

Abstract
Myeloproliferative neoplasms (MPNs) are hematopoietic stem cell disorders with dysregulated myeloid blood cell production and propensity for transformation to acute myeloid leukemia, thrombosis, and bleeding. Acquired mutations in JAK2, MPL, and CALR converge on hyperactivation of Janus kinase 2 (JAK2) signaling as a central feature of MPN. Accordingly, JAK2 inhibitors have held promise for therapeutic targeting. After the JAK1/2 inhibitor ruxolitinib, similar JAK2 inhibitors as fedratinib are entering clinical use. While patients benefit with reduced splenomegaly and symptoms, disease-modifying effects on MPN clone size and clonal evolution are modest. Importantly, response to ruxolitinib may be lost upon treatment suggesting the MPN clone acquires resistance. Resistance mutations, as seen with other tyrosine kinase inhibitors, have not been described in MPN patients suggesting that functional processes reactivate JAK2 signaling. Compensatory signaling, which bypasses JAK2 inhibition, and other processes contribute to intrinsic resistance of MPN cells restricting efficacy of JAK2 inhibition overall. Combinations of JAK2 inhibition with pegylated interferon-α, a well-established therapy of MPN, B-cell lymphoma 2 inhibition, and others are in clinical development with the potential to enhance therapeutic efficacy. Novel single-agent approaches targeting other molecules than JAK2 are being investigated clinically. Special focus should be placed on myelofibrosis patients with anemia and thrombocytopenia, a delicate patient population at high need for options. The extending range of new treatment approaches will increase the therapeutic options for MPN patients. This calls for concomitant improvement of our insight into MPN biology to inform tailored therapeutic strategies for individual MPN patients.
Pathogenesis of myeloproliferative neoplasms
Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell disorders characterized by excessive generation of mature myeloid blood cells. Three subtypes are recognized, including essential thrombocythemia (ET) presenting with isolated thrombocytosis, polycythemia vera (PV) primarily with polyglobulia, as well as primary myelofibrosis (PMF) with progressive bone marrow fibrosis-inducing cytopenias.1,2 ET and PV can progress to myelofibrosis (MF) and all 3 forms have a propensity to transform to acute myeloid leukemia (AML). There is an increased risk for thrombotic and hemorrhagic events relevantly contributing to morbidity and mortality.
On the molecular level, hyperactivation of the Janus kinase 2 (JAK2) signaling pathway is a central feature of MPN.3 JAK2, a nonreceptor tyrosine kinase, is essential for hematopoietic cytokine signaling4 by propagating activation of erythropoietin, thrombopoietin (myeloproliferative leukemia virus [MPL]), and granulocyte-macrophage colony-stimulating factor receptors. The JAK2 V617F mutation is present in 95% of PV and 50%-60% of ET and PMF patients5-8 and induces activation of the JAK2 kinase domain by relieving inhibitory effects of the pseudokinase domain.9 JAK2 exon 12 mutations induce JAK2 activation in the majority of JAK2 V617F unmutated PV.10 In ET and MF, the chaperone calreticulin (CALR) and the thrombopoietin receptor MPL are mutated in 30% and 10% of patients, respectively. Activating mutations of MPL such as W515L converge on JAK2 signaling enhancing analogous pathways as JAK2 V617F.11,12 In CALR13 multiple mutations in exon 9 were identified, which fall in 2 broad categories of type 1 with a 52 base pair deletion or type 2 with a 5 base pair insertion. Altered charge of the CALR C-terminus promotes retention of the mutant CALR in the endoplasmic reticulum, where it can stabilize MPL, thus enhancing MPL-JAK2 signaling.14 As all known driver mutations converge on activated JAK2, the JAK2 signaling pathway represents a key target for therapy. Genetic deletion of Jak2 in a MPLW515L-driven MPN murine model consistently ablated the MPN clone, suggesting an essential role of activated JAK2 signaling.15 There remains a small group of patients with “triple-negative” (ie, JAK2 V617F, CALR, and MPL unmutated) ET or PMF. While triple-negative PMF associates with adverse prognosis,16 this is less clear in ET where low burden canonical driver mutations as well as atypical JAK2 or MPL mutations or polyclonal hematopoiesis have been described.17,18
Additional mutations prevalently observed across myeloid malignancies, frequently occur also in MPN, particularly in MF, and impact on disease dynamics and prognosis. Mutations in ASXL1, EZH2, SRSF2, IDH1, and IDH2 have an adverse prognostic impact, therefore, termed “high molecular risk” mutations.19 Negative prognosis has also been attributed to higher numbers of concomitant mutations.20 The order of acquisition of specific mutation seems to play a significant role influencing progenitor cell proliferation, clinical presentation, and risk of thrombosis.21,22 Several mutations such as TET2 and DNMT3A may occur before or after JAK2 V617F during MPN development. While “JAK2-first patients” seem to predominantly develop PV-phenotype,21 patients with DNMT3A or TET2 mutations acquired before JAK2V617F may mostly present with an ET-phenotype.22,23 At the time of transformation to secondary AML, TP53, and IDH mutations are frequently detected.
JAK2 inhibitors to target MPN pathogenesis
The central role of activated JAK2 signaling in MPN has fueled the development of JAK2 inhibitors. Ruxolitinib, a JAK1/JAK2 inhibitor approved for the treatment of MF24 and hydroxyurea resistant or intolerant PV25, has initiated a new era of molecularly targeted therapy for MPN. JAK1/2 inhibition by ruxolitinib excels by effectively reducing splenomegaly and constitutional symptoms which impact on MPN patients’ quality of life. The need for phlebotomies is lowered in PV.25 Based on the COMFORT studies in MF and the RESPONSE studies in PV, ruxolitinib represents a current standard in these entities, whereas ruxolitinib was not found beneficial so far for high-risk ET (MAJIC-ET).26 Upon longer follow-up, overall survival of MF patients is extended by ruxolitinib therapy.27 The basis for this benefit is not conclusively understood, as ruxolitinib decreases the malignant MPN clone only in a limited number of patients.28,29 Clonal evolution is not halted suggesting a rather limited disease-modifying potential.30,31
Ruxolitinib represents a type I JAK2 inhibitor targeting JAK2 in its active, phosphorylated form.32 Cell-based investigations suggest that the new type I JAK2 inhibitors, which are currently entering clinical use, may share limitations of ruxolitinib and may not be able to decrease the size or the mutational evolution of the MPN clone.33,34 Other principles of JAK2 inhibition like type II JAK2 inhibitors, which target the kinase in the inactive conformation, or mutant selective JAK2 inhibition need to be evaluated for their potential to suppress oncogenic JAK2 signaling more profoundly. Genetic ablation of mutant Jak2 in MPLW515L-driven MPN mouse model abrogates the MPN clone which implies JAK2 dependence of the MPN clone, confirming JAK2 as a central therapeutic target, at least in the mouse models.15
Given current JAK2 inhibitors are not mutant selective, ruxolitinib represents a treatment option both for JAK2 mutated and unmutated MPN patients but relates to anemia and thrombocytopenia. These cytopenias are most prominent at treatment initiation and may stabilize in the further course. Another limitation increasingly evident in clinical practice is loss of responses to ruxolitinib which may occur over time. As shown by long-term follow-up of COMFORT-I, 50% of MF patients loose response over 5 years27 suggesting the MPN clone acquires resistance. It is an active area of research which underlying mechanisms drive emergence of resistance to ruxolitinib, as discussed in detail below. As indicated by in vitro studies, cross-resistance extending to new type I JAK2 inhibitors may also occur. Attention has been raised to an increased incidence of herpes zoster as well as nonmelanoma skin cancer in MPN patients treated with ruxolitinib.35,36 It is hypothesized that these rare side effects relate to reduced immune surveillance upon JAK1 inhibition with ruxolitinib. In contrast, ruxolitinib-mediated JAK1 inhibition is increasingly utilized for treatment of graft versus host disease to mitigate alloimmune effects.
Despite its limitations, ruxolitinib brings relevant clinical benefit to MPN patients, and similar type I JAK2 inhibitors are currently being developed (Table 1). Fedratinib, a JAK2/FMS-like tyrosine kinase 3 (FLT3) inhibitor, has recently been approved for the treatment of intermediate or high-risk MF. As demonstrated in the JAKARTA trials, its main benefit is in reducing splenomegaly, while the most prevalent adverse events are anemia and thrombocytopenia.39 Gastrointestinal side effects relate to its FLT3 inhibitory activity. The approval of fedratinib gives us a second JAK2 inhibitor at hand and broadens our options for targeted therapy of MPN. Of note, the risk of Wernicke’s encephalopathy needs to be considered as highlighted in a black box warning, as clinical development was intermittently halted due to cases of this serious complication. Interference of fedratinib with intestinal thiamine uptake has been reported, and although not conclusive, thiamine levels require monitoring upon fedratinib treatment. The upcoming clinical use of fedratinib in ruxolitinib pretreated patients will inform us on its benefit in patients who lost response to ruxolitinib, even though cell-based studies suggest cross-resistance among these type I JAK2 inhibitors.33,34
Table 1.
Selected Clinical Studies With JAK Inhibitors in MPN.
| JAK Inhibitors | Clinical Trial | Design | Clinical Impact | Main Side Effects |
|---|---|---|---|---|
| Ruxolitinib | MF: COMFORT-I phase 324,27 | vs Placebo | Spleen volume reduction Reduction of constitutional symptoms Survival benefit | Anemia, thrombocytopenia |
| MF: COMFORT-II phase 335,37 | vs BAT | |||
| PV: RESPONSE phase 325,36 | vs BAT | Spleen volume reduction Hematocrit control Reduction of constitutional symptoms | ||
| PV: RESPONSE-2 phase 338 | vs BAT | |||
| ET: MAJIC-ET phase 226 | vs BAT | Nonsignificant benefit (complete hematologic response) | ||
| Fedratinib | MF: JAKARTA phase 339 | vs Placebo | Spleen volume reduction | Anemia, gastrointestinal, encephalopathy (black box warning!) |
| Rux naive | ||||
| MF: JAKARTA-2 phase 240 | vs Placebo | Reduction of constitutional symptoms | ||
| Rux refractory | ||||
| Momelotinib | MF: SIMPLIFY-1 phase 341 | vs Rux | Spleen volume reduction (n.s.) Reduction of constitutional symptoms (n.s.) Decreased transfusion dependence | Peripheral neuropathy, anemia, thrombocytopenia |
| Rux naive | ||||
| MF: SIMPLIFY-2 phase 242 | vs BAT | |||
| Rux refractory | ||||
| Pacritinib | MF: PERSIST-1 phase 343 | vs BAT | Spleen volume reduction Reduction of constitutional symptoms Decreased transfusion dependence | Anemia, thrombocytopenia, gastrointestinal |
| MF: PERSIST-2 phase 344 | vs BAT, in thrombocytopenia |
Key studies on JAK2 inhibitors in clinical development are indicated including design, main clinical impact, and most important side effects.
BAT = best available therapy; ET = essential thrombocythemia; JAK = Janus kinase; MF = myelofibrosis; MPNs = myeloproliferative neoplasms; n.s. = non significant; PV = polycythemia vera; Rux = ruxolitinib.
Momelotinib and pacritinib are JAK2 inhibitors with promising profiles for treatment of MF patients with anemia and thrombocytopenia. As they represent a delicate population at high need for options, JAK2 inhibitors tolerable in this setting are desirable. Momelotinib is a JAK1/JAK2 inhibitor similar to ruxolitinib. Additionally, it seems to mitigate anemia via inhibition of type I activin A receptor and decreased hepcidin production.45 Momelotinib has shown benefits in promoting transfusion independence in MF in the SIMPLIFY trials. However, it has not met the endpoint of ≥35% splenomegaly reduction.41,42 Low-grade peripheral neuropathy appeared in almost half of the patients and was mostly irreversible highlighting that benefits and risks need to be well weighed. Pacritinib is a JAK2/FLT3 inhibitor with a particularly nonmyelosuppressive profile.46 In patients with thrombocytopenia ≤100 × 109/L, pacritinib was tolerable and effective in reducing spleen volume and symptom burden.44 After a clinical hold on the PERSIST studies has been lifted, a renewed phase 3 study in MF with thrombocytopenia is ongoing (PACIFICA). Other type I JAK2 inhibitors such as NS-018 targeting JAK2 and SRC kinases are also in clinical development in line with the central role of JAK2 activation in MPN.
Resistance to JAK inhibitors
Resistance to tyrosine kinase inhibitors represents a key challenge for the therapy of hematological malignancies and solid tumors47 and also affects JAK2 inhibitors. It has been shown that response to ruxolitinib is lost in 50% of patients who initially benefit in a 5-year period.27,35 Several mechanisms for occurrence of acquired resistance have been proposed.48 Furthermore, JAK2 inhibition is also hampered by intrinsic resistance of MPN cells interfering with therapeutic efficacy of ruxolitinib overall (Figure 1 and Table 2). The molecular basis of intrinsic resistance to JAK2 inhibition is increasingly understood and proposes targets for combination therapy approaches to enhance therapeutic efficacy.
Figure 1.
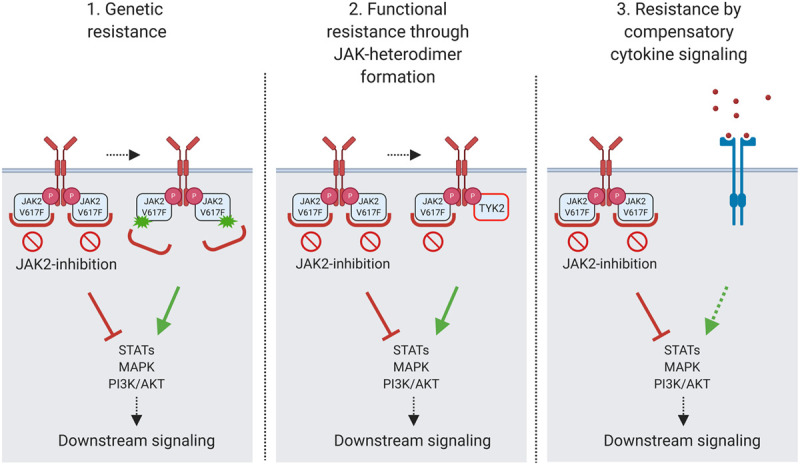
Mechanisms of resistance to Janus kinase 2 (JAK2) inhibitors. Resistance to JAK2 inhibition may develop via acquisition of JAK2 resistance mutations (genetic resistance) or via the formation of JAK family heterodimers of JAK2 with JAK1 or tyrosine kinase 2 (TYK2), which functionally sustain downstream signaling (adaptive resistance). Intrinsic resistance to JAK2 inhibition may relate to compensatory activation of downstream signaling pathways as, for example, the mitogen-activated protein kinase (MAPK) pathway due to bypass activation via receptor tyrosine kinases as, for example, platelet-derived growth factor receptor. The figure was created with BioRender. PI3K = phosphoinositide 3-kinase; STAT = signal transducer and activator of transcription.
Table 2.
Mechanisms of Resistance to JAK Inhibitors.
| Type of Resistance | Resistance Mechanism | In Vitro/In Vivo | Reversible | References |
|---|---|---|---|---|
| Primary (intrinsic) resistance | Preexisting resistance mutations | Reported in hereditary thrombocytosis | No | Marty et al49 |
| Bypass signaling via RTK (eg, PDGFR) | +/+ | No | Stivala et al50 | |
| Protective effects of cytokines | +/+ | Not known | Manshouri et al51 | |
| Bone marrow-fibrocyte-resistance in myelofibrosis | +/+ | Not known | Verstovsek et al52 | |
| Secondary (acquired) resistance | Acquired resistance mutations | +/– | No | Weigert et al53 |
| Deshpande et al54 | ||||
| Functional resistance by formation of JAK-heterodimers | +/+ | Yes | Koppikar et al34 | |
| Meyer et al33 |
Resistance to JAK inhibitors in myeloproliferative neoplasm may occur via intrinsic (primary) mechanisms or may be acquired (secondary) upon prolonged exposure to JAK inhibitors.
JAK = Janus kinase; PDGFR = platelet-derived growth factor receptor; RTK = receptor tyrosine kinase.
Acquired resistance to JAK2 inhibition
Genetic resistance
Acquisition of second-site mutations interfering with inhibitor binding have been described in several tyrosine kinases targeted by inhibitors such as BCR-ABL. Of note, second-site mutations in JAK2, which mediate resistance to JAK2 inhibition have not been detected in MPN patients so far.55 In vitro mutagenesis screens detected resistance mutations in JAK2 including Y931C, G935R, and E864K, which induced partial cross-resistance with other type I JAK2 inhibitors as fedratinib and momelotinib (Figure 1). Interestingly, the JAK2 “gate-keeper“ mutation M929I conferred resistance to ruxolitinib, but not to other JAK2 inhibitors.53,54 In rare patients with hereditary thrombocytosis, germline JAK2 kinase domain mutations mediate insensitivity to JAK2 inhibitors.49 However, these mutations are not acquired under treatment but are preexistent. It is not completely understood why JAK2 resistance mutations are not observed in patients on ruxolitinib although occurring in vitro. Insufficient selective pressure of JAK2 inhibition may play a role.
Adaptive resistance by JAK heterodimer formation
MF patients losing response to ruxolitinib were reported to regain sensitivity after pausing JAK2 inhibition with renewed benefit upon reexposure.56 This observation suggests that acquired resistance relates to adaptive, reversible processes in MPN cells. Long-term exposure to JAK2 inhibitors, including ruxolitinib, fedratinib, and momelotinib, induces resistance of MPN cells via formation of heterodimers of JAK2 with other JAK family members such as JAK2-JAK1 and JAK2-tyrosine kinase 2 (TYK2) able to reactivate JAK2 signaling in presence of JAK2 inhibitors33,34 (Figure 1). This adaptation at the signaling level has been confirmed in primary samples from MPN patients on ruxolitinib as well as in murine models. Resistant MPN cells remain dependent on JAK2 and are still affected by JAK2 knockdown.34 Adaptive resistance via JAK heterodimer formation was shown to be reversible, which fits the clinical observation of renewed sensitivity to ruxolitinib after a drug holiday.56 Intermittent treatment with JAK2 inhibition may represent an appealing approach. However, cytokine rebound upon cessation of ruxolitinib poses a challenge to this strategy and therapeutic effects may be impeded by reduced target inhibition.57
It has recently been shown that JAK2V617F, unlike wildtype JAK2, has the ability to induce ligand-independent dimerization of the homodimer type I cytokine receptors, which cannot be prevented by ruxolitinib. JAK2 pseudokinase domain seems to be indispensable for the ligand-independent dimerization.58 Besides further elucidating the mechanism of pathogenesis in MPN, this phenomenon may also contribute to the occurrence of resistance to JAK2 inhibitors.
Intrinsic resistance to JAK2 inhibition
Resistance mechanisms intrinsic to MPN cells which moderate the impact of JAK2 inhibitors, are increasingly understood and propose additional factors, which also need to be targeted to increase therapeutic efficacy of JAK2 inhibitor therapy.
Compensatory mitogen-activated protein kinase pathway activation
Bypass signaling via cell surface tyrosine kinase receptors is involved in resistance to kinase inhibitors in several cancers. Platelet-derived growth factor receptor alpha (PDGFRα) remains activated in MPN in vivo settings upon treatment with ruxolitinib. While JAK2 inhibitors effectively suppress the mitogen-activated protein kinase (MAPK) signaling pathway in vitro, platelet-derived growth factor-PDGFRα signaling is able to bypass JAK2 inhibition in MPN mouse models and mediates compensatory activation of the MAPK pathway (Figure 1). Combined targeting of JAK2 and MAPK/ERK kinase 1/2 (MEK1/2), intermediate kinases in the MAPK pathway, improves therapeutic efficacy of ruxolitinib, suggesting that compensatory MAPK pathway activation is limiting the effects of JAK2 inhibitor therapy.50 These findings propose the MAPK pathway as a mediator of resistance to JAK2 inhibition, which needs to be targeted to improve efficacy of JAK inhibitor therapy.
Resistance mediated by the bone marrow microenvironment
Mechanisms of resistance to JAK2 inhibition may also relate to the bone marrow (BM) microenvironment constituted by multiple components including hematopoietic cells as well as stroma with mesenchymal stem cells, endothelium, osteoblasts, neurons, and Schwann cells. Increased levels of inflammatory cytokines are characteristic of the BM microenvironment in MPN patients and murine models.59-61 Paracrine effects from the inflammatory milieu of the BM microenvironment on the MPN clone have been implicated in JAK2 inhibitor resistance. Primary JAK2V617F mononuclear cells from a PMF patient co-cultured with stromal cells were spared from inhibitory effects of a JAK2/JAK3 inhibitor, while sensitivity was restored upon neutralization of inflammatory cytokines suggesting protective effects of the inflammatory niche on the MPN clone.51 Cellular components could also interfere with JAK2 inhibitor efficacy. JAK2V617F positive fibrocytes in bone marrow are not decreased by ruxolitinib suggesting they could contribute to JAK2 inhibitor resistance.52 Of note, an impact of the sympathetic nervous system on protective nestin+ mesenchymal stem cells,62 which support expansion of human HSCs,63 has also been reported. The loss can be restored by treatment with β3-adrenergic agonist, but not with ruxolitinib,62 suggesting this aspect of MPN pathogenesis could also contribute to the ruxolitinib resistance. Further studies are warranted to understand the specific roles of BM microenvironment components in resistance to JAK2 inhibition.
Ruxolitinib and clonal evolution in MPN
The order by which mutations are acquired by the MPN clone impact on proliferation of hematopoietic progenitors, clinical presentation, and risk of thrombotic complications, and in addition, could also be important for sensitivity to JAK2 inhibitor therapy. It has been observed that cells from JAK2/TET2 double mutant MPN patients responded better to ruxolitinib when JAK2V617F was acquired first.21 Overall, current JAK2 inhibitors only modestly impact on MPN clone size, although the MPN clone remains JAK2-dependent in the resistance setting.28 In concordance with the notion of limited disease-modifying potential, ruxolitinib does not prevent clonal evolution of MPN with acquisition of additional mutations.30,31 In PMF, 35% of patients develop at least one additional mutation during ruxolitinib treatment, mostly in ASXL1, TET2, EZH2, and TP53. Notably, of these, ASXL1, EZH2, and TP53 mutations associate with adverse prognosis. Clonal evolution with mutations in IDH1, IDH2, and DNMT3A has also been described.20 It becomes evident that clonal evolution in MPN, also when occurring on JAK2 inhibitor treatment, directly impacts on prognosis with shortened survival. This highlights the limited disease-modifying potential of JAK2 inhibitor single-agent therapy calling for enhanced treatment strategies.
Novel therapeutic approaches in MPN treatment
To improve therapeutic efficacy, combination therapy approaches addressing additional targets besides JAK2 as well as innovative single-agent therapies need to be explored. Insight into MPN biology has deepened in recent years, and mechanisms of resistance are increasingly understood. Many new targets for therapeutic intervention have been proposed and clinical and preclinical investigations are under way (Tables 3 and 4 and Figure 2).
Table 3.
Combination Therapies With Ruxolitinib for MPN in Clinical Development.
| Co-Target | Drug Combination | Eligible Diagnosis (MPN) | Study Phase | Clinical Trial Identifier |
|---|---|---|---|---|
| Pegylated IFN-α-2a | Peg-IFN-α-2a + ruxolitinib | MF | 2 | NCT02742324 |
| PV, MF | 2 | EudraCT2013003295-12 | ||
| Hypomethylating agent | Azacitidine + ruxolitinib | MF | 2 | NCT01787487 |
| Decitabine + ruxolitinib/fedratinib | AP/BP MPN | 1/2 | NCT04282187, NCT02076191 | |
| IDH2 | Enasidenib + ruxolitinib | AP/BP MPN, MF | 2 | NCT04281498 |
| IMiDs | Thalidomide + ruxolitinib | MF | 2 | NCT03069326 |
| Pomalidomide + ruxolitinib | MF | 1/2 | NCT01644110 | |
| HDAC | Panobinostat + ruxolitinib | MF | 1 | NCT01693601, NCT01433445 |
| BCL-2/BCL-xL | Navitoclax +/– ruxolitinib | PV, ET, MF | 1 | NCT04041050 |
| MF | 2 | NCT03222609 | ||
| PI3K | Idelalisib + ruxolitinib | MF | 1 | NCT02436135 |
| Parsaclisib + ruxolitinib | MF | 2 | NCT02718300 | |
| Umbralisib + ruxolitinib | PV, MF | 1 | NCT02493530 | |
| JAK1 | Itacitinib + ruxolitinib | MF | 2 | NCT03144687 |
| BET | CPI-0610 +/– ruxolitinib | MF | 1/2 | NCT02158858 |
| GSK-3β | 9-ING-41 +/– ruxolitinib | MF | 2 | NCT04218071 |
| NFκB | Pevonedistat + ruxolitinib | MF | 1 | NCT03386214 |
| HSP90 | PU-H71 + ruxolitinib | MF | 1 | NCT03373877 |
| PIM or CDK4/6 | PIM447/LEE011 + ruxolitinib | MF | 1 | NCT02370706 |
| MDM2/P-selectin/TGF-β | Siremadlin, crizanlizumab, or MBG453 + ruxolitinib | MF | 1/2 | NCT04097821 |
Overview of currently active clinical studies evaluating combination therapies of ruxolitinib plus another agent targeting molecules involved in the pathogenesis of MPNs.
AP/BP = accelerated phase/blast phase; BCL-2/BCL-xL = B-cell lymphoma 2/B-cell lymphoma-extra large; BET = bromodomain and extraterminal domain; CDK4/6 = cyclin-dependent kinase 4/6; ET = essential thrombocythemia; GSK-3β = glycogen synthase kinase-3β; HDAC = histone deacetylase; HSP90 = heat shock protein 90; IMiDs = immunomodulatory imide drugs; IDH2 = isocitrate dehydrogenase 2; IFN-α-2a = interferon-alpha-2a; JAK1 = Janus kinase 1; MDM2 = mouse double minute 2 homolog; MF = myelofibrosis; MPNs = myeloproliferative neoplasms; NFκB = nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K = phosphoinositide 3-kinase; PIM = proviral integration site for Moloney murine leukemia virus; PV = polycythemia vera; TGF-β = transforming growth factor beta.
Table 4.
Novel Single-Agent Therapies for MPN in Clinical Development.
| Target Category | Target | Drug | Eligible Diagnosis (MPN) | Study Phase | Clinical Trial Identifier |
|---|---|---|---|---|---|
| JAK kinases | JAK1/2 | Momelotinib vs ruxolitinib | MF | 3 | NCT01969838 |
| Momelotinib vs danazol | MF | 3 | NCT04173494 | ||
| NS-018 | MF | 1/2 | NCT01423851 | ||
| JAK2 | Fedratinib | MF | 3 | NCT03755518 | |
| Pacritinib | MF | 2,3 | NCT03165734 | ||
| NCT03645824 | |||||
| LY2784544 | PV, ET, MF | 2 | NCT01594723 | ||
| JAK1 | Itacitinib | MF | 2 | NCT01633372 | |
| Interferon-α | Pegylated interferon-α | Peg-IFN-α-2b vs IFN-α | ET | 4 | NCT04226950 |
| Peg-IFN-α-2a/-2b vs HU | PV, ET, MF | 3 | NCT01387763 | ||
| Peg-IFN-α-2a | PV, ET, MF | 2 | NCT00452023 | ||
| Pegylated-proline-interferon-α-2b | Ropeg-IFN-α-2b (AOP2014) vs BAT | PV | 2,3 | NCT02218047, NCT03003325 | |
| Ropeg-IFN-α-2b (P1101) | PV | 2 | NCT04182100 | ||
| ET | 3 | NCT04285086 | |||
| MF | 2 | NCT02370329 | |||
| Telomerase | Telomerase | Imetelstat | PV, ET | 2 | NCT01243073 |
| MF | 2 | NCT02426086 | |||
| Cell cycle | MDM2 | KRT-232 | PV, MF | 2 | NCT03662126 |
| NCT03669965 | |||||
| PIM kinase | TP-3654 | MF | 1 | NCT04176198 | |
| Aurora kinase | Alisertib | MF | 1 | NCT02530619 | |
| Exportin 1 | Selinexor | MF | 2 | NCT03627403 | |
| Epigenetics | HDAC | Givinostat | PV, ET, MF | 2 | NCT01761968 |
| LSD1 | IMG-7289 | PV, ET | 2 | NCT04262141 | |
| ET | 2 | NCT04254978 | |||
| MF | 2 | NCT03136185 | |||
| BET | INCB057643 | MF | 1 | NCT04279847 | |
| Fibrosis | TGF-β signaling | Sotatercept | MF | 2 | NCT01712308 |
| Luspatercept | MF | 2 | NCT03194542 | ||
| TGF-β trap (AVID200) | MF | 1/2b | NCT03895112 | ||
| SAP/pentraxin 2 | PRM-151 | MF | 2 | NCT01981850 | |
| Other targets | PD-1 | Pembrolizumab | MF | 2 | NCT03065400 |
| (TIM-3 + TGF-β) ± PD-1 | MBG453 + NIS793 ± spartalizumab/decitabine | MF | 1 | NCT04283526 | |
| CD123 | Tagraxofusp (SL-401) | MF | 2 | NCT02268253 | |
| SMAC mimetic | LCL161 | MF | 2 | NCT02098161 | |
| HSP90 | PU-H71 | MF, MPN | 1 | NCT03935555, NCT01393509 |
Overview of ongoing clinical studies investigating a therapeutic potential of single-agent therapies in MPNs.
BAT = best available therapy; BET = bromodomain and extraterminal domain; ET = essential thrombocythemia; HDAC = histone deacetylase; HSP90 = heat shock protein 90; HU = hydroxyurea; IFN-α = interferon-alpha; JAK = Janus kinase; LSD1 = lysine-specific histone demethylase 1; MDM2 = mouse double minute 2 homolog; MF = myelofibrosis; MPNs = myeloproliferative neoplasms; PIM = proviral integration site for Moloney murine leukemia virus; PD-1 = programmed cell death protein 1; PV = polycythemia vera; SAP = serum amyloid P component; SMAC = second mitochondria-derived activator; TGF-β = transforming growth factor beta; TIM-3 = T cell immunoglobulin and mucin domain-containing protein 3.
Figure 2.
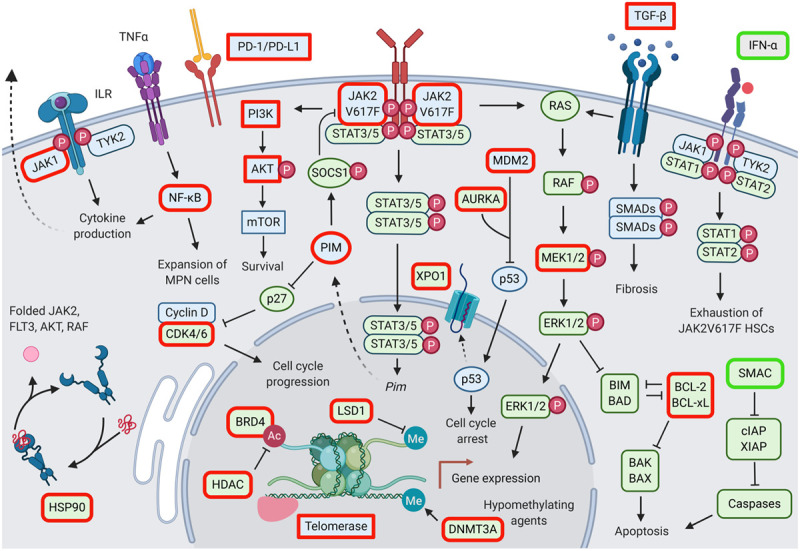
Potential therapeutic targets in myeloproliferative neoplasms. Targeted molecules highlighted in red are inhibited, targeted molecules highlighted in green are activated. Ac = acetyl group; BAD = BCL2-associated agonist of cell death; BAK = BCL2 antagonist/killer; BAX = BCL2 associated X; BCL-2 = B-cell lymphoma 2; BCL-xL = B-cell lymphoma-extra large; BIM = BCL2-interacting mediator of cell death; BRD4 = bromodomain-containing protein 4; CDK4/6 = cyclin-dependent kinase 4/6; cIAP = cellular inhibitor of apoptosis; DNMT3A = DNA methytransferase 3A; ERK1/2 = extracellular signal-regulated kinase 1/2; FLT3 = FMS-like tyrosine kinase 3; HDAC = histone deacetylase; HSCs = hematopoietic stem cells; HSP90 = heat shock protein 90; IFN-α = interferon-alpha; ILR = interleukin receptor; JAK = Janus kinase; LSD1 = lysine-specific histone demethylase 1; MDM2 = mouse double minute 2 homolog; Me = methyl group; MEK1/2 = MAPK/ERK kinase 1/2; MPNs = myeloproliferative neoplasms; mTOR = mammalian target of rapamaycin; NFκB = nuclear factor kappa-light-chain-enhancer of activated B cells; PD-1/PD-L1 = programmed cell death protein 1/programmed death ligand-1; PI3K = phosphoinositide 3-kinase; PIM = proviral integration site for Moloney murine leukemia virus; RAF = rapidly accelerated fibrosarcoma; RAS = rat sarcoma viral oncogene homolog; SMAC = second mitochondria-derived activator; SMADs = SMA- and MAD-related proteins; SOCS1 = suppressor of cytokine signaling 1; STAT = signal transducer and activator of transcription; TGF-β = transforming growth factor beta; TNF-α = tumor necrosis factor alpha; TYK2 = tyrosine kinase 2; XIAP = X-linked inhibitor of apoptosis.
Combination therapies with JAK2 inhibitors
Genetic studies in MPLW515L-driven MPN mouse model show that the MPN clone remains dependent on activated JAK2 signaling suggesting JAK2 should be targeted. Thus, JAK2 inhibitors represent a rational constituent of MPN therapy.15 Simultaneous targeting of additional factors involved in MPN development or JAK2 inhibitor resistance has the potential to synergize and enhance specific therapeutic aspects (Table 3).
Targeting parallel signaling pathways: phosphoinositide 3-kinase/AKT and mitogen-activated protein kinase
Activated JAK2 signaling in MPN promotes proliferation, differentiation, and cell survival via activation of phosphoinositide 3-kinase (PI3K)/AKT, signal transducer and activator of transcription 3/5, and MAPK signaling pathways. Both PI3K/AKT and MAPK signaling pathways have been implicated in limiting response to JAK2 inhibitors.64 Synergism of JAK2 and PI3K/AKT inhibition has been reported in MPN cells,65 and both AKT and PI3K inhibitors are active in MPN murine models.66,67 Preclinical studies demonstrated improved efficacy of combined PI3K inhibition by BEZ235 and ruxolitinib, leading to phase I/II clinical studies evaluating PI3K inhibitors such as idelalisib, parsaclisib, and umbralisib in combination with ruxolitinib in MF (Table 3).
MAPK signaling has been implicated in JAK2 inhibitor resistance in vitro.64 For in vivo settings, we have shown that PDGFRα signaling bypasses JAK2 inhibition mediating compensatory activation of MEK/extracellular signal-regulated kinases (ERKs). Thus, the MAPK pathway represents an additional therapeutic target in MPN which should be addressed to improve therapeutic efficacy of JAK2 inhibition. Combined JAK2 and MEK inhibition by ruxolitinib and binimetinib enhances treatment effects both in JAK2 and MPL mutant settings evident in murine models and clinical isolates from MPN patients.50 Combined JAK2/MEK inhibition was particularly effective in reducing bone marrow fibrosis.
Targeting epigenetic regulation
MPN associate with global perturbations of DNA methylation68 and mutations in epigenetic regulators as DNMT3A, ASXL1, EZH2, IDH1, IDH2, and TET2, which are frequent in other myeloid malignancies, are also prevalent in MPN.20 The hypomethylating agents (HMAs) azacytidine and decitabine are well-established for treatment of AML and myelodysplastic syndrome and are evaluated for beneficial effects in advanced MPN. Combined decitabine/ruxolitinib in accelerated or blast-phase MPN induced responses in a phase I study with median overall survival of 7.9 months.69 Azacytidine/ruxolitinib was studied as a combination in intermediate to high-risk MF showing reduction of splenomegaly and fibrosis.70 The combinations of ruxolitinib and HMA were generally well tolerated apart from cytopenias. Thus, combined HMA/ruxolitinib could provide an intensified targeted approach for patients with advanced MF and leukemic transformation, a population at a high need for options.
Pan-histone deacetylase inhibition is also explored in MPN to interfere at epigenetic levels in combination with JAK2 inhibition. Histone acetylation determines accessibility of DNA and transcriptional activity and histone deacetylase inhibitors such as panobinostat promote acetylation of histones H3 and H4 interfering with JAK2 signaling via increased acetylation of the chaperone heat shock protein 90 (HSP90). JAK2 represents a HSP90 client protein and preclinical studies showed superior effects of combined panobinostat/ruxolitinib in JAK2 mutant MPN.71 Subsequent phase I/II studies reported responses of splenomegaly and anemia in MF patients.72 A phase 2 study on long-term tolerability and efficacy of givinostat in MPN is currently active (NCT01761968).
Epigenetic mechanisms are specifically at play to promote the inflammatory milieu in MPN characterized by increased levels of inflammatory cytokines. Inflammation in MPN contributes to constitutional symptoms of patients60,61 and relates to activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling.68 Chromatin changes involving the histone lysine reader bromodomain-containing protein 4 (BRD4), a member of the bromodomain and extraterminal domain (BET) family of proteins, enhances NF-κB signaling, while BRD4/BET inhibition was able to reduce NF-κB pathway activity in MPN mouse models. Combined BRD4/JAK2 inhibition showed promising results in reducing fibrosis and mutant allele burden.73 Currently, the combination of the BRD4/BET inhibitor CPI-0610 and ruxolitinib is investigated in ruxolitinib pretreated and naïve MF patients in a phase 1/2 study (Table 3). Positive effects, particularly in regard to reduced constitutional symptoms, have been reported.74 Furthermore, combined BRD4/BET and JAK2 inhibition effectively induced apoptosis in blasts of patients with secondary AML progressed from MPN.75 Pevonedistat, which interferes with NF-κB signaling via inhibition of neuronal percursor cell-expressed developmentally down-regulated protein 8 activating enzyme, is also being tested in combination with ruxolitinib in a phase 1 study in MF (NCT03386214).
Promoting apoptosis
Inhibition of B-cell lymphoma 2 (BCL-2) family members, which are essential regulators of apoptotic cell death, was primarily developed for the treatment of lymphoid malignancies,76 but also holds potential for myeloid malignancies. The BCL-2 inhibitor venetoclax has shown important benefits in the treatment of AML77 and BCL-2 inhibition is increasingly explored in MPN. Preclinical studies in JAK2 mutant acute leukemia showed increased efficacy by combined BCL-2/JAK2 inhibition, and BCL-2 inhibition could overcome resistance to JAK2 inhibition in MPN cells.78 Clinical studies of navitoclax in combination with ruxolitinib or as a single agent are evaluating the potential in MF (Table 3) and encouraging results have been reported in regard to leukocytosis control in ruxolitinib pretreated MF patients.79
Targeting chaperone proteins
HSP90 is a chaperon protein responsible for correct folding of many signaling proteins including BCR-ABL, FLT3, AKT, and rapidly accelerated fibrosarcoma. PU-H71, a HSP90 inhibitor, induces degradation of JAK2 suggesting also JAK2 as a HSP90 client. Treatment with PU-H71 showed promising results in preclinical MPN models normalizing cytoses and improving survival without myelosuppression.80 HSP90 inhibition was also able to overcome acquired resistance to JAK2 inhibition.34,53 Several early clinical studies on combined PU-H71/ruxolitinib or PU-H71 as single agent in MF are currently active (Tables 3 and 4). Studies of the HSP90 inhibitor luminespib (AUY922) were halted due to occurrence of gastrointestinal bleeding.
Targeting cell cycle regulators
Proviral integration site for Moloney murine leukemia virus (PIM) kinases are implicated as oncogenes in several cancers and PIM activation enhances activation of cyclin-dependent kinase 4 (CDK4) and CDK6 in MPN. Given the excessive proliferation of hematopoietic cells in MPN, the PIM-CDK4/6 axis has been proposed as a therapeutic target.81 PIM inhibition showed synergistic effects with ruxolitinib in preclinical MPN models82 and triple inhibition of PIM, CDK4/6, and JAK2 is under evaluation in MF.83
Immune modulation
Interferon α (INFα) has long been used to treat MPN given its anti-proliferative and immunomodulatory effect. Furthermore, INFα is able to induce molecular remissions suggesting it as a modality with disease-modifying potential. IFNα appears to activate dormant HSCs in mice inducing exhaustion84 and elimination of Jak2V617F+ MPN-propagating stem cells.85 Despite these benefits, INFα therapy is hampered by flu-like side effects often leading to treatment discontinuation. Pegylated forms of IFNα have mitigated side effects and increased tolerability and adherence. A combination therapy approach of pegylated IFNα with ruxolitinib represents a promising approach and is explored in several studies. While IFNα has disease-modifying activity with reduced MPN clone size, JAK1/JAK2 inhibition with ruxolitinib effectively reduces proliferation and inflammatory symptoms including IFNα-induced side effects, which could lead to improved efficacy, disease modification, and tolerability. Clinical evaluation of pegylated IFNα-2a and ruxolitinib in a phase II study of PV and MF showed substantial reductions of JAK2V617F allele burden with 41% of patients showing a molecular response along with improved cytosis and fibrosis as well as acceptable toxicity (COMBI Study, Table 3).86 The ongoing RUXOPeg study so far showed efficacy in terms of splenomegaly and cytoses along with a favorable tolerability profile and JAK2V617F allele burden reductions at 6 months of treatment.87 These findings demonstrate that pegylated IFNα adds substantial clone suppression to ruxolitinib therapy and that the combination with ruxolitinib improves tolerability of IFNα therapy.
New approaches in MF patients with anemia and thrombocytopenia
Anemia and thrombocytopenia are predictors of shortened survival in MF60 and remain the most common adverse events in patients on ruxolitinib.24 MF patients with these conditions remain the most challenging group to treat. Sotatercept and luspatercept are chimeric trap molecules that prevent ligand binding to activin receptor IIa and IIb, respectively, promoting terminal erythroid differentiation.88,89 Thus, they represent promising anti-anemia agents and are currently being clinically evaluated in MF patients with anemia, both as a monotherapy and in combination with ruxolitinib (Table 4). Six out of 17 (35%) MF patients with anemia on sotatercept showed an anemia response.90 In addition, immunomodulatory imide drugs are investigated for anemia management in MF as single agents and in combination with JAK2 inhibition. Thalidomide and ruxolitinib improved platelet counts in 75% of the MF patients with thrombocytopenia and studies on pomalidomide combinations are ongoing.91 Novel type I JAK2 inhibitors, specifically momelotinib and pacritinib, appear less myelosuppressive and could provide valuable options for MF patients with anemia or thrombocytopenia.41-44
Single-agent therapies
Increasing insight into MPN biology is proposing a growing number of innovative targets for therapy. Several approaches are being tested as an alternative to JAK2 inhibition, rather than a combination with JAK2 inhibitors (Figure 2 and Table 4).
Pegylated interferon-α
Interferon-α has been utilized for MPN therapy with promising results decades before the advent of JAK2 inhibitors, although the specific mechanistic effects relating to interferon-α-mediated immunomodulation are still not entirely dissected. However, flu-like side effects have limited a more extensive use of interferon-α. Modified forms as pegylated IFN-α-2a (Peg-IFN-α-2a) have substantially improved tolerability and adherence to interferon-α in patients with ET and PV. Once weekly, instead of daily application enabled by a longer half-life and reduced side effects has lowered treatment-related burden and improved the net benefit of interferon-α therapy. Peg-IFN-α-2a as a single agent is able to induce molecular responses along with hematologic remissions and limited toxicity in a majority of PV and ET patients.92-94 There is increasing evidence that Peg-IFN-α-2a also represents a valid treatment approach in MF mediating decreased mutant allele burden and increased overall survival.95 Interestingly, JAK2V617F mutant MPN cells may preferentially respond to Peg-IFN-α-2a as compared to a CALR mutant setting. Higher molecular responses in JAK2V617F than CALR mutant patients have been described.96 Further studies are needed to elucidate the basis for disease modification by interferon-α in MPN.
Pegylated-proline-interferon-α-2b (Ropeg-IFN-α-2b) represents another pegylated form of interferon-α with biweekly application and favorable tolerability profile. Direct comparison of Ropeg-IFN-α-2b versus best available therapy, including hydroxyurea in PV, showed that Ropeg-IFN-α-2b induces meaningful molecular remissions after 3 years of treatment correlating with hematologic response, while tolerability was similar to best available therapy (PROUD-PV, CONTINUATION-PV).97 Ropeg-IFN-α-2b has been approved in Europe as monotherapy in PV and is investigated in a phase 2 study for MF. Direct comparison of Ropeg-IFN-α-2b to standard therapy in ET is ongoing (Table 4).
Telomerase inhibition
Innovative approaches have focused on interfering with telomere function by inhibition of telomerase. Imetelstat is a 13-mer oligonucleotide telomerase inhibitor covalently modified with lipid extensions. Imetelstat has been successfully tested in ET and MF patients, although patient numbers were small (n = 18 and 33, respectively). In ET, imetelstat induced complete hematologic response in 89%,98 while in MF, 21% of patients achieved hematologic remissions.99 Larger studies are awaited to consolidate these findings. The association of telomerase inhibition with significant myelosuppression poses a caveat for its broad use in ET patients, who are often oligo- or asymptomatic, as well as in MF patients given their prevalent preexisting cytopenias. A potential benefit of imetelstat in MF patients refractory to JAK2 inhibition is of interest and under evaluation (Table 4).
Anti-fibrotic agents
Bone marrow fibrosis represents a key feature of progressed MPN, particularly MF, mediating cytopenia. Cellular components, as well as soluble factors, have been implicated in fibrogenesis in MPN. Atypical megakaryocytes abundant in MF are thought to promote bone marrow fibrosis via secreted cytokines. Heterozygous deletion as well as pharmacologic inhibition of Aurora kinase A reduced fibrosis in MPN in vivo models.100 Alisertib, an Aurora kinase inhibitor evaluated in a phase 1 study in MF, normalized the megakaryocyte lineage and reduced bone marrow fibrosis validating Aurora kinase A as a therapeutic target in MF.101 In addition, clonal monocyte-derived fibrocytes have been implicated in bone marrow fibrosis in MF. Inhibition of fibrocyte differentiation by recombinant serum amyloid P component/pentraxin-2, reduced fibrosis in preclinical models.52 Clinical investigations of PRM-151, a recombinant human pentraxin-2 molecule, have shown good tolerability and improved bone marrow fibrosis so far (Table 4). Additional approaches to interfere with bone marrow fibrosis include direct targeting of the fibrogenic cytokine transforming growth factor beta (TGF-β). AVID200 is a recombinant TGF-β-receptor-Fc fusion protein acting as a TGF-β1/-β3 trap currently investigated in MF. Inhibition of glycogen synthase kinase-3β by a selective inhibitor 9-ING-41 has also shown potential to decrease TGF-β-induced fibrosis102 in MF.
Additional approaches for therapeutic targeting
Therapeutic approaches with evident benefit in AML or other hematologic malignancies are evaluated for efficacy in MPN. They may enhance apoptosis as the mouse double minute 2 homolog (MDM2) inhibitor idasanutlin, the nuclear export inhibitor selinexor, or second mitochondria-derived activator (SMAC) mimetics. MDM2 represents a negative regulator of the tumor suppressor P53 and shows increased expression in MPN.103 The MDM2 inhibitor KRT-232 is under investigation in MF (Table 4). Selective inhibitors of nuclear export processes have shown proapoptotic effects in leukemia settings.104 A clinical study in MF with the nuclear export inhibitor selinexor inhibitor is ongoing. Second mitochondria-derived activator mimetics interfere with regulators of caspases and beyond AML and myelodysplastic syndrome they hold promise in MF.105 Targeting epigenetic modification processes as with inhibitors of lysine-specific histone demethylase 1 as well as interference with cytokine signaling via interleukin 3 receptor CD123 by tagraxofusp106 has shown promising effects in hematologic malignancies as AML and is under investigation in MPN (Table 4). Immune checkpoint inhibition, which is a promising strategy in solid tumors and hematologic malignancies is explored also for MPN.107 Programmed death-ligand 1/programmed cell death protein 1 signaling seems to play an important role in immune escape in Jak2V167-driven MPN,108 while pembrolizumab, an anti-PD1 monoclonal antibody, is evaluated as monotherapy in MF (Table 4).
Alternative JAK2 inhibition
While novel targets involved in apoptosis, cell cycle, epigenetic regulation, and fibrosis are explored, JAK2 remains an important target for the therapy of MPN. Alternative modes to inhibit JAK2, which could provide enhanced efficacy selectively addressing the MPN clone while sparing wildtype hematopoiesis, are of ultimate need. JAK2 inhibitors in current clinical development represent type I JAK2 inhibitors binding to the adenosine triphosphate pocket in the active form of the kinase. Type II JAK2 inhibitors (CHZ868, BBT594) have recently been explored. They target the inactive conformation of JAK2 and are able to overcome resistance to ruxolitinib. CHZ868 has shown enhanced efficacy in JAK2 driven malignancies including MPN33 and B-cell acute lymphoblastic leukemia.109 Of note, type II JAK2 inhibition with CHZ868 preferentially inhibited mutant hematopoietic cells and decreased MPN clone size in preclinical models.33 However, due to their potency, type II JAK inhibitors could be associated with more pronounced cytopenias.110 Much attention is placed on the development of JAK2V617F mutation-specific inhibitors which could spare normal hematopoiesis and selectively target the MPN clone. Strategies to achieve mutation-specificity may include the development of allosteric inhibitors targeting the pseudokinase JH2 (JAK homology 2) domain rather than kinase JH1 domain.111 The successful development of tyrosine kinase 2 inhibitors targeting the pseudokinase domain is encouraging and will hopefully instruct development of JAK2V617F-specific inhibitors.112
Perspective
The finding of activated JAK2 signaling as a hallmark of MPN has set the stage for the development and broad use of JAK2 inhibitors. JAK2 inhibition has advanced MPN therapy to a new, mechanism-based level of molecularly targeted therapy. Ruxolitinib, as the first in class JAK2 inhibitor, stands for important advantages as convincing symptom control and effective correction of splenomegaly. We expect additional type I JAK2 inhibitors to consolidate these successes and to extend the benefits of JAK2 inhibitor therapy also to subsets of patients with specific vulnerabilities such as anemia or thrombocytopenia. The approval of additional type I JAK2 inhibitors will enable the choice of specific compounds for specific patients according to their profiles. However, the use of ruxolitinib has revealed important shortcomings of JAK2 inhibitors and has made clear that they are not comparable to tyrosine kinase inhibitors of, for example, BCR-ABL in chronic myeloid leukemia. Reductions in mutant allele burden are modest and clonal evolution progresses. Also, resistance mechanisms differ from BCR-ABL inhibitors with adaptive changes in oncogenic signaling being most prominent, while second-site resistance mutations upon JAK2 inhibitor therapy have not been reported in MPN patients so far.
With the development of JAK2 inhibitors, several key questions remain open: First, how to handle loss of response; second, how to address bone marrow fibrosis; third, how to reduce the MPN clone; and forth, how to control progressed MPN. Type II JAK2 inhibition represents a novel mode to target JAK2, which holds the potential to overcome resistance and to reduce the MPN clone. However, type II JAK2 inhibition has not entered clinical development yet. HMAs and BCL-2 inhibitors, which have shown benefits in acute myeloid leukemia, seem to enhance efficacy of JAK2 inhibition and could represent a valid option to control advanced MPN. Aurora kinase inhibition has shown potent fibrosis reducing effects via acting on MPN megakaryocytes, and the further development is urgently awaited. So far, we still rely on modified forms of interferon-α and allogeneic hematopoietic stem cell transplantation for MPN disease modification. However, given the intense efforts for novel combination and single-agent therapies in early clinical stages, the armamentarium for MPN therapy is expected to expand substantially in the next years. This will bring the challenge and the opportunity to learn how to tailor targeted single agents as well as combination therapies with JAK2 inhibitors to specific subsets and to individual patients in order to maximize therapeutic benefit. This prospect urges further efforts for an improved understanding of MPN biology in regard to resistance, fibrogenesis, clonal evolution, and leukemic transformation. Deepened biological insights should support and inform our clinical choices of therapeutics to address as specifically as possible an individual MPN patient’s most pressing needs.
Sources of funding
We acknowledge research support to Dr Meyer by the Swiss National Science Foundation (PBBEP3-144806, PCEFP3_181357), the Swiss Cancer League/Swiss Cancer Research (KFS-3858-02-2016), the Swiss BRIDGE Foundation (PSB-4066-06-2016), the Cancer League Basel (KLbB-4784-02-2019), the Basel Region Childhood Cancer Foundation, the Swiss Group for Clinical Cancer Research Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung, the Foundation for the Fight against Cancer, the Nora van Meeuwen-Häfliger Foundation, the Foundation Peter-Anton, and Anna-Katharina Miescher and the Swiss Society of Hematology.
Disclosures
SCM has consulted for and received honoraria from Celgene/BMS. SB has no conflicts of interest to disclose.
References
- 1.Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017; 376:2168–2181. [DOI] [PubMed] [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127:2391–2405. [DOI] [PubMed] [Google Scholar]
- 3.Rampal R, Al-Shahrour F, Abdel-Wahab O, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014; 123:e123–e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neubauer H, Cumano A, Müller M, et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998; 93:397–409. [DOI] [PubMed] [Google Scholar]
- 5.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005; 352:1779–1790. [DOI] [PubMed] [Google Scholar]
- 6.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365:1054–1061. [DOI] [PubMed] [Google Scholar]
- 7.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005; 7:387–397. [DOI] [PubMed] [Google Scholar]
- 8.James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005; 434:1144–1148. [DOI] [PubMed] [Google Scholar]
- 9.Bandaranayake RM, Ungureanu D, Shan Y, et al. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol. 2012; 19:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007; 356:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006; 3:e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milosevic Feenstra JD, Nivarthi H, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016; 127:325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013; 369:2391–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elf S, Abdelfattah NS, Chen E, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016; 6:368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhagwat N, Koppikar P, Keller M, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014; 123:2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014; 28:1472–1477. [DOI] [PubMed] [Google Scholar]
- 17.Angona A, Fernández-Rodríguez C, Alvarez-Larrán A, et al. Molecular characterisation of triple negative essential thrombocythaemia patients by platelet analysis and targeted sequencing. Blood Cancer J. 2016; 6:e463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cabagnols X, Favale F, Pasquier F, et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. 2016; 127:333–342. [DOI] [PubMed] [Google Scholar]
- 19.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013; 27:1861–1869. [DOI] [PubMed] [Google Scholar]
- 20.Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014; 123:2220–2228. [DOI] [PubMed] [Google Scholar]
- 21.Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015; 372:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nangalia J, Nice FL, Wedge DC, et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica. 2015; 100:e438–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todorova R, Passweg J, Lundberg P, Tzankov A. Does the order of mutational acquisition in myeloproliferative neoplasms matter? Evidence from JAK2 exon 12 and DNMT3A co-mutant polycythemia vera. J Hematop. 2020; 13:105–107. [Google Scholar]
- 24.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012; 366:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015; 372:426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamide. Blood. 2017; 130:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017; 10:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deininger M, Radich J, Burn TC, et al. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood. 2015; 126:1551–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marti-Carvajal AJ, Anand V, Sola I. Janus kinase -1 and Janus kinase-2 inhibitors for treating myelofibrosis. Cochrane Database Syst Rev. 2015:CD010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newberry KJ, Patel K, Masarova L, et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood. 2017; 130:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mylonas E, Yoshida K, Frick M, et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat Commun. 2020; 11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andraos R, Qian Z, Bonenfant D, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012; 2:512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer SC, Keller MD, Chiu S, et al. CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell. 2015; 28:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koppikar P, Bhagwat N, Kilpivaara O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012; 489:155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016; 30:1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiladjian JJ, Zachee P, Hino M, et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. 2020; 7:e226–e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012; 366:787–798. [DOI] [PubMed] [Google Scholar]
- 38.Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017; 18:88–99. [DOI] [PubMed] [Google Scholar]
- 39.Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015; 1:643–651. [DOI] [PubMed] [Google Scholar]
- 40.Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017; 4:e317–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mesa RA, Kiladjian JJ, Catalano JV, et al. SIMPLIFY-1: a phase III randomized trial of momelotinib versus ruxolitinib in Janus kinase inhibitor-naïve patients with myelofibrosis. J Clin Oncol. 2017; 35:3844–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018; 5:e73–e81. [DOI] [PubMed] [Google Scholar]
- 43.Mesa RA, Vannucchi AM, Mead A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. 2017; 4:e225–e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018; 4:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asshoff M, Petzer V, Warr MR, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. 2017; 129:1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singer JW, Al-Fayoumi S, Ma H, et al. Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor. J Exp Pharmacol. 2016; 8:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008; 18:73–79. [DOI] [PubMed] [Google Scholar]
- 48.Meyer SC. Mechanisms of resistance to JAK2 inhibitors in myeloproliferative neoplasms. Hematol Oncol Clin North Am. 2017; 31:627–642. [DOI] [PubMed] [Google Scholar]
- 49.Marty C, Saint-Martin C, Pecquet C, et al. Germ-line JAK2 mutations in the kinase domain are responsible for hereditary thrombocytosis and are resistant to JAK2 and HSP90 inhibitors. Blood. 2014; 123:1372–1383. [DOI] [PubMed] [Google Scholar]
- 50.Stivala S, Codilupi T, Brkic S, et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J Clin Invest. 2019; 129:1596–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manshouri T, Estrov Z, Quintás-Cardama A, et al. Bone marrow stroma-secreted cytokines protect JAK2(V617F)-mutated cells from the effects of a JAK2 inhibitor. Cancer Res. 2011; 71:3831–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verstovsek S, Manshouri T, Pilling D, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp Med. 2016; 213:1723–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weigert O, Lane AA, Bird L, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012; 209:259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deshpande A, Reddy MM, Schade GO, et al. Kinase domain mutations confer resistance to novel inhibitors targeting JAK2V617F in myeloproliferative neoplasms. Leukemia. 2012; 26:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreoli A, Verger E, Robin M, et al. Clinical resistance to ruxolitinib is more frequent in patients without MPN-associated mutations and is rarely due to mutations in the JAK2 kinase drug-binding domain. Blood. 2013; 122:1591. [Google Scholar]
- 56.Gisslinger H, Schalling M, Gisslinger B, et al. Restoration of response to ruxolitinib upon brief withdrawal in two patients with myelofibrosis. Am J Hematol. 2014; 89:344–346. [DOI] [PubMed] [Google Scholar]
- 57.Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc. 2011; 86:1188–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilmes S, Hafer M, Vuorio J, et al. Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science. 2020; 367:643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res. 2013; 37:214–220. [DOI] [PubMed] [Google Scholar]
- 60.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011; 29:392–397. [DOI] [PubMed] [Google Scholar]
- 61.Tefferi A, Vaidya R, Caramazza D, et al. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011; 29:1356–1363. [DOI] [PubMed] [Google Scholar]
- 62.Arranz L, Sánchez-Aguilera A, Martín-Pérez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014; 512:78–81. [DOI] [PubMed] [Google Scholar]
- 63.Isern J, Martín-Antonio B, Ghazanfari R, et al. Self-renewing human bone marrow mesenspheres promote hematopoietic stem cell expansion. Cell Rep. 2013; 3:1714–1724. [DOI] [PubMed] [Google Scholar]
- 64.Winter PS, Sarosiek KA, Lin KH, et al. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal. 2014; 7:ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choong ML, Pecquet C, Pendharkar V, et al. Combination treatment for myeloproliferative neoplasms using JAK and pan-class I PI3K inhibitors. J Cell Mol Med. 2013; 17:1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khan I, Huang Z, Wen Q, et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia. 2013; 27:1882–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bartalucci N, Tozzi L, Bogani C, et al. Co-targeting the PI3K/mTOR and JAK2 signalling pathways produces synergistic activity against myeloproliferative neoplasms. J Cell Mol Med. 2013; 17:1385–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pérez C, Pascual M, Martín-Subero JI, et al. Aberrant DNA methylation profile of chronic and transformed classic Philadelphia-negative myeloproliferative neoplasms. Haematologica. 2013; 98:1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Masarova L, Verstovsek S, Hidalgo-Lopez JE, et al. A phase 2 study of ruxolitinib in combination with azacitidine in patients with myelofibrosis. Blood. 2018; 132:1664–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in accelerated and blast-phase myeloproliferative neoplasms. Blood Adv. 2018; 2:3572–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Evrot E, Ebel N, Romanet V, et al. JAK1/2 and Pan-deacetylase inhibitor combination therapy yields improved efficacy in preclinical mouse models of JAK2V617F-driven disease. Clin Cancer Res. 2013; 19:6230–6241. [DOI] [PubMed] [Google Scholar]
- 72.Mascarenhas J, Marcellino BK, Lu M, et al. A phase I study of panobinostat and ruxolitinib in patients with primary myelofibrosis (PMF) and post–polycythemia vera/essential thrombocythemia myelofibrosis (post–PV/ET MF). Leuk Res. 2020; 88:106272. [DOI] [PubMed] [Google Scholar]
- 73.Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018; 33:29–43.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mascarenhas J, Kremyanskaya M, Hoffman R, et al. MANIFEST, a phase 2 study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), as monotherapy or “add-on” to ruxolitinib, in patients with refractory or intolerant advanced myelofibrosis. Blood. 2019; 134(supplement_1)670. [Google Scholar]
- 75.Saenz DT, Fiskus W, Manshouri T, et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia. 2017; 31:678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson WH, O’Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010; 11:1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018; 19:216–228. [DOI] [PubMed] [Google Scholar]
- 78.Waibel M, Solomon VS, Knight DA, et al. Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep. 2013; 5:1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harrison CN, Garcia JS, Mesa RA, et al. Results from a phase 2 study of navitoclax in combination with ruxolitinib in patients with primary or secondary myelofibrosis. Blood. 2019; 134(supplement_1):671. [Google Scholar]
- 80.Marubayashi S, Koppikar P, Taldone T, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010; 120:3578–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Uras IZ, Maurer B, Nivarthi H, et al. CDK6 coordinates JAK2 V617F mutant MPN via NF-κB and apoptotic networks. Blood. 2019; 133:1677–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mazzacurati L, Collins RJ, Pandey G, et al. The pan-PIM inhibitor INCB053914 displays potent synergy in combination with ruxolitinib in models of MPN. Blood Adv. 2019; 3:3503–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rampal RK, Maria PO, Varshini HSA, Levine RL, Cao A. Synergistic therapeutic efficacy of combined JAK1/2, Pan-PIM, and CDK4/6 inhibition in myeloproliferative neoplasms. Blood. 2016; 128:634. [Google Scholar]
- 84.Essers MA, Offner S, Blanco-Bose WE, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009; 458:904–908. [DOI] [PubMed] [Google Scholar]
- 85.Mullally A, Bruedigam C, Poveromo L, et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood. 2013; 121:3692–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sorensen AL, Mikkelsen SU, Knudsen TA, et al. Ruxolitinib and interferon-alpha2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica. 2019; 105:235648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kiladjian JJ, Soret-Dulphy J, Resche-Rigon M, et al. Ruxopeg, a multi-center Bayesian phase 1/2 adaptive randomized trial of the combination of ruxolitinib and pegylated interferon alpha 2a in patients with myeloproliferative neoplasm (MPN)-associated myelofibrosis. Blood. 2018; 132(supplement 1):581. [Google Scholar]
- 88.Iancu-Rubin C, Mosoyan G, Wang J, et al. Stromal cell-mediated inhibition of erythropoiesis can be attenuated by Sotatercept (ACE-011), an activin receptor type II ligand trap. Exp Hematol. 2013; 41:155–166.e17. [DOI] [PubMed] [Google Scholar]
- 89.Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014; 20:408–414. [DOI] [PubMed] [Google Scholar]
- 90.Bose P, Daver N, Pemmaraju N, et al. Sotatercept (ACE-011) alone and in combination with ruxolitinib in patients (pts) with myeloproliferative neoplasm (MPN)-associated myelofibrosis (MF) and anemia. Blood. 2017; 130(supplement 1):255. [Google Scholar]
- 91.Rampal RK, Verstovsek S, Devlin SM, et al. Safety and efficacy of combined ruxolitinib and thalidomide in patients with myelofibrosis: a phase II study. Blood. 2019; 134:4163. [Google Scholar]
- 92.Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008; 112:3065–3072. [DOI] [PubMed] [Google Scholar]
- 93.Masarova L, Patel KP, Newberry KJ, et al. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017; 4:e165–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yacoub A, Mascarenhas J, Kosiorek H, et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood. 2019; 134:1498–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ianotto JC, Chauveau A, Boyer-Perrard F, et al. Benefits and pitfalls of pegylated interferon-α2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: a French Intergroup of Myeloproliferative neoplasms (FIM) study. Haematologica. 2018; 103:438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Czech J, Cordua S, Weinbergerova B, et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia. 2019; 33:995–1010. [DOI] [PubMed] [Google Scholar]
- 97.Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020; 7:e196–e208. [DOI] [PubMed] [Google Scholar]
- 98.Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015; 373:920–928. [DOI] [PubMed] [Google Scholar]
- 99.Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015; 373:908–919. [DOI] [PubMed] [Google Scholar]
- 100.Wen QJ, Yang Q, Goldenson B, et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat Med. 2015; 21:1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gangat N, Marinaccio C, Swords R, et al. Aurora kinase A inhibition provides clinical benefit, normalizes megakaryocytes, and reduces bone marrow fibrosis in patients with myelofibrosis: a phase I trial. Clin Cancer Res. 2019; 25:4898–4906. [DOI] [PubMed] [Google Scholar]
- 102.Jeffers A, Qin W, Owens S, et al. Glycogen synthase kinase-3β inhibition with 9-ING-41 attenuates the progression of pulmonary fibrosis. Sci Rep. 2019; 9:18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakatake M, Monte-Mor B, Debili N, et al. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene. 2012; 31:1323–1333. [DOI] [PubMed] [Google Scholar]
- 104.Etchin J, Sun Q, Kentsis A, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013; 27:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boddu P, Carter BZ, Verstovsek S, et al. SMAC mimetics as potential cancer therapeutics in myeloid malignancies. Br J Haematol. 2019; 185:219–231. [DOI] [PubMed] [Google Scholar]
- 106.Pemmaraju N, Gupta V, Ali H, et al. Results from a phase 1/2 clinical trial of tagraxofusp (SL-401) in patients with intermediate, or high risk, relapsed/refractory myelofibrosis. Blood. 2019; 134(Supplement_1):558. [Google Scholar]
- 107.Masarova L, Bose P, Verstovsek S. The rationale for immunotherapy in myeloproliferative neoplasms. Curr Hematol Malig Rep. 2019; 14:310–327. [DOI] [PubMed] [Google Scholar]
- 108.Prestipino A, Emhardt AJ, Aumann K, et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med. 2018; 10:eaam7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu SC, Li LS, Kopp N, et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell. 2015; 28:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vainchenker W, Leroy E, Gilles L, et al. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res. 2018; 7:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leroy E, Constantinescu SN. Rethinking JAK2 inhibition: towards novel strategies of more specific and versatile Janus kinase inhibition. Leukemia. 2017; 31:2853. [DOI] [PubMed] [Google Scholar]
- 112.Moslin R, Gardner D, Santella J, et al. Identification of imidazo[1,2-b]pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. Medchemcomm. 2017; 8:700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]