

Abstract
Tumor necrosis factor-α-induced protein 8 (TNFAIP8) is a member of TIPE/TNFAIP8 family, has been involved in the development and progression of various human cancers. We hypothesized that TNFAIP8 promotes prostate cancer (PCa) progression via regulation of oxidative phosphorylation (OXPHOS) and glycolysis. Ectopic expression of TNFAIP8 increased PCa cell proliferation/migration/spheroid formation by enhancing cell metabolic activities. Mechanistically, TNFAIP8 activated the PI3K-AKT pathway and up-regulated PCa cell survival. TNFAIP8 was also found to regulate the expression of glucose metabolizing enzymes, enhancing glucose consumption, and endogenous ATP production. Treatment with a glycolysis inhibitor, 2-deoxyglucose (2-DG), reduced TNFAIP8 mediated glucose consumption, ATP production, spheroid formation, and PCa cell migration. By maintaining mitochondrial membrane potential, TNFAIP8 increased OXPHOS and glycolysis. Moreover, TNFAIP8 modulates the production of glycolytic metabolites in PCa cells. Collectively, our data suggest that TNFAIP8 exerts its oncogenic effects by enhancing glucose metabolism and by facilitating metabolic reprogramming in PCa cells. Therefore, TNFAIP8 may be a biomarker associated with prostate cancer and indicate a potential therapeutic target.
Keywords: Tumor necrosis factor-α-induced protein 8 (TNFAIP8), Cell survival, PI3K-AKT pathway, Metabolic reprogramming, Glycolysis, OXPHOS, Prostate cancer
1. Introduction
Tumor necrosis factor-α-induced protein 8 (TNFAIP8) is a member of the TNFAIP8/TIPE family, which also includes other three members, TNFAIP8-like protein 1 (TIPE1), TNFAIP8-like protein 2 (TIPE2), and TNFAIP8-like protein 3 (TIPE3) (Freundt et al., 2008; Kumar et al., 2000; Niture et al., 2018a; Sun et al., 2008). TNFAIP8 was first identified as a differentially expressed transcript by comparing two matched primary and metastatic head and neck squamous cell carcinomas (Patel et al., 1997). The expression of TNFAIP8 is induced by pro-inflammatory cytokines- TNFα (Day et al., 2017; Horrevoets et al., 1999; Niture et al., 2018b) and involved in the regulation of cellular inflammation, signaling, and immunity-related human diseases (Niture et al., 2019). In human cancers, TNFAIP8 acts as an oncogenic molecule that promotes cancer cell proliferation, invasion, metastasis, drug resistance, autophagy, and tumorigenesis by inhibition of cell apoptosis (Niture et al., 2018a). The expression of TNFAIP8 is controlled by several transcriptional factors such as nuclear factor-kB (NF-kB), androgen receptor (AR), p53, and orphan nuclear receptor chicken ovalbumin upstream promoter transcription factor I (COUP-TFI) (Cheng et al., 2015; Day et al., 2017; Monteith et al., 2016; Niture et al., 2018a; Zhang et al., 2009). Previous reports have suggested that in prostate cancer (PCa), TNFAIP8 acts as a potential biomarker (Romanuik et al., 2009) and is associated with cancer cell proliferation and apoptosis (Day et al., 2017). In several cancer cell lines, TNFAIP8 knockdown increased the expression of anti-proliferative and apoptotic genes such as IL24, FAT3, LPHN2, and EPHA3 and downregulated the expression of several oncogenes such as FOXA1, KRAS, S100P, NFAT5, MALAT1, MET, and OSTF1 (Day et al., 2017). Recently, using microarray analysis, we also showed that the expression of TNFAIP8 in PCa cells downregulated the expression of cell cycle-related genes (CCNB2, CCNE2, CDK2, CHEK, and PCNA). We also demonstrated that TNFAIP8 induces autophagy and drug resistance in prostate and liver cancer cells by inhibiting apoptosis (Niture et al., 2020; Niture et al., 2018b).
In tumor cells, even under aerobic conditions, there is a bias towards glycolysis over the oxidative phosphorylation pathway (Liberti and Locasale, 2016; Vander Heiden et al., 2009). Variability and heterogeneity of glycolysis and overall respiration rate exists in tumor cells (Crabtree, 1929). Higher metabolism can also be attributed to variations in genetic and/or environmental factors (tumor microenvironment) surrounding the tumors (Crabtree, 1929). The role that specific genes, such as oncogenes play in the observed variability in tumor metabolism is not fully understood (Dai et al., 2018). Tumor heterogeneity is also evident at the level of oncometabolites, whose abnormal accumulation in cells promotes oncogenesis (Thompson, 2009; Ward and Thompson, 2012; Yang et al., 2013). Most oncometabolites are produced in cells by dysregulation/mutation of metabolic enzymes, with examples including fumarate hydratase (Tomlinson et al., 2002), succinate dehydrogenase (Baysal et al., 2000; Hao et al., 2009; Niemann and Muller, 2000), and isocitrate dehydrogenase 1/2 (Green and Beer, 2010; Parsons et al., 2008; Yan et al., 2009). In gliomas and acute myeloid leukemia (AML), mutation of isocitrate dehydrogenase I render the enzyme unable to convert isocitrate into α-ketoglutarate and hence results in the production of the oncometabolite, R-2-hydroxyglutarate (Dang et al., 2009; Frezza et al., 2010). Accumulation of R-2-hydroxyglutarate enhances DNA hypermethylation and increases cell proliferation/differentiation in human cancer by activation/upregulation of the TGF-β, WNT, and Notch pathways (Dang et al., 2009; Figueroa et al., 2010; Sasaki et al., 2012; Shim et al., 2014; Terunuma et al., 2014). Additional oncometabolites that have been reported include L-2-hydroxyglutarate, D-2-hydroxyglutarate, fumarate, and succinate, all of which have been shown to regulate tumorigenesis (Collins et al., 2017; Ye et al., 2013).
Metabolic reprogramming refers to a change in the bioenergetics of tumor/cancer cells (Yoshida, 2015). Generally, cancer cells are surrounded by stromal cells, which like the tumor cells are heterogeneous with highly variable genetic background, which can produce altered oxygen and nutrient conditions in the tumor microenvironment. Tumor cells can adapt to hypoxia and hypo-nutrient conditions through metabolic reprogramming (Yoshida, 2015). Studies suggest that alteration of metabolic reprogramming and the accumulation of abnormal oncometabolites in tumor cells facilitate malignant transformation and tumor development (Faubert et al., 2020; Yoshida, 2015). Interestingly, several oncogenes, including MYC, BRCA1, and KRAS regulate metabolic reprogramming and increase cell survival/proliferation in tumor cells by enhancing glycolysis, lipid synthesis, and nucleotide synthesis (Gaglio et al., 2011; Privat et al., 2014; Stine et al., 2015).
Since TNFAIP8 modulates the expression of several oncogenes and is involved in cancer progression, we assessed the specific pathways and associate phenotypic changes brought about by TNFAIP8 in PCa cells. We also studied whether TNFAIP8 plays a role in metabolic reprogramming in PCa cells by measuring glucose metabolizing enzyme expression, OXPHOS, glucose consumption, ATP production, and glucose metabolism. For the first time, we demonstrate that TNFAIP8 regulates PCa cell metabolic reprogramming.
2. Materials and methods
2.1. Cell culture
Prostate cancer PC3, DU145, and LNCaP cells were obtained from the Georgetown University Lombardi Comprehensive Cancer Center cell culture repository. Cells were grown in RPMI medium (Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (FBS; Access Biologicals, Vista, CA), 2 mM glutamine, and 25 μg/ml gentamicin (Invitrogen). RWPE1 normal prostate cells were obtained from ATCC, and cultured in keratinocyte-serum-free medium (K-SFM) supplemented with 0.05 mg/ml bovine pituitary extract, epidermal growth factor (5 ng/ml), and 1% penicillin/streptomycin. PCa MDA-PCa-2b cells (African American donor) were obtained from ATCC and cultured in F-12K medium supplemented with 5% FBS. We generated PC3 and LNCaP TNFAIP8-Myc-tagged protein-expressing stable cell lines. For this, PC3 and LNCaP cells were transfected with an empty vector (EV) or TNFAIP8-Myc-tagged plasmid for 30h. Cells were trypsinized, re-plated, and treated with G418 (600 μg/ml) for 2 weeks. Cell colonies were selected, expanded, and maintained in presence of G418 (400 μg/ml). The stable expression of TNFAIP8-Myc-tagged protein was confirmed by immunoblotting. We also generated a TNFAIP8 isoform II knockdown engineered PC3 mini-pool cell line by CRISPR technology (Synthego, Menlo Park, CA SO # 10733-2). All cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2, grown for at least 24h, and used for experiments once they reached 70–80% confluence.
2.2. Western blotting
Western blotting was performed as described previously (Niture et al., 2018b). Briefly, after transfections and treatments, PCa cells were lysed in cell lysis buffer (Cell Signaling Technology, Danvers, MA) containing a protease inhibitor cocktail (Roche, Indianapolis, IN) and protein concentrations were measured using the Bio-Rad protein assay regent (Bio-Rad, Hercules, CA). Cell lysates (40–50 μg) were separated on NuPAGE 4–12% Bis-Tris-SDS gels (Invitrogen) and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). The membranes were placed in blocking buffer (1×) (Sigma-Aldrich, St. Louis, MO) and incubated with primary antibodies overnight at 4°C. The following antibodies were obtained from Cell Signaling Technology (Danvers, MA): anti-AKT, anti-pS473-AKT, anti-mTOR, anti-pS2448-mTOR, anti-Myc-tagged, anti-GAPDH, anti-β-tubulin, anti-Hexokinase I (HKI), anti-GLUT1, anti-AMPKα, and anti-β-actin antibodies. Anti- Glucose 6 phosphatase (G6P) catalytic subunit antibody was obtained from Abcam and anti-Survivin antibody from Santa Cruz Biotechnology. We used anti-TNFAIP8 antibody from Proteintech Group (Rosemont, IL). All primary antibodies were used as per the manufacturer’s suggestions (mostly at 1:1000 dilution). After washing the membranes three times, the membranes were incubated in the appropriate secondary antibody (1:10000 dilution) (Jackson ImmunoResearch, PA) for 1h at room temperature, and immunoreactive bands were visualized using ECL chemiluminescence detection reagents (Signagen Laboratories, Rockville, MD). The western blots were exposed to X-ray films or developed using Azure C-500 Biosystem.
2.3. Plasmid and siRNA transfection
Empty vector (EV) pcDNA3.1 and human tumor necrosis factor-alpha-induced protein 8 transcript variant 1- TNFAIP8-Myc-DDK-tagged (Origene; Rockville, MD Cat # RC202729) ORF cDNA plasmids were used for transfection. RWPE1, PC3, and LNCaP cells were grown in 6-well plates (1×105 cells/wells) for 24h before transfection, and 1–2 μg of EV or TNFAIP8- Myc tagged plasmid DNA was transfected using Lipofectamine LTX Plus transfection reagent (Invitrogen) according to the manufacturer’s instructions. After 30–40h of transfection, cells were harvested, and the expression of the TNFAIP8-Myc tagged protein was examined by immunoblotting. For siRNA transfection, control siRNA and human TNFAIP8 siRNA was obtained from Dharmacon (Lafayette, CO). RWPE1, PC3, and LNCaP cells were grown in an antibiotic-free medium overnight and transfected with 100 nM control or TNFAIP8 siRNA using Lipofectamine RNAiMAX reagent (Invitrogen). After transfection for 30h, cells were harvested and TNFAIP8 knockdown was confirmed by Western blotting or RT/qPCR.
2.4. RT/qPCR
Total RNAs from RWPE1, PC3, LNCaP, DU145, and MDA-PCa-2B cells were isolated using TRIZOL reagent (Life Technology). In other experiments, RWPE1, PC3, LNCaP cells were transfected with empty vector (EV) pcDNA3.1 and TNFAIP8-Myc plasmids or transfected with control siRNA or TNFAIP8 siRNA separately for 30h. Cells were harvested, and RNAs were isolated using TRIZOl reagent. Equal amounts of RNA (1 μg) was reverse transcribed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems), cDNA was incubated with Power SYBR Green PCR master mix (Applied Biosystems) and combined with forward and reverse primers representing glucose metabolizing genes or TNFAIP8 gene (Supplementary Table S1). GAPDH primers were used as an internal control. The PCR reactions were run on a QuantStudio-3 PCR system (Applied Biosystems) and relative quantitation was determined according to the manufacturer’s protocols.
2.5. ActivSignal IPAD assay
PC3-EV and PC3-TNFAIP8 stable or LNCaP-EV or LNCaP-TNFAIP8 stable cells were grown in RPMI medium containing 400 μg/ml G418 for 30h. Cells were lysed in PBS containing 1% NP-40 with 1X protease and phosphatase inhibitor (Thermo Scientific) and submitted to ActivSignal (https://www.activsignal.com/) for further processing. The effect of stable TNFAIP8 expression on the expression of PI3K-AKT proteins and phosphorylation of key PI3K-AKT pathway proteins was determined by Immuno-Paired-Antibody Detection assay (IPAD). The signal was generated using antibodies with high specificity and sensitivity and using two distinct antibodies per target. The signal was normalized to the expression of the housekeeping gene β-actin. The experiments were performed twice using triplicate samples.
2.6. Cell metabolic/survival/proliferation assay
Normal and PCa cells (1×104 cells/well) were grown in 96 plates, and relative cell survival/proliferation was analyzed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (MP Biochemicals, Santa Ana, CA). In other experiments, cells (1 ×105) were seeded onto 6-well plates overnight and transfected with EV or TNFAIP8-Myc plasmid for 24h. The transfected cells were harvested and 1×104 cells seeded onto 96-well plates for 48h. Cells were incubated with 5μl/well of MTT reagent (5 mg/ml) for 1 h at 37°C in a cell culture incubator. Cells were washed with PBS, formazan crystals were dissolved in DMSO, and cell survival was quantified by reading the plates at 570 nm using a Fluostar Omega plate reader. Similarly, PC3 and LNCaP cells were transfected with the TNFAIP8-Myc plasmid or EV for 24h and treated with 1 nM of docetaxel (DTX) for 30h. Transfected and treated cells (1×104 cells/well) were then trypsinized and re-plated in 96-well plates and incubated for 48h. Cell proliferation was measured by adding 10 μl of WST-1 reagent according to the manufacturer’s instructions (Roche Applied Science, Indianapolis, IN). Cell proliferation was measured by reading the plates at 450 nm using Fluostar Omega plate reader (BMG Lab tech, Cary, NC).
2.7. Cell Colony formation assay
RWPE1, PC3, and LNCaP cells were transiently transfected with TNFAIP8-Myc plasmid or pcDNA3.1-EV plasmid (2 μg/well in 6-well plates). After 24h of transfection, cells were trypsinized and counted, and live cells were re-plated in 6-well plates (3000 cells/well) in triplicate and allowed to grow for 7–10 days. Cells were washed with PBS, fixed with cold methanol, and stained with 0.1% crystal violet for 1h. Cells were washed with distilled water and allowed to dry. Blue colonies were counted and plotted.
2.8. Cell migration assay
The effect of TNFAIP8 overexpression or the effect of 2-DG on the migratory ability of normal RWPE1 and PCa PC3 cells were analyzed by wound healing migration assay as described previously (Cagle et al., 2019). Cells were transfected with EV or TNFAIP8-Myc plasmid for 24 or treated with 2-DG as indicated, and transfected/treated cells were plated. The cell monolayer was scraped using a micropipette tip (A0). At 24h post-wounding (A24), cells were photographed, and the migration gap length was calculated using ImageJ software (https://imagej.nih.gov/ij/). The percent wound closure was calculated using the formula [(A0 – A24)/A0] × 100 and plotted.
2.9. ATP assay
Normal prostate and PCa cells were grown in an appropriate medium for 30h, cells were lysed in cell lysis buffer, and cell lysates were used for ATP qualification. In other experiments, RWPE1, PC3, and LNCaP cells were transfected with TNFAIP8-Myc plasmid or pcDNA3.1-EV for 40h, and cells were lysed. Endogenous ATP production was measured by using a Luminometric ATP Assay Kit (ATP determination kit; Life Technologies). Equal amounts of the cell lysates from control or transfected cells were mixed with ATP reaction mixture and incubated for 15 min in the dark at room temperature. The luminescence intensity was quantified using a Fluostar Omega plate reader (BMG Lab tech, Cary, NC). The endogenous ATP concentrations were quantified by generating a standard curve as per the manufacturer’s instructions. Each experiment was performed in triplicate and repeated two to three times.
2.10. Glucose consumption assay
The relative glucose consumption in RWPE1 or PCa cells was determined by a glucose assay kit (Glucose Determination kit GAHK-20, Sigma-Aldrich) according to the manufacturer’s instructions. In brief, cells were grown in 6 well plates at (5×105 cells/well) in 2 ml with the appropriate culture medium and transfected with EV and TNFAIP8-Myc tagged plasmids or control siRNA and TNFAIP8 siRNA separately for 40h. In other experiments, cells were pretreated with 2-deoxyglucose (2-DG) for 8h, and then cells were transfected with EV or TNFAIP8-myc plasmid for 40h. Then the media were removed from control or transfected/treated cells and the concentrations of glucose in the media were measured. The glucose concentrations were quantified by generating a standard curve as per manufacturer’s instructions. The glucose consumption was calculated by the original medium concentration minus the glucose concentration in the medium after 40h. The experiments were performed in triplicate and repeated two to three times.
2.11. Seahorse Bio-analyzer
The Cell Mito-Stress Assay was used to characterize the effect of stable TNFAIP8 overexpression or TNFAIP8 knockdown on cell energy phenotype and oxidative phosphorylation (OXPHOS) in PCa PC3 and LNCaP cells. TNFAIP8 overexpressing or TNFAIP8 knockdown PC3 cells were seeded at a density of 2 × 104 cells/well and LNCaP cells at a density of 4×104 cells/well in XFp plates and allowed to stabilize overnight. XF media was supplemented with 10 mM D-glucose and 2 mM L-glutamine, as described in the manufacturer’s instructions for the XF Cell Mito-Stress assay. Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured using a Seahorse XFp analyzer. For PC3 cells, 1 μM Oligomycin, 0.5 μM FCCP, and 0.5 μM Rotenone were simultaneously applied to measure glycolysis and OXPHOS in the cells. Injections of 2 μM Oligomycin, 0.5 μM FCCP, and 0.5 μM Rotenone were simultaneously applied to measure the same metabolic parameters in LNCaP cells. Specific conditions were determined by cell characterization methods stated in the manufacturer’s instructions for the XF Cell Mito-Stress assay. TC20 Automated Cell Counter (Bio-Rad) and Incucyte® (Sartorius) technology were used to determine cell number in each well. The results were analyzed using Wave software (Seahorse/Agilent).
2.12. Mitochondrial membrane potential assay
A TMRE-mitochondrial membrane potential assay kit (Cat # ab113852) was used for quantifying changes in mitochondrial membrane potential in stable EV and TNFAIP8 expressing PC3 and LNCaP cells. Ten thousand cells were seeded in 96 well plates, and cells were treated with 200 μM of CoCl2. After 24h, cells were further treated with the TMRM reagent (100 μl of 500 nM) for 30 min at 37°C. Cells were washed with PBS twice, and fluorescence intensities were measured by using a PHERAstar Microplate reader (BMG Lab tech, Cary, NC) using the 590–50 675–50 filter. The experiments were performed in triplicate and repeated two times.
2.13. Metabolomic analysis
Stable EV and TNFAIP8 expressing PC3 cells were grown in three independent experiments, and cell pellets were submitted for metabolomic analysis at the ‘metabolomics core facility’ at Baylor College of Medicine (Houston TX). In other experiments, we used wild type and TNFAIP8 isoform II knockdown PC3 cell line generated by CRISPR technology to carry out glucose-related targeted metabolic profiling. Metabolic profiling was analyzed by LC-MS Single Reaction Monitoring (SRM). For each metabolite in the normalized dataset, a two-tailed t-test was used to compare their expression levels between two groups. Differentially expressed metabolites were identified after adjusting p-values for multiple hypothesis testing using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) and a False Discovery Rate (FDR) of < 0.25. A hierarchical cluster of the differentially expressed metabolites was generated using the R statistical system (https://www.r-project.org/).
2.14. Statistical analysis
Results from duplicate or triplicate independent experiments are presented as Mean ± SEM. Differences between groups were analyzed using either two-tailed Student’s t-test. A P value of < 0.05 was considered statistically significant. Statistical analyses were performed using the IBM SPSS Statistics 25 software (Armonk, NY).
3. Results
3.1. PCa cells exhibit altered metabolism, glucose consumption, and ATP production.
To study the basal metabolic activity of prostate cancer cells, we carried out a comparative analysis of metabolic activities, rate of glucose consumption, and endogenous ATP production in four PCa cell lines (PC3, LNCaP, DU145, and MDA-PCa-2b) and one cell line representing normal prostate cells (RWPE1). As shown in Fig. 1A, logarithmically growing PCa cell lines showed higher (1.5–2.2 fold) cell metabolic/viability activities when compared with normal prostate cells RWPE1 (Fig. 1A). Next carried out a more detailed analysis of the rate of glucose consumption and ATP production. Compared with normal prostate cells RWPE1, the PCa cell lines (PC3, LNCaP, and DU145) displayed higher glucose consumption rates (Fig. 1B). Interestingly, PCa DU145 and MDA-PCa-2b cell lines had reduced ATP production compared with normal prostate cells RWPE1 (Fig. 1C). Among the PCa cell lines, ATP production was higher in LNCaP cells compared with PC3 and DU145, while dramatically lower glucose consumption and ATP production was observed in PCa MDA-PCa-2b cells of African American origin (Fig. 1B & C). We also analyzed spheroid formation in PCa cell lines and the data indicated that, although DU145 and MDA-PCa-2b cells had lower relative ATP production, these cells produced larger spheroids in vitro compared with LNCaP and PC3 or normal prostate cells RWPE1 (Supplementary Fig. 1) lthough, higher glucose consumption and larger spheroids formation were observed in DU145 cells these cells produce a low amount of ATP. Similarly, a lower glucose consumption and ATP production was observed in MDA-PCa-2b, and these cells produced larger and intact spheroids compared with PC3 or normal RWPE1 cells (Fig. 1 B &C, Supplementary Fig. S1). These results indicate that a larger and intact spheroids formation in DU145 and MDA-PCa-2b cells may limit oxygen availability, oxidative phosphorylation (OXY/PHOS), and glycolysis that may result in lower ATP production. The heterogeneity between these PCa cells with respect to OXY/PHOS, glycolysis, and cellular energetics needs to be further investigated. Next, we analyzed the expression of the oncogenic AKT-mTOR pathway in PCa cells since this pathway plays a major role in PCa aggressiveness, cell proliferation/survival, and metabolic activities (Aoki and Fujishita, 2017; Edlind and Hsieh, 2014; Kaarbo et al., 2010). Immunoblotting data showed that, compared with normal RWPE1 cells, a higher level of endogenous AKT-S473 phosphorylation was detected in PC3, LNCaP, and DU145 cells and a higher level of mTOR-S2448 phosphorylation in MDA-PCa-2B cells (Fig. 1D, left and right panels). Also, the higher expression of survivin (cell survival factor) was observed in PCa cells compared with RWPE1 cells (Fig. 1D, left and right panels). These studies highlight the differences among PCa cell lines and normal prostate cells in metabolic activities and regulation of the AKT-mTOR signaling.
Fig. 1.
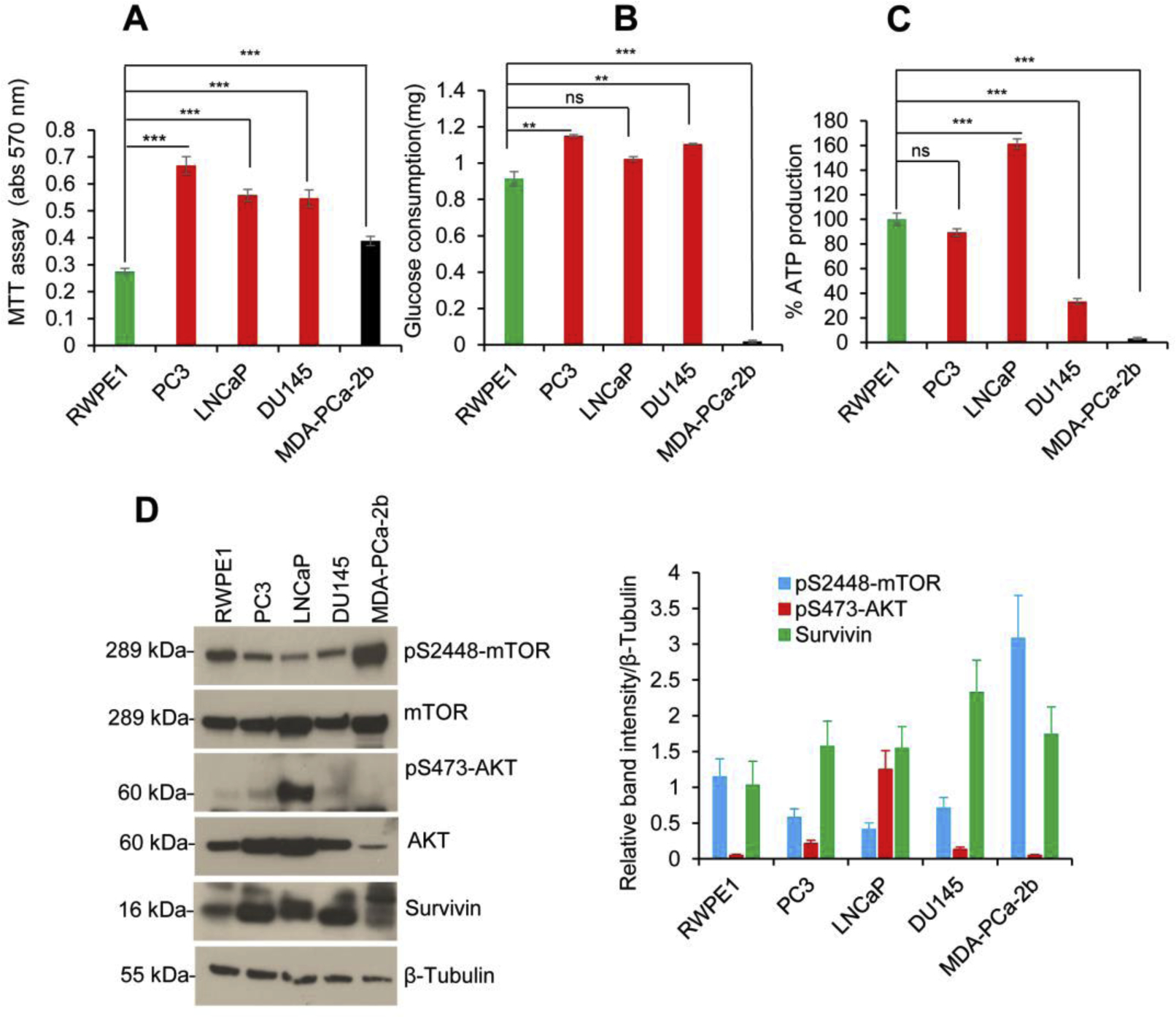
PCa cells show altered cell survival, glucose consumption, and ATP production. (A) RWPE1, PC3, LNCaP, DU145, and MDA-PCa-2b cells (1×104) were grown in 96 well plates for 72h, and relative cell metabolic/cell survival activity was analyzed by MTT assay. (B) Normal prostate RWPE1 and PCa cells (1×105) were grown in six-well plates in regular medium for 40h, and the relative glucose consumption was analyzed using a glucose determination kit GAHK-20 as described in the materials and methods section. (C) RWPE1 and PCa cells (1×105) were grown in six-well plates in regular medium for 40h and cell extracts were prepared. The relative endogenous ATP production was measured by using a Luminometric ATP Assay Kit as described in the materials and methods section. (D) RWPE1, PC3, LNCaP, DU145, and MDA-PCa-2b cells (1×105) were grown in six-well plates for 40h and cell lysates (50 μg) were immunoblotted with indicated antibodies (left panel). Indicated protein levels from (D) were quantified using ImageJ software (https://imagej.nih.gov/ij/) and plotted (right panel). Data represent mean ± SEM. **P<0.01 ***P<0.001, compared to normal prostate RWPE1 cells. ns - not significant.
3.2. TNFAIP8 as an oncogenic molecule in PCa.
TNFAIP8 protein is overexpressed in multiple cancers (Niture et al., 2018a; Niture et al., 2018b). To test the role of TNFAIP8 in PCa cell growth and proliferation, first, we analyzed the expression of TNFAIP8 protein and mRNA expression in PCa cells. Immunoblotting and RT/qPCR data demonstrated that the expression of both TNFAIP8 protein and mRNA was observed in normal prostate RWPE1 cells as well as PCa cell lines (Fig. 2A & B). To confirm the proliferative role of TNFAIP8 we transiently overexpressed TNFAIP8 in RWPE1, PC3, and LNCaP cells to study cell viability and survival. Ectopic expression of TNFAIP8 increased cell viability/survival by 2.75-fold in RWPE1, 1.88- fold in PC3 cells, and 1.22-fold in LNCaP cells compared with EV transfected cells (Fig. 2C & D). Also, TNFAIP8 overexpression increased cell colony formation by 23% in RWPE1, 16% in PC3, and 18% in LNCaP cells (Fig. 2E, left and right panels) and significantly increased cell migration in RWPE1 and PC3 cells as determined by a wound-healing assay (Fig. 2F, left and right panels). Docetaxel (DTX) is an anticancer drug often used in metastatic castrate-resistant PCa, however half of the patients do not respond, and those who respond eventually become resistant to Docetaxel (Seruga et al., 2011). Mechanisms of DTX resistance are not completely understood. We asked whether Docetaxel would be effective in TNFAIP8 overexpressing PCa. As reported earlier, PC3 cells are less sensitive to DTX than LNCaP or DU145 cells (Tamaki et al., 2014) and our data suggest that treatment of low concentration of DTX (1nM) and transfected with EV in PC3 or LNCaP cells showed no significant effect on cell proliferation compared with EV alone transfected cells. Moreover, expression of TNFAP8 in DTX treated PC3 cells increased 4.41fold cell proliferation whereas, TNFAIP8 expression in DTX treated LNCaP cells increased 1.31 fold cell proliferation compared with DTX and EV transfected cells suggesting that expression of TNFAIP8 enhances drug-resistant more efficiently in PC3 (androgen receptor-negative cells) compared with LNCaP (androgen receptor-positive) cells (Fig. 2G). The exact role and mechanism of TNFAIP8 in DTX resistance in these cell lines needs to be further investigated.
Fig. 2.
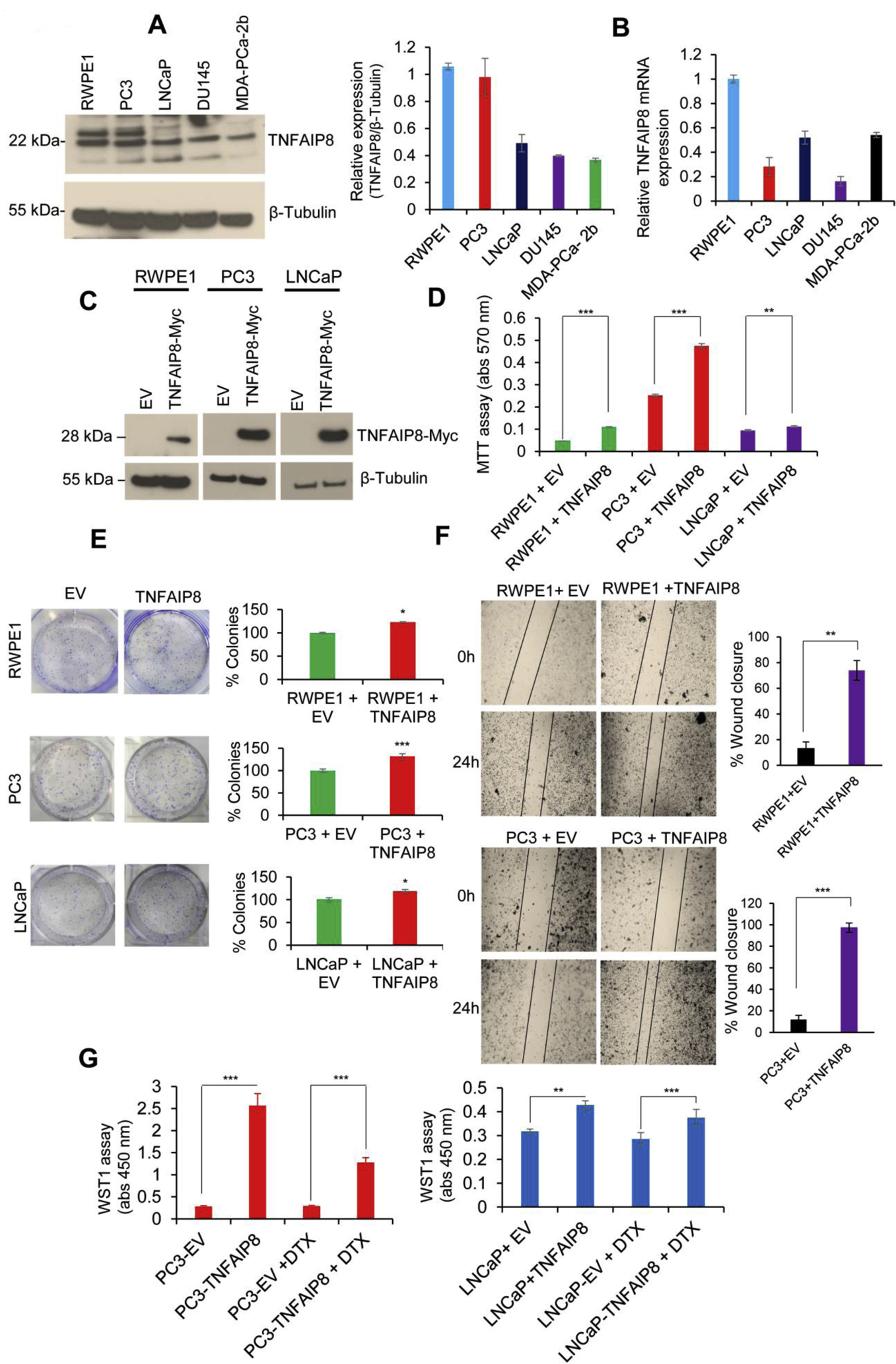
TNFAIP8 enhances cell metabolic activity, cell viability, and colony formation in PCa cells. (A) RWPE1, PC3, LNCaP, DU145, and MDA-PCa-2b cells (1×105) were grown in six-well plates for 40h, and cell lysates (50 μg) were immunoblotted with TNFAIP8 and β-tubulin antibodies. TNFAIP8 protein levels were quantified using ImageJ software (https://imagej.nih.gov/ij/) and plotted (left panel). (B) Normal and PCa cells (1×105) were grown in six-well plates for 72h, and expression of relative TNFAIP8 mRNA levels was analyzed by RT/qPCR as described in the materials and methods section. (C) RWPE1, PC3, LNCaP cells were transfected with EV or TNFAIP8-Myc tagged plasmid (2 μg) for 30h, and expression of TNFAIP8-Myc tagged protein in RWPE1 and PCa cells were analyzed by western blotting. (D) Similarly, EV and TNFAIP8-Myc transfected cells (1×104) were re-plated in 96 well plates, and the effect of TNFAIP8 expression on cell survival was analyzed by MTT assay. (E) RWPE1, PC3, LNCaP cell were transfected with EV or TNFAIP8-Myc plasmids and transfected cells (3 × 103) were re-plated in six-well plates for 7 to 10 days and effect of TNFAIP8 expression on cell colony formation were measured and plotted (left and right panels). (F) Wound healing assay: RWPE1 and PC3 cells were transfected with EV or TNFAIP8-Myc plasmid for 30 h. The effect of the TNFAIP8-Myc expression on cell migration was analyzed by wound-healing assay. Representative images of the wound healing assay (left panels) and the calculated scratch area are shown (right panels). (G) PC3 and LNCaP cells were transfected with EV or TNFAIP8-Myc plasmid for 24h and treated with DTX (1nM) for an additional 30h, and cell proliferation was analyzed by WST1 assay. Data represent mean ± SEM from three independent experiments. *P<0.05, **P<0.01, ***P<0.001 compared to EV transfected cells.
3.3. TNFAIP8 activates the PI3K-AKT pathway and increases cell proliferation in PCa cells.
An oncogenic PI3K-AKT signaling pathway is known to promote cell proliferation/survival (Aoki and Fujishita, 2017; Edlind and Hsieh, 2014; Kaarbo et al., 2010). We analyzed the effect of TNFAIP8 on the modulation of the PI3K-AKT pathway. We generated stable TNFAIP8-expressing PC3 and LNCaP PCa cell lines (Fig. 3A). Stable expression of TNFAIP8 in PC3 cells increased the expression of pS473-AKT, p-S2448-mTOR compared with EV stable expressing cells (Fig. 3B). To investigate in more detail the role of TNFAIP8 in the regulation of PI3K-AKT signaling pathway and related targets in PCa cells, we used these stable cell lines to assess the impact of TNFAIP8 on the PI3K-AKT pathway using the ActivSignal IPAD assay (Fig. 3C, upper and lower panels). The data suggested that stable expression of TNFAIP8 significantly increased the phosphorylation of S473-AKT, p-4EBP1, p-β-Catenin, p-Mek1, and p-p44 in PC3 cells and also pS473-AKT in LNCaP cells (Fig. 3C, upper and lower panels).
Fig. 3.
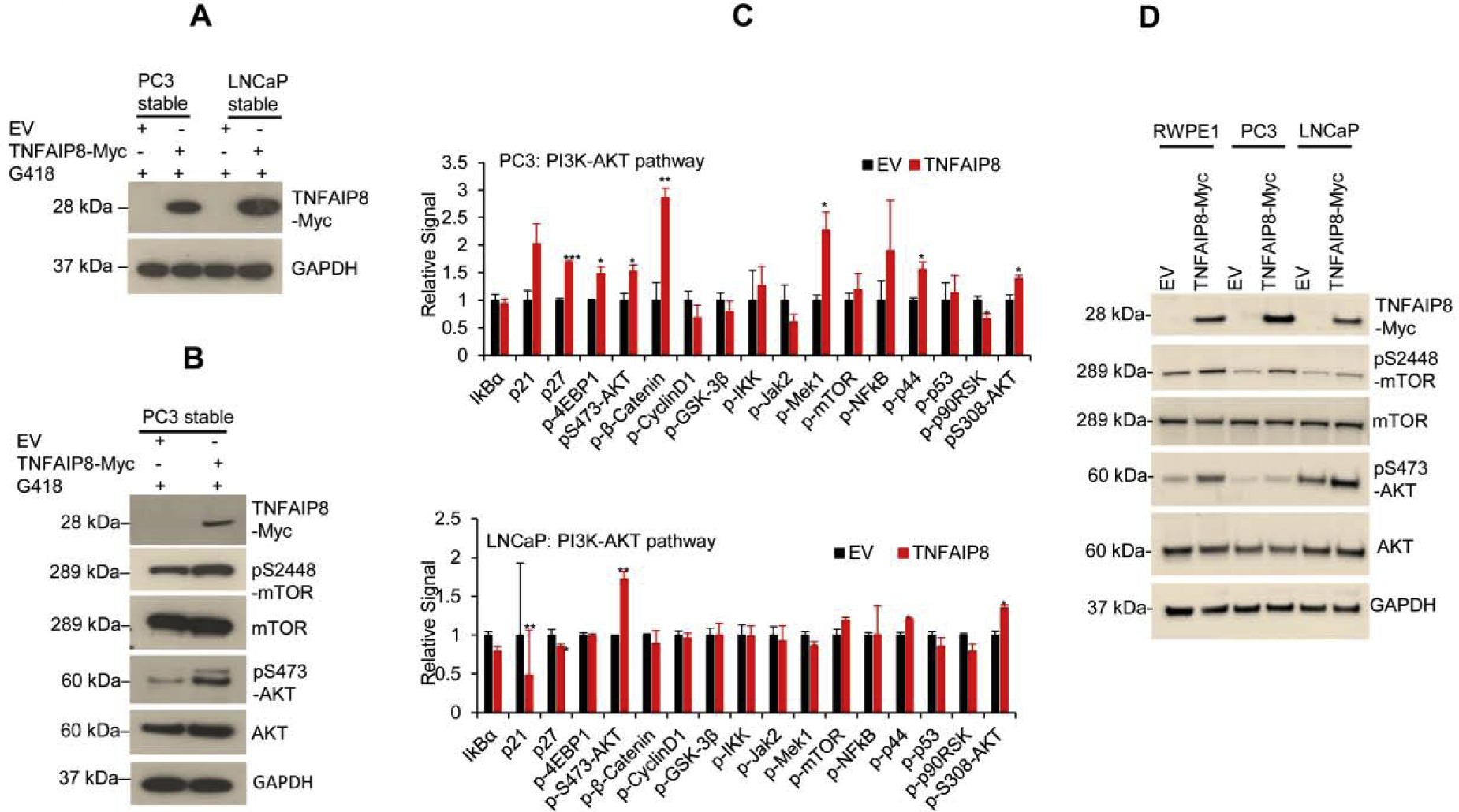
TNFAIP8 activates the PI3K-AKT pathway and induces cell proliferation. (A) Stable expression of TNFAIP8-Myc tagged protein in PC3 and LNCaP cells were analyzed by western blotting. (B) The effects of TNFAIP8 stable expression on AKT-mTOR pathway proteins were analyzed by western blotting. (C) ActivSignal IPAD assay. The impact of stable EV or TNFAIP8 expression on the regulation of PI3K-AKT pathway proteins and phosphorylation of key PI3K-AKT pathway proteins were analyzed from PC3 and LNCaP cells. The signal was normalized to the expression of housekeeping gene β-actin and plotted. *P<0.05, **P<0.01, ***P<0.001 compared to EV stable transfected cells. (D) RWPE1, PC3, LNCaP cells were transfected with EV or TNFAIP8-Myc tagged plasmid (2 μg) for 30h, and expression of TNFAIP8-Myc tagged protein and pS2448-mTOR, mTOR, pS473-AKT and AKT were analyzed by western blotting.
Transiently overexpressed TNFAIP8 significantly increased AKT-S473 and mTOR-S2448 phosphorylation in PC3, LNCaP, and RWPE1 normal prostate cells compared with EV transfected cells (Fig. 3D). To test whether TNFAIP8-mediated activation of AKT promotes PCa cell survival, we inactivated AKT using AKT inhibitor MK-2206. Expression of TNFAIP8 in PC3 and LNCaP cells increased AKT-S473 phosphorylation and cell proliferation in both cell lines (Supplementary Fig. S2A-D). However, pretreatment with AKT inhibitor MK-2206 completely abolished AKT-S473 phosphorylation in EV or TNFAIP8-transfected cells and significantly reduced TNFAIP8-mediated cell survival suggesting that TNFAIP8-mediated activation of AKT enhanced cell proliferation in PCa cells (Supplementary Fig. S2A-D). Collectively, the data indicate that by activation of the PI3K-AKT pathway, TNFAIP8 enhances cell metabolic/cell survival activities in PCa cells.
3.4. TNFAIP8 increases glucose consumption/ATP production by regulating glucose metabolizing enzymes.
We earlier showed that TNFAIP8 increases cell proliferation by activation of AKT in PCa cells as seen by MTT metabolic assay. We asked whether TNFAIP8 could also modulate glucose metabolism/energetics and the expression of glycolytic enzymes in PCa cells. Overexpression of TNFAIP8 in RWPE1, PC3, and LNCaP cells increased the expression of TNFAIP8 mRNA compared with EV transfected cells (Fig. 4A). We demonstrated that overexpression of TNFAIP8 significantly increased mRNA expression of glucose metabolism enzymes, HK1, HK2, GLUT1, G6PD, LDHA, and PEPCK in RWPE1 normal prostate cells (Fig. 4B, left panel). In PCa cells, overexpression of TNFAIP8 increased mRNA levels of HK1, GLUT1, LDHA in PC3 cells, and HK2, G6PD, AMPK, LDHA, and PEPCK expression in LNCaP cells (Fig. 4B, middle and right panels). Interestingly, the data also demonstrated that expression of TNFAIP8 increased expression of IL-6, a pro-inflammatory cytokine in PC3 and RWPE1 cells but not in LNCaP cells. We also analyzed the expression HK1, GLUT1, G6P (catalytic subunit), and AMPKα proteins after overexpressing TNFAIP8 in RWPE1, PC3, and LNCaP cells (Fig. 4C). Overexpression of TNFAIP8 increased expression of HK1, GLUT1, AMPKα, and G6P in PCa cells (Fig. 4C).
Fig. 4.
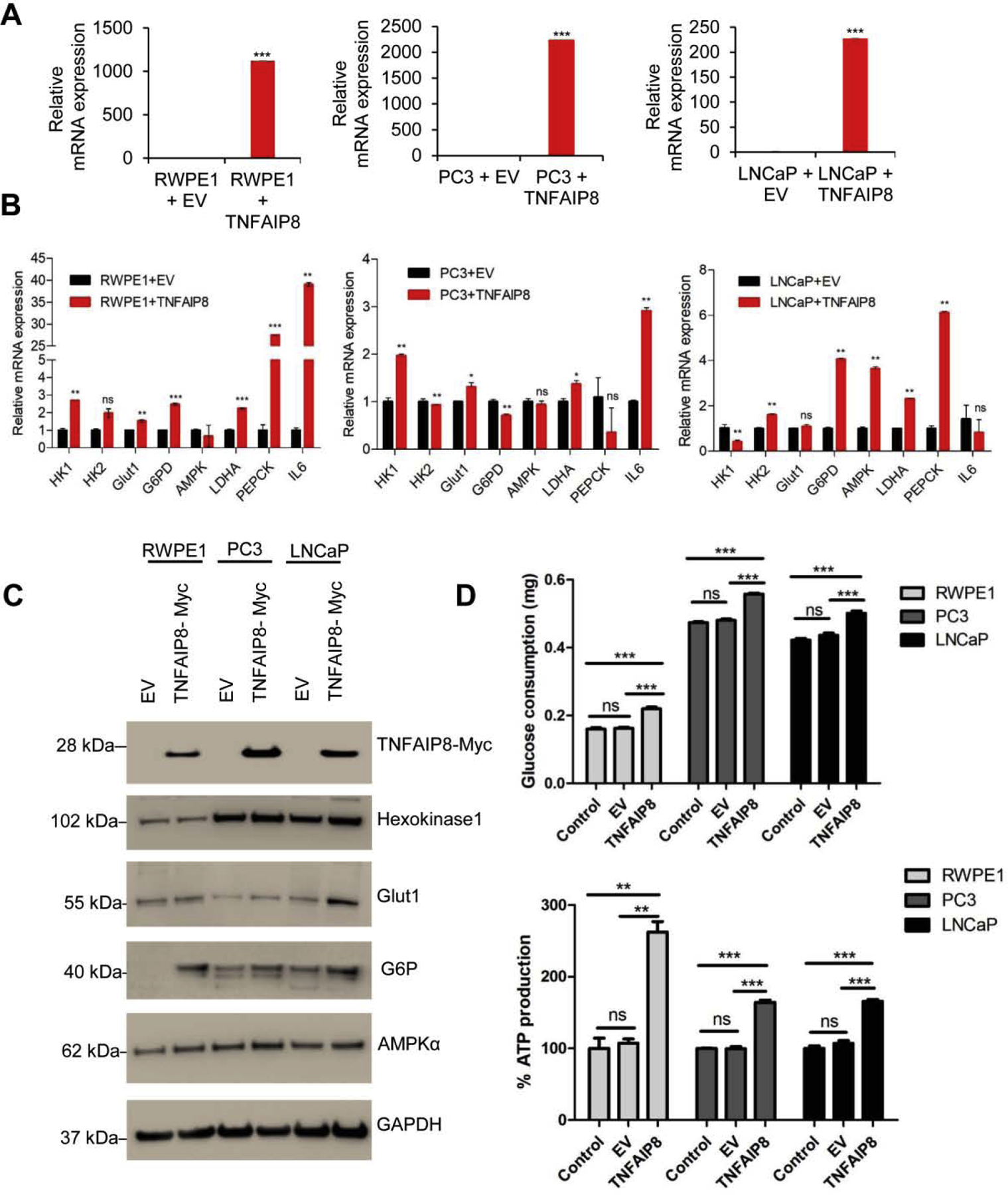
TNFAIP8 regulates glucose metabolizing enzyme expression and glucose consumption in PCa cells. (A) RWPE1 and PCa cells (1×105) were grown in six-well plates 24h and transfected with EV or TNFAIP8 plasmid for 30h. The expression of TNFAIP8 mRNA levels was analyzed by RT/qPCR. (B) Similarly, the effect of TNFAIP8 expression on the regulation of glucose metabolizing enzymes (HK1, HK2, GLUT1, G6PD, AMPK, LDHK, PEPCK, and IL-6) mRNA expression was analyzed by RT/qPCR as described in the materials and methods section. (C) RWPE1, PC3, and LNCaP (1×105) cells were grown in six-well plates 24h and transfected with EV or TNFAIP8-myc plasmid for 30h. The cell lysates were immunoblotted with Hexokinase 1 (HK1), Glut1, G6P, AMPKα, and GAPDH antibodies. (D) RWPE1, PC3, and LNCaP (1×105) cells were grown in 6-well plates for 24h and transfected with EV or TNFAIP8-myc plasmid for 40h and the effect of TNFAIP8 expression on glucose consumption and endogenous ATP production was determined (upper and lower panels). Data represent mean ± SEM from three independent experiments. *P<0.05, **P<0.01,***P<0.001 compared to EV transfected or control cells.
We asked whether TNFAIP8 could modulate glucose consumption and ATP production in prostate cells. As shown in Fig. 4D (upper and lower panels), overexpression of TNFAIP8 significantly increased glucose consumption and endogenous ATP production in RWPE1 and PCa cells compared with EV transfected cells. No significant impact on glucose consumption and endogenous ATP production was observed when cell transfected with EV or treated with LTX plus transfection reagents compared with control/untreated cells (Fig. 4D and Supplementary Fig. S3A, upper panel).
In a complementary approach, TNFAIP8 knockdown by siRNA was carried out in RWPE1, PC3, and LNCaP cells, and the effect on enzymes involved in glucose metabolism was analyzed (Fig. 5A). TNFAIP8 knockdown decreased HK1 and AMPKα protein expression in PC3 and LNCaP cells, but no change was observed in RWPE1 cells (Fig. 5A). Next, the effect of TNFAIP8 knockdown on glucose consumption and ATP production was analyzed (Fig. 5B, upper and lower panels). TNFAIP8 knockdown significantly decreased glucose consumption in RWPE1, LNCaP, and PC3 cells (Fig. 5B, upper panel). A significant decrease in ATP production was also observed in PC3 and LNCaP cells (Fig. 5B, lower panel) upon TNFAIP8 knockdown. No significant difference in glucose consumption/ATP production was observed when cells transfected with EV or treated with Lipofectamine 2000 siRNA transfection reagent compared with control/untreated cells (Fig. 5B and Supplementary Fig. S3A, lower panel). Importantly, RWPE1 and PCa cells pretreated with 2-deoxyglucose (2-DG), a glycolysis inhibitor, significantly reduced glucose consumption as observed and TNFAIP8 was unable to increase glucose consumption compared with EV transfected cells (Fig 5C).
Fig. 5.
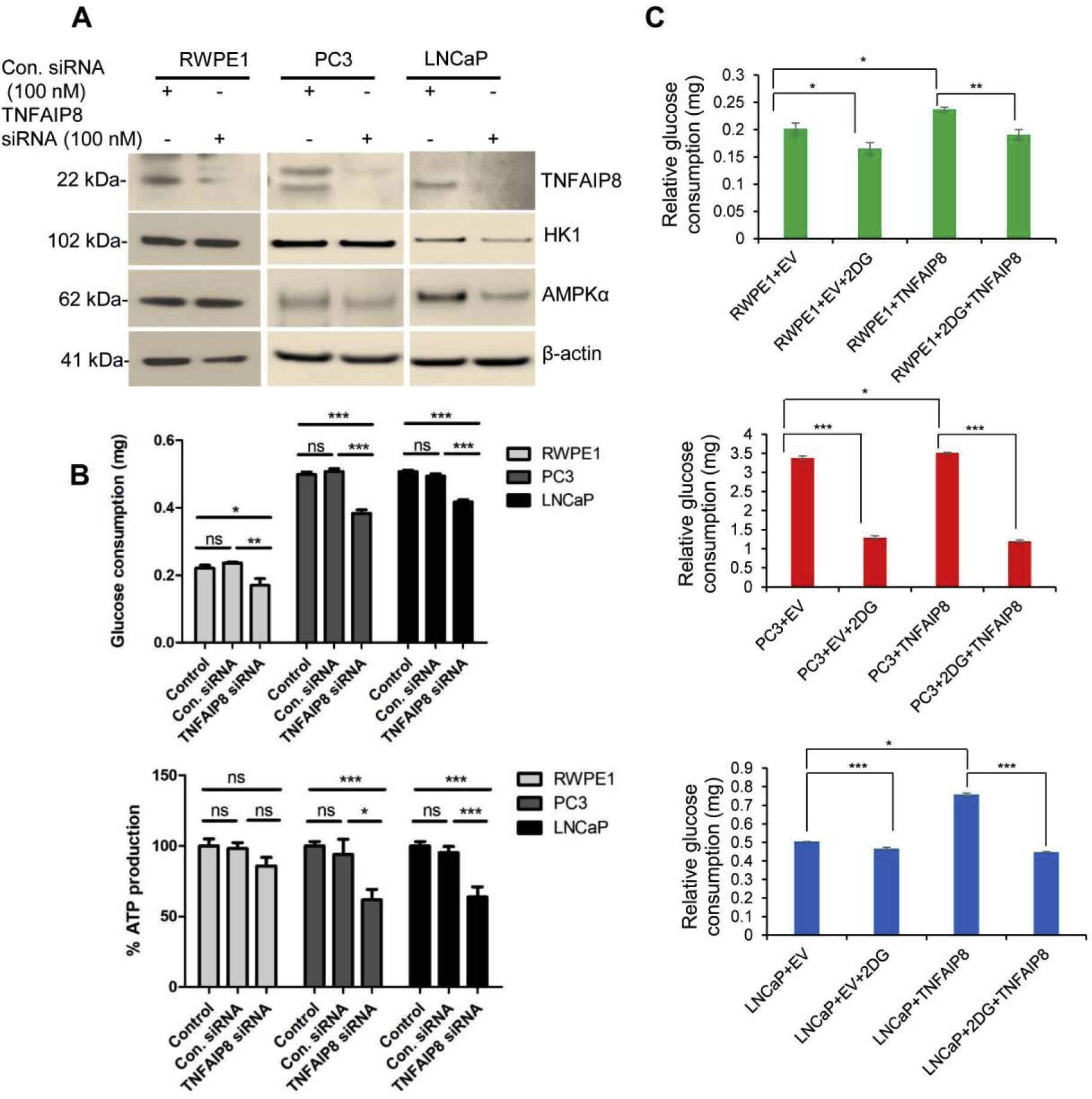
TNFAIP8 knockdown reduced glucose consumption and ATP production in PCa cells. (A) RWPE1, PC3, and LNCaP (1×105) cells were grown in six-well plates 24h and transfected with control siRNA or TNFAIP8 siRNA. Cell lysates from control siRNA or TNFAIP8 siRNA transfected cells were immunoblotted with TNFAIP8, HK1, AMPKα, and β-actin antibodies. (B) RWPE1, PC3, and LNCaP (1×105) cells were grown in six-well plates 24h and transfected with control siRNA or TNFAIP8 siRNA for 40h. The effect of TNFAIP8 knockdown on relative glucose consumption (upper panel) and endogenous ATP production (lower panel) was measured as described in the materials and methods sections. Data represent mean ± SEM from two independent experiments in triplicates. *P<0.05, **P<0.01, ***P<0.01 compared to control siRNA transfected or control cells. ns- not significant. (C) RWPE1, PC3, and LNCaP (1×105) cells were grown in six-well plates 24h and pre-treated with 2-DG (5mM) for 8h, and then cells were transfected with EV or TNFAIP8 plasmid for 40h and relative cellular glucose consumption was analyzed as described in material and methods section. Data represent mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 compared to EV or TNFAIP8 transfected cells.
Moreover, TNFAIP8 overexpression also promoted ATP production as well as TNFAIP8 mediated spheroid formation/ cell migration (Supplementary Fig. S4 A&B, Supplementary Fig. S5). Pretreatment and in presence of 2-DG, TNFAIP8 was unable to increase ATP production as well as TNFAIP8-mediated spheroid formation/cell migration in PCa cells (Supplementary Fig. S4 A&B, Supplementary Fig. S5). Collectively our data suggested that TNFAIP8 enhances glucose consumption /ATP production, spheroid formation, and cell migration in PCa cells.
3.5. TNFAIP8 increases OXPHOS and glycolysis.
TNFAIP8 increase glucose consumption and ATP production in PCa cells therefore we further examined the overall rate of OXPHOS and glycolysis in PC3 and LNCaP cells stably expressing TNFAIP8 protein by using the Seahorse Bio-analyzer. Since these stable TNFAIP8 expressing cells maintained in presence of G418 (geneticin) antibiotic, first we analyzed the effect of G418 on TNFAIP8 expression and glucose consumption (Supplementary Fig. S3B, upper and lower panels). Immunoblotting data suggest that when these cells grow in absence of G418 for 40h showed TNFAIP8 expression, as well as an, increased in glucose consumption similar to when cells grown in presence of G418 compared with EV transfected cells indicate that, under these control conditions G418 has no significant role in TNFAIP8 mediated glucose consumption (Supplementary Fig. S3B, upper and lower panels). Similarly using G418 free XF medium we analyzed OXPHOS and glycolysis in PC3 and LNCaP cells stably expressing TNFAIP8 protein. In stably transfected cells, compared with EV, TNFAIP8 expressing PC3 and LNCaP cells showed increased glycolysis and mitochondrial respiration (~2.2–2.5 fold) (Fig. 6 A&B, left and right panels).
Fig. 6.
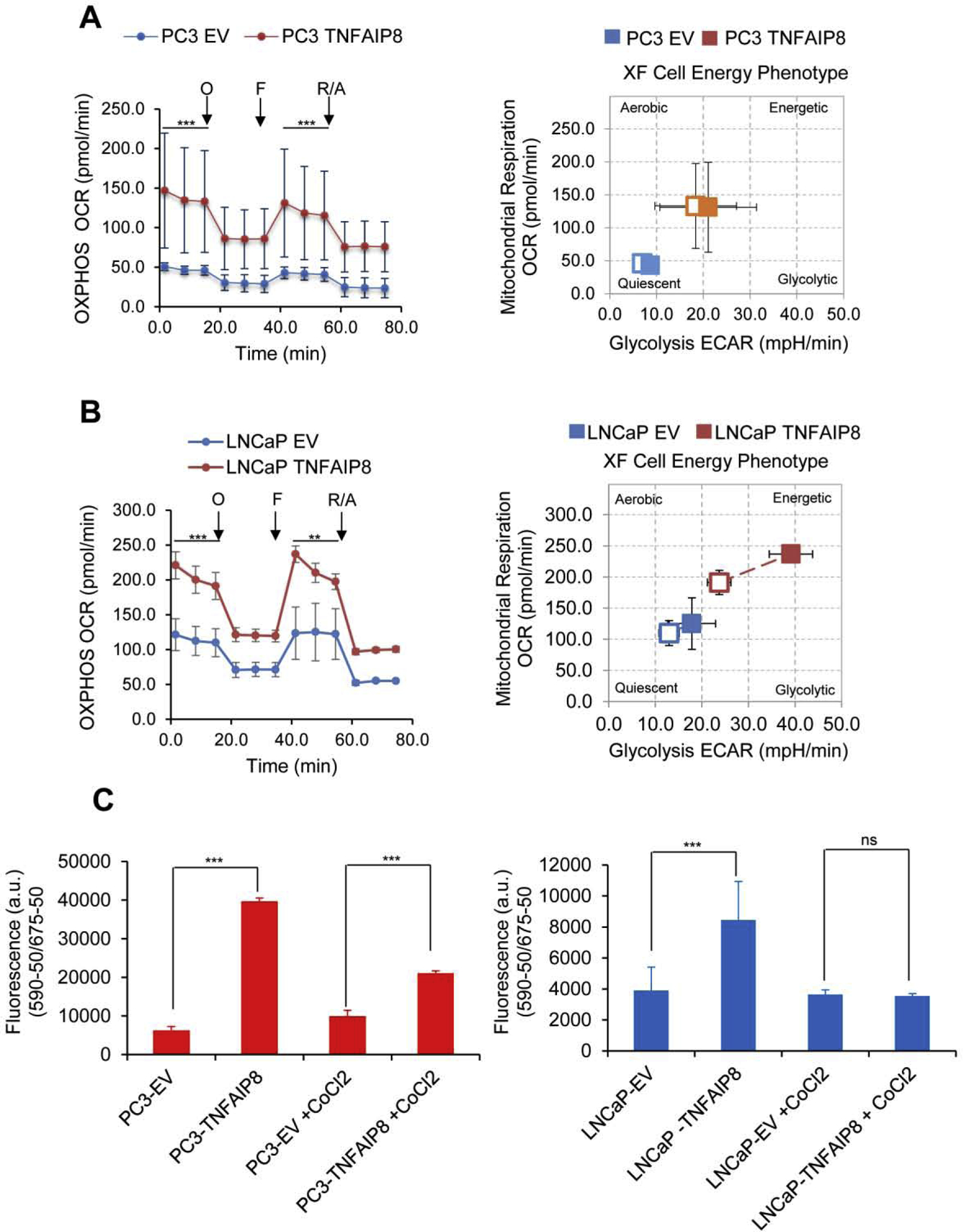
TNFAIP8 increased mitochondrial OXPHOS, glycolysis, and cell energy phenotype in PCa cells. (A & B) TNFAIP8 stable expressing PC3 and LNCaP cells or EV stable expressing cells were subjected to Seahorse Bio-analyzer, and relative OXPHOS and glycolysis were analyzed as described in the materials and methods section (Left and right panels). **P<0.01, ***P<0.001 compared to EV expressing PCa cells. O- Oligomycin. F- FCCP. R/A- Rotenone and Antimycin. (C) TMRE-mitochondrial membrane potential assay. Stable EV and TNFAIP8 expressing PC3 and LNCaP (1×104) cells were seeded in 96 well plates and treated with 200 μM of CoCl2 for 24h as indicated, and mitochondrial membrane potentials were analyzed by exposing the cells with TMRM reagent as described in materials and methods sections. Data represent mean ± SEM. ***P<0.001 compared to EV stable expression cells. ns- not significant. All the experiments were performed in triplicate and repeated twice.
We earlier demonstrated that TNFAIP8 activated AKT signaling and promoted cell proliferation. We asked if AKT signaling plays a role in TNFAIP8 modulated metabolic changes. We analyzed the effect of AKT inhibitor MK-2206 on TNFAIP8 mediated OXPHOS and glycolysis in PC3 and LNCaP cells. Pre-treatment with MK-2206 modestly reduced TNFAIP8 induced OXPHOS and glycolysis in PC3 and LNCaP cells suggesting a role for AKT signaling (Fig. 6A & B and Supplementary Fig. S6 A & B). On the other hand, TNFAIP8 knockout in PC3 and LNCaP cells also significantly decreased OXPHOS and glycolysis compared with control siRNA transfected cells (Supplementary Fig. S7 A&B, left and right panels)
Since the integrity of the mitochondrial membrane is important for OXPHOS and mitochondrial respiration, we further analyzed the role of TNFAIP8 in regulating mitochondrial membrane potential. Stable TNFAIP8 expressing PC3 and LNCaP cells were treated with CoCl2, which is widely used as hypoxia mimetic and known to induce mitochondrial membrane dysfunction and mitochondrial fragmentation (He et al., 2018; Lee et al., 2013). The effect of TNFAIP8 expression and CoCl2 treatment on mitochondrial membrane changes were analyzed by the post-treatment of the cells with TMRE-reagent (Abcam). The binding of TMRE to mitochondrial membranes was significantly increased (as indicated by fluorescence arbitrary units) in TNFAIP8-expressing PC3 and LNCaP cells compared with EV transfected cells (Fig. 6C, left and right panels). On the other hand, treatment with CoCl2 decreased the TMRE binding to mitochondrial membranes in both PC3 and LNCaP cells, whereas treatment with CoCl2 in TNFAIP8-expressing PC3 cells increased TMRE binding to mitochondrial membranes indicating that TNFAIP8 facilitates mitochondrial membrane potential at least in PC3 cells (Fig. 6C, left and right panels). Our data collectively indicates that TNFAIP8 enhanced glycolysis is associated with alteration in the levels of glycolytic enzymes and further, it modulates mitochondrial respiration by increasing mitochondrial membrane potential.
3.5. TNFAIP8 modulates the glycolytic metabolic profile in PCa cells.
Since TNFAIP8 increased OXPHOS and glycolysis, we further interrogated the metabolic alterations due to TNFAIP8 by global profiling. We performed metabolic profiling of PC3 cells overexpressing TNFAIP8 and cells with TNFAIP8 knockdown. Overexpression of TNFAIP8 was confirmed by immunoblotting (data not shown). PC3 cells stably expressing TNFAIP8 exhibited accumulation/endogenous production of 36 metabolites and a decrease in ~ 23 metabolites compared with EV stable cells (Fig. 7A and Supplementary Table S2). Among changed metabolites, TNFAIP8-expressing PC3 cells showed higher accumulation of several glycolytic metabolites, including glucose-6-phosphate, fructose-6-phosphate, 2-phosphoglyceric acid, 3-phosphoglyceric acid, pyruvic acid, and lactic acid (Fig. 7A). Higher accumulation of several other metabolites such as lactate, D-gluconate 6-phosphate, UDP-D-glucose, hexose-phosphate, trehalose 6-phosphate, and amino acids like glutamine, methionine, aspartic acid, 2-methylbutyroylcarnitine were also observed in TNFAIP8 expressing cells (Fig. 7A and Supplementary Table S2).
Fig. 7.
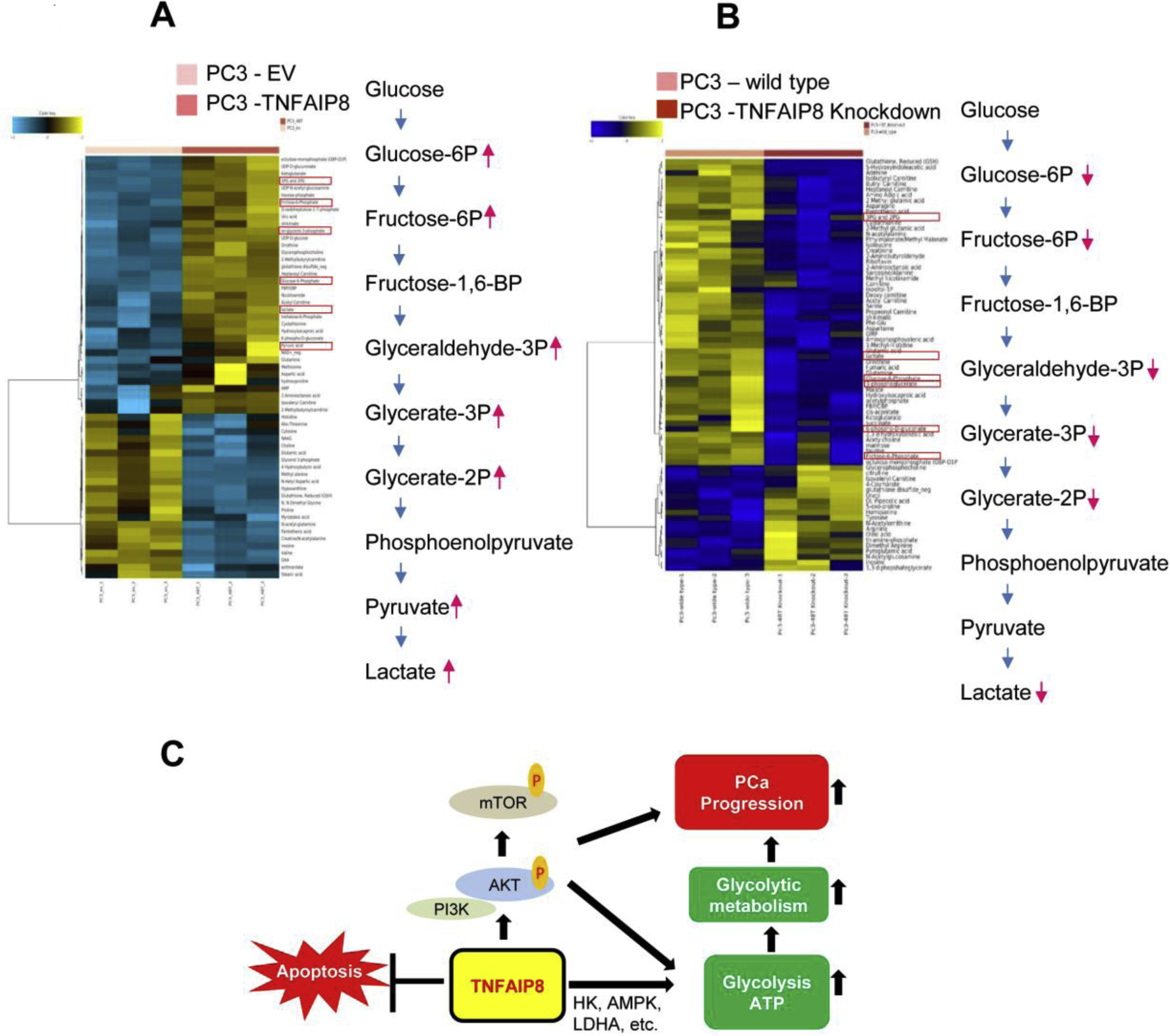
TNFAIP8 facilitates glycolytic metabolic profile in PCa PC3 cells. (A) Stable EV and TNFAIP8 expressing PC3 cells were grown in three independent samples, and glucose-related targeted metabolic profiling was analyzed by LC-MS Single Reaction Monitoring (SRM) as described in the materials and methods section and a heat map of differentially regulated key glycolytic metabolites was presented. (B) TNFAIP8 isoform II knockdown PC3 cells were grown in three independent experiments and metabolic profiling was analyzed by LC-MS Single Reaction Monitoring (SRM). The heat map of down-regulated key glycolytic metabolites was presented. (C) The model represents the possible roles of TNFAIP8 in AKT activation, metabolic reprogramming and PCa progression.
As a complementary approach, we knocked down TNFAIP8 isoform II in PC3 cells, which is the predominant isoform in many cancer cells including prostate cancer cells, by CRISPR editing (Supplementary Fig. S8A). Knockdown was confirmed by analyzing the expression of TNFAIP8 isoform II by RT/PCR and immunoblotting (Supplementary Fig. S8 B&C). TNFAIP8-II knockdown cells also showed decreased expression HK1 and G6P when compared with wild-type PC3 cells (Supplementary Fig. S8C). Metabolic profiling of TNFAIP8 knockout PC3 cells showed downregulation of several glycolytic metabolic products such as glucose-6-phosphate, fructose-6-phosphate, 2-phosphoglyceric acid, 3-phosphoglyceric acid, glyceraldehyde-3P, and lactic acid production when compared with wild type PC3 cells (Fig. 7B and Supplementary Table S3). The metabolic profiling data strengthens and supports the role of TNFAIP8 in facilitating glycolytic metabolic reprogramming in PCa cells.
4. Discussion
Although the distinct roles of the TIPE/TNFAIP8 family members are still being investigated, the oncogenic role of TNFAIP8 is now well established. TNFAIP8 contains a death effector domain, negatively regulates apoptosis (Kumar et al., 2000), increases drug resistance (Wu et al., 2019; Xing et al., 2018), induces autophagy (Niture et al., 2018b), and promotes cancer cell growth/proliferation in several cancers (Dong et al., 2017; Han et al., 2018; Monteith et al., 2016; Wang et al., 2014; Zhang et al., 2006). However, the role of TNFAIP8 protein in PCa progression is not yet clear. In the current study, we demonstrated that the expression of TNFAIP8 activated the PI3-AKT pathway and enhanced cell metabolic activities in PCa cells. We further studied the role of TNFAIP8 in metabolic reprogramming by analyzing TNFAIP8-mediated changes in cell metabolic activity, glucose consumption, cellular energetics, mitochondrial respiration, and changes in the glycolytic metabolomic profile in PCa cells. Our data demonstrated that TNFAIP8 modulates glycolysis and increases glucose consumption probably by modulating the expression of glycolytic enzymes and enhances endogenous ATP production in PCa cells. Further, TNFAIP8 enhanced mitochondrial respiration/OXPHOS and also increased glycolytic metabolite production in PCa cells.
PCa tumors consist of a highly heterogeneous cell population, and therefore resistance to most therapeutics is inevitable (Dagogo-Jack and Shaw, 2018; van Duijn et al., 2018). The heterogeneity of PCa and related drug resistance is strongly associated with genomic or epigenetic alterations in oncogene/tumor suppressor gene expression (Brocks et al., 2014) or differential regulation of PCa key cell-signaling cascades, for example, AR and PI3K/AKT/mTOR signaling (Ciccarese et al., 2017) that can create a change in overall metabolic phenotype (Eidelman et al., 2017). Studies demonstrated that K-Ras and p53 mutations occasionally occur in small foci of prostate tumors, and regions of the tumor with mutations in p53 are associated with invasive growth (Konishi et al., 1995). PCa tumor heterogeneity is also associated with metabolic reprogramming and bioenergetics of tumor cells (Gentric et al., 2017; Yoshida, 2015), and several oncogenes, including MYC, BRCA1, and KRAS regulate metabolic activity as well as metabolic reprogramming in tumor cells. These changes typically lead to increases in glycolysis, lipid synthesis, and nucleotide synthesis (Gaglio et al., 2011; Privat et al., 2014; Stine et al., 2015). Our data suggested that the rates of glucose consumption and endogenous ATP production within PCa cells are highly variable and the metabolic variability may at least be partially associated with TNFAIP8 expression. As depicted Fig. 7C, apart from activation of PI3K-AKT signaling, TNFAIP8 increased metabolic activity not only by increasing mitochondrial respiration, but also by enhancing glucose consumption/endogenous ATP production by modulation of glucose metabolizing enzyme expression. We have earlier shown the role of TNFAIP8 in drug resistance (Niture et al., 2020; Niture et al., 2018b). Our data suggests that these changes in cell metabolic activities by TNFAIP8 may lead to drug resistance in PCa (Fig. 7C).
Metabolic reprogramming in tumor cells is exhibited by several oncogenes. We analyzed the TNFAIP8-induced alteration of the global metabolomic profile in PCa cells, including metabolite profiles associated with glycolysis and mitochondrial respiration (Bertram et al., 2007; Tan et al., 2015). Our data suggested that TNFAIP8 upregulates aerobic respiration and glycolytic/ OXPHOS in PC3 cells. Increased production of several metabolites associated with glycolytic pathways, including glucose-6-phosphate, fructose-6-phosphate, 2-phosphoglyceric acid, 3-phosphoglyceric acid, pyruvic acid, and lactic acid was observed. Multiple studies have shown that abnormal accumulation of metabolites/oncometabolites facilitates tumor development/progression in various cancers (Ward and Thompson, 2012; Yang et al., 2013; Yoshida, 2015). Reports also suggest that dysregulation or mutation of metabolic enzymes in glycolysis or in the Krebs cycle (Baysal et al., 2000; Green and Beer, 2010; Hao et al., 2009; Niemann and Muller, 2000; Parsons et al., 2008; Sreedhar and Zhao, 2018; Thompson, 2009) dysregulates/upregulates the production and accumulation of several oncometabolites and these oncometabolites are involved in alteration of cancer cell metabolism as well as cell signaling. For example, the oncometabolite R-2-hydroxyglutarate produced by the mutation of isocitrate dehydrogenase I (citric cycle enzyme) regulates DNA hypermethylation in acute myeloid leukemia, activates the TGF-β, WNT/ Notch pathway, and promotes cell proliferation/differentiation (Collins et al., 2017; Dang et al., 2009; Figueroa et al., 2010; Sasaki et al., 2012; Shim et al., 2014; Terunuma et al., 2014; Ye et al., 2013). The exact role of TNFAIP8 mediated accumulation of these metabolites in PCa cells/tumors remains unknown and needs further investigation. In conclusion, our data suggest that TNFAIP8 facilitates the glycolytic metabolic reprogramming in PCa cells and increases the production of several metabolites related to glycolysis and other pathways.
Supplementary Material
Highlights.
TNFAIP8 activates PI3K-AKT signaling and promotes prostate cancer progression.
TNFAIP8 regulates glucose consumption and ATP production.
TNFAIP8 increases glycolysis/oxidative phosphorylation.
TNFAIP8 facilitates glycolytic metabolic reprogramming in prostate cancer cells.
Acknowledgments
We gratefully acknowledge the grants 1U01CA194730, 1U54MD012392 and 1R01MD012767 from the National Institutes of Health to DK. The authors would like to thank all members of our laboratory for their help and suggestions during this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Aoki M, and Fujishita T. 2017. Oncogenic Roles of the PI3K/AKT/mTOR Axis. Curr Top Microbiol Immunol. 407:153–189. [DOI] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, and Devlin B. 2000. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 287:848–851. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y. 1995. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B. 57:289–300. [Google Scholar]
- Bertram R, Satin LS, Pedersen MG, Luciani DS, and Sherman A. 2007. Interaction of glycolysis and mitochondrial respiration in metabolic oscillations of pancreatic islets. Biophys J. 92:1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, Oakes C, Zucknick M, Lipka DB, Weischenfeldt J, Feuerbach L, Cowper-Sal Lari R, Lupien M, Brors B, Korbel J, Schlomm T, Tanay A, Sauter G, Gerhauser C, Plass C, and Project IEOPC. 2014. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 8:798–806. [DOI] [PubMed] [Google Scholar]
- Cagle P, Niture S, Srivastava A, Ramalinga M, Aqeel R, Rios-Colon L, Chimeh U, Suy S, Collins SP, Dahiya R, and Kumar D. 2019. MicroRNA-214 targets PTK6 to inhibit tumorigenic potential and increase drug sensitivity of prostate cancer cells. Sci Rep. 9:9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Yu P, Duan X, Liu C, Xu S, Chen Y, Tan Y, Qiang Y, Shen J, and Tao Z. 2015. Genome-wide analysis of androgen receptor binding sites in prostate cancer cells. Exp Ther Med. 9:2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarese C, Massari F, Iacovelli R, Fiorentino M, Montironi R, Di Nunno V, Giunchi F, Brunelli M, and Tortora G. 2017. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat Rev. 54:68–73. [DOI] [PubMed] [Google Scholar]
- Collins RRJ, Patel K, Putnam WC, Kapur P, and Rakheja D. 2017. Oncometabolites: A New Paradigm for Oncology, Metabolism, and the Clinical Laboratory. Clin Chem. 63:1812–1820. [DOI] [PubMed] [Google Scholar]
- Crabtree HG 1929. Observations on the carbohydrate metabolism of tumours. Biochem J. 23:536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagogo-Jack I, and Shaw AT. 2018. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 15:81–94. [DOI] [PubMed] [Google Scholar]
- Dai C, Arceo J, Arnold J, Sreekumar A, Dovichi NJ, Li J, and Littlepage LE. 2018. Metabolomics of oncogene-specific metabolic reprogramming during breast cancer. Cancer Metab. 6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, and Su SM. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 462:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TF, Mewani RR, Starr J, Li X, Chakravarty D, Ressom H, Zou X, Eidelman O, Pollard HB, Srivastava M, and Kasid UN. 2017. Transcriptome and Proteome Analyses of TNFAIP8 Knockdown Cancer Cells Reveal New Insights into Molecular Determinants of Cell Survival and Tumor Progression. Methods Mol Biol. 1513:83–100. [DOI] [PubMed] [Google Scholar]
- Dong Q, Fu L, Zhao Y, Xie C, Li Q, and Wang E. 2017. TNFAIP8 interacts with LATS1 and promotes aggressiveness through regulation of Hippo pathway in hepatocellular carcinoma. Oncotarget. 8:15689–15703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlind MP, and Hsieh AC. 2014. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 16:378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidelman E, Twum-Ampofo J, Ansari J, and Siddiqui MM. 2017. The Metabolic Phenotype of Prostate Cancer. Front Oncol. 7:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Solmonson A, and DeBerardinis RJ. 2020. Metabolic reprogramming and cancer progression. Science. 368:eaaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li YS, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun ZX, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJM, Thompson CB, Levine RL, and Melnick A. 2010. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell. 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt EC, Bidere N, and Lenardo MJ. 2008. A different TIPE of immune homeostasis. Cell. 133:401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Tennant DA, and Gottlieb E. 2010. IDH1 Mutations in Gliomas: When an Enzyme Loses Its Grip. Cancer Cell. 17:7–9. [DOI] [PubMed] [Google Scholar]
- Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, and Chiaradonna F. 2011. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Molecular Systems Biology. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentric G, Mieulet V, and Mechta-Grigoriou F. 2017. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid Redox Sign. 26:462–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A, and Beer P. 2010. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med. 362:369–370. [DOI] [PubMed] [Google Scholar]
- Han Y, Tang Z, Zhao Y, Li Q, and Wang E. 2018. TNFAIP8 regulates Hippo pathway through interacting with LATS1 to promote cell proliferation and invasion in lung cancer. Mol Carcinog. 57:159–166. [DOI] [PubMed] [Google Scholar]
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CWRJ, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, and Rutter J. 2009. SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, Is Mutated in Paraganglioma. Science. 325:1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YT, Gan XQ, Zhang L, Liu BL, Zhu ZL, Li T, Zhu JF, Chen JS, and Yu HY. 2018. CoCl2 induces apoptosis via a ROS-dependent pathway and Drp1-mediated mitochondria fission in periodontal ligament stem cells. Am J Physiol-Cell Ph. 315:C389–C397. [DOI] [PubMed] [Google Scholar]
- Horrevoets AJ, Fontijn RD, van Zonneveld AJ, de Vries CJ, ten Cate JW, and Pannekoek H. 1999. Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood. 93:3418–3431. [PubMed] [Google Scholar]
- Kaarbo M, Mikkelsen OL, Malerod L, Qu S, Lobert VH, Akgul G, Halvorsen T, Maelandsmo GM, and Saatcioglu F. 2010. PI3K-AKT-mTOR pathway is dominant over androgen receptor signaling in prostate cancer cells. Cell Oncol. 32:11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi N, Hiasa Y, Matsuda H, Tao M, Tsuzuki T, Hayashi I, Kitahori Y, Shiraishi T, Yatani R, Shimazaki J, and et al. 1995. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am J Pathol. 147:1112–1122. [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Whiteside TL, and Kasid U. 2000. Identification of a novel tumor necrosis factor-alpha-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin- 1beta-converting enzyme-inhibitory protein. J Biol Chem. 275:2973–2978. [DOI] [PubMed] [Google Scholar]
- Lee JH, Choi SH, Baek MW, Kim MH, Kim HJ, Kim SH, Oh SJ, Park HJ, Kim WJ, and Jung JY. 2013. CoCl2 induces apoptosis through the mitochondria- and death receptor-mediated pathway in the mouse embryonic stem cells. Molecular and Cellular Biochemistry. 379:133–140. [DOI] [PubMed] [Google Scholar]
- Liberti MV, and Locasale JW. 2016. The Warburg Effect: How Does it Benefit Cancer Cells? (vol 41, pg 211, 2016). Trends Biochem Sci. 41:287–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteith JA, Mellert H, Sammons MA, Kuswanto LA, Sykes SM, Resnick-Silverman L, Manfredi JJ, Berger SL, and McMahon SB. 2016. A rare DNA contact mutation in cancer confers p53 gain-of-function and tumor cell survival via TNFAIP8 induction. Mol Oncol. 10:1207–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann S, and Muller U. 2000. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 26:268–270. [DOI] [PubMed] [Google Scholar]
- Niture S, Dong X, Arthur E, Chimeh U, Niture SS, Zheng W, and Kumar D. 2018a. Oncogenic Role of Tumor Necrosis Factor alpha-Induced Protein 8 (TNFAIP8). Cells. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niture S, Gyamfi MA, Lin M, Chimeh U, Dong X, Zheng W, Moore J, and Kumar D. 2020. TNFAIP8 regulates autophagy, cell steatosis, and promotes hepatocellular carcinoma cell proliferation. Cell Death Dis. 11:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niture S, Moore J, and Kumar D. 2019. TNFAIP8: Inflammation, Immunity and Human Diseases. J Cell Immunol. 1:29–34. [PMC free article] [PubMed] [Google Scholar]
- Niture S, Ramalinga M, Kedir H, Patacsil D, Niture SS, Li J, Mani H, Suy S, Collins S, and Kumar D. 2018b. TNFAIP8 promotes prostate cancer cell survival by inducing autophagy. Oncotarget. 9:26884–26899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr., Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, and Kinzler KW. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science. 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Wang FH, Whiteside TL, and Kasid U. 1997. Identification of seven differentially displayed transcripts in human primary and matched metastatic head and neck squamous cell carcinoma cell lines: implications in metastasis and/or radiation response. Oral Oncol. 33:197–203. [DOI] [PubMed] [Google Scholar]
- Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, Sapin V, Bignon YJ, and Morvan D. 2014. BRCA1 Induces Major Energetic Metabolism Reprogramming in Breast Cancer Cells. Plos One. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanuik TL, Ueda T, Le N, Haile S, Yong TM, Thomson T, Vessella RL, and Sadar MD. 2009. Novel biomarkers for prostate cancer including noncoding transcripts. Am J Pathol. 175:2264–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, You-Ten A, Li WY, Schalm S, Su SM, Virtanen C, Reifenberger G, Ohashi PS, Barber DL, Figueroa ME, Melnick A, Zuniga-Pflucker JC, and Mak TW. 2012. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 488:656-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seruga B, Ocana A, and Tannock IF. 2011. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol. 8:12–23. [DOI] [PubMed] [Google Scholar]
- Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S, Dutta S, Chenna B, Rea SL, Mishur RJ, Li QH, Johnson-Pais TL, Guo L, Bae S, Wei S, Block K, and Sudarshan S. 2014. L-2-Hydroxyglutarate: An Epigenetic Modifier and Putative Oncometabolite in Renal Cancer. Cancer Discov. 4:1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedhar A, and Zhao Y. 2018. Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomed Rep. 8:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV. 2015. MYC, Metabolism, and Cancer. Cancer Discov. 5:1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, Kong L, Xu L, Hilliard B, Hu S, Shen H, Yang X, and Chen YH. 2008. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 133:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki H, Harashima N, Hiraki M, Arichi N, Nishimura N, Shiina H, Naora K, and Harada M. 2014. Bcl-2 family inhibition sensitizes human prostate cancer cells to docetaxel and promotes unexpected apoptosis under caspase-9 inhibition. Oncotarget. 5:11399–11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Xiao H, Li FN, Zeng LM, and Yin YL. 2015. The profiles of mitochondrial respiration and glycolysis using extracellular flux analysis in porcine enterocyte IPEC-J2. Anim Nutr. 1:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, Karoly ED, Chan K, Samanta S, Prieto D, Hsu TYT, Kurley SJ, Putluri V, Sonavane R, Edelman DC, Wulff J, Starks AM, Yang YM, Kittles RA, Yfantis HG, Lee DH, Loffe OB, Schiff R, Stephens RM, Meltzer PS, Veenstra TD, Westbrook TF, Sreekumar A, and Ambs S. 2014. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. Journal of Clinical Investigation. 124:398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB 2009. Metabolic Enzymes as Oncogenes or Tumor Suppressors. New Engl J Med. 360:813–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA, and Multiple Leiomyoma C. 2002. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 30:406–410. [DOI] [PubMed] [Google Scholar]
- van Duijn PW, Marques RB, Ziel-van der Made ACJ, van Zoggel HJAA, Aghai A, Berrevoets C, Debets R, Jenster G, Trapman J, and van Weerden WM. 2018. Tumor heterogeneity, aggressiveness, and immune cell composition in a novel syngeneic PSA-targeted Pten knockout mouse prostate cancer (MuCaP) model. Prostate. 78:1013–1023. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Song Y, and Men X. 2014. Variance of TNFAIP8 expression between tumor tissues and tumor-infiltrating CD4+ and CD8+ T cells in non-small cell lung cancer. Tumour Biol. 35:2319–2325. [DOI] [PubMed] [Google Scholar]
- Ward PS, and Thompson CB. 2012. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 21:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SX, Li WH, Wu ZH, Cheng TR, Wang P, Li N, Liang XN, Chi MM, Zhang SM, Ma YF, Li YY, and Chai LH. 2019. TNFAIP8 promotes cisplatin resistance in cervical carcinoma cells by inhibiting cellular apoptosis. Oncology Letters. 17:4667–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Liu YC, Liu TB, Meng QW, Lu HL, Liu W, Hu J, Li CH, Cao MR, Yan S, Huang J, Wang T, and Cai L. 2018. TNFAIP8 promotes the proliferation and cisplatin chemoresistance of non-small cell lung cancer through MDM2/p53 pathway. Cell Communication and Signaling. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin GL, McLendon R, Rasheed BA, Yuan WS, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, and Bigner DD. 2009. IDH1 and IDH2 Mutations in Gliomas. New Engl J Med. 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Soga T, and Pollard PJ. 2013. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 123:3652–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Ma SH, Xiong Y, and Guan KL. 2013. R-2-Hydroxyglutarate as the Key Effector of IDH Mutations Promoting Oncogenesis. Cancer Cell. 23:274–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida GJ 2015. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Canc Res. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Chakravarty D, Sakabe I, Mewani RR, Boudreau HE, Kumar D, Ahmad I, and Kasid UN. 2006. Role of SCC-S2 in experimental metastasis and modulation of VEGFR-2, MMP-1, and MMP-9 expression. Mol Ther. 13:947–955. [DOI] [PubMed] [Google Scholar]
- Zhang LJ, Liu X, Gafken PR, Kioussi C, and Leid M. 2009. A chicken ovalbumin upstream promoter transcription factor I (COUP-TFI) complex represses expression of the gene encoding tumor necrosis factor alpha-induced protein 8 (TNFAIP8). J Biol Chem. 284:6156–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.