

Abstract
Despite its great success, cancer immune therapy is still of limited efficacy in the majority of cancer patients1,2. Many efforts are underway to identify novel approaches to enhance immune checkpoint therapy3–5. Here we show that inhibition of PCSK9, a key protein in regulating cholesterol metabolism6–8, can boost tumor response to immune checkpoint therapy, albeit through a mechanism independent of its cholesterol regulating functions. Deletion of the PCSK9 gene in murine cancer cells significantly attenuated or prevented their growth in mice in a cytotoxic T-cell-dependent manner. It also enhanced the efficacy of anti-PD1 immune checkpoint therapy significantly. Furthermore, clinically approved PCSK9-neutralizing antibodies could synergize with anti-PD1 therapy in suppressing tumor growth in murine tumor models. PCSK9 inhibition, either through genetic deletion or PCSK9 antibodies, caused a significant increase in tumor cell surface major histocompatibility protein class I (MHC I) expression, which promoted robust intratumoral infiltration of cytotoxic T-cells. Mechanistically, we discovered that PCSK9 could disrupt the recycling of MHC I to the cell surface by promoting its relocation and degradation in the lysosome through physical association. Taken together, we believe PCSK9 inhibition is a promising strategy to enhance cancer immune checkpoint therapy.
The importance of cholesterol metabolism in cancer immunotherapy was highlighted recently by the finding that inhibition of ACAT1, a cholesterol esterification enzyme, could potentiate CD8+ T cells’ anti-tumor activities by enhancing the clustering of T cell receptors9. It was also reported that lowering blood cholesterol levels could boost adoptive T cell cancer immunotherapy10. Cholesterol in the cellular membrane has also been shown to play key roles in MHC I recycling11. Because of those findings, we hypothesized that PCSK9 might play a role in regulating anti-tumor immunity. PCSK9’s capacity to regulate cholesterol levels in the body lies in its ability to down-regulate the cell surface level of low-density lipoprotein receptor (LDLR) by redirecting it to the lysosome for degradation instead of recycling back to the surface through both extracellular and intracellular routes12–16, thereby reducing cholesterol metabolism. In addition to LDLR, PCSK9 was also shown to regulate the cell surface levels of other receptors such as very low density lipoprotein receptor (VLDLR), apolipoprotein E receptor 2 (ApoeER2)17, low density lipoprotein-related protein 1 (LRP-1)18, CD3619, and beta secretase 1 (BACE1)20. The ability of PCSK9 to regulate a diverse group of cell surface proteins gave us hints it might also be able to influence additional membrane proteins that are important in anti-tumor immune response. Targeting PCSK9 for tumor treatment is also attractive because two neutralizing antibodies against it, evolocumab and alirocumab, have already been approved for human clinical use to lower cholesterol levels21,22.
PCSK9 deficiency and tumor growth rate
To assess the roles of PCSK9 on tumor growth, we knocked out the PCSK9 gene in four malignant murine cancer cell lines (B16F10, 4T1, MC38, and CT26) by use of the CRISPR/Cas9 technology (Extended Data Fig. 1a)23,24. PCSK9 knockout (PCSK9KO) did not alter the morphology or the in vitro growth rates of tumor cells (Extended Data Fig. 1b–d). When PCSK9-deficient cells were inoculated into syngeneic mouse hosts, however, their abilities to form tumors were significantly attenuated in comparison to vector controls (Fig 1a–h). Preferential growth suppression of PCSK9-deficient cells was further confirmed through in vivo competition experiments with fluorescently labeled tumor cells (Extended Data Fig. 1e–g). Furthermore, reintroduction of PCSK9 into the PCSK9KO B16F10 cells rescued tumorigenic abilities of the PCSK9KO B16F10 cells (Extended Data Fig. 2a–c), thereby ruling out potential off-target CRISPR/Cas9 knockouts being responsible for observed tumor growth delay.
Fig1. depletion attenuates tumor growth in syngeneic mice.
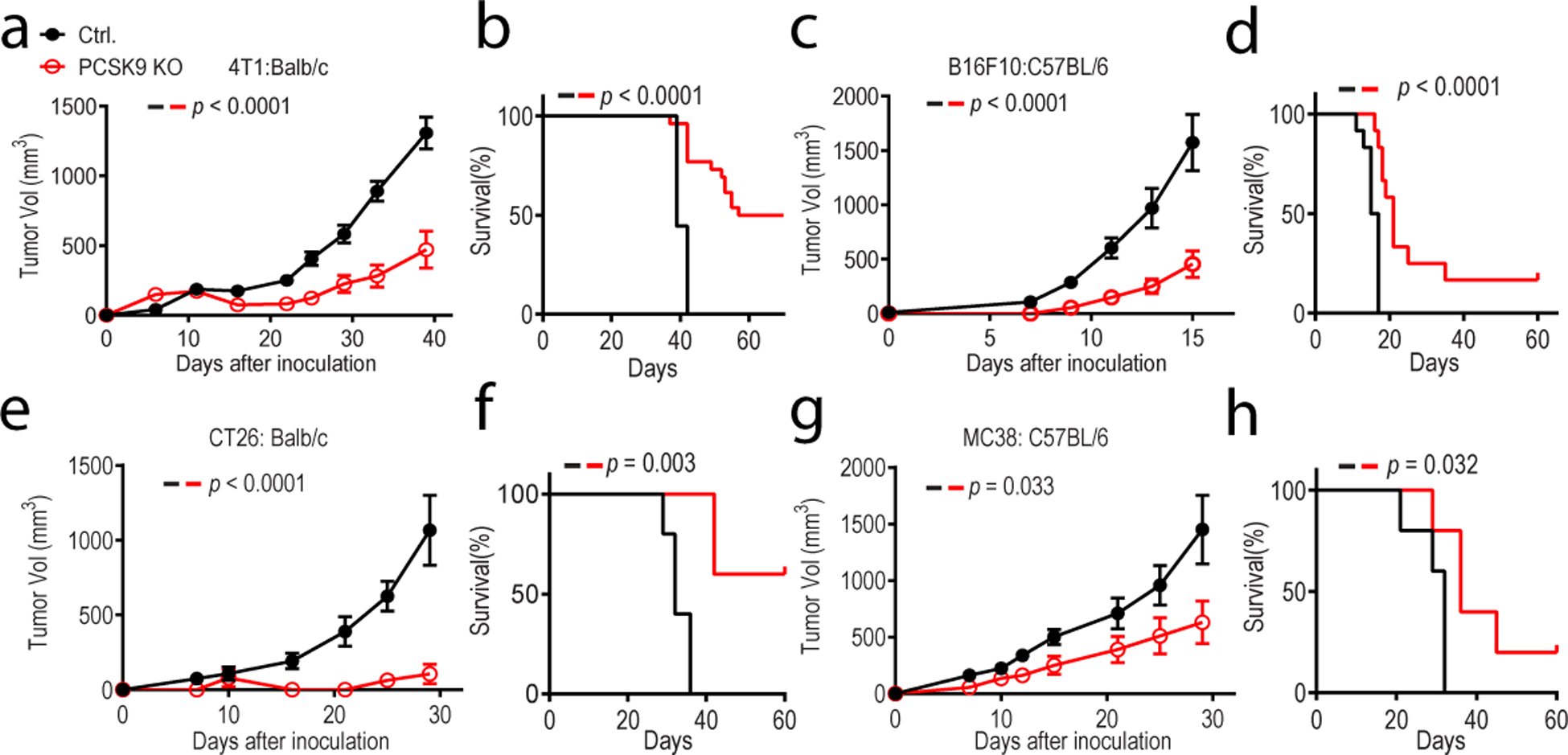
About 1 × 105 vector control and PCSK9 knockout murine tumor cells were inoculated subcutaneously into syngeneic mice and observed for tumor formation. Both tumor size and overall survival were monitored. a-b. 4T1 breast cancer line grown in Balb/c mice. n=9 and 20 mice for control and PCSK9KO tumor cells, respectively. c-d. B16F10 melanoma line grown in C57BL/6 mice. n=12 mice for both groups. e-f. CT26 colon cancer line grown in Balb/c mice. n=5 mice for both groups. g-h. MC38 colon cancer line grown in C57BL/6 mice. n=5 mice for both groups. Error bars: mean ± S.E.M. P values were calculated by two-way ANOVA in a, c, e, g and log-rank test in b, d, f, h, respectively.
To determine involvement of the immune system, PCSK9-deficient and vector control 4T1 and B16F10 tumor cells were inoculated into NCG mice deficient in T cells, B cells, and NK cells. Our results showed that PCSK9 deficiency had no effect on tumor growth in NCG mice (Extended Data Fig. 2d–i). Furthermore, we also show that PCSK9 deficiency did not influence B16F10 tumor formation in Rag1-deficient mice, which do not have mature T or B cells (Extended Data Fig. 2j, k).
PCSK9 inhibition and immunotherapy
Because PCSK9 is known to regulate cholesterol levels by promoting LDLR degradation in the lysosome13,14, we also examined their involvement regulating tumorigenicity of murine tumors. We generated LDLR deficient B16F10 cells (Extended Data Fig. 3a) and evaluated the tumor forming abilities of these cells in C57BL/6 mice. Our results indicated LDLR deficiency in the tumor cells did not have any significant effect on tumor growth (Extended Data Fig. 3b–c). We further evaluated tumor growth from control and PCSK9KO B16F10 melanoma cells in syngeneic wild type and LDLR−/− mice that were fed a high-fat diet, which should cause cholesterol levels 10–20 fold higher in the latter25. Our results indicated that the attenuation in tumor growth from PCSK9 deficiency was not affected by host LDLR status or cholesterol levels (Extended Data Fig.3d, e).
To evaluate if PCSK9-deficiency could synergize immune checkpoint blockade therapy1,26, we carried out tumor growth delay experiments with a murine anti-PD1 immune checkpoint inhibitor in syngeneic mice inoculated with PCSK9-deficient B16F10, MC38, 4T1, and CT26 tumor cells. Our results indicate that anti-PD1 antibody synergized with PCSK9 deficiency in all four models (Fig. 2a–f, and Extended Data Fig. 4a–f) in suppressing tumor growth.
Fig.2. inhibition overcomes tumor resistance to anti-PD1 therapy.
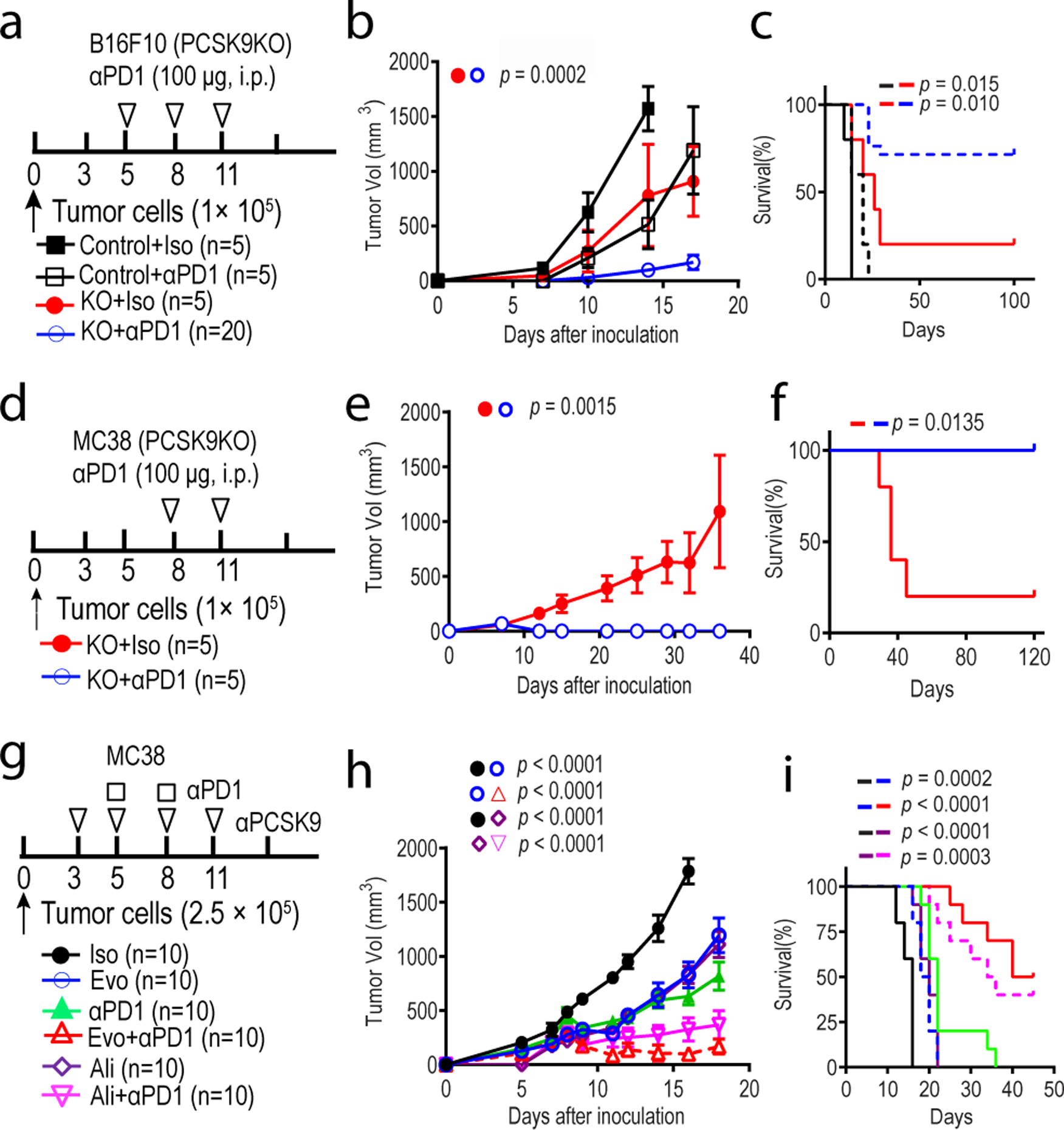
a-c. Treatment of vector control and PCSK9 knockout B16F10 melanoma with an anti-PD1 antibody in syngeneic mice. Experimental protocol (a), tumor growth curve (b), and overall survival (c) were shown. n=5, 5, 5, and 20 mice in the four groups, respectively. d-f. Treatment of vector control and PCSK9KO MC38 colon cancer with an anti-PD1 antibody in syngeneic mice. Experimental protocol (d), tumor growth curve (e), as well as overall survival (f) were shown. n=5 mice per group. g-i. Treatment of MC38 colon cancer with combined anti-PCSK9 (evolocumab, Evo; or alirocumab, Ali) and anti-PD1 antibodies in mice. Experimental protocol (g), tumor growth curve (h), as well as overall survival (i) were shown. n=10 mice per group. Shown were combined results from two separate experiments. Error bars represent mean ± S.E.M. P values were calculated by two-way ANOVA in b, e, h and logrank test in c, f, i, respectively.
We next examined if the observed synergy between PCSK9 deficiency and anti-PD1 antibody treatment could be recapitulated by use of evolocumab or alirocumab, two PCSK9 neutralizing antibodies that are approved to treat hyperlipidemia in human patients21,22. Both were shown to be effective in lowering cholesterol in mice27,28. While administration of the anti-PCSK9 antibodies alone delayed tumor growth of MC38 tumors, their efficacies were enhanced significantly when combined with an anti-PD1 antibody, with long-term survival of some host mice (Fig. 2g–i). Thus our results showed that antibody-mediated PCSK9 neutralization could enhance anti-PD1 therapy in murine tumor models.
We next assessed if anti-PCSK9 antibodies could work in tumors that had developed resistance to immune checkpoint therapy. We first developed anti-PD1 resistant MC38R colon cancer model through three rounds of in vivo/in vitro selection (Extended Data Fig. 4g). Tumors established in syngeneic mice from the MC38R tumor cells were then treated with an anti-PD1 and/or evolocumab. Evolocumab treatment was effective either alone or in combination with anti-PD1 treatment for MC38R tumors (Extended Data Fig. 4h–j). Furthermore, evolocumab had no effect on tumor growth in PCSK9-deficient MC38 tumors (Extended Data Fig. 4k–m), thereby indicating tumor cell intrinsic rather than host PCSK9 was important for antitumor efficacy of evolocumab.
In further experiments, we examined if those mice that remain tumor-free after initial tumor cell inoculation and treatment could resist a re-challenge with parental tumor cells. Lethal doses of wild type tumor cells with an intact Pcsk9 gene were injected into those mice that remained tumor free after the initial challenge. Our results showed that a significant fraction of long-term survivors of 4T1-PCSK9KO tumors (Extended Data Fig. 5a–c), B16F10-PCSK9KO tumors (Extended Data Fig. 5d–f), and MC38-PCSK9KO tumors (Extended Data Fig. 5g–i), rejected the WT tumor cell re-challenge, indicating an anti-tumor immune memory.
PCSK9 and lymphocyte infiltration
We next attempted to quantify immune effector cells in control and PCSK9-deficient B16F10 tumors by immunofluorescence staining and flow cytometry (see Extended Data Fig. 5j for flow cytometry gating strategy). Immunofluorescence staining indicated that PCSK9 depletion caused an overall increase in intratumoral infiltration of CD45+ leukocytes (Extended Data Fig. 6a) and CD8+ cells (Extended Data Fig. 6b). Of particular interest was the observation of CD8+ cells staying mostly in the periphery in control tumors (Extended Data Fig. 6b, top panels) but moving into tumor cell-rich areas in the PCSK9-deficient B16F10 tumors (Extended Data Fig. 6b, bottom panels).
Flow cytometry analyses confirmed significant increases in intratumoral CD8+ cytotoxic T cells (CTLs) (Fig. 3a), CD4+ T helper (Th) cells (Fig. 3b), γδT cells (Fig. 3c), and NK cells (Fig. 3d) in PCSK9-deficient tumors. In contrast, no significant increases in CD4+Foxp3+ regulatory T (Treg) cells were observed (Fig. 3e). Moreover, no increases in CD4+ or CD8+ T-cells were observed in the spleen of host mice (Extended Data Fig. 6c). The ratios of CTLs vs Tregs in the PCSK9-deficient tumors were significantly increased (Fig. 3f). Consistently, both the numbers and percentages of IFNγ+, or Granzyme B+ (GZMB+) CTLs also increased significantly in PCSK9-deficient tumors (Fig. 3g, h, Extended Data Fig. 6d), as well as intratumoral expression of IFNG and GZMB (qRT-PCR, Extended Data Fig. 6e, f). On the other hand, measurement of T cell exhaustion markers showed no significant changes (Extended Data Fig. 6g–i).
Fig.3. depletion enhances intratumoral T-cell infiltration.
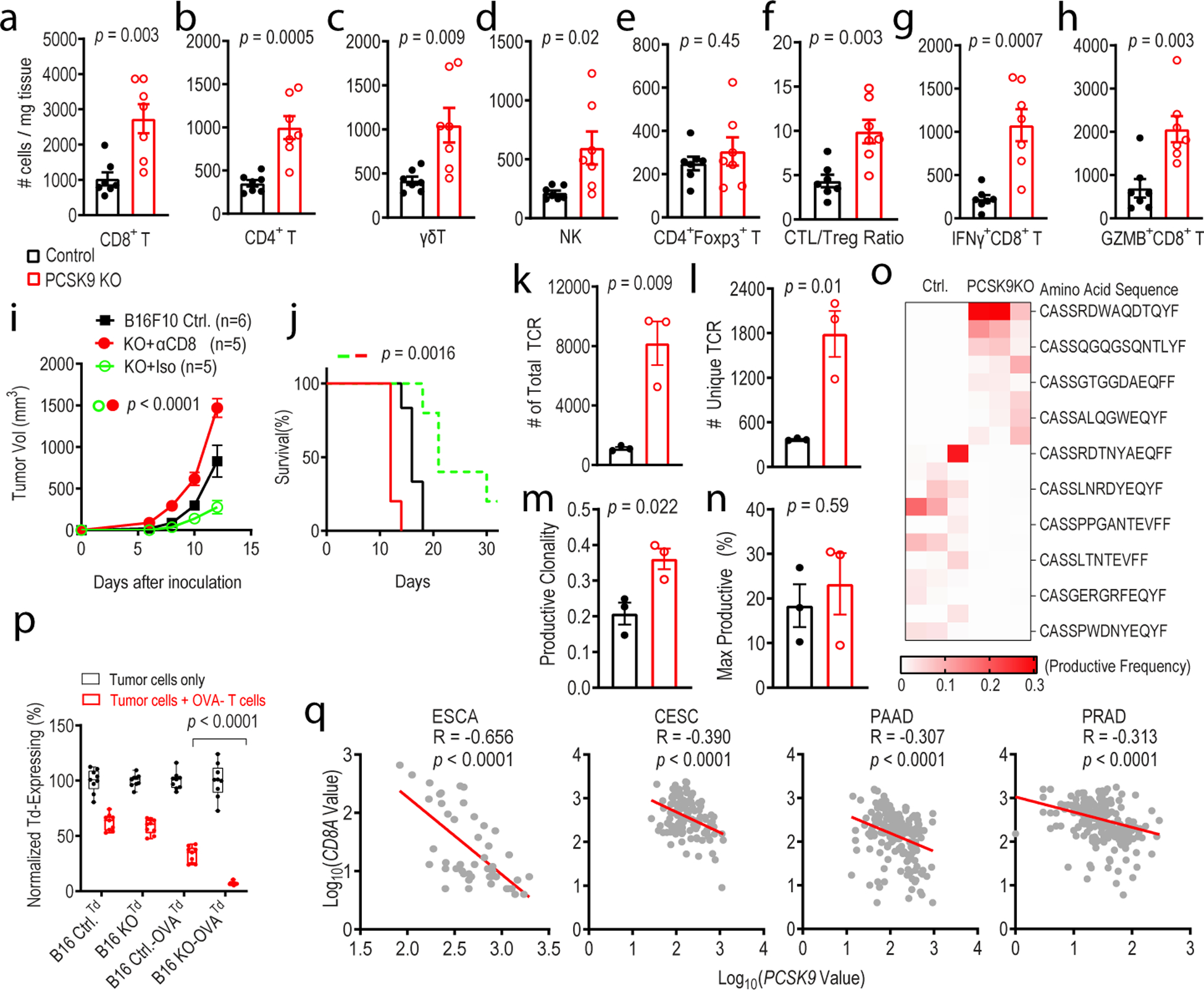
a-e. Quantitative estimate of various immune effector cells per mg of tumor tissue in vector control and PCSK9KO B16F10 tumors as determined by flow cytometry. n=7 tumors per group. f. Ratio of CD8+ T /CD4+Foxp3+ Treg cells in control and PCSK9KO B16F10 tumors. n=7 tumors per group. g-h. Average numbers of tumor-infiltrating IFNγ+ CD8+ T (g) and Gzmb+CD8+T (h) cells per mg of tumor tissue in control or PCSK9 KO tumors. n=7 tumors per group. i-j. Tumor growth (i) and host survival (j) from control and PCSK9KO B16F10 tumor cells in C57BL/6 mice depleted of CD8+ T cells. n=6, 5, 5 tumors in the three groups. k-o. Total TCR (k), unique TCR (l), productive clonality (m), and max productive frequency (n), and heatmap of top 5% TCR (o) in control and PCSK9 knockout tumors by use of TCRB CDR3 sequencing. n=3 tumors per group. p. Quantitative estimates of the fraction of live control or PCSK9KO B16F10-tdTomato tumor cells remaining after 24 hrs of incubation with activated OVA-specific T cells. n=3 biologically independent samples per group. Three representative fields from each sample were counted. q. Negative correlation of PCSK9 mRNA levels with that of CD8A mRNA levels in human esophageal carcinoma (ESCA, 47 samples), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC,112 samples), pancreatic adenocarcinoma (PAAD,173 samples), prostate adenocarcinoma (PRAD, 313 samples). Data from the GENT database. R represents Pearson correlation coefficient. Error bars represent mean ± S.E.M throughout the figure. P values in a-h, k-n, and p were calculated by unpaired two-sided t-test. P values in i and j determined by two-way ANOVA and log-rank test, respectively. P values in q were calculated by F test.
We next examined the effect of evolocumab on the tumor immune microenvironment in the 4T1 murine breast cancer model. Our results indicated that it also had clear anti-tumor efficacy in this model (Extended Data Fig. 6j–l). However, anti-PD1 therapy was ineffective, as reported before29. When intratumoral CTLs were analyzed, it was discovered that both anti-PD1 and anti-PCSK9 antibodies could boost the numbers of CTLs inside the tumor (Extended Data Fig. 6m). However, only the presence of evolocumab could boost the number of active IFNγ+ CTLs (Extended Data Fig. 6n).
In order to determine the relative importance of different immune effector cells, we used a well-established antibody-based approach30 to deplete CD4+ T cells, CD8+ T cells, or NK cells to assess their relative importance in regulating growth of PCSK9-deficient tumors. Our data indicate that depletion of CD8+ T cells completely abolished the tumor growth delay of PCSK9-deficient tumors (Fig. 3i, j). In contrast, depletion of CD4+ T cells or NK cells only had marginal effects (Extended Data Fig. 7a–e).
To further characterize the effects of PCSK9 deficiency on intratumoral T-cells, we carried out molecular analysis of the T-cell receptor (TCR) repertoire. Our analysis suggests that total TCR counts (Fig. 3k) and the number of unique TCRs (Fig. 3l) were significantly increased in PCSK9-deficient tumors. These results suggest that both the number and diversity of mature T-cells were significantly elevated in PCSK9-deficient tumors. Further analysis showed that productive clonality, a measurement of the dominance of individual T-cell clones, was elevated in PCSK9-deficient tumors (Fig. 3m), indicating significant expansion and dominance of a subset of T-cell clones in addition to the overall increase in the diversity of mature T-cells. Closer examination revealed that the maximum dominance of individual T-cell clones in both control and PCSK9-deficient tumors was close to 20–30%, which suggests that a few T-cell clones accounted for 30% of all intratumoral T-cell population (Fig. 3n). Of interest is the fact that the dominant clones in PCSK9-deficient tumors were different from those in control tumors (Fig.3o).
We then determined the influence of PCSK9 deficiency on CTL-mediated cell killing of tumor cells by using the OT-1 transgenic mouse model31 where T-cells engineered to express TCR specific for the chicken ovalbumin (OVA) antigen (SIINFEKL) were isolated. Their cytotoxic effects against OVA-transduced, tdTomato-labeled vector control and PCSK9-deficient B16F10 melanoma cells were evaluated in vitro. Our results indicated that PCSK9 more susceptible to killing by CTLs (Fig. 3p and Extended Data Fig. 7f). Consistent with our preclinical data, PCSK9 mRNA expression was negatively correlated with expression of CTL marker CD8A in several human malignancies (Fig. 3q).
PCSK9 and MHC I regulation
We next focused on the molecular mechanism(s) involved in the enhanced CTL killing of PCSK9-deficient tumor cells. Because of known roles of PCSK9 in regulating cell surface protein levels such as LDLR17,32,33, we hypothesized that PCSK9-deficiency may influence antigen presentation on the surface of tumor cells. Indeed, our results indicated that H2-Kb/SIINFEKL staining was significantly enhanced on the surface of IFNγ-stimulated PCSK9-deficient B16F10-OVA cells when compared with control B16F10-OVA cells (Fig. 4a and Extended Data Fig. 7g). These results demonstrated that PCSK9 had a strong influence on MHC I presentation of peptide antigens. We further examined the effect of PCSK9 knockout on H2b MHC I alloantigen levels on the surface of B16F10 tumor cells grown in vivo. Our analysis indicated that tumor cell surface MHC I expression was significantly increased in PCSK9-deficient tumors when compared with control (Fig. 4b and c). Similarly, PCSK9 deficiency also caused a significant increase in H2d MHC I alloantigens in IFNγ-treated 4T1 cells in vitro (Fig. 4d). In contrast, PCSK9 deficiency failed to show any effect on MHC II expression and only had a marginal effect on PD-L1 expression in B16F10 cells (Extended Data Fig. 7h and i). We further observed increased human leukocyte antigen (HLA)-A2 levels in human breast cancer line MDA-MB-231 with PCSK9KO (Extended Data Fig. 7j, Fig. 4e) or incubated with anti-PCSK9 antibodies (Extended Data Fig. 7k). Similarly, evolocumab treatment of murine 4T1 tumors also boosted MHC-I levels on the surface of 4T1 cells (Extended Data Fig. 7l). On the other hand, incubation of exogenous PCSK9 protein with PCSK9-deficient MDA-MB-231 cells caused a decrease in surface HLA-A2 levels (Fig. 4f).
Fig. 4. PCSK9 promotes lysosome-mediated degradation of MHC I in tumor cells.
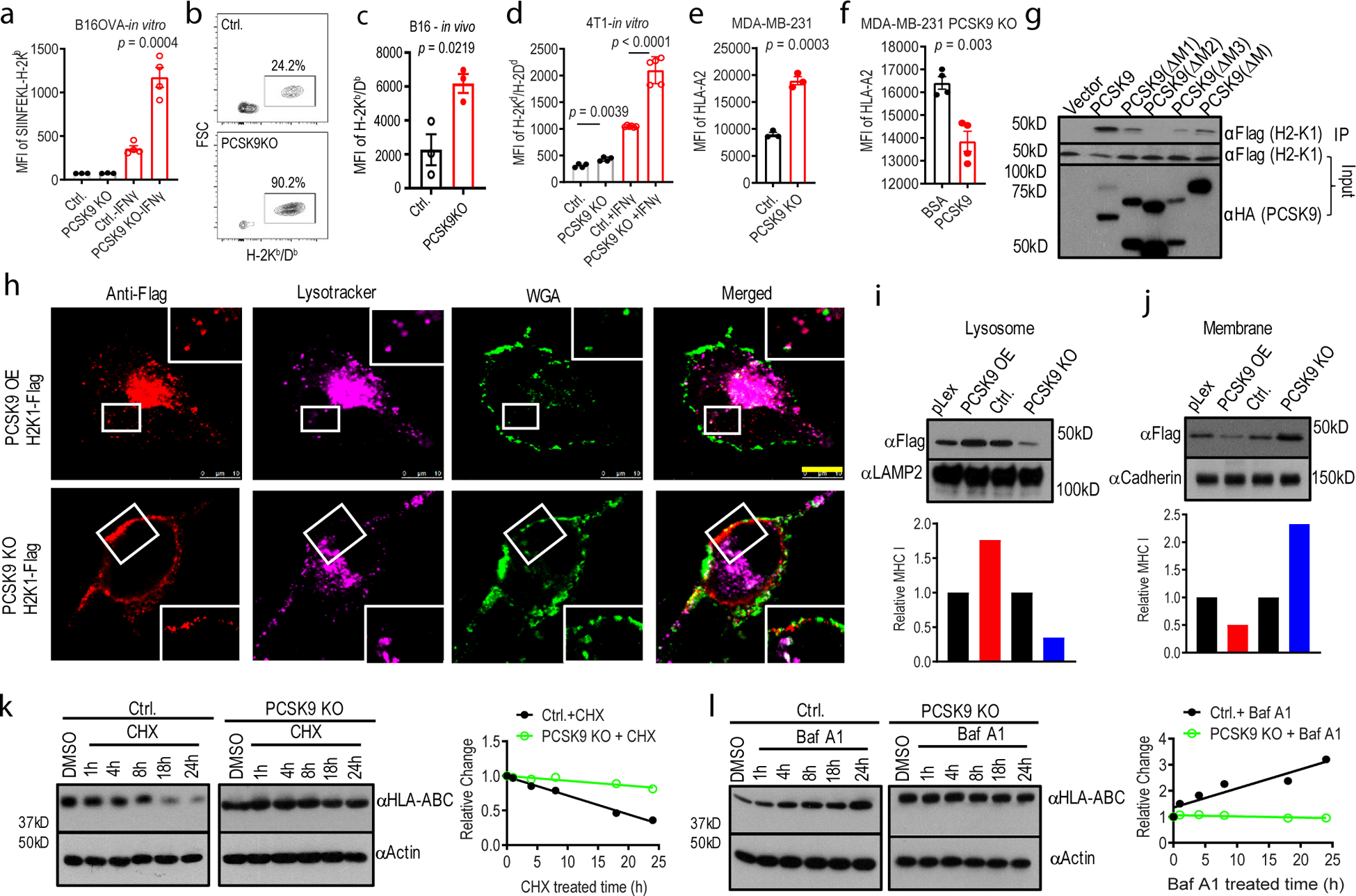
a. Quantitative estimates of SIINFEKL-H-2Kb levels in B16F10-OVA cells with or without IFNγ treatment in control and PCSK9KO B16F10 cells. n=4,4,6,5 biologically independent samples, respectively. b-c. Flow cytometry estimates of H-2Kb/Db levels on the surface of subcutaneously grown control and PCSK9-deficient B16F10 cells. n=3 tumors per group. d. Quantitative estimate of MHC I levels on the surface of in tissue cultured control and PCSK9-deficient 4T1 cells treated with or without IFNγ. n=5 biologically independent samples. e. The effect of PCSK9 deficiency on HLA-A2 expression on the surface of control and PCSK9KO MDA-MB-231 cells. n=3 biologically independent samples. f. The effect of exogenous PCSK9 protein on HLA-A2 degradation on the surface of MDA-MB-231 PCSK9KO cells. n=4 biologically independent samples. g. Interaction of mouse PCSK9 and H2-K1 in 293T cells were transduced with FLAG-tag labeled H2-K1 gene in combination with HA-tag labeled full-length or various deleted PCSK9 genes. h. PCSK9-promoted H2-K1 migration into the lysosome. Representative fluorescence confocal images of MHC I (H2-K1Flag) distribution in PCSK9 (PCSK9-HA) over-expressing (top panels) or PCSK9 KO (lower panels) B16 F10 cells. Scale bar: 10 μm. Insets showed magnified areas with additional details of co-localization. i-j. Western blot quantification of H2-K1-Flag in the lysosome (i) and plasma membrane (j) fractions of PCSK9 overexpressing (PCSK9-OE) and PCSK9KO B16F10 cells. Bottom panels are the quantitative estimates of MHC I (H2-K1-Flag) expression levels based on data in the top panels. k-l. Western blot analysis of HLA-ABC expression in cycloheximide (CHX, k)- and bafilomycin A1 (BafA1, l)-treated vector control and PCSK9KO MDA-MB-231 cells. In k and l, the right panels showed the quantitative estimates of HLA-ABC levels based on WB analysis. Error bars in a-f, mean ± S.E.M. P values in a-f were determined by unpaired two-sided t test. Two independent experiments were done with similar results for g-l.
We next over-expressed H2-K1 in B16F10 cells (Extended Data Fig. 8a) and evaluated its tumor-forming abilities and response to anti-PD1 therapy. Our result indicated that H2-K1 expression could significantly attenuate tumor growth alone or in combination with anti-PD1 (Extended Data Fig. 8b–c). In contrast, genetic depletion of H2-K1 abrogated tumor growth delay induced by PCSK9 inhibition (Extended Data Fig. 8d, e). Our results therefore established H2-K1 as an essential downstream factor of PCSK9 in regulating tumor growth.
How does PCSK9 regulate cell surface MHC I expression? Previously it was reported that cholesterol levels may influence MHC I recycling11. Therefore, it is possible that PCSK9 could regulate MHC I indirectly through LDLR, the key cholesterol regulator that is also an established downstream target of PCSK9. In order to determine if LDLR levels could regulate MHC I levels downstream of PCSK9, we generated B16F10 cells with LDLR knockdown alone or in combination with PCSK9KO (Extended Data Fig. 8f). We subsequently showed that LDLR knockdown did not diminish the growth suppression caused by PCSK9 deficiency in B16F10 tumors (Extended Data Fig. 8g and h). Neither did it have any influence on MHC I expression on the surface of B16F10 cells (Extended Data Fig. 8i). Furthermore, LDLR knockout did not affect the up-regulation of surface MHC I expression induced by PCSK9 deficiency (Extended Data Fig. 8j).
Because of PCSK9’s known ability to down-regulate LDLR protein through physical interaction and re-direct its transport into the lysosome for degradation12–16, we examined if PCSK9 could regulate MHC I surface levels by direct interaction. We first created recombinant mouse PCSK9 mutants with various internal deletions or truncations based on domain structure (Extended Data Fig. 9a). Each of those deleted PCSK9 genes was then co-transduced into B16F10 cells together with a full-length H2-K1 gene. Our analysis showed that deletion of M2 domain within the C-terminal region PCSK9 showed almost complete loss of binding to H2-K1 while deletions of the other two domains (M1 & M3) and the aa367–386 domain, which was involved in LDLR binding34, in the catalytic region, also showed reduced H2-K1 binding (Fig. 4g, Extended Data Fig. 9b). On the other hand, deletion mapping of H2-K1 showed that absence of the α1 region (aa66–100) and in particular, of amino acids aa68–70, which sits in the loop right before the helix structure of the α1 domain35, completely abolished binding to PCSK9 (Extended Data Fig. 9c, d). To demonstrate the functional importance of the interacting domains in PCSK9 and H2-K1, we re-introduced WT or PCSK9ΔM2 (PCSK9 with deletion of the M2 region, aa535–608) into PCSK9KO B16F10 cells. In tumor growth experiments, re-introduction of the wild type PCSK9 abolished the tumor growth delay while that of PCSK9ΔM2 had no effect (Extended Data Fig. 9e, f). In comparison, re-introduction of WT H2-K1 gene attenuated the tumor-forming abilities of both H2-K1 deficient and PCSK9/H2-K1DKO cells. On the other hand, re-introduction of the H2-K1Δ68–70 did not slow down tumor growth (Extended Data Fig.9g, h).
To understand how PCSK9 regulate MHC I surface expression, immunofluorescence co-staining of exogenously expressed H2-K1 and/or PCSK9 were carried out in B16F10 cells with PCSK9 knockout or PCSK9 over-expression (OE). In PCSK9OE cells, more H2-K1 was localized in the lysosome and not in the plasma membrane (Fig. 4h, top panels). On the other hand, in PCSK9KO cells, H2-K1 staining indicated a more plasma membrane localization (Fig. 4h, lower panels). Western blot analysis of fractionated cellular lysates confirmed the immunofluorescence staining results. In the lysosome fraction, PCSK9 OE caused an increase in H2-K1 protein while PCSK9KO induced a decrease (Fig. 4i). In contrast, in the membrane fraction, PCSK9OE reduced the relative abundance of the H2-K1 protein while PCSK9 knockout increased it (Fig. 4j).
The functional significance of PCSK9/H2-K1 physical association and localization in the lysosome was further analyzed by WB analysis. In the absence of de novo protein synthesis (cycloheximide), HLA-ABC levels in PCSK9KO MDA-MB-231 cells declined significantly slower compared to control cells (Fig. 4k). However, when the cells were exposed to bafilomycin to inhibit lysosome function, the levels of HLA-ABC increased significantly in control cells but did not change much in PCSK9KO cells (Fig. 4l), thereby suggesting a key role for PCSK9 in regulating HLA-ABC levels through lysosome-mediated degradation. Our results thus suggest that PCSK9 down-regulates MHC I surface levels in a manner similar to its negative regulation of LDLR by lysosome-mediated degradation. Consistent with the preclinical findings, patients with high tumor PCSK9 mRNA expression had worse overall survival than those with low expression in 9 different TCGA (The Cancer Genome Atlas) patient cohorts (Extended Data Fig. 9i).
Discussion
The finding that PCSK9 plays important roles in regulating cell surface MHC I levels (see Extended Data Fig. 10 for a summary schema) and thus influences intratumoral immune infiltration is novel mechanistically and has great translational potential. It suggests that PCSK9 neutralization may promote intratumoral T cell infiltration and thus render tumors more responsive to immune checkpoint therapy. Previous studies have demonstrated that the amount of active T-cells is positively correlated with the success of immune checkpoint blockade therapy36,37. Our results, demonstrating potent tumor-suppressing efficacy of combined anti-PD1 antibodies with either evolocumab or alirocumab, provide compelling rationales for conducting future clinical trials in human cancer patients. Given the well-known safety profiles of the anti-PCSK9 antibodies, PCSK9 inhibitors may enhance immune checkpoint blockade therapy without additional side effects.
Methods
Cell lines.
B16F10 mouse melanoma cells, CT26 mouse colon carcinoma, 4T1 mouse breast carcinoma cells, MDA-MB-231 human breast cancer cells were purchased from the Cell Culture Facility of Duke University School of Medicine. Their identities were verified by the STR method. 293T cells were purchased from ATCC (Manassas, VA). MC38 mouse colon adenocarcinoma cells was purchased from Kerafast (Boston, MA). B16F10, CT26, 4T1, MC38, MDA-MB-231 cells were all grown in DMEM (Sigma) with 10% fetal bovine serum (FBS) and 100 units/ml penicillin and 100 μg/ml streptomycin antibiotics. All cell lines were subjected to mycoplasma test periodically by use of the Universal Mycoplasma Detection Kit (ATCC).
CRISPR/Cas9-mediated gene knockout.
Knockout cells were generated by use of lentivirus mediated CRISPR/Cas9 technology. Single guided RNA (sgRNA) sequences were designed with the use of a public domain online CRISPR design tool (www.chopchop.cbu.uib.no)38. SgRNA sequences targeting mouse PCSK9: #1, AGAACCACGAGTGGCCCCGA, #2, CGGCTATACCCACCGGCCAG; #3, CATGCTTCATGTCACAGAGT; #4, TCATTTGACGCTGTCTGGGG; human PCSK9: #1,CAGATGGGGGTCTTACCGGG, #2,TCTTGGTGAGGTATCCCCGG; mouse LDLR: #1, ACAGTCGACATCCCCGTCGC, #2,CCGCGGATCTGATGCGTCGC; mouse H2-K1:#1,CGAATCGCCGACAGGTGCGA, #2, CGAGATATGAGCCGCGGGCG. Double stranded oligos encoding the sgRNA sequences were cloned into BsmB1(Thermal Fisher Scientific) digested plasmid LentiCRISPRv2 (deposited by Dr. Feng Zhang of MIT to Addgene, Cambridge, MA)24,39, which co-expresses Cas9 and sgRNA in the same vector. The sgRNA-encoding CRISPR lentivirus vectors were then produced according an established protocol by the Trono lab (https://www.epfl.ch/labs/tronolab/laboratory-of-virology-and-genetics/lentivectors-toolbox/). To generate the knockout cell lines, target cells were infected with sgRNA-encoding CRISPR lentivirus and cultured in DMEM with 10% FBS and selected in puromycin (1μg/ml for B16F10, CT26, MC38, MDA-MB-231 and 4μg/ml for 4T1) for 7–10 days. They were then seeded into 96-well plates. Clones emerging from the plates were then expanded and expression of target protein (PCSK9) in infected cells were detected by western blot to verify the knockdown. For generating PCSK9KO/LDLR-KD or PCSK9KO/H2-K1KD B16F10 cell lines, mouse LDLR or H2-K1 sgRNA were cloned into LentiCRISPRv2 vector with neomycin gene. PCSK9 knockout cells were infected with sgRNA-encoding CRISPR lentivirus and selected in G418 for 10 days. Expression of H2-K1 and LDLR protein in mixed infected cells were then detected by western blot to verify the knockdown.
Soft agar colony formation assay.
To measure the ability of PCSK9 deficient tumor cells to grow in 3D, soft agar assay was performed according to an established protocol40. Cells were seeded at a density of 10,000 cells per well in 6-well plates with soft agar. The colonies were fixed and stained with 0.005% crystal violet after 3 weeks of culture. The number of colonies per well were then counted. Two independent experiments were carried out.
Antibodies and related reagents.
Antibodies and reagents used in this study western blot (WB), immunofluorescence (IF), and immunoprecipitation (IP). The list of antibodies, their source, and dilution information for various applications are as follows. FITC anti-Mouse CD45(Clone 30-F11, Cat No. #103108, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). Pacific blue anti-Mouse CD3ε (Clone 145–2c11, Cat No. #100334, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). AF647 anti-Mouse CD4 (Clone GK1.5, Cat No. #100424, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). APC/Fire750 anti-Mouse CD8a (Clone 53–6.7, Cat No. #100766, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-Mouse NK1.1 (Clone PK136, Cat No. #108707, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-Mouse Foxp3 (Clone MF-14, Cat No. #126403, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). APC anti-Mouse TCR g/d (Clone GL3, Cat No. #118115, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-Mouse Gzmb (Clone QA16A02, Cat No. #372207, 5 μl per 10e6 cells in 100 μl dilution buffer, Biolegend). AF647 anti-Mouse IFNγ, (Clone XMG1.2, Cat No. #505816, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). FITC anti-Human HLA-A2 (Clone BB7.2, Cat No. #343322, 0.5 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). FITC anti-Mouse H-2Kb/Db (Clone 28-8-6, Cat No. #114605, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-Mouse H-2Kb/Db (Clone 28-8-6, Cat No. #114608, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). FITC anti-Mouse H-2Kd/Dd (Clone 34-1-2S, Cat No. #114706, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE/Cy7 anti-Mouse H-2Kb bound to SIINFEKL (Clone 25-D1.16, Cat No. #141607, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). AF647 anti-mouse PD1 (Clone 29F.1A12, Cat No. #135229, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-mouse TIGIT (Clone 1G9, Cat No. #142103, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-mouse CTLA4, (Clone UC10–4B9, Cat No. #106305, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE anti-mouse TNFα (Clone MP6-XT22, Cat No. #506305, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). APC anti-mouse I-A/I-E (Clone M5/114.15.2, Cat No. #107613, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE/Cy7 anti-mouse PD-L1 (Clone 10F.9G2, Cat No. #124313, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). TruStain FcX™ (anti-mouse CD16/32) (Clone 93, Cat No. #101319, 1 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). APC anti-Mouse TCRV_5.1/5.2 (Clone MR9–4, Cat No. #139505, 0.25 μg per 10e6 cells in 100 μl dilution buffer, Biolegend). PE/Cy7 anti-Cas9 monoclonal antibody (Clone 6H4, Cat No. #25-6499-82, 0.06 μg per 10e6 cells in 100 μl dilution buffer, ThermoFisher). Purified anti-Mouse CD45 (Clone 30-F11, Cat No. #103101, IF, 1:50 dilution, Biolegend). Purified rat anti-Mouse CD8a (Clone 53–6.7, Cat No. #550281, IF, 1:50 dilution, BD). Mouse/Rat PCSK9 Antibody (Cat No.#AF3985, WB, 1 μg/mL dilution, R&D Systems). Mouse/human/rat PCSK9 antibody (Cat No. #55206–1-AP, WB, 1:1000 dilution, Proteintech). Mouse LDLR antibody (Cat No. #AF2255, WB, 0.1 μg/ml dilution, R&D). Anti-HLA Class 1 ABC antibody (Cat No. #15240–1-AP, WB, 1:10000 dilution, Proteintech). Anti-Flag antibody (Cat No. #F1804, IP, 2.5 μg/ml dilution; WB, 0.5 μg/ml dilution; Sigma). Anti-HA-Tag antibody (Clone C29F4, Cat No. #3724T, IP, 1:50; WB, 1:1000, Cell signaling Technology). LAMP2 antibody (Clone 2D3B9, Cat No. #66301–1-lg, WB, 1:1000, Proteintech). Pan-cadherin (Cat No. #NB200–592, WB, 1:200, Novus Biologicals). GAPDH Antibody (Cat No. #60004–1-Ig, WB, 1:20000 dilution, Proteintech). Actin Monoclonal Antibody (ACTN05 (C4)) (Clone ACTN05 (C4), Cat No. #MA5–11869, WB, 0.5 μg/ml dilution, ThermoFisher).
Western blot analysis.
Cells were washed with cold PBS, then lysed in RIPA buffer supplemented with protease inhibitors (Sigma). Equal amounts of lysates were separated by SDS-PAGE and transferred to PVDF membrane. Proteins were then probed with specific antibodies followed by secondary antibodies conjugated with HRP. The HRP signal was developed by use of ECL. Quantification of interested proteins was analyzed using Image J (NIH). Original uncropped images highlighting the cropped areas were shown in Supplementary Data Fig. 1.
Molecular cloning.
A series of mouse PCSK9 full-length and deletion mutants were generated by PCR from cDNA derived from the B16F10 cells. These mutants were then cloned into the pLEX-MCS recombinant lentiviral vector (Thermo-Fisher) to be used for gene expression. An HA tag was attached to the 3’prime end of different PCSK9 mutants. The same was done for mouse H2-K1 gene with a Flag tag.
SpeI-mPCSK9-F, 5’-CCGACTCTACTAGAGGATCCACTAGTGCCACCatgggcacccactgctctgc-3’;
mPCSK9-NotI-R, 5’-caaaggcctcctgggttcagGCGGCCGCTTATCCGTATGATGTTCCTG-3’;
mPCSK9–184-NotI-R (Δ184–693), 5’-atggaagcagccaggtggagGCGGCCGCTTATCCGTATGATGTTCCTG-3’;
mPCSK9–184-NotI-R (Δ453–693), 5’-cactgccccccagcacccatGCGGCCGCTTATCCGTATGATGTTCCTG-3’;
mPCSK9-(ΔM1 456–534) F, 5’-gttgtggatgctgcagttggcgcctgtctcatgggtgctggggggC-3’;
mPCSK9-(ΔM2 535–608) F, 5’-ctccttgattttgcattccagacggggaaccaggcagcatctcgcg-3’;
mPCSK9-(ΔM3 609–694) R-NotI, 5’-CctgctgccatgccccagggGCGGCCGCTTATCCGTATGATGTTCCTG-3’.
The primers used to clone H2-K1 are:
H2-K1-Flag-F, 5’-CCGACTCTACTAGAGGATCCACTAGTGCCACCatggtaccgtgcacgctgctcct-3’;
H2-K1-Flag-R, 5’-AGAGGGGCGACCGGTGGCCAGACGCGTtcaCTTGTCGTCATCGTCTTTGTAGTCCTCGAGcgctagagaatgagggtcat-3’;
H2K1–67-R, 5’-cggctcatatctcggattctccgc-3’;
H2K1-(Δ66–100) F 5’-gcggagaatccgagatatgagccgaccctgctcggctactacaa-3’;
H2K1-(Δ68–70) F 5’- gcggagaatccgagatatgagccgtggatggagcaggaggggcccgagtat −3’.
Ectopic gene expression.
PCR primers with sequences listed in the previous section were used to obtain full length and various deleted mouse PCSK9 with HA tag and full length and deleted H2-K1 with Flag tag. DNA fragments encoding PCSK9-HA were then cloned into the pLEX-MCS vector with a HA tag and H2-K1-Flag was cloned into pLEX-MCS vector with a Flag tag by use of the Gibson assembly kit following manufacturer’s instructions (New England Biolabs). Vectors encoding the genes were co-transfected into 293T cells for 24 hours and the cells were harvested for IP-western analyses.
Tumor growth in mice.
All animal experiments conducted in this study were approved by Duke University Institutional Animal Use and Care Committee (IACUC). C57BL/6J, Balb/c mice, Rag1 knockout mice, LDLR knockout mice and OT-1 transgenic mice (in the C57BL/6J background) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). NOD CRISPR PrkdcIl2rGamma (NCG) triple-immunodeficient mice were purchased from Charles River Laboratory (Wilmington, MA). Female LDLR KO mice were fed with 60% high fat diet (Research Diets, Inc; New Brunswick, NJ). Mice were housed at an ambient temperature of 72 degree Fahrenheit, with a humidity of 30–70%, and a light cycle of 12 hour on/12 hour off that is set from 7am to 7pm. Prior to tumor cell injection, age-matched, 6–8 weeks old mice were shaved at flank. Tumor cells were then injected into the shaved flank subcutaneously with at 1.0 × 105 CRISPR/Cas9 modified control or target gene knockout tumor cells. In experiments involving PCSK9 neutralizing antibodies, 6–8 weeks old syngeneic female C57BL/6J or Balb/c mice were inoculated subcutaneously with MC38 cells (2.5 × 105/mouse) or 4T1 cells (1 × 105/mouse), respectively. About 200 μg of anti-PCSK9 monoclonal antibodies (evolocumab from Amgen, or alirocumab from Sanofi/Regeneron) or 200 μg human IgG2 isotype control (BioXcell) were injected (intraperitoneally) on days 3, 5, 8, and 11. In addition 100 μg of anti-PD1 (clone RMP1–14, BioXcell) antibody were injected on days 5, 8 in some mice in combination with the anti-PCSK9 antibodies. At least 5 mice per treatment group were included. No randomization or blinding was used in our tumor growth delay experiments. Mice were monitored for tumor growth every 2 days afterwards. Tumor size were measured by use of a caliper and calculated using the formula Volume = (length)(width)2/2. Endpoint was defined as the time at which a progressively growing tumor reached 1.5 cm in the longest dimension or 2,000 mm3 in diameter. Mice were also euthanized when they experienced open skin lesions, weight loss >15% total body weight, or failed to thrive.
Lymphocyte depletion.
To evaluate the role of specific subsets of immune effector cells in mice, CD4+ T cells, CD8+ T cells, or NK cells were depleted with 150μg of i.p. injected anti-CD4 (BioXcell, clone GK1.5), 100 μg of anti-CD8β (BioXcell, clone 53–5.8), and 200 μg of anti-NK1.1 (BioXcell, clone PK136), respectively, on days −3, 0, 3, 8. Equal amounts of IgG isotype antibodies (BioXcell) were injected as a control.
In vivo competition assay.
B16F10 cells stably expressing EGFP or tdTomato were infected with PCSK9-targeting sgRNA or control lentiviral vectors, respectively, and selected with 1μg/ml puromycin for 10 days. Subsequently, about 5 × 104 PCSK9 knockout cells (EGFP expressing) and 5×104 control cells (tdTomato expressing) were mixed and inoculated subcutaneously to C57BL/6J female mice. Tumors were excised 12–14 days after inoculation. They were then minced and incubated in DNase I (50 μg/ml, Sigma) and collagenase P (2mg/ml, Sigma) for 20 min at 37 °C. The dissociated tumor cells were passed through 70 μm cell strainer (BD). Tumor cells were washed and re-suspended in ice-cold PBS with 2% FBS. The ratio of GFP and tdTomato tumor cells were analyzed by use of BD Canto II flow cytometry system (Flow Cytometry Shared Facility, Duke University School of Medicine).
Analysis of cell surface MHC I expression.
For in vitro experiments, mouse B16F10 and 4T1 cells were treated with interferon gamma (IFNγ, 4T1: 4ng/ml; B16F10,1 ng/ml) for 12 hours to stimulate MHC I expression and stained with FITC labeled anti-H-2Kb/Db, or anti-H-2Kd/Dd antibodies, respectively, for 20 minutes on ice. Human MDA-MB-231 cells were treated with 1 mg/ml evolocumab or alirocumab, or human IgG2 isotype control (BioXcell) for 72 hours, or 1 μg/ml recombinant PCSK9 protein (Prospec-Tany TechoGene Ltd) or bovine serum albumin (BSA) (Sigma) for 6 hours and stained with a FITC labeled HLA-ABC antibody for 30 minutes on ice. After washing with PBS-2%FBS, the surface expression of MHC I was analyzed by BD Canto II flow cytometer (Flow Cytometry Shared Facility, Duke University School of Medicine). For in vivo experiments, vector control or PCSK9-deficient tumor cells transduced with exogenous markers (EGFP/tdTomato/Cas9) expression were inoculated separately into mice, and tumors were harvested 10–12 days later. Tumor tissues were then disaggregated and processed as described above and subjected to flow cytometry analysis using an anti-H-2Kb/Db antibody.
Analysis of tumor-infiltrating lymphocytes.
About 1 × 105 control and PCSK9 KO or control cells were inoculated subcutaneously into C57BL/6 mice. Tumors were collected on day 12 after inoculation, weighted, and mechanically minced and incubated in DNase I (50 μg/ml, Sigma) and collagenase P (2mg/ml, Sigma) for 20 minutes at 37°C. The dissociated cells were passed through a 70 μm cell strainer (BD). The filtered cells were then blocked with an anti-CD16/32 antibody and stained with indicated surface antibodies for 20 minutes on ice. Dead cells were marked using Live/Dead Fixable Aqua dye (Thermo Fisher Scientific). Intracellular antibodies were added after fixation and permeabilization as per the manufacturer’s instructions. The anti-mouse fluorochrome-conjugated antibodies were listed above in antibodies and related agent section. BD Canto II flow cytometer (Flow Cytometry Shared Facility, Duke University School of Medicine) was used for our analysis. Collection of the cells were carried out by using the FACS DIVA software (version 8.01) and data analysis were carried out by using FlowJo (V10).
Quantitative RT-PCR of CTL-related gene expression.
Total RNA was extracted from CRISPR/Cas9 control or PCSK9KO B16F10 tumors (around 200–300 mm3 in volume) from tumor-bearing mice using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA was subjected to cDNA synthesis with random hexamer primers using Superscript II reverse transcriptase (Invitrogen). Real-time quantitative RT-PCR (qRT-PCR) was performed using QuantiTestSYBR Green PCR Master Mix Kit (Qiagen). Primers used are: mouse GZMB, forward 5’-CCACTCTCGACCCTACATGG-3’, and reverse 5’-GGCCCCCAAAGTGACATTTATT-3’; mouse IFNG, forward, 5’-ATGAACGCTACACACTGCATC-3’, and reverse, 5’-CCATCCTTTTGCCAGTTCCTC-3’.
Tumor infiltrating lymphocyte TCR sequencing.
Control and PCSK9KO B16F10 cells were inoculated as described above and on day 10 after inoculation they were collected for genomic DNA extraction. Genomic DNA was extracted using DNeasy Blood & Tissue kit (Qiagen) and submitted to Adaptive Biotechnologies for mouse TCRB CDR3 survey sequencing. About 2.6 μg of initial DNA was used as input for PCR reaction. Data were analyzed using Adaptive Biotechnologies online analysis platform ImmunoSEQ Analyzer 3.0.
OT-1 T cell culture.
OT-1 CD8+ T cells expressing a transgene encoding a TCR specifically recognizing SIINFEKL peptide bound to mouse H-2Kb were harvested from spleens of OT-1 C57BL/6 mice.31 Activated OT-1 T cells were generated by incubation of 5 × 106 cells/ml OT-1 SIINFEKL-pulsed mouse splenocytes in vitro for 5–7 days in the presence of mouse recombinant IL2.41,42 Briefly, an OT-1 mouse spleen was harvested and homogenized using aseptic techniques. The released cells were pelleted and re-suspend in 3 ml ACK buffer (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM EDTA) for 2 minutes to lyse red blood cells at room temperature. The splenocytes were then pelleted, washed, and re-suspended at 5 × 106 cells/ml in complete growth medium (RPMI1640 Sigma-Aldrich) with 10% FBS (Corning), 1× penicillin-streptomycin (ThermoFisher Scientific), 1× sodium pyruvate (ThermoFisher Scientific), and 1× 2-Mercaptoethanol (ThermoFisher Scientific) containing 0.75 μg/ml SIINFEKL peptide (GenScript), and incubated at 37°C in a 95% air/5% CO2 humidified environment. Mouse recombinant IL2 (ThermoFisher Scientific) was added on days 3 and 5 at 30 U/ml with fresh complete growth medium. On day 7, the cells were harvested for assays. The specificity was determined by flow cytometry analysis using APC/Fire750-labeled anti-mouse CD8 and APC-labeled anti-mouse TCRVβ5.1/5.2 antibodies (BioLegend).
Tumor cell and OT-1 specific T cell co-culture analysis.
B16F10 Control-Td (expressing tdTomato fluorescent protein), B16F10 PCSK9KO-Td, B16F10 Control-OVA-Td (expressing ovalbumin), and B16F10 PCSK9 KO-OVA-Td cells were first stimulated by incubation with mouse recombinant IFNγ at 1 ng/ml for 12 hours. The stimulated tumor cells were then cultured with OVA-specific T cells at 1:1 ratio or without OVA-specific T cells in T cell complete growth medium with mouse recombinant IL2 (30 U/ml) for 24 hours. Subsequently, tdTomato fluorescence were captured by Zeiss Axio Observer.Z1 fluorescence microscope imaging station using ZEN imaging software (2012, blue edition, Carl Zeiss Microscopy GmbH). NIH ImageJ (version 1.52h) was used for counting tdTomato-expressing tumor cells.
Cell fractionation and quantification of MHC I in different compartments.
To investigate the distribution of MHC I in the lysosome or the plasma membrane, 5 × 107 H2-K1-Flag transduced PCSK9 over-expressing or knockout B16F10 cells were used to isolate lysosome and membrane fractions. For lysosome isolation, a density gradient ultracentrifugation method was used following manufacturer’s instructions (Lysosome Enrichment kit, Thermo Scientific). The membrane fraction was separated by use of membrane separation buffer A (50 mM Tris-HCl, pH7.5, 450 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1 mM EGTA, 1 mM DTT) and buffer B (buffer A plus 1% NP40, 0.1% SDS). MHC I expression in lysosome and membrane fraction was detected by western blot by use of a mouse anti-Flag antibody. Mouse anti-LAMP2 and rabbit anti-pan-cadherin were used as makers of lysosome and membrane, respectively.
Immunofluorescence and immunohistochemistry analysis.
For immunofluorescence analysis, tumors from mice were fixed in 10% neutral-buffered formalin, embedded into paraffin, sectioned and then mounted onto slices. They were then stained according to standard procedures by use of antibodies against mouse CD45 or CD8a (see antibodies and related agents section). For lysosome co-localization experiments, PCSK9 over-expression or knockout B16F10 cells were incubated with Lyso-Tracker (deep red, Thermo Fisher) at 37°C for 20 min and then washed with HBSS for twice. The cells were stained with 5 μg/ml fluorescent conjugated Wheat Germ Agglutinin (CF488 WGA) at 37 °C for 10 min, which selectively binds to N-acetyglucosamine and sialic acid residues to indicate the plasma membrane. Cells were then washed twice with PBS, fixed with 4% paraformaldehyde at room temperature for 15 min, and permeabilized with blocking buffer (1% BSA, 5% Donkey serum, 0.1% digitonin) at room temperature for 30 min. The cells were then incubated with anti-Flag primary antibody overnight at 4°C, followed by incubation with fluorescence-labeled secondary antibodies for 1 hour at room temperature. After washing with PBS, the stained slices were mounted with mounting medium (Vector Laboratories) containing DAPI. Images were captured by use of confocal microscopy.
Protein co-immunoprecipitation (CO-IP) and western blot analysis of PCSK9 and H2-K1 interaction.
293T cell were transduced with HA-tagged PCSK9 and Flag-tagged H2-K1 constructs and cultured for about 24 hrs and then processed for co-IP analysis. Cultured cells in 10-cm Petri dish were washed with ice-cold PBS twice and directly lysed with 500 μl IP lysis buffer (150 mM NaCl, 50 mM Tris, 0.1% NP-40) supplemented with protease inhibitors (Sigma) on ice. Cell lysates were transferred to 1.7 ml Eppendorf tubes and end-to-end rotated for 15 min at 4°C. Protein concentration in lysates was measured by use of the Bio-Rad protein assay. For IP of HA tagged PCSK9 protein, 500 μl of cell lysates were incubated with 3 μl anti-HA antibody (Cell signaling technology) on a rotator at 4°C overnight. Lysates with HA antibodies were then incubated with 20 μl Protein A/G agarose beads (Santa Cruz Biotechnology) for 2 hours at 4°C. After washing with IP lysis buffer three times, the pull-down complex was boiled in 2X SDS loading buffer for SDS-PAGE and western blot analysis.
Cycloheximide chase Assay.
To determine PCSK9’s influence on lysosomal degradation of the MHC I protein, vector control or PCSK9 knockout MDA-MB-231 cells were treated with 20 μg/ml cycloheximide (CHX, Sigma) to inhibit protein biosynthesis for 1, 4, 8, 18, 24 hrs. For lysosome inhibition, MDA-MB-231 cells were treated with 20 nM Bafilomycin A1 (Baf A1, Sigma) for 1, 4, 8, 18, 24 hrs. The cells were then harvested and MHC I proteins were detected by western blot by use of anti-HLA-ABC antibody.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 6 software and statistical significance was defined as p value less than 0.05. Two-way ANOVA was used for multiple comparisons in tumor growth delay experiments. Log-rank test was used for mouse survival analysis. In other experiments, comparisons between two groups were conducted by use of unpaired two-sided Student`s t test. The Gene Expression across Normal and Tumor tissue database (GENT)43 was used to analyze the relationship between PCSK9 and CD8A in indicated patient cohorts. Data on PCSK9 gene expression and patient survival were obtained from cBioportal website44,45. In particular, the TCGA datasets were used. Overall survivals of PCSK9 high and low groups were evaluated using log-rank test and a value of p <0.05 was considered statistically significant.
Data availability.
PCSK9 and CD8A mRNA expression data of various human cancers were downloaded from the GENT (http://gent2.appex.kr/gent2/) database43. PCSK9 mRNA expression and overall survival data were from the TCGA datasets that were included in cBioportal44,45 (https://www.cbioportal.org/) in November, 2018. Western blot source data are provided in Supplementary Fig. 1. Source data for the quantitative graphs are provided. Other data in support of this study are available from the corresponding author upon reasonable request.
Extended Data
Extended Fig.1. CRISPR-Cas9 mediated knockout of PCSK9 gene and its effect on tumor cell growth in vitro and in vivo.
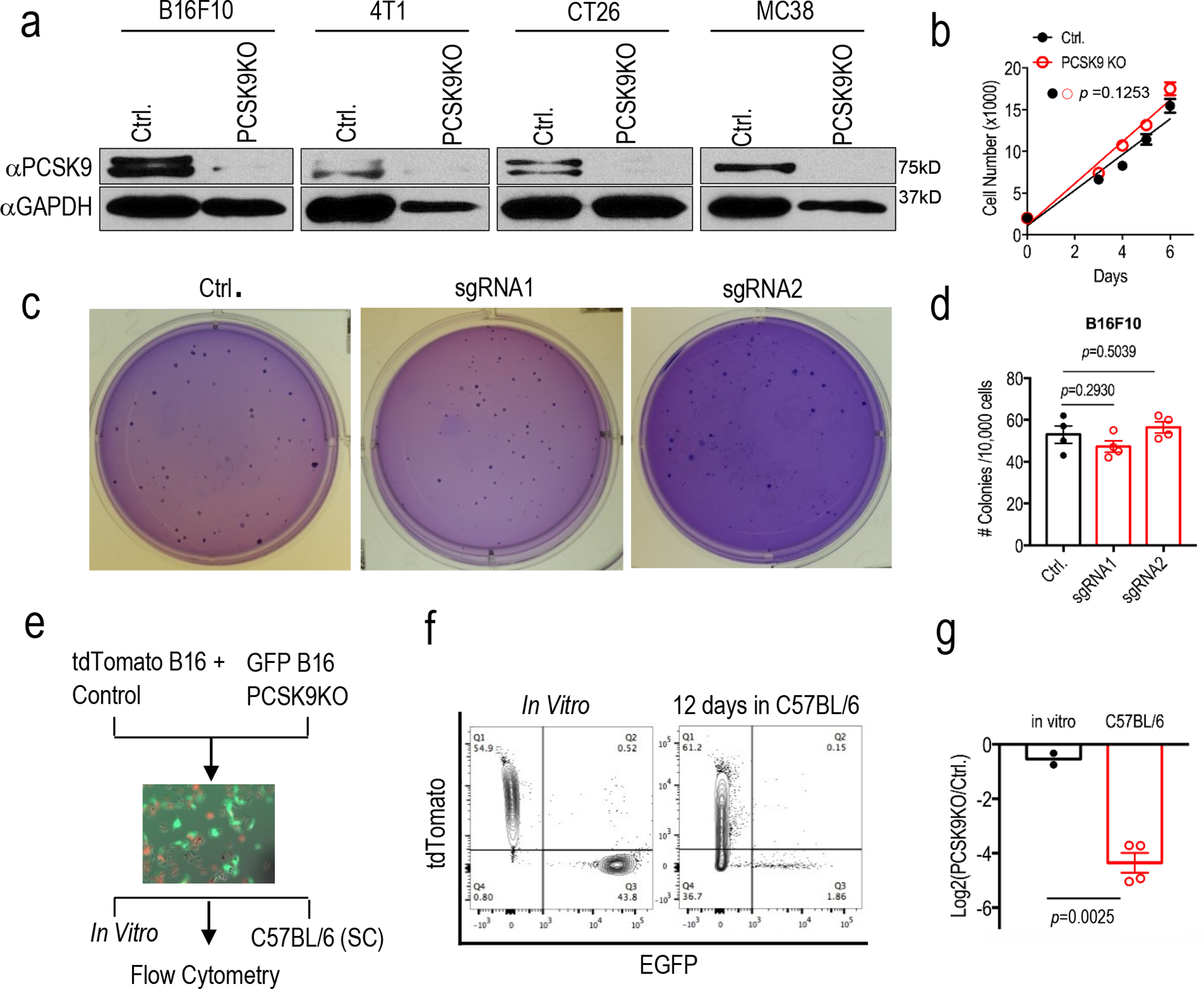
a. Western blot analysis of the expression of PCSK9 in murine tumor lines with PCSK9 knockout. GAPDH was used as protein loading control. Analysis was done twice with biologically independent samples. b. Cell growth of vector control or PCSK9 KO B16F10 tumor cells. Results from 5 biologically independent samples. Error bars, mean ± S.E.M. P value calculated by unpaired two-sided t test. c. Soft agar analysis of the colony formation ability of vector control or PCSK9 sgRNA-transduced B16F10 tumor cells. d. Quantitative representation of soft agar formation in c. n=4 biologically independent samples. Error bars, mean± S.E.M. P values calculated by unpaired two-sided t test. e. Diagram of in vivo competition assay. f. Change in ratios of mixed control-tdTomato and PCSK9 KO-EGFP B16F10 cells after 12 days grown in vivo (subcutaneously) in C57BL/6 mice, as determined by flow cytometry. g. Quantitative representation of the flow analysis in f. Error bar, mean± S.E.M. n=2 and 4 biologically independent tumor samples for the in vitro and in vivo groups, respectively. P value determined by unpaired two-sided t test.
Extended Data Fig. 2. The effect of PCSK9 re-expression and the host immune system on tumor formation of PCSK9KO tumor cells.
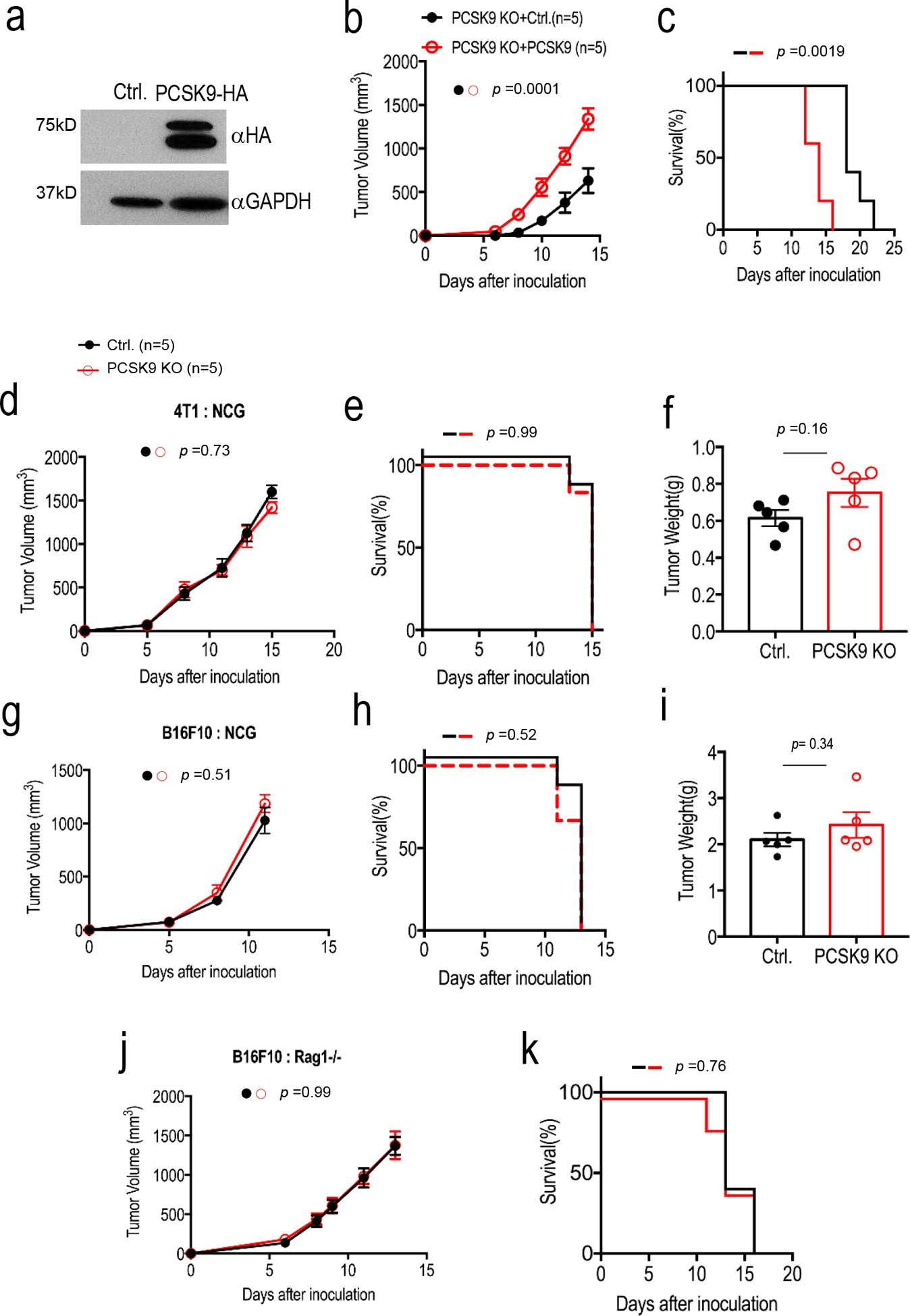
a. Western blot analysis of the expression of exogenously transduced, HA-tagged PCSK9 in PCSK9KO B16F10 cells. The analysis was done once. b-c. Tumor formation (b) from B16F10-PCSK9KO cells transduced with either the vector control or the PSCK9 gene and Kaplan-Meier survival curve of host mice (c). About 2 × 105 tumor cells were injected subcutaneously into C57BL/6 mice and observed for tumor formation. n=5 tumors per group. Error bars, mean ± S.E.M. P values determined by two-way ANOVA in b and log-rank test in c. d-i. Growth rate, host survival and endpoint tumor weight of vector control and PCSK9KO 4T1 (d-f) and B16F10 (g-i) tumors. In each case, about 1 × 105 tumor cells were injected subcutaneously and observed for tumor formation in NCG mice. n=6 mice for d, e, g, and h and n=5 tumors for f and i. Error bars in d,f g, and i represent mean ± S.E.M. ns: not significant, as determined by two-way ANOVA (d,g), log-rank test (e,h), or unpaired two- sided t test (f, i). j-k. tumor growth from vector control and PCSK9KO B16F10 cells (j) and Kaplan-Meier survival curve of tumor-bearing host mice (k) in Rag1−/− C57BL/6 mice. About 1 × 105 vector 381 control or PCSK9KO B16F10 tumor cells were injected into Rag1−/− C57BL/6 mice and observed for tumor formation. ns: not significant. n=5 tumors per group. Error bars in j, mean ± S.E.M. P values calculated by two-way ANOVA in j and by log-rank in k.
Extended Data Fig. 3. The influence of tumor or host cell LDLR and host cholesterol levels on tumor growth from control or PCSK9KO tumor cells in immunocompetent hosts.
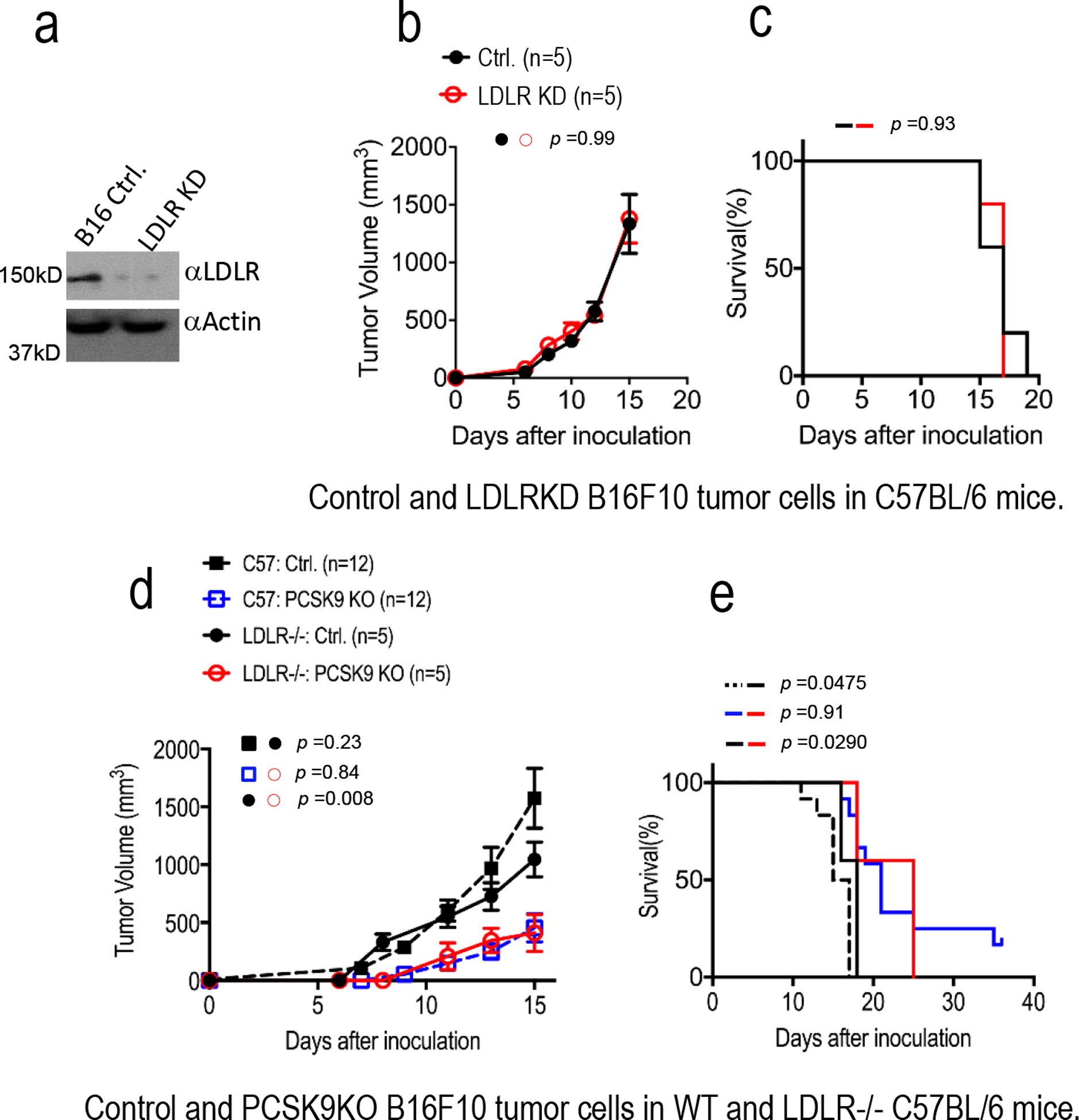
a. Western blot analysis of CRISPR-Cas9 mediated knockdown (KD) of LDLR in B16F10 cells. The analysis was done once. b. Tumor growth from vector control and LDLR KD B16F10 cells in C57BL/6 mice. n=5 tumors per group. Error bars, mean ± S.E.M. P value calculated by two-way ANOVA. c. Kaplan Meier survival curve of mice (from b) bearing control and LDLR KD B16F10 tumors. n=5 mice per group. P value calculated by log-rank test. d. Tumor growth from vector control and PCSK9 knockout B16F10 cells in WT and LDLR−/− mice fed with high-fat diet. n=12, 12, 5, and 5 tumors in C57BL/6 mice inoculated with control and PCSK9KO tumor cells, and LDLR−/− mice inoculated with control and PCSK9 KO tumor cells. Error bars, mean ± S.E.M. P values calculated by two-way ANOVA with multiple comparisons. e. Kapan-Meier survival curve of wild type and LDLR−/− mice (from d) bearing vector control and PCSK9 knockout tumors. P value calculated by log-rank test.
Extended Data Fig. 4. Additional data on anti-PD1 treatment in murine tumors.
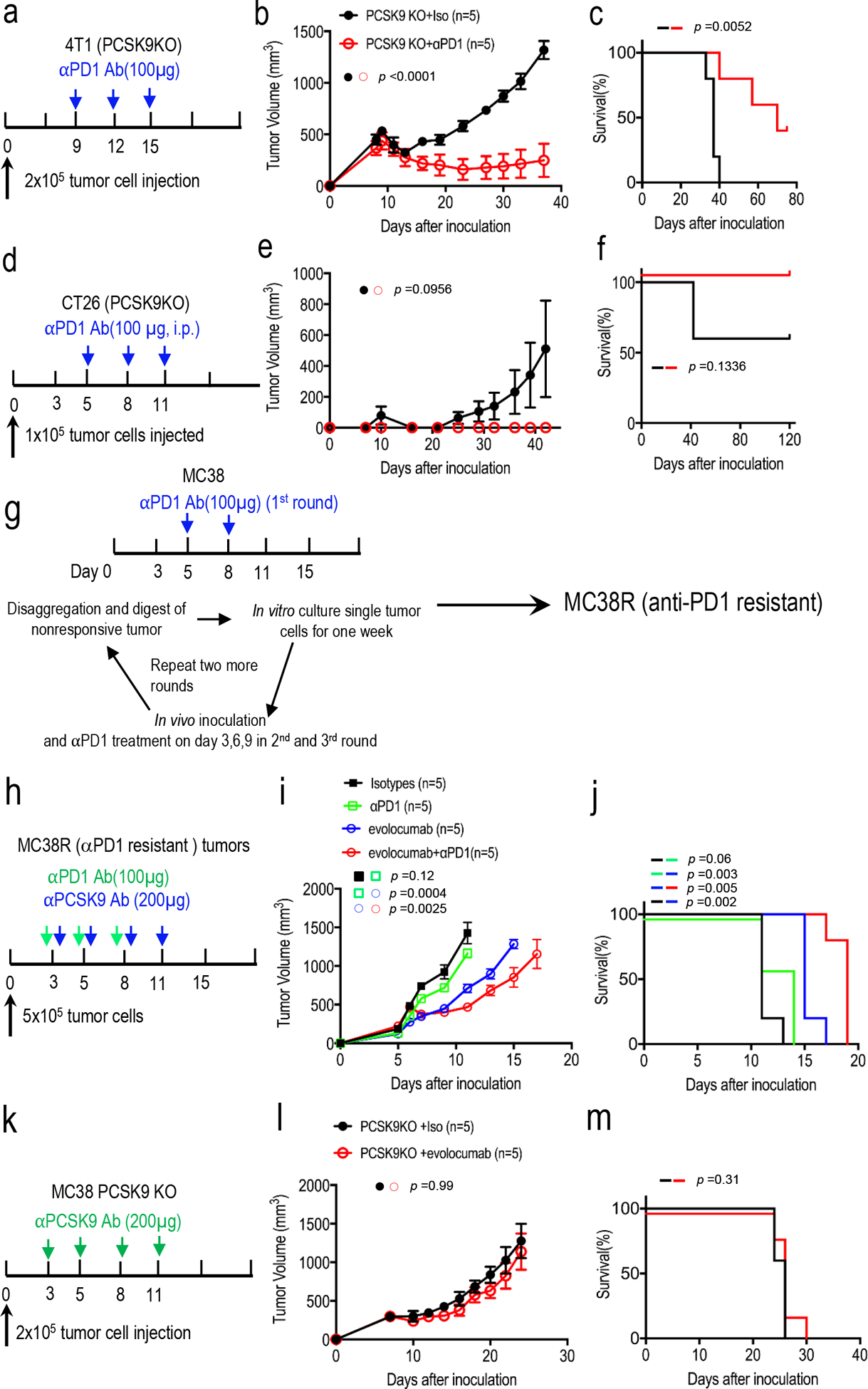
a. Treatment schedule for PCSK9KO 4T1 tumors. Balb/c mice implanted with PCSK9KO 4T1 tumor cells but did not form visible tumors on day 9 after inoculation were excluded for treatment. b. Tumor growth delay in mice bearing PCSK9KO 4T1 tumors with or without anti- PD1 treatment. n=5 tumors per group. Error bars, mean ± S.E.M. P values calculated by two-404 way ANOVA test. c. Kaplan-Meier survival curve of tumor-bearing mice in b. P value calculated by log rank test. d. Treatment schedule for PCSK9KO CT26 tumors. Balb/c mice were implanted subcutaneously with PCSK9KO CT26 tumor cells and treated with an anti-PD1 antibody and observed for tumor formation. e. Tumor growth delay in mice bearing PCSK9KO CT26 tumors with or without anti-PD1 treatment. n=5 tumors per group. Error bars, mean ± SEM. P values were determined by two-way ANOVA test. f. Kaplan-Meier survival curve of tumor-bearing mice in e. Error bars, mean ± SEM. P value was determined by log-rank test. g. A scheme to develop anti-PD1 resistant MC38R tumor cells. h. Treatment scheme of anti-PD1 resistant MC38R tumors with evolocumab and an anti-PD1 antibody. i. Tumor growth kinetics from anti-PD1 resistant MC38R tumor treated with anti-PD1 and/or evolocumab. n=5 tumors per 414 group. Error bars: mean ± SEM. P values were determined by two-way ANOVA test. j. Kaplan-Meier survival curve for mice bearing MC38R tumors in i. P values determined by log-rank test. k. Treatment schedule for PCSK9KO MC38 tumors. l-m. Tumor growth delay (l) and host mice survival (m) among isotype- (iso) or evolocumab-treated mice bearing MC38-PCSK9KO tumors. n=5 tumors per group. P values were calculated by two-way ANOVA test in l and log-rank test in m.
Extended Data Fig.5. Re-challenge of mice that were tumor free after initial tumor 4inoculation and gating strategy of intratumoral immune effector cells.
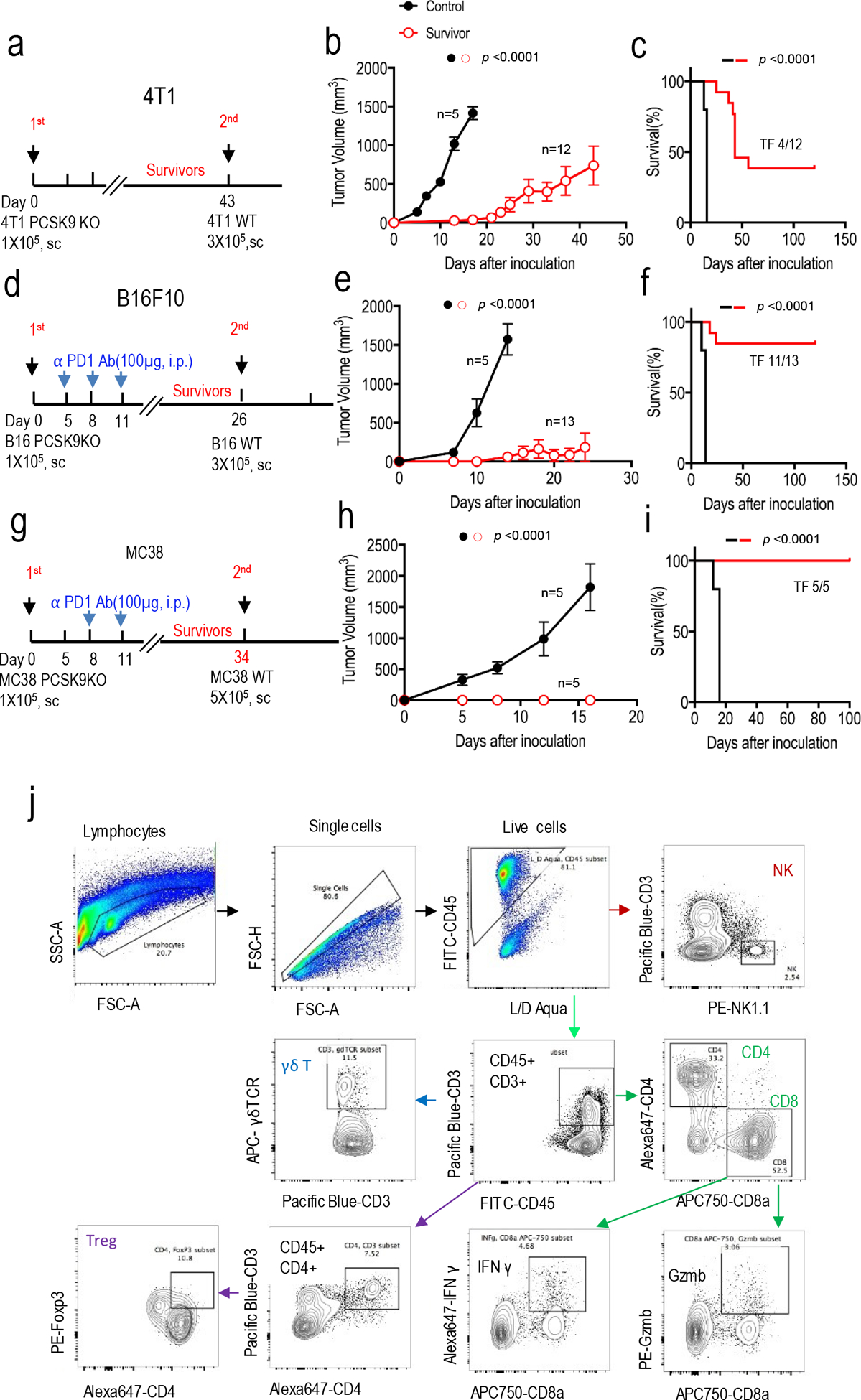
a-c. Treatment scheme (a), tumor growth (b), and survival of host mice (c) after re-challenge with wild type 4T1 tumor cells in Balb/c mice that remained tumor-free 43 days after initial challenge with PCSK9 deficient 4T1 cells. The control group consists of tumor-naive Balb/c mice challenged with wild type 4T1 cells. n=5 and 12 mice for naïve and re-challenged group, respectively. Error bars in b, mean ± S.E.M. P values in b and c calculated by two-way ANOVA test and log-rank test, respectively. d-f. Treatment scheme (d), tumor growth (e), and survival of host mice (f) after re-challenge with wild type B16F10 tumor cells in C57BL/6 mice that remained tumor-free 26 days after initial challenge with PCSK9 deficient B16F10 cells and the treatment with anti-PD1 antibody. The control group consisted of tumor-naïve C57BL/6 mice challenged with wild type B16F10 cells. n=5 and 13 mice for control and re-challenge groups, respectively. Error bars in e, mean ± S.E.M. P values in e and f calculated by two-way ANOVA test and log-rank test, respectively. g-i. Treatment scheme (g), tumor growth (h), and survival of host mice (i) after re-challenge with parental MC38 tumor cells in C57BL/6 mice that remained tumor-free 34 days after initial challenge with PCSK9-deficient MC38 cells and the treatment with anti-PD1 antibody. The control group consisted of tumor-naïve C57BL/6 mice challenged with wild type MC38 cells. n=5 mice per group. Error bars in h, mean ± S.E.M. P values calculated by two-way ANOVA test in h and log-rank test in i. j. Representative flow cytometry gating strategy to quantitate the numbers of various immune effector cell subsets in murine tumors.
Extended Data Fig. 6. Additional data on the characterization of lymphocyte infiltration into murine tumors.
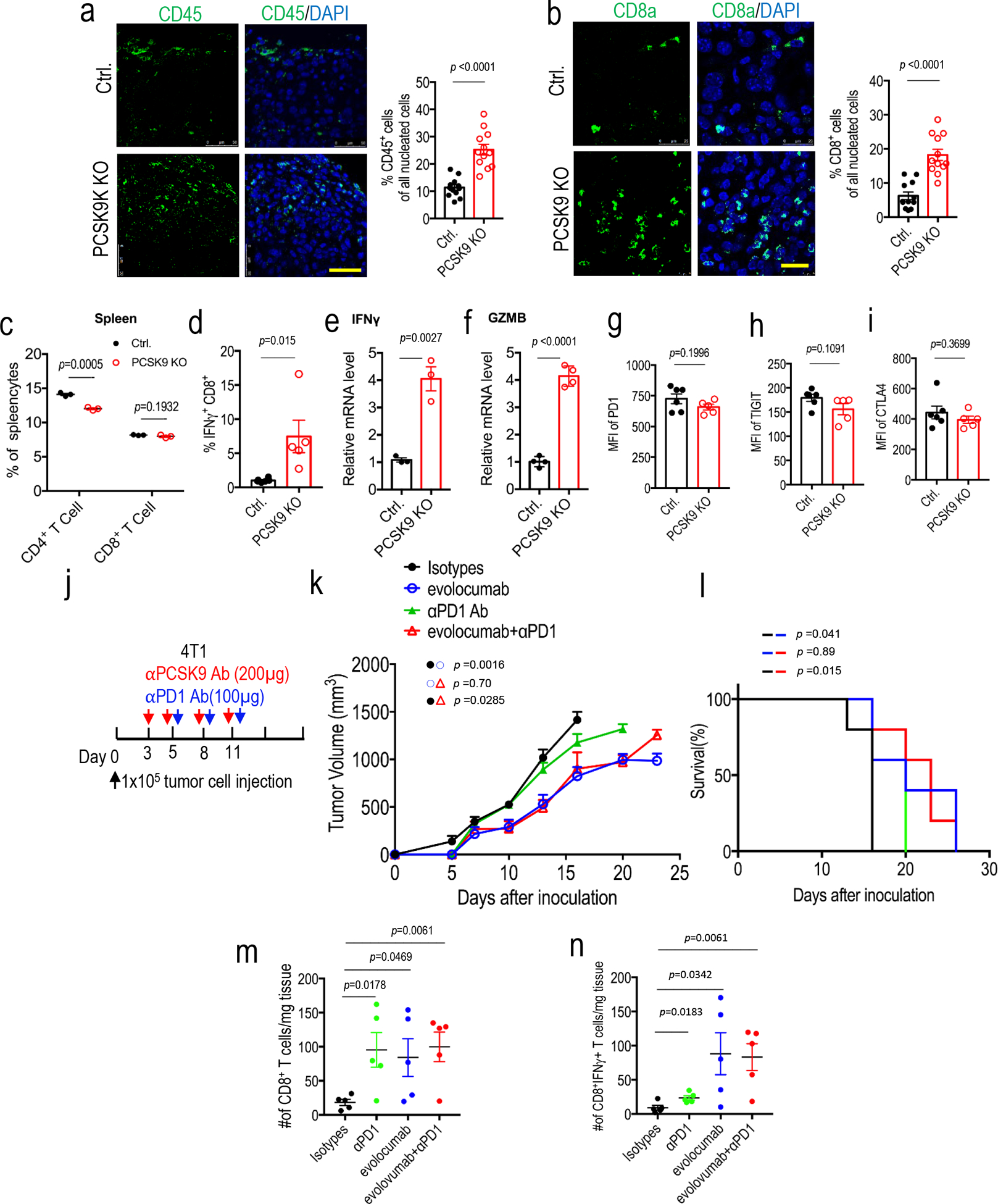
a. Immunofluorescence staining (left panel) and quantitative estimate (right panel) of CD45+ leukocytes in control and PCSK9KO tumors grown in syngeneic C57BL/6 mice. Scale bar = 50 μm. n=3 biologically independent samples. Four fluorescent fields for each of the three samples were counted. Error bars, mean ± S.E.M. P value calculated using unpaired two-sided t-test. b.Immunofluorescence staining (left panel) and quantitative estimate (right panel) of CD8a+ cells in control and PCSK9KO B16F10 tumors. Scale bar = 20 μm. n=3 biologically independent samples. Four fluorescent fields for each of the three samples were counted. Error bars, mean ± S.E.M. P value calculated using unpaired two-sided t test. c. Quantitative estimates of CD4+ and 452 CD8+ T cells in the spleens of mice bearing control and PCSK9KO B16F10 tumors as determined by flow cytometry. n=3 mice per group. Error bars, mean ± SEM. P values calculated using unpaired two-sided t-test. d. Flow cytometry determination of the percentage of intratumoral CD8+ T cells that were IFNγ+. n=6,5 tumors in the two groups. Error bars, mean ± S.E.M. P value calculated by unpaired two-sided t-test. e, f. Q-RT-PCR analysis of intratumoural IFNG (e) and GZMB (f) mRNA levels in control and PCSK9KO tumors. n=3 and 4 tumors for INFG and GZMB groups, respectively. Error bars, mean ± S.E.M. P values were determined by unpaired two-sided t test. g-i. Flow cytometry characterization of the cell surface expression levels of exhaustion markers for intratumoural CD8+ T cells in vector control and PCSK9KO tumors. n=6, 5 tumors. Error bars, mean ± S.E.M. P values were determined by unpaired two-sided t test. j. Evolocumab and anti-PD1 treatment schedule for syngeneic 4T1 tumor model. k. Growth of 4T1 tumors treated with anti-PD1 and/or evolocumab. n=5 mice per group. P values were determined by two-way ANOVA test. l. Kaplan-Meier survival curve for mice in k. P values were determined by log-rank test. m. Frequency of CD8+ T cells in 4T1 tumors treated with anti-PD1 and/or evolocumab. n=5 tumors per group. Error bars, mean ± SEM. P values were determined by unpaired two-sided t test. n. Frequency of IFNγ+CD8+ T cells in 4T1 tumors treated with anti-PD1 and/or evolocumab. n=5 tumors per group. Error bars, mean 469 ± SEM. P values were determined by unpaired two-sided t test.
Extended Data Fig. 7. Additional data on the effect of PCSK9 inhibition on immune effector function and antigen presentation.
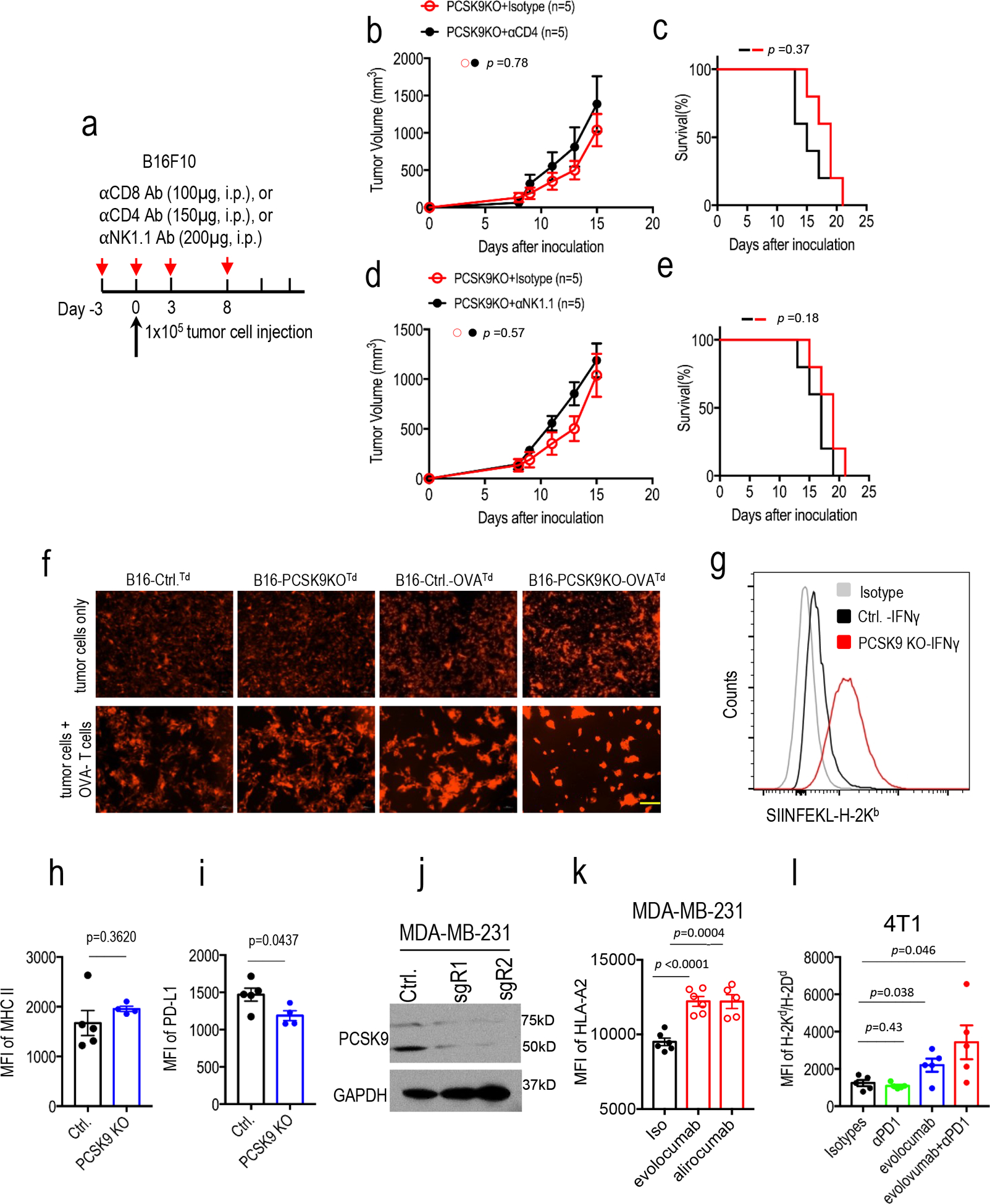
a. Injection schedule for antibody-mediated immune cell depletion of CD4+, CD8+, and NK cells. b-c. Growth rates (b) and host mice survival (c) of PCSK9KO tumors in mice administered with control or anti-CD4 antibody. n=5 tumors per group. Error bars, mean ± S.E.M. P values determined by two-way ANOVA test in b and log-rank test in c. d-e. Growth rates (d) and host mice survival (e) of PCSK9KO tumors in mice administered with control or anti-NK1.1 antibody. n=5 tumors per group. Error bars, mean ± S.E.M. P values in d and e determined by two-way ANOVA test and logrank test, respectively. f. Fluorescence images of the tdTomato-labeled tumor cells with or without the OVA antigen in the presence or absence of OVA-specific T cells. The experiments were completed twice with similar results. Scale bar = 200 μm for all image panels. g. Enhanced presentation of OVA antigen (SIINFEKL) by MHC-I in cultured B16F10 cells with PCSK9 deficiency. Control and PCSK9KO B16F10 cells transduced with the OVA gene were treated with IFNγ and assayed for the amount of cell surface H-2Kb-SIINFEKL complex using flow cytometry. Shown were representative results from analyses of 4 sets of biologically independent samples. h-i. Flow cytometry analysis of MHC II (h) and PD-L1(i) expression in control and PCSK9KO B16F10 cells. n=5 and 4 biologically independent samples, respectively. P values determined by unpaired two-sided t test. j. Western blot of PCSK9 expression in control or PCSK9KO MDA-MB-231 cells. The analyses were done twice. k. The effect of evolocumab and alirocumab on HLA-ABC expression on the surface of MDA-MB-231 human breast cancer cells. n=6, 6, and 5 biologically independent samples. Data represent mean ± S.E.M, P values were determined by unpaired two-sided t test. l. H2-Kd/Dd expression levels of 4T1 tumor cells that were exposed to anti-PD1and/or evolocumab in vivo. n=5 mice per group. Error bars, mean ± S.E.M. P values were determined by unpaired two-sided t test.
Extended Data Fig. 8. Additional data on the analysis of PCSK9, H2-K1, and LDLR in murine tumor cells.
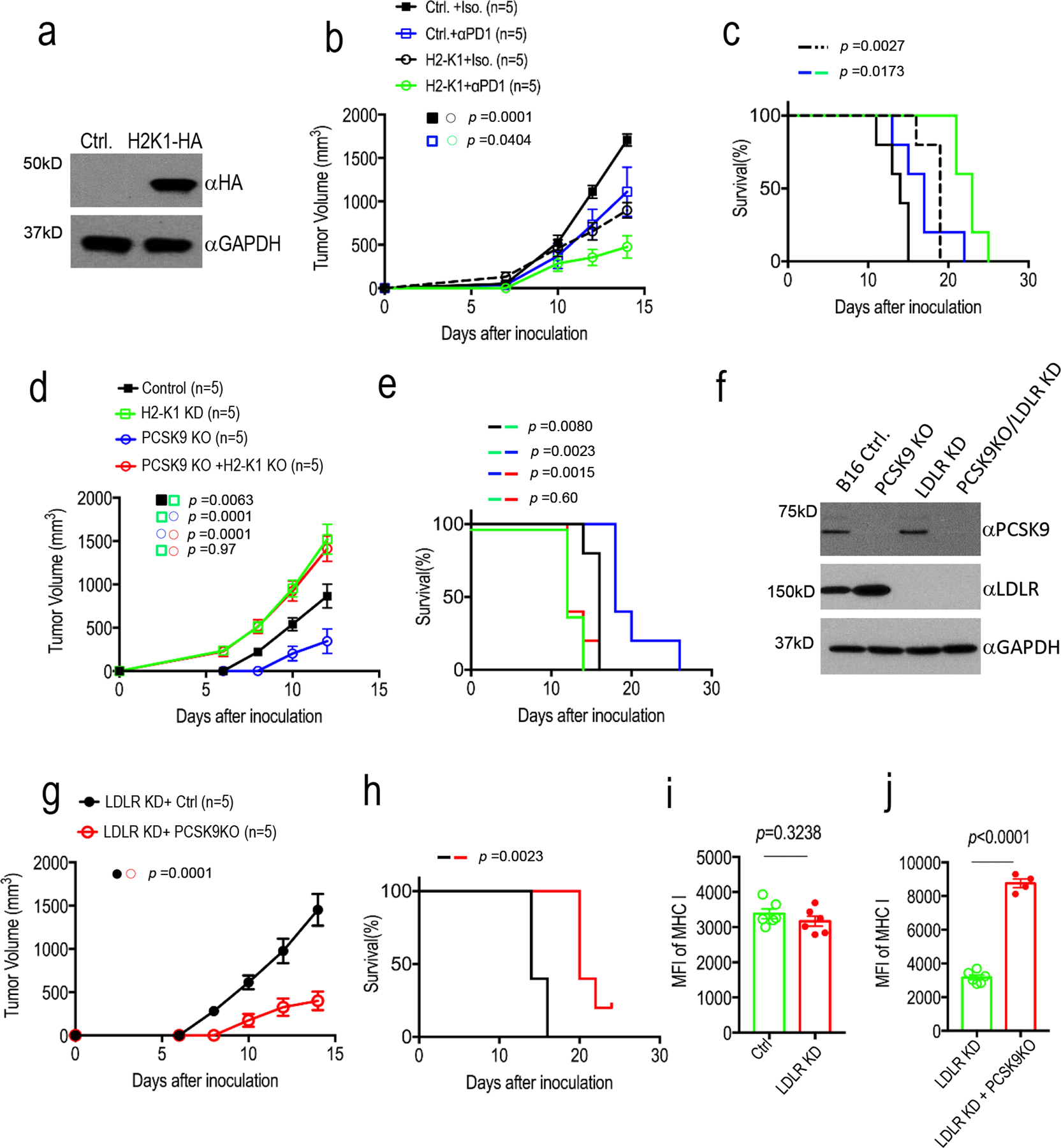
a. Lentivirus mediated over-expression of HA-tagged H2-K1 gene in B16F10 cells as determined by western blot analysis. The analysis was done once. b-c. Tumor growth delay (b) and Kaplan-Meier survival curve (c) of tumor-bearing C57BL/6 mice implanted with vector control or H2-K1 over-expression B16F10 cells. Error bars, mean ± S.E.M. n=5 tumors in each group. P values in b and c were determined by two-way ANOVA and log-rank test, respectively. d-e. Tumor growth delay (d) and Kaplan-Meier survival curves (c) in mice injected with vector control, H2-K1 KD, PCSK9 KO, or H2-K1KD/PCSK9KO B16F10 cells. n=5 mice per group. Error bars, mean ± S.E.M. P values were determined by two-way ANOVA and log-rank test, respectively. f. Western blot analysis of and LDLR knockdown in control and PCSK9KO B16F10 tumor cells. The analysis was done once. g-h. Tumor growth delay (g) and Kaplan-Meier survival curves (h) from LDLRKD and LDLRKD/PCSK9KO B16F10 tumors. n=5 mice per group. Error bars, mean ± S.E.M. P values were determined by two-way ANOVA and log-rank test, respectively. i. Flow cytometry analysis of MHC I expression in tumors formed from tdTomato-labeled control and LDLRKD B16F10 cells. n=6 biologically independent tumors. Error bars, mean ± S.E.M. P value calculated by unpaired two-sided t-test. j. Flow cytometry analysis of MHC I expression in tumors formed from tdTomato-labeled LDLRKD (n=6) and LDLRKD/PCSK9KO cells (n=4). Error bars, mean ± S.E.M. P value calculated by unpaired two-sided t-test.
Extended Data Fig. 9. Additional data on mapping and functional characterization of interacting domains in PCSK9 and MHC I and the association of PCSK9 expression and the prognosis of TCGA cancer cohorts.
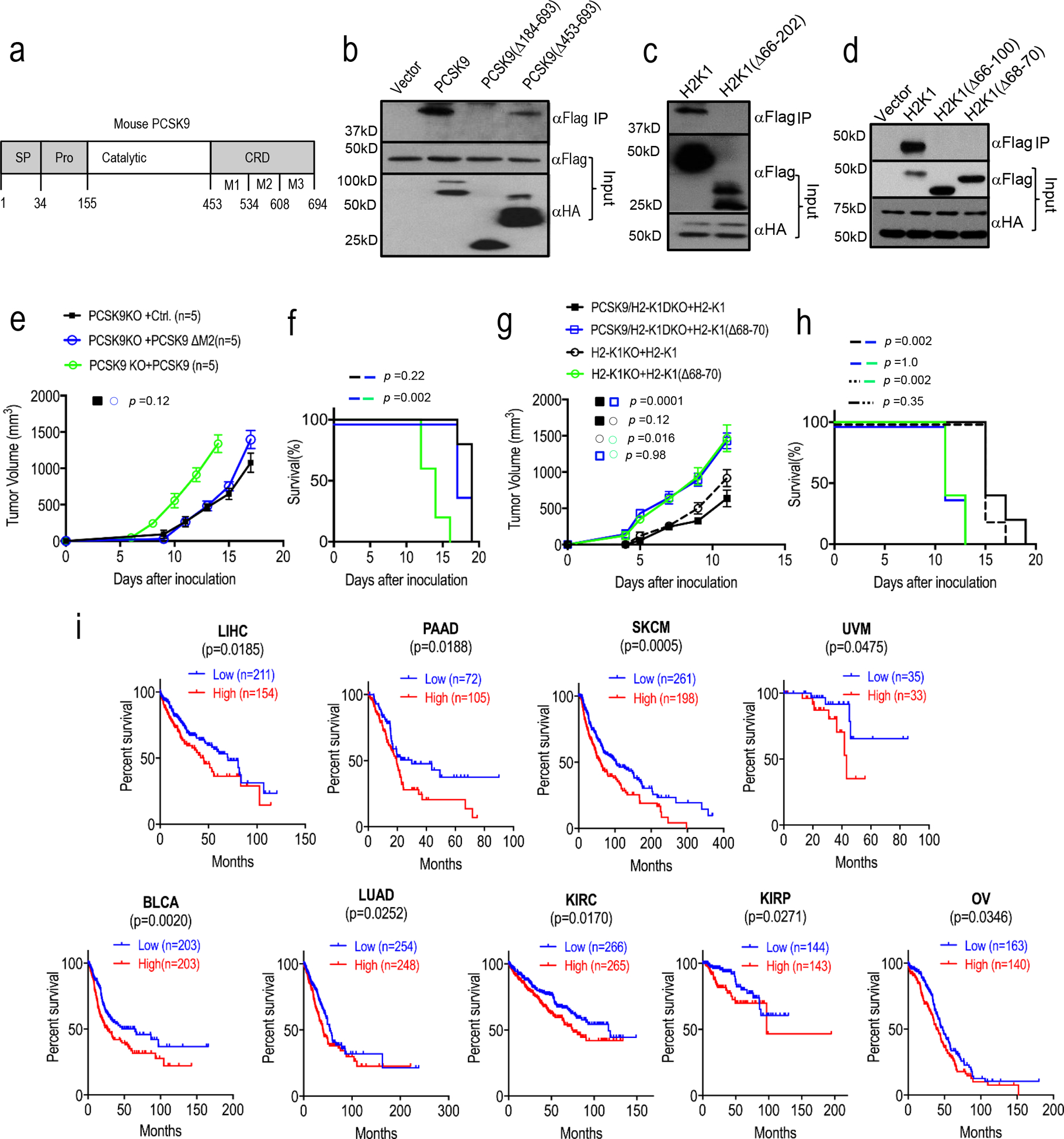
a. Domain structure of the mouse PCSK9 protein. SP, signal peptide; Pro, propeptide; Catalytic, catalytic domain; CRD; C-terminal domain. b. IP-Western blot analysis of the interaction between full-length Flag-labeled H2-K1 and full length or partially deleted mouse PCSK9-HA. Plasmids encoding the two genes were transfected into 293T cells in pairs and lysates from transduced cells were immunoprecipitated first with an anti-HA antibody and probed with an anti-Flag antibody by western blot analysis. The analyses were repeated twice with biologically independent samples with similar results. c. IP-Western blot analysis of the interaction between full-length HA-labeled mouse PCSK9-HA and full-length or partially deleted H2-K1-Flag (aa66–202) (α1-α2 domains). The analyses were repeated twice with biologically independent samples with similar results. d. IP-Western blot analysis of the interaction of HA-labeled mouse PCSK9-HA to full-length H2-K1 or H2-K1 with more limited deletions (aa 66–100, α1 domain; or aa68–70). The analyses were repeated twice with biologically independent samples with similar results. e-f. Tumor growth rates (e) and Kaplan-Meier survival curve (f) of mice inoculated with PCSK9KO B16F10 tumor cells re-expressed with wild type or partially (ΔM2) deleted PCSK9. n=5 tumors per group. Error bars, mean ± S.E.M. P values were determined by two-way ANOVA and log-rank test in e and f, respectively. g-h. Tumor growth rates (g) and Kaplan-Meier survival curve (h) of mice inoculated with H2-K1KO or H2-K1/PCSK9 DKO B16F10 tumor cells re-expressed with wild type or partially deleted (Δ68–70) H2-K1 gene. n=5 tumors per group. Error bars, mean ± S.E.M. P values were determined by two-way ANOVA and log-rank test, respectively. i. Higher levels of PCSK9 expression correlated to worse survival in 9 cancer patient cohorts including liver hepatocellular carcinoma (LIHC), pancreatic adenocarcinoma (PAAD), skin cutaneous melanoma (SKCM), uveal melanoma (UVM), bladder urothelial carcinoma (BLCA), lung adenocarcinoma (LUAD), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), and ovarian carcinoma (OV). P values calculated by log-rank test. Data from TCGA datasets.
Extended Data Fig. 10. A schematic diagram illustrating PCSK9-mediated degradation of MHC I in the lysosome.
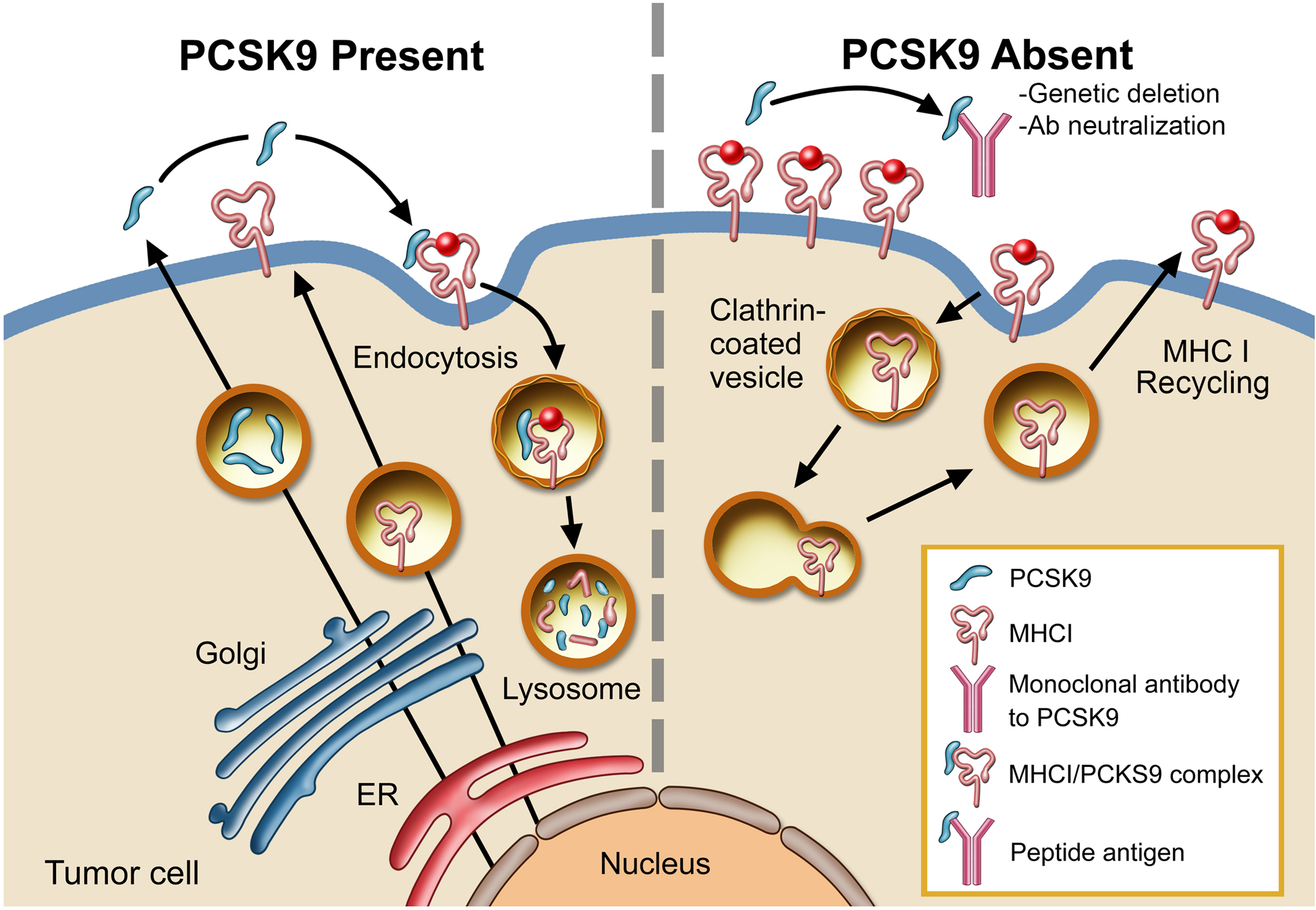
In the presence of PCSK9, MHC I is transported into the lysosome and degraded (left panel). In the absence of PCSK9, either because of genetic deletion or antibody neutralization, MHC I levels on the surface remains high and is thus able to present tumor-specific peptic antigens more efficiently to T cells (right panel). Illustration by Stan Coffman.
Supplementary Material
Acknowledgments
We thank J. Michael Cook and colleagues at the Flow Cytometry Facility of Duke University School of Medicine for their assistance. We also thank the Duke University Light Microscopy Core Facility for their professional help in our use of confocal microscopy. We thank Isabella Li for proof-reading our manuscript. We also thank Stan Coffman of MedMedia Solutions for his help with illustrations. C-Y Li is supported by US National Institutes of Health grants ES024015, CA208852, CA216876, and the Cancer Center Support Grant (CCSG) CA014236 to Duke University. X Liu is supported by Guangdong Basic and Applied Basic Research Foundation grant 2020B1515020054 and Shenzhen Science and Technology Program grant JCYJ20190807154813511.
Footnotes
Competing interest X.L. and C. L. are inventors on a patent application filed by Duke University that covers the use of anti-PCSK9 antibody in cancer immunotherapy. The other authors declare no competing interest.
References
- 1.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454, doi: 10.1056/NEJMoa1200690 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmer JR et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455–2465, doi: 10.1056/NEJMoa1200694 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manguso RT et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418, doi: 10.1038/nature23270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan D et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775, doi: 10.1126/science.aao1710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel SJ et al. Identification of essential genes for cancer immunotherapy. Nature 548, 537–542, doi: 10.1038/nature23477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abifadel M et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 34, 154–156, doi: 10.1038/ng1161 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Cohen J et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 37, 161–165, doi: 10.1038/ng1509 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Cohen JC, Boerwinkle E, Mosley TH Jr. & Hobbs HH Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 354, 1264–1272, doi: 10.1056/NEJMoa054013 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Yang W et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655, doi: 10.1038/nature17412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma X et al. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J Exp Med 215, 1555–1569, doi: 10.1084/jem.20171576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naslavsky N, Weigert R & Donaldson JG Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol Biol Cell 15, 3542–3552, doi: 10.1091/mbc.e04-02-0151 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benjannet S et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 279, 48865–48875, doi: 10.1074/jbc.M409699200 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Maxwell KN, Fisher EA & Breslow JL Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A 102, 2069–2074, doi: 10.1073/pnas.0409736102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang DW et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem 282, 18602–18612, doi: 10.1074/jbc.M702027200 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Lagace TA et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest 116, 2995–3005, doi: 10.1172/JCI29383 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poirier S et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem 284, 28856–28864, doi: 10.1074/jbc.M109.037085 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poirier S et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 283, 2363–2372, doi: 10.1074/jbc.M708098200 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Canuel M et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 8, e64145, doi: 10.1371/journal.pone.0064145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demers A et al. PCSK9 Induces CD36 Degradation and Affects Long-Chain Fatty Acid Uptake and Triglyceride Metabolism in Adipocytes and in Mouse Liver. Arterioscler Thromb Vasc Biol 35, 2517–2525, doi: 10.1161/ATVBAHA.115.306032 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Jonas MC, Costantini C & Puglielli L PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep 9, 916–922, doi: 10.1038/embor.2008.132 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blom DJ et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med 370, 1809–1819, doi: 10.1056/NEJMoa1316222 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Robinson JG et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 372, 1489–1499, doi: 10.1056/NEJMoa1501031 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, doi: 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishibashi S et al. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest 92, 883–893, doi: 10.1172/JCI116663 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolchok JD et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369, 122–133, doi: 10.1056/NEJMoa1302369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhnast S et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res 55, 2103–2112, doi: 10.1194/jlr.M051326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan JC et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A 106, 9820–9825, doi: 10.1073/pnas.0903849106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A 111, 11774–11779, doi: 10.1073/pnas.1410626111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandramohan V et al. Improved efficacy against malignant brain tumors with EGFRwt/EGFRvIII targeting immunotoxin and checkpoint inhibitor combinations. J Immunother Cancer 7, 142, doi: 10.1186/s40425-019-0614-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogquist KA et al. T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Shan L et al. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun 375, 69–73, doi: 10.1016/j.bbrc.2008.07.106 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Fasano T, Sun XM, Patel DD & Soutar AK Degradation of LDLR protein mediated by ‘gain of function’ PCSK9 mutants in normal and ARH cells. Atherosclerosis 203, 166–171, doi: 10.1016/j.atherosclerosis.2008.10.027 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Duff CJ et al. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J 419, 577–584, doi: 10.1042/BJ20082407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu YY et al. Definition and transfer of a serological epitope specific for peptide-empty forms of MHC class I. Int Immunol 11, 1897–1906, doi: 10.1093/intimm/11.12.1897 (1999). [DOI] [PubMed] [Google Scholar]
- 36.Zhang L et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348, 203–213, doi: 10.1056/NEJMoa020177 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Carstens JL et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 8, 15095, doi: 10.1038/ncomms15095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labun K et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47, W171–W174, doi: 10.1093/nar/gkz365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87, doi: 10.1126/science.1247005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borowicz S et al. The soft agar colony formation assay. J Vis Exp, e51998, doi: 10.3791/51998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore MW, Carbone FR & Bevan MJ Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 54, 777–785 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Curtsinger JM, Lins DC & Mescher MF CD8+ memory T cells (CD44high, Ly-6C+) are more sensitive than naive cells to (CD44low, Ly-6C-) to TCR/CD8 signaling in response to antigen. J Immunol 160, 3236–3243 (1998). [PubMed] [Google Scholar]
- 43.Park SJ, Yoon BH, Kim SK & Kim SY GENT2: an updated gene expression database for normal and tumor tissues. BMC Med Genomics 12, 101, doi: 10.1186/s12920-019-0514-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404, doi: 10.1158/2159-8290.CD-12-0095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1, doi: 10.1126/scisignal.2004088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PCSK9 and CD8A mRNA expression data of various human cancers were downloaded from the GENT (http://gent2.appex.kr/gent2/) database43. PCSK9 mRNA expression and overall survival data were from the TCGA datasets that were included in cBioportal44,45 (https://www.cbioportal.org/) in November, 2018. Western blot source data are provided in Supplementary Fig. 1. Source data for the quantitative graphs are provided. Other data in support of this study are available from the corresponding author upon reasonable request.