

Abstract
Aim
The tools that have been used to assess the function of the vagus nerve lack specificity. This could explain discrepancies about the role of vagal gut‐brain signalling in long‐term control of energy balance. Here we use a validated approach to selectively ablate sensory vagal neurones that innervate the gut to determine the role of vagal gut‐brain signalling in the control of food intake, energy expenditure and glucose homoeostasis in response to different diets.
Methods
Rat nodose ganglia were injected bilaterally with either the neurotoxin saporin conjugated to the gastrointestinal hormone cholecystokinin (CCK), or unconjugated saporin as a control. Food intake, body weight, glucose tolerance and energy expenditure were measured in both groups in response to chow or high‐fat high‐sugar (HFHS) diet. Willingness to work for fat or sugar was assessed by progressive ratio for orally administered solutions, while post‐ingestive feedback was tested by measuring food intake after an isocaloric lipid or sucrose pre‐load.
Results
Vagal deafferentation of the gut increases meal number in lean chow‐fed rats. Switching to a HFHS diet exacerbates overeating and body weight gain. The breakpoint for sugar or fat solution did not differ between groups, suggesting that increased palatability may not drive HFHS‐induced hyperphagia. Instead, decreased satiation in response to intra‐gastric infusion of fat, but not sugar, promotes hyperphagia in CCK‐Saporin‐treated rats fed with HFHS diet.
Conclusions
We conclude that intact sensory vagal neurones prevent hyperphagia and exacerbation of weight gain in response to a HFHS diet by promoting lipid‐mediated satiation.
Keywords: fat, obesity, palatability, post‐ingestive, sugar, vagus nerve
Abbreviations
- CCK
cholecystokinin
- HFHS
high‐fat high‐sugar
- NG
nodose ganglia
- NTS
nucleus tractus solitarius
- SAP
saporin
1. INTRODUCTION
The vagus nerve is the longest of the 12 cranial nerves. It broadly innervates many peripheral organs, underscoring its critical role in several physiological processes necessary for survival. 1 Vagal efferent neurones located in the hindbrain send motor information from the brain to peripheral organs. 2 Vagal sensory neurones, located in the nodose ganglia (NG), convey interoceptive feedback from each organ to the brain. 3 Thus, vagal neurones located in anatomically distinct sites provide bidirectional information between peripheral organs and the brain. In both mice and rats, sensory vagal fibres extensively outnumber motor fibres in transverse sections of the vagus nerve, 4 , 5 suggesting a primary role of the vagus nerve in providing continuous homoeostatic feedback to the brain. At the level of NG, vagal sensory neurones can be categorized by organ innervation at least in mice, 3 but no discernable anatomical organization for these ensembles has been identified. Within organs, the structural morphology of sensory vagal terminals distinguishes mechanosensitive and chemosensitive neurones, 6 suggesting that vagal afferent neurones are involved in sensing multiple physiological events in peripheral organs.
The gastrointestinal tract is particularly densely innervated by the vagus nerve. 6 In addition to its role in digestion and absorption, there is substantial evidence indicating that the vagus nerve controls food intake control in rodents and humans. 7 , 8 , 9 , 10 , 11 Receptors capable of sensing fats, 12 , 13 carbohydrates, 14 gut hormones 3 , 15 , 16 , 17 , 18 and stretch 19 are all expressed by NG neurones. Furthermore, each of these stimuli increases firing activity in whole vagus nerve electrophysiological recordings. 3 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 Post‐prandial signalling is blunted in response to chemical or physical lesioning, as demonstrated by reduced neuronal activity in the nucleus tractus solitarius (NTS), the central site of vagal sensory termination. 31 Individual stimuli such as lipids, 32 carbohydrates 33 and gastric distension 34 also require an intact vagus nerve to activate NTS neurones. Failure to activate vagal afferent neurones, by removal of the ingesta during a meal, results in continuous eating in rats, 35 underlying the crucial role of post‐ingestive gut‐brain signalling in meal termination. Furthermore, optogenetic and chemogenetic stimulation of gastric and duodenal innervating vagal neurone populations acutely reduce food intake in mice. 7 , 36 Thus, the vagus nerve is anatomically positioned within the gut and is necessary and sufficient to convey moment‐to‐moment feedback to the brain about the quantity and makeup of a meal to trigger satiation.
There is evidence that repeated inhibition of satiation at each meal by disrupting vagal gut‐brain signalling results in long‐term metabolic consequences. Eating palatable calorie‐dense diets rich in fat and/or sugar for a prolonged period reduces the sensitivity of vagal afferent neurones to tension, 37 , 38 satiation hormones (eg Cholecystokinin [CCK]) 37 , 39 , 40 , 41 , 42 , 43 , 44 and intestinal nutrients 12 , 45 , 46 , 47 in both mice and rats. In rats, reduced vagal sensing coincides with the onset of hyperphagia and is associated with increased body weight. 48 In a genetic mouse study, inhibition of vagal signalling resulted in a reduction of vagal afferent sensitivity to CCK and was sufficient to induce hyperphagia and weight gain. 49 , 50 Conversely, chronic vagal stimulation reduces food intake and body weight in many different animal species, 51 , 52 , 53 , 54 , 55 , 56 including humans. 57 These data support a role for the vagus nerve in long‐term control of food intake by reducing postprandial feedback from the gut to the brain at every meal that could promote overeating in obesity. Based on this hypothesis it would be anticipated that inhibition of the vagus nerve would cause hyperphagia and increased body weight. In contradiction, previous studies in which vagal signalling is inhibited using surgical, 58 , 59 chemical 60 , 61 or genetic 62 , 63 , 64 , 65 approaches have little effect on daily cumulative chow intake. We hypothesize that the lack of specificity of the lesioning approaches to target sensory information for the gut and aggregation of studies using different diets has led to the erroneous conclusion that the vagus nerve only has a short‐term role in the control of food intake.
The majority of what is known about the function of the sensory vagal neurones relies on its anatomy, or techniques that lack precision and have several pitfalls. Vagus nerve stimulation is primarily applied unilaterally to the cervical vagus nerve which non‐selectively activates both sensory and motor arms of the vagus nerve with little consensus regarding stimulation parameters. Furthermore, chronic cervical vagal stimulation cause elevated fasting blood glucose levels and impairs glucose tolerance in rats. 66 Inhibition achieved by vagotomy abolishes both the sensory and motor arms of the vagus. Capsaicin is not necessarily specific to the vagus nerve and only abolishes a subpopulation of unmyelinated sensory neurones and partially ablates motor neurones. 67 Subdiaphragmatic vagal deafferentation approach improves on vagotomy and capsaicin by leaving 50% of motor control intact, but similarly lacks organ specificity. 68 While more targeted viral‐mediated RNA interference approaches have been employed, 50 , 69 , 70 this requires a priori knowledge of the signalling receptor to be knocked down. To address these issues, we have recently validated a novel pharmacotoxin approach, in which the neurotoxin saporin (SAP) is conjugated to the gastrointestinal hormone CCK (CCK‐SAP), for selectively deleting CCK receptor‐expressing sensory vagal neurones that innervate the stomach and small intestine. 71 This approach ablates approximately 80% of sensory neurones innervating the proximal gut, while sparing motor neurones of the vagus nerve. 71 It is important to note that ablating CCK‐A receptor‐expressing neurones with this approach, will delete the majority of vagal‐gut‐brain signalling rather than exclusively impairing CCK signalling. Because CCK receptor‐expressing neurones are involved in multimodal response including mechanosensation, anorectic signalling and orexigenic signalling, 36 CCK‐SAP injections in the NG causes vagal deafferentation rather than just CCK‐receptor blockade. We utilize this novel gut‐specific vagal deafferentation approach to evaluate the role of gut‐brain signalling in the control of energy balance in rats fed either chow or high‐fat high‐sugar (HFHS) diets. We hypothesized that selective vagal deafferentation of the gut would differentially impair energy balance, glucose tolerance and gut nutrient sensing.
2. RESULTS
2.1. Vagal deafferentation increases number of meals in chow‐fed rats
To determine the role of vagal sensory neurones in controlling energy homoeostasis, we generated two groups of animals either lacking vagal gut‐brain signalling followed by bilateral NG injection of CCK‐SAP (400 ng/side, n = 16) or with normal vagal signalling that received bilateral NG injection of the control SAP (400 ng/side, n = 21). Pre‐surgical body weight was not significantly different (SAP: 277 ± 3.8 g, CCK‐SAP: 275 ± 6.6 g). We verified CCK‐SAP induced lesioning by performing a CCK‐induced satiation test 6 weeks after the surgery. CCK8S (4 μg/kg; IP) reduced food intake at 30‐120 minutes in SAP‐treated rats (Figure 1A), but not in CCK‐SAP‐treated rats (Figure 1B). Over the 6 weeks post‐op during which rats were maintained on the chow diet, we observed no differences in body weight gain between groups (Figure 1C). CCK‐SAP had no effect on weekly chow intake over the 6 weeks after surgery compared to SAP controls (Figure 1D). We performed an IP glucose tolerance test (IPGTT, Figure 1E) 5 weeks after surgery and found that vagal deafferentation of the gut did not affect glucose tolerance. In a separate cohort of rats, we found that 5 weeks after surgery, the average energy expenditure over 3 days was not different between chow‐fed SAP and CCK‐SAP‐treated rats (Figure 1F,G) irrespective of whether the animals were in the light (Figure 1H) or dark (Figure 1I) phase.
FIGURE 1.
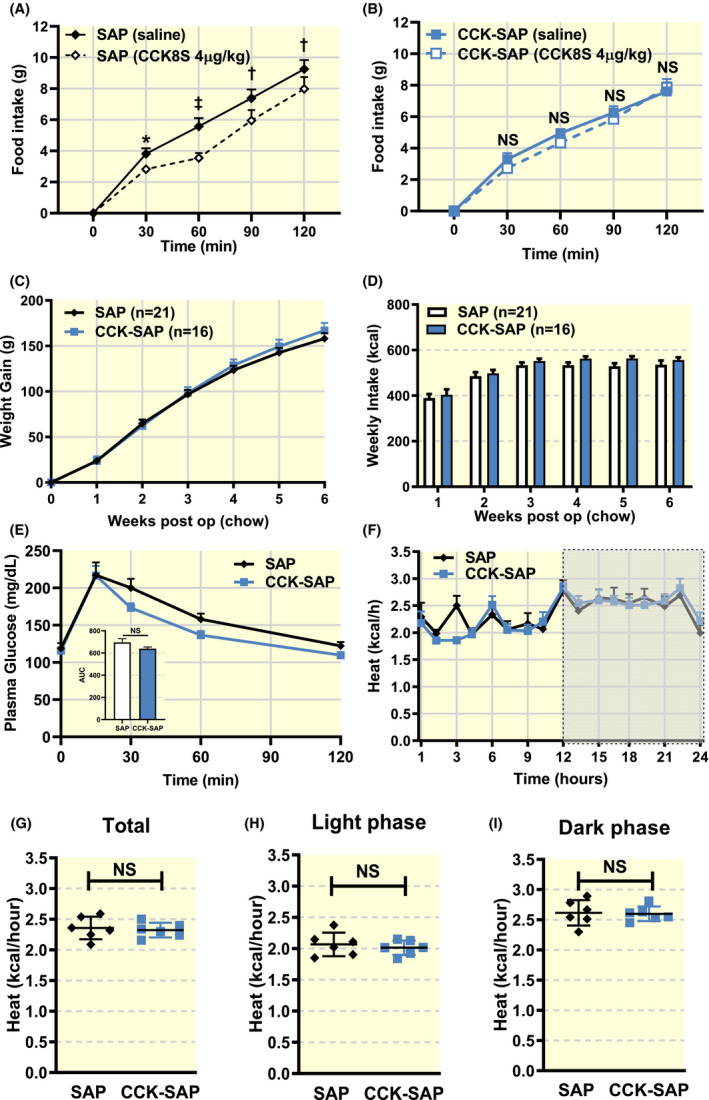
Gut‐specific vagal deafferentation has no long‐term effect on metabolism in lean, chow‐fed rats. Rats received bilateral NG injection of SAP (400 ng/side, n = 21) or CCK‐SAP (400 ng/side, n = 16). A‐B, CCK8S (IP 4 µg/kg) reduces food intake at 30‐120 minutes in (A) SAP injected( (n = 9, two‐way ANOVA, interaction F(4,32) = 3.662, P = .0145, Holm‐Sidak post hoc analysis, *P < .05, † P < .01, ‡ P < .0001) but not (B) CCK‐SAP injected rats (n = 7, two‐way ANOVA, interaction F(4,24) = 1.904, P = .1424, NS). C, Post‐op,CCK‐SAP had no effect on body weight gain (n = 21/16, two‐way ANOVA, interaction F(6,210) = 1.0, P = .4263; Holm‐Sidak post hoc analysis NS). D, In chow‐fed conditions, there was no difference in weekly caloric intake in either group. (n = 21/16, two‐way ANOVA, interaction F(5,175) = 0.4263, P = .83; Holm‐Sidak post hoc analysis NS). E, There is no difference in glucose tolerance between groups (n = 6, two‐way ANOVA, interaction F(4,52) = 0.9778, P = .4277; Holm‐Sidak post hoc analysis NS). Insert shows no difference in AUC between groups (two‐tailed unpaired t‐test, P = .1289). F‐I, Energy expenditure was measured in a metabolic chamber. F, Energy expenditure (kcal/hour) is not significantly different between SAP or CCK‐SAP (treatment (n = 6, two‐way ANOVA, interaction F(16,160) = 1.106, P = .3540; Holm‐Sidak post hoc analysis NS). G, Average kcal expended per hour during 24th day (n = 6, two‐tailed unpaired t‐test, P = .7057), H light phase (n = 6, two‐tailed unpaired t‐test, P = .5673), and I dark phase (n = 6, two‐tailed unpaired t‐test, P = .8642) were not affected by vagal deafferentation
Based on previous work highlighting a role for vagal signalling in the short‐term control of food intake, we monitored continuous ad libitum chow intake using a BioDAQ system. There was a shift in the time of day that food was consumed between groups; SAP‐treated rats started eating before dark onset and stopped before light onset, while CCK‐SAP‐treated rats started eating once the dark phase had started and continued eating until the end of the dark phase (Figure 2A). In addition, meal patterns were significantly altered in CCK‐SAP rats; meal number was increased compared to control SAP rats and this was associated with a reduction in meal size and ingestion rate, suggesting reduced satiety (Figure 2B). These effects were observed in the dark phase (Figure 2C) but were absent in the light phase (Figure 2D).
FIGURE 2.
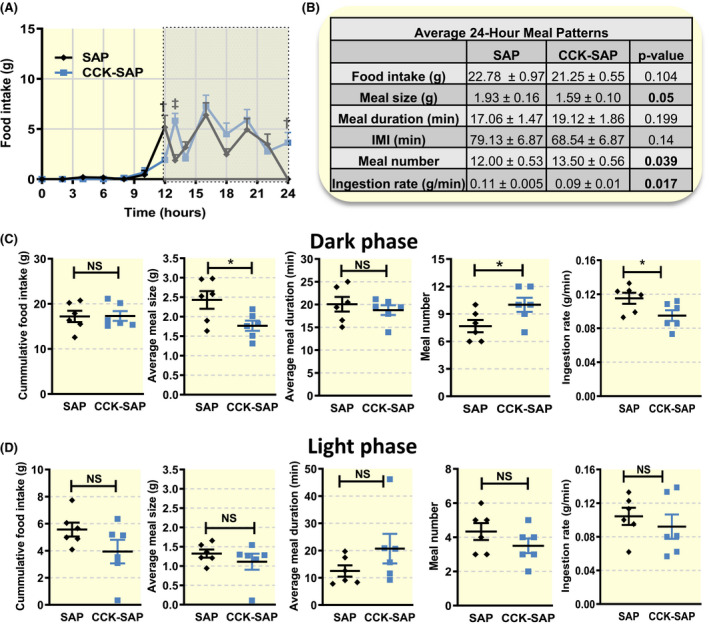
Gut‐specific vagal deafferentation alters meal patterns in lean chow‐fed rats. Food intake patterns were measured in a subset of rats receiving bilateral NG injection of SAP (400 ng/side, n = 6) or CCK‐SAP (400 ng/side, n = 6). A, Vagal deafferentation shifted the anticipatory circadian cycle evoked changes in food intake (n = 6, two‐way ANOVA, interaction F(13,182) = 3.823, P < .0001, Holm‐Sidak post hoc analysis, † P ≤ .01, ‡ P < .001). B‐D, Meal patterns differed between groups. B, Over the course of the full day and (C) in the dark phase CCK‐SAP‐treated rats ate slower and smaller meals with no net change in daily food intake due to greater number of meals (two‐tailed unpaired t‐test, *P < .05). D, In the light phase there were no differences in meal patterns between groups (two‐tailed unpaired t‐test, NS)
2.2. Vagal deafferentation exacerbates weight gain following exposure to HFHS diet
At 6 weeks post‐op, a subset of rats (n = 8/group) was switched from chow to HFHS diet. CCK‐SAP‐treated rats trended (P < .06) to gain more weight in the first 2 weeks of HFHS diet. Body weight gain was significantly elevated in CCK‐SAP rats from 3 weeks onwards compared to SAP controls (Figure 3A). Interestingly, despite significant weight gain differences between groups at 3 weeks, IPGTT was not different (Figure 3B). However, weekly food intake was significantly greater at each time point compared to controls (Figure 3C). The time of day at which food intake occurred was similar between groups (Figure 3D). The increased cumulative food intake resulted from increases in ingestion rate, meal size, meal duration and meal number (Figure 3E). The altered meal patterns were restricted primarily to the dark phase (Figure 3F), although the ingestion rate was also increased in the light phase (Figure 3F).
FIGURE 3.
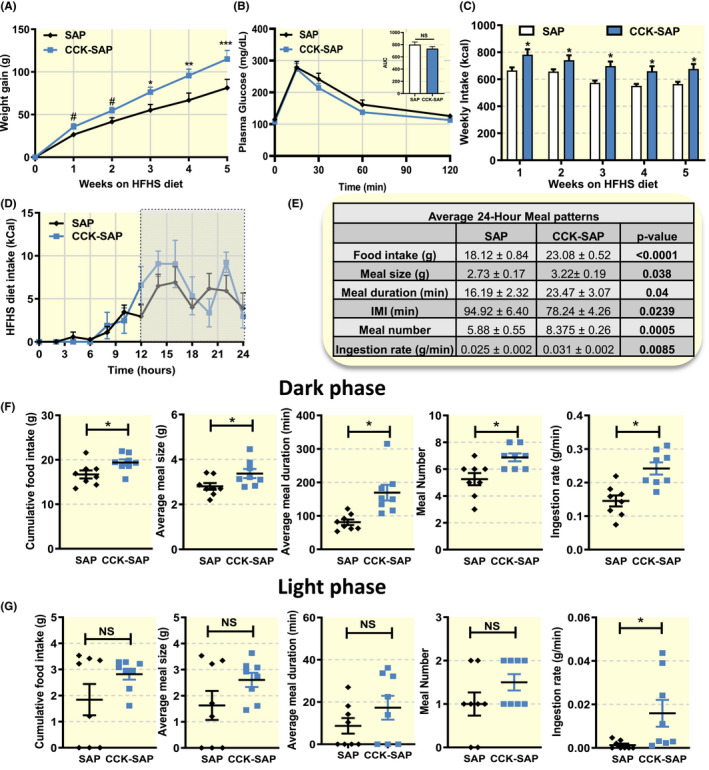
Vagal deafferentation causes weight gain and hyperphagia in rats fed high‐fat high‐sugar (HFHS) diet. 6 weeks post‐op, chow‐fed rats were switched to HFHS diet (A) CCK‐SAP rats gained more weight (n = 7/8, two‐way ANOVA, interaction F(5,60) = 5.636, P = .0003, Holm‐Sidak post hoc analysis, # P = .06 *P < .05, † P < .01, ‡ P < .001). B, There was no effect of dietary change on glucose tolerance between group (n = 7/8, two‐way ANOVA, interaction F(4,48) = 0.3731, P = .8267, Holm‐Sidak post hoc analysis, NS). Insert shows no difference in AUC (two tailed unpaired t‐test, P = .2406). C, In response to HFHS diet, CCK‐SAP‐treated rats ate more calories than SAP rats. (n = 7/8, two‐way ANOVA, interaction F(4,52) = 0.9384, P = .4491, Holm‐Sidak post hoc analysis, *P < .05). D‐G, Meal patterns of CCK‐SAP and SAP rats were recorded at 9 weeks post‐op averaging three consecutive days of food intake in rats acclimated to BioDAQ. D Both groups of animals consume food at the same time of day (n = 7/8, two‐way ANOVA, interaction F(12,144) = 1.012, P = .4410, Holm‐Sidak post hoc analysis, NS). E, Daily food intake was increased as a results of increased meal size, duration, ingestion rate and increased meal number (n = 8, two‐tailed unpaired t‐test). These effects were mediated altered meal patterns in (F) the dark phase, rather than (G) the light phase (n = 8, two‐tailed unpaired t‐test, *P < .05)
To determine whether the increased body weight gain and food intake in the CCK‐SAP‐treated animal was the direct consequence of dietary change, rather than a coincidence related to the age of the animals or a delayed effect of the surgery, we repeated the experiment in a new cohort of rats. As before rats received a bilateral injection of CCK‐SAP (n = 8) or SAP (n = 8) at the same age, but this time were maintained on chow for 11 weeks post‐op before switching to HFHS diet for an additional 5 weeks. SAP‐ and CCK‐SAP‐treated rats weighed the same (Figure 4A) and consumed the same amount of food (Figure 4C) at every time point over 11 weeks while maintained on chow. Similar to the CCK‐SAP‐treated rats in Figure 3, the CCK‐SAP‐treated rats gained more weight once switched to HFHS diet (Figure 4B). We also found that the CCK‐SAP rats consumed more calories while on HFHS diet compared to SAP controls (Figure 4D).
FIGURE 4.
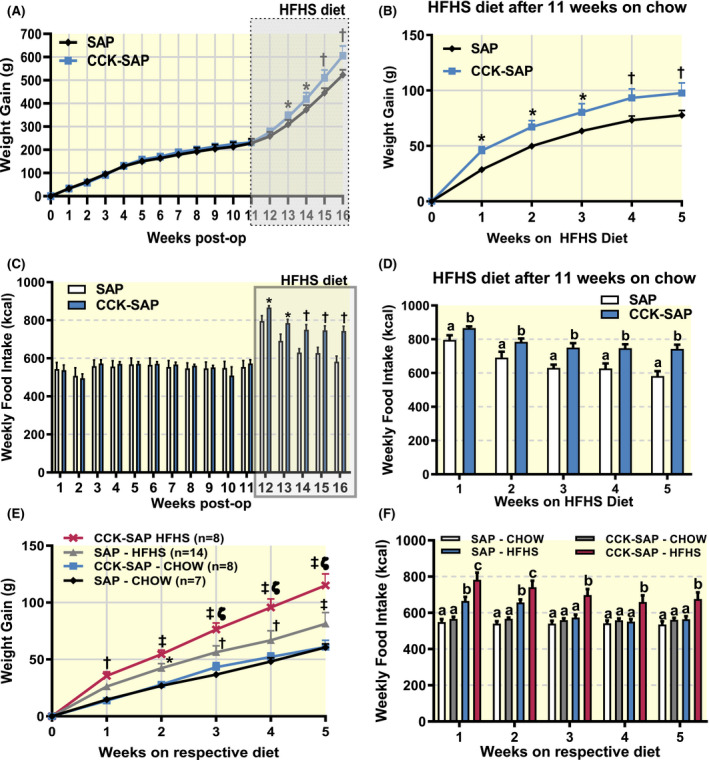
HFHS diet causes hyperphagia and weight gain in CCK‐SAP‐treated rats. Rats received bilateral NG injection of SAP (400 ng/side) or CCK‐SAP (400 ng/side) and maintained on chow for 11 weeks post‐op before switching to HFHS diet (n = 8/group). A, There was no post‐op body weight gain during 11 weeks on chow, but CCK‐SAP‐treated rats started gaining more weight upon transitioning to HFHS diet (n = 8, two‐way ANOVA, interaction F(16,112) = 2.585, P = .0019, Benjamini, Krieger and Yekutieli post hoc analysis with FDR 0.05, *P < .05, † P < .01). B, Normalizing weight gain to the body weight upon dietary switch, confirms greater weight gain in CCK‐SAP‐treated rats compared to SAP controls (n = 8, two‐way ANOVA, interaction F(5,60) = 3.381, P = .0093, Holm Sidak post hoc analysis, *P < .05, † P < .01). C, Weekly food intake remained similar between groups for the rats on chow for 11. Caloric intake increased in response to HFHS diet in both groups compared to chow (n = 8, two‐way ANOVA, interaction F(15, 210) = 4.457, P < .0001, Benjamini, Krieger and Yekutieli post hoc analysis, *P < .05, † P < .01). D, Caloric intake was greater in CCK‐SAP‐treated, compared to SAP‐treated, rats (n = 8, two‐way ANOVA, interaction F(4,52) = 2.176, P = .0845, Holm Sidak, different lettersa,b indicate P < .05). E‐F, Analysis of age‐matched 6 weeks post‐surgery of bilateral SAP or CCK‐SAP in NG on respective diets. E, HFHS diet increases body weight gain more rapidly in CCK‐SAP‐treated compared to SAP‐treated rats (n = 7‐14, two‐way ANOVA, interaction F(15,165) = 10.53, P < .001, Holm‐Sidak post hoc analysis, *P < .05, † P < .01, ‡ P < .001 compared to chow, and ζp < 0.01 compared to SAP‐HFHS group). F, HFHS‐fed CCK‐SAP‐treated rats consume more calories per week than all other groups at time point, while HFHS‐fed SAP‐treated rats normalize intake compared to chow‐fed rats in the last 3 weeks of the study (n = 7‐14, two‐way ANOVA, interaction F(15,165) = 10.53, P < .001, Holm‐Sidak post hoc analysis, different lettersa,b,c indicate P < .05 between groups)
We combined the weight gain and food intake data from the age‐matched rats fed HFHS diet (Figure 3) or chow (Figure 4) at 6‐11 weeks after NG surgery. Although both SAP and CCK‐SAP‐treated rats gained more weight when fed HFHS diet, it is clear that vagal deafferentation exacerbated weight gain in response to the diet (Figure 4E). Notably, the CCK‐SAP‐treated animals weighed more than lean animals from week 1 while SAP‐treated animals started to weigh more than lean controls from week 2, and HFHS‐fed CCK‐SAP rats weighed more that HFHS‐fed SAP rats (Figure 4E). In addition, CCK‐SAP animals consumed more calories at each time point when fed HFHS compared to CCK‐SAP rats fed chow (Figure 4F); while SAP animals consumed more calories in the first couple of weeks before reducing their food consumption to match the caloric intake of chow‐fed rats after 3 weeks on HFHS diet (Figure 4F).
2.3. Vagal deafferentation blocks post‐ingestive lipid‐mediated meal termination
We hypothesized that the increased intake of HFHS diet in CCK‐SAP‐treated rats was either a consequence of reduced post‐ingestive feedback from the gut, and/or increased wanting for the highly palatable diet. Both these putative mechanisms were tested in lean chow‐fed rats exposed to equicaloric fat or sugar. Importantly, the rats were tested while fed chow diet to prevent the well‐characterized confounds from reduction in vagal sensory neurone sensitivity to nutrients, 12 , 45 , 46 , 47 hormones 37 , 39 , 40 , 41 , 42 , 43 , 44 and tension 37 , 38 in animals chronically fed HFHS diet. The rationale for opting to study the mechanism of CCK‐SAP‐induced hyperphagia in chow‐fed rats that are not hyperphagic is supported by the fact that CCK‐SAP rats started to overconsume immediately upon first exposure to HFHS diet (Figures 3C and 4D,F). Confirming our previous results in Figures 1 and 4, we observed no group differences in body weight gain (Figure 5A) or average food intake (Figure 5B) in this cohort of chow‐fed animals over the 13 weeks of the study (n = 7/group). We verified vagal deafferentation of the gut in this group of rats using a CCK (IP, 4 µg/kg) satiation test. SAP animals significantly decrease 1 hour food intake in response to exogenous CCK‐8, while CCK‐SAP animals exhibited no change in food intake after CCK administration (Figure 5C).
FIGURE 5.
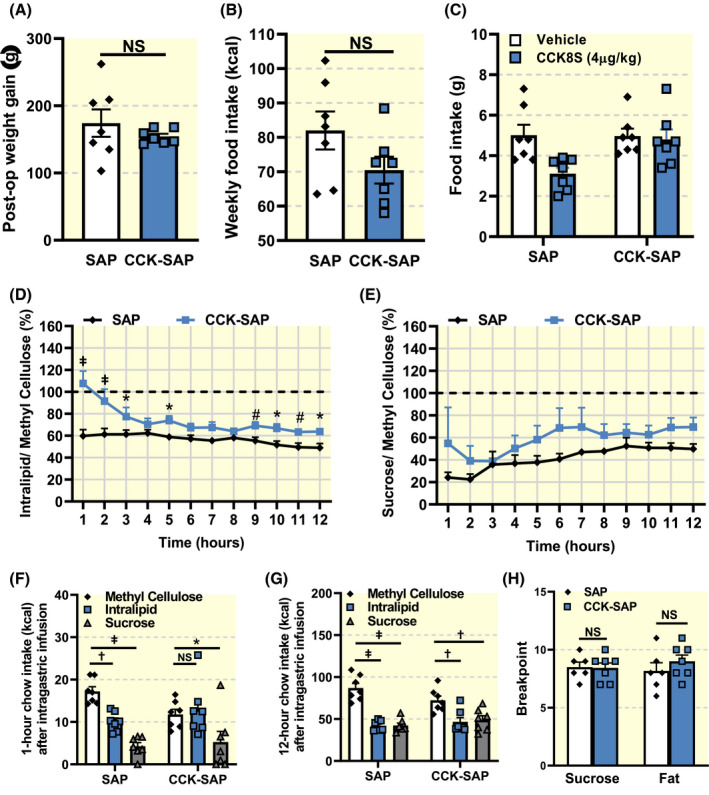
Vagal deafferentation causes inhibition of lipid‐mediated satiation. A, Weight gain was similar between groups (n = 7, two tailed unpaired t‐test, P = .3612). B, Chow intake was not significantly different between groups (n = 7, two tailed unpaired t‐test, P = .1149). C, 1 hour food intake in response to i.p. CCK (4 µg/kg) was reduced in SAP, but not in CCK‐SAP‐treated rats (n = 7, two way RM ANOVA, Interaction F(1,12) = 6.392, P = .0265, Holm‐Sidak post hoc analysis † P < .01). D, SAP but not CCK‐SAP animals reduce food intake following intra‐gastric pre‐load of lipids (n = 7, two way ANOVA Interaction F(11,132) = 4.003, P < .0001, Benjamini and Hochberg post hoc analysis with FDR of 0.05 # P < .06, *P < .05, ‡ P < .001). E, Both SAP and CCK‐SAP animals reduce food intake following intra‐gastric pre‐load of sucrose (n = 7, two way ANOVA Interaction F(11,132) = 0.6730, P = .7619, Benjamini and Hochberg post hoc analysis with FDR of 0.05, NS). F, Caloric intake 1 hour after intra‐gastric pre‐load of methyl cellulose, fat or sugar (n = 7, two way ANOVA, Interaction F(2,24) = 3.878, P = .0347, Dunnett's post hoc analysis, *P < .05, † P < .01, ‡ P < .001). G, Caloric intake 12 hours after intra‐gastric pre‐load of methyl cellulose, fat or sugar (n = 7, two way ANOVA, Interaction F(2,24) = 4.944, P = .0159, Dunnett's post hoc analysis, † P < .01, ‡ P < .001). H Breakpoint in the progressive ratio licking test was unchanged across groups irrespective of macronutrient reward (n = 7, Two‐way RM ANOVA, Interaction F(1,8) = 0.8037, P < .05, Holm‐Sidak post hoc analysis, NS)
To determine the role of post‐ingestive feedback in HFHS diet‐induced hyperphagia, chow‐fed SAP or CCK‐SAP‐treated rats were infused 10 mL of either 37.5% fat, 75% sucrose or 2% methyl cellulose into the stomach at a rate of 1 mL/min. To account for the individual and group differences in meal patterns, we designed the experiments to be a within‐animal design in which each animal acted as its own control. Furthermore, we controlled for distension effects by using methyl cellulose as a baseline. A 2% methyl cellulose pre‐load was used as a control as it is composed of non‐metabolizable nutrients and has a similar viscosity to fat and sugar solutions. A saline pre‐load was also performed, and similar results were observed (data not shown). Importantly, chow intake in response to a pre‐load of 2% methyl cellulose was similar between SAP‐ and CCK‐SAP‐treated animals (Figure 5E,G). In response to an intra‐gastric pre‐load of a fat solution, SAP‐treated rats markedly decreased their food intake for 12 hours, when compared to a methyl cellulose pre‐load (Figure 5D,F,G). By contrast, CCK‐SAP‐treated animals did not reduce intake in the first 2 hours following the fat pre‐load compared to a methyl cellulose pre‐load, and maintained significantly higher food intake than SAP controls at the majority time points over 12 hours (Figure 5D,F,G). Furthermore, both CCK‐SAP and SAP rats significantly decreased food intake following an intra‐gastric pre‐load of an equicaloric sucrose solution (Figure 5E‐G). Together, these data suggest that CCK‐SAP animals have dampened lipid‐mediated, but not sugar‐mediated, reduction in food intake.
To determine whether CCK‐SAP‐treated rats are more motivated to consume palatable HFHS diet, we tested the willingness of the animals to work for the taste of fat or sugar. The same lean chow‐fed SAP or CCK‐SAP‐treated rats were trained to lick for equicaloric solutions of 10% fat or 20% sucrose before performing a progressive ratio licking task, 72 in which exponentially increasing numbers of licks were required for either fat or sugar delivery. The breakpoint, the point at which the animals ceased licking the dry sipper to get the reward, presented as the last reward achieved, was used to determine the willingness of the animals to work for fat or sugar. Similar breakpoints were recorded for both groups of rats licking for either fat or sugar reward (Figure 5H), suggesting that gut deafferentation failed to increase the willingness to work for oral intake of either fat or sugar.
3. DISCUSSION
The study aimed to determine the role of sensory vagal neurones that innervate the gut in the control of energy balance. On utilizing a novel highly selective approach for vagal deafferentation of the gut, we found that rats lacking gut‐brain signalling did not affect energy expenditure or glucose homoeostasis, but significantly altered meal patterns in chow‐fed conditions. When the same animals were switched to a HFHS diet, cumulative food intake was markedly increased over multiple weeks resulting in increased body weight. These data suggest a role for the gut‐brain axis in both short‐term and long‐term control of food intake depending on nutrient availability. By directly infusing nutrients into the gut we demonstrate that vagal deafferentation impairs fat, but not sugar satiation, suggesting that impaired post‐ingestive feedback of fat is sufficient to promote hyperphagia and weight gain in response to a HFHS diet.
CCK receptors are G‐protein coupled receptors that are expressed in NG neurones. 15 , 73 Upon binding of CCK‐SAP to CCK receptors, the internalization of the ligand‐receptor complex results in the saporin‐mediated ablation of the cell. Thus this approach not only abolishes CCK signalling but actually deletes these neurones and therefore prevents all signalling by NG cells that express CCK receptors. Using a retrograde tracing approach, we previously reported that the population of vagal sensory neurones that are ablated in response to CCK‐SAP injection primarily innervate the mucosal and muscular layers of the stomach and at least the upper part of the small intestine. 71 Previous work supports a role for CCK sensitive vagal neurones in both gastric and duodenal mechanoreceptive fibre firing 74 and that CCK‐sensitive neurones are required for mediating the satiating effect of nutrients. 75 CCK receptor‐expressing vagal sensory neurones co‐express hormone receptors associated with anorectic, 18 , 76 as well orexigenic function, 77 , 78 , 79 suggesting that these neurones can convey conflicting sensory signals. Furthermore, a recent RNA profiling study in mice confirm that CCK receptor‐expressing NG neurones innervate the length of the gastrointestinal tract and that these neurones co‐express (a) Glucagon‐like peptide‐1 receptor (GLP1R), a marker of gastric mechanosensitive neurones, (b) Oxytocin receptor (OxtR), a marker of intestinal mechanosensitive neurones as well as (c) vasoactive intestinal peptide (VIP) a marker of intestinal chemosensitive neurones. 36 In line with this, we previously demonstrated that a dose of exogenous GLP1 that causes satiation in SAP control rats fails to inhibit food intake in CCK‐SAP‐treated rats. 71 Importantly, Bai et al found that stimulation of subpopulations of CCKR neurones in the NG, specifically those that co‐express with GLP1R or OxtR, decreased food intake. 36 Thus, the ablation of CCK receptor‐expressing NG neurones prevents multimodal signalling from the gut to the brain, and is therefore a useful tool for vagal deafferentation of the gut.
CCK‐SAP ablated rats have altered meal patterns on a chow diet, characterized by increased meal number and frequency, and decreased meal size and ingestion rate. This may suggest that vagal gut‐brain circuits primarily control satiety, which prevents eating between meals, rather than satiation, which terminates a meal. Consistent with our results implicating a role of the vagus nerve in satiety rather than satiation, subdiaphragmatic vagotomy causes smaller, more frequent meals in both rats fed with liquid chow diet 80 and mice fed chow. 81 In addition, i.p. capsaicin‐induced deafferentation in chow‐fed rats increased meal frequency. 82 Thus, irrespective of the mechanism by which vagal signalling is disrupted, the consistent finding in each of these studies is that the total number of meals is increased in chow‐fed animals and this is usually accompanied by a reduced meal size. The current work significantly improves on the lack of organ specificity following vagotomy and capsaicin treatment, and provides direct evidence that gut‐specific sensory signalling is necessary for satiety. Notably, these results conflict with the CCK signalling literature. CCK administration decreases meal size followed by a compensatory increase in meal number, 83 while inhibition of CCK receptor by genetic 84 , 85 , 86 or pharmacological 87 , 88 methods increases meal size and decreases meal number. Our data suggest that gut‐brain vagal sensory signalling cannot be defined as an aggregate of individual signalling components.
Interestingly, when these same rats were given access to a palatable energy‐dense diet high in fat and sugar, vagal deafferentation of the gut increased cumulative food intake. Similar to that observed in lean rats, CCK‐SAP increased the number of meals; however, it also increased meal size, duration and ingestion rate, resulting in hyperphagia compared to SAP‐treated HFHS‐fed controls. The meal patterns changes in rats were restricted to the dark phase irrespective of diet, presumably because rodents consume the majority of their food in the dark. Interestingly, other feeding studies in which vagal sensory signalling was impaired also reported disruption of meal patterns only in the dark, 49 , 50 , 70 and is therefore a conserved phenomenon. In HFHS‐fed animals, vagal gut‐brain signalling conveys signals that affect both satiation and satiety, resulting in pronounced hyperphagia. This result was very robust, occurring in all rats with confirmed desensitization to CCK‐induced satiation in multiple batches of rats irrespective of when the HFHS diet was introduced post‐op, suggesting that CCK‐SAP‐induced hyperphagia was a result of the diet rather than age or time after surgery. Similar increases in meal size have been reported in capsaicin‐treated animals fed novel high‐fat foods in the first few days post‐surgery compared to controls. 9 , 10 , 11 Similarly, subdiaphragmatic vagal deafferentation causes an increase in meal size in rats fed a calorically dense liquid diet, 58 although this was accompanied by a decreased meal frequency. The difference between these previous studies and our current findings are likely caused by retaining intact vagal motor function. In mice lacking Nav 1.8 neurones, which include a subpopulation of vagal afferent neurones, 89 there was no effect on meal patterns in mice fed with HFHS diet compared to littermate controls. 90 It is likely that these mice develop compensatory mechanisms from lacking Nav1.8 neurones from birth, or that there is sufficient vagal innervation of the gut remaining to communicate nutrient information to the brain, especially since fat‐induced cFos signalling in the NTS was not different between ablated and control mice. 90 We find that highly specific vagal deafferentation of the gut in adult rats increases food intake and impairs fat sensing, suggesting that a fully functional vagus prevents overeating and excessive weight gain when challenged with a high‐calorie density diet.
The potential protective role of the intact vagus nerve in preventing exacerbation of weight gain in response to high‐calorie diets may be relevant in the progression of obesity. Chronic exposure to HFHS diet results in reduced sensitivity of NG neurones to tension, 37 , 38 satiation hormones (eg, CCK), 37 , 39 , 40 , 41 , 42 , 43 , 44 and intestinal nutrients. 12 , 45 , 46 , 47 Furthermore, post‐prandial neuronal activation in the NTS is significantly lower in obese, compared to lean, rats, 47 , 91 suggesting that gut‐brain signalling is severely blunted. Our previous work demonstrates that disrupted vagal signalling coincides with the onset of hyperphagia. 44 , 50 The exact mechanism by which vagal signalling is reduced in obesity remains unclear but may involve blunted receptor expression, 44 , 92 , 93 , 94 , 95 impaired neuropeptide release 50 and/or altered biophysical properties. 37 Irrespective of mechanism it is clear that in obesity, loss of vagal signalling reduces gut‐brain signalling, and our new data demonstrate that this is sufficient for long‐term dysregulation of energy homoeostasis.
To determine the mechanism by which vagal deafferentation increased food intake and body weight, we tested the relative importance of post‐ingestive nutrient sensing as a feedback mechanism for meal termination. Intra‐gastric administration of fat pre‐load significantly inhibited food intake in SAP‐treated rats but failed to acutely reduce food intake in CCK‐SAP rats lacking gut‐brain signalling. Baseline food intake was slightly, but not significantly, lower following methyl cellulose pre‐load in the CCK‐SAP compared to control rats; however, it is unlikely that this accounts for the blunted response to fat for three reasons. Firstly, the study is a within‐animal design which accounts for baseline intake of individual animals, and controls for the group differences in meal patterns we report in Figure 2. Secondly, we observed the same blunted response to fat when normalized to saline. Finally, sugar pre‐loads continue to be reduced compared to methyl cellulose (or saline), suggesting that fat signalling was selectively inhibited. Together these data suggest that (a) vagal gut‐brain signalling is critical for fat sensing and monitoring, and (b) impaired fat‐specific post‐ingestive feedback is sufficient to trigger hyperphagia at least acutely. Certainly, the biased loss of fat sensing in response to targeted ablation of CCK receptor‐expressing sensory vagal neurones is consistent with previous data showing that CCK is released preferentially in response to fat and amino acids compared to carbohydrates. 96 , 97 There is extensive evidence that infusion of fats into the gut results in both meal termination 98 , 99 and appetition. 100 , 101 Previous work demonstrates that CCK‐SAP prevents intra‐gastric fat‐induced dopamine release, the ability of the animals to form a flavour conditioned preference for fat, or learn to self administer intra‐gastric fat. 7 CCK, chemogenetic or optogenetic stimulation of this gut‐innervating vagal pathway causes a reduction of food intake, and many hallmark reward behaviours including conditioned flavour learning, conditioned place preference and self‐stimulation. 7 Thus, in mice, both CCK and fat reduce food intake via a reward circuit that requires CCK receptor‐expressing vagal sensory neurones. In rats, nutrient‐induced satiation signals influence motivation to work for food. 102 These data suggest an overlap in post‐ingestive fat mediated reward and meal termination through a vagal gut‐brain circuit. Based on these data we hypothesize that reduced post‐ingestive signalling of fat is sufficient to prevent appropriate meal termination and that chronic overeating at every meal promotes weight gain.
Intra‐gastric pre‐load of sucrose reduced subsequent chow intake similarly in both CCK‐SAP‐treated rats and SAP controls. In support of these findings, subdiaphragmatic deafferentation failed to inhibit glucose intake after a small gastric pre‐load of sugar, while fat had no effect. 58 Interestingly, we found that intra‐gastric sucrose pre‐load was a more potent inhibitor of food intake than lipid pre‐load in SAP‐treated animals. Similar observations were previously reported in humans, where carbohydrate supplementation was a more potent appetite suppressor than fat supplementation at breakfast. 103 The fact that sugar pre‐load equally reduced food intake in both SAP and CCK‐SAP‐treated rats suggests that the CCK‐receptor expressing vagal sensory neurones are not necessary for the appetite suppressant effects of intra‐gastric sucrose. A rapid rise in circulating glucose may act through a vagally independent mechanism via the hindbrain or hypothalamus, or an insulin‐mediated mechanism that is released in response to glucose that acts though a population of vagal afferent neurones that express insulin receptors 104 but not CCK receptor. Alternatively, the high osmolarity from the very concentrated sucrose solution infused in the gut may cause aversion. We observed no difference in the rats’ willingness to work for either orally administered fat or sugar. Since small volumes were administered in the progressive ratio task, we expect negligible vagal signalling from the gut and thus no post‐ingestive reward. Therefore we conclude from the data that increased preference for the taste of the HFHS diet was not driving motivation to overeat. Little work has been done to address the role of the vagus nerve in taste perception. One study indicates that after subdiaphragmatic vagotomy rats no longer considered oral palatability when deciding how much food to eat. 105 Specifically, vagotomized rats ate the same amount of three test meals in which one was regular chow, one was chow sweetened with sodium cyclamate and one was chow adulterated with quinine hydrochloride. 105 Altogether, this suggests that although the CCK‐SAP rats consumed more of the HFHS diet, the increased palatability of the diet is not likely to be an important factor in the hyperphagia. However, future work of directly testing the taste profiles of different fat and sugar concentrations is warranted following vagal deafferentation of the gut.
While some studies involving the gut‐vagus‐brain axis indicate an ability of gut nutrient sensing vagal afferents to control glucose homoeostasis 70 , 106 and modulate energy expenditure in rodents, 107 , 108 , 109 the specific vagal branch(es) mediating these effects remain unclear. In our current study, despite changes in meal patterns, the ablation of CCK‐sensitive vagal afferent neurones in rats did not affect glucose tolerance or whole‐body energy expenditure under chow‐fed conditions. Although our IPGTT data suggest that gut‐brain vagal signalling is not necessary for glucose metabolism after 3 weeks of HFHS diet, it is important to note that previous studies have reported that carbohydrate‐induced incretin release is sufficient for insulin release via a vagally mediated mechanism. 70 Therefore, monitoring incretin release and additional time points should be tested alongside when conducting hyperglycaemic clamp experiments to fully assess the role of gut‐brain signalling in diet‐induced glucose homoeostasis. Duodenal infusions of lipid emulsions at doses that reduce meal size have been reported to increase the temperature in brown adipose tissue in rats, indicative of increased thermogenesis. 107 Notably, the effect of BAT activation was reported to be (a) decreased with chronic high‐fat diet consumption, (b) abrogated by cervical vagotomy 108 or (c) blocked by local intestinal application of the anaesthetic tetracaine, as well as by (d) peripheral administration of the CCK1R devazepide. 107 BAT thermogenesis accounts for a small fraction of total energy expenditure, it is therefore possible that by measuring whole‐body energy expenditure, we missed a small change in thermogenesis and/or that this was compensated by a reduction in activity or basal metabolic rate. Furthermore, we did not measure whole‐body energy expenditure in the animals fed HFHS diet, therefore, whether vagal deafferentation of the gut affects energy expenditure in response to diet‐induced obesity needs further investigation.
A variety of diets ranging in physical form (solid vs liquid) and fat composition have been used in the past to assess the role of vagal signalling in the control of food intake. The distinction in the outcomes of vagal disruption in response to these different diets has been unclear. This combined with the lack of specificity in targeting selective organs or sensory arm of the vagus nerve has caused discordance about the function of gut‐to‐brain signalling in food intake. Here we have used a method for rapid and long‐lasting ablation of a targeted subpopulation of vagal sensory neurones that broadly sense both mechanical and chemical signals along the length of the gastrointestinal tract. We find that CCK receptor‐expressing vagal sensory neurones are necessary to control short‐term satiety. Interestingly, this gut‐brain circuit is necessary to prevent excessive overconsumption of fat, and inhibiting this post‐ingestive feedback mechanism promotes overeating and weight. Thus, a fully functioning vagus nerve prevents against long‐term weight gain caused by an obesogenic diet. Altogether these data identify a putative mechanism by which targeting the vagus nerve can be a useful therapeutic strategy for obesity.
4. MATERIALS AND METHODS
4.1. Animals and Housing
Adult male Wistar rats (220‐250 g starting bodyweight, Harlan, San Diego, CA and Envigo, Tampa, FL) were individually housed at 22°C under a 12‐h light‐dark cycle with ad libitum access to water and either chow (3.1 kcal/g, Teklad 2018, Envigo, Sommerset, NJ) or HFHS diet (45% calories from fat; 4.7 kcal/g, Research Diets D12451, New Brunswink, NJ), unless stated otherwise. Animals were allowed for 1 week to acclimate before any experiments were started. All experiments were approved by the University of Florida, and John B. Pierce Laboratory Institutional Animal Care and Use Committees (IACUC).
4.2. Peptides and drugs
Saporin (SAP) and CCK‐Saporin were obtained from Advanced Targeting Systems (San Diego, Ca). CCK‐8 was obtained from Bachem BioScience Inc (King of Prussia, PA).
4.3. Surgery
4.3.1. Nodose ganglia injection
Before surgery, rats were fasted overnight with ad libitum access to water. Twenty minutes before surgery, rats received a subcutaneous injection of atropine sulphate (0.05 mg/kg; Henry Schein, Wallingford, CT) and carprofen (5.0 mg/kg; Henry Schein). Rats were anaesthetized with isoflurane (2% Isoflurane; Henry Schein). The rat was shaved from the chin to thorax and placed supine on a heated pad with a nose cone ventilator (SomnoSuite; Kent Scientific, Torrington, CT). A midline incision was made with a scalpel along the length of the neck; salivary glands and lymph nodes were retracted away from the midline. The sternohyoid and omohyoid were separated and retracted to expose the carotid artery and vagus nerve. The vagus nerve was separated from surrounding fascia and the carotid artery with fine tip forceps and retractors until the NG became accessible. A glass capillary (30 μm tip, beveled 30 degree angle) attached to a micromanipulator was used to position and puncture the NG and 1.5 µL volume of CCK‐SAP (250 ng/µL) or SAP (250 ng/µL) was injected with a Picospritzer III injector (Parker Hannifin, North Haven, CT) in the centre of the NG. The same procedure was repeated contralaterally before the skin was closed with sterile suture. Rats were allowed to recover under infrared heat until they chose to reside in the unheated side of the cage, at which point they were returned to their home cage, deprived of water for 6 hours and food overnight. We found that the previously used post‐op feeding regimen 71 , 110 did not improve recovery or survival, therefore post‐op rats received carprofen (5.0 mg/kg; SQ) and were returned to ad libitum access to chow from day 1.
4.3.2. Gastric catheter implant
Rats were fasted overnight and received carprofen (5 mg/kg; SQ) 20 minutes prior to surgery. The back of the neck and the abdomen were shaved around the midline and rat was placed supine on a heated pad with a nose cone ventilator (SomnoSuite; Kent Scientific, Torrington, CT). A 15‐mm midline incision was made with a scalpel in the skin of the abdomen beginning at the sternum, followed by a similar incision in the linea alba of the muscle layer. The stomach was externalized and a 5‐mm purse suture was placed in the greater curvature near the fundus. Fine tip forceps were used to form a puncture in the centre of the purse suture and a catheter made from silicone tubing (SIL 047; Braintree Scientific, Braintree, MA) was inserted into the stomach. The purse suture was then tightened around the catheter and tied off to secure it in place. The stomach was returned to the abdominal cavity and a second 5‐mm purse suture was placed in the muscle layer of the abdomen 1 cm lateral to the midline incision, punctured with fine tip forceps and the open end of the catheter pulled through and secured by tying off the suture. The abdominal muscle layer was then closed with suture and the animal turned on its side. A small incision was made at the base of the skull and haemostats were used to blunt dissect the skin away from the muscle layer between the incision at the back of the neck and the abdominal incision, behind the shoulder, forming a tract for the catheter. The open end of the catheter was then pulled through to the back of the neck and secured using a purse suture around the opening. The skin of the abdomen was closed with suture and the cathether flushed with sterile saline prior to being capped. Rats were allowed to recover under infrared heat until they chose to reside in the unheated side of the cage, at which point they were returned to their home cage with ad libitum access to food and water. Day 1 post‐op rats received Carprofen (5.0 mg/kg; SQ) and moistened chow on the cage floor.
4.4. Phenotyping
4.4.1. Timeline
After NG surgery (CCK‐SAP n = 16, SAP n = 21), animals were kept on a chow diet (Figures 1 and 2). After 6 weeks, half of the animals from each group were switched to a HFHS diet for 5 weeks (HFHS n = 8/group; Figure 3) while the rest were maintained on a chow diet (Chow n = 8/group).After 11 weeks of chow, these rats were switched to HFHS diet for 5 weeks (n = 8/group; Figure 4). Throughout the experiment, body weight and food intake were measured twice weekly. CCK satiety tests were performed at 6 weeks post op. IPGTT was performed 3 weeks post‐op in chow fed, and 3 weeks after switching to HFHS diet. A separate cohort of rats received bilateral NG injections of SAP or CCK‐SAP (n = 7/group; Figure 5), had gastric catheter implanted and were maintained on chow diet.
4.4.2. Feeding studies
CCK
To validate CCK‐SAP‐induced ablation of CCK receptor expressing NG neurones, we tested the hypophagic responses to CCK in all rats from each cohort. Within subject design was used with each rat receiving counterbalanced vehicle (500 μL; saline) or sulphated CCK octapeptide (CCK‐8S, 4 μg/kg body weight, IP, Bachem, Torrance, CA, USA). 50 Animals were fasted for 16 hours on wire bottom cages and injected 1 hours into the dark cycle. The injections were administered in a counterbalanced fashion with half animals receiving the drug on first day followed by vehicle injection on second day, and others receiving vehicle on first day followed by the drug injection on second day. Food weight and spillage were manually recorded for 2 hours or in an automated episodic food intake monitoring system (BioDAQ). Rats with greater than 25% reduction in food intake in response to CCK (4 μg/kg; IP) compared to saline were included in the SAP group, and rats with less than 5% satiation after CCK compared to saline were included in CCK‐SAP group. Three rats were excluded based on these parameters.
Pre‐load intra‐gastric infusions
Rats were fasted overnight starting at light onset. Beginning at dark onset on the following day, animals received a 10 mL intra‐gastric infusion at 1 mL/minute rate, 30 minutes before refeeding. Infusates were 75% sucrose (Sigma‐Aldrich, MO, USA), 37.5% fat (Microlipid®, Nestle SA, Switzerland) or 2% methyl cellulose (viscosity:15 cP, Sigma‐Aldrich, MO, USA) with three consecutive replicate days and food intake was averaged across the 3 days for each condition.
Food intake microstructure
Meal patterns were analysed in a BioDAQ (Research diets Inc, New Brunswick, NJ) episodic food intake monitoring system (n = 8/group). Meals were defined by at least 0.2 g consumed without interruption by a pause of >10 minutes. The system consists of a low spill enlarged opening food hopper placed on an electronic balance mounted together on the animals’ home cage. Chow intake was continuously measured from weeks 3‐6 in chow‐fed rats (Figure 1), or weeks 6‐11 in HFHS (Figure 3).
4.4.3. Energy expenditure
In a separate cohort of chow‐fed rats (n = 6/group), the volume of oxygen consumed (VO2, mL/kg body weight/h) and carbon dioxide produced (VCO2, mL/kg body weight/h) were continuously recorded at 12‐minutes intervals by indirect calorimetry (CLAMS setup; 2‐L/min flow) following a previously published protocol. 111 The total energy expenditure was computed using the following equation: 3.815 × VO2 (L/h) + 1.232 × VCO2 (L/h), and data were represented as kcal/h. A week before entering CLAMS, rats were provided with powdered chow instead of pellets in a food cup to mimic the CLAMS cage set up. At 6 weeks post‐op, rats were placed in the CLAMS cages. Rats were allowed to acclimatize for 3 days, and indirect calorimetry was performed during days 4 and 5.
4.4.4. Intraperitoneal glucose (IPGTT) tolerance test
After overnight fasting (~16 hours), an IP injection of 50% glucose solution at a dose of 2 g/kg body weight was administered. Glucose concentrations were determined from the tail venous blood using a hand‐held glucometer (Accu‐Chek™ glucose meter, Roche Diagnostics, QC, Canada) at 0, 30, 60 and 120 minutes after glucose injection.
4.4.5. Progressive ratio test
Rats were food restricted to 90% at the starting body weight prior to the behavioural task. They were then conditioned to lick for 20% sucrose or 10% fat solutions (Microlipid®, Nestle SA, Switzerland) in a lickometer box that dispensed <3 µL per lick on a fixed ratio 1 (FR1) schedule for 1 hour per day for 5 days. Once conditioned to the FR1 schedule, rats were trained to lick on a fixed ratio 5 (FR5) schedule for 3 days before they were tested using a progressive ratio breakpoint test.
4.5. Statistics
Statistical analysis for the experiments is described in each figure legend and was determined using GraphPad Prism 8.3 software. Two‐tailed unpaired Student's t tests were used for comparing two groups; one‐way ANOVA, with or without repeated‐measures, was used for comparing three groups; two‐way ANOVA, with or without repeated‐measures, was used for comparing more than one factor between groups as performed for food intake, energy expenditure, weight gain and blood glucose during IPGTT. Data are presented as mean ± SEM and statistical significance is declared at P < .05.
CONFLICT OF INTEREST
None of the authors declare any conflict of interest.
AUTHORS’ CONTRIBUTIONS
MM: acquisition of data, analysis and interpretation of data, drafting of the manuscript and statistical analysis. AS, CD, DQ: acquisition of data, analysis and interpretation of data and editing of the manuscript CS: analysis and interpretation of data, editing of the manuscript, obtained funding. GL: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, obtained funding and study supervision.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grant R00DK094871 (GL), R01 DK116004 (GL) and R21DK110511 (CdlS).
McDougle M, Quinn D, Diepenbroek C, Singh A, de la Serre C, de Lartigue G. Intact vagal gut‐brain signalling prevents hyperphagia and excessive weight gain in response to high‐fat high‐sugar diet. Acta Physiol.2021;231:e13530. 10.1111/apha.13530
Molly McDougle and Danielle Quinn contributed equally to this work.
Funding information
This work was supported by National Institutes of Health Grant R00DK094871, R01 DK116004 and R21DK110511
REFERENCES
- 1. de Lartigue G, Diepenbroek C. Novel developments in vagal afferent nutrient sensing and its role in energy homeostasis. Curr Opin Pharmacol. 2016;31:38‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Browning KN, Travagli RA. Central nervous system control of gastrointestinal motility and secretion and modulation of gastrointestinal functions. Compr Physiol. 2014;4(4):1339‐1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams Erika K, Chang Rui B, Strochlic David E, Umans Benjamin D, Lowell Bradford B, Liberles SD. Sensory neurons that detect stretch and nutrients in the digestive system. Cell. 2016;166(1):209‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang Rui B, Strochlic David E, Williams Erika K, Umans Benjamin D, Liberles SD. Vagal sensory neuron subtypes that differentially control breathing. Cell. 2015;161(3):622‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Agostoni E, Chinnock JE, De Daly MB, Murray JG. Functional and histological studies of the vagus nerve and its branches to the heart, lungs and abdominal viscera in the cat. J Physiol. 1957;135(1):182‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Powley TL, Spaulding RA, Haglof SA. Vagal afferent innervation of the proximal gastrointestinal tract mucosa: chemoreceptor and mechanoreceptor architecture. J Comp Neurol. 2011;519(4):644‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Han W, Tellez LA, Perkins MH, et al. A neural circuit for gut‐induced reward. Cell. 2018;175(3):665‐678.e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gortz L, Bjorkman AC, Andersson H, Kral JG. Truncal vagotomy reduces food and liquid intake in man. Physiol Behav. 1990;48(6):779‐781. [DOI] [PubMed] [Google Scholar]
- 9. Chavez M, Kelly L, York DA, Berthoud HR. Chemical lesion of visceral afferents causes transient overconsumption of unfamiliar high‐fat diets in rats. Am J Physiol. 1997;272(5 Pt 2):R1657‐R1663. [DOI] [PubMed] [Google Scholar]
- 10. Kelly L, Morales S, Smith BK, Berthoud HR. Capsaicin‐treated rats permanently overingest low‐ but not high‐concentration sucrose solutions. Am J Physiol Regul Integr Comp Physiol. 2000;279(5):R1805‐R1812. [DOI] [PubMed] [Google Scholar]
- 11. Kelly LA, Chavez M, Berthoud HR. Transient overconsumption of novel foods by deafferentated rats: effects of novel diet composition. Physiol Behav. 1999;65(4‐5):793‐800. [DOI] [PubMed] [Google Scholar]
- 12. Duca FA, Swartz TD, Sakar Y, Covasa M. Decreased intestinal nutrient response in diet‐induced obese rats: role of gut peptides and nutrient receptors. Int J Obes (Lond). 2013;37(3):375‐381. [DOI] [PubMed] [Google Scholar]
- 13. Darling RA, Zhao H, Kinch D, Li AJ, Simasko SM, Ritter S. Mercaptoacetate and fatty acids exert direct and antagonistic effects on nodose neurons via GPR40 fatty acid receptors. Am J Physiol Regul Integr Comp Physiol. 2014;307(1):R35‐R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grabauskas G, Zhou SY, Lu Y, Song I, Owyang C. Essential elements for glucosensing by gastric vagal afferents: immunocytochemistry and electrophysiology studies in the rat. Endocrinology. 2013;154(1):296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zarbin MA, Wamsley JK, Innis RB, Kuhar MJ. Cholecystokinin receptors: presence and axonal flow in the rat vagus nerve. Life Sci. 1981;29(7):697‐705. [DOI] [PubMed] [Google Scholar]
- 16. Ronveaux CC, de Lartigue G, Raybould HE. Ability of GLP‐1 to decrease food intake is dependent on nutritional status. Physiol Behav. 2014;135:222‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakagawa A, Satake H, Nakabayashi H, et al. Receptor gene expression of glucagon‐like peptide‐1, but not glucose‐dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton. Neurosci. 2004;110(1):36‐43. [DOI] [PubMed] [Google Scholar]
- 18. Burdyga G, de Lartigue G, Raybould HE, et al. Cholecystokinin regulates expression of Y2 receptors in vagal afferent neurons serving the stomach. J Neurosci. 2008;28(45):11583‐11592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Umans BD, Liberles SD. Neural sensing of organ volume. Trends Neurosci. 2018;41(12):911‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Page AJ, Martin CM, Blackshaw LA. Vagal mechanoreceptors and chemoreceptors in mouse stomach and esophagus. J Neurophysiol. 2002;87(4):2095‐2103. [DOI] [PubMed] [Google Scholar]
- 21. Page AJ, Blackshaw LA. An in vitro study of the properties of vagal afferent fibres innervating the ferret oesophagus and stomach. J Physiol. 1998;512(Pt 3):907‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Niijima A. Glucose‐sensitive afferent nerve fibres in the hepatic branch of the vagus nerve in the guinea‐pig. J Physiol. 1982;332:315‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niijima A. The effect of endogenous sugar acids on the afferent discharge rate of the hepatic branch of the vagus nerve in the rat. Physiol Behav. 1988;44(4‐5):661‐664. [DOI] [PubMed] [Google Scholar]
- 24. Niijima A. Electrophysiological study on nervous pathway from splanchnic nerve to vagus nerve in rat. Am J Physiol. 1983;244(6):R888‐R890. [DOI] [PubMed] [Google Scholar]
- 25. Randich A, Tyler WJ, Cox JE, Meller ST, Kelm GR, Bharaj SS. Responses of celiac and cervical vagal afferents to infusions of lipids in the jejunum or ileum of the rat. Am J Physiol Regul Integr Comp Physiol. 2000;278(1):R34‐R43. [DOI] [PubMed] [Google Scholar]
- 26. Tome D, Schwarz J, Darcel N, Fromentin G. Protein, amino acids, vagus nerve signaling, and the brain. Am J Clin Nutr. 2009;90(3):838s‐843s. [DOI] [PubMed] [Google Scholar]
- 27. Kentish SJ, O'Donnell TA, Isaacs NJ, et al. Gastric vagal afferent modulation by leptin is influenced by food intake status. J Physiol. 2013;591(7):1921‐1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koda S, Date Y, Murakami N, et al. The role of the vagal nerve in peripheral PYY3–36‐induced feeding reduction in rats. Endocrinology. 2005;146(5):2369‐2375. [DOI] [PubMed] [Google Scholar]
- 29. Li Y, Zhu J, Owyang C. Electrical physiological evidence for highand low‐affinity vagal CCK‐A receptors. Am J Physiol. 1999;277(2):G469‐G477. [DOI] [PubMed] [Google Scholar]
- 30. Blackshaw LA, Grundy D. Effects of cholecystokinin (CCK‐8) on two classes of gastroduodenal vagal afferent fibre. J Auton Nerv Syst. 1990;31(3):191‐201. [DOI] [PubMed] [Google Scholar]
- 31. Timofeeva E, Baraboi ED, Richard D. Contribution of the vagus nerve and lamina terminalis to brain activation induced by refeeding. Eur J Neurosci. 2005;22(6):1489‐1501. [DOI] [PubMed] [Google Scholar]
- 32. Monnikes H, Lauer G, Bauer C, Tebbe J, Zittel TT, Arnold R. Pathways of Fos expression in locus ceruleus, dorsal vagal complex, and PVN in response to intestinal lipid. Am J Physiol. 1997;273(6):R2059‐R2071. [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto T, Sawa K. c‐Fos‐like immunoreactivity in the brainstem following gastric loads of various chemical solutions in rats. Brain Res. 2000;866(1‐2):135‐143. [DOI] [PubMed] [Google Scholar]
- 34. Traub RJ, Sengupta JN, Gebhart GF. Differential c‐fos expression in the nucleus of the solitary tract and spinal cord following noxious gastric distention in the rat. Neuroscience. 1996;74(3):873‐884. [DOI] [PubMed] [Google Scholar]
- 35. Gibbs J, Young RC, Smith GP. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature. 1973;245(5424):323‐325. [DOI] [PubMed] [Google Scholar]
- 36. Bai L, Mesgarzadeh S, Ramesh KS, et al. Genetic identification of vagal sensory neurons that control feeding. Cell. 2019;179(5):1129‐1143.e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daly DM, Park SJ, Valinsky WC, Beyak MJ. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol. 2011;589(Pt 11):2857‐2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kentish S, Li H, Philp LK, et al. Diet‐induced adaptation of vagal afferent function. J Physiol. 2012;590(Pt 1):209‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Covasa M, Ritter RC. Rats maintained on high‐fat diets exhibit reduced satiety in response to CCK and bombesin. Peptides. 1998;19(8):1407‐1415. [DOI] [PubMed] [Google Scholar]
- 40. Covasa M, Ritter RC. Adaptation to high‐fat diet reduces inhibition of gastric emptying by CCK and intestinal oleate. Am J Physiol Regul Integr Comp Physiol. 2000;278(1):R166‐R170. [DOI] [PubMed] [Google Scholar]
- 41. Savastano DM, Covasa M. Adaptation to a high‐fat diet leads to hyperphagia and diminished sensitivity to cholecystokinin in rats. J Nutr. 2005;135(8):1953‐1959. [DOI] [PubMed] [Google Scholar]
- 42. Swartz TD, Savastano DM, Covasa M. Reduced sensitivity to cholecystokinin in male rats fed a high‐fat diet is reversible. J Nutr. 2010;140(9):1698‐1703. [DOI] [PubMed] [Google Scholar]
- 43. Duca FA, Sakar Y, Covasa M. The modulatory role of high fat feeding on gastrointestinal signals in obesity. J Nutr Biochem. 2013;24(10):1663‐1677. [DOI] [PubMed] [Google Scholar]
- 44. de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PLoS One. 2012;7(3):e32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Covasa M, Marcuson JK, Ritter RC. Diminished satiation in rats exposed to elevated levels of endogenous or exogenous cholecystokinin. Am J Physiol Regul Integr Comp Physiol. 2001;280(2):R331‐R337. [DOI] [PubMed] [Google Scholar]
- 46. Duca FA, Swartz TD, Sakar Y, Covasa M. Increased oral detection, but decreased intestinal signaling for fats in mice lacking gut microbiota. PLoS One. 2012;7(6):e39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Covasa M, Grahn J, Ritter RC. Reduced hindbrain and enteric neuronal response to intestinal oleate in rats maintained on high‐fat diet. Auton Neurosci. 2000;84(1‐2):8‐18. [DOI] [PubMed] [Google Scholar]
- 48. de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet‐induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab. 2011;301(1):E187‐E195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Lartigue G, Ronveaux CC, Raybould HE. Deletion of leptin signaling in vagal afferent neurons results in hyperphagia and obesity. Mol Metab. 2014;3(6):595‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee SJ, Krieger JP, Vergara M, et al. Blunted vagal cocaine‐ and amphetamine‐regulated transcript promotes hyperphagia and weight gain. Cell Rep. 2020;30(6):2028‐2039.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Lartigue G. Role of the vagus nerve in the development and treatment of diet‐induced obesity. J Physiol. 2016;594(20):5791‐5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yao G, Kang L, Li J, et al. Effective weight control via an implanted self‐powered vagus nerve stimulation device. Nat Commun. 2018;9(1):5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matyja A, Thor PJ, Sobocki J, et al. Effects of vagal pacing on food intake and body mass in pigs. Folia Med Cracov. 2004;45(3‐4):55‐62. [PubMed] [Google Scholar]
- 54. Sobocki J, Thor PJ, Uson J, et al. Microchip vagal pacing reduces food intake and body mass. Hepatogastroenterology. 2001;48(42):1783‐1787. [PubMed] [Google Scholar]
- 55. Roslin M, Kurian M. The use of electrical stimulation of the vagus nerve to treat morbid obesity. Epilepsy Behav. 2001;2(3):S11‐S16. [Google Scholar]
- 56. Krolczyk G, Zurowski D, Sobocki J, et al. Effects of continuous microchip (MC) vagal neuromodulation on gastrointestinal function in rats. J Physiol Pharmacol. 2001;52(4 Pt 1):705‐715. [PubMed] [Google Scholar]
- 57. Burneo JG, Faught E, Knowlton R, Morawetz R, Kuzniecky R. Weight loss associated with vagus nerve stimulation. Neurology. 2002;59(3):463‐464. [DOI] [PubMed] [Google Scholar]
- 58. Schwartz GJ, Salorio CF, Skoglund C, Moran TH. Gut vagal afferent lesions increase meal size but do not block gastric preload‐induced feeding suppression. Am J Physiol. 1999;276(6 Pt 2):R1623‐R1629. [DOI] [PubMed] [Google Scholar]
- 59. Walls EK, Phillips RJ, Wang FB, Holst MC, Powley TL. Suppression of meal size by intestinal nutrients is eliminated by celiac vagal deafferentation. Am J Physiol. 1995;269(6 Pt 2):R1410‐R1419. [DOI] [PubMed] [Google Scholar]
- 60. van de Wall EH, Pomp ER, Strubbe JH, Scheurink AJ, Koolhaas JM. Deafferentation affects short‐term but not long‐term control of food intake. Physiol Behav. 2005;84(4):659‐667. [DOI] [PubMed] [Google Scholar]
- 61. Castonguay TW, Bellinger LL. Capsaicin and its effects upon meal patterns, and glucagon and epinephrine suppression of food intake. Physiol Behav. 1987;40(3):337‐342. [DOI] [PubMed] [Google Scholar]
- 62. Gautron L, Sakata I, Udit S, Zigman JM, Wood JN, Elmquist JK. Genetic tracing of Nav1.8‐expressing vagal afferents in the mouse. J Comp Neurol. 2011;519(15):3085‐3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fox EA, Phillips RJ, Baronowsky EA, Byerly MS, Jones S, Powley TL. Neurotrophin‐4 deficient mice have a loss of vagal intraganglionic mechanoreceptors from the small intestine and a disruption of short‐term satiety. J Neurosci. 2001;21(21):8602‐8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Biddinger JE, Fox EA. Reduced intestinal brain‐derived neurotrophic factor increases vagal sensory innervation of the intestine and enhances satiation. J Neurosci. 2014;34(31):10379‐10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fox EA, Biddinger JE, Baquet ZC, Jones KR, McAdams J. Loss of neurotrophin‐3 from smooth muscle disrupts vagal gastrointestinal afferent signaling and satiation. Am J Physiol Regul Integr Comp Physiol. 2013;305(11):R1307‐R1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stauss HM, Stangl H, Clark KC, Kwitek AE, Lira VA. Cervical vagal nerve stimulation impairs glucose tolerance and suppresses insulin release in conscious rats. Physiol Rep. 2018;6(24):e13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Browning KN, Babic T, Holmes GM, Swartz E, Travagli RA. A critical re‐evaluation of the specificity of action of perivagal capsaicin. J Physiol. 2013;591(Pt 6):1563‐1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Norgren R, Smith GP. A method for selective section of vagal afferent or efferent axons in the rat. Am J Physiol. 1994;267(4):R1136‐R1141. [DOI] [PubMed] [Google Scholar]
- 69. Iwasaki Y, Sendo M, Dezaki K, et al. GLP‐1 release and vagal afferent activation mediate the beneficial metabolic and chronotherapeutic effects of D‐allulose. Nat Commun. 2018;9(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ. Knockdown of GLP‐1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes. 2016;65(1):34‐43. [DOI] [PubMed] [Google Scholar]
- 71. Diepenbroek C, Quinn D, Stephens R, et al. Validation and characterization of a novel method for selective vagal deafferentation of the gut. Am J Physiol Gastrointest Liver Physiol. 2017;313(4):G342‐G352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kissileff HR, Herzog M. Progressive ratio (PR) schedules and the sipometer: Do they measure wanting, liking, and/or reward? A tribute to Anthony Sclafani and Karen Ackroff. Appetite. 2018;122:44‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moran TH, Smith GP, Hostetler AM, McHugh PR. Transport of cholecystokinin (CCK) binding sites in subdiaphragmatic vagal branches. Brain Res. 1987;415(1):149‐152. [DOI] [PubMed] [Google Scholar]
- 74. Schwartz GJ, Moran TH. Sub‐diaphragmatic vagal afferent integration of meal‐related gastrointestinal signals. Neurosci Biobehav Rev. 1996;20(1):47‐56. [DOI] [PubMed] [Google Scholar]
- 75. Schwartz GJ, McHugh PR, Moran TH. Pharmacological dissociation of responses to CCK and gastric loads in rat mechanosensitive vagal afferents. Am J Physiol. 1994;267(1 Pt 2):R303‐R308. [DOI] [PubMed] [Google Scholar]
- 76. Egerod KL, Petersen N, Timshel PN, et al. Profiling of G protein‐coupled receptors in vagal afferents reveals novel gut‐to‐brain sensing mechanisms. Mol Metab. 2018;12:62‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Expression of cannabinoid CB1 receptors by vagal afferent neurons: kinetics and role in influencing neurochemical phenotype. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G63‐G69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Ghrelin receptors in rat and human nodose ganglia: putative role in regulating CB‐1 and MCH receptor abundance. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1289‐G1297. [DOI] [PubMed] [Google Scholar]
- 79. Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Feeding‐dependent depression of melanin‐concentrating hormone and melanin‐concentrating hormone receptor‐1 expression in vagal afferent neurones. Neuroscience. 2006;137(4):1405‐1415. [DOI] [PubMed] [Google Scholar]
- 80. Snowdon CT, Epstein AN. Oral and intragastric feeding in vagotomized rats. J Comp Physiol Psychol. 1970;71(1):59‐67. [DOI] [PubMed] [Google Scholar]
- 81. Powley TL, Chi MM, Baronowsky EA, Phillips RJ. Gastrointestinal tract innervation of the mouse: afferent regeneration and meal patterning after vagotomy. Am J Physiol Regul Integr Comp Physiol. 2005;289(2):R563‐R574. [DOI] [PubMed] [Google Scholar]
- 82. Reidelberger R, Haver A, Anders K, Apenteng B. Role of capsaicin‐sensitive peripheral sensory neurons in anorexic responses to intravenous infusions of cholecystokinin, peptide YY‐(3–36), and glucagon‐like peptide‐1 in rats. Am J Physiol Endocrinol Metab. 2014;307(8):E619‐E629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. West DB, Fey D, Woods SC. Cholecystokinin persistently suppresses meal size but not food intake in free‐feeding rats. Am J Physiol. 1984;246(5 Pt 2):R776‐R787. [DOI] [PubMed] [Google Scholar]
- 84. Bi S, Chen J, Behles RR, Hyun J, Kopin AS, Moran TH. Differential body weight and feeding responses to high‐fat diets in rats and mice lacking cholecystokinin 1 receptors. Am J Physiol Regul Integr Comp Physiol. 2007;293(1):R55‐R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Moran TH. Unraveling the obesity of OLETF rats. Physiol Behav. 2008;94(1):71‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Moran TH, Katz LF, Plata‐Salaman CR, Schwartz GJ. Disordered food intake and obesity in rats lacking cholecystokinin A receptors. Am J Physiol. 1998;274(3):R618‐R625. [DOI] [PubMed] [Google Scholar]
- 87. Moran TH, Ameglio PJ, Peyton HJ, Schwartz GJ, McHugh PR. Blockade of type A, but not type B, CCK receptors postpones satiety in rhesus monkeys. Am J Physiol. 1993;265(3 Pt 2):R620‐R624. [DOI] [PubMed] [Google Scholar]
- 88. Moran TH, Ameglio PJ, Schwartz GJ, McHugh PR. Blockade of type A, not type B, CCK receptors attenuates satiety actions of exogenous and endogenous CCK. Am J Physiol. 1992;262(1 Pt 2):R46‐R50. [DOI] [PubMed] [Google Scholar]
- 89. Kupari J, Haring M, Agirre E, Castelo‐Branco G, Ernfors P. An atlas of vagal sensory neurons and their molecular specialization. Cell Rep. 2019;27(8):2508‐2523.e2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Udit S, Burton M, Rutkowski JM, et al. Nav1.8 neurons are involved in limiting acute phase responses to dietary fat. Mol Metab. 2017;6(10):1081‐1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Covasa M, Grahn J, Ritter RC. High fat maintenance diet attenuates hindbrain neuronal response to CCK. Regul Pept. 2000;86(1‐3):83‐88. [DOI] [PubMed] [Google Scholar]
- 92. Paulino G, Barbier de la Serre C, Knotts TA, et al. Increased expression of receptors for orexigenic factors in nodose ganglion of diet‐induced obese rats. Am J Physiol Endocrinol Metab. 2009;296(4):E898‐E903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kentish SJ, Ratcliff K, Li H, Wittert GA, Page AJ. High fat diet induced changes in gastric vagal afferent response to adiponectin. Physiol Behav. 2015;152(Pt B):354‐362. [DOI] [PubMed] [Google Scholar]
- 94. Kentish SJ, Wittert GA, Blackshaw LA, Page AJ. A chronic high fat diet alters the homologous and heterologous control of appetite regulating peptide receptor expression. Peptides. 2013;46:150‐158. [DOI] [PubMed] [Google Scholar]
- 95. Nefti W, Chaumontet C, Fromentin G, Tome D, Darcel N. A high‐fat diet attenuates the central response to within‐meal satiation signals and modifies the receptor expression of vagal afferents in mice. Am J Physiol Regul Integr Comp Physiol. 2009;296(6):R1681‐R1686. [DOI] [PubMed] [Google Scholar]
- 96. Gibbs J, Falasco JD, McHugh PR. Cholecystokinin‐decreased food intake in rhesus monkeys. Am J Physiol. 1976;230(1):15‐18. [DOI] [PubMed] [Google Scholar]
- 97. Anika SM, Houpt TR, Houpt KA. Satiety elicited by cholecystokinin in intact and vagotomized rats. Physiol Behav. 1977;19(6):761‐766. [DOI] [PubMed] [Google Scholar]
- 98. Booth DA. Postabsorptively induced suppression of appetite and the energostatic control of feeding. Physiol Behav. 1972;9(2):199‐202. [DOI] [PubMed] [Google Scholar]
- 99. Maljaars J, Romeyn EA, Haddeman E, Peters HP, Masclee AA. Effect of fat saturation on satiety, hormone release, and food intake. Am J Clin Nutr. 2009;89(4):1019‐1024. [DOI] [PubMed] [Google Scholar]
- 100. Yeomans MR. Flavour‐nutrient learning in humans: an elusive phenomenon? Physiol Behav. 2012;106(3):345‐355. [DOI] [PubMed] [Google Scholar]
- 101. Lucas F, Sclafani A. Flavor preferences conditioned by intragastric fat infusions in rats. Physiol Behav. 1989;46(3):403‐412. [DOI] [PubMed] [Google Scholar]
- 102. Maske CB, Loney GC, Lilly N, Terrill SJ, Williams DL. Intragastric nutrient infusion reduces motivation for food in male and female rats. Am J Physiol Endocrinol Metab. 2018;315(1):E81‐E90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Blundell JE, Burley VJ, Cotton JR, Lawton CL. Dietary fat and the control of energy intake: evaluating the effects of fat on meal size and postmeal satiety. Am J Clin Nutr. 1993;57(5 Suppl):772S‐778S; discussion 777S‐778S. [DOI] [PubMed] [Google Scholar]
- 104. Iwasaki Y, Shimomura K, Kohno D, et al. Insulin activates vagal afferent neurons including those innervating pancreas via insulin cascade and Ca(2+) influx: its dysfunction in IRS2‐KO mice with hyperphagic obesity. PLoS One. 2013;8(6):e67198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Louis‐Sylvestre J, Giachetti I, Le Magnen J. Vagotomy abolishes the differential palatability of food. Appetite. 1983;4(4):295‐299. [DOI] [PubMed] [Google Scholar]
- 106. Breen DM, Rasmussen BA, Côté CD, Jackson VM, Lam TK. Nutrient‐sensing mechanisms in the gut as therapeutic targets for diabetes. Diabetes. 2013;62(9):3005‐3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Blouet C, Schwartz GJ. Duodenal lipid sensing activates vagal afferents to regulate non‐shivering brown fat thermogenesis in rats. PLoS One. 2012;7(12):e51898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Madden CJ, Santos da Conceicao EP, Morrison SF. Vagal afferent activation decreases brown adipose tissue (BAT) sympathetic nerve activity and BAT thermogenesis. Temperature (Austin). 2017;4(1):89‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu C, Bookout AL, Lee S, et al. PPARgamma in vagal neurons regulates high‐fat diet induced thermogenesis. Cell Metab. 2014;19(4):722‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Suarez AN, Hsu TM, Liu CM, et al. Gut vagal sensory signaling regulates hippocampus function through multi‐order pathways. Nat Commun. 2018;9(1):2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Singh A, Zapata RC, Pezeshki A, Workentine ML, Chelikani PK. Host genetics and diet composition interact to modulate gut microbiota and predisposition to metabolic syndrome in spontaneously hypertensive stroke‐prone rats. Faseb J. 2019;33(6):6748‐6766. [DOI] [PubMed] [Google Scholar]