

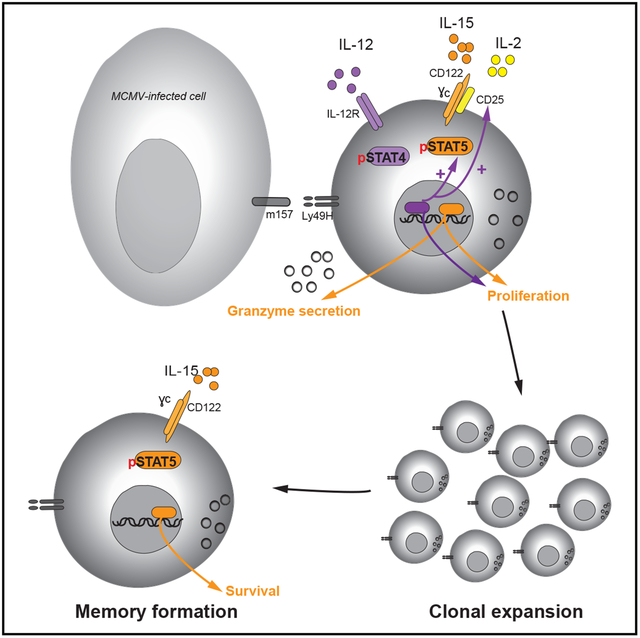
SUMMARY
Natural killer (NK) cells are innate lymphocytes with the capacity to elicit adaptive features, including clonal expansion and immunological memory. Because signal transducer and activator of transcription 5 (STAT5) is essential for NK cell development, the roles of this transcription factor and its upstream cytokines interleukin-2 (IL-2) and IL-15 during infection have not been carefully investigated. In this study, we investigate how STAT5 regulates transcription during viral infection. We demonstrate that STAT5 is induced in NK cells by IL-12 and STAT4 early after infection and that partial STAT5 deficiency results in a defective capacity of NK cells to generate long-lived memory cells. Furthermore, we find a functional dichotomy of IL-2 and IL-15 signaling outputs during viral infection, whereby both cytokines drive clonal expansion, but only IL-15 is required for memory NK cell survival. We thus highlight a role for STAT5 signaling in promoting an optimal anti-viral NK cell response.
In Brief
Wiedemann et al. demonstrate that Stat5a and Stat5b are induced by IL-12 and STAT4 signaling in NK cells following MCMV infection. They further provide evidence that the cytokines IL-2 and IL-15 upstream of STAT5 differentially promote the early and late stages of the adaptive NK cell response to MCMV infection.
Graphical Abstract
INTRODUCTION
Natural killer (NK) cells, traditionally classified as lymphoid cells of the innate immune system, have in recent years been demonstrated to elicit a number of adaptive immune features (Rapp et al., 2018). In response to mouse cytomegalovirus (MCMV) infection, NK cells expressing the activating surface receptor Ly49H have the capacity to recognize the virally encoded glycoprotein m157 on MCMV-infected cells. This recognition leads to activation and robust clonal expansion, followed by contraction of effector NK cells, and ultimately results in the formation of a pool of long-lived memory NK cells capable of undergoing a rapid, antigen-specific recall response (Sun et al., 2009a). In addition to antigen receptor signaling, this process is driven by a complex interplay of a range of pro-inflammatory cytokines and downstream transcription factors, resulting in epigenetic and transcriptional changes within the NK cells (Lau et al., 2018).
Signal transducer and activator of transcription (STAT) factors are key regulators of NK cell functions. STAT1 and STAT2 (downstream of type 1 interferon [IFN] signaling) and STAT4 (downstream of interleukin-12 [IL-12] signaling) promote adaptive NK cell responses to MCMV in a non-redundant manner, with deficiency in one of these factors resulting in defective NK cell expansion and memory formation (Rapp et al., 2018). STAT5 is the downstream transcription factor shared by all common gamma chain (γc) cytokines and is required for NK cell development, maturation, survival, and cytotoxicity (Eckelhart et al., 2011; Friedmann et al., 1996; Gotthardt and Sexl, 2016; Imada et al., 1998; Johnston et al., 1995). NK-cell-intrinsic STAT5 deficiency results in severely reduced NK cell numbers and NK-cell-mediated tumor control in mice (Gotthardt et al., 2016; Imada et al., 1998; Villarino et al., 2017). Because of the detrimental impact of STAT5 deficiency on NK cells, functional studies on STAT5-deficient NK cells are limited, and the role of STAT5 during anti-viral NK cell responses remains to be investigated.
The two most functionally relevant γc cytokines for NK cells are thought to be IL-2 and IL-15. Both cytokines signal through the γc and the shared intermediate affinity heterodimeric receptor consisting of the IL-2 receptor (IL-2R) β chain (CD122), although each cytokine preferentially binds to this receptor complex in the presence of its own distinct high-affinity α chain (IL-2Rα [CD25] and IL-15Rα) (Sugamura et al., 1996). CD25 is expressed together with the γc and CD122 to form the high-affinity IL-2R on the NK cell surface upon activation, whereas IL-15Rα is expressed by neighboring cells, including dendritic cells and macrophages, and predominantly trans-presents IL-15 to cells that express the γc and CD122 chains (Leonard et al., 2019). Despite their shared receptor subunits and downstream signaling pathways, IL-2 and IL-15 are thought to elicit distinct functions in NK cells. IL-15 is required for NK cell development and survival (Kennedy et al., 2000; Koka et al., 2003), whereas deficiency in IL-2 signaling does not affect NK cell numbers but impairs NK cell activation and target cell killing (Gasteiger et al., 2013; Kündig et al., 1993). During infection, T-cell-derived IL-2 has been implicated in aiding NK cell responses in a mouse model of Leishmania infection (Bihl et al., 2010), and early upregulation of the high-affinity IL-2R during MCMV infection suggests a functional requirement in the early response (Lee et al., 2012). However, despite decades of culturing mouse and human NK cells using IL-2, an in vivo role for this cytokine in NK cell responses has not been stringently investigated using models of genetic ablation. Here, we provide a detailed analysis of the role of STAT5 and upstream IL-2 versus IL-15 signaling in the early and adaptive phases of NK cell response to viral infection and describe divergent transcriptional changes driven by these distinct signaling pathways.
RESULTS AND DISCUSSION
STAT5 Is Transcriptionally and Epigenetically Regulated by IL-12 and STAT4 Early after MCMV Infection
We first investigated mRNA levels of STAT5 in NK cells throughout the course of MCMV infection. STAT5 is encoded by two adjacent genes in mammals, namely, Stat5a and Stat5b, of which Stat5b has a greater impact on NK cells (Imada et al., 1998; Villarino et al., 2017). During the course of infection, we found that both Stat5a and Stat5b mRNA peaked on day 2 post-infection (PI) (Figure 1A), with transcript levels returning to initial levels by day 4 PI. Interestingly, Stat5b mRNA then increased at later time points and was found at significantly higher levels in memory NK cells than in naive NK cells (Figure 1A), suggesting a functional requirement for STAT5 in both early and late NK cell responses against MCMV infection.
Figure 1. IL-12- and STAT4-Dependent Induction of STAT5 in NK Cells during MCMV Infection.
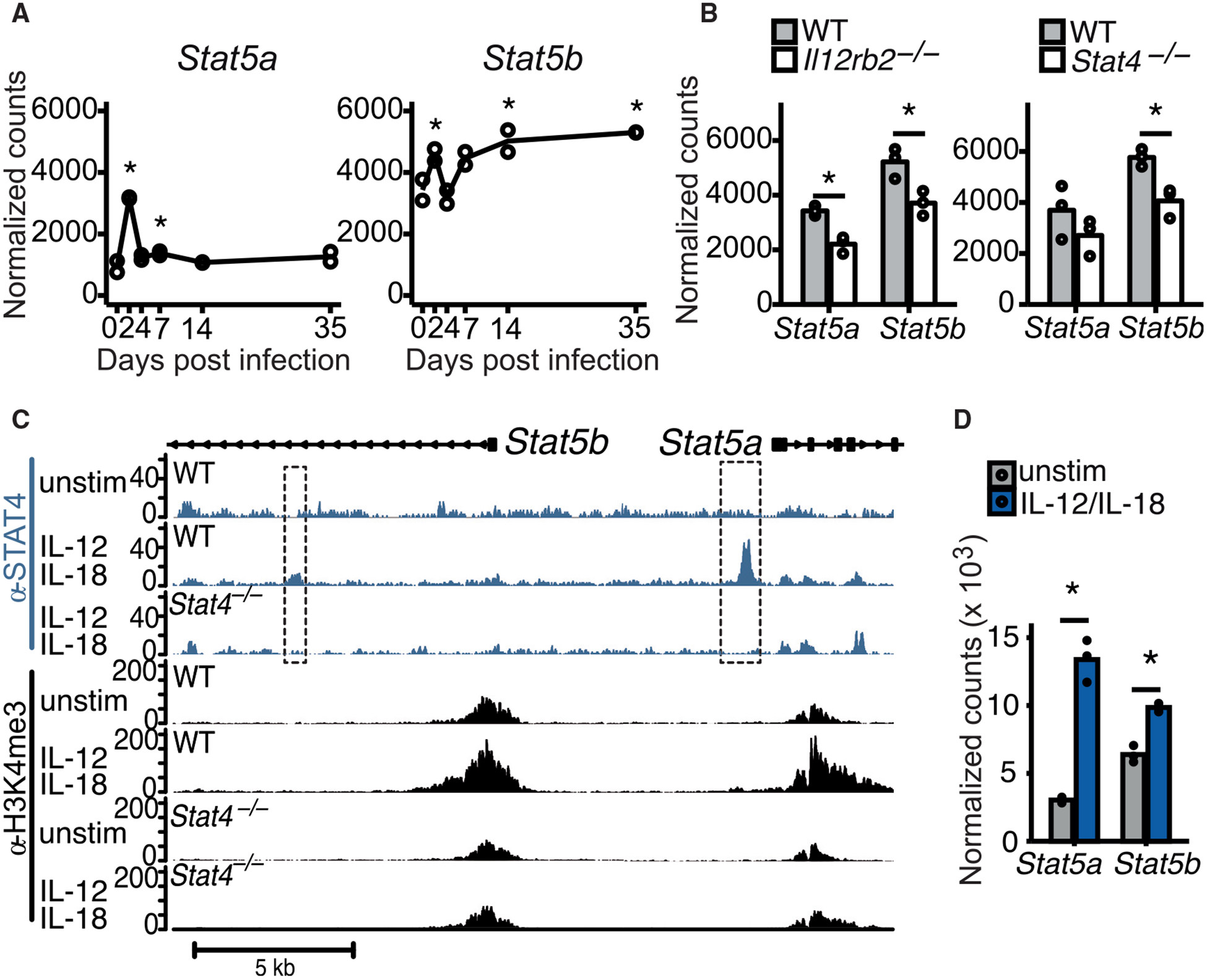
(A) RNA-seq was performed on Ly49H+ NK cells on days 0, 2, 4. 7, 14, and 35 of MCMV infection. Normalized counts of Stat5a and Stat5b are displayed.
(B) RNA-seq on WT versus Il12rb2−/− or Stat4−/− from mixed BMC mice on day 2 PI. Normalized counts of Stat5a and Stat5b are displayed.
(C) ChIP-seq was performed for STAT4 and H3K4me3 on WT and Stat4−/− NK cells cultured in media alone (unstim) or with IL-12 and IL-18. Representative gene tracks of mapped STAT4 ChIP (blue tracks) or H3K4me3 ChIP (black tracks) are displayed.
(D) NK cells were cultured in media (unstim) or with IL-12 + IL-18, and RNA-seq was performed after 3 h of culture. Normalized counts of Stat5a and Stat5b are displayed.
Error bars indicate SEM.
To determine whether exposure to pro-inflammatory cytokines led to the induction of Stat5 in NK cells after MCMV infection, we performed RNA sequencing (RNA-seq) on NK cells isolated from wild-type (WT), IL-12-receptor-deficient (Il12rb2−/−), or STAT4-deficient (Stat4−/−) mice at day 2 PI. Interestingly, Stat5 induction was significantly reduced in NK cells lacking IL-12R or STAT4 on day 2 PI (Kaplan et al., 1996; Wu et al., 2000; Figure 1B), whereas IL-12 receptor deficiency did not affect Stat5 levels in uninfected NK cells (Figure S1A). No decreases in Stat5 were observed in NK cells lacking interferon α/β receptor (IFNAR) or STAT1, with the Stat5a transcript even increased in Stat1−/− NK cells (Meraz et al., 1996; Figure S1B), suggesting that IL-12 signaling by STAT4 is the main inducer of STAT5 and that STAT1 inhibits Stat5a induction. To further support our hypothesis, we observed STAT4 binding at the Stat5a promoter and at a Stat5b intronic region by chromatin immunoprecipitation sequencing (ChIP-seq) in WT but not STAT4-deficient NK cells stimulated with IL-12 and IL-18 (Figure 1C). We also found an increased deposition of the permissive histone mark H3K4me3 at the Stat5a and Stat5b promoter regions of NK cells during IL-12 and IL-18 stimulation, which was abrogated in Stat4−/− NK cells receiving the same stimuli (Figure 1C). Moreover, stimulation with IL-12 and IL-18 also led to a significant upregulation of Stat5a and Stat5b transcripts in NK cells (Figure 1D). Altogether, these data suggest that IL-12 signaling in NK cells causes STAT4 to directly target the Stat5 loci, thus influencing the epigenetic landscape and enabling increased transcription of Stat5 in response to viral infection.
STAT5 Is Required for Optimal NK Cell Responses to MCMV Infection
To investigate a functional role for STAT5 in NK cells during MCMV infection, we generated mice with an NK-cell-selective deletion of Stat5a and Stat5b (NKCre × Stat5fl/fl). When we infected Stat5fl/fl, NKCre × Stat5fl/+, and NKCre × Stat5fl/fl mice with MCMV, we observed that NKCre × Stat5fl/fl mice containing Stat5-deficient NK cells showed significantly greater mortality and viral load (Figure 2A). Furthermore, using NKCre × Stat5fl/+:WT mixed bone marrow chimeric (mBMC; Figure S2A) mice (because NKCre × Stat5fl/fl lack NK cells [Eckelhart et al., 2011] and mBMC mice allow for investigation of WT and STAT5-deficient NK cells in the same environment), we observed that even NK cells with heterozygous STAT5 levels had reduced granzyme A and B production at day 2 PI (Figure 2B) and were less mature both at steady state and on day 2 PI (Figures 2C, S2B, and S2C). Thus, we conclude that STAT5 in NK cells is required for proper NK cell maturation under steady-state conditions and for optimal NK cell effector functions during MCMV infection.
Figure 2. STAT5-Dependent Anti-viral NK Cell Response.
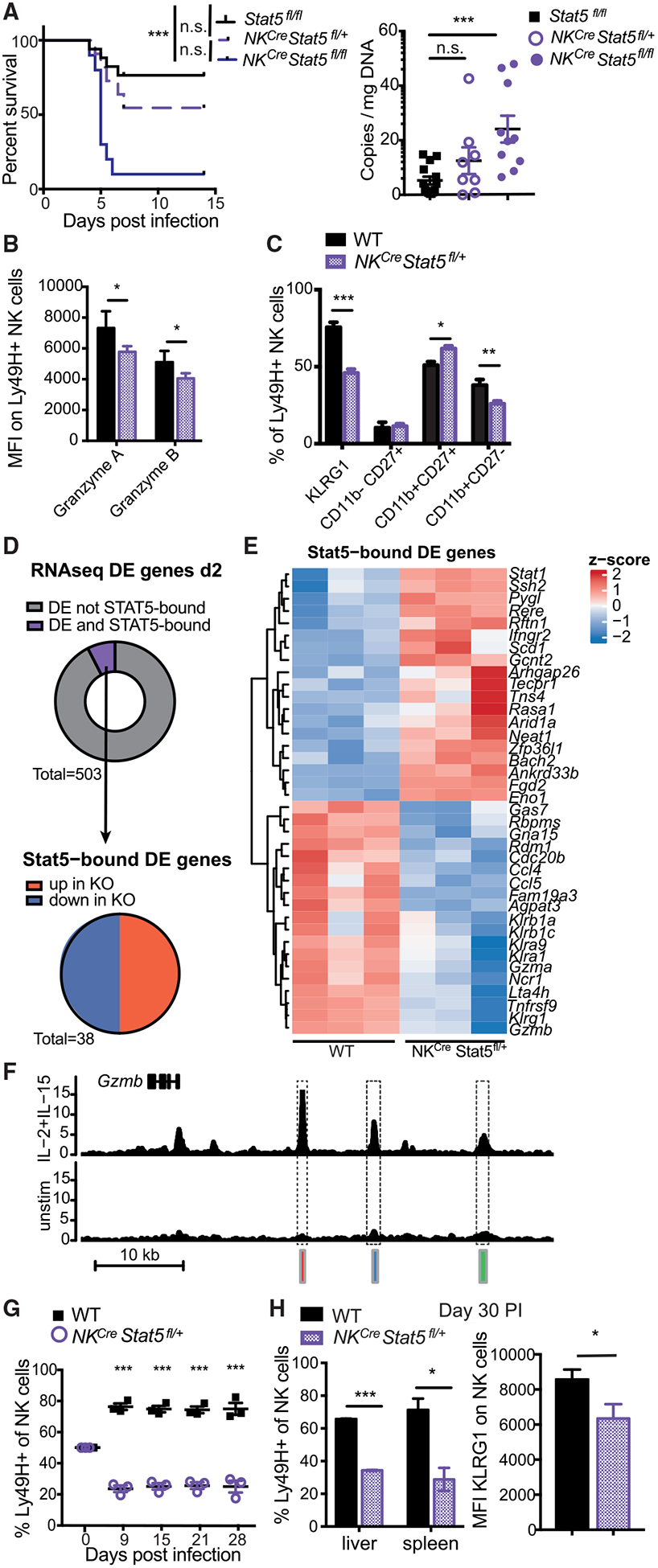
(A) Stat5fl/fl, NKCre × Stat5fl/+, and NKCre × Stat5fl/fl mice were infected with high-dose MCMV. Viral titers in the serum were measured on day 4 PI. Data are representative of 2 independent experiments (n = 4–6).
(B and C) Mixed WT:NKCre × Stat5fl/+ BMC mice were infected with MCMV. Ly49H+ WT or NKCre × Stat5fl/+ NK cells from spleen were analyzed on day 2 PI for granzyme A and B expression (B) or indicated activation/maturation markers (C). Data are representative of at least 2 independent experiments (n = 4).
(D) RNA-seq was performed on Ly49H+ WT or NKCre × Stat5fl/+ NK cells at day 2 PI (n = 3). ChIP-seq was performed on NK cells stimulated with IL-2 and IL-15 for 3 h. Donut graph displays the proportion of DE genes (adjusted p value [padj] < 0.05) that are STAT5 bound (purple). Pie graph displays STAT5-bound DE genes (padj < 0.05) categorized by direction of gene expression.
(E) Heatmap shows Z scores of all DE and STAT5-bound genes described in (D).
(F) Representative gene tracks of mapped STAT5 ChIP.
(G and H) Splenocytes from mixed WT:NKCre × Stat5fl/+ BMC mice were adoptively transferred into Rag2−/− × Ly49h−/− mice and infected with MCMV. (G) Graph shows the relative WT to NKCre × Stat5fl/+ NK cell percentages that were measured over the course of infection. (H) Bar graphs show percentage of memory Ly49H+ WT and NKCre × Stat5fl/+ NK cells in spleen and liver and KLRG1 surface expression (liver) on day 30 PI. Data are representative of 2 independent experiments (n = 3–4).
All error bars indicate SEM.
To determine the direct downstream targets of STAT5 during MCMV infection, we performed RNA-seq on WT and NKCre × Stat5fl/+ NK cells from mBMC mice on day 2 PI and overlapped the genes differentially expressed (DE) with STAT5 target genes defined by ChIP-seq. Interestingly, only a small number of genes DE in NKCre × Stat5fl/+ NK cells was found to be STAT5 bound (38 out of 503 DE genes in total) (Figure 2D). The high percentage of DE non-STAT5 target genes might be attributable to a compensatory activation of other pathways, such as the tumor necrosis factor receptor superfamily pathways (TNFRSFs) (Villarino et al., 2017). Among the STAT5 target genes DE at day 2 PI, the number of genes upregulated and downregulated were similar (Figure 2D). Importantly, STAT5-bound genes with significantly lower expression in NKCre × Stat5fl/+ NK cells on day 2 PI included Gzma, Gzmb, Klrg1, and Ly49 genes (Klra1 and Klra9), whereas RNA-seq of uninfected NK cells revealed no changes in granzyme levels in STAT5-deficient NK cells (Figures 2E, 2F, and S2C). These findings underscore the requirement of STAT5 for proper NK cell maturation and cytotoxicity during infection.
Finally, we tested whether STAT5 was required for the adaptive features of anti-viral NK cells. Using an adoptive transfer model in which WT and NKCre × Stat5fl/+ NK cells were derived from mBMC mice (Figure S2D), we observed that NK cells with heterozygous STAT5 levels had a pronounced defect in clonal expansion compared with WT NK cells following MCMV infection (Figures 2G and S2D), resulting in a reduced pool of memory NK cells in liver and spleen at 4 weeks PI (Figure 2H). Moreover, NKCre × Stat5fl/+ memory NK cells showed a diminished expression of the maturation marker KLRG1 compared to WT controls (Figure 2H). Altogether, these findings suggest that STAT5 in NK cells mediates a multitude of critical effector functions that underlie protective host responses against viral infection.
Non-redundant Requirement for IL-2 and IL-15 during NK Cell Clonal Expansion
In order to elucidate the importance and contribution of specific signaling pathways upstream of STAT5, we used mice with NK cells containing a tamoxifen-inducible deficiency in the shared IL-2/IL-15Rβ chain CD122 (UbcCre-ERT−2 × Il2rbfl/fl) or high-affinity IL-2Rα chain CD25 (NKCre × Cd25fl/fl). Reduced expression of CD122 and CD25 on NK cells was confirmed by flow cytometry (Figures S3A and S3B). In adoptive co-transfer experiments of WT and CD25- or CD122-deficient NK cells, both CD25-deficient and CD122-deficient NK cells were significantly outcompeted by WT NK cells during the first week after MCMV infection and continued to show diminished numbers at memory time points (Figure 3A). To understand how these signaling pathways drive NK cell expansion during MCMV exposure, we measured the proliferation of adoptively transferred WT or knockout (KO) NK cells labeled with cell tracer violet (CTV) (Figure S3E). Both CD25- and CD122-deficient NK cells displayed reduced proliferation compared to WT NK cells as early as day 3 PI (Figure 3B), indicating a requirement of IL-2 and possibly IL-15 during clonal expansion.
Figure 3. Both IL-2 and IL-15 Drive NK Cell Expansion In Vivo.
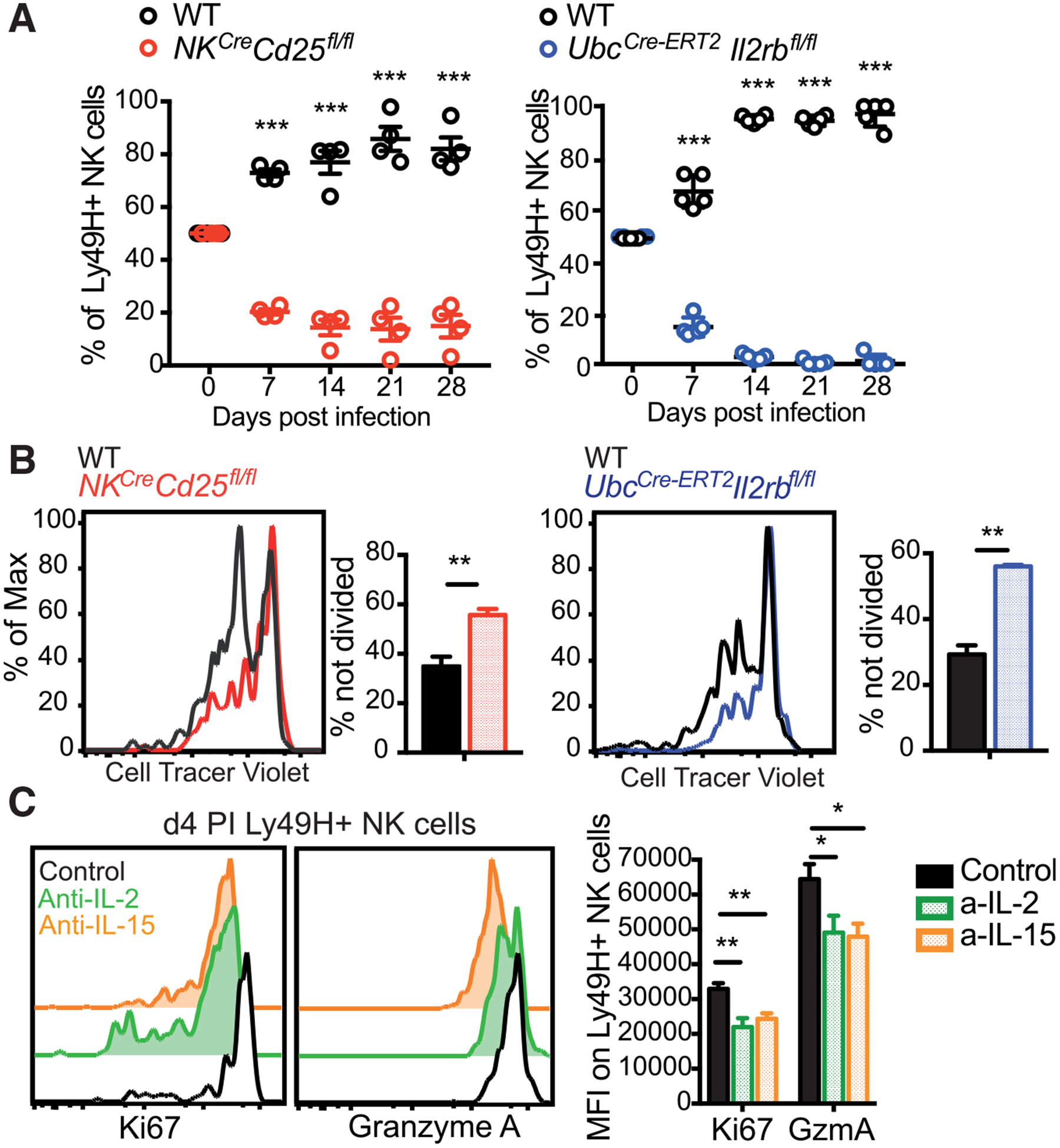
(A) Equal numbers of WT and UbcCre-ERT2 × Il2rbfl/fl or NKCre × CD25fl/fl NK cells were transferred into Ly49h−/− mice, followed by infection with MCMV. Donor UbcCre-ERT2 × Il2rbfl/fl mice were treated with tamoxifen on days −3, −2, and −1 prior to adoptive transfer. Following MCMV infection, relative percentages of Ly49H+ WT and KO NK cells are displayed (n = 4–5).
(B) NK cells from WT mice, NKCre × CD25fl/fl mice, or UbcCre-ERT2 × Il2rbfl/fl mice treated with tamoxifen on days −3, −2, and −1 were labeled with CTV and transferred into Ly49h−/− mice, followed by infection with MCMV. Representative flow plots and graphs show amounts of undivided WT and KO NK cells from the liver at day 3 PI.
(C) WT Ly49H+ NK cells were transferred into Ly49h−/− mice treated followed by infection with MCMV and treatment with PBS, anti-IL-2, or anti-IL-15 on days −1 to 4 PI. Flow plots and graph show amount of Ki67 and granzyme A on transferred Ly49H+ NK cells at day 4 PI. Data are pooled from 2 independent experiments (n = 4–5). All error bars indicate SEM.
In order to further delineate the requirement of IL-2 versus IL-15 in promoting NK cell expansion, we neutralized either IL-2 or IL-15 by using antibodies from day −1 to 4 PI in mice adoptively transferred with WT NK cells and infected with MCMV (Figure S3F). Interestingly, neutralization of either cytokine resulted in decreased NK cell proliferation, measured by Ki67 expression on day 4 PI (Figure 3C). Similarly, blockade of either IL-2 or IL-15 resulted in decreased granzyme A production (Figure 3D), highlighting a non-redundant role for both cytokines in driving NK cell expansion and cytotoxicity during MCMV infection.
Memory NK Cell Survival during the Contraction Phase Is STAT5 Dependent
Finally, we investigated how STAT5 and IL-2 versus IL-15 shape the contraction and memory phases of the NK cell response. To this end, we generated UbcCre-ERT2 × Stat5fl/fl mice that allow for inducible deletion of STAT5 in NK cells upon tamoxifen treatment. Following adoptive transfer of WT and UbcCre-ERT2 × Stat5fl/fl NK cells and infection with MCMV, recipient mice were treated with tamoxifen or oil at days 8 to 10 PI, immediately following the peak of the clonal expansion of NK cells (Figure 4A). Within a week after tamoxifen treatment, STAT5-deleted NK cells were outcompeted by WT NK cells, resulting in a reduced memory NK cell pool in tamoxifen-treated mice (Figure 4B). We determined the level of activated caspases by fluorochrome inhibitor of caspases (FLICA) staining (as a measure of apoptosis) in NK cells on day 13 PI and found increased FLICA incorporation in STAT5-deleted NK cells (Figure 4C). In order to discriminate between the function of IL-2 versus IL-15 during the maintenance of memory NK cells, we treated mice with antibodies against IL-2 or IL-15 between days 8 and 18 PI (Figure S3). In contrast to our findings during the expansion phase, only IL-15 neutralization resulted in diminished numbers of memory NK cells (Figure 4D), suggesting a differential role for these γc cytokines during the distinct phases of the NK cell response against viral infection.
Figure 4. IL-15 and STAT5 Are Required for the Survival of Memory NK Cells.
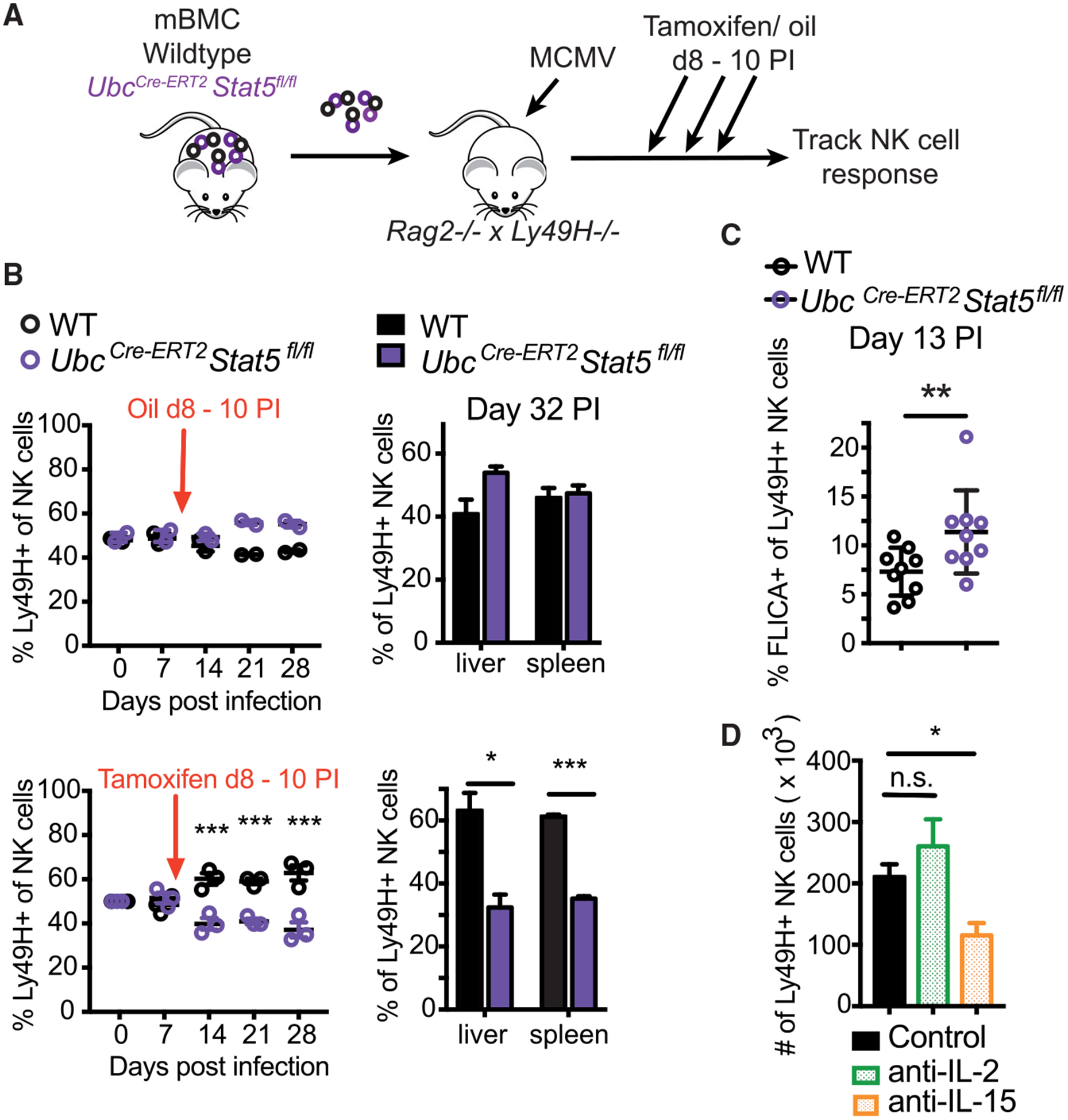
(A–C) Splenic NK cells from WT:UbcCre-ERT2 × Stat5fl/fl mBMC mice were transferred into Rag2−/− × Ly49h−/− mice. Tamoxifen or oil (as a control) was administered on days 8 to 10 following MCMV infection. (B) Graphs show relative percentages of Ly49H+ WT and KO NK cells in the peripheral blood at indicated time points and in spleen and liver at day 32 PI. Data are representative of 2 independent experiments (n = 2–4). (C) Graph shows percentage of NK cells from spleen staining positive for FLICA on day 13 PI. Data are pooled from 2 independent experiments (n = 4–5).
(D) WT Ly49H+ NK cells were transferred into Ly49h−/− mice infected with MCMV, and recipient mice were treated with PBS, anti-IL-2, or anti-IL-15 from days 8 to 18 PI. Graph shows the number of NK cells in the spleen on day 30 PI. Data are representative of 2 independent experiments (n = 4–5).
All error bars indicate SEM.
In summary, our data suggest that STAT5 is required throughout the course of MCMV infection for optimal NK cell responses. During the expansion phase, STAT5 drives NK cell maturation and cytotoxicity, and STAT5 deficiency in NK cells resulted in an increased susceptibility to MCMV. Our data suggest that STAT5 is induced by IL-12 through direct STAT4 binding at the Stat5 loci early after MCMV infection, which is a mechanism similar to the IL-12-mediated and STAT4-dependent induction of its upstream receptor CD25 (Lee et al., 2012). Interestingly, STAT5 and CD25 expression peak at nearly the same time following infection (~day 2 PI), and here, we demonstrate a functional impact for IL-2 signaling during the early anti-viral NK cell response. This apparent cooperative interaction between STAT4 and STAT5 transcription factors represents the mechanistic opposite of the previously reported cross-regulatory antagonism that occurs with STAT4 and STAT1 (Lau et al., 2018). Interestingly, because the Stat5a transcript was upregulated in Stat1−/− NK cells early after infection, we may be observing an antagonism between STAT4 and STAT1 in the regulation of Stat5, as has been previously described for the induction of IFN-γ (Nguyen et al., 2000).
During the proliferative burst of NK cells in response to MCMV infection, both IL-2 and IL-15 and their respective receptors CD25 and CD122 are required in a non-redundant manner. Although IL-2 has been used for decades to grow NK cells in vitro, and a putative role for IL-2 during NK cell proliferation had been suggested (Lee et al., 2012; Nandagopal et al., 2014), it was previously reported that IL-15- and γc-independent NK cell expansion could occur during MCMV infection (Sun et al., 2009b). However, in a competitive setting in vivo, we now observe a reduced capacity of NK cells deficient in STAT5, CD25, or CD122 to generate a robust anti-viral response or long-lived memory pool. Although IL-12 may be sufficient to drive some expansion in the absence of γc signaling (Sun et al., 2009b), optimal NK cell responses require non-redundant IL-2 and IL-15 signaling. In contrast to the expansion phase, IL-15 signaling by STAT5 is sufficient to sustain memory NK cell survival during the contraction phase in the absence of IL-2 signaling. Because IL-2 and IL-15 use the same receptor β and γ chain and the intracellular signaling cascade driven by either cytokine is very similar (Ring et al., 2012), the observed differences in memory NK cell generation may be due to both a lack of CD25 expression on NK cells during the late stages of infection and lower local and systemic IL-2 levels (data not shown). Our findings thus reveal a functional dichotomy between IL-2 and IL-15 in anti-viral NK cells—one that is shared by CD8+ T cells (Becker et al., 2002; Mitchell et al., 2010; Schluns et al., 2002), whereby IL-2 predominantly drives proliferation and effector differentiation, but IL-15 alone promotes survival during the contraction to memory transition.
Because the phosphatidylinositol 3-kinase (PI3K)-mammalian target of rapamycin (mTOR) signaling pathway is another major pathway downstream of IL-2 and IL-15 receptors (Ring et al., 2012) and has been implicated in both MCMV-induced NK cell expansion and mitophagy during the contraction phase (Nandagopal et al., 2014; O’Sullivan et al., 2015), we cannot exclude that abrogation of this pathway (in our receptor ablation or cytokine neutralization studies) may contribute to our findings. Nonetheless, we clearly demonstrate that STAT5 signaling is required for the formation of effector and memory NK cells during all stages of MCMV infection. Overall, our study highlights the distinct effects of two closely related cytokines and their major downstream transcription factor on NK cell functions during MCMV infection and uncovers another aspect of the complex cytokine signaling network that facilitates the anti-viral NK cell response.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph Sun (sunj@mskcc.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data generated in this study have been deposited in the Gene Expression Omnibus. Accession numbers for RNA-seq and ChIP-seq are GSE142821 and GSE140043, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice used in this study were housed and bred under specific pathogen-free conditions at Memorial Sloan Kettering Cancer Center (MSKCC) in accordance with all guidelines of the Institutional Animal Care and Use Committee (IACUC). The following strains were used, all on the C57BL/6 genetic background: C57BL/6 (CD45.2), B6.SJL (CD45.1), NKp46iCre (Narni-Mancinelli et al., 2011), UbcERT2-Cre, NKp46Cre-ERT2, Rosa26flox.tdTom, Stat5fl/fl (Cui et al., 2004), Il2rbfl/fl (JAX stock #029657; Chinen et al., 2016), Il2rafl/fl (CD25fl/fl) (Chinen et al., 2016), Rag2−/− × Il2rg−/− (Jackson), Rag2−/− × Klra8−/− (Ly49h−/−), and Klra8−/− (Ly49h−/−) (Fodil-Cornu et al., 2008). Experiments were conducted using age- and gender-matched mice in accordance with approved institutional protocols. Animals were typically 6–10 weeks old at the time of use and consisted of males and females.
Mixed bone marrow chimeric mice were generated by lethally irradiating (900 cGy) host CD45.1×CD45.2 mice, which were then reconstituted with a 1:1 or 5:1 (for NKCre × Stat5fl/+ donors, which possess lower numbers of NK cells) KO:WT mixture of bone marrow cells from WT (CD45.1) and knockout (CD45.2) donor mice. Hosts were co-injected with anti-NK1.1 (clone PK136) to deplete residual mature NK cells.
METHOD DETAILS
Virus infection
MCMV (Smith strain) was serially passaged through BALB/c hosts twice, then viral stocks were prepared by dissociating salivary glands 3 weeks after infection with a dounce homogenizer. Intraperitoneal infections with MCMV were performed as previously described (Rapp et al., 2017).
Adoptive transfer studies
Adoptive transfer experiments were performed by mixing splenocytes from WT (CD45.1) and knockout (CD45.2) mice (or from mixed bone marrow chimeric mice) to achieve equal numbers of Ly49H+KLRG1lo NK cells, and injecting i.v. into adult Klra8−/− or Rag2−/− × Il2rg−/− mice. Recipient mice were then infected by i.p. injection of 7.5 × 102 PFU of MCMV. Experimental mixed bone marrow chimera mice were infected by i.p. injection of 7.5 × 103 PFU of MCMV. For survival studies, mice were infected by i.p. injection of 4 × 104 PFU of MCMV (Johnson et al., 2016).
Tamoxifen and antibody treatment
Mice were administered 4 mg tamoxifen dissolved in 200 μL corn oil by oral gavage. Control mice received 200 μL corn oil. Anti-IL-2 treatment was administered every second day with 300 μg anti-IL-2 (150 μg each of clone JES6–1A12 and S4B6–1, BioXCell). Anti-IL15 treatment was performed daily with 25 μg anti-IL-15 antibody (clone AIO.3, BioXCell).
Flow cytometry and cell sorting
Single cell suspensions were prepared and stained with indicated surface or intracellular antibodies (BD Biosciences, eBioscience, BioLegend, Tonbo, and R&D Systems), as previously described (Adams et al., 2019). Apoptosis was evaluated by caspase activity staining using the FLICA poly caspase assay kit (Immunochemistry Technologies). NK cell proliferation was analyzed by labeling cells with 5 μM Cell Trace Violet (CTV, Invitrogen) prior to transfer and infection. Flow cytometry was performed on an LSR II (BD). Cell sorting was performed on an Aria II cytometer (BD). All data were analyzed with FlowJo software (TreeStar). Flow cytometry of purified lymphocytes was performed using the following fluorophore-conjugated antibodies: CD3ε (17A2), TCRβ (H57–597), CD19 (ID3), F4/80 (BM8.1), NK1.1 (PK136), Ly49H (3D10), CD45.1 (A20), CD45.2 (104), CD11b (M1/70), CD27 (LG.3A10), KLRG1 (2F1), Ly49D (4E5), Ly49A (YE1/48.10.6), Ly49C/I (5E6), CD69 (H1.2F3), Granzyme A (3G8.5), Granzyme B (GB11), IFN-γ (XMG1.2), CD107a (1D4B), and CD49b (DX5).
ChIP-seq and analysis
Chromatin immunoprecipitation sequencing (ChIP-seq) for STAT4 and H3K4me3 were performed and described previously by our lab (Lau et al., 2018; Rapp et al., 2017). For the STAT5 ChIP, 4–5 × 106 NK cells (CD3ε− TCRb− CD19− F4/80− NK1.1+) were stimulated with or without 20 ng/mL recombinant mouse IL-2 (Fisher Scientific) plus 20 ng/ml recombinant mouse IL-15 (R&D Systems). DNA and proteins were cross-linked for 8 min at room temperature using 1% formaldehyde. Cross-linked cells incubated in cell lysis buffer (25 mM HEPES, 1.5 mM MgCl2, 10mM KCl, 0.1% NP-40, 1mM DTT, 1× proteinase inhibitor) and nuclei were isolated by centrifugation at 5400 rpm for 5.5 min. Chromatin was sheared using a Covaris ultrasonicator. ChIP was performed using 8 μg anti-Stat5 (#AF2168, R&D Systems) and Dynabeads Protein G (Invitrogen). Immunoprecipitated DNA was quantified by PicoGreen and the size was evaluated by Agilent BioAnalyzer. Whenever possible, fragments between 100 and 600 bp were size selected using aMPure XP beads (Beckman Coulter catalog # A63882) and Illumina libraries were prepared using the KAPA HTP Library Preparation Kit (Kapa Biosystems KK8234) according to the manufacturer’s instructions with up to 10ng input DNA and 8–11 cycles of PCR. Bar-coded libraries were run on a HiSeq 4000 in a 50bp/50bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina).
STAT5 ChIP-seq reads generated in this study and those generated elsewhere (GSE100674; Villarino et al., 2017) were trimmed to remove low quality reads using Trimmomatic (v.0.36) (Bolger et al., 2014). Trimmed reads were mapped to the Mus musculus genome (mm10 assembly) using Bowtie2 (v2.2.9) (Langmead and Salzberg, 2012), and MACS2 (v.2.1.1.20160309) (Zhang et al., 2008) was used with arguments “-f BAM -p 0.05 -m 2 50” for single-end peak calling. For each condition, irreproducible discovery rate (IDR) calculations using scripts provided by the ENCODE project (https://www.encodeproject.org/software/idr/; v2.0.3) (Li et al., 2011) were performed on all pairs of replicates using an oracle peak list derived from all samples within that condition, keeping reproducible peaks showing an IDR value of 0.05 or less in all pairs. A union of IDR-thresholded peaks generated from both unstimulated and stimulated conditions comprised the final peak list 1,201 peaks, annotated with ChipPeakAnno (v.3.8.9) (Zhu et al., 2010) using the UCSC Known Gene model (Speir et al., 2016).
RNA-seq and analysis
RNA-seq processing and analyses for MCMV time course and STAT4-deficient Ly49H+ NK cells were performed and described previously (Lau et al., 2018). RNA-seq processing and analyses from IL12R-deficient NK cells was performed in the same manner as STAT4-deficient NK cells described above. RNA was isolated from sorted CD45.1+ or CD45.2+ NK cells (TCRb− CD3− NK1.1+ Ly49H+) from WT versus NKCre × Stat5fl/+ splenocytes using the PicoPure RNA Isolation Kit (Thermo Fisher). After RiboGreen quantification and quality control by Agilent BioAnalyzer, 0.705–2ng total RNA with RNA integrity numbers ranging from 6.5 to 10 underwent amplification using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech catalog # 63488), with 12 cycles of amplification. Subsequently, 10ng of amplified cDNA was used to prepare libraries with the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) using 8 cycles of PCR. Samples were bar-coded and run on a HiSeq 4000 in a 50bp/50bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina).
RNA-seq sample reads were trimmed as above. Transcript quantification was based on the mm10 UCSC Known Gene model and performed using the quasi-mapping-based mode of Salmon (v.0.10.2) (Patro et al., 2017) correcting for potential GC bias. Transcripts were summarized to gene level using tximport (v.1.8.0) (Soneson et al., 2015). Differential analyses were executed with DESeq2 (v1.14.1) (Love et al., 2014). Normalized counts are raw counts scaled by per-sample size factors calculated by DESeq2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Data are shown as mean ± SEM in all graphs and statistical differences were calculated using a two-tailed unpaired Student’s t test, unless otherwise indicated. p values < 0.05 were considered significant. All statistical analyses and plots were produced in GraphPad Prism or R (v3.3.3).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD3ε (clone 17A2) | Tonbo Biosciences | Cat#25–0032; RRID: AB_2621619 |
| Anti-Mouse TCRβ (clone H57–597) | BioLegend | Cat#109220; RRID: AB_893624 |
| Anti-Mouse CD19 (clone 6D5) | BioLegend | Cat#115530; RRID: AB_830707 |
| Anti-Mouse F4/80 (clone BM8.1) | BioLegend | Cat#123117; RRID: AB_893489 |
| Anti-Mouse NK1.1 (clone PK136) | Tonbo Biosciences | Cat#65–5941; RRID: AB_2621910 |
| Anti-Mouse NKp46 (clone 29A1.4) | BioLegend | Cat#137604; RRID: AB_2235755 |
| Anti-Mouse Ly49H (clone 3D10) | eBioscience | Cat#11-5886-81; RRID: ABJ257160 |
| Anti-Mouse CD45.1 (clone A20) | BioLegend | Cat#110729; RRID: AB_1134170 |
| Anti-Mouse CD45.2 (clone 104) | BioLegend | Cat#109821; RRID: AB_493730 |
| Anti-Mouse CD49b (clone Dx5) | BioLegend | Cat#108918; RRID: AB_2265144 |
| Annexin V | BioLegend | Cat# 640943, RRID: AB_2616658 |
| Anti-Mouse/Human CD11b (clone M1/70) | BioLegend | Cat#101223; RRID: AB_755985 |
| Anti-CD27 (clone LG.7F9) | eBioscience | Cat#14-0271-81; RRID: AB_467182 |
| Anti-Mouse KLRG1 (clone 2F1) | Tonbo Biosciences | Cat#50–5893; RRID: AB_2621800 |
| Anti-Mouse Ly49D (clone 4E5) | BioLegend | Cat#138308; RRID: AB_10639939 |
| Anti-Mouse Ly49A (clone YE1/48.10.6) | BioLegend | Cat#116810; RRID: AB_572013 |
| Anti-Mouse Ly49C and Ly49l (clone 5E6) | BD Biosciences | Cat#553277; RRID: AB_394751 |
| Anti-Mouse CD69 (clone H1.2F3) | BioLegend | Cat#104524; RRID: AB_2074979 |
| Anti-Mouse Granzyme A (clone 3G8.5) | BioLegend | Cat#149704 RRID:AB_2565310 |
| Anti-Human/Mouse Granzyme B (clone GB11) | BioLegend | Cat#515403; RRID: AB_2114575 |
| Anti-Mouse IFN gamma (clone XMG1.2) | Tonbo Biosciences | Cat#20–7311; RRID: AB_2621616 |
| Anti-Mouse CD107a (clone 1D4B) | BioLegend | Cat#121611; RRID: AB_1732051 |
| Anti-Mouse Ki67 (Clone 16A8) | BioLegend | Cat# 652422; RRID: AB_2564490 |
| InVivoMab Anti-Mouse CD8α (NK cell enrichment, clone 2.43) | Bio X Cell | Cat#BE0061; RRID: AB_1125541 |
| InVivoMab Anti-Mouse CD4 (NK cell enrichment, clone GK1.5) | Bio X Cell | Cat#BE0003–1; RRID: AB_1107636 |
| InVivoMab Anti-Mouse CD19 (NK cell enrichment, clone 1D3) | Bio X Cell | Cat#BE0150; RRID: AB_10949187 |
| InVivoMab Anti-Mouse Ter-119 (NK cell enrichment, clone TER-119) | Bio X Cell | Cat#BE0183; RRID: AB_10949625 |
| Human/Mouse STAT5a/b Pan Specific Antibody (ChlP, polyclonal) | R&D Systems | Cat#AF2168; RRID: AB_355174 |
| Anti-STAT1 (M-22) (ChlP, polyclonal) | Santa Cruz Biotechnology | Cat#sc-592; RRID: AB_632434 |
| Bacterial and Virus Strains | ||
| Murine Cytomegalovirus (MCMV) | Johnson et al., 2016 | Smith Strain |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IL-12 Protein | R&D Sytems | Cat#419-ML |
| Recombinant Mouse IL-18 | MBL | Cat#B002–5 |
| Recombinant Mouse IL-2 Protein | R&D Systems | Cat#402-ML |
| Recombinant Mouse IL-15 Protein | R&D Systems | Cat#447-ML |
| Recombinant Mouse IFNa 1 Protein | R&D Systems | Cat#12105–1 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat#P8139 |
| lonomycin calcium salt from Streptomyces conglobatus (lonomycin) | Sigma-Aldrich | Cat#I0634 |
| Critical Commercial Assays | ||
| QIAamp DNA Blood Mini Kit | QIAGEN | Cat#51106 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#15596026 |
| PicoPure RNA Isolation Kit | Thermo Fisher Scientific | Cat# KIT0204 |
| Foxp3 Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat#00-5523-00 |
| iQ SYBR Green Supermix | Bio-Rad | Cat#1708880 |
| BioMag Goat Anti-Rat IgG (NK cell enrichment) | QIAGEN | Cat#310107 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat#C34557 |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat#65-0866-18 |
| FAM FLICA Poly Caspase Kit | Bio-Rad | Cat#ICT092 |
| Deposited Data | ||
| Raw Data Files for RNA and ChIP Sequencing | NCBI Gene | GSE106139 |
| Raw Data Files for RNA and ChIP Sequencing | Expression Omnibus | GSE142821 |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT or CD45.2: C57BL/6J | The Jackson Laboratory | Stock#000644; RRID:IMSR_JAX:000664 |
| Stat5fl/fl | Cui et al., 2004 | N/A |
| M2rbfl/fl | The Jackson Laboratory | Stock#029657 |
| H2rafi/fi | Chinen et al., 2016 | N/A |
| Mouse: Nkp46iCre | Narni-Mancinelli et al., 2011 | N/A |
| Mouse: IfnarT^: B6(Cg)-lfnar1tmr2Ees/J | The Jackson Laboratory | Stock#028288; RRID: IMSR_JAX:028288 |
| Mouse: Stati ~’- | Meraz et al., 1996 | N/A |
| Mouse: Rag2−/− IL2rg~’~: C;129S4- Rag2trnl.1Flv il>ml-IFIv/j |
The Jackson Laboratory | Stock#014593; RRID:IMSR_JAX:014593 |
| Mouse: M12rt)2 ^ | Wu et al., 2000 | N/A |
| Mouse: Stat4”^ | Kaplan et al., 1996 | N/A |
| y|-)CCre-ERT2. ^dori T9(UBC-Cre/ERT2)1 Eib | The Jackson Laboratory | Stock#007179 |
| Mouse: Klra8~’~ or Ly49H-deficient | Fodil-Cornu et al., 2008 | N/A |
| Oligonucleotides | ||
| Primers Forward MCMV IE-1: TCGCCCATCGTTTCGAGA | Johnson et al., 2016 | N/A |
| Primer Reverse MCMV IE-1: TCTCGTAGGTCCACTGACGGA | Johnson et al., 2016 | N/A |
| Software and Algorithms | ||
| DESeq2(v.1.14.1) | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Trimmomatic (v.0.36) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Bowtie2 (v.2.2.9) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Salmon (v.0.10.2) | Patro et al., 2017 | https://salmon.readthedocs.io/en/latest/salmon.html |
| tximport(v.1.8.0) | Soneson et al., 2015 | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| R (v.3.3.3) | https://www.r-project.org/ | https://www.r-project.org/ |
| MACS2(v.2.1.1.20160309) | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| ChipPeakAnno (v.3.8.9) | Zhu et al., 2010 | https://bioconductor.org/packages/release/bioc/html/ChlPpeakAnno.html |
| UCSC mm10 Known Gene Annotation Package(v.9) | Speir et al., 2016 | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Mmusculus.UCSC.mm10.knownGene.html |
| Irreproduoible Discovery Rate (IDR) (v.2.0.3) | Li et al., 2011 | https://www.encodeprojeot.org/software/idr/ |
Highlights.
STAT15 is induced in NK cells by IL-12 via STAT4 during MCMV infection
NK cells require STAT5 for protection against viral infection
IL-2 and IL-15 non-redundantly drive clonal proliferation of anti-viral NK cells
IL-15 and STAT5 promote NK cell survival during the memory phase
ACKNOWLEDGMENTS
G.M.W. was supported by a DFG (Deutsche Forschungsgemeinschaft) research fellowship (WI4927/1-1 and WI4927/2-1). C.M.L. was supported by the Cancer Research Institute as a Cancer Research Institute-Carson Family Fellow. M.R. was supported by a fellowship from the German Academic Exchange Service (DAAD). G.G. was supported by the DFG priority programme ILCs (GA2129/2-1), the DFG Emmy Noether programme (GA2129/2-1), and the European Research Council (ERCStG 759176). J.C.S. was supported by the Ludwig Center for Cancer Immunotherapy, the American Cancer Society, the Burroughs Wellcome Fund, and the NIH (AI100874, AI130043, and P30CA008748).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108498.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams NM, Geary CD, Santosa EK, Lumaquin D, Le Luduec J-B, Sot-tile R, van der Ploeg K, Hsu J, Whitlock BM, Jackson BT, et al. (2019). Cytomegalovirus Infection Drives Avidity Selection of Natural Killer Cells. Immunity 50, 1381–1390.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, and Ahmed R (2002). Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med 195, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihl F, Pecheur J, Bréart B, Poupon G, Cazareth J, Julia V, Glaichenhaus N, and Braud VM (2010). Primed antigen-specific CD4+ T cells are required for NK cell activation in vivo upon Leishmania major infection. J. Immunol 185, 2174–2181. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD, and Rudensky AY (2016). An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol 17, 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, and Hennighausen L (2004). Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol. Cell. Biol 24, 8037–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, Rülicke T, Mueller M, Casanova E, and Sexl V (2011). A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 117, 1565–1573. [DOI] [PubMed] [Google Scholar]
- Fodil-Cornu N, Lee S-H, Belanger S, Makrigiannis AP, Biron CA, Buller RM, and Vidal SM (2008). Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol 181, 6394–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann MC, Migone TS, Russell SM, and Leonard WJ (1996). Different interleukin 2 receptor beta-chain tyrosines couple to at least two signaling pathways and synergistically mediate interleukin 2-induced proliferation. Proc. Natl. Acad. Sci. USA 93, 2077–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, and Rudensky AY (2013). IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J. Exp. Med 210, 1167–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotthardt D, and Sexl V (2016). STATs in NK-Cells: The Good, the Bad, and the Ugly. Front. Immunol 7, 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotthardt D, Putz EM, Grundschober E, Prchal-Murphy M, Straka E, Kudweis P, Heller G, Bago-Horvath Z, Witalisz-Siepracka A, Cumaraswamy AA, et al. (2016). STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 6, 414–429. [DOI] [PubMed] [Google Scholar]
- Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, and Leonard WJ (1998). Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J. Exp. Med 188, 2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LR, Weizman OE, Rapp M, Way SS, and Sun JC (2016). Epitope-Specific Vaccination Limits Clonal Expansion of Heterologous Naive T Cells during Viral Challenge. Cell Rep. 17, 636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Bacon CM, Finbloom DS, Rees RC, Kaplan D, Shibuya K, Ortaldo JR, Gupta S, Chen YQ, Giri JD, et al. (1995). Tyrosine phosphorylation and activation of STAT5, STAT3, and Janus kinases by interleukins 2 and 15. Proc. Natl. Acad. Sci. USA 92, 8705–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, and Grusby MJ (1996). Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382, 174–177. [DOI] [PubMed] [Google Scholar]
- Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, et al. (2000). Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med 191, 771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koka R, Burkett PR, Chien M, Chai S, Chan F, Lodolce JP, Boone DL, and Ma A (2003). Interleukin (IL)-15R[α]-deficient natural killer cells survive in normal but not IL-15R[α]-deficient mice. J. Exp. Med 197, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kündig TM, Schorle H, Bachmann MF, Hengartner H, Zinkernagel RM, and Horak I (1993). Immune responses in interleukin-2-deficient mice. Science 262, 1059–1061. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, Adams NM, Geary CD, Weizman O-E, Rapp M, Pritykin Y, Leslie CS, and Sun JC (2018). Epigenetic control of innate and adaptive immune memory. Nat. Immunol 19, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Fragoso MF, and Biron CA (2012). Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL-12 induction of CD25 to form high-affinity IL-2 receptors on NK cells. J. Immunol 189, 2712–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard WJ, Lin JX, and O’Shea JJ (2019). The γc Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 50, 832–850. [DOI] [PubMed] [Google Scholar]
- Li QH, Brown JB, Huang HY, and Bickel PJ (2011). Measuring Reproducibility of High-Throughput Experiments. Ann. Appl. Stat 5, 1752–1779. [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, et al. (1996). Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84, 431–442. [DOI] [PubMed] [Google Scholar]
- Mitchell DM, Ravkov EV, and Williams MA (2010). Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J. Immunol 184, 6719–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandagopal N, Ali AK, Komal AK, and Lee S-H (2014). The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front. Immunol 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. (2011). Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. USA 108, 18324–18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KB, Cousens LP, Doughty LA, Pien GC, Durbin JE, and Biron CA (2000). Interferon alpha/beta-mediated inhibition and promotion of interferon gamma: STAT1 resolves a paradox. Nat. Immunol 1, 70–76. [DOI] [PubMed] [Google Scholar]
- O’Sullivan TE, Johnson LR, Kang HH, and Sun JC (2015). BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 43, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp M, Lau CM, Adams NM, Weizman O-E, O’Sullivan TE, Geary CD, and Sun JC (2017). Core-binding factor β and Runx transcription factors promote adaptive natural killer cell responses. Sci. Immunol 2, eaan3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp M, Wiedemann GM, and Sun JC (2018). Memory responses of innate lymphocytes and parallels with T cells. Semin. Immunopathol 40, 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Lin J-X, Feng D, Mitra S, Rickert M, Bowman GR, Pande VS, Li P, Moraga I, Spolski R, et al. (2012). Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat. Immunol 13, 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluns KS, Williams K, Ma A, Zheng XX, and Lefrançois L (2002). Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J. Immunol 168, 4827–4831. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. (2016). The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 44, D717–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Ohbo K, Nakamura M, and Takeshita T (1996). The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu. Rev. Immunol 14, 179–205. [DOI] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, and Lanier LL (2009a). Adaptive immune features of natural killer cells. Nature 457, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Ma A, and Lanier LL (2009b). Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J. Immunol 183, 2911–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino AV, Sciumè G, Davis FP, Iwata S, Zitti B, Robinson GW, Hennighausen L, Kanno Y, and O’Shea JJ (2017). Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J. Exp. Med 214, 2999–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Wang X, Gadina M, O’Shea JJ, Presky DH, and Magram J (2000). IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J. Immunol 165, 6221–6228. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study have been deposited in the Gene Expression Omnibus. Accession numbers for RNA-seq and ChIP-seq are GSE142821 and GSE140043, respectively.