

Abstract
Background.
The acute respiratory distress syndrome (ARDS) is a lung inflammatory process mainly caused by sepsis. Most previous studies that identified genetic risks for ARDS were focused on biological candidates. We aimed to identify novel genetic variants associated with ARDS susceptibility and to provide complementary functional evidence.
Methods.
We conducted a case-control genome-wide association study (GWAS) in 1,935 European subjects, using sepsis-associated ARDS patients as cases and sepsis patients without ARDS as controls. The discovery stage included 672 patients admitted into a network of Spanish intensive care units. The replication stage comprised 1,345 individuals from two independent datasets involving the MESSI cohort study (U.S.A.) and the VISEP/MAXSEP trials of the SepNet study (Germany). We used RNAseq-based gene expression data from lung biopsies, in silico analyses, and luciferase reporter assays to assess functionality.
Findings.
We identified a novel genome-wide significant association with sepsis-associated ARDS susceptibility (rs9508032, odds ratio [OR]=0·61 [95% CI=0·41-0·91], p-value=5·18×10−8) located within the Fms Related Tyrosine Kinase 1 (FLT1) gene encoding the vascular endothelial growth factor (VEGF) receptor 1 (VEGFR-1). The region containing the sentinel variant and its best proxies acted as a silencer for FLT1 promoter, and alleles with protective effects in ARDS further reduced promoter activity (p=4·66×10−3). A literature mining of all previously described ARDS genes validated the association of VEGFA (p=4·69×10−5; OR=0·55 [95%CI = 0·41-0·73]).
Interpretation.
A common variant within FLT1 gene is associated with sepsis-associated ARDS. Our findings support the central role of VEGF signaling pathway in ARDS pathogenesis and provides a potential therapeutic target.
Introduction
Acute respiratory distress syndrome (ARDS) is a serious complication of sepsis of pulmonary or non-pulmonary origin.1 This syndrome is defined as an acute inflammatory process of the lung caused by injury to the alveolar-capillary barrier, resulting in increased alveolar-capillary permeability and protein-rich pulmonary edema. This leads to severe hypoxemia (assessed by PaO2/FiO2 ratio), bilateral pulmonary infiltrates, and decreased lung compliance.
The annual incidence of ARDS ranges from five to 80 cases per 100,000 individuals,2,3 with an overall hospital mortality of approximately 40%.4 In fact, ARDS is a cause of morbidity and mortality in adult intensive care units (ICUs) worldwide. Survivors often develop physical and cognitive impairments, including neuropsychiatric disorders, that diminish their quality of life.5,6 At present, there are no available methods to treat or rapidly rehabilitate the lungs of affected patients. Effective therapeutic options remain elusive, likely due to the heterogeneity of the syndrome. Currently, the only available interventions that impact patient survival involve specific strategies for mechanical ventilation (MV) and patient position to minimize ventilator-induced lung injury.7,8
Given the limited therapeutic options, there is a strong interest in identifying genetic factors that modify ARDS risks and which may serve as potential therapeutic targets. Several studies have reviewed the implication of genetic factors in ARDS susceptibility and outcomes.9 Overall, most genetic studies have focused on biologically motivated candidate genes mainly involved in the immune response, vascular permeability and metabolism.9 In addition, two small whole-exome sequencing studies in ARDS patients revealed that the MYLK gene was associated with ARDS severity as measured by ventilator-free days,10 and that three other genes (ARSD, XKR3, and ZNF335) were associated with ARDS susceptibility, severity and mortality outcomes.11
Two genome-wide association studies (GWAS) of ARDS have been published to date, one used trauma-associated ARDS cases of European ancestry12 and the other used all-cause ARDS African-American cases.13 These studies revealed two potential ARDS genes, PPFIA1 and SELPLG, although the reported variants both failed to reach genome-wide significance. Despite the marginal associations, prior GWAS results were paired with functional analyses either based on expression quantitative trait loci (eQTL) or on animal models to reinforce the role of prioritized genes in ARDS susceptibility.
Nonetheless, the genetics of ARDS susceptibility remains largely elusive. Thus, further studies on a genomic scale and larger sample sizes are needed. To our knowledge, here we performed the first GWAS of susceptibility to ARDS in 1,935 individuals of European ancestry, using sepsis-associated ARDS patients as cases and sepsis patients without ARDS as controls. With the hypothesis that frequent genetic variants in the population associate with disease risk, we aimed to identify genetic variants associated with ARDS susceptibility and provide complementary functional evidence using in silico analyses, gene expression data, and luciferase reporter assays.
Methods
Study design and sample description
We performed a case-control GWAS of ARDS in sepsis patients of European ancestry. A discovery stage was designed to prioritize variants based on their suggestive association. At the conclusion of this stage (during November of 2017), investigators from two independent cohorts were contacted and their data were used to validate the associations in the replication stage. Finally, a meta-analysis combining the discovery and replication association results was performed during September of 2018 to identify variants significantly associated with ARDS.
The GEN-SEP cohort was used in the discovery stage (Figure 1 and Appendix). It consisted of 672 unrelated adult patients with sepsis14 who were followed for the development of ARDS according to the Berlin definition criteria.15 Controls constituted patients with all-cause sepsis who did not develop ARDS during their ICU stay. DNA was extracted from peripheral blood of all patients (Appendix).
Figure 1. Flow chart of quality control steps for the samples and genotyped SNPs in the discovery and replication stages.
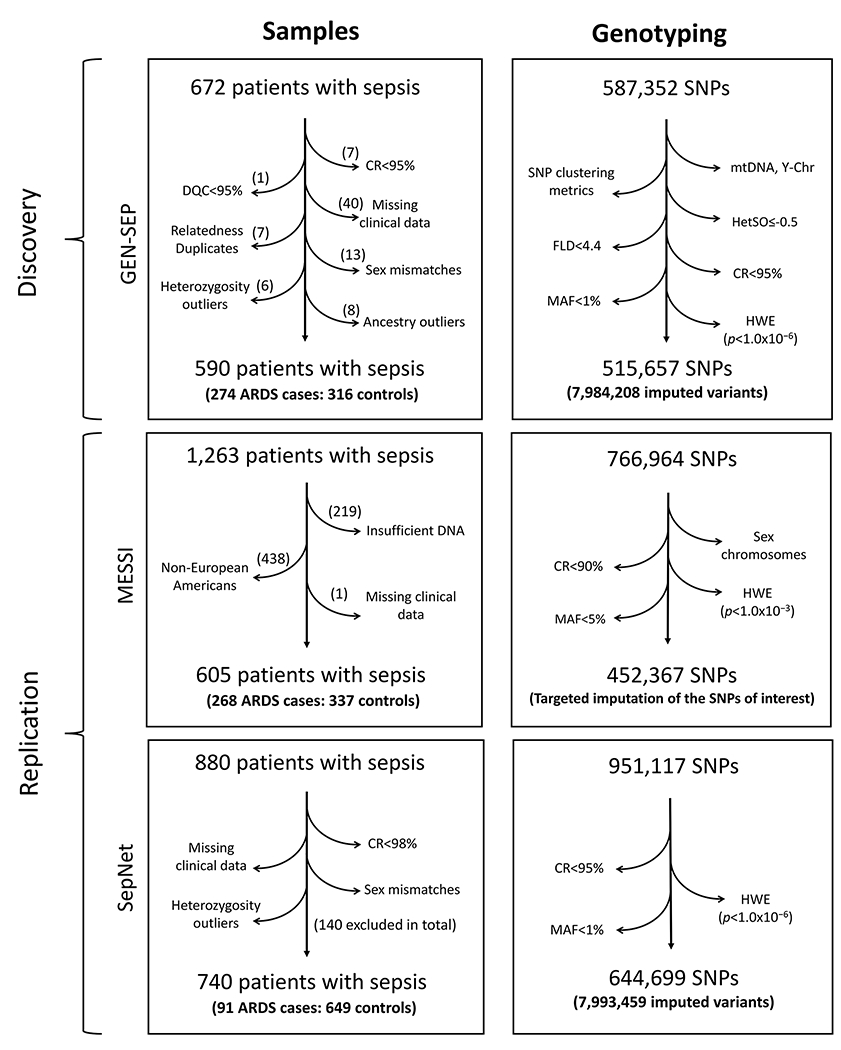
CR, Call rate; DQC, Affymetrix dish quality control; FLD, Fisher’s Linear Discriminant; HetSO, Heterozygous Cluster Strength Offset; HWE, Hardy-Weinberg Equilibrium; MAF, minor allele frequency; mtDNA, mitochondrial DNA; Y-chr, Y chromosome.
The replication stage was conducted on two independent datasets from European-ancestry ICU patients, where sepsis-associated ARDS were used as cases and sepsis without ARDS were considered as at-risk controls (Figure 1). The first dataset was derived from 605 patients (268 cases and 337 controls) out of 1,263 patients of multiple ancestries from the “Molecular Epidemiology of Sepsis in the ICU” (MESSI) cohort study (U.S.A.). The second dataset was obtained from 740 patients (91 cases and 649 controls) out of 880 patients from the VISEP and MAXSEP trials of the SepNet study group (Germany). Patients in both studies meet the Berlin definition criteria for ARDS.15 These datasets, thereafter referred to as MESSI and SepNet datasets, have been previously described.16,17
Genotyping and statistical analyses
For the discovery stage, a total of 587,352 SNPs were genotyped using the Axiom Genome-Wide Human CEU 1 Array (Affymetrix). Additionally, a principal component (PC) analysis (PCA) was conducted to reduce the effects of population stratification in the analysis (Appendix and Supplementary Figure 1). SNPs were genotyped using the Affymetrix Axiom TxArray v.1 (Affymetrix) in the MESSI study, while HumanOmniExpressExome arrays (Illumina, Inc.) were used in the SepNet study. Genotyping procedures are detailed in the Appendix.
After variant imputation in GEN-SEP data, logistic regression models assumed an additive inheritance. Sex, age, and the Acute Physiology and Chronic Health Evaluation II (APACHE II) score were included as covariates to address potential bias. Variants with low allele frequency (MAF<1%) or with a low imputation quality (Rsq<0·3) were excluded from the analysis. Details of imputation and association procedures are described in Appendix. Independent variants showing a p<5·0×10−5 were followed up in the replication stage.
In the MESSI study, logistic regression models were performed assuming additive inheritance considering the first two PCs, age, and sex as covariates. For the SepNet study, logistic regressions were performed including the first three PCs, sex, age, and APACHE II as covariates. Meta-analysis was performed on the results of these two studies. For variants that showed a nominal association (p<0·05) with ARDS susceptibility in the replication stage, a meta-analysis including discovery and replication stages was also performed. Genome-wide significance was declared with a meta-analysed significance of p<9·26×10−8 according to the most recent empirical estimations in European populations.18
FLT1 and VEGFA gene expression and functional annotation of genetic variants
In silico and in vitro approaches were used to investigate potential biological consequences of variants associated with ARDS. First, FLT1 and VEGFA expressions were assessed in nine lung biopsies from healthy individuals by means of RNA-sequencing (Appendix). In parallel, we accessed public gene expression data (GSE32707) from 88 critically-ill adult patients that were evaluated for sepsis and ARDS (Appendix). Next, to highlight the functional role of the associated variant and of SNPs that were LD proxies in Europeans (r2=1·0), we applied several in silico tools for variant prioritization [DeepSea, DSNetwork, Open Targets Genetics] and to predict potential regulatory genomic regions including epigenetic modifications [DeepSea, HaploReg, RegulomeDB], long-distance physical interactions [Capture Hi-C Plotter (CHiCP)], and tissue specific local expression quantitative trait loci (cis eQTLs) [GTEx, TIVAN]. Additional tools [VEP, SNPdelScore] were used to predict the likelihood of deleteriousness of each SNP. See Appendix for further details.
Dual-luciferase reporter assays
The potential regulatory effect of the ARDS-associated variant on promoter activity was investigated using a Dual-Luciferase Reporter Assay System® (Promega, Madison, WI). Experiments were performed using human lung epithelial (A549) and peripheral blood monocyte (THP-1) cell lines, both known to have an active FLT1 promoter activity and expressing VEGFR-1.19 Two types of constructs were generated: 1) a reporter construct including a fragment of the FLT1 promoter inserted into a promoterless pGL4.10 [luc2] luciferase reporter vector, and 2) two regulatory constructs including a region of intron 10 containing either the reference or alternative alleles of the most significantly associated variant within FLT1 and its perfect LD proxies, which were inserted into the reporter construct. Promoter activities were expressed as a relative response ratio of Firefly luciferase/Renilla luciferase signals. See Appendix for further details.
Literature mining of previously reported ARDS-associated genes
A literature search for all studies reporting genes which were significantly associated with ARDS was conducted. Association results in the discovery stage were extracted and an effective number of independent signals per gene was measured in order to adjust for multiple testing. See Appendix for further details.
Role of the funding source
The funders had no role in the study design, data collection, analysis, interpretation of data, decision to publish, or preparation of the manuscript. CF was involved in all stages of study development and delivery, had full access to all data in the study, and had final responsibility for the decision to submit for publication.
Results
GWAS of sepsis-associated ARDS
After filtering steps and the quality control, a total of 515,657 SNPs from 590 patients (274 sepsis-associated ARDS cases and 316 controls with sepsis) were used for the discovery stage (Figure 1). Demographic and clinical features of these patients are shown in Table 1. Genotype imputation on the HRC r.1.1 allowed us to perform the association testing of this stage on 7·98 million variants with MAF≥1%. The genomic inflation factor (λ=0·98) did not show signs of inflation of the results (Supplementary Figure 2). Suggestive associations (p<5·0×10−5) were detected for 229 variants residing in 53 independent loci (lowest p=2·6×10−7) (Figure 2, Supplementary Table 1).
Table 1.
Demographic and clinical features of the GEN-SEP study.
| Controls (n=316) | Cases (n=274) | p-value† | |
|---|---|---|---|
| Sex (n males/N) | 197/316 (62·3%) | 194/274 (70·8%) | 0·04 |
| Mean age (years)* | 63·0 ± 15·0 | 62·5 ± 14·1 | 0·47 |
| Hypertension (n/N) | 60/144 (41·7%) | 73/160 (45·6%) | 0·56 |
| Smokers (n/N) | 61/188 (32·4%) | 59/175 (33·7%) | 0·88 |
| Previous surgery (%) | 32/127 (25·2%) | 35/137 (25·5%) | 1·00 |
| Ischemic cardiac disease (n/N) | 31/285 (10·9%) | 19/210 (9·0%) | 0·61 |
| Pulmonary sepsis (n/N) | 83/267 (31·1%) | 128/252 (50·8%) | 7·5×10−6 |
| APACHE II (median) (P25–P75)* | 20 (15-24) | 22 (18-27) | 2·2×10−5 |
| ICU mortality (n/N) | 79/310 (25·5%) | 115/268 (42·9%) | 1·5×10−5 |
| Pathogen (n/N) | |||
| Gram-positive | 48/178 (27·0%) | 58/162 (35·8%) | 0·09 |
| Gram-negative | 74/178 (41·6%) | 59/162 (36·4%) | 0·41 |
| Gram-positive and Gram-negative | 26/178 (14·6%) | 17/162 (10·5%) | 0·34 |
| Fungi | 5/178 (2·8%) | 3/162 (1·9%) | 0·83 |
| Virus | 2/178 (1·1%) | 9/162 (5·6%) | 0·04 |
| Polymicrobial | 16/178 (9·0%) | 11/162 (6·8%) | 0·45 |
| Organ dysfunction (n/N) | |||
| Circulatory | 232/270 (85·9%) | 238/255 (93·3%) | 0·01 |
| Coagulation | 62/270 (23·0%) | 68/255 (26·7%) | 0·38 |
| Hepatic | 48/269 (17·8%) | 41/255 (16·1%) | 0·67 |
| Neurologic | 54/270 (20·0%) | 59/254 (23·2%) | 0·43 |
| Renal | 124/316 (39·2%) | 108/274 (39·4%) | 1·00 |
n=number of individuals with data available, N=group size.
All individuals have age and APACHE II data. Percentages refer only to the individuals with available data for each clinical feature.
Mean age and APACHE II comparisons were conducted by the Wilcoxon signed-rank test; the other variables were compared by a chi-square test. APACHE II, Acute Physiology and Chronic Health Evaluation II; ARDS, acute respiratory distress syndrome; ICU, intensive care unit; P25, percentile 25; P75, percentile 75.
Figure 2. Manhattan plot of GWAS results for the discovery stage.
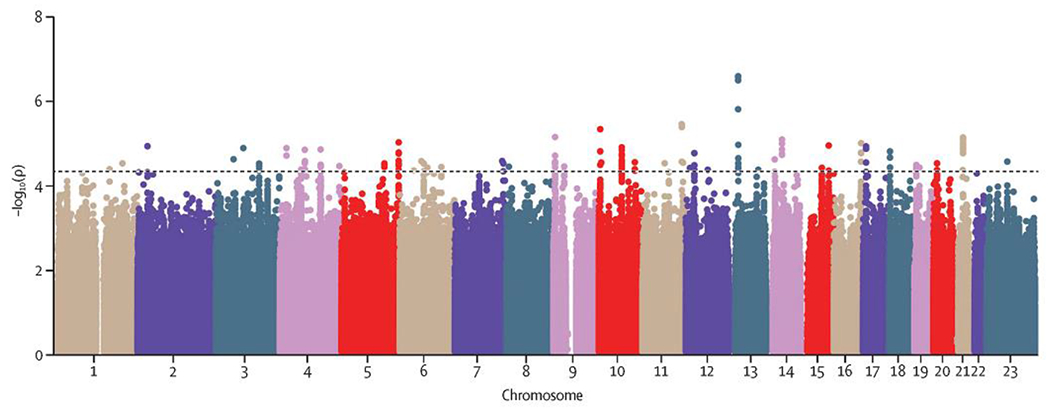
Axes display the -log10 transformed p-values by position in each chromosome. The horizontal line indicates the threshold considered for prioritizing variants for the replication stage (p=5·0×10−5).
The replication stage in a total of 359 patients with sepsis-associated ARDS and 986 controls with sepsis focused on the sentinel variants (variants with the smallest p-values) of 52 autosomal loci (Figure 1, Supplementary Table 2). Because of the difficulties in accessing data, we did not follow up the X chromosome variants in the replication stage. Association testing in the replication stage revealed four SNPs that were nominally significant (uncorrected p<0·05; Table 2), although none of them was significantly associated with ARDS susceptibility after a Bonferroni correction (threshold p=9·62×10−4). The first signal is an intronic variant (rs9508032) of the FLT1 gene (Figure 3), encoding the transmembrane receptor known as the VEGFR-1. The other three SNPs were located intergenic (rs11195238) to the genes encoding the structural maintenance of chromosomes 3 (SMC3) and the RNA binding motif protein 20 (RBM20); intergenic (rs8001184) to the genes encoding slit and neurotrophic tyrosine kinase (NTRK) like family member 5 (SLITRK5) and glypican 5 (GPC5); and in intron one (rs2734600) of the gene encoding serine protease 3 (PRSS3). Meta-analysis of results from the discovery and replication stages for these four SNPs revealed that the sentinel variant rs9508032, located intronic to the FLT1 gene, was the only SNP that reached genome-wide significance (Table 2). The FLT1 variant showed consistent direction of effects, with an odds ratio (OR) for the T allele of 0·61 (95% confidence interval (CI) = 0·41-0·91), and a p-value of 5·18×10−8. A sensitivity analysis of the association of rs9508032 at FLT1 supported that the association was robust to adjustment for comorbidities, isolated pathogen, and disease severity (Supplementary Table 3), though clinical data was missing for a significant proportion of subjects for some variables (up to 55%). The rs9508032-ARDS association demonstrated similar effect sizes and directions even when the sample size was significantly reduced due to missing clinical data. Furthermore, there were 16 additional variants residing in FLT1 among the 226 SNPs with suggestive associations in the discovery stage (Supplementary Table 4). All but one was nominally significant in the replication stage, and five achieved genome-wide significance after meta-analysis of discovery and replication stages. In ad hoc analyses, we evaluated if the association of the sentinel variant persisted when unselected population controls were used instead of the at-risk controls. Based on the genotypes from 927 unrelated Spanish individuals that were genotyped with the same array in previous studies,20,21 results also supported a significant association of rs9508032 with ARDS (OR= 0·73, 95% CI= 0·58-0·90, p-value=3·86×10−3). We also evaluated if the sentinel variant predicted ICU mortality. However, our results indicated that it did not predict ICU survival among sepsis or ARDS patients from the GEN-SEP cohort (Supplementary Table 5). This evidence further supports that the FLT1 association with sepsis-associated ARDS was genuine. Finally, at this stage, we assessed if the sentinel variant (and perfect LD proxies) of the FLT1 also associated with ARDS after trauma, but none of them was present in the GWAS of Christie and colleagues (Appendix).12
Table 2.
Results for the SNPs that were nominally significant in the replication stage.
| Discovery (274:316)* | Replication (359:986)* |
Meta-analysis (633:1,302)* |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Chr | Position | Gene | A1/A2 | MAF | OR [95% CI] | p-value | MAF† | OR [95% CI] | p-value | OR [95% CI] | p-value |
| rs9508032 | 13 | 28995940 | FLT1 | T/C | 0·29 | 0·49 [0·38, 0·65] | 2·62×10−7 | 0·25 | 0·74 [0·60, 0·92] | 5·98×10−3 | 0·61 [0·41, 0·91] | 5·18×10−8 |
| rs11195238 | 10 | 112388857 | SMC3-RBM20 | T/C | 0·15 | 0·47 [0·33, 0·67] | 2·97×10−5 | 0·13 | 0·69 [0·52, 0·91] | 9·55×10−3 | 0·60 [0·48, 0·74] | 3·73·10−6 |
| rs2734600 | 9 | 33753355 | PRSS3 | T/C | 0·15 | 0·48 [0·33, 0·68] | 3·76×10−5 | 0·12 | 0·85 [0·36, 1·97] | 0·02 | 0·52 [0·37, 0·72] | 7·76×10−5 |
| rs8001184 | 13 | 90603540 | SLITRK5-GPC5 | A/C | 0·48 | 1·65 [1·30, 2·10] | 4·48×10−5 | 0·49 | 1·26 [1·04, 1·52] | 0·02 | 1·40 [1·20, 1·62] | 1·19×10−5 |
Cases:Controls;
For rs9508032, MAF was 0·27 in MESSI and 0·22 in SepNet; For rs11195238, MAF was 0·14 in MESSI and 0·12 in SepNet. Fort the other two SNPs, MAFs were identical in the two replication cohorts. A1, Effect allele; A2, Non-effect allele; CI, Confidence Interval; MAF, Minor Allele Frequency; OR, Odd Ratio. Top hit is indicated in bold. Alleles referred to the positive strand of hg19. The imputation quality (Rsq/INFO) for the four variants ranged from 0·93 to 1·00 in all studies.
Figure 3. Regional plot of association results for the genome-wide significant locus.
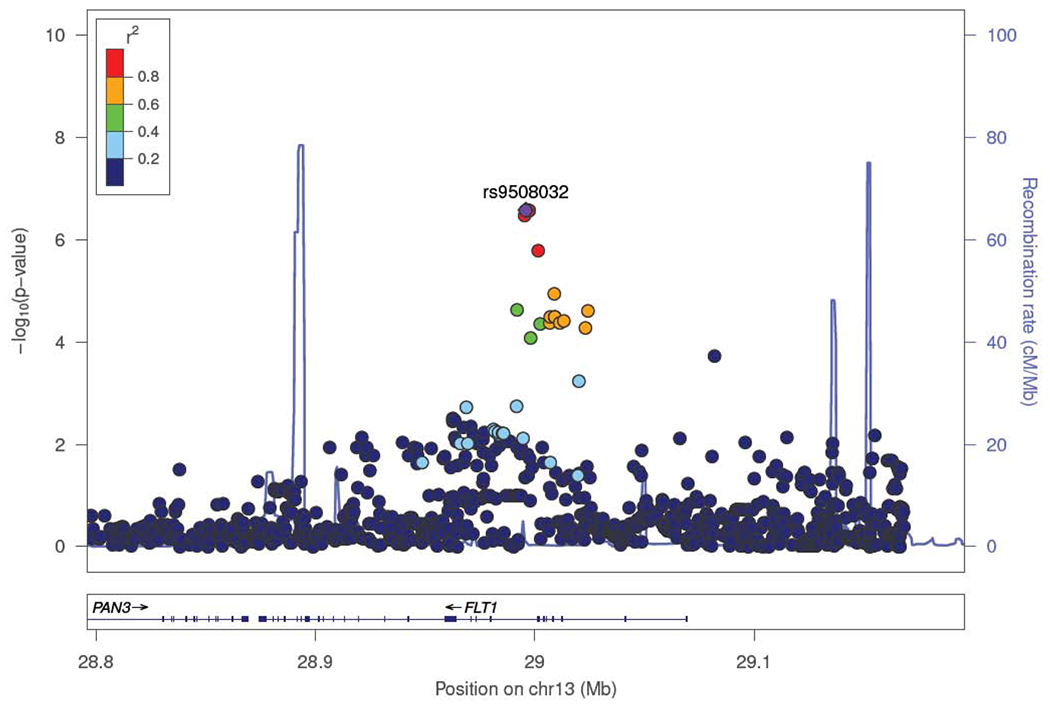
The -log10 transformed p-values for association tests are plotted by position. The SNP rs number indicated on the plot denotes the sentinel SNP. The remaining SNPs are color coded to reflect their degree of linkage disequilibrium with the indicated SNP based on pairwise r2 values from the European population from The 1000 Genomes Project. Estimated recombination rates (light blue line) are plotted on the right y-axis.
Gene expression and functional impact predictions at variant sites
Transcriptomic data from lung biopsies obtained from non-ARDS control subjects revealed a high expression of FLT1 (9,977 counts per million on average ± 5,228) and VEGFA (19,221 counts per million on average ± 16,165) in lung tissues, which is in agreement with GTEx information supporting a prominent expression of these genes in the lung. Among the eight FLT1 isoforms that were evaluated on the RNAseq dataset, the canonical isoform encoding a membrane-spanning protein (FLT1-201, ENST00000282397) and the next one in size (FLT1-207, ENST00000615840), which encodes a secreted VEGF-binding protein of 687 amino acids,22 accumulated more than 10 times more reads on average than the rest of the gene isoforms (Supplementary Table 6). Among the 29 VEGFA isoforms we assessed, those that had higher expression in the lungs were VEGFA-205 (ENST00000372067), VEGFA-229 (ENST00000621747), VEGFA-227 (ENST00000615393), VEGFA-222 (ENST00000520948), VEGFA-206 (ENST00000372077), VEGFA-212 (ENST00000480614), and VEGFA-215 (ENST00000497139) (Supplementary Table 6). We also accessed array expression data from peripheral blood obtained from a cohort of critically-ill patients that included donors with sepsis, with and without ARDS, as well as non-sepsis patients. These data strongly supported that the mean FLT1 expression level in peripheral blood varied significantly among patient groups (ANOVA, p=0·002), with a higher average FLT1 gene expression among ARDS patients than in ICU controls without sepsis or systemic inflammatory response syndrome (t-test, p=0·001) (Supplementary Figure 3). On the contrary, the expression levels for the three available probes of VEGFA did not vary significantly among ICU patient groups (ANOVA; ILMN_2375879, p=0·638; ILMN_1693060, p=0·435; and ILMN_1803882, p=0·214) (Supplementary Figure 3). Next, we performed an in silico bioinformatic approach to explore the functional features of rs9508032 and the other five variants of FLT1 that reached genome-wide significance after meta-analysis (Supplementary Table 4). Relevant functional information was found for rs9508032 and two of its proxies (rs722503 and rs8002446), all of them from intron 10, as these three SNPs were located in enhancer and promoter histone marks, in DNase I hypersensitive sites (DHS) of many cell types, and were related to the alteration of regulatory motifs (Supplementary Table 7). Additionally, rs722503 and rs8002446 have predicted effects on transcription factor binding. The algorithmic framework of DeepSEA predicted a significant functional effect for rs722503 (p=0·045). DSNetwork predicted similar results where rs722503 was prioritized as the best candidate variant for further functional analysis in this region. In contrast, Open Targets Genetics prioritized rs8002446 as potentially functional based on information of DHS and enhancer- transcription start sites data. Using GTEx, no significant eQTLs were identified for rs9508032 or its proxies, although we did observe that both rs9508032 and rs722503 had high CellulAr dePendent dEactivating (CAPE) scores for eQTL and DNase I QTLs in human umbilical vein endothelial cells, fibroblasts, epithelial and immune (monocyte) cells (Supplementary Table 7). Using the CHiCP to visualize capture Hi-C experiments conducted by Mifsud and colleagues,23 we observed the existence of physical interactions between the region containing the three variants and the FLT1 promoter region in a lymphoblastoid cell line (GM12878).
In vitro luciferase assays
Based on the above evidence, we then performed luciferase promoter assays to assess the effect of the intron 10 region containing the genome-wide significance SNPs on FLT1 promoter activity (Figure 4A). Our results showed that the FLT1 intron 10 region containing these variants repressed gene promoter activity with a consistent effect on both peripheral blood monocytes (65·1 ± 10·7% reduction) and human lung epithelial cells (48·7 ± 4·1% reduction) (Wilcoxon test, p=4·10×10−4 and p=0·02, respectively) (Figure 4B). When we compared the constructs with reference vs. alternative alleles for all positions within intron 10 of FLT1, we found that the presence of alternative alleles (protective for ARDS) were associated with a further decrease (48·6 ± 7·2% reduction) of the FLT1 promoter activity in peripheral blood monocytes (Wilcoxon test, p=4·66×10−3) (Figure 4C). No significant reduction of the FLT1 promoter activity was found for pneumocytes (Wilcoxon test, p=0·89).
Figure 4. Luciferase reporter assay to assess the role of intron 10 and of rs9508032 and its perfect LD proxies on FLT1 promoter activity.
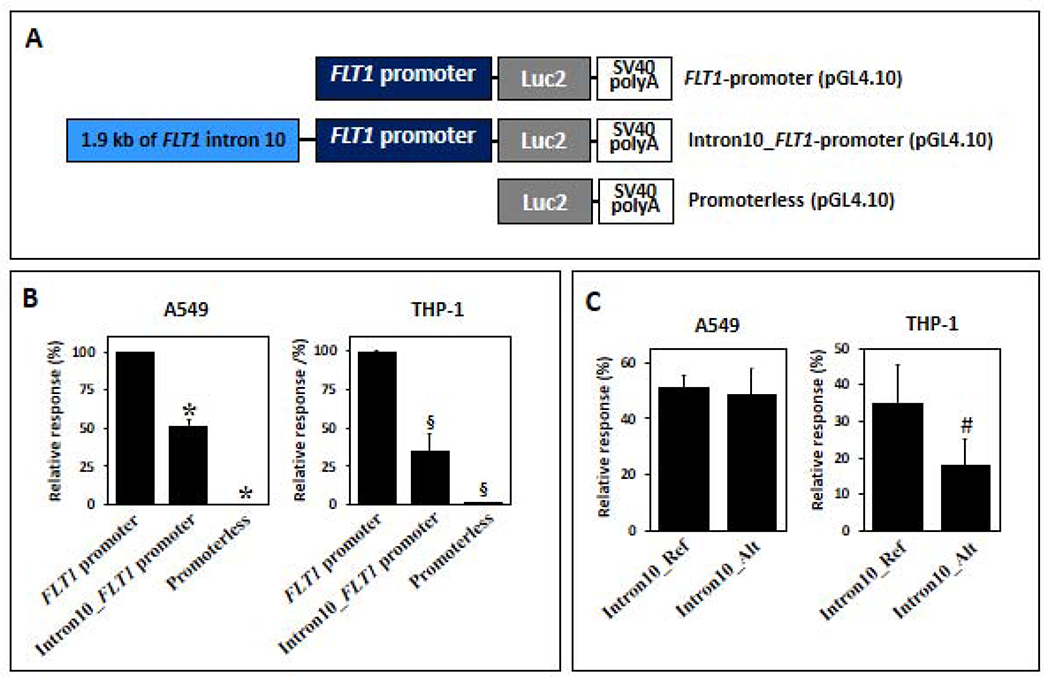
A) Scheme of vector constructs. B) Experimental data showing that the intron 10 fragment harboring the reference alleles suppresses the FLT1 promoter activity in A549 and THP-1 cells. C) Experimental data showing that the intron 10 fragment harboring the alternative alleles further decreased the FLT1 promoter activity, showing a significant difference in THP-1 cells. Significance was assessed by Wilcoxon signed-rank tests (*p<0·05, #p<0·005, §p<0·0005). Ref and Alt indicate risk and protective alleles, respectively.
Association of previously reported ARDS genes
Finally, we performed a thorough literature mining on genes previously associated with ARDS in our discovery stage. Results of our search merged with previous reviews identified 96 genes with prior reported association with ARDS susceptibility or outcome (Supplementary Table 8). Although none of the 96 genes surpassed a study-wise Bonferroni-corrected threshold in the discovery (p=2·18×10−6), the VEGFA gene reached a gene-wise significance after Bonferroni correction in the discovery study (top signal: OR= 0·55, 95% CI = 0·41-0·73; p=4·69×10−5) (Supplementary Table 8). Not surprisingly, VEGF-A is one of the main ligands of VEGFR-1.19
Discussion
To our knowledge, here we reported the results of the first GWAS of sepsis-associated ARDS completed to date, where we identified a locus located in FLT1 associated with ARDS that reached genome-wide significance in a combined meta-analysis of all cohorts. Of note, the sentinel SNP of FLT1 (rs9508032) and the perfect LD proxies were all located in close proximity within intron 10, a region which we observed acting as a silencer of the FLT1 promoter activity in monocyte and human lung epithelial cell lines. In conjunction with human transcriptomics data, we also determined that different FLT1 isoforms are expressed in lung tissues, and that its expression in peripheral blood is positively correlated with the severity of illness, with the highest levels detected in ARDS patients. Evidence from our studies suggested a possible functional role of the sentinel SNP (rs9508032) and two of its perfect LD proxies in Europeans. Findings also revealed allelic effects of intron 10 on the FLT1 promoter activity, which was particularly significant on monocytes. Interestingly, FLT1 and other nearby genes (FLT3 and PAN3) were strongly associated with monocyte counts in the UK Biobank.24 All these findings reinforce the concept that monocytes are also crucial in the VEGF-mediated lung response.19 Variants from FLT1 had never been associated with ARDS susceptibility or outcomes in previous independent studies, although Kim and colleagues25 have reported the association of FLT1 with all-cause pulmonary complications. Additionally, there is evidence of association of FLT1 with other complex diseases, such as coronary arterial disease26 and preeclampsia,27,28 where the endothelium plays an important pathophysiological role.
FLT1 encodes VEGFR-1, a tyrosine-protein kinase that acts as a transmembrane receptor of VEGF-A, other VEGF family members, and the placental growth factor (PLGF). VEGF was originally identified as a vascular permeability factor,19 although it has diverse and pleiotropic activities beyond the regulation of the alveolar-capillary barrier.29 VEGF has been involved in the fibroproliferative phase of ARDS30, as well as in resolution of ventilator-associated pneumonia.31 However, Ware and colleagues found that levels of VEGF were similar in undiluted edema fluid from hydrostatic and ARDS patients.32 Although its role remains unclear, abundant evidence supports a negative regulatory role of an alternatively spliced soluble form of VEGFR-1 (sFLT-1) sequestering part of VEGF bioactivity.22 High levels of sFLT-1 in the alveolar space are associated in humans with the occurrence of late ARDS in trauma,33 as well as with sepsis severity, organ dysfunction, and ICU survival.34,35,36 In parallel, we have found that FLT1 expression varied between ARDS and other ICU patients in peripheral blood, while VEGFA expression did not show differences. Taken together, this suggests that disease-related VEGF bioavailability could be dependent on the receptor isoforms. Interestingly, the array-based transcriptomics experiment specifically targeted exon 30 of FLT1 (Supplementary Figure 3), which critically involves the canonical receptor (FLT1-201), one of the few highly expressed isoforms. These observations offer a potential mechanistic link between the GWAS results and ARDS pathophysiology, suggesting that the FLT1 SNPs could be linked to the expression of the VEGFR-1 transmembrane isoform. The decrease of FLT1 promoter activity in vitro in the presence of intron 10 alleles associated with ARDS protection may translate in a reduction of the canonical VEGFR-1 expression and, thus, in a decrease of VEGF signalling. This hypothesis reconciles with the attenuation of many of the VEGF-mediated pathophysiologic effects in ARDS, including the formation of pulmonary edema. However, given the limitations to distinguish expression levels from gene isoforms in array-based transcriptomics experiments and that the cell type(s) that mechanistically link FLT1 SNPs with the ARDS pathophysiology remains unknown, this scenario is purely speculative.
Despite the central role of VEGF in ARDS and the availability of VEGF-targeting drugs, clinical trials using drugs directed towards VEGF pathways for ARDS patients are scarce. There is one entry of clinical trial of the efficacy of bevacizumab (anti-VEGF antibody) to prevent sepsis-associated ARDS (ClinicalTrials.gov identifier: NCT01314066). However, it was withdrawn without a single patient enrolled due to a lack of funding. A search in DrugBank37 and additional in silico explorations in Gene2drug38 allowed us to systematically identify available drugs targeting this pathway. Although most of them are currently in use for cancer treatment (none of them under evaluation in ARDS patients), nintedanib constitutes one of the few effective antifibrotic therapies as it targets VEGFR-1 and slows the rate of forced vital capacity decline of idiopathic pulmonary fibrosis.39 In addition, the antifungal drug itraconazole is known to inhibit the glycosylation of VEGFR-1 and VEGFR-2, affecting their migration pattern and signaling activity.40 Based on this and our findings, nintedanib and itraconazole potentially might be repurposed as ARDS drugs and warrant further investigation.
We acknowledge there are strengths and limitations of our study. The main strength is that, to our knowledge, our study is the first GWAS of sepsis-associated ARDS, a complex acute syndrome with a high morbidity and mortality in ICUs worldwide. We contrast our ARDS cases with similarly well-characterized critically ill sepsis patients that did not develop ARDS to address the heterogeneity of the syndrome. This approach allowed us to identify reproducible associations at one locus. We provide strong evidence (transcriptomics data, functional annotations and in vitro experiments) to sustain a functional implication of FLT1 variants in ARDS physiopathology. However, this study also has some limitations. The main weakness is the small sample size overall, limiting the power for detecting variants of smaller effects or of lower frequency. The limited sample size can be attributed to the low incidence and high heterogeneity of the syndrome, which makes sample collection difficult and slow. In this respect, it is plausible that rare variants in or near the identified regions remain undetected because of technological limitations. Whole-exome and genome sequencing analyses would offer better resolution to achieve that aim. Therefore, more ARDS loci are to be expected as the genomic studies of ARDS increase in size and marker resolution. Additionally, this study focused only on European ancestry patients. Further studies are needed to identify whether FLT1 variation also impacts ARDS risk in non-European populations. We used the A549 cell line as a model for human alveolar epithelial cell, which inherently entails experimental limitations because of its cancerous nature. Further experiments should evaluate primary human alveolar type 2 cells to assess the impact of this choice in our observations. Finally, because the X chromosome is usually filtered out from most GWAS because it adds a level of difficulty to the analyses, we were unable to follow-up a variant in OPHN1 gene (encoding a Rho-GTPase-activating protein) in the replication stage.
In summary, we describe the results of a GWAS of sepsis-associated ARDS. We report one novel locus located in FLT1 involved in ARDS susceptibility. Based on these results and the accumulated evidence, this study provides an orthogonal demonstration of the genuine central role of VEGF signalling pathway in ARDS susceptibility and strongly favours that VEGFR-1 is a therapeutic target for preventing ARDS. Independent studies should aim to validate our findings, including independent association studies in non-sepsis ARDS patients.
Supplementary Material
Research in context.
Evidence before this study:
We conducted a literature search on PubMed for all studies reporting genes which were significantly associated with ARDS up to November 2018. Most previous genetic studies in ARDS have focused on biological candidate genes mainly involved in the immune response, vascular permeability, and metabolism. Two small whole-exome sequencing studies and two genome-wide association studies (GWAS) of ARDS have been published to date, although none of them focused exclusively on sepsis-associated ARDS.
Added value of this study:
To our knowledge, we report the results of the first GWAS of sepsis-associated ARDS susceptibility conducted on 1,935 European patients with sepsis. We reveal a novel protective genome-wide significant association with sepsis-associated ARDS within the Fms Related Tyrosine Kinase 1 (FLT1) gene, encoding the vascular endothelial growth factor receptor 1 (VEGFR-1). We also report that SNP alleles with protective effects in ARDS reduce FLT1 promoter activity. These findings reinforce the need to target VEGF signaling in ARDS pathogenesis, a pathway linked to vascular permeability and immune and inflammatory responses.
Implications of all the available evidence:
Our results support the central role of VEGF signaling in ARDS pathogenesis and suggest VEGFR-1 as a potential therapeutic target. There are effective drugs targeting this protein that are being used in other diseases and they could be potentially repurposed for ARDS.
Acknowledgments
We would like to thank all patients for their willingness to participate in the study. We thank all the investigators of GENetics of SEPsis-induced ARDS (GEN-SEP) Network and the nurses that assisted in the collection of donor samples. Similarly, we thank all members of the SepNet and MESSI study groups for their willingness to support data sharing. This study was funded by the Instituto de Salud Carlos III (CB06/06/1088; PI10/0393; PI14/00844; PI16/00049; PI16/00759; PI17/00610; FI17/00177) and co-financed by the European Regional Development Funds, “A way of making Europe” from the European Union; and by the agreement OA17/008 with Instituto Tecnológico y de Energías Renovables (ITER) to strengthen scientific and technological education, training, research, development and innovation in Genomics, Personalized Medicine and Biotechnology. BGG was supported by a fellowship from Agencia Canaria de Investigación, Innovación y Sociedad de la Información (TESIS2015010057) co-funded by European Social Fund. The genotyping service of GEN-SEP was carried out at CEGEN-PRB3-ISCIII; it is supported by grant PT17/0019, of the PE I+D+i 2013-2016, funded by ISCIII and ERDF. The SepNet GWAS project was supported by the Paul-Martini-Sepsis Research Group, funded by the Thuringian Ministry of Education, Science and Culture (ProExcellence; grant PE 108-2); the public funded Thuringian Foundation for Technology, Innovation and Research (STIFT) and the German Sepsis Society (GSS); the Jena Center of Sepsis Control and Care (CSCC), funded by the German Ministry of Education and Research (BMBF; 01 EO 1002, 01 EO 1502). The VISEP and MAXSEP trials from the SepNet Study Group had been supported by a BMBF grant (01 KI 0106) and by unrestricted grants from B. Braun, HemoCue, Novo Nordisk, Astra Zeneca GmbH, Wedel, Germany and Bayer Healthcare, Leverkusen, Germany. Finally, AS was supported by the BMBF (01 ZZ 1803C) within the SMITH (“Smart Medical Information Technology for Healthcare”) consortium. The MESSI cohort was funded by NIH HL137006 and HL137915 (Meyer) and by an American Thoracic Society Foundation award. Finally, we would like to thank the six anonymous reviewers for their critical advice to improve this manuscript.
Funding. Instituto de Salud Carlos III, European Regional Development Funds, Instituto Tecnológico y de Energías Renovables, Agencia Canaria de Investigatión, Innovatión y Sociedad de la Informatión, European Social Fund, Thuringian Ministry of Education, Science and Culture, the public funded Thuringian Foundation for Technology, Innovation and Research, the German Sepsis Society, German Ministry of Education and Research, NIH, and the American Thoracic Society Foundation.
Conflict of interests
JPR reports grants from NIH, during the conduct of the study. MK reports that his institution has granted patents (EP2592421, CN104204808A, EP2780719) and pending patent applications (US20140248631, JP2014533368, EP3239712, WO2017186842). MS receives funding from Pfizer Inc. for a project not related to this research. NJM reports grants from GlaxoSmithKline, Inc, advisory board membership from SOBI, Inc, and consulting fees from Competitive Drug Development International on behalf of Bayer, Inc, outside the submitted work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics committee approval
All participating studies were performed according to The Code of Ethics of the World Medical Association (Declaration of Helsinki), and informed consent was obtained from all subjects or their representatives. The Research Ethics Committees at participating centers approved this study.
References
- 1.Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994; 149: 818–24. [DOI] [PubMed] [Google Scholar]
- 2.Villar J, Blanco J, Añón JM, et al. The ALIEN study: incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med 2011; 37: 1932–41. [DOI] [PubMed] [Google Scholar]
- 3.Buregeya E, Fowler RA, Talmor DS, Twagirumugabe T, Kiviri W, Riviello ED. Acute respiratory distress syndrome in the global context. Glob Heart 2014; 9: 289–95. [DOI] [PubMed] [Google Scholar]
- 4.Bellani G, Laffey JG, Pham T, et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016; 315: 788–800. [DOI] [PubMed] [Google Scholar]
- 5.Spragg RG, Bernard GR, Checkley W, et al. Beyond mortality: future clinical research in acute lung injury. Am J Respir Crit Care Med 2010; 181: 1121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang M, Parker AM, Bienvenu OJ, et al. Psychiatric Symptoms in Acute Respiratory Distress Syndrome Survivors. Crit Care Med 2016; 44: 954–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acute Respiratory Distress Syndrome Network, Brower RG, Matthay MA, et al. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000; 342: 1301–8. [DOI] [PubMed] [Google Scholar]
- 8.Guérin C, Reignier J, Richard J-C, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013; 368: 2159–68. [DOI] [PubMed] [Google Scholar]
- 9.Guillén- Guío B, Acosta- Herrera M, Villar J, Flores C. Genetics of Acute Respiratory Distress Syndrome. eLS. John Wiley Sons 2016. Doi: 10.4046/trd.2001.51.1.5 [DOI] [Google Scholar]
- 10.Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012; 91: 224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shortt K, Chaudhary S, Grigoryev D, et al. Identification of novel single nucleotide polymorphisms associated with acute respiratory distress syndrome by exome-seq. PLoS One 2014; 9: e111953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christie JD, Wurfel MM, Feng R, et al. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS One 2012; 7: e28268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bime C, Pouladi N, Sammani S, et al. Genome-Wide Association Study in African Americans with Acute Respiratory Distress Syndrome Identifies the Selectin P Ligand Gene as a Risk Factor. Am J Respir Crit Care Med 2018; 197: 1421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016; 315: 801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012; 307: 2526–33. [DOI] [PubMed] [Google Scholar]
- 16.Scherag A, Schöneweck F, Kesselmeier M, et al. Genetic Factors of the Disease Course after Sepsis: A Genome-Wide Study for 28Day Mortality. EBioMedicine 2016; 12: 239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reilly JP, Wang F, Jones TK, et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med 2018; 44: 1849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanai M, Tanaka T, Okada Y. Empirical estimation of genome-wide significance thresholds based on the 1000 Genomes Project data set. J Hum Genet 2016; 61: 861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barratt S, Medford AR, Millar AB. Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respiration 2014; 87: 329–42. [DOI] [PubMed] [Google Scholar]
- 20.Barreto-Luis A, Pino-Yanes M, Corrales A, et al. Genome-wide association study in Spanish identifies ADAM metallopeptidase with thrombospondin type 1 motif, 9 (ADAMTS9), as a novel asthma susceptibility gene. J Allergy Clin Immunol 2016; 137:964–6. [DOI] [PubMed] [Google Scholar]
- 21.Guillen-Guio B, Lorenzo-Salazar JM, González-Montelongo R, et al. Genomic analyses of human European diversity at the southwestern edge: isolation, African influence and disease associations in the Canary Islands. Mol Biol Evol 2018; 35:3010–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A 1993; 90: 10705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mifsud B, Tavares-Cadete F, Young AN, el al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 2015; 47: 598–606. [DOI] [PubMed] [Google Scholar]
- 24.Astle WJ, Elding H, Jiang T, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016; 167: 1415–1429.el9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JY, Hildebrandt MAT, Pu X, et al. Variations in the vascular endothelial growth factor pathway predict pulmonary complications. Ann Thorac Surg 2012; 94: 1079–84; discussion 1084–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The CARDIoGRAMplusC4D Consortium. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013; 45: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGinnis R, Steinthorsdottir V, Williams NO, et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat Genet 2017; 49:1255–1260. [DOI] [PubMed] [Google Scholar]
- 28.Gray KJ, Saxena R, Karumanchi SA. Genetic predisposition to preeclampsia is conferred by fetal DNA variants near FLT1, a gene involved in the regulation of angiogenesis. Am J Obstet Gynecol 2018;218:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becker PM, Verin AD, Booth MA, Liu F, Birukova A, Garcia JG. Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 2001; 281: L1500–11. [DOI] [PubMed] [Google Scholar]
- 30.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A(164/165) and P1GF: roles in angiogenesis and arteriogenesis. Trends Cardiovasc Med 2003; 13: 169–75. [DOI] [PubMed] [Google Scholar]
- 31.Strouvalis I, Routsi C, Adamopoulou M, et al. Early increase of VEGF-A is associated with resolution of ventilator-associated pneumonia: Clinical and experimental evidence. Respirology 2018; 23:942–949. [DOI] [PubMed] [Google Scholar]
- 32.Ware LB, Kaner RJ, Crystal RG, et al. VEGF levels in the alveolar compartment do not distinguish between ARDS and hydrostatic pulmonary oedema. Eur Respir J 2005; 26:101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo J, Yan W, Yang Y, Wang Z, Tian F. Monitoring of vascular endothelial growth factor and its soluble receptor levels in early trauma. J Trauma Acute Care Surg 2017; 82: 766–70. [DOI] [PubMed] [Google Scholar]
- 34.Shapiro NI, Schuetz P, Yano K, et al. The association of endothelial cell signaling, severity of illness, and organ dysfunction in sepsis. Crit Care 2010; 14: R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skibsted S, Jones AE, Puskarich MA, et al. Biomarkers of endothelial cell activation in early sepsis. Shock 2013; 39: 427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou PC, Filbin MR, Wang H, et al. Endothelial Permeability and Hemostasis in Septic Shock: Results From the ProCESS Trial. Chest 2017; 152: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018; 46: D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Napolitano F, Carrella D, Mandriani B, et al. gene2drug: a computational tool for pathway-based rational drag repositioning. Bioinformatics 2018; 34: 1498–505. [DOI] [PubMed] [Google Scholar]
- 39.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–82. [DOI] [PubMed] [Google Scholar]
- 40.Nacev BA, Grassi P, Dell A, Haslam SM, Liu JO. The Antifungal Drag Itraconazole Inhibits Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) Glycosylation, Trafficking, and Signaling in Endothelial Cells. J Biol Chem 2011; 286: 44045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.