

Abstract
The localization of NQO1 near acetylated microtubules has led to the hypothesis that NQO1 may work in concert with the NAD+-dependent deacetylase SIRT2 to regulate acetyl α-tubulin (K40) levels on microtubules. NQO1 catalyzes the oxidation of NADH to NAD+ and may supplement levels of NAD+ near microtubules to aid SIRT2 deacetylase activity. While HDAC6 has been shown to regulate the majority of microtubule acetylation at K40, SIRT2 is also known to modulate microtubule acetylation (K40) in the perinuclear region. In this study we examined the potential roles NQO1 may play in modulating acetyl α-tubulin levels. Knock-out or knock-down of NQO1 or SIRT2 did not change the levels of acetyl α-tubulin in 16HBE human bronchial epithelial cells and 3T3-L1 fibroblasts; however, treatment with a mechanism-based inhibitor of NQO1 (MI2321) led to a short-lived temporal increase in acetyl α-tubulin levels in both cell lines without impacting the intracellular pools of NADH or NAD+. Inactivation of NQO1 by MI2321 resulted in lower levels of NQO1 immunostaining on microtubules, consistent with redox-dependent changes in NQO1 conformation as evidenced by the use of redox-specific, anti-NQO1 antibodies in immunoprecipitation studies. Given the highly dynamic nature of acetylation-deacetylation reactions at α-tubulin K40 and the crowded protein environment surrounding this site, disruption in the binding of NQO1 to microtubules may temporally disturb the physical interactions of enzymes responsible for maintaining the microtubule acetylome.
Keywords: NQO1, SIRT2, α-Tubulin, Acetylation, Pyridine nucleotides, Microtubules
Graphical abstract
Highlights
-
•
NQO1which produces NAD and Sirt2 which uses NAD are located in the perinuclear region.
-
•
Depleting cellular NAD+ led to increased levels of acetyl α-tubulin.
-
•
Knockout or knockdown of NQO1 did not change perinuclear acetyl α-tubulin levels.
-
•
Pharmacological inhibition of NQO1 by MI2321 increased α-tubulin acetylation.
-
•
Redox changes in NQO1 conformation and binding modulate microtubule acetyltubulin.
1. Introduction
The cellular redox environment plays a critical role in many biological processes including cancer, metabolic diseases, diabetes, and aging. Proteins that influence the redox environment have become of interest in an attempt to target these diseases for therapeutic intervention. NQO1 (NAD(P)H:quinone oxidoreductase 1) is a flavoenzyme that utilizes reduced pyridine nucleotide cofactors (NADH, NADPH) to catalyze the reduction of a broad range of substrates from superoxide to complex quinones such as coenzyme Q10 [1]. NQO1 exists in the cell as two interlocking monomers that form a homodimeric protein as shown by crystal structure studies [2]. NQO1 is expressed in many different cell types, including endothelial and epithelial cells, adipocytes, and multiple tumor cell types [3,4]. NQO1 is mostly located in the cytoplasm but low levels of NQO1 have been found in the nucleus under normal conditions [5] and under conditions of stress, NQO1 has been shown to migrate to the nucleus [6] and the plasma membrane [7]. NQO1 expression in these cells is transcriptionally regulated primarily by the KEAP1/NRF2 and Ah receptor-mediated pathways, leading to upregulation in the expression of many enzymes, including NQO1, to confer cellular protection against a wide range of metabolic and environmental insults [1,8]. In addition to its role in reductive metabolism, NQO1 has been characterized as a RNA binding protein [9] and was found to enhance the translation of SERPINA1 mRNA [10]. NQO1 has also been shown to bind to a large number of proteins and protect these targets from proteasomal degradation [[11], [12], [13], [14]]. In many cases, protection against degradation by NQO1 was dependent upon pyridine nucleotide levels suggesting that the interaction of NQO1 with target proteins may be influenced directly by the NAD(P)H:NAD(P)+ ratio. Recently, it has been shown that NQO1 undergoes a conformational change upon binding of reduced pyridine nucleotides, resulting in loss of immunoreactivity to antibodies that bind to helix 7 of the catalytic core domain or the C-terminus [15]. This has led to interest in NQO1 as a potential redox sensor that can respond to changes in the pyridine nucleotide redox environment by altering its conformation. A number of recent studies have also shown the importance of flexibility and mobility of NQO1 with respect to its function which can be influenced by FAD and either ligands or inhibitors [16,17].
NQO1 is unique in that it can influence cellular redox through its ability to rapidly generate oxidized pyridine nucleotides via metabolism and, at the same time, can respond to alterations in the NAD(P)H:NAD(P)+ ratio by changing its conformation to modulate protein-protein binding. In double immunostaining studies, NQO1 has been shown to co-localize with α-tubulin and acetyl α-tubulin (K40) on microtubules including the mitotic spindle and midbody [15,18,19]. α-Tubulin K40 acetylation has been shown to increase protein binding on microtubule and microtubule stability [20,21]. The levels of acetyl α-tubulin K40 are regulated in part by the NAD+-dependent deacetylase SIRT2, which has also been shown to bind to microtubules [22,23]. The close physical association on microtubules of both NQO1, which can generate NAD+, and SIRT2, which consumes NAD+ to catalyze deacetylation reactions, suggests that these enzymes may work in concert to regulate α-tubulin acetylation. In this study, we utilized the non-malignant human bronchial epithelial cell line 16HBE and mouse embryonic 3T3-L1 fibroblasts to examine the relationship between pyridine nucleotides, NQO1, SIRT2, and the levels of acetyl α-tubulin K40.
2. Materials and methods
2.1. Cell lines
16HBE human lung epithelial cells were provided by Dr. Brian Day, Department of Medicine, National Jewish Health (Denver, CO). 3T3-L1 fibroblasts were purchased from Sigma-Aldrich (St. Louis, MO). Both cell lines were grown in Dulbecco's Modified Eagles Medium (4.5 g glucose/L, plus glutamine, without sodium pyruvate, Corning Life Sciences, Corning NY), supplemented with 10% fetal bovine serum and 100 μg/ml streptomycin and 100 units/ml penicillin (complete medium). Both cell lines were maintained in a humidified incubator at 37 °C with 5% carbon dioxide.
2.2. Reagents
The NQO1 mechanism-based inhibitor MI2321 and non-inhibiting analog MI3190 were synthesized in the laboratory of Dr. Christopher Moody, Department of Chemistry, University of Nottingham, U.K. β-lapachone and trichostatin A (TSA) were purchased from Santa Cruz Biotechnology (Dallas, TX) while olaparib and FK866 (Daporinad) were obtained from the Cayman Chemical Company (Ann Arbor, MI). The above reagents were dissolved in sterile DMSO. Butyric acid, NADH, FAD, dicumarol, 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI), and 2,6-dichloroindophenol (DCPIP), were purchased from Sigma-Aldrich.
2.3. Antibodies
Rabbit anti-acetyl α-tubulin (K40, #5335), rabbit anti-SIRT2 (#12672) and mouse anti-α tubulin (#3873) were obtained from Cell Signaling Technology (Danvers, MA). Mouse anti-NQO1 (A180, NB200-209) was obtained from Novus Biologicals (Centennial, CO) and rabbit anti-NQO1 (C-terminus, N5288) and mouse anti-β actin (#A2228) were purchased from Sigma-Aldrich. Mouse anti-SIRT2 (#66410-1-lg) was obtained from Proteintech (Rosemont, IL) and used in studies with 3T3-L1 fibroblasts. Horseradish peroxidase-, Alexa fluor 488- or rhodamine Red-X conjugated goat anti-mouse IgG or goat anti-rabbit IgG secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
2.4. CRISPR/cas9 knockout of NQO1
NQO1 protein expression was deleted in 16HBE cells using CRISPR/cas9 gene editing as described in Supplementary Material. NQO1-deficient 16HBE clones D7 and D19 were grown as described above.
2.5. siRNA mediated knockdown of NQO1 and Sirt2
16HBE cells (2 × 105 cells) were seeded into each well of a 6-well plate in 3 ml of antibiotic-free complete medium. On-target plus non-targeting siRNA (control) and on-target plus human NQO1 and human Sirt2 siRNA smartpools were purchased from Horizon (Lafayette, CO). siRNAs and DharmaFect 4 transfection reagent (Horizon) were prepared for transfection (6-well plate) as described by the manufacturer (see: https://horizondiscovery.com/-/media/Files/Horizon/resources/Protocols/basic-dharmafect-protocol.pdf). siRNA-mediated knockdown of NQO1 and Sirt2 was carried out independently. For these studies, each transfection was carried out using 50 nM of siRNA in combination with 4 μl/well of DharmaFect 4 transfection reagent. Cells were treated with siRNA/transfection reagent diluted in 3 ml of antibiotic-free complete medium. After 48 h, the medium was aspirated, cells were washed twice with 3 ml of phosphate buffered saline (PBS), and 100 μl of cell lysis buffer (1X cell lysis buffer; Cell Signaling Technologies) supplemented with protease and phosphatase inhibitors, 5 μM TSA and 5 mM butyric acid was added. Lysates from 2 wells were combined into a single tube, centrifuged at 13 K for 3 min at 4 °C, and the supernatants were collected and stored at −80 °C.
3T3L1 fibroblasts (1 × 105 cells) were seeded into 60 mm plates plate in 4 ml of antibiotic-free complete medium. On-target plus non-targeting siRNA (control) and on-target plus mouse NQO1and mouse Sirt2 siRNA smartpools were purchased from Horizon (Lafayette, CO). siRNAs and DharmaFect 1 transfection reagent (Horizon) were prepared for transfection as described by the manufacturer (see: https://horizondiscovery.com/-/media/Files/Horizon/resources/Protocols/basic-dharmafect-protocol.pdf). siRNA mediated knockdown of NQO1 and Sirt2 was carried out independently. For these studies each transfection was carried out using 25 nM of siRNA (NQO1) or 50 nM siRNA (Sirt2) in combination with 8 μl/plate of DharmaFect 1 transfection reagent. Cells were treated with siRNA/transfection reagent diluted in 4 ml antibiotic-free complete medium. After 48 h the medium was aspirated, cells washed twice with 3 ml of PBS then 100 μl of cell lysis buffer was added. Lysates were centrifuged at 13 K for 3 min at 4 °C and the supernatants were collected and stored at −80 °C.
2.6. Drug treatments
16HBE cells or 3T3-L1 fibroblasts were grown to approximately 80% confluency in 100-mm plates in 10 ml of complete medium. Drug treatments were initiated by replacing growth medium with 10 ml of fresh complete medium containing drugs. At the indicated times, the spent medium was aspirated, the cells washed twice with 10 ml of PBS, then collected in 500 μl (16HBE) or 200 μl (3T3-L1) cell lysis buffer (above). Lysates were centrifuged at 13 K for 3 min at 4 °C and the supernatants were collected and stored at −80 °C. Where indicated, NQO1 catalytic activity was measured in supernatants prior to freezing using the dicumarol-inhibitable reduction of DCPIP as previously described [24].
2.7. SDS-PAGE/immunoblot analysis
Protein concentrations were determined on supernatant fractions from cell lysates using the method of Lowry [25]. For SDS-PAGE, samples were diluted in 2X Laemmli buffer containing 2-mercaptoethanol, heated to 80 °C for 10 min, then centrifuged at 13, for 1 min and 20–40 μg protein/lane was separated by 12% SDS-PAGE (Bio-Rad precast minigel, Bio-Rad Laboratories, Hercules, CA). Proteins were then transferred to 0.2 μm polyvinylidene fluoride membranes in 25 mM Tris, containing 192 mM glycine and 20% (v/v) methanol at 100 V for 1 h at 4 °C. Membranes were blocked in 10 mM Tris-HCl, pH 7.8, 150 mM NaCl and 0.2% (v/v) Tween-20 (TBST) containing 5% (w/v) non-fat dry milk for 1 h at 22 °C. All primary antibodies were diluted in TBST containing 5% non-fat dry milk then used to probe membranes overnight at 22 °C. After incubation with primary antibodies, membranes were washed extensively in TBST followed by the addition of horseradish peroxidase-conjugated secondary antibodies diluted in TBST containing 5% non-fat dry milk for 30 min at 22 °C. Membranes were washed extensively with TBST and protein bands were visualized and quantified using enhanced chemiluminescence (GE Healthcare Life Sciences, Pittsburgh, PA) using a Bio-Rad Gel-Doc imager.
2.8. Immunoprecipitation of NQO1
16HBE cells were grown to 80% confluency in 10 ml complete medium in 100-mm plates. Cells were pretreated with DMSO, MI2321 (1 μM) or MI3190 (1 μM) for 30 min after which the medium was aspirated and 10 ml of fresh complete medium containing 2 mM H2O2 was added. After 2 h the medium was aspirated, the cells washed twice with 10 ml PBS, then collected in 500 μl/plate of cell lysis buffer (see above). Protein concentrations were determined using the method of Lowry and 1 mg/sample of lysate was transferred to a new 1.7 ml tube and the volume was brought up to 500 μl with cell lysis buffer. Rabbit anti-NQO1 (C-terminus) antibody (2 μg) was added for 1 h at 22 °C while gently rocking followed by 50 μl of protein A-conjugated magnetic beads (SureBeads, Bio-Rad) for an additional 30 min at 22 °C. The beads were collected using the magnet rack (Bio-Rad) and the unbound proteins were removed by aspiration. The beads were then washed twice with 0.5 ml of cell lysis buffer, then once with 0.5 ml of 25 mM Tris-HCl, pH 7.4. Beads were then diluted in 25 μl/sample of 2X Laemmli buffer containing 2-mercaptoethanol, heated to 70 °C for 5 min, then centrifuged at 13 K for 1 min and 20 μl/sample was analyzed for NQO1 by SDS-PAGE/immunoblot analysis (see above) using the mouse anti-NQO1 (A180) antibody. Immunoprecipitation of purified recombinant human NQO1 was carried out as described previously [15].
2.9. Non-denaturing PAGE
Recombinant human NQO1 (rhNQO1) was purified as described previously [26]. Reactions (30 μl) containing rhNQO1 (2.5 μg) were incubated in 62.5 mM Tris-HCl, pH 7.0 containing 1.2 M sucrose, 0.01% bromophenol blue and 5 μM FAD in the presence and absence of 500 μM NADH and/or 50 μM MI2321 for 30 min at room temperature. Samples were then loaded on to a 4–20% gradient non-denaturing polyacrylamide gel (Mini-protean TGX, Bio-Rad) and electrophoresed in 25 mM Tris, 192 mM glycine and 5 μM FAD at 150 V for 18 h at 4 °C. Since NQO1 has an overall positive charge the electrodes were in reverse orientation and the protein was attracted towards the negative electrode. Following electrophoreses the gel was stained for 1 h with Coomassie R-250 (Imperial Protein Stain, ThermoFisher Scientific, Waltham, MA).
2.10. Immunocytochemistry/confocal microscopy
16HBE cells or 3T3-L1 fibroblasts were grown on 18-mm glass coverslips in complete medium. Coverslips were transferred to 6-well plates (1 coverslip/well) for drug treatments in 2 ml of complete medium. Following drug treatments, cells were washed once with 2 ml of PBS then fixed with 2 ml of freshly prepared 4% (v/v) methanol-free formaldehyde (Ted Pella Inc., Redding, CA) diluted in PBS for 12 min at 22 °C. Coverslips were rinsed 3 times with PBS then permeabilized with 0.1% (v/v) Triton X-100 diluted in PBS for 12 min at 22 °C. Coverslips were rinsed 3 times with PBS then coverslips were blocked in complete medium diluted 1:1 with PBS (blocking buffer) for 1 h at 22 °C. Primary antibodies were diluted in 1 ml blocking buffer and added to each coverslip for 60–90 min at 22 °C while gently rocking. Coverslips were rinsed 3 times with TBST then fluorescent-labeled secondary antibodies and DAPI (1 μg/ml) diluted in 1 ml blocking buffer were added for an additional 30 min. Coverslips were washed extensively in TBST, dipped in ddH2O, inverted, then mounted on a glass microscope slide containing one drop of Vectashield anti-fade mounting media (Vector Laboratories, Burlingame, CA). Coverslips were then covered with SuperMount (BioGenex, Fremont, CA) and allowed to dry overnight in the dark at 22 °C. Cells were visualized on a Nikon TI2-E microscope (600X) equipped with a1-plus confocal system with lasers at 402 nm, 488 nm and 561 nm (Nikon, Melville, NY).
2.11. Quantitative metabolomic analysis for reduced and oxidized pyridine nucleotides
16HBE cells or 3T3-L1 fibroblasts were grown in 100-mm plates in 10 ml of complete medium and treated with drugs as described above. Following drug treatment, the medium was aspirated, the cells washed once in 10 ml PBS, and then cells were scrapped off into 1 ml PBS. The cells were transferred to a 2 ml tube then pelleted at 2.5 K for 1 min at 22 °C. The PBS was carefully aspirated and the cell pellet was dissolved in 120 μl of deoxygenated ethanoic solution heated to 80 °C. U13-Labeled internal standard and nucleotide internal standards were then added and the samples were placed on a heating block at 80 °C for 10 min while gently vortexing each sample every 3 min. Sample were then centrifuged at 13 for 10 min at 4 °C then 100 μl of supernatant was transferred to a glass autosampler vial for analysis. Samples were analyzed for reduced and oxidized pyridine nucleotides using LC/MS as described in Supplementary Material.
3. Results
The relationship between intracellular NAD+ concentrations, the NAD+-dependent deacetylase SIRT2, and NQO1 on the levels of acetyl α-tubulin (K40) were examined in 16HBE human lung epithelial cells and mouse 3T3-L1 embryonic fibroblasts. To investigate the association between intracellular NAD+ concentrations and acetyl α-tubulin levels we treated 16HBE cells or 3T3-L1 fibroblasts with the nicotinamide phosphoribosyl transferase inhibitor FK866. Treatment with FK866 gradually decreased NAD+ levels in both cell lines over 72 h, while during the same time course the levels of acetyl α-tubulin gradually increased (Fig. 1A and B). Immunostaining for acetyl α-tubulin confirmed the immunoblot data and showed that following treatment with FK866 there was a gradual increase in immunostaining for acetyl α-tubulin in both cell lines (Fig. 1C). These data also showed that the increase in immunostaining for acetyl α-tubulin initially appeared in perinuclear regions, which is consistent with previous studies that have suggested that SIRT2 may be responsible for regulating acetylation of perinuclear microtubules [27]. We have confirmed the perinuclear localization of SIRT2 and NQO1 in 16HBE cells (Fig. 1D). Double immunostaining for NQO1 and α-tubulin is shown in Supplementary Material, Fig. S1 and colocalization of NQO1 with microtubules was shown in previous work using immunocytochemistry and proximate ligation assays [15].
Fig. 1.

NAD+depletion increased acetylated α-tubulin levels in 16HBE and 3T3-L1 cells. (A) Intracellular pyridine nucleotide concentrations in 16HBE cells (n = 3) treated with DMSO or FK866 (500 nM) for the indicated times. DMSO controls were treated for 72 h. (B) Immunoblot analysis for acetyl α-tubulin in 16HBE and 3T3-L1 cells treated with DMSO or FK866 (500 nM) for the indicated times. (C) Immunostaining for acetyl α-tubulin in 16HBE and 3T3-L1 cells treated with DMSO or FK866 for the indicated times. (D) Immunostaining for SIRT2 and NQO1 in 16HBE cells demonstrating the perinuclear localization of SIRT2 and NQO1.
To investigate whether NQO1 has a role in maintaining basal levels of acetyl α-tubulin we utilized the CRISPR/cas9 gene editing technique. Immunoblot analysis of parental 16HBE cells and two CRISPR/cas9 modified clones (D7 and D19) confirmed knockout of NQO1 without significant change in the basal levels of acetyl α-tubulin/α-tubulin ratios in NQO1-deficient clones (Fig. 2A). In addition, siRNA-mediated knocked-down of NQO1 in 16HBE cells did not significantly alter the acetyl α-tubulin levels compared to cells treated with non-targeting siRNA controls (Fig. 2B). The latter results were also observed in 3T3-L1 fibroblasts (Fig. 2C). These data demonstrate that loss of NQO1 did not change the basal levels of acetyl α-tubulin in the two cell lines.
Fig. 2.

Stable knock-out or transient knock-down of NQO1 did not increase the levels of acetyl α-tubulin in 16HBE or 3T3-L1 cells. (A) Acetyl α-tubulin levels were measured in parental 16HBE cells and two stable NQO1-null clones (D7 and D19) generated using CRISPR/cas9 directed gene silencing. β-actin was included as a loading control. (B) Acetyl α-tubulin levels were measured in (B) 16HBE cells and (C) 3T3-L1 fibroblasts after treatment with non-targeting control siRNA or siRNA targeted to NQO1 for 48 h.
In similar studies we examined whether loss of SIRT2 would affect the basal levels of acetyl α-tubulin in 16HBE cells and 3T3-L1 fibroblasts. siRNA targeted to SIRT2 significantly reduced SIRT2 protein expression in both cell lines compared to non-targeting siRNA controls, and yet no changes in the levels of acetyl α-tubulin were observed (Fig. 3A and B).
Fig. 3.

Transient knock-down of SIRT2 did not increase the levels of acetyl α-tubulin in 16HBE and 3T3-L1 cells. Acetyl α-tubulin levels were measured in (A) 16HBE cells and (B) 3T3-L1 fibroblasts after treatment with non-targeting control siRNA or siRNA targeted to SIRT2 for 48 h (SIRT2 isoform I, SIRT2 isoform II). NQO1 protein levels were not altered by transient SIRT2 knockdown in either cell line. Raw image of the SIRT2 immunoblot in 16HBE cells is shown in Supplementary Material, Fig. S3.
One drawback from using knockout or knockdown strategies is that these methods take relatively long periods of time (days) to significantly lower NQO1 or SIRT2 protein levels. To examine the relationship between acetyl α-tubulin, SIRT2 and NQO1 at shorter times (min to h) we utilized a potent and specific mechanism-based inhibitor of NQO1 (MI2321). Inactivation of NQO1 by MI2321 results from the bioreduction of MI2321 to its hydroquinone form followed by the release of a high reactive indolequinone-derived iminium ion, which rapidly alkylates critical amino acid residues in the active site of the enzyme [28]. The covalent binding of MI2321 in the active site permanently inactivates NQO1 and prevents the enzyme from catalyzing the oxidation of reduced pyridine nucleotides and the reduction of co-substrates. We examined the effects of NQO1 inactivation by MI2321 on basal acetyl α-tubulin levels in 3T3-L1 fibroblasts. For these studies, 3T3-L1 fibroblasts were exposed to MI2321 (1 μM) for the indicated times and then acetyl α-tubulin levels were measured by immunoblot analysis. A modest increase in acetyl α-tubulin levels was observed between 15 min and 1 h following exposure to MI2321 (Fig. 4A). While the increase in acetyl α-tubulin levels was short-lived, inactivation of NQO1 by MI2321 occurred rapidly following treatment (<1 min) and was prolonged (>8 h, Fig. 4B). The reasons underlying the temporal nature of increased acetyl α-tubulin are unclear but may reflect the activity of acetylases/deacetylases not affected by a conformational change in NQO1.
Fig. 4.

Inactivation of NQO1 results in a temporal increase in acetyl α-tubulin levels in 3T3-L1 fibroblasts. (A) Acetyl α-tubulin levels were measured in 3T3-L1 fibroblasts after treatment with MI2321 (1 μM) for the indicated times. (B) NQO1 catalytic activity was measured in 3T3-L1 lysates obtained from fibroblasts treated with DMSO or MI2321 (1 μM) for the indicated times.
Similar studies in 16HBE cells also showed a temporal increase in acetyl α-tubulin levels following treatment with MI2321 (Fig. 5A). In 16HBE cells the maximal increase in acetyl α-tubulin was observed between 1-2 h following exposure to MI2321 and, similar to 3T3-L1 fibroblasts, inactivation of NQO1 by MI2321 occurred rapidly following treatment (<1 min) and was prolonged (>8 h, Fig. 5B). We undertook a more detailed study of the relationship between NQO1 inactivation by MI2321 and acetyl α-tubulin levels in 16HBE cells. In these studies, we utilized a non-inhibiting analog of MI2321 (MI3190, Fig. 6A) as a control since quinones are redox-reactive and have the potential to alter the NAD(P)H:NAD(P)+ redox balance. The treatment of 16HBE cells with MI2321 for 2 h resulted in a significant increase in acetyl α-tubulin levels compared to either DMSO or MI3190 controls (Fig. 6B). To investigate whether the intracellular concentrations of NADH or NAD+ were affected by the inactivation of NQO1 we measured the levels of these two molecules in 16HBE cells by LC-MS following a 2 h exposure to MI2321. Neither inactivation of NQO1 by MI2321 or treatment with the non-inhibiting analog MI3190 significantly affected pyridine nucleotide levels compared to the DMSO control (Fig. 6C). In these experiments, we also included as positive controls treatment with the redox-active quinone β-lapachone, which rapidly depletes cells of both NADH and NAD+, and treatment with β-lapachone plus the PARP inhibitor olaparib, which selectively depletes cells of NADH (Fig. 6C) [15].
Fig. 5.

Inactivation of NQO1 results in a temporal increase in acetyl α-tubulin levels in 16HBE cells. (A) Acetyl α-tubulin levels were measured in 16HBE cells after treatment with MI2321 (1 μM) for the indicated times. (B) NQO1 catalytic activity was measured in 16HBE cell lysates obtained from cells treated with DMSO or MI2321 (1 μM) for the indicated times.
Fig. 6.

Inactivation of NQO1 increased acetyl α-tubulin levels but did not alter the NADH or NAD+concentrations in 16HBE cells. (A) Chemical structures of MI2321 and the non-inhibiting control indolequinone MI3190. (B) Acetyl α-tubulin levels and NQO1 catalytic activity were measured in 16HBE cells after treatment with MI2321 or MI3190 (1 μM) for 2 h. (C) Intracellular NADH and NAD+ concentrations were measured in 16HBE cells (n = 3) following treatment with MI2321 or MI3190 (1 μM) for 2 h. Treatment of 16HBE cells (n = 3) with 10 μM β-lapachone (β-LAP) or 10 μM β-LAP plus 5 μM olaparib (β-LAP + OLA) for 2 h were included as positive controls.
We have recently demonstrated that non-denatured, catalytically active NQO1 displays redox-dependent immunoreactivity (Fig. 7A), [15]. Anti-NQO1 antibody (A180) that specifically targets helix 7 of the catalytic core domain binds to the oxidized but not the reduced form of NQO1. Similarly, anti-NQO1 antibodies that target the C-terminus of NQO1 bind also to the oxidized form of NQO1 but not to its reduced form. The redox-dependent immunoreactivity of NQO1 may be due to a change in the conformation of NQO1 following transfer of a hydride from reduced pyridine nucleotides to the flavin co-factor in the active site [15]. Using redox-dependent anti-NQO1 antibodies to distinguish between oxidized and reduced forms of NQO1, we examined whether inactivation of NQO1 by MI2321 locked NQO1 into a more oxidized or reduced-like conformation. For these studies, we pretreated 16HBE cells with DMSO or MI2321 (1 μM × 2 h) then exposed cells to hydrogen peroxide (2 mM × 2 h) to deplete cells of pyridine nucleotides via DNA damage and PARP-mediated NAD+ consumption. Following treatments, NQO1 was immunoprecipitated from lysates using the redox-dependent anti-NQO1 antibody that targets the C-terminus. In the absence of hydrogen peroxide, the majority of NQO1 is not immunoprecipitated due to its reduced conformation (Fig. 7B, lane 1); however, in samples pretreated with MI2321 in the absence of hydrogen peroxide, moderate levels of NQO1 could be pulled down (Fig. 7B, lane 2) suggesting MI2321 induces a change in the conformation of NQO1. In samples treated with hydrogen peroxide alone, large amounts of NQO1 could be immunoprecipitated due to the conversion of NQO1 to an oxidized conformation (Fig. 7B, lane 3). Pretreatment with MI2321 before hydrogen peroxide decreased the amount of NQO1 immunoprecipitated (Fig. 7B, lane 4, compare to lane 3). In contrast, M3190, a non-inhibitory analog of MI2321, had little effect on the amount of NQO1 pulled down (Fig. 7B, lanes 6 and 7, compare to lanes 1 and 3). β-lapachone was used as a positive control for oxidized NQO1 (Fig. 7B, lane 5 [15]). Taken together these data suggest that binding of MI2321 (but not the inactive analog of the inhibitor, M3190) alters the structure of NQO1 and locks the protein into an inactivated conformation which is non-responsive to pyridine nucleotides (Fig. 7A and B, lanes 2 and 4).
Fig. 7.

Inactivation by MI2321 alters the structure of NQO1. (A) Schematic representation of the three conformations of NQO1 and their immunoreactivity towards the redox-specific anti-NQO1 C-terminus antibody. (B) 16HBE cells were pretreated with DMSO, MI2321 (1 μM) or MI3190 (1 μM) for 30 min followed by exposure to 2 mM H2O2 for 2 h. NQO1 was then immunoprecipitated from lysates (1 mg) using the anti-NQO1 C-terminus antibody. β-Lapachone (β-Lap), 10 μM × 2 h was included as a positive control for oxidized NQO1. (C) Purified recombinant human NQO1 (0.2 μg) was incubated in the absence and presence of NADH (200 μM) for 15 min then hydrogen peroxide (5 mM) was added for an additional 1 h. Following incubations, catalase (800 μg/ml) was added to each tube and NQO1 was immunoprecipitated using the anti-NQO1 C-terminus antibody. (D) Migration of purified rhNQO1 following inactivation by MI2321. Purified rhNQO1 (2.5 μg) was incubated with 500 μM NADH and 50 μM MI2321 for 30 min then analyzed by non-denaturing PAGE.
In control experiments using purified recombinant human NQO1, the addition of hydrogen peroxide did not directly catalyze the oxidation of the reduced form of NQO1 (Fig. 7C). The addition of NADH generated the reduced form of NQO1, which was not pulled down by anti-NQO1 C-terminus antibodies (Fig. 7C, lanes 2 and 3), and exposure to hydrogen peroxide (5 mM) did not stimulate the direct oxidation of reduced NQO1 (Fig. 7C, lanes 4 and 5) demonstrating that the effects observed in Fig. 7 occur via the intermediacy of pyridine nucleotides. To demonstrate that inactivation by MI2321 alters the structure of NQO1 purified rhNQO1 was incubated with NADH and MI2321 then analyzed by non-denaturing polyacrylamide gel electrophoresis (Fig. 7D). Inactivation of NQO1 by MI2321 (Fig. 7D, lane 4) resulted in slower migration of NQO1 compared to non-inactivated proteins (Fig. 7D, lanes 1–3).
Previously, we have shown that NQO1 co-localizes to microtubules/acetylated microtubules by immunostaining for NQO1 using a redox-specific, anti-NQO1 antibody A180 [15]. We extended these studies to 16BE cells and 3T3-L1 fibroblasts treated with DMSO or MI2321, then immunostained for NQO1 using anti-NQO1 antibody A180. In these studies, we observed a significant decrease in the intensity of immunostaining for NQO1 on the microtubules of cells treated with MI2321 compared to DMSO controls (Fig. 8, left panels, fluorescence quantification, Supplementary Material, Fig. S2), while, at the same time, we observed a significant increase in staining for acetylated tubulin (Fig. 8, middle panels, fluorescence quantification, Supplementary Material, Fig. S2). The merged analysis confirmed that the increased acetylated tubulin immunostaining was perinuclear (Fig. 8, right panels). These results confirm the immunoprecipitation studies that suggest inactivation of NQO1 by MI2321 induces a conformational change in NQO1, which may either inhibit its binding to microtubules or result in the binding of NQO1 to microtubules in an altered conformation.
Fig. 8.

Inactivation of NQO1 by MI2321 results in lower levels of immunostaining for NQO1 and higher levels of perinuclear acetyl α-tubulin in intact cells. 16HBE cells (top panels) were treated with DMSO or MI2321 (1 μM) for 2 h while 3T3-L1 fibroblasts (bottom panels) were treated with DMSO or MI2321 (1 μM) for 30 min. Following treatments the cells were fixed then immunostained for NQO1 and acetyl α-tubulin. NQO1 immunostaining was performed using the redox-specific anti-NQO1 A180 antibody.
4. Discussion
Microtubules are the result of the highly dynamic process of polymerization and depolymerization of heterodimers of α- and β-tubulin. The growth and shrinkage of microtubules is regulated, in part, by post-translational protein modifications of which acetylation has emerged as a potential regulatory mechanism [29]. An increasing number of lysine residues on α- and β-tubulin have been found to be acetylated [30,31], but lysine 40 (K40) in α-tubulin has received the most attention. Currently, it is not clear what role this modification plays in microtubule dynamics. Some studies suggest a relationship between acetyl α-tubulin K40 levels and microtubule stability [20,21], while other studies suggest it influences microtubule flexibility [32]. The acetyl α-tubulin K40 modification is unique in that it resides in the internal lumen of the microtubule and how this site is accessed by acetyltransferase and deacetylase enzymes is not well-understood. Tubulin acetyltransferase aTAT1 and the deacetylase HDAC6 regulate a large majority of the microtubule acetylome including α-tubulin K40 but smaller roles for other deacetylases have emerged [33]. A role for SIRT2 in regulating acetyl α-tubulin K40 levels in a subset of perinuclear microtubules has been described [27].
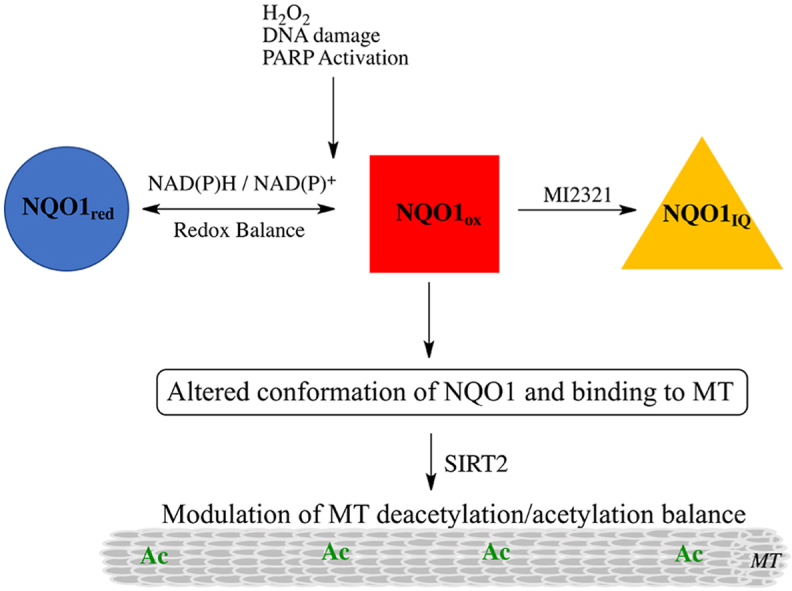
In addition to posttranslational modifications, microtubule binding proteins (MAPs) also influence microtubule dynamics [34]. The cytosolic quinone reductase NQO1 has been shown to co-localize with microtubules especially in structures containing high levels of α-tubulin K40 including the mitotic spindles and midbody [15]. The binding of NQO1 to microtubules and acetylated microtubules was confirmed using proximity ligation assay [15]. NQO1 and SIRT2 have been shown to interact via co-immunoprecipitation studies [19] and our immunostaining studies showed the localization of NQO1 and SIRT2 in perinuclear regions, raising the possibility that these two proteins may act together due to their complementary biochemistry. The ability of NQO1 to generate NAD+ for SIRT2-dependent deacetylase activity is an attractive testable hypothesis supported by previous studies in MCF-7 tumor cells [19]. However, it has been recently shown that the binding of NQO1 to microtubules may be redox-dependent since NQO1 can change conformation depending upon intracellular reduced pyridine nucleotide concentrations [15]. Under normal conditions where NAD(P)H levels are plentiful, NQO1 adopts a reduced conformation following hydride transfer from NAD(P)H to the FAD co-factor in NQO1. There have been a number of stopped-flow studies examining the catalytic cycle of NQO1 in the presence of efficient electron acceptors such as quinones [[35], [36], [37]]. The early work of Hosoda (1974) is particularly useful when considering the stability of the reduced flavin form of NQO1 in the absence of efficient electron acceptors [36]. Hosoda et al. demonstrated that there was only a slow rate of reaction (1.5 nmol O2/min/mg protein) between O2 and reduced flavin in NQO1 with a first order rate constant for this reaction of 4.4 × 10−2 s−1 suggesting that the reduced form of the enzyme has appreciable stability. X-ray crystallography, biochemical and computational studies performed by Amzel, and colleagues suggested that the stability of the reduced form of NQO1 occurred in part as a result of stabilization of charge separation in the reduced flavin by the protein [[38], [39], [40]]. Our study is in agreement with the work of Hosoda et al. and Amzel and colleagues. The reduced and oxidized forms of NQO1 can be distinguished by using redox-dependent antibodies that bind to oxidized but not reduced NQO1 [15]. In the presence of NAD(P)H but in the absence of an efficient quinone electron acceptor, very little NQO1 could be immunoprecipitated demonstrating the presence of the reduced form of NQO1. As NAD(P)H levels are depleted, NQO1 can no longer maintain a reduced conformation and the protein reverts to its oxidized form enabling immunoprecipitation. Thus, instead of directly providing NAD+ for SIRT2 activity NQO1 may be responding to the depleted levels of pyridine nucleotides in regions surrounding SIRT2 by altering its conformation in a redox dependent manner which in turn modulates the acetylation/deacetylation balance of microtubules (Fig. 9).
Fig. 9.

Proposed mechanism for the roles of the redox environment and mechanism-based inhibitor MI2321 on NQO1 structure and binding to microtubules. Reduced pyridine nucleotides regulate the redox conformation of NQO1 and its binding to microtubules. Inactivation of NQO1 by the indolequinone (IQ) component of the mechanism-based inhibitor MI2321 alters the conformation of NQO1, resulting in decreased binding of NQO1 to microtubules. Lower levels of NQO1 bound to microtubules result in either increased acetylation or decreased deacetylation of lysine40 of α-tubulin in the lumen of the microtubule.
While the binding of NQO1 to microtubules has not been characterized in detail, we hypothesize that dimeric NQO1 binds to microtubules via a pair of positively charged C-terminal tails. The C-terminal domains play an important role in many aspects of NQO1 biochemistry including the binding of FAD, NAD(P)H and inhibitors through their ability to communicate conformational and dynamic information between distal regions in NQO1 [16,17,41,42]. Evidence to support a role for the C-terminal tails binding to microtubules comes from studies which show that the C-terminal domains are not exposed to the external environment when NQO1 is in the reduced form but are exposed when NQO1 is in the oxidized form [15,42]. In addition, antibodies which target the C-terminus of NQO1 do not show NQO1 immunostaining on microtubules suggesting that the C-terminals are not accessible to antibodies when oxidized NQO1 is bound to microtubules. The C-terminus of NQO1 carries an overall positive charge (pI 10.29) which would promote its binding to negatively charged microtubules. A similar interaction has been demonstrated for the binding of HDAC6 to microtubules where the positively charged N-terminus is attracted to the negatively charged microtubule [43].
Data presented herein demonstrates that the knock-out or knock-down of NQO1 or SIRT2 did not affect basal levels of acetyl α-tubulin K40; however, inactivation of NQO1 by the mechanism-based inhibitor MI2321 did result in a short-lived temporal increase. This apparent discrepancy may be explained by the highly dynamic nature and crowded environment of the microtubule lumen. The time required to generate cells with reduced protein expression is in most cases relatively long (days), potentially allowing microtubules and accompanying MAPs to re-equilibrate after the loss of NQO1 or SIRT2. Inactivation of NQO1 by MI2321 on the other hand is relatively rapid in comparison. The change in NQO1 conformation following inactivation by MI2321 appears to reduce the amount of NQO1 bound to microtubules, disrupting the complex balance of microtubule-binding proteins (Fig. 9). We speculate that decreased binding of NQO1 to microtubules could either enable access of acetyltransferases to acetyl α-tubulin K40 or result in inhibition of the activity of deacetylases at that specific site in the microtubule lumen. In support of these data, significant differences in the in-vivo acetylome have also been observed in mice after genetic manipulation of NQO1 levels [44].
In summary, using the nicotinamide phosphoribosyl transferase inhibitor FK866, we confirmed the association between decreased intracellular NAD+ concentrations and increased acetyl α-tubulin K40 levels. CRISPR/cas9 knockout or siRNA-mediated knockdown of NQO1 had little effect on acetyl α-tubulin K40 levels. However, use of the mechanism-based inhibitor MI2321, which alters the conformation of NQO1, resulted in decreased immunostaining for NQO1 on microtubules and led to a rapid but transient increase in the levels of acetyl α-tubulin K40. Taken together these data suggest an alternative role for NQO1 in the regulation of the microtubule acetylome where the binding of NQO1 to microtubules is regulated by the conformation of the protein which is, in turn, controlled primarily by the surrounding redox environment.
Declaration of competing interest
None.
Acknowledgments
We are grateful to Dr. C. J. Moody, University of Nottingham for the supply of the NQO1 inhibitor MI2321 and analog MI3190 and the University of Colorado Skaggs School of Pharmacy and Pharmaceutical Sciences Mass Spectrometry Facility for analyses.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101840.
Funding
The work was supported by NIH grant R01-DK109964-04 and, in part, by the Intramural Research Program of the National Institute on Aging, NIH.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Ross D. Quinone reductases multitasking in the metabolic world. Drug Metab. Rev. 2004;36(3–4):639–654. doi: 10.1081/dmr-200033465. [DOI] [PubMed] [Google Scholar]
- 2.Li R., Bianchet M.A., Talalay P., Amzel L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. U. S. A. 1995;92(19):8846–8850. doi: 10.1073/pnas.92.19.8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlager J.J., Powis G. Cytosolic NAD(P)H:(quinone-acceptor)oxidoreductase in human normal and tumor tissue: effects of cigarette smoking and alcohol. Int. J. Canc. 1990;45(3):403–409. doi: 10.1002/ijc.2910450304. [DOI] [PubMed] [Google Scholar]
- 4.Siegel D., Ross D. Immunodetection of NAD(P)H:quinone oxidoreductase 1 (NQO1) in human tissues. Free Radic. Biol. Med. 2000;29(3–4):246–253. doi: 10.1016/s0891-5849(00)00310-5. [DOI] [PubMed] [Google Scholar]
- 5.Winski S.L., Koutalos Y., Bentley D.L., Ross D. Subcellular localization of NAD(P)H:quinone oxidoreductase 1 in human cancer cells. Canc. Res. 2002;62(5):1420–1424. [PubMed] [Google Scholar]
- 6.Milkovic L., Tomljanovic M., Cipak Gasparovic A., Novak Kujundzic R., Simunic D., Konjevoda P. Nutritional stress in head and neck cancer originating cell lines: the sensitivity of the NRF2-NQO1 Axis. Cells. 2019;8(9) doi: 10.3390/cells8091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyun D.H., Emerson S.S., Jo D.G., Mattson M.P., de Cabo R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc. Natl. Acad. Sci. U. S. A. 2006;103(52):19908–19912. doi: 10.1073/pnas.0608008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaiswal A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000;29(3–4):254–262. doi: 10.1016/s0891-5849(00)00306-3. [DOI] [PubMed] [Google Scholar]
- 9.Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149(6):1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 10.Di Francesco A., Di Germanio C., Panda A.C., Huynh P., Peaden R., Navas-Enamorado I. Novel RNA-binding activity of NQO1 promotes SERPINA1 mRNA translation. Free Radic. Biol. Med. 2016;99:225–233. doi: 10.1016/j.freeradbiomed.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asher G., Tsvetkov P., Kahana C., Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19(3):316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asher G., Bercovich Z., Tsvetkov P., Shaul Y., Kahana C. 20S Proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol Cell. 2005;17(5):645–655. doi: 10.1016/j.molcel.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Garate M., Wong R.P., Campos E.I., Wang Y., Li G. NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of the tumour suppressor p33(ING1b) EMBO Rep. 2008;9(6):576–581. doi: 10.1038/embor.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adamovich Y., Shlomai A., Tsvetkov P., Umansky K.B., Reuven N., Estall J.L. The protein level of PGC-1 alpha, a key metabolic regulator, is controlled by NADH-NQO1. Mol. Cell Biol. 2013;33(13):2603–2613. doi: 10.1128/MCB.01672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel D., Dehn D.D., Bokatzian S.S., Quinn K., Backos D.S., Di Francesco A. Redox modulation of NQO1. PloS One. 2018;13(1) doi: 10.1371/journal.pone.0190717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medina-Carmona E., Palomino-Morales R.J., Fuchs J.E., Padin-Gonzalez E., Mesa-Torres N., Salido E. Conformational dynamics is key to understanding loss-of-function of NQO1 cancer-associated polymorphisms and its correction by pharmacological ligands. Sci. Rep. 2016;6:20331. doi: 10.1038/srep20331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pey A.L., Megarity C.F., Timson D.J. NAD(P)H quinone oxidoreductase (NQO1): an enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 2019;39(1) doi: 10.1042/BSR20180459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siegel D., Kepa J.K., Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) localizes to the mitotic spindle in human cells. PloS One. 2012;7(9) doi: 10.1371/journal.pone.0044861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang H.J., Song H.Y., Ahmed M.A., Guo Y., Zhang M., Chen C. NQO1 regulates mitotic progression and response to mitotic stress through modulating SIRT2 activity. Free Radic. Biol. Med. 2018;126:358–371. doi: 10.1016/j.freeradbiomed.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maruta H., Greer K., Rosenbaum J.L. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J. Cell Biol. 1986;103(2):571–579. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piperno G., LeDizet M., Chang X.J. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J. Cell Biol. 1987;104(2):289–302. doi: 10.1083/jcb.104.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.North B.J., Marshall B.L., Borra M.T., Denu J.M., Verdin E. The human Sir 2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003;11(2):437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 23.North B.J., Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PloS One. 2007;2(8):e784. doi: 10.1371/journal.pone.0000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lind C., Cadenas E., Hochstein P., Ernster L. DT-diaphorase: purification, properties, and function. Methods Enzymol. 1990;186:287–301. doi: 10.1016/0076-6879(90)86122-c. [DOI] [PubMed] [Google Scholar]
- 25.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 26.Sharkis D.H., Swenson R.P. Purification by cibacron blue F3GA dye affinity chromatography and comparison of NAD(P)H:quinone reductase (E.C.1.6.99.2) from rat liver cytosol and microsomes. Biochem. Biophys. Res. Commun. 1989;161(2):434–441. doi: 10.1016/0006-291x(89)92617-x. [DOI] [PubMed] [Google Scholar]
- 27.Skoge R.H., Ziegler M. SIRT2 inactivation reveals a subset of hyperacetylated perinuclear microtubules inaccessible to HDAC6. J. Cell Sci. 2016;129(15):2972–2982. doi: 10.1242/jcs.187518. [DOI] [PubMed] [Google Scholar]
- 28.Winski S.L., Faig M., Bianchet M.A., Siegel D., Swann E., Fung K. Characterization of a mechanism-based inhibitor of NAD(P)H:quinone oxidoreductase 1 by biochemical, X-ray crystallographic, and mass spectrometric approaches. Biochemistry. 2001;40(50):15135–15142. doi: 10.1021/bi011324i. [DOI] [PubMed] [Google Scholar]
- 29.Janke C. The tubulin code: molecular components, readout mechanisms, and functions. J. Cell Biol. 2014;206(4):461–472. doi: 10.1083/jcb.201406055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 31.Liu N., Xiong Y., Ren Y., Zhang L., He X., Wang X. Proteomic profiling and functional characterization of multiple post-translational modifications of tubulin. J. Proteome Res. 2015;14(8):3292–3304. doi: 10.1021/acs.jproteome.5b00308. [DOI] [PubMed] [Google Scholar]
- 32.Xu Z., Schaedel L., Portran D., Aguilar A., Gaillard J., Marinkovich M.P. Microtubules acquire resistance from mechanical breakage through intralumenal acetylation. Science. 2017;356(6335):328–332. doi: 10.1126/science.aai8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janke C., Montagnac G. Causes and consequences of microtubule acetylation. Curr. Biol. 2017;27(23):R1287–R1292. doi: 10.1016/j.cub.2017.10.044. [DOI] [PubMed] [Google Scholar]
- 34.Brouhard G.J., Rice L.M. Microtubule dynamics: an interplay of biochemistry and mechanics. Nat. Rev. Mol. Cell Biol. 2018;19(7):451–463. doi: 10.1038/s41580-018-0009-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tedeschi G., Chen S., Massey V., Dt-diaphorase Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 1995;270(3):1198–1204. doi: 10.1074/jbc.270.3.1198. [DOI] [PubMed] [Google Scholar]
- 36.Hosoda S., Nakamura W., Hayashi K. Properties and reaction mechanism of DT diaphorase from rat liver. J. Biol. Chem. 1974;249(20):6416–6423. [PubMed] [Google Scholar]
- 37.Anoz-Carbonell E., Timson D.J., Pey A.L., Medina M. The catalytic cycle of the antioxidant and cancer-associated human NQO1 enzyme: hydride transfer, conformational dynamics and functional cooperativity. Antioxidants. 2020;9(9) doi: 10.3390/antiox9090772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cavelier G., Amzel L.M. Mechanism of NAD(P)H:quinone reductase: ab initio studies of reduced flavin. Proteins. 2001;43(4):420–432. [PubMed] [Google Scholar]
- 39.Faig M., Bianchet M.A., Talalay P., Chen S., Winski S., Ross D. Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. U. S. A. 2000;97(7):3177–3182. doi: 10.1073/pnas.050585797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bianchet M.A., Faig M., Amzel L.M. Structure and mechanism of NAD[P]H:quinone acceptor oxidoreductases (NQO) Methods Enzymol. 2004;382:144–174. doi: 10.1016/S0076-6879(04)82009-3. [DOI] [PubMed] [Google Scholar]
- 41.Lienhart W.D., Gudipati V., Uhl M.K., Binter A., Pulido S.A., Saf R. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:quinone oxidoreductase(1) FEBS J. 2014;281(20):4691–4704. doi: 10.1111/febs.12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen S., Deng P.S., Bailey J.M., Swiderek K.M. A two-domain structure for the two subunits of NAD(P)H:quinone acceptor oxidoreductase. Protein Sci. : a publication of the Protein Society. 1994;3(1):51–57. doi: 10.1002/pro.5560030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ustinova K., Novakova Z., Saito M., Meleshin M., Mikesova J., Kutil Z. The disordered N-terminus of HDAC6 is a microtubule-binding domain critical for efficient tubulin deacetylation. J. Biol. Chem. 2020;295(9):2614–2628. doi: 10.1074/jbc.RA119.011243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Francesco Andrea, Choi Youngshim, Bernier Michel, Zhang Yingchun, Diaz-Ruiz Alberto, Aon Miguel A. NQO1 protects obese mice through improvements in glucose and lipid metabolism. NPJ Aging Mech. Dis. 2020;6(1):1–17. doi: 10.1038/s41514-020-00051-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.