

Abstract
The molecular mechanisms of cellular insulin action have been the focus of much investigation since the discovery of the hormone 100 years ago. Insulin action is impaired in metabolic syndrome, a condition known as insulin resistance. The actions of the hormone are initiated by binding to its receptor on the surface of target cells. The receptor is an α2β2 heterodimer that binds to insulin with high affinity, resulting in the activation of its tyrosine kinase activity. Once activated, the receptor can phosphorylate a number of intracellular substrates that initiate discrete signaling pathways. The tyrosine phosphorylation of some substrates activates phosphatidylinositol-3-kinase (PI3K), which produces polyphosphoinositides that interact with protein kinases, leading to activation of the kinase Akt. Phosphorylation of Shc leads to activation of the Ras/MAP kinase pathway. Phosphorylation of SH2B2 and of Cbl initiates activation of G proteins such as TC10. Activation of Akt and other protein kinases produces phosphorylation of a variety of substrates, including transcription factors, GTPase-activating proteins, and other kinases that control key metabolic events. Among the cellular processes controlled by insulin are vesicle trafficking, activities of metabolic enzymes, transcriptional factors, and degradation of insulin itself. Together these complex processes are coordinated to ensure glucose homeostasis.
While the 100th anniversary of insulin’s discovery is a reminder of the astounding progress toward elucidating the molecular basis of insulin action, it also highlights the numerous gaps in understanding of the signaling pathways used by this important hormone. A major complexity is the pleiotropic nature of insulin’s effects, which depend critically on tissue target, time course, and the presence of other hormones and biogenic amines. All of insulin’s actions are mediated by its receptor, IR (encoded by INSR), a cell surface protein that signals via multiple pathways involving protein and lipid phosphorylation, activation of small G protein molecular switches, control of trafficking events, and regulation of a network of enzymes and transcriptional factors that together define insulin’s unique actions (1–3). A central component of metabolic syndrome’s pathophysiology is insulin resistance, produced by reduced responsiveness to insulin in fat, liver, muscle, and other tissues (4). Numerous longitudinal studies demonstrate that insulin resistance is an early step in the development of type 2 diabetes (T2D) and is closely linked to other health problems such as obesity, fatty liver, polycystic ovarian syndrome, hypertension, and atherosclerosis (5–7). Its central role in so many aspects of metabolic syndrome makes understanding insulin resistance, and hence insulin action, of great importance.
Insulin is a potent anabolic agent, promoting the cellular uptake, storage, and synthesis of nutrients, while blocking nutrient breakdown and release into the circulation. Insulin stimulates nutrient transport into cells, acutely regulates metabolic enzyme activity, controls transcription of metabolic genes, regulates cellular growth and differentiation, and controls its own clearance, all through activation of its receptor. I will review current understanding of the insulin receptor, its interacting proteins, the proximal events responsible for signal initiation, and the specific pathways governing different aspects of insulin action. Throughout, I will comment on the negative regulation of insulin signaling that may be responsible for cellular insulin resistance, and review genetic evidence for the importance of specific pathways, while highlighting outstanding questions that remain enigmatic.
The insulin receptor
The insulin receptor is a glycosylated, disulfide-linked α2β2 tetramer that belongs to a subfamily of receptor tyrosine kinases, including insulin-like growth factor 1 (IGF-1) receptor and insulin receptor–related receptor (IRR) (8, 9) (Figure 1A). The receptor’s two subunits derive from a single precursor processed by a furin-like enzyme to produce an α/β subunit complex, which then undergoes disulfide linkage to form the tetramer (8, 10, 11). β Subunits in the mature receptor traverse the membrane via a helical structure, and the receptor is mostly found in the plasma membrane (12), although a fraction may be found in the nucleus (13, 14). However, the location of the insulin receptor and signal initiation remains controversial.
Figure 1. The insulin receptor, its substrates, and its activation of kinase cascades.

(A) The insulin receptor is a disulfide-linked, α/β heterodimer glycoprotein that resides largely on the cell surface. The α subunit binds to insulin with high affinity, alleviating PTP-mediated repression of the β subunit’s tyrosine kinase activity by inducing close proximity between the β subunits, permitting transphosphorylation on tyrosines in three β subunit domains. Phosphorylation of three crucial tyrosines leads to full activation of the receptor kinase. Once activated, the receptor kinase can phosphorylate exogenous substrates that act as adaptors: IRS-1–IRS-4 and Shc. Both are recruited to the juxtamembrane region via their PTB domains. SH2B2 is recruited to the kinase region’s triple phosphorylation motif via its SH2 domain, serving as an adaptor protein for the substrate Cbl. (B) Activation of kinase cascades. Once phosphorylated, IRS and Shc activate lipid and protein kinases. IRS proteins are phosphorylated on tyrosines within specific motifs, recruiting the p85 subunit of PI3K, which binds to IRS through its SH2 domain. This results in activation of the p110 catalytic domain to generate polyphosphoinositides such as PI-(3,4,5)trisphosphate (PIP3). These phosphoinositides can be degraded by the PI phosphatases PTEN and SHIP2. PIP3 interacts with proteins containing PH domains, notably PDK1 and Akt. Once recruited to the plasma membrane, PDK1 and mTORC2 phosphorylate and activate Akt, which can phosphorylate a number of substrates, including the GAP proteins RalGAPA, AS160, and TSC2, as well as Foxo proteins, GSK3, and others. Upon phosphorylation, Shc interacts with the SH2/SH3 adaptor protein Grb2, which is constitutively associated with the GEF SOS. SOS is thus recruited to the plasma membrane, and catalyzes the exchange of GTP for GDP on Ras. In its active, GTP-bound state, Ras interacts with the protein kinase Raf, leading to activation of the MAPK cascade through sequential phosphorylation of MEK and ERK.
The receptor is activated upon binding of insulin to the α subunit, which derepresses the tyrosine kinase activity of the β subunit. Full activation requires one subunit to phosphorylate the other, causing a conformational change that further increases kinase activity, producing phosphorylation of exogenous substrates (15–18). Because α/β heterodimers of insulin, IGF-1, and IRR receptors undergo activation by transphosphorylation, a dominant-negative mutant form of one receptor subtype can inhibit the other’s activity, explaining why individuals with insulin receptor mutations exhibit both metabolic insulin resistance and growth retardation (17).
Recent insights from x-ray crystallography and cryo–electron microscopy (cryo-EM) illustrate precisely how insulin binding induces the conformational changes that lead to tyrosine kinase activation (19). Crystallography of the unbound receptor/ligand-binding domain dimer revealed an inverted U structure with about 115 Å distance between membrane insertion regions (20, 21). Importantly, single-particle cryo-EM of full-length insulin receptor corroborated this finding (22). Insulin binding converts the receptor extracellular domain into a T-like shape that draws the membrane-proximal regions together, leading to transphosphorylation in the β subunit (22).
The β subunit’s cytoplasmic region consists of a juxtamembrane domain, a tyrosine kinase domain, and a carboxy-terminal tail. Tyrosine autophosphorylation sites have been mapped in all three regions (23, 24). While substrates can interact with each domain, phosphorylation of the three sites within the kinase domain are key for activation, as revealed by crystal structures of the tyrosine kinase domain determined in several different phosphorylation states and with bound substrates (25, 26). Crystallographic, biophysical, and biochemical evidence shows that the β subunits’ phosphorylated kinase domains bring together the juxtamembrane region proximal to the kinase domain, cooperatively increasing transphosphorylation and activation.
To date, over 100 INSR mutations have been discovered (27) that cause reduced biosynthesis of the receptor, impaired transport or recycling to the plasma membrane, decreased binding affinity, or reduced tyrosine kinase activity. Rare INSR mutations cause inherited insulin-resistant syndromes such as Donohue syndrome and Rabson-Mendenhall syndrome. These recessive conditions are characterized by restricted intrauterine and postnatal growth, dysmorphic features, altered glucose homeostasis, and early death (28, 29). Mutations in the kinase domain can also alter insulin action and produce disease with a dominant pattern of inheritance. A Gly996Val mutation in a conserved Gly-X-Gly-X-X-Gly motif impairs tyrosine kinase activity and is associated with insulin resistance and acanthosis nigricans, suggesting a dominant-negative effect on the tyrosine kinase (30). Gain-of-function mutations in the kinase domain leading to familial hyperinsulinemic hypoglycemia have also been identified (31).
Insulin receptor substrates
Insulin-stimulated autophosphorylation of its receptor recruits several proteins for phosphorylation to initiate signaling pathways. At least nine intracellular substrates of insulin and IGF-1 receptor tyrosine kinases have been identified (Figure 2). Four belong to the insulin/IGF-1 receptor substrate (IRS) protein family (32–34). Other direct substrates include Gab-1 (35), DOK1 (36), Cbl (37), SH2B2 (APS) (38), SHP2 (39), and the various isoforms of Shc (40), each of which initiates a signaling pathway. The tyrosine residues phosphorylated in each substrate occur in specific sequence motifs; once phosphorylated, they serve as docking sites for intracellular molecules containing SH2 (Src homology 2) domains (1). The best-characterized substrates are the IRS proteins Shc, SH2B2, and Cbl. Each contains either a phosphotyrosine-binding (PTB) domain (IRS proteins, Shc) or an SH2 domain (SH2B2, Cbl) that mediates receptor interaction. IRS protein and Shc PTB domains bind to the juxtamembrane autophosphorylation site pY972 within a canonical PTB domain binding site (NPXpY). SH2B2’s SH2 domain binds to the phosphorylated receptor kinase activation loop; once phosphorylated, SH2B2 binds to Cbl, permitting its phosphorylation on three tyrosines (38, 41).
Figure 2. Regulation of glycogen metabolism by compartmentalized phosphorylation.
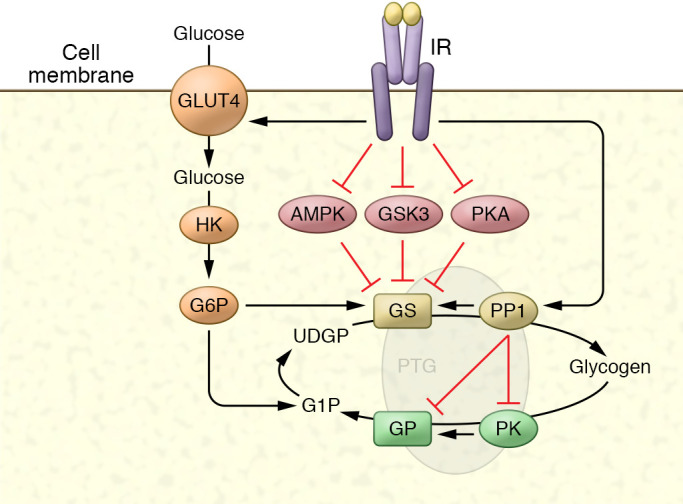
Like other metabolic enzymes, control of glycogen metabolism is mediated by changes in phosphorylation of the enzymes glycogen synthase (GS) and glycogen phosphorylase (GP) through inhibition of kinases and activation of phosphatases. GS is inhibited by phosphorylation on up to nine amino acids, and insulin activates the enzyme by reversing this phosphorylation through a combination of kinase inhibition and phosphatase activation, primarily through protein phosphatase 1 (PP1). Similarly, GP is activated by phosphorylation, and insulin inhibits the enzyme by reducing phosphorylation. These events occur in discrete cellular compartments owing to the presence of scaffolding proteins such as PTG (Ppp1R3C) and others, by binding to GS, GP, phosphorylase kinase (PK), and AMPK, and targeting these proteins to glycogen itself. GS is also regulated by the binding of glucose-6-phosphate (G6P) to an allosteric site that increases activity.
Insulin receptor–mediated phosphorylation of IRS proteins occurs on at least nine tyrosines within sequence motifs that recognize and activate phosphatidylinositol-3-kinase (PI3K) and downstream protein kinases. IRS-1 and IRS-2 are widely distributed, whereas IRS-3 and IRS-4 expression is more limited (42). Although the IRS proteins are homologous and possess similar tyrosine phosphorylation motifs, knockout studies suggest complementary roles. IRS-1–deficient mice exhibit pre- and postnatal growth retardation due to IGF-1 resistance, as well as insulin resistance and impaired glucose tolerance, primarily in muscle and fat (43, 44). IRS-2–deficient mice exhibit hepatic insulin resistance, with some growth defects in the brain, β cells, and retinal cells (45).
Shc isoforms also undergo tyrosine phosphorylation by binding to the insulin receptor through their PTB domain. Upon tyrosine phosphorylation, Shc interacts with the SH2/SH3 adaptor protein Grb2 to activate the Ras pathway (40, 46). SH2B2 is the insulin receptor’s highest-affinity substrate (38, 47). This adaptor protein interacts directly with the triad of phosphotyrosines in the activation loop as a homodimer, in which each member interacts with a separate receptor β subunit, then undergoes phosphorylation on a single tyrosine (47). SH2B2 thereupon serves as an adaptor for c-Cbl phosphorylation through the adaptor protein CAP (SORBS1), leading to downstream activation of G proteins including TC10 (RhoQ) (see below). This pathway occurs largely in lipid raft domains of the adipocyte plasma membrane (38).
Control of glucose homeostasis requires a rapid on/off response to insulin to maintain blood glucose within a narrow range. Following dissociation of insulin, phosphorylation of the insulin receptor and its substrates is rapidly reversed by protein tyrosine phosphatases (PTPases). Although the substrate specificity of PTPases has proven difficult to evaluate, several identified PTPases can catalyze IR dephosphorylation, and some are upregulated in insulin-resistant states (48–50). Most attention has focused on the phosphatase PTP-1b (encoded by Ptpn1). Disrupting Ptpn in mice increases insulin-dependent tyrosine phosphorylation of the insulin receptor and IRS proteins, leading to improved insulin sensitivity (51, 52). PTP-1b–deficient mice are resistant to diet-induced obesity, suggesting that PTP-1b deletion in the brain may influence energy uptake and expenditure via leptin signaling (52, 53).
IRS proteins and the insulin receptor also undergo serine phosphorylation that is generally associated with reduced insulin action (54). Serine phosphorylation of the receptor or substrates blocks insulin action by decreasing tyrosine phosphorylation and sequestering tyrosines by promoting interaction with 14-3-3 proteins (55). Multiple intracellular kinases are implicated in this serine phosphorylation (7, 56), including some activated by insulin, such as Akt (57), JNK (58), ERK (59), and PI3K (60), which potentially provide feedback inhibition. Moreover, serine kinases activated in obesity or by inflammation, especially PKCε (7), can phosphorylate and inhibit substrate tyrosine phosphorylation (61). However, the physiological relevance of these negative phosphorylation events remains uncertain (62, 63).
Several polymorphisms in human IRS-1 (G971R and A513P) observed in T2D produce decreased associated PI3K activity (64). These polymorphisms are associated with insulin resistance, hyperinsulinemia, adiposity, dyslipidemia, and risk of coronary disease, along with reduced IRS-1 protein levels and decreased IRS-1–associated PI3K activity (65). Other polymorphisms associated with obesity or T2D have been detected for SH2B2, SORBS1, and Cbl (66).
Control of phosphoinositide metabolism
The enzyme PI3K is pivotal in the metabolic and mitogenic actions of insulin (67) (Figure 3), and PI3K inhibitors or targeted deletion block insulin’s metabolic actions. The class 1 form of PI3K consists of a p85 regulatory unit (encoded by PIK3R1) and a p110 catalytic subunit (PIK3CA) and is activated by the two SH2 domains in the regulatory subunit interacting with tyrosine-phosphorylated IRS proteins (68, 69). The eight identified isoforms of regulatory subunits derive from three genes that undergo alternative splicing (70). p85α and p85β contain an SH3 domain, a BCR homology domain flanked by two proline-rich domains, two SH2 domains, and an inter-SH2 domain containing the p110 binding region (70). The shorter splicing variants of regulatory subunits p55α and p50α lack the N-terminal half. p85α is ubiquitously expressed, while p55α and p50α play specific roles (71). In mice, disruption of all three Pik3r1 isoforms is lethal within a few weeks of birth, indicating the enzyme’s importance in normal growth and metabolism (72).
Figure 3. Transcriptional control of metabolism by insulin.
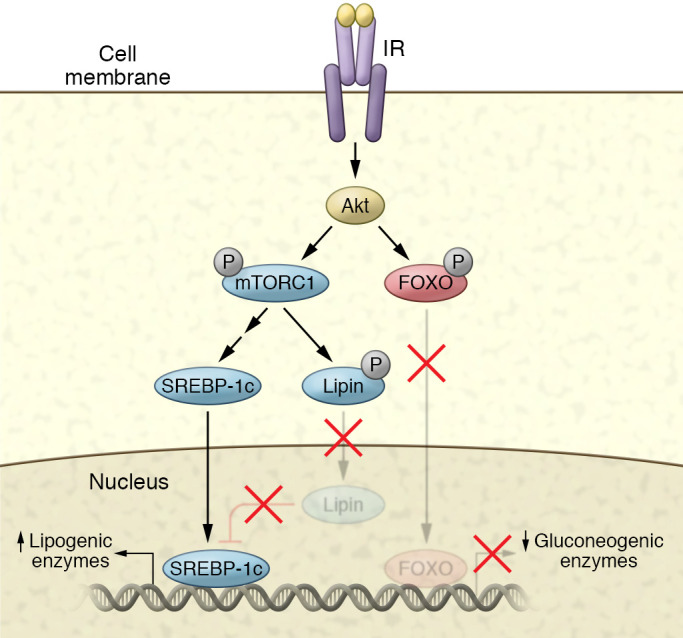
Insulin increases the expression of lipogenic genes while inhibiting the expression of gluconeogenic genes in hepatocytes. Akt phosphorylates the transcription factor FOXO1, leading to the exclusion of the protein from the nucleus, and thus reducing transcription of gluconeogenic genes such as PEPCK, G6P, and others. Akt can also phosphorylate mTORC1, which in turn phosphorylates S6K. S6K activation leads to the activation of the SREBP pathway. mTORC1 also phosphorylates lipin, which inhibits SREBP action. Phosphorylation of this protein maintains a cytoplasmic localization, thus preventing its inhibitory activity.
PI3K catalyzes the phosphorylation of phosphoinositides on the 3-position to generate PI-(3)P, PI-(3,4)P2, and PI-(3,4,5)P3. These lipids bind to the pleckstrin homology (PH) domains of target proteins, altering activity or subcellular localization. This pathway can be terminated by phosphoinositide phosphatases (73), such as PTEN (74) and SHIP2 (encoded by Inppl1) (75). PTEN dephosphorylates phosphoinositides on the 3′-position, while SHIP2 is a 5′-phosphoinositide phosphatase. Disrupting Inppl1 yields mice with increased insulin sensitivity (75). Polymorphisms in INPPL1 are associated with increased incidence of hypertension, obesity, T2D, and metabolic syndrome (76). A polymorphism in INPPL1’s catalytic domain identified in a Japanese population of diabetic subjects suggests possible protection from insulin resistance (77).
Regulation of serine kinase cascades
Serine phosphorylation events are initiated downstream of insulin receptor substrate tyrosine phosphorylation via PI3K and small GTPase activation (Figure 1B). PI-3 phosphates (PI3Ps) regulate three major classes of signaling molecules: the AGC superfamily of serine/threonine protein kinases, guanine nucleotide exchange proteins targeting the Rho family of GTPases, and the TEC family of tyrosine kinases, including BTK and ITK. The best-characterized pathway in insulin signaling involves the AGC kinase Akt. Once recruited to the plasma membrane by PI3P, Akt is activated by a multistep process that requires phosphorylation of both Thr308 in the kinase domain’s activation loop and Ser473 in the regulatory domain. PI-(3,4,5)P3 recruits the serine/threonine kinase phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane through its PH domain and phosphorylates Thr308 on Akt (78, 79). Ser473 appears to be phosphorylated mainly by the rapamycin-insensitive mTOR complex mTORC2 (80). S473 phosphorylation may be partially redundant but stabilizes Akt’s active conformation state (81). The mechanism of mTORC2 regulation remains uncertain.
Akt amplifies multiple pathways in insulin action (82). Targeted deletion of Akt isoforms produces insulin resistance and glucose intolerance (83, 84), and Akt mutations have been identified in patients with severe insulin resistance (85), whereas an activating mutation produced hypoglycemia (86). However, studies using Akt inhibitors and activators have not uniformly inhibited or mimicked insulin actions (87). In part, variability may reflect the presence of three Akt isoforms (88). Although Akt1 impacts cell survival and growth, Akt2 appears to play a more prominent role in the liver (84). Stable expression of a constitutively active, membrane-bound form of Akt in 3T3L1 murine adipocytes resulted in increased glucose transport and persistent localization of GLUT4 to the plasma membrane (89–91), but did not fully reproduce insulin action. Conversely, expression of a dominant-interfering Akt mutant inhibited insulin-stimulated GLUT4 translocation. Parenthetically, full expression of insulin action likely requires other signaling pathways (92). Other AGC kinases activated downstream of PI3K include the protein kinase C (PKC) family, particularly PKC-ζ. Overexpressing PKC-ζ or PKC-λ resulted in GLUT4 translocation (93, 94), whereas expressing a dominant-interfering PKC-λ blocked insulin action (95).
Akt phosphorylates a variety of substrates, including glycogen synthase kinase-3 (GSK3) (96), the forkhead (FOXO) transcription factors, cAMP regulatory element–binding protein (CREB) (87, 97, 98), and the GAP proteins TSC2, AS160, and RalGAPA (99). Once activated at the plasma membrane, phosphorylated Akt can translocate to the cytoplasm or nucleus, depending on cell type (100).
Although PI3K activity is clearly necessary for insulin action, several lines of evidence suggest that additional signals may also be required. Activation of PI3K with other hormones, such as PDGF and IL-4, does not stimulate glucose transport in adipocytes (92). Likewise, adding a PI-(3,4,5)trisphosphate (PIP3) analog alone did not effect glucose transport (101), and two insulin receptor mutants that produced complete PI3K activation failed to mediate full insulin action (102). As mentioned above, insulin initiates additional pathways by recruiting other adaptor proteins, particularly SH2B2, which binds to the activated insulin receptor (47). Upon phosphorylation, SH2B2 recruits a complex of SORBS1 and c-Cbl (38, 103), triggering insulin receptor–catalyzed tyrosine phosphorylation of c-Cbl, which then interacts with the adaptor protein Crk in complex with C3G, a guanyl nucleotide exchange factor (GEF) (104). C3G in turn activates the small GTPase TC10 (RhoQ) (105, 106). SORBS1 expression correlates well with insulin responsiveness and increases when cells are treated with insulin-sensitizing thiazolidinediones (66, 107).
Insulin receptor–mediated tyrosine phosphorylation of Shc isoforms produces Ras/MAP kinase (Ras/MAPK) pathway activation (108). Phosphorylated Shc interacts with the SH2/SH3 domain–containing adaptor protein Grb2, which is constitutively associated with the Ras nucleotide exchange factor SOS, leading to Ras activation. Full activation of Ras by insulin also requires stimulation of the tyrosine phosphatase SHP2, which interacts with insulin receptor substrates including Gab-1 and IRS-1/2 (109, 110). Once activated, Ras operates as a molecular switch, converting upstream tyrosine phosphorylation into a serine kinase cascade via stepwise activation of Raf and the MAPKs MEK, ERK1, and ERK2 (111, 112). The MAPKs can phosphorylate cytoplasmic substrates or translocate into the nucleus and catalyze the phosphorylation of transcription factors (Elk1, p62TCF, and others), initiating a transcriptional program that commits the cell to a proliferative or differentiative cycle. Blocking the Ras/MAPK pathway with dominant-negative mutants or pharmacologic inhibitors prevents insulin stimulation of cell growth but does not affect any metabolic actions of the hormone (113).
Insulin also controls protein synthesis via a process closely linked with nutrient sensing, involving the protein kinase mTORC1. mTORC1 is a PI3K family member but appears to serve primarily as a protein kinase. Insulin-mediated mTOR stimulation involves PI3K and other inputs (114–119). Akt phosphorylates and inhibits the GTPase-activating protein (GAP) tuberosclerosis complex 2 (TSC2), which forms a complex with the scaffolding protein TSC1 that negatively controls the small GTPase Rheb, a key regulator of the mTORC1 complex (120, 121). mTOR regulates mRNA translation via phosphorylation and activation of the p70 ribosomal S6 kinase, as well as the phosphorylation of the eIF-4E inhibitor PHAS1 (also called 4E-BP1; EIF4EBP1). p70 S6 kinase phosphorylates ribosomal S6 protein, activating ribosome biosynthesis and increasing translation of mRNAs with a 5′-terminal oligopyrimidine tract. mTOR’s phosphorylation of PHAS1 induces its dissociation from eIF-2, allowing cap-dependent translation of mRNAs with a highly structured 5′-untranslated region (122).
Small GTPases control vesicle trafficking and nutrient transport
Controlling transport processes, especially the uptake of nutrients into cells for storage, is a key aspect of insulin action. The rate-limiting step in insulin’s control of glucose homeostasis is stimulation of glucose transport in fat and muscle (99, 123, 124). This occurs via the translocation of facilitative GLUT4 glucose transporters from intracellular sites to the plasma membrane. The GLUT4 protein consists of 12 transmembrane helices, with C- and N-terminal tails both oriented on the cytoplasmic side of the vesicle or plasma membrane. GLUT4 continuously recycles between the cell surface and various intracellular compartments in the basal state. Insulin markedly increases the rate of GLUT4 vesicle exocytosis (99, 125). After endocytosis, GLUT4 returns to the plasma membrane via sorting endosomes or intracellular compartments. In the basal state, at least half of the GLUT4 population is found in a specialized vesicle compartment, and stimulation with insulin depletes a proportion of these GLUT4-enriched vesicles storage (GSVs), directing them to the plasma membrane.
Control of GLUT4 sorting and GSV trafficking relies on activity of several small GTPases that assemble effectors mediating vesicle budding, transport, tethering, and fusion. Small GTPases are active in the GTP-bound state, and inactive upon hydrolysis of GTP to GDP due to the intrinsic activity of the proteins. GTPases are activated by GEF recruitment and inhibited by GAPs. As a general rule, upstream GEFs and GAPs regulate GTPases that control different steps in GLUT4 sorting in adipocytes and muscle cells (126). Insulin activates TC10 via recruitment of the GEF C3G (106). The Akt substrate AS160 is a RabGAP that targets Rab8 and Rab14 in muscle cells, and Rab10 in adipocytes (126, 127). These Rabs have a positive role in GLUT4 translocation, suggesting that they may regulate GSV formation and/or intracellular retention (127–129). Insulin stimulates AS160 phosphorylation via Akt, relieving AS160’s inhibitory effect on target Rabs (130, 131). Insulin-mediated activation of Rab8 and Rab14 was observed in muscle cells, but Rab10 activation has not been detected (129, 132). Nevertheless, Rab10 is a bona fide target of AS160 (132), and necessary for maximal GLUT4 exocytosis in response to insulin. Several lines of evidence indicate that Rab10 cycling may increase glucose uptake (127).
A tethering/docking step targets GSVs to regions of the plasma membrane that contain the fusion machinery. GLUT4 tethering relies on the exocyst, an evolutionarily conserved octameric complex that assembles at sites of exocytosis and tethers exocytic vesicles on the plasma membrane (133, 134). The exocyst mediates initial contact between exocytic vesicles and the plasma membrane and can thus tether GSVs before the final membrane fusion step. Inhibiting exocyst assembly in adipocytes disrupts GSV fusion without affecting their translocation, demonstrating that this complex is necessary for vesicle targeting to the plasma membrane (135).
Insulin regulates exocyst-mediated targeting through exocyst assembly, recognition of the exocyst by GSVs, and disengagement to enable fusion (135–137). Once activated, TC10 binds to the exocyst scaffolding subunit Exo70, which assembles the complex at the plasma membrane (135, 136, 138, 139). GSVs recognize the exocyst via the small GTPase RalA, which is present on GLUT4-containing vesicles. Insulin controls RalA activity primarily by inhibiting the RalGAP complex, comprising a regulatory subunit (RalGAPB) and a catalytic subunit (RalGAPA) that specifically inactivates Ral GTPases. RalGAP function requires RalGAPB, and deleting RalGAPB leads to RalGAPA instability (140). Akt-catalyzed phosphorylation of RalGAPA on three residues inhibits the complex and allows for RalA-GTP binding (140). Knockdown or overexpression of a dominant-negative RalA mutant blocks insulin-stimulated glucose uptake and GLUT4 insertion into the plasma membrane, while constitutively active RalA mutants increase insulin’s effect (137). Moreover, targeted knockout of Exo70 blocks glucose uptake in vivo (138). Conditional knockout of RalGAPB leads to RalA activation in both adipocytes (141) and muscle (142), along with a dramatic increase in glucose uptake and improved glucose tolerance (142). Once activated, RalA interacts with exocyst subunits Sec5 and Exo84 (137, 143, 144). Although the precise role of these two RalA-binding proteins remains uncertain, both are required for insulin-stimulated glucose uptake (137).
In addition to suppressing lipolysis, insulin stimulates fatty acid uptake (145). The fatty acid transporters CD36 (146) and FATP1 (147) are both implicated, and studies show that insulin can increase the translocation of these transporters from intracellular vesicles to the cell surface (146) to enhance fatty acid uptake in fat and muscle cells (142, 147). Likewise, although the pathways involved remain unclear, insulin can increase amino acid uptake in these cells, potentially reflecting increased protein synthesis downstream of mTORC1 activation (148).
Acute regulation of metabolic enzymes
Insulin acutely controls metabolic enzyme activity through a combination of changes in phosphorylation, gene expression, and interaction with allosteric regulators to coordinate an increase in energy storage and decrease in utilization. Upon entering the cell, glucose is rapidly phosphorylated by hexokinase and either stored as glycogen via the activity of glycogen synthase or oxidized to generate ATP synthesis via enzymes such as pyruvate kinase. In muscle, liver, and adipose tissue, glucose is stored as glycogen and triglycerides. In general, insulin regulates the rate-limiting enzymes involved in glycolysis and lipolysis, as well as in glycogen and lipid synthesis, by decreasing their serine/threonine phosphorylation state via a combination of protein kinase inhibition and phosphatase activation (149–151).
Insulin stimulates glycogen accumulation through coordinated increases in glucose transport and glycogen synthesis (Figure 2). Hormonal activation of glycogen synthase involves both allosteric interaction with glucose-6-phosphate (152) and dephosphorylation promoted by kinase inhibition (including PKA, AMPK, or GSK3; refs. 149, 153) and phosphatase activation (primarily protein phosphatase 1 [PP1]; ref. 154). Insulin’s reduction in glycogen synthase phosphorylation is downstream of PI3K; Akt phosphorylates GSK3 (82, 155) and AMPK (156) to inactivate the kinases, resulting in decreased phosphorylation of glycogen synthase and an increase in its activity state. GSK3 inhibition is not sufficient for full activation of glycogen, since GSK3 does not phosphorylate all of the residues of glycogen synthase that are dephosphorylated in response to insulin (157), and knockins of GSK3 mutants have failed to support this enzyme’s role in stimulating glycogen synthesis (158, 159). Insulin also reduces the activities of other GSKs, notably PKA and AMPK (156).
PP1 activation correlates well with changes in glycogen synthase activity (160). However, insulin does not appear to globally activate PP1, but rather targets specific pools of the phosphatase localized on the glycogen particle. The compartmentalized, insulin-mediated activation of PP1 is due to glycogen-targeting subunits that serve as molecular scaffolds, incorporating the enzyme with its substrates in a macromolecular complex (149). Four different proteins (GM, GL, PTG, and R6) reportedly target PP1 to glycogen. Overexpressing these scaffolding proteins dramatically increases glycogen levels (161). Overexpressing PTG makes glycogen stores refractory to breakdown by agents that raise intracellular cAMP levels, suggesting that PTG locks the cell into a glycogenic mode, whereas PTG knockouts dramatically reduce glycogen levels (162). The mechanism by which insulin activates glycogen-associated PP1 remains unknown. Although it was proposed that MAPK activation leads to phosphorylation of the targeting protein GM to activate PP1 and dephosphorylate GS, blocking the pathway did not affect insulin’s activation of glycogen synthase, and mutating the identified phosphorylation sites did not impair insulin action (113). However, PI3K inhibitors can block insulin’s activation of PP1 (163), indicating that PIP3-dependent protein kinases are involved. Moreover, the genes encoding some of the scaffolding proteins are regulated, raising the possibility that transcriptional control constitutes a portion of glycogen synthesis regulation. Importantly, despite years of investigation, insulin’s mechanism of regulating glycogen synthase remains uncertain.
In adipocytes, glucose is stored primarily as lipid, the result of increased uptake of glucose and activation of lipid synthetic enzymes, including pyruvate dehydrogenase, fatty acid synthase, and acetyl-CoA carboxylase, through dephosphorylation. Although insulin undoubtedly promotes dephosphorylation of these enzymes, the pathways mediating these effects are not well understood. Insulin also inhibits lipolysis in adipocytes, primarily by inhibiting the enzyme hormone-sensitive lipase (HSL) (164). PKA (150) and AMPK (165) also regulate HSL activation via phosphorylation, while insulin inhibits HSL via a combination of kinase inhibition and phosphatase activation, and a major pathway involves reductions in cAMP levels due to the activation of the cAMP-specific phosphodiesterases PDE4 and PDE3B in fat cells (150, 151). Although these phosphodiesterases can be phosphorylated and presumably regulated via Akt (166), Akt knockout did not compromise the insulin-mediated inhibition of HSL in adipocytes (167), indicating that the pathways responsible for this important action of insulin remain unknown.
Regulation of gluconeogenesis via changes in gene expression
Insulin inhibits hepatic and renal glucose production and release by blocking gluconeogenesis and glycogenolysis through phosphorylation and dephosphorylation (as described above), controlling substrates via crosstalk with other tissues, and regulating expression of genes encoding key hepatic enzymes (168–170). Controversy remains regarding the importance of direct insulin action in controlling hepatic glucose output. Several studies demonstrate that Akt2 is required for insulin’s direct effect on glycogenolysis and gluconeogenesis (159). Insulin dramatically inhibits the transcription of PEPCK, encoding phosphoenolpyruvate carboxylase, the rate-limiting step in gluconeogenesis. Insulin also counteracts glucagon action by decreasing transcription of FBP1 (encoding bisphosphatase) and G6P (glucose-6-phosphatase) and increases transcription of genes encoding glycolytic enzymes such as glucokinase and pyruvate kinase and lipogenic enzymes such as fatty acid synthase and acetyl-CoA carboxylase (169, 171).
Several transcription factors play a role in insulin’s control of hepatic gene expression (Figure 3). Hepatic nuclear factor-3 (HNF3) and HNF4 both appear to be involved in regulating PEPCK expression (169, 172). Insulin regulates sterol regulatory element–binding protein-1c (SREBP-1c), which may also contribute to insulin’s negative effect on PEPCK transcription (173). However, the primary mechanism controlling expression of hepatic gluconeogenic genes appears to involve FOXO1 (174, 175). Both PEPCK and G6P promoter sequences contain Foxo binding sites, and FOXO1 overexpression markedly increases expression of glucose-6-phosphatase’s catalytic subunit. Akt-mediated phosphorylation of FOXO1 inhibits its activity by retaining FOXO1 in the cytoplasm (176, 177). Both HNF4 and FOXO1 are modified by the coactivator PGC-1, which may also be a target of insulin action (178).
Interestingly, hepatic FoxO1 deletion can normalize glucose tolerance and peripheral insulin sensitivity in mice lacking the insulin receptor, its substrates, or Akt isoforms in liver (179–181). Conversely, deleting FoxO1 in liver eliminates the requirement for direct insulin action to suppress glucose output (179, 181, 182). In global insulin receptor–knockout mice, restoring expression of the insulin receptor in liver failed to correct systemic insulin resistance and glucose intolerance, despite restoring insulin signaling in hepatocytes (183). Thus, the relative importance of direct versus indirect control of hepatic glucose metabolism remains uncertain.
While insulin plays a key role in regulating the enzymes of gluconeogenesis, it can also indirectly influence glucose metabolism. This occurs via changes in the availability of gluconeogenesis substrates released from muscle and fat (184). Thus, when insulin levels are low, hydrolysis of muscle protein and adipocyte triglycerides increases levels of gluconeogenic substrates such as alanine and free fatty acids. In humans, the indirect pathway may contribute to diabetes pathogenesis, especially in individuals with central obesity, since visceral fat is less sensitive than subcutaneous fat to insulin inhibition of lipolysis, resulting in direct flux of fatty acids through the portal vein to the liver (185).
Transcriptional regulation of lipogenesis
Insulin action plays a key role in controlling hepatic lipid metabolism and the development of steatosis during insulin resistance (186). Despite pronounced insulin resistance, hepatocyte-specific knockout of the insulin receptor protects mice from fatty liver (187, 188). These mice have reduced serum triglycerides and increased fatty acid oxidation and fail to induce de novo lipogenesis and the lipogenic gene program. Observations that patients with insulin receptor loss-of-function mutations display extreme insulin resistance and hyperglycemia and are protected from developing fatty liver (189) support the essential role of direct insulin action in regulating overall hepatic lipid accumulation.
Insulin’s stimulation of lipogenesis is largely mediated through the transcription factors SREBP-1c (190–192) and ChREBP (193). Dominant-negative forms of SREBP-1 can block lipogenic gene expression (191, 192), while SREBP-1 overexpression can increase it (194). Hepatic SREBP-1 levels are increased in rodent models of lipodystrophy, which is associated with coordinated increases in fatty acid synthesis and gluconeogenesis, mimicking the phenotype observed in genetic and dietary models of obesity-induced diabetes. These observations led Shimomura et al. (194) to speculate that increased expression of the isoform SREBP-1c might lead to “mixed insulin resistance” observed in the diabetic liver, with increased rates of both gluconeogenesis and lipogenesis. The pathways accounting for changes in SREBP-1c expression lie downstream of the IRS/PI3K pathway. However, the molecular basis for mixed insulin resistance remains uncertain. It remains possible that additional signals generated in obesity, such as nutritional factors (amino acids, sugars), ER stress, inflammation, or other hormones or transcriptional pathways (including LXR), drive the lipogenic pathway in a manner that still requires insulin (195).
mTORC1 is the major downstream effector of Akt in the control of lipogenic gene expression (196); the mTORC1 inhibitor rapamycin blocks insulin-stimulated SREBP activation and lipogenesis. Inhibiting S6 kinase, a downstream target of mTORC1, prevents SREBP-1 processing in TSC1/2-null MEFs, but fails to block insulin-induced SREBP-1c activation in primary rat hepatocytes, suggesting an additional S6 kinase–independent SREBP-1c step. The phosphatidic acid phosphatase and transcriptional coactivator lipin-1 is a direct substrate of mTORC1 and regulator of nuclear SREBP activity (197). mTORC1-mediated lipin-1 phosphorylation blocks its nuclear localization, thus permitting activation of both SREBP-1 and SREBP-2. Conversely, expression of a nonphosphorylated form of lipin-1 results in nuclear localization of lipin-1, reduced nuclear SREBP levels, and altered SREBP localization. Knockdown of lipin-1 in liver-specific Raptor-deficient mice restores SREBP-1 activation (197).
Finally, insulin also regulates SREBP activity by controlling SREBP ER-to-Golgi transport and proteolytic activation. Insulin stimulates ER-to-Golgi transport of SREBP-1c by promoting SREBP-1c phosphorylation and association with coat protein complex II (COPII) vesicles. In mouse liver, Akt also regulates SREBP transport through insulin-induced gene-2 (INSIG-2) levels. Insulin negatively regulates Insig-2a mRNA expression in an Akt-dependent manner, resulting in decreased INSIG-2 protein (198).
The role of ChREBP downstream of hepatic insulin action is less well defined. ChREBP is a glucose-responsive transcription factor that activates a lipogenic program similar to SREBP-1c, controlling Fasn, Scd1, and Elovl6 gene expression (193). Dietary fructose administration induces hepatic steatosis, an effect dependent on ChREBP (199). Hepatic overexpression of ChREBP is sufficient to induce fatty liver in mice, and increased ChREBP has been correlated with nonalcoholic fatty liver disease in humans (200). Targeted deletion of ChREBP reduces fatty acid synthesis independently of changes in SREBP (200). Moreover, liver-specific knockdown of ChREBP reduces de novo lipogenesis and hepatic triglyceride in ob/ob mice (201). Although it is now well established that de novo lipogenesis and fat accumulation are increased in the livers of insulin-resistant people (202, 203), the precise activation status of SREPB-1c and ChREBP across a broad population remains to be determined. Future studies will be needed to better understand the specific effects of these key lipogenic transcription factors and how these molecules interact in regulating lipogenic gene expression and de novo lipogenesis in humans.
Regulation of insulin degradation
Once insulin is released into the abdominal portal vein via the pancreas, it immediately enters the liver. Interestingly, the liver clears approximately half of this insulin, and it never enters systemic circulation. Reduced hepatic clearance has been observed in patients with insulin resistance (204), and may correlate with ethnic differences in diabetes risk (203, 205), suggesting an important regulatory role. This is potentially important, since it remains possible that the hyperinsulinemia resulting from decreased clearance can potentiate or even cause insulin resistance through classic homologous desensitization pathways (206).
Several studies demonstrated that insulin is degraded in liver via a receptor-mediated mechanism that involves tyrosine phosphorylation (207). Although the precise details remain uncertain, this process is thought to involve tyrosine phosphorylation of CEACAM1 (207), a single-transmembrane protein highly expressed on the sinusoidal surface membrane of hepatocytes (208). This glycoprotein undergoes phosphorylation on a single tyrosine in response to insulin, and in turn forms complexes with other signaling molecules such as Shc and SHP1 (208). Insulin receptor activation is required for the rapid internalization of the insulin-receptor complex into clathrin-coated vesicles to be ultimately targeted for degradation (209–211). Whereas insulin undergoes degradation, the insulin receptor may either recycle back to the plasma membrane or translocate into lysosomes for degradation. CEACAM1 phosphorylation appears to accelerate receptor internalization and insulin degradation by increasing the complex’s interactions with AP1-containing complexes in coated pits (207).
Final comments
The complexities of insulin action indicate the requirement for multiple signaling pathways. Indeed, as insulin maintains blood sugar within a narrow range and controls the transport and metabolism of other nutrients in multiple tissues, the fine-tuning of metabolic changes in energy metabolism are unlikely to be explained by a single phosphorylation or protein interaction event. Years of investigations indicate that insulin controls a complex series of intertwining pathways that together instruct cells how and when to store energy. While extensive progress has been made, numerous gaps remain in our understanding of the precise molecular events involved in insulin action, and will remain the focus of much investigation for years to come.
Acknowledgments
I recognize that this is an incomplete review of the many signaling molecules and pathways that are controlled by insulin, and there are several important areas not covered in great depth, including the control of lipoprotein metabolism, amino acid transport and metabolism, and cellular growth and differentiation, involving additional signaling molecules, kinases, phosphatases, G proteins, and transcriptional factors; and the effects of insulin in additional tissues such as brain, kidney, vasculature, and others. I also apologize to the numerous investigators whose work was not mentioned or cited owing to space limitations. I thank members of my laboratory for useful discussions. The work of my laboratory is supported by NIH grants P30DK063491 and R01DK124496, -076906, -125820, -122804, and -117551.
Version 1. 01/04/2021
Print issue publication
Footnotes
Conflict of interest: ARS is a cofounder of Elgia Therapeutics, and receives funding for his laboratory from Dong-A ST Co.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(1):e142241.https://doi.org/10.1172/JCI142241.
References
- 1.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 3.Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol. 2018;19(1):31–44. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saltiel AR. Insulin resistance in the defense against obesity. Cell Metab. 2012;15(6):798–804. doi: 10.1016/j.cmet.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Saltiel AR. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 2001;104(4):517–529. doi: 10.1016/S0092-8674(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 6.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23(7):804–814. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kido Y, Nakae J, Accili D. Clinical review 125: The insulin receptor and its cellular targets. J Clin Endocrinol Metab. 2001;86(3):972–979. doi: 10.1210/jcem.86.3.7306. [DOI] [PubMed] [Google Scholar]
- 9.Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30(6):586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 10.Hedo JA, Kahn CR, Hayashi M, Yamada KM, Kasuga M. Biosynthesis and glycosylation of the insulin receptor. Evidence for a single polypeptide precursor of the two major subunits. J Biol Chem. 1983;258(16):10020–10026. [PubMed] [Google Scholar]
- 11.Lane MD, Ronnett G, Slieker LJ, Kohanski RA, Olson TL. Post-translational processing and activation of insulin and EGF proreceptors. Biochimie. 1985;67(10–11):1069–1080. doi: 10.1016/S0300-9084(85)80104-8. [DOI] [PubMed] [Google Scholar]
- 12.Freychet P, Roth J, Neville DM., Jr Insulin receptors in the liver: specific binding of ( 125 I)insulin to the plasma membrane and its relation to insulin bioactivity. Proc Natl Acad Sci U S A. 1971;68(8):1833–1837. doi: 10.1073/pnas.68.8.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldfine ID, Smith GJ. Binding of insulin to isolated nuclei. Proc Natl Acad Sci U S A. 1976;73(5):1427–1431. doi: 10.1073/pnas.73.5.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hancock ML, et al. Insulin receptor associates with promoters genome-wide and regulates gene expression. Cell. 2019;177(3):722–736. doi: 10.1016/j.cell.2019.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasuga M, Zick Y, Blithe DL, Crettaz M, Kahn CR. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature. 1982;298(5875):667–669. doi: 10.1038/298667a0. [DOI] [PubMed] [Google Scholar]
- 16.Kahn CR, et al. The insulin receptor and its substrate: molecular determinants of early events in insulin action. Recent Prog Horm Res. 1993;48:291–339. doi: 10.1016/b978-0-12-571148-7.50015-4. [DOI] [PubMed] [Google Scholar]
- 17.Ablooglu AJ, Kohanski RA. Activation of the insulin receptor’s kinase domain changes the rate-determining step of substrate phosphorylation. Biochemistry. 2001;40(2):504–513. doi: 10.1021/bi002292m. [DOI] [PubMed] [Google Scholar]
- 18.Ebina Y, et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell. 1985;40(4):747–758. doi: 10.1016/0092-8674(85)90334-4. [DOI] [PubMed] [Google Scholar]
- 19.De Meyts P. Insulin/receptor binding: the last piece of the puzzle? What recent progress on the structure of the insulin/receptor complex tells us (or not) about negative cooperativity and activation. Bioessays. 2015;37(4):389–397. doi: 10.1002/bies.201400190. [DOI] [PubMed] [Google Scholar]
- 20.McKern NM, et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006;443(7108):218–221. doi: 10.1038/nature05106. [DOI] [PubMed] [Google Scholar]
- 21.Croll TI, et al. Higher-resolution structure of the human insulin receptor ectodomain: multi-modal inclusion of the insert domain. Structure. 2016;24(3):469–476. doi: 10.1016/j.str.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutmann T, Kim KH, Grzybek M, Walz T, Coskun U. Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J Cell Biol. 2018;217(5):1643–1649. doi: 10.1083/jcb.201711047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohanski RA. Insulin receptor autophosphorylation. II. Determination of autophosphorylation sites by chemical sequence analysis and identification of the juxtamembrane sites. Biochemistry. 1993;32(22):5773–5780. doi: 10.1021/bi00073a008. [DOI] [PubMed] [Google Scholar]
- 24.White MF, Shoelson SE, Keutmann H, Kahn CR. A cascade of tyrosine autophosphorylation in the beta-subunit activates the phosphotransferase of the insulin receptor. J Biol Chem. 1988;263(6):2969–2980. [PubMed] [Google Scholar]
- 25.Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16(18):5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372(6508):746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 27.Taylor SI. Lilly Lecture: Molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin-receptor gene. Diabetes. 1992;41(11):1473–1490. doi: 10.2337/diab.41.11.1473. [DOI] [PubMed] [Google Scholar]
- 28.Longo N, Langley SD, Griffin LD, Elsas LJ. Two mutations in the insulin receptor gene of a patient with leprechaunism: application to prenatal diagnosis. J Clin Endocrinol Metab. 1995;80(5):1496–1501. doi: 10.1210/jcem.80.5.7538143. [DOI] [PubMed] [Google Scholar]
- 29.Kahn CR, Goldstein BJ. Molecular defects in insulin action. Science. 1989;245(4913):13. doi: 10.1126/science.2662406. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto R, et al. Defect in tyrosine kinase activity of the insulin receptor from a patient with insulin resistance and acanthosis nigricans. J Clin Endocrinol Metab. 1990;70(4):869–878. doi: 10.1210/jcem-70-4-869. [DOI] [PubMed] [Google Scholar]
- 31.Hojlund K, et al. A novel syndrome of autosomal-dominant hyperinsulinemic hypoglycemia linked to a mutation in the human insulin receptor gene. Diabetes. 2004;53(6):1592–1598. doi: 10.2337/diabetes.53.6.1592. [DOI] [PubMed] [Google Scholar]
- 32.Sun XJ, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352(6330):73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- 33.Lavan BE, Lane WS, Lienhard GE. The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J Biol Chem. 1997;272(17):11439–11443. doi: 10.1074/jbc.272.17.11439. [DOI] [PubMed] [Google Scholar]
- 34.Sun XJ, et al. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377(6545):173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 35.Lehr S, et al. Identification of major tyrosine phosphorylation sites in the human insulin receptor substrate Gab-1 by insulin receptor kinase in vitro. Biochemistry. 2000;39(35):10898–10907. doi: 10.1021/bi000982k. [DOI] [PubMed] [Google Scholar]
- 36.Wick MJ, Dong LQ, Hu D, Langlais P, Liu F. Insulin receptor-mediated p62dok tyrosine phosphorylation at residues 362 and 398 plays distinct roles for binding GTPase-activating protein and Nck and is essential for inhibiting insulin-stimulated activation of Ras and Akt. J Biol Chem. 2001;276(46):42843–42850. doi: 10.1074/jbc.M102116200. [DOI] [PubMed] [Google Scholar]
- 37.Baumann CA, et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407(6801):202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Kimura A, Baumann CA, Saltiel AR. APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol Cell Biol. 2002;22(11):3599–3609. doi: 10.1128/MCB.22.11.3599-3609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lechleider RJ, Freeman RM, Neel BG. Tyrosyl phosphorylation and growth factor receptor association of the human corkscrew homologue, SH-PTP2. J Biol Chem. 1993;268(18):13434–13438. [PubMed] [Google Scholar]
- 40.Gustafson TA, He W, Craparo A, Schaub CD, O’Neill TJ. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol Cell Biol. 1995;15(5):2500–2508. doi: 10.1128/MCB.15.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, DeYoung SM, Hwang JB, O’Leary EE, Saltiel AR. The roles of Cbl-b and c-Cbl in insulin-stimulated glucose transport. J Biol Chem. 2003;278(38):36754–36762. doi: 10.1074/jbc.M300664200. [DOI] [PubMed] [Google Scholar]
- 42.Myers MG, White MF. New frontiers in insulin receptor substrate signaling. Trends Endocrinol Metab. 1995;6(6):209–215. doi: 10.1016/1043-2760(95)00092-V. [DOI] [PubMed] [Google Scholar]
- 43.Araki E, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372(6502):186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 44.Yamauchi T, et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol Cell Biol. 1996;16(6):3074–3084. doi: 10.1128/MCB.16.6.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Withers DJ, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 46.Sasaoka T, Rose DW, Jhun BH, Saltiel AR, Draznin B, Olefsky JM. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem. 1994;269(18):13689–13694. [PubMed] [Google Scholar]
- 47.Hu J, Liu J, Ghirlando R, Saltiel AR, Hubbard SR. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol Cell. 2003;12(6):1379–1389. doi: 10.1016/S1097-2765(03)00487-8. [DOI] [PubMed] [Google Scholar]
- 48.Goldstein BJ, Bittner-Kowalczyk A, White MF, Harbeck M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J Biol Chem. 2000;275(6):4283–4289. doi: 10.1074/jbc.275.6.4283. [DOI] [PubMed] [Google Scholar]
- 49.Sugimoto S, Wandless TJ, Shoelson SE, Neel BG, Walsh CT. Activation of the SH2-containing protein tyrosine phosphatase, SH-PTP2, by phosphotyrosine-containing peptides derived from insulin receptor substrate-1. J Biol Chem. 1994;269(18):13614–13622. [PubMed] [Google Scholar]
- 50.Cheng A, Dube N, Gu F, Tremblay ML. Coordinated action of protein tyrosine phosphatases in insulin signal transduction. Eur J Biochem. 2002;269(4):1050–1059. doi: 10.1046/j.0014-2956.2002.02756.x. [DOI] [PubMed] [Google Scholar]
- 51.Zinker BA, et al. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc Natl Acad Sci U S A. 2002;99(17):11357–11362. doi: 10.1073/pnas.142298199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elchebly M, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 53.Klaman LD, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20(15):5479–5489. doi: 10.1128/MCB.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. 2005;2005(268):pe4. doi: 10.1126/stke.2682005pe4. [DOI] [PubMed] [Google Scholar]
- 55.Craparo A, Freund R, Gustafson TA. 14-3-3 (epsilon) interacts with the insulin-like growth factor I receptor and insulin receptor substrate I in a phosphoserine-dependent manner. J Biol Chem. 1997;272(17):11663–11669. doi: 10.1074/jbc.272.17.11663. [DOI] [PubMed] [Google Scholar]
- 56.Birnbaum MJ. Turning down insulin signaling. J Clin Invest. 2001;108(5):655–659. doi: 10.1172/JCI13714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, DeFea K, Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J Biol Chem. 1999;274(14):9351–9356. doi: 10.1074/jbc.274.14.9351. [DOI] [PubMed] [Google Scholar]
- 58.Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 59.De Fea K, Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J Biol Chem. 1997;272(50):31400–31406. doi: 10.1074/jbc.272.50.31400. [DOI] [PubMed] [Google Scholar]
- 60.Tanti JF, Gremeaux T, Van Obberghen E, Le Marchand-Brustel Y. Insulin receptor substrate 1 is phosphorylated by the serine kinase activity of phosphatidylinositol 3-kinase. Biochem J. 1994;304(Pt 1):17–21. doi: 10.1042/bj3040017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogihara T, et al. 14-3-3 protein binds to insulin receptor substrate-1, one of the binding sites of which is in the phosphotyrosine binding domain. J Biol Chem. 1997;272(40):25267–25274. doi: 10.1074/jbc.272.40.25267. [DOI] [PubMed] [Google Scholar]
- 62.Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010;11(1):84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morino K, et al. Muscle-specific IRS-1 Ser->Ala transgenic mice are protected from fat-induced insulin resistance in skeletal muscle. Diabetes. 2008;57(10):2644–2651. doi: 10.2337/db06-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Almind K, Bjorbaek C, Vestergaard H, Hansen T, Echwald S, Pedersen O. Aminoacid polymorphisms of insulin receptor substrate-1 in non-insulin-dependent diabetes mellitus. Lancet. 1993;342(8875):828–832. doi: 10.1016/0140-6736(93)92694-O. [DOI] [PubMed] [Google Scholar]
- 65.Kilpeläinen TO, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43(8):753–760. doi: 10.1038/ng.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang TJ, et al. Genetic variation of SORBS1 gene is associated with glucose homeostasis and age at onset of diabetes: A SAPPHIRe Cohort Study. Sci Rep. 2018;8(1):10574. doi: 10.1038/s41598-018-28891-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol. 1994;14(7):4902–4911. doi: 10.1128/MCB.14.7.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Backer JM, et al. Phosphatidylinositol 3’-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11(9):3469–3479. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Myers MG, et al. Insulin receptor substrate-1 mediates phosphatidylinositol 3’-kinase and p70S6k signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. J Biol Chem. 1994;269(46):28783–28789. [PubMed] [Google Scholar]
- 70.Carpenter CL, Cantley LC. Phosphoinositide kinases. Curr Opin Cell Biol. 1996;8(2):153–158. doi: 10.1016/S0955-0674(96)80060-3. [DOI] [PubMed] [Google Scholar]
- 71.Kerouz NJ, Horsch D, Pons S, Kahn CR. Differential regulation of insulin receptor substrates-1 and -2 (IRS-1 and IRS-2) and phosphatidylinositol 3-kinase isoforms in liver and muscle of the obese diabetic (ob/ob) mouse. J Clin Invest. 1997;100(12):3164–3172. doi: 10.1172/JCI119872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fruman DA, et al. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat Genet. 2000;26(3):379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- 73.Lazar DF, Saltiel AR. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat Rev Drug Discov. 2006;5(4):333–342. doi: 10.1038/nrd2007. [DOI] [PubMed] [Google Scholar]
- 74.Nakashima N, Sharma PM, Imamura T, Bookstein R, Olefsky JM. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3-L1 adipocytes. J Biol Chem. 2000;275(17):12889–12895. doi: 10.1074/jbc.275.17.12889. [DOI] [PubMed] [Google Scholar]
- 75.Clement S, et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001;409(6816):92–97. doi: 10.1038/35051094. [DOI] [PubMed] [Google Scholar]
- 76.Kaisaki PJ, et al. Polymorphisms in type II SH2 domain-containing inositol 5-phosphatase (INPPL1, SHIP2) are associated with physiological abnormalities of the metabolic syndrome. Diabetes. 2004;53(7):1900–1904. doi: 10.2337/diabetes.53.7.1900. [DOI] [PubMed] [Google Scholar]
- 77.Kagawa S, et al. Impact of SRC homology 2-containing inositol 5’-phosphatase 2 gene polymorphisms detected in a Japanese population on insulin signaling. J Clin Endocrinol Metab. 2005;90(5):2911–2919. doi: 10.1210/jc.2004-1724. [DOI] [PubMed] [Google Scholar]
- 78.Alessi DR, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–269. doi: 10.1016/S0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 79.Stephens L, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279(5351):710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 80.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 81.Yang J, et al. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002;9(6):1227–1240. doi: 10.1016/S1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- 82.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garofalo RS, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112(2):197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cho H, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292(5522):1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 85.George S, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hussain K, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334(6055):474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114(Pt 16):2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- 88.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13(22):2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 89.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271(49):31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 90.Hill MM, Clark SF, Tucker DF, Birnbaum MJ, James DE, Macaulay SL. A role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol. 1999;19(11):7771–7781. doi: 10.1128/MCB.19.11.7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3(4):267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 92.Wiese RJ, Mastick CC, Lazar DF, Saltiel AR. Activation of mitogen-activated protein kinase and phosphatidylinositol 3’-kinase is not sufficient for the hormonal stimulation of glucose uptake, lipogenesis, or glycogen synthesis in 3T3-L1 adipocytes. J Biol Chem. 1995;270(7):3442–3446. doi: 10.1074/jbc.270.7.3442. [DOI] [PubMed] [Google Scholar]
- 93.Bandyopadhyay G, et al. Activation of protein kinase C (alpha, beta, and zeta) by insulin in 3T3/L1 cells. Transfection studies suggest a role for PKC-zeta in glucose transport. J Biol Chem. 1997;272(4):2551–2558. doi: 10.1074/jbc.272.4.2551. [DOI] [PubMed] [Google Scholar]
- 94.Standaert ML, Galloway L, Karnam P, Bandyopadhyay G, Moscat J, Farese RV. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem. 1997;272(48):30075–30082. doi: 10.1074/jbc.272.48.30075. [DOI] [PubMed] [Google Scholar]
- 95.Kotani K, et al. Requirement of atypical protein kinase clambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18(12):6971–6982. doi: 10.1128/MCB.18.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rommel C, et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3(11):1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 97.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376(6541):599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 98.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10(2):262–267. doi: 10.1016/S0955-0674(98)80149-X. [DOI] [PubMed] [Google Scholar]
- 99.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13(6):383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 100.Andjelkovic M, Maira SM, Cron P, Parker PJ, Hemmings BA. Domain swapping used to investigate the mechanism of protein kinase B regulation by 3-phosphoinositide-dependent protein kinase 1 and Ser473 kinase. Mol Cell Biol. 1999;19(7):5061–5072. doi: 10.1128/MCB.19.7.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang T, Sweeney G, Rudolf MT, Klip A, Traynor-Kaplan A, Tsien RY. Membrane-permeant esters of phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(18):11017–11024. doi: 10.1074/jbc.273.18.11017. [DOI] [PubMed] [Google Scholar]
- 102.Krook A, Moller DE, Dib K, O’Rahilly S. Two naturally occurring mutant insulin receptors phosphorylate insulin receptor substrate-1 (IRS-1) but fail to mediate the biological effects of insulin. Evidence that IRS-1 phosphorylation is not sufficient for normal insulin action. J Biol Chem. 1996;271(12):7134–7140. doi: 10.1074/jbc.271.12.7134. [DOI] [PubMed] [Google Scholar]
- 103.Ribon V, Printen JA, Hoffman NG, Kay BK, Saltiel AR. A novel, multifuntional c-Cbl binding protein in insulin receptor signaling in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18(2):872–879. doi: 10.1128/MCB.18.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ribon V, Hubbell S, Herrera R, Saltiel AR. The product of the cbl oncogene forms stable complexes in vivo with endogenous Crk in a tyrosine phosphorylation-dependent manner. Mol Cell Biol. 1996;16(1):45–52. doi: 10.1128/MCB.16.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Knudsen BS, Feller SM, Hanafusa H. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. J Biol Chem. 1994;269(52):32781–32787. [PubMed] [Google Scholar]
- 106.Chiang SH, et al. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature. 2001;410(6831):944–948. doi: 10.1038/35073608. [DOI] [PubMed] [Google Scholar]
- 107.Ribon V, Johnson JH, Camp HS, Saltiel AR. Thiazolidinediones and insulin resistance: peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc Natl Acad Sci U S A. 1998;95(25):14751–14756. doi: 10.1073/pnas.95.25.14751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McCormick F. Signal transduction. How receptors turn Ras on. Nature. 1993;363(6424):15–16. doi: 10.1038/363015a0. [DOI] [PubMed] [Google Scholar]
- 109.Milarski KL, Saltiel AR. Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J Biol Chem. 1994;269(33):21239–21243. [PubMed] [Google Scholar]
- 110.Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc Natl Acad Sci U S A. 1995;92(3):664–668. doi: 10.1073/pnas.92.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 112.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4(5):373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 113.Lazar DF, et al. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J Biol Chem. 1995;270(35):20801–20807. doi: 10.1074/jbc.270.35.20801. [DOI] [PubMed] [Google Scholar]
- 114.Ozes ON, et al. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci U S A. 2001;98(8):4640–4645. doi: 10.1073/pnas.051042298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(pt 20):3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Alessi DR, Pearce LR, Garcia-Martinez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal. 2009;2(67):pe27. doi: 10.1126/scisignal.267pe27. [DOI] [PubMed] [Google Scholar]
- 118.Mori H, et al. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009;9(4):362–374. doi: 10.1016/j.cmet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107(8):3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Demetriades C, Doumpas N, Teleman AA. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell. 2014;156(4):786–799. doi: 10.1016/j.cell.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bar-Peled L, et al. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gingras AC, Raught B, Sonenberg N. mTOR signaling to translation. Curr Top Microbiol Immunol. 2004;279:169–197. doi: 10.1007/978-3-642-18930-2_11. [DOI] [PubMed] [Google Scholar]
- 123.Dugani CB, Klip A. Glucose transporter 4: cycling, compartments and controversies. EMBO Rep. 2005;6(12):1137–1142. doi: 10.1038/sj.embor.7400584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. J Biol Chem. 2019;294(30):11369–11381. doi: 10.1074/jbc.REV119.008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Haney PM, Levy MA, Strube MS, Mueckler M. Insulin-sensitive targeting of the GLUT4 glucose transporter in L6 myoblasts is conferred by its COOH-terminal cytoplasmic tail. J Cell Biol. 1995;129(3):641–658. doi: 10.1083/jcb.129.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miinea CP, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391(pt 1):87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sano H, et al. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 2007;5(4):293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 128.Randhawa VK, et al. GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J Biol Chem. 2008;283(40):27208–27219. doi: 10.1074/jbc.M804282200. [DOI] [PubMed] [Google Scholar]
- 129.Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2008;411(1):89–95. doi: 10.1042/BJ20071318. [DOI] [PubMed] [Google Scholar]
- 130.Kane S, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277(25):22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 131.Sano H, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278(17):14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 132.Karunanithi S, et al. A Rab10:RalA G protein cascade regulates insulin-stimulated glucose uptake in adipocytes. Mol Biol Cell. 2014;25(19):3059–3069. doi: 10.1091/mbc.e14-06-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ahmed SM, Nishida-Fukuda H, Li Y, McDonald WH, Gradinaru CC, Macara IG. Exocyst dynamics during vesicle tethering and fusion. Nat Commun. 2018;9(1):5140. doi: 10.1038/s41467-018-07467-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.He B, Guo W. The exocyst complex in polarized exocytosis. Curr Opin Cell Biol. 2009;21(4):537–542. doi: 10.1016/j.ceb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422(6932):629–633. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 136.Inoue M, Chiang SH, Chang L, Chen XW, Saltiel AR. Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol Biol Cell. 2006;17(5):2303–2311. doi: 10.1091/mbc.e06-01-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell. 2007;13(3):391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 138.Wu H, Turner C, Gardner J, Temple B, Brennwald P. The Exo70 subunit of the exocyst is an effector for both Cdc42 and Rho3 function in polarized exocytosis. Mol Biol Cell. 2010;21(3):430–442. doi: 10.1091/mbc.e09-06-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sano H, Peck GR, Blachon S, Lienhard GE. A potential link between insulin signaling and GLUT4 translocation: association of Rab10-GTP with the exocyst subunit Exoc6/6b. Biochem Biophys Res Commun. 2015;465(3):601–605. doi: 10.1016/j.bbrc.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen XW, et al. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell. 2011;22(1):141–152. doi: 10.1091/mbc.e10-08-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Skorobogatko Y, et al. RalA controls glucose homeostasis by regulating glucose uptake in brown fat. Proc Natl Acad Sci U S A. 2018;115(30):7819–7824. doi: 10.1073/pnas.1801050115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen Q, et al. Targeting RalGAPα1 in skeletal muscle to simultaneously improve postprandial glucose and lipid control. Sci Adv. 2019;5(4):eaav4116. doi: 10.1126/sciadv.aav4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4(1):66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- 144.Moskalenko S, et al. Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem. 2003;278(51):51743–51748. doi: 10.1074/jbc.M308702200. [DOI] [PubMed] [Google Scholar]
- 145.Ramos-Roman MA, Lapidot SA, Phair RD, Parks EJ. Insulin activation of plasma nonesterified fatty acid uptake in metabolic syndrome. Arterioscler Thromb Vasc Biol. 2012;32(8):1799–1808. doi: 10.1161/ATVBAHA.112.250019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Abumrad NA, Sfeir Z, Connelly MA, Coburn C. Lipid transporters: membrane transport systems for cholesterol and fatty acids. Curr Opin Clin Nutr Metab Care. 2000;3(4):255–262. doi: 10.1097/00075197-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 147.Stahl A, Evans JG, Pattel S, Hirsch D, Lodish HF. Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes. Dev Cell. 2002;2(4):477–488. doi: 10.1016/S1534-5807(02)00143-0. [DOI] [PubMed] [Google Scholar]
- 148.Walker DK, et al. Insulin increases mRNA abundance of the amino acid transporter SLC7A5/LAT1 via an mTORC1-dependent mechanism in skeletal muscle cells. Physiol Rep. 2014;2(3):e00238. doi: 10.1002/phy2.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes. 2000;49(12):1967–1977. doi: 10.2337/diabetes.49.12.1967. [DOI] [PubMed] [Google Scholar]
- 150.Stralfors P, Bjorgell P, Belfrage P. Hormonal regulation of hormone-sensitive lipase in intact adipocytes: identification of phosphorylated sites and effects on the phosphorylation by lipolytic hormones and insulin. Proc Natl Acad Sci U S A. 1984;81(11):3317–3321. doi: 10.1073/pnas.81.11.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bjorgell P, Rosberg S, Isaksson O, Belfrage P. The antilipolytic, insulin-like effect of growth hormone is caused by a net decrease of hormone-sensitive lipase phosphorylation. Endocrinology. 1984;115(3):1151–1156. doi: 10.1210/endo-115-3-1151. [DOI] [PubMed] [Google Scholar]
- 152.Roach PJ. Multisite and hierarchal protein phosphorylation. J Biol Chem. 1991;266(22):14139–14142. [PubMed] [Google Scholar]
- 153.Brady MJ, Pessin JE, Saltiel AR. Spatial compartmentalization in the regulation of glucose metabolism by insulin. Trends Endocrinol Metab. 1999;10(10):408–413. doi: 10.1016/S1043-2760(99)00201-5. [DOI] [PubMed] [Google Scholar]
- 154.Brady MJ, Printen JA, Mastick CC, Saltiel AR. Role of protein targeting to glycogen (PTG) in the regulation of protein phosphatase-1 activity. J Biol Chem. 1997;272(32):20198–20204. doi: 10.1074/jbc.272.32.20198. [DOI] [PubMed] [Google Scholar]
- 155.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 156.Valentine RJ, Coughlan KA, Ruderman NB, Saha AK. Insulin inhibits AMPK activity and phosphorylates AMPK Ser485/491 through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch Biochem Biophys. 2014;562:62–69. doi: 10.1016/j.abb.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lawrence JC, Roach PJ. New insights into the role and mechanism of glycogen synthase activation by insulin. Diabetes. 1997;46(4):541–547. doi: 10.2337/diab.46.4.541. [DOI] [PubMed] [Google Scholar]
- 158.von Wilamowitz-Moellendorff A, et al. Glucose-6-phosphate-mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes. 2013;62(12):4070–4082. doi: 10.2337/db13-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wan M, et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab. 2013;18(1):99–105. doi: 10.1016/j.cmet.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brady MJ, Bourbonais FJ, Saltiel AR. The activation of glycogen synthase by insulin switches from kinase inhibition to phosphatase activation during adipogenesis in 3T3-L1 cells. J Biol Chem. 1998;273(23):14063–14066. doi: 10.1074/jbc.273.23.14063. [DOI] [PubMed] [Google Scholar]
- 161.Berman HK, O’Doherty RM, Anderson P, Newgard CB. Overexpression of protein targeting to glycogen (PTG) in rat hepatocytes causes profound activation of glycogen synthesis independent of normal hormone- and substrate-mediated regulatory mechanisms. J Biol Chem. 1998;273(41):26421–26425. doi: 10.1074/jbc.273.41.26421. [DOI] [PubMed] [Google Scholar]
- 162.Lu B, et al. Metabolic crosstalk: molecular links between glycogen and lipid metabolism in obesity. Diabetes. 2014;63(9):2935–2948. doi: 10.2337/db13-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shepherd PR. Mechanisms regulating phosphoinositide 3-kinase signalling in insulin-sensitive tissues. Acta Physiol Scand. 2005;183(1):3–12. doi: 10.1111/j.1365-201X.2004.01382.x. [DOI] [PubMed] [Google Scholar]
- 164.Holm C, Osterlund T, Laurell H, Contreras JA. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Annu Rev Nutr. 2000;20:365–393. doi: 10.1146/annurev.nutr.20.1.365. [DOI] [PubMed] [Google Scholar]
- 165.Kim SJ, Tang T, Abbott M, Viscarra JA, Wang Y, Sul HS. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol Cell Biol. 2016;36(14):1961–1976. doi: 10.1128/MCB.00244-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kitamura T, et al. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol. 1999;19(9):6286–6296. doi: 10.1128/MCB.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.DiPilato LM, Ahmad F, Harms M, Seale P, Manganiello V, Birnbaum MJ. The role of PDE3B phosphorylation in the inhibition of lipolysis by insulin. Mol Cell Biol. 2015;35(16):2752–2760. doi: 10.1128/MCB.00422-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- 169.Lin HV, Accili D. Hormonal regulation of hepatic glucose production in health and disease. Cell Metab. 2011;14(1):9–19. doi: 10.1016/j.cmet.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Leavens KF, Birnbaum MJ. Insulin signaling to hepatic lipid metabolism in health and disease. Crit Rev Biochem Mol Biol. 2011;46(3):200–215. doi: 10.3109/10409238.2011.562481. [DOI] [PubMed] [Google Scholar]
- 171.Wong RH, Sul HS. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr Opin Pharmacol. 2010;10(6):684–691. doi: 10.1016/j.coph.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Edgerton DS, et al. Effects of insulin on the metabolic control of hepatic gluconeogenesis in vivo. Diabetes. 2009;58(12):2766–2775. doi: 10.2337/db09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chakravarty K, et al. Sterol regulatory element-binding protein-1c mimics the negative effect of insulin on phosphoenolpyruvate carboxykinase (GTP) gene transcription. J Biol Chem. 2001;276(37):34816–34823. doi: 10.1074/jbc.M103310200. [DOI] [PubMed] [Google Scholar]
- 174.Farmer SR. The forkhead transcription factor Foxo1: a possible link between obesity and insulin resistance. Mol Cell. 2003;11(1):6–8. doi: 10.1016/S1097-2765(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 175.Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108(9):1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rena G, Prescott AR, Guo S, Cohen P, Unterman TG. Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem J. 2001;354(Pt 3):605–612. doi: 10.1042/0264-6021:3540605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27(16):2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 178.Puigserver P, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423(6939):550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 179.Lu M, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18(3):388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.O-Sullivan I, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dong XC, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8(1):65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Titchenell PM, et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 2016;23(6):1154–1166. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Okamoto H, Obici S, Accili D, Rossetti L. Restoration of liver insulin signaling in Insr knockout mice fails to normalize hepatic insulin action. J Clin Invest. 2005;115(5):1314–1322. doi: 10.1172/JCI23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bradley DC, Poulin RA, Bergman RN. Dynamics of hepatic and peripheral insulin effects suggest common rate-limiting step in vivo. Diabetes. 1993;42(2):296–306. doi: 10.2337/diab.42.2.296. [DOI] [PubMed] [Google Scholar]
- 185.Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab. 2000;11(9):351–356. doi: 10.1016/s1043-2760(00)00323-4. [DOI] [PubMed] [Google Scholar]
- 186.Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015;16(11):678–689. doi: 10.1038/nrm4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Haas JT, et al. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15(6):873–884. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Biddinger SB, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7(2):125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Semple RK, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119(2):315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim JB, et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest. 1998;101(1):1–9. doi: 10.1172/JCI1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Foretz M, Guichard C, Ferre P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A. 1999;96(22):12737–12742. doi: 10.1073/pnas.96.22.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Foretz M, et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19(5):3760–3768. doi: 10.1128/MCB.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab. 2013;24(5):257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 194.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96(24):13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kim JY, et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell. 2018;175(1):133–145. doi: 10.1016/j.cell.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bakan I, Laplante M. Connecting mTORC1 signaling to SREBP-1 activation. Curr Opin Lipidol. 2012;23(3):226–234. doi: 10.1097/MOL.0b013e328352dd03. [DOI] [PubMed] [Google Scholar]
- 197.Peterson TR, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Dobrosotskaya IY, Goldstein JL, Brown MS, Rawson RB. Reconstitution of sterol-regulated endoplasmic reticulum-to-Golgi transport of SREBP-2 in insect cells by co-expression of mammalian SCAP and Insigs. J Biol Chem. 2003;278(37):35837–35843. doi: 10.1074/jbc.M306476200. [DOI] [PubMed] [Google Scholar]
- 199.Kim MS, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126(11):4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101(19):7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dentin R, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55(8):2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 202.Petersen KF, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104(31):12587–12594. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Jones CN, Pei D, Staris P, Polonsky KS, Chen YD, Reaven GM. Alterations in the glucose-stimulated insulin secretory dose-response curve and in insulin clearance in nondiabetic insulin-resistant individuals. J Clin Endocrinol Metab. 1997;82(6):1834–1838. doi: 10.1210/jcem.82.6.3979. [DOI] [PubMed] [Google Scholar]
- 205.Bergman RN, Piccinini F, Kabir M, Kolka CM, Ader M. Hypothesis: role of reduced hepatic insulin clearance in the pathogenesis of type 2 diabetes. Diabetes. 2019;68(9):1709–1716. doi: 10.2337/db19-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lesniak MA, Roth J. Regulation of receptor concentration by homologous hormone. Effect of human growth hormone on its receptor in IM-9 lymphocytes. J Biol Chem. 1976;251(12):3720–3729. [PubMed] [Google Scholar]
- 207.Najjar SM, Perdomo G. Hepatic insulin clearance: mechanism and physiology. Physiology (Bethesda) 2019;34(3):198–215. doi: 10.1152/physiol.00048.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hinoda Y, et al. Molecular cloning of a cDNA coding biliary glycoprotein I: primary structure of a glycoprotein immunologically crossreactive with carcinoembryonic antigen. Proc Natl Acad Sci U S A. 1988;85(18):6959–6963. doi: 10.1073/pnas.85.18.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Carpentier JL, Fehlmann M, Van Obberghen E, Gorden P, Orci L. Insulin receptor internalization and recycling: mechanism and significance. Biochimie. 1985;67(10-11):1143–1145. doi: 10.1016/S0300-9084(85)80112-7. [DOI] [PubMed] [Google Scholar]
- 210.Gorden P, Carpentier JL, Freychet P, LeCam A, Orci L. Intracellular translocation of iodine-125-labeled insulin: direct demonstration in isolated hepatocytes. Science. 1978;200(4343):782–785. doi: 10.1126/science.644321. [DOI] [PubMed] [Google Scholar]
- 211.Yamada K, Carpentier JL, Cheatham B, Goncalves E, Shoelson SE, Kahn CR. Role of the transmembrane domain and flanking amino acids in internalization and down-regulation of the insulin receptor. J Biol Chem. 1995;270(7):3115–3122. doi: 10.1074/jbc.270.7.3115. [DOI] [PubMed] [Google Scholar]