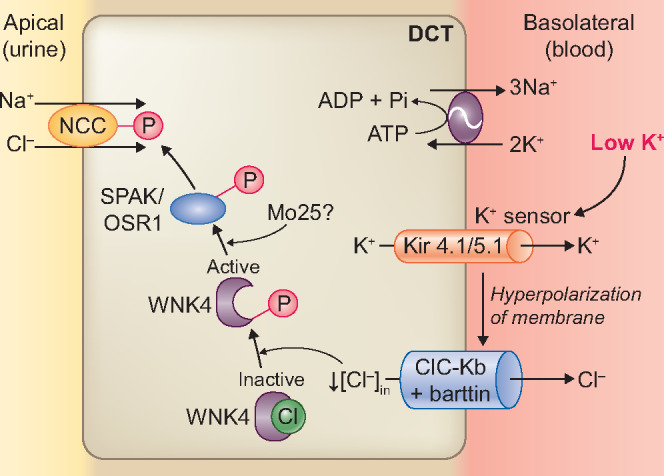
Molecular pathways involved in the effects of a low K+ diet on the NCC. A low extracellular K+ concentration is sensed by the K+ channel Kir 4.1/5.1, resulting in efflux of K+ through Kir4.1/5.1. This leads to membrane hyperpolarization and chloride (Cl−) efflux through a basolateral voltage-gated Cl−channel (ClC-Kb/barttin). A reduction in intracellular Cl− concentration relieves the inhibition of WNK4 autophosphorylation. In turn, phosphorylated WNK4 activates SPAK–OSR1, which phosphorylates NCC. In Drosophila melanogaster kidney tubules, WNK4 phosphorylation of SPAK–OSR1 depends on the scaffold protein Mo25.
HIGH POTASSIUM DIET AND NATRIURESIS
Acute potassium-induced natriuresis
Extracellular K+ is a small fraction of total body K+. In order to prevent large fluctuations in extracellular K+ after an acute K+ load, our body is equipped with efficient mechanisms to respond to changes in extracellular K+, namely, by translocating K+ into cells (‘shift’) and by K+ excretion via the kidneys or the gut. These regulations maintain extracellular K+ within a safe range (between 3.5 and 5.5 mmol/L) [32]. In the kidneys, urinary K+ excretion is regulated by aldosterone. A rise in plasma K+ stimulates aldosterone secretion, which acts through the mineralocorticoid receptor to regulate the transcription of multiple genes that control ENaC activity [33, 88, 89]. However, this response occurs relatively late in the kidneys because it involves genomic effects. The kaliuretic effect of dietary K+ intake precedes the increase in plasma aldosterone and is accompanied by natriuresis [90–92]. Recent insights suggest that this aldosterone-independent mechanism is mediated by acute NCC dephosphorylation in the DCT. NCC inhibition secondary to a high K+ load induces natriuresis that serves to facilitate kaliuresis. This phenomenon is called K+-induced natriuresis. Vallon et al. [47] were one of the first to report a reduction of NCC abundance and pNCC in mice on a high K+ diet despite an increase in plasma aldosterone. This indicates that K+ can dissociate the stimulatory effect of aldosterone on NCC [93–95]. Sorensen et al. further characterized that an acute K+ load dephosphorylated NCC within 15 min and linked this to the natriuresis and kaliuresis starting 30–60 min after the K+ load. While the kaliuretic response persists ∼6 h, the natriuretic response declines earlier (after ∼3 h). This decline in natriuresis was likely caused by upregulation of ENaC. Compared to wild-type mice, the K+-induced early natriuretic effect was blunted in NCC knockout mice, indicating that acute K+-induced natriuresis primarily depends on a functional downregulation of NCC [55]. This study also supports that K+-induced NCC dephosphorylation is aldosterone-independent because K+-loading suppressed NCC phosphorylation in mice lacking the enzyme aldosterone synthase similarly to wild-type mice [55]. Jensen et al. found that urinary K+ excretion rates were attenuated when ENaC was downregulated by a high Na+ diet or pharmacologically blocked by ENaC inhibition with benzamil [53]. Thus, this study further supports that acute K+-induced kaliuresis is ENaC dependent. Interestingly, Hunter et al. showed that acute NCC inhibition by thiazide diuretics does not always result in kaliuresis [96]. Thus, the effect of thiazide diuretics is not completely similar to the effect of an acute K+ load. Simply increasing distal Na+ delivery by NCC inhibition may not be sufficient for the rapid effect that is observed after acute K+ loading.
Potassium inactivates NCC
The pathways linking the effect of a high K+ diet to reduced NCC activity are subject of ongoing research. Veiras et al. [97] showed that acute K+ ingestion failed to dephosphorylate NCC in angiotensin II-infused rats that exhibited hypokalaemia secondary to angiotensin II-mediated ENaC upregulation. Normalizing plasma K+ by K+ supplementation relieved this. This suggests that plasma K+ acts as the predominant driver of NCC activity. In contrast to a low K+ diet, the effect of a high K+ diet on NCC activity does not only depend on intracellular chloride. Penton et al. [58] used kidney slices to test the effect of extracellular K+ on NCC. This experiment supported the previous observation that the effect of low extracellular K+ on NCC depends on intracellular chloride. However, rapid dephosphorylation of NCC by high extracellular K+ was neither blocked in the presence of a Cl− channel blocker nor by low extracellular Cl−, and NCC dephosphorylation occurred in the absence of significant changes in SPAK/OSR1 phosphorylation. This suggests that NCC dephosphorylation by high K+ may occur independent of changes in intracellular chloride. Because of the rapidity of the response, Penton et al. [58] hypothesized that activation of protein phosphatases (PPs) may mediate NCC dephosphorylation in response to high extracellular K+ (Figure 4). Shoda et al. [56] further found that an acute K+ load acutely dephosphorylated NCC in mice independently of the WNK-SPAK/OSR1 cascade. This dephosphorylation of NCC was prevented by a calmodulin inhibitor or a calcineurin inhibitor, suggesting that the effect of high K+ on the dephosphorylation of NCC is mediated by the calcium-binding protein calmodulin and its downstream PPs such as calcineurin. Whether a high K+ load increases intracellular calcium and increases the activity of calmodulin and calcineurin requires further study. Studies by Chen et al. [84] and Ishizawa et al. [98] do support a role for the WNK4-SPAK/OSR1 pathway in modifying NCC phosphorylation after a K+ load (Figure 4). Chen et al. [84] found different responses in acute and chronic K+ loading in Cl−-insensitive WNK4 knock-in mice. Acute K+ loading through oral gavage increased plasma K+ and decreased pNCC in 30 min in wild-type mice, the decrease in pNCC was not observed in WNK4 knock-in mice. However, chronic K+ loading deactivated NCC in both wild-type and WNK4 knock-in mice, indicating mechanisms other than Cl−-sensing by WNK4. Whether this is associated with activation of PPs was not reported in this study. Ishizawa et al. [98] showed that high extracellular K+ in HEK cells activated calcineurin-mediated dephosphorylation of KLHL3 at serine 433, which in turn decreased WNK4 activity by its ubiquitination to WNK4 and thus limiting NCC phosphorylation. In addition to the downregulation of NCC, experimental studies and mathematical modelling have shown that high K+ also reduces Na+ reabsorption in the proximal tubule [62, 63, 99, 100] and thick ascending limb [62, 101–105]. All these processes serve to enhance Na+ delivery to the ASDN, where Na+ reabsorption leads to K+ secretion. A recent finding in vivo and in CCD cells showed that K+ directly influences K+ secretion via ENaC stimulation in the principal cells, independent of plasma aldosterone [106]. This was mediated via basolateral Kir4.1 channels leading to possible changes in intracellular chloride similar to the effects in the DCT. Subsequently, WNK1 stimulates the Type 2 mammalian target of rapamycin (mTOR) complex and serum and glucocorticoid inducible kinase 1 (SGK1), resulting in ENaC activation and thereby K+ secretion. This mechanism acts in concert with aldosterone-dependent mechanisms for K+ excretion [106].
FIGURE 4.
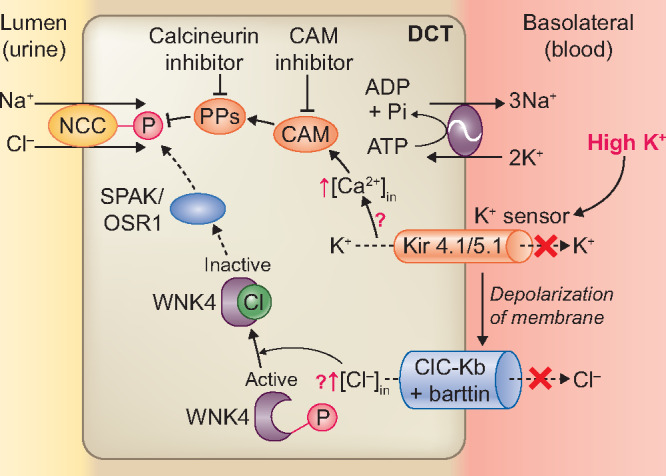
Molecular pathways involved in the effects of a high K+ diet on the NCC. High extracellular K+ concentration is sensed by K+ channel Kir4.1/5.1 and causes membrane depolarization. This may lead to an increase in intracellular calcium (Ca2+) through unknown mechanisms. Ca2+ stimulates calcium-binding protein calmodulin (CaM) and downstream PPs such as PP3 (calcineurin). This dephosphorylates NCC. In acute K+ loading, mechanisms that depend on intracellular chloride ([Cl−]in) may also be stimulated through effects dependent on the K+ channel Kir4.1/5.1. An increase in [Cl−]in may inhibit WNK4 autophosphorylation. This would prevent SPAK–OSR1 phosphorylation and ultimately, NCC phosphorylation.
Chronic adaptation to a high potassium diet
Chronically, a high K+ diet intake leads to compensatory changes that attenuate the initial natriuresis and help to preserve Na+ balance. This decline in natriuresis is ENaC dependent and could be partially explained by the delay in reaching the peak levels of aldosterone after an acute K+ load [55, 107]. Interestingly, NCC knockout mice exhibit a blunted K+-induced early natriuretic response compared with wild-type mice [55]. Structural adaptation was observed in mouse models of persistent NCC inhibition or activation. For example, Grimm et al. identified the activation of an α-ketoglutarate paracrine signalling system and distal nephron remodelling process in SPAK-deficient mice (a model for persistent NCC inhibition) [108]. Both mechanisms act together to induce Na+ reabsorption in the CNT and CCD to compensate for the loss of NCC function. In constitutively active SPAK mice (a model for persistent NCC activation), the same investigators found delayed restoration of urinary K+ excretion and plasma K+ to the levels of wild-type mice upon thiazide treatment within 3 days [109]. They concluded that NCC hyperactivity drives the cellular remodelling in the ASDN with a reduction in CNT mass and attenuation in ENaC and ROMK, whereas NCC inhibition could reverse this remodelling. Therefore, during chronic K+ loading, NCC was persistently inhibited and compensatory mechanisms were activated. This adaptation attenuates K+-induced natriuresis by an acute K+ load [110]. Evaluating cellular remodelling of nephron segments in other genetically modified animals (e.g. WNK4 knockout, Cl−-insensitive WNK4 knock-in mice or ENaC knockout mice) may explain alterations in Na+ and K+ handling in acute and chronic responses to dietary K+ [86].
GUT–KIDNEY KALIURETIC SIGNALLING
How K+ is ‘sensed’ remains an unanswered question, as well as how the signal is conveyed from the gut to the kidney and whether this depends on plasma K+ (Figure 5). Rabinowitz et al. [111] challenged the traditional view of feedback control that is that high plasma K+ during a high K+ diet is the major factor to trigger aldosterone-mediated transcellular K+ shift and kaliuresis. In studies in sheep, they found that meal-induced kaliuresis was not accompanied by a change in plasma aldosterone and only subtle changes in plasma K+. The authors proposed the idea of feedforward control by proposing that kaliuresis is initiated before the rise in plasma K+, by a mechanism controlled by K+ sensing in the gut. Lee et al. [112] further provided evidence for this ‘gut factor’ in rats by finding that intra-gastric K+ infusion with meal provokes a significant increase in urinary K+ excretion without a rise in plasma K+, while intra-portal and systemic K+ infusion do increase plasma K+. Complementing these experimental data, Preston et al. [113] conducted a physiological study in healthy human volunteers by comparing the kaliuretic effect in three conditions: oral K+ load only, K+-deficient meal only, or an oral K+ load plus a K+-deficient meal. Results showed that a significant increase in kaliuresis, without changes in plasma K+, was observed in the group with K+ plus meal. This kaliuresis remained intact following pharmacological blockade of the mineralocorticoid receptor with eplerenone, supporting the existence of a gut–kidney kaliuretic signalling that regulates urinary K+ excretion by a feedforward mechanism, independently of plasma K+ and plasma aldosterone. Although these studies support a ‘gut factor’, another possibility to explain feedforward control could be that a minor rise in the K+ concentration in peritubular capillaries by dietary K+ may be sufficient to mediate NCC inhibition [114]. Possible signalling molecules for the ‘gut factor’ remain to be determined. Recent studies provide new insights into gut–kidney cross-talk by showing that gut microbiota-derived short chain fatty acids can affect BP and kidney function (Figure 5). The G protein-coupled receptor Gpr41 and olfactory receptor Olfr78 in the afferent arteriole of the kidney respond to these short chain fatty acids to regulate BP via vascular resistance or renin secretion [115, 116]. Similarly, dietary sodium has been shown to influence BP regulation through direct effects on the microbiome [117]. In summary, although the ‘gut factor’ remains unclear, the physiological implication would be that both feedback control and feedforward control work in concert to maintain K+ and Na+ homoeostasis.
FIGURE 5.

Working hypothesis of how dietary K+ may confer cardiovascular and kidney protection. The effects of dietary K+ may either be relayed through its effect on the microbiome and its metabolites (gut–vessel and gut–kidney signalling) or through plasma K+. The protective effects of K+ can be mediated both through BP-dependent and -independent mechanisms.
DIETARY POTASSIUM AND BP
Besides NCC regulation, dietary K+ may also exert effects on blood vessels, nerves and the intra-kidney renin–angiotensin system that may contribute to BP effects (Figure 5). Studies have shown that K+ depletion activates renin, angiotensin II and endothelin-1 in the kidney independent of the systemic renin–angiotensin system. The activated intra-kidney renin–angiotensin system is associated with structural changes in the kidney and salt-sensitive hypertension [118–120]. For example, Suga et al. [118] showed that rats made hypokalaemic by a low K+ diet exhibited tubulointerstitial injury, hypoxia and an imbalance in local vasoactive mediators that favours vasoconstriction. There was evidence for upregulation of angiotensin-converting enzyme at sites of tubulointerstitial injury, increased cortical-to-plasma angiotensin II ratio, increased endothelin-1 in both the cortex and the medulla, reduced urinary prostaglandin E2, and a decrease in kallikrein. Moreover, these rats demonstrated salt-sensitive hypertension that was not corrected after normalizing plasma K+. Beneficial effects of K+ are also mediated through neural effects. For example, K+ may regulate Na+ excretion and BP by modulating the activity of the kidney’s sympathetic nervous system (SNS). Fujita and Sato [121] evaluated the effect of K+ supplementation on the kidney’s SNS in rats treated with deoxycorticosterone acetate (DOCA), a model of salt-sensitive hypertension. They found that the norepinephrine turnover rate in the kidney was significantly increased in DOCA-salt rats, and that K+-supplemented DOCA-salt rats decreased but not completely inhibited this effect. K+-supplemented DOCA-salt rats also showed attenuated Na+ retention and an antihypertensive response. These results confirmed previous reports showing that kidney SNS activation impairs natriuresis, and that kidney denervation increases urinary Na+ excretion [122, 123]. Studies also support that kidney SNS activation is an important factor influencing salt-sensitive hypertension in obese animal models [124–126]. Mu et al. [127] linked SNS activity and salt-sensitive hypertension to NCC activation in the DCT, which was mediated by stimulation of β1-adrenergic receptors. The β1-adrenergic stimulation of NCC possibly involves the WNK4–OSR1 pathway [128] and the Kir4.1 channel in the DCT [129]. A recent study showed that β1-adrenergic stimulation of NCC involves PP1 inhibitor-1 (I-1), which is an endogenous inhibitor of PP1. Stimulation of β1-adrenergic receptors results in changes in cyclic adenosine monophosphate (cAMP) and stimulates a protein kinase A-dependent phosphorylation of I-1, which in turn inhibits PP1-dependent NCC dephosphorylation [130]. K+ supplementation may decrease but not completely inhibit this pathway through decreasing SNS activity. Aside from modifying the activity of the SNS in the kidney, Zicha et al. [131] found that a high K+ diet may attenuate sympathetic vasoconstriction and result in an antihypertensive effect in immature salt-sensitive Dahl rats; however, this mechanism is absent in adult salt-loaded rats and in rats with established salt-sensitive hypertension. Similar findings were reported by Dietz et al. [132] in stroke-prone spontaneously hypertensive rats. They showed that a high K+ diet partially reversed salt-induced changes in noradrenaline metabolism, resulting in improved neuronal uptake of noradrenaline, attenuated sensitivity of vascular smooth muscle to noradrenaline and reduced release of noradrenaline into the plasma. All these changes attenuate sympathetic vasoconstriction. Direct effects of K+ on vascular tone may be mediated through endothelium-dependent vasodilation. In patients with essential hypertension, local K+ infusion facilitates endothelium-dependent vasodilation induced by acetylcholine. This effect appears to be mediated by nitric oxide production from endothelial cells and was independent of mean arterial BP [133]. This finding is consistent with experimental data. For instance, aortic endothelium-dependent relaxation was reduced in salt-sensitive Dahl rats, whereas a high K+ diet significantly enhanced endothelium-dependent relaxation to acetylcholine independent of changes in BP [134]. Zhou et al. [135] also showed that K+ supplementation improves endothelium-dependent relaxation by increasing endothelial nitric oxide in the carotid arteries of salt-loaded Dahl salt-sensitive rats. K+ can also influence endothelial cell structure. For example, in various studies, Oberleithner et al. [136–138] showed that high extracellular K+ significantly reduces the stiffness of vascular endothelial cells by changing endothelial cell structure and increasing the release of nitric oxide, whereas high extracellular Na+ and aldosterone prevented these changes. One clinical study also supported the notion that K+ has effects on vascular tone independent of the kidney. In this study, Dolson et al. [139] studied 11 anuric haemodialysis patients who initially received haemodialysis with a dialysate containing 2.0 mmol/L K+ and were then randomly allocated to groups with a dialysate K+ of either 3.0 or 1.0 mmol/L for at least 1 month. All groups showed a reduction in BP during haemodialysis because of fluid removal. However, patients with a 1.0 mmol/L K+ dialysate showed significantly increased BP 1-h post-dialysis, whereas patients with a 3.0 mmol/L K+ dialysate did not exhibit this ‘rebound hypertension’.
DIETARY POTASSIUM AND CARDIOVASCULAR RISK
A high Na+ and low K+ diet contribute to hypertension, which increases the burden of cardiovascular and kidney disease. K+ supplementation is proposed to block this vicious cycle by BP reduction via natriuresis, attenuation of SNS activity or direct vascular effects, which in turn, improve cardiovascular and kidney health (Figure 5). A high K+ diet may also provide BP-independent protective effects such as anti-inflammatory, antifibrotic and antioxidant effects, improvement of endothelial function and prevention of atherosclerosis [140]. The beneficial effects of increasing dietary K+ intake on BP and cardiovascular outcomes are becoming increasingly evident from epidemiological, clinical and experimental studies.
Epidemiological studies
Higher dietary K+ intake is associated with reduced BP and a lower risk of stroke. However, Aburto et al. [12] found no significant associations with incident cardiovascular disease. This may be attributable to the lack of adequately powered randomized trials. Subsequent data from the large-scale PURE study did show inverse associations between urinary K+ excretion, major cardiovascular events and mortality. Compared with a urinary K+ excretion <1.5 g/day, individuals with higher urinary K+ excretion had a decreased risk of cardiovascular events and death [13]. A more recent analysis of the PURE cohort showed that although Na+ intake has a positive association with BP across communities, the association between dietary Na+ intake and cardiovascular events revealed a J-shaped relationship. There was a linear association between dietary K+ intake and cardiovascular outcomes [14]. This supports the role of dietary K+ in cardiovascular protection but also indicates that the effects of dietary Na+ and K+ on cardiovascular events are complex and cannot be explained solely by the BP-lowering effects.
Clinical trials
The potential cardioprotective effects of high K+ intake are also supported by clinical intervention studies. For example, a long-term intervention study in Taiwan analysed the effect of K+-enriched salt on cardiovascular mortality. Five kitchens were randomized to prepare meals with K+-enriched salt (experimental group, n = 768) or regular salt (control group, n = 1213). After a follow-up of 31 months, the incidence of cardiovascular deaths reduced by nearly 40% and the life expectancy was longer among participants in the experimental group [141]. These effects may be primarily attributable to K+ because the increase in urinary K+-to-creatinine ratio was greater than the decrease in urinary Na+-to-creatinine ratio (76 versus 17%, respectively). However, it is unclear whether this cardiovascular benefit was dependent on BP due to a lack of BP measurements. In a recent population-level salt-substitution study in Peru (involving a total of 2376 individuals from six villages), regular table salt (sodium chloride) was replaced by K+-enriched salt (25% potassium chloride) using a stepped-wedge cluster randomized trial design [142]. This intervention decreased SBP and DBP by 1.29 mmHg and 0.76 mmHg, respectively. Again, the BP-lowering effect was most pronounced in subjects with hypertension and in older individuals. In addition, the incidence of hypertension was reduced by 51% among participants without hypertension at baseline. This BP-lowering effect was likely attributable to K+, because of higher 24-h urinary K+ and similar Na+ excretion at the end of the intervention. Currently, a large-scale cluster-randomized controlled trial in 600 villages (20 996 participants at high cardiovascular risk) in northern rural mainland China is investigating the effect of long-term potassium-salt substitution (70% sodium chloride and 30% potassium chloride) on cardiovascular outcomes (follow-up of 5 years). In this study, the primary outcomes are fatal and non-fatal stroke and the secondary outcomes are total major vascular events (non-fatal stroke, non-fatal acute coronary syndrome or death from cardiovascular or cerebrovascular causes) and total mortality [143]. Pending these results, potassium-enriched salt substitutes are increasingly recognized as a potential means of lowering BP at population scale [144]. There is also evidence that dietary K+ can exert BP-independent effects on vascular function. He et al. [145] conducted a placebo-controlled crossover trial comparing K+ chloride with K+ bicarbonate in 42 pre-hypertensive individuals. Compared with placebo, both K+ chloride and K+ bicarbonate had significant beneficial effects on cardiovascular parameters (improved endothelial function, increased arterial compliance, decreased left ventricular mass and improved left ventricular diastolic function) despite the fact that the BP-lowering effect was modest during the K+ treatment (possibly because they consumed a relatively low Na+-high K+ diet at baseline). A recent meta-analysis explored which vascular functions are improved by dietary K+ supplementation and found that this was primarily the case for pulse pressure [146]. Overall, the cardiovascular benefits of dietary K+ are likely attributable to both BP-dependent and BP-independent effects (Figure 5).
Experimental studies
Experiments in stroke-prone spontaneously hypertensive rats showed that a high K+ diet reduces the size of cerebral infarcts, decreases vessel wall thickness and increases vascular compliance independently of changes in BP [147]. Several studies have shown that K+ supplementation may attenuate inflammation and oxidative stress. For example, in salt-loaded Dahl salt-sensitive rats, K+ supplementation attenuated salt-induced acceleration of neointima proliferation, adventitial macrophage infiltration and generation of reactive oxygen species in cuffed arteries despite the small reduction in BP. This indicates a potential vascular protective effect of K+ via its antioxidant activities [148]. Sun et al. linked dietary K+ intake to vascular smooth muscle cell calcification. In an atherogenic animal model and an ex vivo tissue model, low K+ induced differentiation and calcification of vascular smooth muscle cells and promoted vascular calcification, whereas a high K+ inhibited these responses [149]. Compared with low K+-fed mice, a high K+ diet concurrently reduced aortic pulse wave velocity, which is an indicator of arterial stiffness.
DIETARY POTASSIUM AND CKD
The close relationship between BP, cardiovascular disease and kidney disease has led to the hypothesis that dietary K+ may also protect the kidney. Indeed, such effects are being supported by emerging evidence both from epidemiological studies in humans and experimental data from animal models.
Epidemiological studies
Several observational studies have analysed the association between urinary K+ excretion and kidney outcomes (Figure 6). These studies are heterogeneous in terms of baseline kidney function and method to assess urinary K+ excretion. Below we will discuss these studies; they were also included in a systematic review [150]. Three studies were conducted in populations with preserved kidney function [estimated glomerular filtration rate (eGFR) >60 mL/min/1.73 m2]. First, Araki et al. [151] studied the association of a single baseline 24-h urine K+ excretion with the incidence of kidney and cardiovascular events in 623 Japanese patients with Type 2 diabetes. After a median follow-up period of 11 years, a urinary K+ excretion >1.72 g/day was associated with lower incidence of kidney replacement therapy or cardiovascular events and 50% decline in eGFR or progression to CKD Stage 4 compared with patients who had a urinary K+ excretion of <1.72 g/day. Furthermore, patients in the highest quartile of urinary K+ excretion (>2.9 g/day) had a significantly lower rate of annual eGFR decline. Secondly, Kieneker et al. analysed the association between two 24-h urine K+ excretions with the risk of CKD (defined as newly developed eGFR <60 mL/min/1.73 m2 or albuminuria >30 mg/24 h, or both) in the population-based Prevention of Renal and Vascular End-Stage Disease (PREVEND) cohort (5315 individuals) [152]. During a median follow-up of 10.3 years, they showed that each 21 mmol/day (1 SD) decrement in urinary K+ excretion was independently associated with a 16% higher risk of de novo CKD even after adjustment for confounders and other possible mediators. This association was more pronounced in individuals with hypertension. Thirdly, Olde Engberink et al. conducted a retrospective cohort study (involving a total of 541 patients) in the outpatient setting showing that higher urinary K+ excretion (>82 mmol/day based on two 24-h urine collections) was associated with a lower risk of 60% eGFR decline or start of kidney replacement therapy after a median follow-up time of 16 years [153]. Two studies were conducted in populations including patients with and without CKD. Smyth et al. performed a post hoc analysis of the Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET) and Telmisartan Randomised Assessment Study in ACE-Intolerant Subjects with Cardiovascular Disease (TRANSCEND) cohorts (involving a total of 28 879 participants with a mean eGFR of 68 mL/min/1.73 m2) and found that higher 24-h urinary K+ excretion (estimated from a fasting morning urine sample) was associated with a lower risk of eGFR decline, progression of proteinuria and initiation of dialysis [154]. Of interest, subgroup analysis showed a loss of this association in individuals with more severe CKD (eGFR <45 mL/min/1.73 m2). Nagata et al. [155] evaluated the association of 24-h urinary K+ excretion with kidney outcomes and death in Japanese patients with diabetes (involving a total of 1230 patients with eGFR >30 mL/min/1.73 m2) with a mean follow-up of 5.5 years. They found that higher 24-h urinary K+ excretion (2.0–3.0 g/day) was associated with a lower risk of 30% eGFR decline or death compared with patients with urinary K+ excretion <1.5 g/day. Similarly, a subgroup analysis of patients with eGFR between 30 and 60 mL/min/1.73 m2 showed a loss of statistical significance between urinary K+ excretion and outcomes. Two studies evaluated the association between spot urine Na+/K+ ratio with kidney function decline in the general population. Deriaz et al. conducted a longitudinal population-based study (including a total of 4141 adults with a mean baseline of eGFR 88 ± 15 mL/min/1.73 m2), and their results showed that a high urinary Na+ excretion and high urine Na+/K+ ratio was associated with faster kidney function decline; however, there was no significant association between urine K+ excretion and kidney outcomes [156]. Another study also analysed spot urine Na+/K+ (7063 participants, ∼15% CKD) and revealed a trend similar to that described by Deriaz et al. [156] but concluded that a single determination of the urinary Na+/K+ ratio does not have sufficient prognostic utility in assessing kidney function decline [157]. Three studies mainly focused on patients with CKD. Leonberg-Yoo et al. evaluated the association between time-updated average urinary K+ excretion, kidney failure (defined as start of kidney replacement therapy) and all-cause mortality in a post hoc analysis of the Modification of Diet in Renal Disease cohort [158]. These patients have advanced CKD (812 patients with CKD Stages G3–5, measured GFR 32.6 ± 12.0 mL/min/1.73 m2). The results showed an inverse association between urine K+ excretion and all-cause mortality, but there was no association with kidney failure. Kim et al. investigated the relationship between spot urinary K+-to-creatinine ratio and kidney outcomes in the Korean KNOW-CKD Study (including 1821 patients with CKD Stages G1–G5) [159]. During 5326 person-years of follow-up, the lowest quartile of urinary K+-to-creatinine ratio was associated with increased risk of CKD progression (>50% decrease in eGFR from baseline values and the onset of end-stage kidney disease). These results were also evident in a subgroup analysis in which only patients with an eGFR <45 mL/min/1.73 m2 were included. He et al. [160] conducted a prospective study and evaluated the association of 24-h urinary K+ excretion with kidney outcomes in the Chronic Renal Insufficiency Cohort (CRIC, including 3939 patients with eGFR 20–70 mL/min/1.73 m2). Surprisingly, their data demonstrated that high urinary K+ excretion was associated with a higher risk of incident end-stage kidney disease and halving of eGFR. Other studies focused more generally on the association between healthy dietary patterns (which are typically K+ rich) and kidney outcomes. A recent meta-analysis included 18 cohort studies of non-CKD populations and demonstrated that a healthy dietary pattern was associated with a lower risk of incident CKD, and a lower incidence of albuminuria, but not with a lower incidence of eGFR decline [161]. Another meta-analysis conducted by the same group showed that a plant-based dietary pattern among patients with established CKD was associated with lower mortality, but not with the risk of end-stage kidney disease [162]. A recent review also recommends plant-based diets for both primary and secondary prevention of CKD [163]. Some cohort studies used food frequency questionnaire to evaluate the association between dietary K+ intake and kidney outcomes [164–166]. For example, Mun et al. recently studied the association of dietary K+ with eGFR <60 mL/min/1.73 m2 and >15% decline in eGFR in Korean rural populations with CKD Stage 2 (involving a total of 5064 patients) [166]. Compared with the subjects in the lowest quartile of dietary K+ intake (<1236 mg/day), patients in the highest quartile of dietary K+ intake (>2323 mg/day) had a 40% lower risk of CKD development and patients in all quartiles had a 28–46% lower risk of eGFR decline during the follow-up period of 47 months. However, this association was only statistically significant in subjects with hypertension. Two other studies also showed that higher K+ intake is associated with better kidney outcomes [159, 165], while another study showed no association [164]. Taken together, the majority of these observational studies demonstrate that diets rich in K+ were associated with better kidney outcomes in the overall population or patients with early-stage CKD (Figure 6) [151, 152, 154, 155, 165, 166].
FIGURE 6.

Overview of cohort studies that analysed the association between urinary K+ or urine Na+/K+ (as proxy for dietary intake) and kidney outcomes. The average baseline eGFR is shown for each cohort.
Experimental studies
In rats, a K+-deficient diet leads to tubulointerstitial injury with macrophage infiltration and interstitial collagen deposition, development of salt-sensitive hypertension and decline in kidney function [118, 167]. K+ depletion also caused proximal tubular hypertrophy and a marked increase in angiotensin II Type 1 receptor density in healthy rats and those subjected to subtotal nephrectomy. These changes may explain the mechanisms of the deleterious effects of K+ deficiency on the kidney [168]. Conversely, in prehypertensive Dahl salt-sensitive rats, adding K+ to high salt diets reduced progression of kidney injury (preventing 30–50% of the tubular lesions and 20% of the glomerular lesions) decreased arteriolar wall thickness and diminished the severity of arteriolar lesions, and this occurred independent of BP [169]. Ellis et al. [170] also reported a reduction in albuminuria and improvement in hypertensive kidney lesions in K+-supplemented, salt-loaded spontaneously hypertensive rats. In the 5/6th nephrectomy model of CKD, high dietary K+ intake decreased interstitial fibrosis and suppressed inflammation [171]. More specifically, high dietary K+ intake resulted in less collagen Types I and III deposition in the kidney, less macrophage infiltration, lower expression of the inflammatory cytokines interleukin-1 beta and intercellular adhesion molecule 1, and decreased activation of nuclear factor kappa B (NF-κB). In this rat CKD model, the kidney protective effects were linked to a reduction in transforming growth factor beta (TGF-β), a key mediator of kidney fibrosis, upregulation of Smad7 (a negative regulator of TGF-β and NF-κB) and lower BP. Similar kidney protective effects were observed in another rat CKD model (albumin overload) [172]. A K+-induced upregulation of the kinin–kallikrein system reduced BP, proteinuria and tubulointerstitial fibrosis. These effects were also accompanied by increased Smad7 production and downregulation of the TGF-β pathway [172–174]. In vitro, bradykinin prevented albumin-induced tubular phenotypic changes and the expression of mesenchymal markers, and inhibited TGF-β expression. All of these effects were reversed by pharmacological blockage of the kinin pathway [174]. These results support that the K+-induced upregulation of the kinin–kallikrein system may protect the kidney. Although experimental evidence supports the kidney protective effects of dietary K+, clinical intervention studies are necessary to establish if these findings also extend to humans.
PERSPECTIVES
The evidence for a beneficial effect of dietary K+ on human health is strengthening. Although the ideal dietary K+ content is unknown, data from association studies consistently suggest that a low K+ diet is harmful. This implies that increasing dietary K+ intake to recommended levels (90–120 mmol/day) could be an effective public health strategy to reduce hypertension and cardiovascular disease. Because hypertension and cardiovascular diseases are very prevalent in patients with CKD, there are good reasons to believe that patients with CKD may also benefit from increasing dietary K+. This assumption is now increasingly supported by both experimental and epidemiological data (Figures 5 and 6). However, several caveats apply and several knowledge gaps remain (Table 1). First, association studies do not prove causality. For example, high K+ or healthier diets may not only be rich in K+ but also in alkaline-rich fruits and vegetables. The anion accompanying K+, other nutrients, lower protein, higher fibre content and lower phosphate content in these plant-based diets may have contributed to the outcomes. Indeed, alkali therapy will help to correct the metabolic acidosis of CKD, which in turn may slow CKD progression [175–178]. Also of interest is a potential interaction between K+ and phosphate in patients with CKD. An observational study conducted in healthy individuals showed that a Western diet, which is poor in K+ and rich in phosphate, is characterized by higher fibroblast growth factor 23 and found an inverse association with a low K+ intake [179]. In mice, a high Na+-low K+ diet increases urinary calcium and phosphate excretion [180]. Studies have shown that K+ supplementation increases the tubular phosphate reabsorption capacity in rats [181] and increases serum phosphate in healthy individuals [182], indicating a possible interaction between K+, phosphate and fibroblast growth factor 23. Another important consideration is that it matters how dietary K+ intake is assessed (Figure 6). Although most studies use 24-h urine excretions, weekly or monthly rhythms may influence daily excretions [183, 184]. Indeed, multiple 24-h measurements may better predict outcomes better than a single one [185]. Repeated spot urine Na+/K+ ratios have shown to be strongly correlated to 24-h urine Na+/K+ ratios in patients with CKD Stages G1–3 but not CKD Stages G4–5 [186]. With declining kidney function, more K+ may be excreted by the gastro-intestinal tract or redistributed internally. Racial differences in K+ homoeostasis also make the relationship between K+ excretion and CKD more difficult to interpret [187]. Compared with urinary K+ excretion, food frequency questionnaires suffer from recall bias, inaccuracies in the natural K+ content in food composition tables, and do not account for K+ additives. If more conclusive evidence for a beneficial effect of increasing dietary K+ in patients with CKD becomes available, this must be weighed against the risk of hyperkalaemia [188]. The prevalence of plasma K+ >5.0 mmol/L is estimated to be 12–18% in patients with CKD compared with 3–4% of patients without CKD [189–191]. Based on epidemiological data, the lowest risk of all-cause mortality is estimated at plasma K+ between 4.0 and 4.5 mmol/L and even mild hyperkalaemia has been associated with higher risk of mortality [189]. However, the ideal plasma K+ range appears to be higher in patients with kidney disease [192, 193]. It is also not clear whether the positive effects of dietary K+ are mediated by plasma K+ and, if so, whether this is concentration dependent (Table 1). Put provocatively, one could argue that the beneficial effects of renin–angiotensin inhibitors are partly related to their effect on plasma K+. To address some of these questions, we are currently conducting a randomized clinical trial in which patients with progressive CKD Stage G3b or G4 are being treated with potassium chloride, potassium citrate, or placebo for 2 years [194]. The primary endpoint of this study is eGFR decline, while secondary outcomes include 24-h BP and albuminuria. We hope that such intervention studies will contribute to stronger clinical evidence to support the recent exciting insights in the physiology and epidemiology of potassium homoeostasis.
Table 1.
Knowledge gaps
|
FUNDING
This work is supported by the Dutch Kidney Foundation (grant CP16.01).
CONFLICT OF INTEREST STATEMENT
Dr. Vogt reports support from Vifor Fresenius Medical Care Renal Pharma and AstraZeneca, both outside the submitted work. Dr. De Borst reports support from Vifor Fresenius Medical Care Renal Pharma, Astra Zeneca, Amgen, Sanofi Genzyme, Bayer, Kyowa Kirin, and Pharmacosmos, all outside the submitted work.
This Review was written in collaboration with NDT Educational.
REFERENCES
- 1. Eaton SB, Konner M. Paleolithic nutrition. A consideration of its nature and current implications. N Engl J Med 1985; 312: 283–289 [DOI] [PubMed] [Google Scholar]
- 2. Cogswell ME, Zhang Z, Carriquiry AL et al. Sodium and potassium intakes among US adults: NHANES 2003–2008. Am J Clin Nutr 2012; 96: 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mente A, O’Donnell MJ, Rangarajan S et al. Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med 2014; 371: 601–611 [DOI] [PubMed] [Google Scholar]
- 4. McDonough AA, Veiras LC, Guevara CA et al. Cardiovascular benefits associated with higher dietary K(+) vs. lower dietary Na(+): evidence from population and mechanistic studies. Am J Physiol Endocrinol Metab 2017; 312: E348–E356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kamel KS, Schreiber M, Halperin ML. Renal potassium physiology: integration of the renal response to dietary potassium depletion. Kidney Int 2018; 93: 41–53 [DOI] [PubMed] [Google Scholar]
- 6. DuBose TD., Jr. Inadequate dietary potassium and progression of CKD. Clin J Am Soc Nephrol 2019; 14: 319–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Intersalt Cooperative Research Group. Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ 1988; 297: 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sacks FM, Svetkey LP, Vollmer WM et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-sodium collaborative research group. N Engl J Med 2001; 344: 3–10 [DOI] [PubMed] [Google Scholar]
- 9. Whelton PK, Appel LJ, Espeland MA et al. ; TONE Collaborative Research Group. Sodium reduction and weight loss in the treatment of hypertension in older persons: a randomized controlled trial of nonpharmacologic interventions in the elderly (TONE). JAMA 1998; 279: 839–846 [DOI] [PubMed] [Google Scholar]
- 10. Bovee DM, Visser WJ, Middel I et al. A randomized trial of distal diuretics versus dietary sodium restriction for hypertension in chronic kidney disease. J Am Soc Nephrol 2020; 31: 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.World Health Organization. Guideline: Potassium Intake for Adults and Children. Geneva, Switzerland: World Health Organization, 2012 [PubMed] [Google Scholar]
- 12. Aburto NJ, Hanson S, Gutierrez H et al. Effect of increased potassium intake on cardiovascular risk factors and disease: systematic review and meta-analyses. BMJ 2013; 346: f1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O’Donnell M, Mente A, Rangarajan S et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med 2014; 371: 612–623 [DOI] [PubMed] [Google Scholar]
- 14. Mente A, O’Donnell M, Rangarajan S et al. Urinary sodium excretion, blood pressure, cardiovascular disease, and mortality: a community-level prospective epidemiological cohort study. Lancet 2018; 392: 496–506 [DOI] [PubMed] [Google Scholar]
- 15. Clase CM, Carrero JJ, Ellison DH et al. Potassium homeostasis and management of dyskalemia in kidney diseases: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) controversies conference. Kidney Int 2019; 97: 42–61 [DOI] [PubMed] [Google Scholar]
- 16. Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med 2013; 369: 448–457 [DOI] [PubMed] [Google Scholar]
- 17. Bashyam H. Lewis Dahl and the genetics of salt-induced hypertension. J Exp Med 2007; 204: 1507–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DiNicolantonio JJ, O’Keefe JH. The history of the salt wars. Am J Med 2017; 130: 1011–1014 [DOI] [PubMed] [Google Scholar]
- 19. Dahl LK. Possible role of salt intake in the development of essential hypertension. Int J Epidemiol 2005; 34: 967–972; discussion 9724 [DOI] [PubMed] [Google Scholar]
- 20. Dahl LK, Heine M, Tassinari L. Effects of chronic excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. J Exp Med 1962; 115: 1173–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dahl LK, Leitl G, Heine M. Influence of dietary potassium and sodium/potassium molar ratios on the development of salt hypertension. J Exp Med 1972; 136: 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Addison WL. The use of sodium chloride, potassium chloride, sodium bromide, and potassium bromide in cases of arterial hypertension which are amenable to potassium chloride. Can Med Assoc J 1928; 18: 281–285 [PMC free article] [PubMed] [Google Scholar]
- 23. Meneely GR, Ball CO. Experimental epidemiology of chronic sodium chloride toxicity and the protective effect of potassium chloride. Am J Med 1958; 25: 713–725 [DOI] [PubMed] [Google Scholar]
- 24. Mancilha-Carvalho Jde J, Souza e Silva NA. The Yanomami Indians in the INTERSALT Study. Arq Bras Cardiol 2003; 80: 289–300 [DOI] [PubMed] [Google Scholar]
- 25. Whelton PK, He J, Cutler JA et al. Effects of oral potassium on blood pressure. Meta-analysis of randomized controlled clinical trials. JAMA 1997; 277: 1624–1632 [DOI] [PubMed] [Google Scholar]
- 26. Cappuccio FP, MacGregor GA. Does potassium supplementation lower blood pressure? A meta-analysis of published trials. J Hypertens 1991; 9: 465–473 [DOI] [PubMed] [Google Scholar]
- 27. Geleijnse JM, Kok FJ, Grobbee DE. Blood pressure response to changes in sodium and potassium intake: a metaregression analysis of randomised trials. J Hum Hypertens 2003; 17: 471–480 [DOI] [PubMed] [Google Scholar]
- 28. Krishna GG, Miller E, Kapoor S. Increased blood pressure during potassium depletion in normotensive men. N Engl J Med 1989; 320: 1177–1182 [DOI] [PubMed] [Google Scholar]
- 29. Krishna GG, Kapoor SC. Potassium depletion exacerbates essential hypertension. Ann Intern Med 1991; 115: 77–83 [DOI] [PubMed] [Google Scholar]
- 30. Institute Of M. Dietary reference intakes for water Potassium, sodium, chloride, and sulfate. Washington, DC: The National Academies Press, 2005 [Google Scholar]
- 31. Appel LJ, Moore TJ, Obarzanek E et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med 1997; 336: 1117–1124 [DOI] [PubMed] [Google Scholar]
- 32. Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis: core curriculum 2019. Am J Kidney Dis 2019; 74: 682–695 [DOI] [PubMed] [Google Scholar]
- 33. Masilamani S, Kim GH, Mitchell C et al. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 1999; 104: R19–R23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rieg T, Vallon V, Sausbier M et al. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int 2007; 72: 566–573 [DOI] [PubMed] [Google Scholar]
- 35. Hoorn EJ, Loffing J, Ellison DH. An integrated view of potassium homeostasis. N Engl J Med 2015; 373: 1786. [DOI] [PubMed] [Google Scholar]
- 36. Terker AS, Zhang C, McCormick JA et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 2015; 21: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoorn EJ, Gritter M, Cuevas CA et al. Regulation of the renal NaCl cotransporter and its role in potassium homeostasis. Physiol Rev 2020; 100: 321–356 [DOI] [PubMed] [Google Scholar]
- 38. Ellison DH, Velazquez H, Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol 1987; 253: F546–F554 [DOI] [PubMed] [Google Scholar]
- 39. Moes AD, van der Lubbe N, Zietse R et al. The sodium chloride cotransporter SLC12A3: new roles in sodium, potassium, and blood pressure regulation. Pflugers Arch Eur J Physiol 2014; 466: 107–118 [DOI] [PubMed] [Google Scholar]
- 40. Wilson FH, Disse-Nicodeme S, Choate KA et al. Human hypertension caused by mutations in WNK kinases. Science 2001; 293: 1107–1112 [DOI] [PubMed] [Google Scholar]
- 41. Simon DB, Nelson-Williams C, Bia MJ et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 1996; 12: 24–30 [DOI] [PubMed] [Google Scholar]
- 42. Louis-Dit-Picard H, Barc J, Trujillano D et al. ; International Consortium for Blood Pressure (ICBP). KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet 2012; 44: 456–460 [DOI] [PubMed] [Google Scholar]
- 43. Boyden LM, Choi M, Choate KA et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012; 482: 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pathare G, Hoenderop JGJ, Bindels RJM et al. A molecular update on pseudohypoaldosteronism type II. Am J Physiol Renal Physiol 2013; 305: F1513–F1520 [DOI] [PubMed] [Google Scholar]
- 45. Pacheco-Alvarez D, Cristóbal PS, Meade P et al. The Na+:Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 2006; 281: 28755–28763 [DOI] [PubMed] [Google Scholar]
- 46. Richardson C, Rafiqi FH, Karlsson HK et al. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 2008; 121: 675–684 [DOI] [PubMed] [Google Scholar]
- 47. Vallon V, Schroth J, Lang F et al. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 2009; 297: F704–F712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vitzthum H, Seniuk A, Schulte LH et al. Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J Physiol 2014; 592: 1139–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takahashi D, Mori T, Nomura N et al. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci Rep 2014; 34: e00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L et al. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 2014; 306: F1507–F1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ferdaus MZ, Barber KW, Lopez-Cayuqueo KI et al. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol 2016; 594: 4945–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang YS, Xie J, Yang SS et al. Differential roles of WNK4 in regulation of NCC in vivo. Am J Physiol Renal Physiol 2018; 314: F999–F1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jensen IS, Larsen CK, Leipziger J et al. Na(+) dependence of K(+) -induced natriuresis, kaliuresis and Na(+)/Cl(-) cotransporter dephosphorylation. Acta Physiol 2016; 218: 49–61 [DOI] [PubMed] [Google Scholar]
- 54. Veiras LC, Girardi ACC, Curry J et al. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 2017; 28: 3504–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sorensen MV, Grossmann S, Roesinger M et al. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 2013; 83: 811–824 [DOI] [PubMed] [Google Scholar]
- 56. Shoda W, Nomura N, Ando F et al. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int 2017; 91: 402–411 [DOI] [PubMed] [Google Scholar]
- 57. Rengarajan S, Lee DH, Oh YT et al. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 2014; 306: F1059–F1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Penton D, Czogalla J, Wengi A et al. Extracellular K(+) rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl(-)-dependent and independent mechanisms. J Physiol 2016; 594: 6319–6331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wade JB, Fang L, Coleman RA et al. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 2011; 300: F1385–F1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 2010; 299: F890–F897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van der Lubbe N, Moes AD, Rosenbaek LL et al. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl- cotransporter. Am J Physiol Renal Physiol 2013; 305: F1177–F1188 [DOI] [PubMed] [Google Scholar]
- 62. Yang L, Xu S, Guo X et al. Regulation of renal Na transporters in response to dietary K. Am J Physiol Renal Physiol 2018; 315: F1032–F1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang L, Frindt G, Lang F et al. SGK1-dependent ENaC processing and trafficking in mice with high dietary K intake and elevated aldosterone. Am J Physiol Renal Physiol 2017; 312: F65–F76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Terker AS, Zhang C, Erspamer KJ et al. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 2016; 89: 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang LE, Sandberg MB, Can AD et al. Effects of dietary salt on renal Na+ transporter subcellular distribution, abundance, and phosphorylation status. Am J Physiol Renal Physiol 2008; 295: F1003–F1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rojas-Vega L, Gamba G. Mini-review: regulation of the renal NaCl cotransporter by hormones. Am J Physiol Renal Physiol 2016; 310: F10–F14 [DOI] [PubMed] [Google Scholar]
- 67. Lourdel S, Paulais M, Cluzeaud F et al. An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol 2002; 538: 391–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Palygin O, Levchenko V, Ilatovskaya DV et al. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight 2017; 2: e92331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Su XT, Ellison DH, Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K(+) excretion. Am J Physiol Renal Physiol 2019; 316: F582–F586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cuevas CA, Su XT, Wang MX et al. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 2017; 28: 1814–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang MX, Cuevas CA, Su XT et al. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 2018; 93: 893–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wu P, Gao ZX, Zhang DD et al. Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol 2019; 30: 1425–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hennings JC, Andrini O, Picard N et al. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol 2017; 28: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zaika O, Tomilin V, Mamenko M et al. New perspective of ClC-Kb/2 Cl- channel physiology in the distal renal tubule. Am J Physiol Renal Physiol 2016; 310: F923–F930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Teulon J, Planelles G, Sepulveda FV et al. Renal chloride channels in relation to sodium chloride transport. Compr Physiol 2018; 9: 301–342 [DOI] [PubMed] [Google Scholar]
- 76. Estevez R, Boettger T, Stein V et al. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature 2001; 414: 558–561 [DOI] [PubMed] [Google Scholar]
- 77. Zhang C, Wang L, Zhang J et al. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 2014; 111: 11864–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Malik S, Lambert E, Zhang J et al. Potassium conservation is impaired in mice with reduced renal expression of Kir4.1. Am J Physiol Renal Physiol 2018; 315: F1271–F1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nomura N, Shoda W, Wang Y et al. Role of ClC-K and barttin in low potassium-induced sodium chloride cotransporter activation and hypertension in mouse kidney. Biosci Rep 2018; 38: BSR20171243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bockenhauer D, Feather S, Stanescu HC et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10. N Engl J Med 2009; 360: 1960–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Piala AT, Moon TM, Akella R et al. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 2014; 7: ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L et al. The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 2015; 26: 1781–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wade JB, Liu J, Coleman R et al. SPAK-mediated NCC regulation in response to low-K+ diet. Am J Physiol Renal Physiol 2015; 308: F923–F931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen JC, Lo YF, Lin YW et al. WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci USA 2019; 116: 4502–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sun Q, Wu Y, Jonusaite S et al. Intracellular chloride and scaffold protein Mo25 cooperatively regulate transepithelial ion transport through WNK signaling in the malpighian tubule. J Am Soc Nephrol 2018; 29: 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rodan AR. Intracellular chloride: a regulator of transepithelial transport in the distal nephron. Curr Opin Nephrol Hypertens 2019; 28: 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ishizawa K, Xu N, Loffing J et al. Potassium depletion stimulates Na-Cl cotransporter via phosphorylation and inactivation of the ubiquitin ligase Kelch-like 3. Biochem Biophys Res Commun 2016; 480: 745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lu M, Wang J, Jones KT et al. mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol 2010; 21: 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gleason CE, Frindt G, Cheng CJ et al. mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J Clin Invest 2015; 125: 117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. van Buren M, Rabelink AJ, Bijlsma JA et al. Natriuretic and kaliuretic response to potassium load: modulation by sodium intake. Nephrol Dial Transplant 1993; 8: 495–500 [DOI] [PubMed] [Google Scholar]
- 91. Calo L, Borsatti A, Favaro S et al. Kaliuresis in normal subjects following oral potassium citrate intake without increased plasma potassium concentration. Nephron 1995; 69: 253–258 [DOI] [PubMed] [Google Scholar]
- 92. Rabinowitz L, Green DM, Sarason RL et al. Homeostatic potassium excretion in fed and fasted sheep. Am J Physiol 1988; 254: R357–R380 [DOI] [PubMed] [Google Scholar]
- 93. Cheng L, Poulsen SB, Wu Q et al. Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 2019; 30: 1454–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Velazquez H, Bartiss A, Bernstein P et al. Adrenal steroids stimulate thiazide-sensitive NaCl transport by rat renal distal tubules. Am J Physiol 1996; 270: F211–F219 [DOI] [PubMed] [Google Scholar]
- 95. Kim GH, Masilamani S, Turner R et al. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 1998; 95: 14552–14557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hunter RW, Craigie E, Homer NZ et al. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol 2014; 306: F457–F467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Veiras LC, Han J, Ralph DL et al. Potassium supplementation prevents sodium chloride cotransporter stimulation during angiotensin II hypertension. Hypertension 2016; 68: 904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ishizawa K, Wang Q, Li J et al. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc Natl Acad Sci USA 2019; 116: 3155–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Brandis M, Keyes J, Windhager EE. Potassium-induced inhibition of proximal tubular fluid reabsorption in rats. Am J Physiol 1972; 222: 421–427 [DOI] [PubMed] [Google Scholar]
- 100. Weinstein AM. A mathematical model of the rat kidney: K(+)-induced natriuresis. Am J Physiol Renal Physiol 2017; 312: F925–F950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Stokes JB. Consequences of potassium recycling in the renal medulla. Effects of ion transport by the medullary thick ascending limb of Henle's loop. J Clin Invest 1982; 70: 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kirchner KA. Effect of acute potassium infusion on loop segment chloride reabsorption in the rat. Am J Physiol 1983; 244: F599–F605 [DOI] [PubMed] [Google Scholar]
- 103. Higashihara E, Kokko JP. Effects of aldosterone on potassium recycling in the kidney of adrenalectomized rats. Am J Physiol 1985; 248: F219–F227 [DOI] [PubMed] [Google Scholar]
- 104. Unwin R, Capasso G, Giebisch G. Potassium and sodium transport along the loop of Henle: effects of altered dietary potassium intake. Kidney Int 1994; 46: 1092–1099 [DOI] [PubMed] [Google Scholar]
- 105. Cheng CJ, Truong T, Baum M et al. Kidney-specific WNK1 inhibits sodium reabsorption in the cortical thick ascending limb. Am J Physiol Renal Physiol 2012; 303: F667–F673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sorensen MV, Saha B, Jensen IS et al. Potassium acts through mTOR to regulate its own secretion. JCI Insight 2019; 5: e126910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hené RJ, Koomans HA, Rabelink AJ et al. Mineralocorticoid activity and the excretion of an oral potassium load in normal man. Kidney Int 1988; 34: 697–703 [DOI] [PubMed] [Google Scholar]
- 108. Grimm PR, Lazo-Fernandez Y, Delpire E et al. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest 2015; 125: 2136–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Grimm PR, Coleman R, Delpire E et al. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 2017; 28: 2597–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rabelink TJ, Koomans HA, Hené RJ et al. Early and late adjustment to potassium loading in humans. Kidney Int 1990; 38: 942–947 [DOI] [PubMed] [Google Scholar]
- 111. Rabinowitz L, Sarason RL, Yamauchi H. Effect of aldosterone on potassium excretion during potassium chloride infusion in sheep. Am J Physiol 1985; 249: R455–R461 [DOI] [PubMed] [Google Scholar]
- 112. Lee FN, Oh G, McDonough AA et al. Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol 2007; 293: F541–F547 [DOI] [PubMed] [Google Scholar]
- 113. Preston RA, Afshartous D, Rodco R et al. Evidence for a gastrointestinal-renal kaliuretic signaling axis in humans. Kidney Int 2015; 88: 1383–1391 [DOI] [PubMed] [Google Scholar]
- 114. Hoorn EJ, Zietse R. Gut-kidney kaliuretic signaling: looking forward to feeding. Kidney Int 2015; 88: 1230–1232 [DOI] [PubMed] [Google Scholar]
- 115. Pluznick JL, Protzko RJ, Gevorgyan H et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA 2013; 110: 4410–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pluznick JL, Caplan MJ. Chemical and physical sensors in the regulation of renal function. Clin J Am Soc Nephrol 2015; 10: 1626–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wilck N, Matus MG, Kearney SM et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017; 551: 585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Suga SI, Phillips MI, Ray PE et al. Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am J Physiol Renal Physiol 2001; 281: F620–F629 [DOI] [PubMed] [Google Scholar]
- 119. Suga S, Mazzali M, Ray PE et al. Angiotensin II type 1 receptor blockade ameliorates tubulointerstitial injury induced by chronic potassium deficiency. Kidney Int 2002; 61: 951–958 [DOI] [PubMed] [Google Scholar]
- 120. Suga S, Yasui N, Yoshihara F et al. Endothelin a receptor blockade and endothelin B receptor blockade improve hypokalemic nephropathy by different mechanisms. J Am Soc Nephrol 2003; 14: 397–406 [DOI] [PubMed] [Google Scholar]
- 121. Fujita T, Sato Y. Changes in renal and central noradrenergic activity with potassium in DOCA-salt rats. Am J Physiol 1984; 246: F670–F675 [DOI] [PubMed] [Google Scholar]
- 122. DiBona GF. Neurogenic regulation of renal tubular sodium reabsorption. Am J Physiol 1977; 233: F73–F81 [DOI] [PubMed] [Google Scholar]
- 123. Katholi RE, Naftilan AJ, Oparil S. Importance of renal sympathetic tone in the development of DOCA-salt hypertension in the rat. Hypertension 1980; 2: 266–273 [DOI] [PubMed] [Google Scholar]
- 124. Lohmeier TE, Iliescu R, Liu B et al. Systemic and renal-specific sympathoinhibition in obesity hypertension. Hypertension 2012; 59: 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nagae A, Fujita M, Kawarazaki H et al. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in obesity-induced hypertension. Circulation 2009; 119: 978–986 [DOI] [PubMed] [Google Scholar]
- 126. Hirohama D, Fujita T. Evaluation of the pathophysiological mechanisms of salt-sensitive hypertension. Hypertens Res 2019; 42: 1848–1857 [DOI] [PubMed] [Google Scholar]
- 127. Mu SYu, Shimosawa T, Ogura S et al. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med 2011; 17: 573–580 [DOI] [PubMed] [Google Scholar]
- 128. Terker AS, Yang CL, McCormick JA et al. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 2014; 64: 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Duan XP, Gu L, Xiao Y et al. Norepinephrine-induced stimulation of Kir4.1/Kir5.1 is required for the activation of NaCl transporter in distal convoluted tubule. Hypertension 2019; 73: 112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Penton D, Moser S, Wengi A et al. Protein phosphatase 1 inhibitor-1 mediates the cAMP-dependent stimulation of the renal NaCl cotransporter. J Am Soc Nephrol 2019; 30: 737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zicha J, Dobesova Z, Behuliak M et al. Preventive dietary potassium supplementation in young salt-sensitive Dahl rats attenuates development of salt hypertension by decreasing sympathetic vasoconstriction. Acta Physiol (Oxf) 2011; 202: 29–38 [DOI] [PubMed] [Google Scholar]
- 132. Dietz R, Schomig A, Dart AM et al. Modulation of sympathetic vasoconstriction by potassium. J Cardiovasc Pharmacol 1984; 6: S230–S235 [DOI] [PubMed] [Google Scholar]
- 133. Taddei S, Mattei P, Virdis A et al. Effect of potassium on vasodilation to acetylcholine in essential hypertension. Hypertension 1994; 23: 485–490 [DOI] [PubMed] [Google Scholar]
- 134. Raij L, Lüscher TF, Vanhoutte PM. Vanhoutte PM. High potassium diet augments endothelium-dependent relaxations in the Dahl rat. Hypertension 1988; 12: 562–567 [DOI] [PubMed] [Google Scholar]
- 135. Zhou MS, Kosaka H, Yoneyama H. Potassium augments vascular relaxation mediated by nitric oxide in the carotid arteries of hypertensive Dahl rats. Am J Hypertens 2000; 13: 666–672 [DOI] [PubMed] [Google Scholar]
- 136. Oberleithner H, Callies C, Kusche-Vihrog K et al. Potassium softens vascular endothelium and increases nitric oxide release. Proc Natl Acad Sci USA 2009; 106: 2829–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Oberleithner H, Kusche-Vihrog K, Schillers H. Endothelial cells as vascular salt sensors. Kidney Int 2010; 77: 490–494 [DOI] [PubMed] [Google Scholar]
- 138. Oberleithner H, Riethmuller C, Schillers H et al. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci USA 2007; 104: 16281–16286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Dolson GM, Ellis KJ, Bernardo MV et al. Acute decreases in serum potassium augment blood pressure. Am J Kidney Dis 1995; 26: 321–326 [DOI] [PubMed] [Google Scholar]
- 140. Gritter M, Rotmans JI, Hoorn EJ et al. Blood pressure reduction, cardiovascular protection, and renoprotection. Hypertension 2019; 73: 15–23 [DOI] [PubMed] [Google Scholar]
- 141. Chang HY, Hu YW, Yue CS et al. Effect of potassium-enriched salt on cardiovascular mortality and medical expenses of elderly men. Am J Clin Nutr 2006; 83: 1289–1296 [DOI] [PubMed] [Google Scholar]
- 142. Bernabe-Ortiz A, Sal Y, Ponce-Lucero V et al. Effect of salt substitution on community-wide blood pressure and hypertension incidence. Nat Med 2020; 26: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Neal B, Tian M, Li N et al. Rationale, design, and baseline characteristics of the Salt Substitute and Stroke Study (SSaSS)-A large-scale cluster randomized controlled trial. Am Heart J 2017; 188: 109–117 [DOI] [PubMed] [Google Scholar]
- 144. Greer RC, Marklund M, Anderson CAM et al. Potassium-enriched salt substitutes as a means to lower blood pressure: benefits and risks. Hypertension 2020; 75: 266–274 [DOI] [PubMed] [Google Scholar]
- 145. He FJ, Marciniak M, Carney C et al. Effects of potassium chloride and potassium bicarbonate on endothelial function, cardiovascular risk factors, and bone turnover in mild hypertensives. Hypertension 2010; 55: 681–688 [DOI] [PubMed] [Google Scholar]
- 146. Tang X, Wu B, Luo Y et al. Effect of potassium supplementation on vascular function: a meta-analysis of randomized controlled trials. Int J Cardiol 2017; 228: 225–232 [DOI] [PubMed] [Google Scholar]
- 147. Dorrance AM, Pollock DM, Romanko OP et al. A high-potassium diet reduces infarct size and improves vascular structure in hypertensive rats. Am J Physiol Regul Integr Comp Physiol 2007; 292: R415–R422 [DOI] [PubMed] [Google Scholar]
- 148. Kido M, Ando K, Onozato ML et al. Protective effect of dietary potassium against vascular injury in salt-sensitive hypertension. Hypertension 2008; 51: 225–231 [DOI] [PubMed] [Google Scholar]
- 149. Sun Y, Byon CH, Yang Y et al. Dietary potassium regulates vascular calcification and arterial stiffness. JCI Insight 2017; 2: e94920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Picard K, B, Silva MI, Mager D et al. Dietary potassium intake and risk of chronic kidney disease progression in predialysis patients with chronic kidney disease: a systematic review. Adv Nutr 2020; 11: 1002–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Araki S, Haneda M, Koya D et al. Urinary potassium excretion and renal and cardiovascular complications in patients with type 2 diabetes and normal renal function. Clin J Am Soc Nephrol 2015; 10: 2152–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kieneker LM, Bakker SJL, de Boer RA et al. Low potassium excretion but not high sodium excretion is associated with increased risk of developing chronic kidney disease. Kidney Int 2016; 90: 888–896 [DOI] [PubMed] [Google Scholar]
- 153. Olde Engberink RHG, van den Born BH, Peters-Sengers H et al. Long-term potassium intake and associated renal and cardiovascular outcomes in the clinical setting. Clin Nutrition 2020: S0261-5614(20)30144-8 [DOI] [PubMed] [Google Scholar]
- 154. Smyth A, Dunkler D, Gao P et al. The relationship between estimated sodium and potassium excretion and subsequent renal outcomes. Kidney Int 2014; 86: 1205–1212 [DOI] [PubMed] [Google Scholar]
- 155. Nagata T, Sobajima H, Ohashi N et al. Association between 24h urinary sodium and potassium excretion and estimated glomerular filtration rate (eGFR) decline or death in patients with diabetes mellitus and eGFR more than 30 ml/min/1.73m2. PLoS One 2016; 11: e0152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Deriaz D, Guessous I, Vollenweider P et al. Estimated 24-h urinary sodium and sodium-to-potassium ratio are predictors of kidney function decline in a population-based study. J Hypertens 2019; 37: 1853–1860 [DOI] [PubMed] [Google Scholar]
- 157. Tabara Y, Takahashi Y, Setoh K et al. ; the Nagahama Study Group. Prognostic significance of spot urine Na/K for longitudinal changes in blood pressure and renal function: the Nagahama study. Am J Hypertens 2017; 30: 899–906 [DOI] [PubMed] [Google Scholar]
- 158. Leonberg-Yoo AK, Tighiouart H, Levey AS et al. Urine potassium excretion, kidney failure, and mortality in CKD. Am J Kidney Dis 2017; 69: 341–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kim HW, Park JT, Yoo TH et al. ; on behalf of the KNOW-CKD Study Investigators. Urinary potassium excretion and progression of CKD. Clin J Am Soc Nephrol 2019; 14: 330–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. He J, Mills KT, Appel LJ et al. ; for the Chronic Renal Insufficiency Cohort Study Investigators. Urinary sodium and potassium excretion and CKD progression. J Am Soc Nephrol 2016; 27: 1202–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Bach KE, Kelly JT, Palmer SC et al. Healthy dietary patterns and incidence of CKD: a meta-analysis of cohort studies. Clin J Am Soc Nephrol 2019; 14: 1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kelly JT, Palmer SC, Wai SN et al. Healthy dietary patterns and risk of mortality and ESRD in CKD: a meta-analysis of cohort studies. Clin J Am Soc Nephrol 2017; 12: 272–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Joshi S, Hashmi S, Shah S et al. Plant-based diets for prevention and management of chronic kidney disease. Curr Opin Nephrol Hypertens 2020; 29: 16–21 [DOI] [PubMed] [Google Scholar]
- 164. Mirmiran P, Nazeri P, Bahadoran Z et al. Dietary sodium to potassium ratio and the incidence of chronic kidney disease in adults: a longitudinal follow-up study. Prev Nutr Food Sci 2018; 23: 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Smyth A, Griffin M, Yusuf S et al. ; The NIH-AARP diet and health study. Diet and major renal outcomes: a prospective cohort study. J Ren Nutr 2016; 26: 288–298 [DOI] [PubMed] [Google Scholar]
- 166. Mun KH, Yu GI, Choi BY et al. Association of dietary potassium intake with the development of chronic kidney disease and renal function in patients with mildly decreased kidney function: the Korean multi-rural communities cohort study. Med Sci Monit 2019; 25: 1061–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ray PE, Suga S, Liu XH et al. Chronic potassium depletion induces renal injury, salt sensitivity, and hypertension in young rats. Kidney Int 2001; 59: 1850–1858 [DOI] [PubMed] [Google Scholar]
- 168. Fryer JN, Burns KD, Ghorbani M et al. Effect of potassium depletion on proximal tubule AT1 receptor localization in normal and remnant rat kidney. Kidney Int 2001; 60: 1792–1799 [DOI] [PubMed] [Google Scholar]
- 169. Tobian L, MacNeill D, Johnson MA et al. Potassium protection against lesions of the renal tubules, arteries, and glomeruli and nephron loss in salt-loaded hypertensive Dahl S rats. Hypertension 1984; 6: I170–I176 [DOI] [PubMed] [Google Scholar]
- 170. Ellis D, Banner B, Janosky JE et al. Potassium supplementation attenuates experimental hypertensive renal injury. J Am Soc Nephrol 1992; 2: 1529–1537 [DOI] [PubMed] [Google Scholar]
- 171. Wang W, Soltero L, Zhang P et al. Renal inflammation is modulated by potassium in chronic kidney disease: possible role of Smad7. Am J Physiol Renal Physiol 2007; 293: F1123–F1130 [DOI] [PubMed] [Google Scholar]
- 172. Ardiles LG, Loyola F, Ehrenfeld P et al. Modulation of renal kallikrein by a high potassium diet in rats with intense proteinuria. Kidney Int 2006; 69: 53–59 [DOI] [PubMed] [Google Scholar]
- 173. Ardiles L, Cardenas A, Burgos ME et al. Antihypertensive and renoprotective effect of the kinin pathway activated by potassium in a model of salt sensitivity following overload proteinuria. Am J Physiol Renal Physiol 2013; 304: F1399–F1410 [DOI] [PubMed] [Google Scholar]
- 174. Cardenas A, Campos J, Ehrenfeld P et al. Up-regulation of the kinin B2 receptor pathway modulates the TGF-beta/Smad signaling cascade to reduce renal fibrosis induced by albumin. Peptides 2015; 73: 7–19 [DOI] [PubMed] [Google Scholar]
- 175. de Brito-Ashurst I, Varagunam M, Raftery MJ et al. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009; 20: 2075–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Mahajan A, Simoni J, Sheather SJ et al. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int 2010; 78: 303–309 [DOI] [PubMed] [Google Scholar]
- 177. Phisitkul S, Khanna A, Simoni J et al. Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int 2010; 77: 617–623 [DOI] [PubMed] [Google Scholar]
- 178. Driver TH, Shlipak MG, Katz R et al. Low serum bicarbonate and kidney function decline: the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis 2014; 64: 534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Eckberg K, Kramer H, Wolf M et al. Impact of westernization on fibroblast growth factor 23 levels among individuals of African ancestry. Nephrol Dial Transplant 2015; 30: 630–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. van der Wijst J, Tutakhel OAZ, Bos C et al. Effects of a high-sodium/low-potassium diet on renal calcium, magnesium, and phosphate handling. Am J Physiol Renal Physiol 2018; 315: F110–F122 [DOI] [PubMed] [Google Scholar]
- 181. Jaeger P, Bonjour JP, Karlmark B et al. Influence of acute potassium loading on renal phosphate transport in the rat kidney. Am J Physiol 1983; 245: F601–F605 [DOI] [PubMed] [Google Scholar]
- 182. Sebastian A, Hernandez RE, Portale AA et al. Dietary potassium influences kidney maintenance of serum phosphorus concentration. Kidney Int 1990; 37: 1341–1349 [DOI] [PubMed] [Google Scholar]
- 183. Gumz ML, Rabinowitz L, Wingo CS. An integrated view of potassium homeostasis. N Engl J Med 2015; 373: 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Rakova N, Juttner K, Dahlmann A et al. Long-term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell Metab 2013; 17: 125–131 [DOI] [PubMed] [Google Scholar]
- 185. Olde Engberink RHG, van den Hoek TC, van Noordenne ND et al. Use of a single baseline versus multiyear 24-hour urine collection for estimation of long-term sodium intake and associated cardiovascular and renal risk. Circulation 2017; 136: 917–926 [DOI] [PubMed] [Google Scholar]
- 186. Okuyama Y, Uchida HA, Iwahori T et al. The relationship between repeated measurement of casual and 24-h urinary sodium-to-potassium ratio in patients with chronic kidney disease. J Hum Hypertens 2019; 33: 286–297 [DOI] [PubMed] [Google Scholar]
- 187. Turban S, Miller ER, Ange B et al. Racial differences in urinary potassium excretion. J Am Soc Nephrol 2008; 19: 1396–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yeung SMH, Vogt L, Rotmans JI et al. Potassium: poison or panacea in chronic kidney disease? Nephrol Dial Transplant 2019; 34: 175–180 [DOI] [PubMed] [Google Scholar]
- 189. Kovesdy CP, Matsushita K, Sang Y et al. ; CKD Prognosis Consortium. Serum potassium and adverse outcomes across the range of kidney function: a CKD prognosis consortium meta-analysis. Eur Heart J 2018; 39: 1535–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Hughes-Austin JM, Rifkin DE, Beben T et al. The relation of serum potassium concentration with cardiovascular events and mortality in community-living individuals. Clin J Am Soc Nephrol 2017; 12: 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Collins AJ, Pitt B, Reaven N et al. Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease, and/or diabetes. Am J Nephrol 2017; 46: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Gasparini A, Evans M, Barany P et al. Plasma potassium ranges associated with mortality across stages of chronic kidney disease: the Stockholm CREAtinine measurements (SCREAM) project. Nephrol Dial Transplant 2019; 34: 1534–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kovesdy CP, Regidor DL, Mehrotra R et al. Serum and dialysate potassium concentrations and survival in hemodialysis patients. Clin J Am Soc Nephrol 2007; 2: 999–1007 [DOI] [PubMed] [Google Scholar]
- 194. Gritter M, Vogt L, Yeung SMH et al. Rationale and design of a randomized placebo-controlled clinical trial assessing the renoprotective effects of potassium supplementation in chronic kidney disease. Nephron 2018; 140: 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]