

SUMMARY
Nascent RNA sequencing has revealed that pre-mRNA splicing can occur shortly after introns emerge from RNA polymerase II (RNA Pol II). Differences in co-transcriptional splicing profiles suggest regulation by cis-and/or trans-acting factors. Here, we use single-molecule intron tracking (SMIT) to identify a cohort of regulators by machine learning in budding yeast. Of these, Nab2 displays reduced co-transcriptional splicing when depleted. Unexpectedly, these splicing defects are attributable to aberrant "intrusive" transcriptional readthrough from upstream genes, as revealed by long-read sequencing. Transcripts that originate from the intron-containing gene’s own transcription start site (TSS) are efficiently spliced, indicating no direct role of Nab2 in splicing per se. This work highlights the coupling between transcription, splicing, and 3′ end formation in the context of gene organization along chromosomes. We conclude that Nab2 is required for proper 3′ end processing, which ensures gene-specific control of co-transcriptional RNA processing.
In Brief
The nuclear poly(A)-binding protein Nab2 is implicated in splicing. Long-read sequencing of nascent RNA molecules reveals that chimeric pre-mRNAs that span multiple genes fail to splice. Alpert et al. identify a role of Nab2 in proper 30′ end cleavage, highlighting the importance of gene organization for RNA processing.
Graphical Abstract
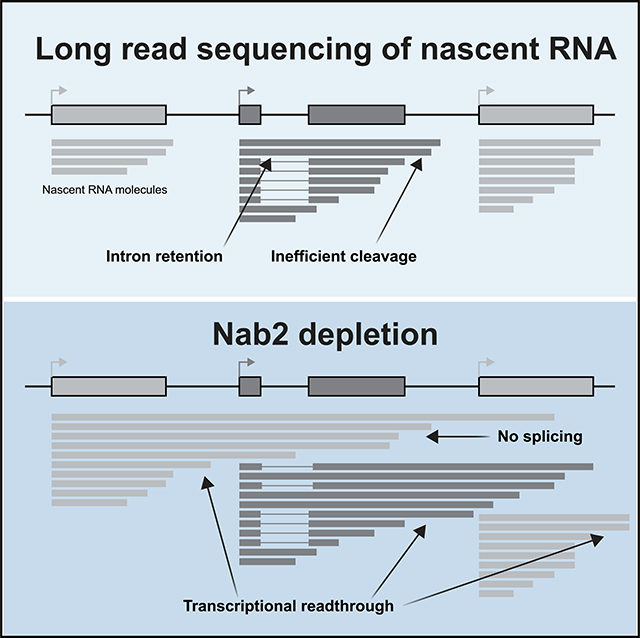
INTRODUCTION
When protein-coding genes are transcribed by RNA polymerase II (RNA Pol II), the nascent RNA undergoes 5′ end capping, splicing within the transcript body, and 3′ end cleavage before a mature mRNA can be exported to the cytoplasm for translation. The majority ofthese processingevents are co-transcriptional and closely coordinated with transcription and chromatin regulation (Alpert et al., 2017; Saldi et al., 2016; Tellier et al., 2020). For example, cleavage at the 3′ end initiates transcription termination. Intron-exon architecture has a dramatic effect on the position of RNA Pol II, general transcription factors, and active chromatin marks such as H3K4me3 along the lengths of genes (Bieberstein et al., 2012). Because of these findings, it is currently thought that coordination of the transcription and chromatin landscape with RNA processing steps is critical for execution of gene expression programs (Herzel et al., 2017; Moore and Proudfoot, 2009).
Previous work by our lab has identified coordination between co-transcriptional splicing and 3′ end cleavage in Schizosaccharomyces pombe (Herzel et al., 2018). This discovery was facilitated by long-read sequencing of nascent RNA, where entire nascent transcripts are sequenced from the 5′ end (defined by the transcription start site [TSS]) to the 3′ end (defined as the position of RNA Pol II at the time of isolation). This method identified a preponderance of "all or none" co-transcriptional splicing, where nascent transcripts had all introns spliced and displayed efficient 3′ end formation or, alternatively, had no introns spliced and displayed readthrough transcription (Herzel et al., 2018). These findings suggest functional coupling between splicing (or retention) of multiple introns and 3′ end cleavage in vivo.
Previous work has indicated that cleavage and polyadenylation (poly(A)) factors help define the terminal exon so that impairment of 3′ end cleavage inhibits splicing (Cooke et al., 1999; Fong and Bentley, 2001; Niwa and Berget, 1991; Rigo and Martinson, 2008). Conversely, mutations in the 3′ splice site (SS) of the last intron in pre-mRNA can inhibit splicing and poly(A) cleavage (Cooke et al., 1999; Davidson and West, 2013; Martins et al., 2011). However, these correlations between splicing and 3′ end cleavage are based on experiments that disrupt these processes and monitor populations of RNA molecules, most often in vitro. Thus, the existing evidence leaves open the question of whether coordination between splicing and poly(A) cleavage occurs in unperturbed cells and/or has a widespread role in normal gene regulation. Moreover, a mechanistic understanding of how splicing and cleavage are coordinated is lacking.
We set out to comprehensively determine which factors in budding yeast provide channels of communication between splicing, transcription, and other RNA processing events. To do so, we took advantage of the co-transcriptional splicing kinetic measurements from our previously published single-molecule intron tracking (SMIT) approach (Oesterreich et al., 2016). These data revealed an intriguingly high level of gene-specific variability in co-transcriptional splicing, which we leveraged to identify regulatory factors. A machine learning model was trained to predict splicing kinetic parameters using gene-to-gene variation in publicly available cross-linking immunoprecipitation (CLIP) and chromatin immunoprecipitation (ChIP) datasets as well as gene sequence and architecture. This unbiased approach led us to further investigate candidate regulators. The SMIT data collected in this study reveal the resilience of co-transcriptional splicing of somegenesandheightenedsensitivity ofotherstoapanelofper-turbations. In addition, long-read sequencing allowed us to identify readthrough transcription as the major defect caused by depletion of the essential protein Nab2, which has been implicated previously in nuclear export and splicing (Leung et al., 2009; Schmid etal., 2015; Souceketal., 2016; Tudeket al.,2018).
RESULTS
SMIT analyses of ~40% of intron-containing genes in budding yeast revealed that co-transcriptional splicing profiles are highly variable from gene to gene (Oesterreich et al., 2016). These profiles were defined by two key parameters that describe co-transcriptional splicing kinetics: saturation value (the mean fraction spliced near the end of the gene) and 1/2 max value (the RNA Pol II position where half of the saturation value is reached) (Figure S1A; STAR Methods). To obtain mechanistic insights into gene-specific variation, we trained a machine learning model to predict saturation (Figure 1A) and 1/2 max (Figures S1E and S1F) as follows. Each gene was characterized by 14 genetic and 398 epigenetic features derived from the budding yeast genome and publicly available genome-wide experiments (e.g., ChIP and CLIP), associating each measured parameter with gene positions, such as 5′ and 3′ SSs. Hierarchical clustering of the features produced 100 feature groups that had similar functions and positions along the gene (Table S1; Figures S1B–S1D). For example, U1, U2, and U5 small nuclear ribonucleoprotein particle (snRNP) proteins were all prominently detected at 3′ SSs by ChIP (Görnemann et al., 2005; Kotovic et al., 2003; Lacadie and Rosbash, 2005; Lacadie et al., 2006; Tardiff and Rosbash, 2006); as expected, high U1, U2, and U5 snRNP ChIP signals at 3′ SSs comprise one of the 100 feature groups identified by clustering. We then determined the relative importance of each feature group for the model’s prediction strength (Figure 1B; Table S2).
Figure 1. Machine Learning Predicts cis- and trans-Acting Factors Associated with Co-transcriptional Splicing.

(A) Observed and predicted saturation values are correlated with the variance explained (R2) as indicated for training (gray) and holdout (black) data.
(B) Feature groups used in the model are plotted according to their regression coefficient (b) and colored according to their cellular process (legend in C). Yellow indicates a feature group with mixed processes.
(C) Feature groups (gray box) are displayed above or below (positive or negative regression coefficient, respectively) the geneannotation(black) accordingto the genetic position where those features were identified as significant. Regression coefficient values (gray) are indicated to the right of the feature group.
(D) Normalized PAR-CLIP signals for Nab2 are aligned to 5′ SSs, 3′ SSs, and poly(A) sites (PASs) of all intron-containing genes in budding yeast (data from Baejen et al., 2014).
A Lasso regression model (Tibshirani, 1994) trained on 80% of the data was able to predict the remaining 20% of saturation values (Figure 1A). The model selected 21 non-genetic factors along with eight genetic features (Table S2) that contribute to prediction performance. Thirteen feature groups were associated positively with co-transcriptional splicing, and eight were associated negatively (Figures 1B and 1C). Several identified feature groups agreed with previous reports; for example, Npl3 and Gbp2, which have been implicated in transcription, splicing, 3′ end formation, mRNA export, and translation (Hackmann et al., 2014; Kress et al., 2008; Windgassen et al., 2004). The alternative histone, H2A.Z, is normally enriched at active promoters and promotes splicing of weak SSs (Neves et al., 2017; Nissen et al., 2017); our model utilized elevated H2A.Z ChIP signals at the poly(A) cleavage site (PAS) as a strong negative predictor of co-transcriptional splicing (β = 0.68).
The presence of a conserved poly(A) binding protein, Nab2, at 5′ and 3′ SSs was the strongest positively correlated feature in our model (β =0.26)(Figure 1C). Alignment of the Nab2 PAR-CLIP (photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation) signal (Baejen et al., 2014) to the relevant gene landmarks revealed enrichment of binding along the intron and downstream of the PAS (Figure 1D), suggesting a potential role in nascent RNA processing in addition to Nab2’s canonical poly(A) tail binding activity. Indeed, Nab2 truncation mutants have been shown to display subtle splicing defects (Soucek et al., 2016). Note that the model identified Nab2 (and H2A.Z, see above) at unusual positions along genes that do not agree with their canonical functions of poly(A) binding and promoter defi-nition, respectively. This can be attributed to the model’s reliance on heterogeneity to identify patterns that correlate with co-transcriptional splicing levels. A signal that is nearly ubiquitous across all intron-containing genes would not be incorporated into the model.
We set out to determine whether we could experimentally identify specific defects in co-transcriptional splicing kinetics associated with depletion of these factors. To perform SMIT on a genome-scale cohort of endogenous genes in the context of factor perturbation, fresh genomic deletion strains were derived for every non-essential factor identified by machine learning (rtt103Δ, gbp2Δ, pub1Δ, npl3Δ, tho2Δ, and htz1Δ [H2A.Z]). In addition, the top hit positively associated with co-transcriptional splicing, the essential protein Nab2, was depleted from the nucleus using a Nab2 Anchor-Away strain (Nab2-AA) (Haruki et al., 2008; Schmid et al., 2015). The SMIT protocol was optimized extensively to improve reproducibility and reduce the length and number of independent steps in the protocol (Figures S1G–S1K). Endogenous heterogeneity among co-transcriptional splicing profiles is what enabled our study; therefore, we expected heterogeneous gene-specific changes in response to different perturbations. For some deletions, we were surprised to observe that nearly all co-transcriptional splicing profiles were indistinguishable from the wild type (WT) (Figure S2), indicating that levels of co-transcriptional splicing were robustly maintained when diverse nuclear pathways were perturbed. The lack of effect on co-transcriptional splicing in the npl3Δ mutant was unexpected, given the previously observed changes in steady-state splicing levels (Kress et al., 2008); in this case, we speculate that post-transcriptional effects of npl3 deletion, such as RNA stability and/or mRNA export changes, could account for differences in the prevalence of introns.
Depletion of Nab2 had the most substantial effect on co-transcriptional splicing profiles. We performed SMIT at 0, 10, and 30 min of rapamycin treatment to trigger cytoplasmic sequestration of Nab2-AA; an isogenic control strain expressed endogenous, untagged Nab2 (Control-AA). Nab2 depletion altered the fraction co-transcriptionally spliced for most genes. Examples in Figure S3A show the full range of gene-specific responses to Nab2 depletion, including instances of reduced splicing, improved splicing, and unchanged splicing. The Euclidean distances of the 10- and 30-min-treated samples from the 0-min sample were used to quantify the difference between SMIT splicing profiles (ΔSMIT). The distribution of ΔSMIT values for Nab2-AA showed a significant change in splicing compared with Control-AA at 10 min (p = 4.28e-05) and 30 min (p = 0.0357) (Mann-Whitney U test) (Figure 2B). There was no significant difference between the 10- and 30-min control samples (p = 0.4517). The observed changes in fraction spliced upon Nab2-depletion were validated by RT-PCR (Figure 2C; Figure S3B). These data suggest that Nab2 is required for proper co-transcriptional splicing of some but not all of the 53 yeast introns analyzed by SMIT.
Figure 2. Nab2 Depletion Variably Affects Co-transcriptional Splicing Profiles.

(A) Co-transcriptional splicing profiles for Control-AA (left) and Nab2-AA (right) for three genes that exemplify the range of variation seen. Data from 0, 10, and 30 min of rapamycin treatment are modeled together (top legend) using a Loess smoothing method (solid line) with a 95% confidence interval. DSMIT values, indicated at the top left of each profile, are calculated as the Euclidean distance between the 0 and 10 min samples for the first 300 bp (bins = 60 bp). The PAS is indicated by a vertical dashed line, if the data extend to the end of the gene.
(B) Distribution of ΔSMIT values from the 0-min time point for all samples with significance (Mann-Whitney U test) as follows: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
(C) RT-PCR validation of splicing changes for two pre-mRNAs from (A). Random hexamers were used to reverse-transcribe nascent RNA, and intron-spanning primers amplify unspliced (top) and spliced (bottom) bands.
The mechanistic role of Nab2 in splicing regulation is unknown. To independently determine the effects of Nab2 depletion on co-transcriptional processing, we performed long-read sequencing of nascent RNA in the Nab2-AA and Control-AA strains after 10 min of rapamycin treatment, when splicing disruption was already determined to be significant by SMIT (Figure 2B). To capture full-length molecules, strand-switching reverse transcription was used to add common sequences to 50 and 30 ends of nascent RNA. Global amplification of the resulting cDNA was followed by blunt ligation of Nanopore barcode adapters, size selection, and sequencing on a minION flow cell (Figure S4A). Approximately 7 million base-called reads (12.7 Gb) were generated.
Unexpectedly, long-read sequencing revealed a role for Nab2 in 3′ end cleavage of nascent RNA instead of a role in splicing. Specifically, depletion of Nab2 led to a disruption in cleavage and termination, resulting in transcriptional readthrough and chimeric reads. Readthrough transcripts that extend past the PAS were occasionally observed under control conditions and were disproportionately unspliced, suggesting that crosstalk between co-transcriptional splicing and 3′ end formation occurs in WT budding yeast (Figure 3A, teal). This coupling between co-transcriptional splicing and cleavage in S. cerevisiae has also been observed previously in WT S. pombe (Herzel et al., 2018). Upon Nab2 depletion, a slight decrease in splicing (Figure S4B) and a large increase in readthrough transcription were observed (Figures 3A and 3B; Figures S4C and S4D). Increased readthrough was observed for spliced and unspliced transcripts (Figure 3A, orange). After filtering for reads that begin near the annotated TSS, the gene-specific fraction spliced was highly correlated between Control-AA and Nab2-AA (Figure 3C; R2 = 0.85). Deviation from the linear fit (Figure 3C, gray) was within the range seen between replicates (Figure S4E) and was likely due to stochastic noise in lowly expressed genes that were spliced inefficiently. We conclude that Nab2 depletion leads to no change in splicing when transcription of the intron is initiated at that gene’s TSS.
Figure 3. Long-Read Sequencing Reveals Transcriptional Readthrough upon Nab2 Depletion.

(A) Nanopore sequencing reads were sorted by 3′ end position for YPL079W (gray) for Control-AA (teal) and Nab2-AA (orange) samples. Reads were filtered for overlap with the intron-containing gene and must start no more than 100 bp downstream of the annotated TSS. Unspliced reads are displayed as a solid lineina darker color, and spliced reads are shown in a lighter color, with a thin line representing missing sequence information. All reads shown arise from the Watson strand. Read count and fraction spliced (percent) are shown.
(B) Coverage of reads downstream of the PAS was normalized to the signal at the PAS.
(C) The fraction spliced per gene is calculated for long reads that start within 50 bp of the annotated TSS and is plotted for Control-AA and Nab2-AA. The adjusted R2 value is displayed for the linear regression fit (gray line) and 95% confidence interval (gray ribbon).
Data from two biological replicates were first analyzed separately and then combined for display upon qualitative agreement between replicates.
Further analysis identified a set of transcripts that initiates in upstream genes and fails to cleave at the 3′ end, reading through into the intron-containing genes of interest, as exemplified in Figure 4A (and Figure S5), which again depicts YPL079W, this time with all overlapping reads beginning no more than 100 bp downstream of the TSS. We classified these reads which initiate in upstream genes as "intrusive transcripts." Although readthrough transcripts and intrusive transcripts are generated by the same phenomenon (failure to cleave), their relation to the intron-containing gene differentiates them. Whether the failed cleavage event occurs before or after transcription of the intron affects splicing efficiency (Figures 4B–4D). We quantified the reads from Figure 4A in a table (Figure 4B) that shows that intrusive and readthrough reads are primarily unspliced. Across the entire genome, readthrough transcripts were frequently unspliced (only 33% and 31% of Control-AA and Nab2-AA reads were spliced, respectively), and the fraction of spliced reads classified as intrusive was reduced further (18% and 10%; Figure 4C, left panel). The levels of readthrough and intrusive transcripts doubled during Nab2 depletion (1.93 and 2.13, respectively; Figure 4C, right panel), indicating that increased readthrough was responsible for the global splicing deficit observed in this study by SMIT as well as in other studies (Soucek et al., 2016).
Figure 4. Intrusive Transcripts Generated by Transcriptional Readthrough Are Poorly Spliced.

(A) Nanopore reads aligned to YPL079W (gray) were filtered to start no more than 100 bp downstream of the TSS. Intrusive reads that began more than 100 bp upstream of the TSS are displayed separately above reads that began near the TSS. Reads that do not span the entire intron of YPL079W are colored gray and were not included in spliced/unspliced values in (B). Reads are colored a darker shade of teal (Control-AA) or orange (Nab2-AA) when the YPL079W intron is unspliced. All reads shown arise from the Watson strand.
(B) Read counts (n =) are displayed for each category diagrammed in (C). The number of spliced and unspliced reads is also indicated alongside the fraction spliced (percent).
(C) Top: gene diagram (black) showing how example reads (gray) are classified according to readthrough status relative to the intron-containing gene. Left: colored bar plots showing the fraction of reads that are spliced or unspliced in each readthrough category. Right: grayscale bar plots showing the fraction of reads for each dataset that belong to the three readthrough categories (see legend).
(D) The fraction spliced is calculated for each gene using all reads or only intrusive reads and plotted for each condition. Values arising from less than 10 reads were removed. Reads that begin more than 50 bp upstream of the annotated TSS are defined as intrusive. The dashed line (gray) is y = x, and the black line is a linear regression model fit to the data with a 95% confidence interval. R2 for the model is displayed on each plot (p < 2.2 × 10−16 for both).
Data from two biological replicates were combined after confirming agreement between replicates for each parameter.
To address whether Nab2’s role was general or gene specific, we determined the fraction spliced of all reads aligned to a given gene or intrusive reads only (Figure 4D; Figure S6A). The majority of genes exhibited lower levels of splicing for intrusive reads, revealing a general trend with occasional outliers. Additionally, we determined how changes in levels of intrusive reads relate to changes in fraction spliced and found that the most populated quadrant was the bottom right, with a positive change in intrusive transcripts and negative change in splicing (Figure S6B). Some heterogeneity in the degree of intrusive transcript induction and the splicing efficiency of intrusive transcripts was apparent, and it remains unclear why certain genes may not require Nab2 for 3′ end cleavage. No significant linear trend was observed between induction of readthrough or intrusive transcripts and gene expression (Figures S6C and S6D), although outlying genes with high levels of readthrough and intrusive transcripts were all lowly expressed. Ribosomal protein genes (RPGs) make up a large fraction (32%) of intron-containing genes in yeast and often exhibit differential splicing and expression levels (Ares et al., 1999; Clark et al., 2002; Pleiss et al., 2007). We found no difference in the induction of readthrough or intrusive transcripts when comparing RPGs with non-RPGs (Figures S6E and S6F). Genes that were most sensitive to Nab2 depletion (Figure S6G) were more likely to contain a non-consensus 5′ SS (Figure S6I), suggesting that SS strength contributes to intron recognition and removal from intrusive transcripts. We conclude that Nab2 has an important role in 3′ end formation and that decreased splicing is a secondary effect of transcriptional readthrough.
DISCUSSION
Although pre-mRNA splicing is a standard step in the biogenesis of eukaryotic mRNAs, the progress of that reaction is surprisingly variable from gene to gene. Previous work in other laboratories has contributed to our understanding of the factors that control overall splicing levels in total or mRNA (Clark et al., 2002; Pleiss et al., 2007). Here we discovered that readthrough transcription, which results from failure of 3′ end cleavage and leads to RNA Pol II transcription into the next gene (Irniger et al., 1991), represses splicing of downstream introns. If factors that affect the efficiency of 3′ end cleavage are perturbed, then classic RNA analysis methods (RNA sequencing [RNA-seq], RT-PCR, northern blotting, etc.) may detect a splicing phenotype and suggest that the factor directly affects splicing. We show, using long-read sequencing, that splicing inhibition can instead be a secondary effect of readthrough. We applied an additional single-molecule RNA-seq strategy (SMIT) to generate gene-specific co-transcriptional splicing profiles on a global scale and tested perturbations of genes identified by machine learning as potential regulators. Nab2, the yeast homolog of ZC3H14 associated with intellectual disability in flies and humans (Pak et al., 2011), emerged as an important candidate. As predicted by our algorithm, rapid (10–30 min) Nab2 depletion led to a reduction of co-transcriptional splicing in some but not all genes. Analysis by long-read sequencing of nascent RNA revealed that the predominant co-transcriptional role of Nab2 is in 3′ end cleavage. The demonstration that co-transcriptional splicing defects are a consequence of readthrough transcription highlights the importance of proper 3′ end cleavage for maintaining gene-autonomous transcription and splicing independent of genome architecture, which can place neighboring genes dangerously close together. Below we discuss this unexpected activity of Nab2 in the context of coordinated transcription and RNA processing.
Nab2 is an essential, predominantly nuclear protein that has been implicated in multiple steps of mRNA expression. Initially identified as an mRNA export factor (Green et al., 2002), Nab2 is known to interact with proteins that associate with nuclear pores (Aibara et al., 2017; Soucek et al., 2016). Nab2’s role in export and stability are likely related to its role in binding to poly(A) tails (Batisse et al., 2009; Tuck and Tollervey, 2013; Viphakone et al., 2008). Nab2 depletion leads to global loss of poly(A)+ mRNA irrespective of whether the gene contains an intron; this effect was attributable to the nuclear exosome, indicating that RNA binding by Nab2 or its role in export prevents the observed mRNA decay (Schmid et al., 2015; Tudek et al., 2018). Nab2 is thought to bind poly(A) tails; however, its RNA binding preference is not limited to poly(A) because various analyses have shown a preference for a run of As followed by G in addition to tolerance for other nucleotides (Kim Guisbert et al., 2005; Riordan et al., 2011). Indeed, CLIP experiments in yeast revealed that Nab2 binds throughout the body of the transcript and that binding is especially high at 3′ ends near the PAS (Baejen et al., 2014). Moreover, a previous study showed elevated nascent RNA density downstream of PASs upon Nab2 depletion, suggesting effects on termination (Tudek et al., 2018); dependency of HFF1 pre-mRNA 3′ end cleavage on Nab2 is consistent with our findings. Our long-read sequencing data and analysis support our speculation that Nab2 binding near PASs could have a widespread role in stimulating 3′ end cleavage, preventing transcriptional readthrough and formation of unspliced chimeric transcripts.
Although we ultimately found that Nab2’s role in splicing was related to intrusive transcription from upstream genes, other interactions predicted in our model (Figure 1C) could be gene autonomous. Cleavage and poly(A) factors are known to contribute to exon definition of terminal exons in metazoans (Fong and Bentley, 2001; Li et al., 2001; Niwa and Berget, 1991; Rigo and Martinson, 2008). For example, Pcf11 is one of few cleavage factors bound to RNA Pol II through the C-terminal domain (CTD) along the entire gene body (Baejen et al., 2017; Licatalosi et al., 2002), and its presence near 3′ SSs has been predicted to contribute positively to splicing. Other poly(A) cleavage factors, like Rna15 and Yth1, were associated negatively. These factors remain to be further investigated.
Our study demonstrates the power of long-read sequencing for identifying coordinated transcription and RNA processing events. Here chimeric readthrough transcripts create intron-containing pre-mRNAs with first exons that are many thousands of nucleotides long. This is unusual because the first exon in all species is usually extremely short (<200 nt). This length distribution influences chromatin architecture and RNA Pol II in human cells (Bieberstein et al., 2012). An obvious suggestion is that pre-mRNA substrates with very long first exons are spliced inefficiently; we speculate that the nuclear cap-binding complex at the transcript’s 5′ end, which typically promotes splicing (Carrocci and Neugebauer, 2019), may be too far awayfrom the intronto perform this role. Indeed, cap-dependent splicing is inhibited when first exons are lengthened (Lewis et al., 1996). Alternatively, the very long first exon could be too highly packaged with proteins and/or RNA secondary structure to allow SS recognition and splicing so far downstream. Intriguingly, we observed intrusive reads that are spliced at an upstream annotated intron but fail to splice at the second annotated intron they encounter (Figure 4A), indicating that these reads are splicing competent and that failure to splice is specific to the intron with a long first exon.
Broadly speaking, long-read sequencing is likely to transform how we analyze and draw conclusions about the effects of mutations that affect transcription and RNA processing in cells. This work clearly illustrates an example where the actual substrates of the splicing reaction are not those inferred by short-read sequencing. This is also clear from the demonstration by long-read sequencing of coordinated splicing among introns in the same transcript (Drexler et al., 2020; Herzel et al., 2018; Tilgner et al., 2018). Furthermore, the correlation between intron retention and transcriptional readthrough was first observed in S. pombe using long-read sequencing (Herzel et al., 2018); the data presented here in WT budding yeast show that this relationship is evolutionarily conserved. Many studies from yeast to humans have perturbed the abundance of regulatory factors and used short-read RNA-seq to quantify the abundance of RNA isoforms. This study reveals that the mechanisms underlying those results may be less straightforward than initially assumed. For example, an RNA-seq approach using fragmentation would observe a large increase in intron retention for YPL079W without revealing that these transcripts originate from the upstream gene (Figure 4A). Finally, our findings underscore the importance of 3′ end formation and transcription termination in ensuring the independent expression of genes. In renal clear cell carcinoma cells, transcriptional readthrough generates aberrant exons, resulting in giant fusion transcripts originating from neighboring genes (Grosso et al., 2015). A high proportion of human diseases are associated with mutations in trans-acting splicing factors or cis-acting splicing-regulatory elements in genes (Manning and Cooper, 2017), making it important to further investigate the mechanisms underlying splicing changes as well as the downstream consequences of splicing inhibition.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karla Neugebauer (karla.neugebauer@yale.edu), Department of Molecular Biophysics and Biochemistry, Yale University, New Haven CT 06520.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
SMIT processing code is available on GitHub: https://github.com/carrillo/SMITproject. All other code including machine learning modeling is available at https://github.com/NeugebauerLab/Alpert2020_Nab2. The accession number for the SMIT and long-read sequencing data reported in this paper is GEO:GSE156133. Original data have been deposited to to Mendeley Data: http://dx.doi.org/10.17632/dddf2vhjyg.1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and treatment
For a list of all strains please refer to Table S5. Yeast cells were grown in YPAD medium at 30°C and shaking at 200 rpm. For SMIT experiments, 50 mL cultures were grown overnight to an OD600 = 0.6–0.8 (logarithmic growth phase). Cells were pelleted at 1100x g for 5 minutes at 4°C. Pellets were washed once with ice-cold 1x PBS and then transferred to an eppendorf tube for a second wash before being snap frozen in liquid nitrogen and stored at -80°C. For Nab2-AA and Control-AA strains (obtained from Torben Jensen; Schmid et al., 2015), rapamycin (Calbiochem) was added at 1 μg/ml final concentration to exponentially growing cells for 10 and 30 minutes of incubation. The same concentration of rapamycin was added to 1x PBS for all washing steps until cells were snap frozen.
METHOD DETAILS
Feature engineering and machine learning
To identify features correlating with saturation values of co-transcriptional splicing and kinetic measurements, we characterized each gene by 412 features (Table S1). These can be broadly divided into genetic and epigenetic features. We define genetic features as features, which can be derived directly from the genome sequence and its annotation. These features include splice sequence strength encoded by its Levenshtein distance to the consensus of 5′ SS, branch point sequence (BPS), 3′ SS, as well as characteristics of exons and introns. Genetic features were derived from annotation files using custom software (https://github.com/NeugebauerLab/Alpert2020_Nab2). We define epigenetic features as features, which cannot be directly inferred from the genome annotation. These features include genome-wide abundance levels of RNA and DNA binding factors as well as profiles of nucleosome organization. These abundance profiles were derived from publicly available datasets (Table S3). Datasets include CLIP, ChIP and MNase digestion experiments analyzed by deep sequencing or microarrays. For microarray data analysis, probe positions were converted from sacCer2 to sacCer3 with the liftOver tool (Hinrichs et al., 2006) and the data were converted into genome coverage tracks (pileup-format) with custom scripts. For deep sequencing analysis reads were mapped to the genome using tophat2 and genome-wide profiles generated using samtools (Li et al., 2009). To represent differential factor abundance over gene regions, we characterize each factor at three well-defined regions per gene: the 5′ SS, 3′ SS and PAS. The mean abundance in windows both 50 nt up- and downstream were determined for each factor. Thus, each gene was characterized by 6 position specific mean abundance values per factor. Engineering for epigenetic features was performed by custom software (https://github.com/NeugebauerLab/Alpert2020_Nab2).
Clustering of Features
Many features were highly correlated: uniform factor abundance across genes will lead to highly correlated feature values for different gene positions of the same factor. Alternatively, factors acting in the same complex or biological process may follow similar gene profiles, thereby leading to a correlation between factors. This correlation represents redundancy in the data, which can lead to undesired effects for machine learning. We therefore reduced dimensionality of the data by grouping correlated features and represent these feature-groups by a single representative meta-feature. Hierarchical clustering of all features was performed, using squared Pearson-correlation as a similarity measurement, to identify groups of correlated features. The hierarchical cluster can be represented by a dendrogram. Cutting the dendrogram at different heights divides the input data into a defined number of clusters or feature groups. This can be understood as compression of the data. A cluster-count of 412 represents uncompressed data, whereas a cluster-count of 1 would represent maximal compressing, by averaging over all dimensions (features). How can we find a feature group count which reduces correlation, but does not lose too much information due to compression? Such a set of input features is expected to lead to a good prediction performance in machine learning. We trained lasso regression models (see below) on iteratively compressed input data, represented by decreasing cluster count. For each iteration we determined the predictive performance of the compressed features by cross-validation. We further hypothesize that correlation clustering should reflect functional clustering. Functional clustering includes the identification of correlated gene position of one factor and correlated gene position values between factors of the same biological function (see above). To measure functional clustering each feature was characterized by gene position (5′ SS, 3′ SS, PAS). We determined how many features in each cluster belong to each position class and quantified the extent of mixing by calculating the Shannon entropy of the position-frequency distribution for each cluster. Separation of positions reduces mixing and thus the Shannon entropy. A perfect separation of positions between clusters would lead to a value of 0. For all cluster counts (1 to 412) we determined the mean Shannon entropy over all clusters. Analogous analysis was performed for i) experiment type, ii) factor identity and iii) function.
Machine learning – Lasso regression
Machine learning was used to train combinations of features and their relative contribution to optimally predict a target value (i.e., co-transcriptional splicing saturation and RNA Pol II position at 50% of the saturation level). Saturation values were defined as the mean fraction spliced of the four bins (30 bp each) immediately preceding the PAS. For genes where data does not extend to this region, the terminal four bins were used. Only bins with ≥ 10 reads were considered. Features are numerical or categorical characteristics of genes or feature groups (see clustering of features). While other machine learning models, like neural networks performed slightly better (data not shown), we used a regularized linear regression model due to its interpretability regarding i) importance and ii) direction of correlation. We divided the available data over all genes into training data (80%) and hold-out data (20%). Preprocessing of input features was not performed prior to training but included into the cross-validation during model selection. Input features were Yeo-Johnson transformed (Yeo and Johnson, 2000), scaled and centered (μ, sd = 1). Saturation values were logit transformed to map probabilities on the real axis. Model selection was performed by 5-fold cross-validation repeated 3 times on shuffled training data. Categorical features were one-hot encoded. Model performance was measured by root mean squared error. Machine learning was performed using caret (Kuhn, 2008) and code can be downloaded ( https://github.com/NeugebauerLab/Alpert2020_Nab2).
Construction of deletion strains
Saccharomyces Genome Deletion Collection strains (Giaever et al., 2002; Winzeler et al., 1999) were the generous gifts of Dr. March Hochstrasser and were used for the gene locus amplification and transformation. Each locus was substituted with the KanMX gene which confers resistance to geneticin or G418, which was used as a selection marker. Deletions were made fresh to limit compensatory mutations (Teng et al., 2013). Genomic DNA was isolated from each deletion strain from 2 mL of saturated overnight yeast culture in YPAD. Cells were resuspended in lysis buffer (final 10 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, 1% SDS, 2% Triton X-100) with equal volume Phenol:Chloroform pH 8 and zirconia beads (BioSpec) for vortexing. After centrifugation, the aqueous layer was collected for ethanol precipitation. Amplification of the KanMX cassette was performed using primers with added homology arms for the genomic locus of choice (Table S4). Purified, linear PCR product was used as the linear insert for transformation.
Budding yeast transformation
Yeast cells were grown in 50 mL YPAD medium at 30°C and shaking at 200 rpm to an OD600 = 0.5 (logarithmic growth phase). Cells were pelleted and washed with sterile water before resuspension in 0.1 M LiAc, 10 mM Tris-HCl, 1 mM EDTA, pH 7.4. One μg linear PCR product was added to cells with 10 μL single-stranded carrier DNA (salmon sperm DNA, Invitrogen). LiAc-TE-PEG buffer (1/10 of 10x TE, 1/10 of 1 M LiAc, 8/10 of 50% PEG 4000) was added to 6x the volume of the cell mixture. Sample was incubated 30 min at room temp. while rotating on wheel. 70 μL of 100% DMSO (prewarmed) was added before heat shocking the samples for 15 min at 42°C. Cells were pelleted at 1,100x g for 5 min at room temperature, resuspended in 300 μL YPAD and incubated on a rotating wheel at room temperature for four hours. Cells were then plated on YPAD plates containing 350 ug/ml G418. After ~48 hours, single colonies were picked for culture growth and strain validation by Sanger sequencing. All primers can be found in Table S4.
Nascent RNA isolation from chromatin
All steps were performed at 4°C if not stated otherwise. This protocol was modified slightly from Carrillo Oesterreich et al. (2010). Frozen cell pellets were resuspended in 1 mL buffer 1(20 mM HEPES pH 8.0, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 0.8% Triton X-100, 0.25 M sucrose, 1 mM DTT, 0.2 mM PMSF, 2.5 mM spermidine, 0.5 mM spermine). Cells were lysed by vortexing with zirconia beads (BioSpec) for 5 × 60 s pulses of beads with 60 s pauses on ice between each pulse. Beads were separated from lysate using a custom bead filter setup at 500x g for 5 min. Supernatant was transferred into a fresh eppendorf tube and centrifuged at 2,000x g for 15 min. The pellet was resuspended in 1 mL of buffer 2 (20 mM HEPES pH 7.6, 450 mM NaCl, 7.5 mM MgCl2, 20 mM EDTA, 10% glycerol, 1% NP-40, 2 M urea, 0.5 M sucrose, 1 mM DTT, 0.2 mM PMSF) and centrifuged for 15 min at 20,000x g. Each of these centrifugations were performed twice with clean buffer. Finally, pellets were resuspended in buffer P (50 mM sodium acetate, 50 mM NaCl, 1% SDS) and phenol:chloroform:IAA (pH 6.0) and incubated at 37°C for 1 hour with shaking (1150 rpm). Samples were spun at 13,000 rpm for 3 min at room temperature and the aqueous phase was transferred to a clean tube with 3 M sodium acetate pH 5.3 and 100% ice cold ethanol. Samples were incubated overnight at -80°C and then spun for 30 min at 20,000x g. The pellet was washed with 1 mL 75% ice cold ethanol and briefly spun again. Pellets were dried at room temperature for 5 minutes and then resuspended in 80 μl water. DNA was removed using two rounds of TurboDNase (Ambion) digestion. RNA samples were then depleted three times of polyA+ RNA by incubation with oligo(dT)-coated beads from Dynabeads mRNA DIRECT Micro Purification Kit (ThermoFisher), each time keeping the supernatant and discarding the beads. For long-read sequencing, ribosomal RNA was removed by up to three digestions with Terminator 5′-Phosphate-Dependent Exonuclease (Lucigen). Samples were cleaned between each step with RNA Clean & Concentrator Kit (Zymo Research).
3′ end adaptor ligation
600 ng DNase-treated, polyA-depleted nascent RNA was combined with 50 pmol 3′ end adaptor (/5rApp/NNNNNCTGTAGGCAC CATCAAT/3ddC/, Integrated DNA Technologies) and denatured at 65°C for 5 min followed by 4°C for 1 min. Buffer (50 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.5, 25% PEG 8000), 40 U RNaseOUT, and 200 U T4 RNA ligase II (truncated K227Q) (NEB) were added to the denatured RNA and incubated for 12 hours at 16°C. Samples were cleaned with RNA Clean & Concentrator Kit (Zymo Research).
Single Molecule Intron Tracking
Adaptor-ligated nascent RNA served as template for reverse transcription using SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer’s protocol with a custom SMIT RT primer. Two PCRs were used to first capture the splicing status and 3′ end position, and then to add the Illumina sequencing adapters. In the first PCR, cDNA samples were amplified with Phusion High-Fidelity polymerase (NEB) with all 62 gene-specific forward primers pooled (1 μM each final) together with an adaptor-specific reverse primer. Samples were cleaned with MinElute PCR purification kit (QIAGEN), and input into the second PCR. Each reaction consisted of 15 cycles (30 cycles total). All primers can be found in Table S4. Samples were submitted to the Yale Center for Genome Analysis (YCGA) for gel-based size selection (250 bp – 1000 bp) and sequencing on Illumina HiSeq 2500 (High-Output Mode V4, paired-end, 2×75 bp read length). Up to 6 different samples were pooled per lane (~50MIO reads/ sample).
SMIT data processing
Fastq files were filtered for read quality with the FASTX toolkit (RRID:SCR_005534) (fastq_quality_filter -Q 33 -q 20 -p 90). 3′ end adaptor sequence along with Illumina adaptor sequence was trimmed from R1 reads with cutadapt (Martin, 2011) (-g CATT GATGGTGCCTACAG -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCACGACCTCATCTCGTATGCCGTCTTCTGCTTG -n 2 -O 18 -m 23 -e 0.11 –match-read-wildcards –discard-untrimmed). Adaptor sequences were also trimmed from R2 reads (-a CTGTAGG CACCATCAATG -a AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT -n 2 -m 28 -M < read length-21 > -e 0.11 –match-read-wildcards). PCR duplicates were removed with Prinseq (Schmieder and Edwards, 2011), followed by removal of a 5 nt random sequences with the FASTX toolkit (fastx_trimmer -Q 33 -f 6). Reads were mapped with paired-end, splicing-sensitive parameters using HISAT2 (Kim et al., 2019) to the S. cerevisiae genome (sacCer3). Scripts are available at https://github.com/carrillo/SMITproject. Custom scripts were written in R to extract splicing status and 3′ end position for plotting. Insert length bias correction was performed as described previously (Oesterreich et al., 2016). The DSMIT parameter was calculated as the Euclidean distance from the respective 0 min sample. Only the first 300 bp after the 3′ SS were considered as those positions have the highest density of data. Data were binned into 60 nt segments to minimize the impact of sequencing noise.
Long read sequencing library preparation
Full length cDNA was generated from the adaptor-ligated nascent RNA with strand-switching reverse transcription (SMARTer PCR cDNA Synthesis Kit, Clontech), replacing the CDS Primer IIA with a custom primer complementary to the 3′ end adaptor (see Table S4). Double-stranded cDNA was amplified using the CDS Primer IIA for 12 cycles (Advantage 2 PCR kit, Clontech) and cleaned up with AMPure XP beads (Beckman Coulter). Purified product was then end-prepped with the NEBNext Ultra II repair/dA-tailing module (NEB) and ligated to Nanopore barcode adapters (Oxford Nanopore Technologies, PCR Barcoding Kit SQK-PBK004) with Blunt TA/ Ligase Master Mix (NEB). A second round of PCR using Nanopore barcode primers from the ONT kit was performed with Advantage 2 for 8 cycles. AMPure XP beads were used to clean up the sample between each reaction with a ratio of 2:1 (beads:sample) until the final PCR where a ratio of 0.6:1 was used for size selection. Barcoded library was eluted in 10 mM Tris-HCl pH 8.0 with 50 mM NaCl and samples were pooled for a total of 25 ng in 10 ul. Library was incubated with 1 ml RAP (ONT) for 5 min at room temp. A MinION FLO-MIN106 flow cell was brought to room temperature from 4 C storage and washed with flow cell priming mix as described in ONT protocol. The pooled library was combined with sequencing buffer and library beads as per the ONT protocol and loaded onto the flow cell and immediately sequenced on the MinION device for 48 hours generating 12.66 gigabases of sequence data.
Genome assembly and annotation
For all experiments, S. cerevisiae genome version 3 (sacCer3) was used. For accurate representation of untranslated regions (UTRs), experimentally derived UTR annotations (Nagalakshmi et al., 2008) were used to supplement the genome annotation.
Nanopore data processing and filtering
Raw fast5 files were basecalled with the high-accuracy model of Guppy 3.3.0 algorithm and demultiplexed with Qcat (https://github.com/nanoporetech/qcat) which also removes nanopore adapters. Sequencing is performed from either end of the amplified DNA product, so Cutadapt (Martin, 2011) was used to identify the location of the 3′ end adaptor sequence CTGTAGGCACCATCAATG on either strand and trim it. Primer IIA sequences from the SMARTer kit are removed from the opposite end of the sequence, and finally the reverse complement is generated for reads which had 3′ end adaptor on the 5′ end of the molecule. Reads without 3′ end adaptor are discarded as we cannot definitively determine whether they were associated with RNA Pol II. Reads which do not have 5′ IIA sequence may not accurately represent the true 5′ end of the molecule and likely arise from falloff of the reverse transcriptase, however, we retain these transcripts because the 3′ end (Pol II position) of these molecules is still reliable and can be used for certain analyses. Filters for read start position applied later in the processing pipeline will filter these reads out when necessary. Trimmed reads were then mapped to the S. cerevisiae sacCer3 genome using Minimap2 (Li, 2018) and the flags -ax splice -k 10 -G 2000–secondary = no. Resulting sam files were converted to bam and bed files using SAMtools (Li et al., 2009) and Bedtools (https://bedtools.readthedocs.io/en/latest/) for downstream analyses. A custom script was written to filter out mapped reads with soft-clipped polyA stretches. Reads with soft-clipped bases shorter than 30 nt were discarded if a stretch of 6 A’s was identified while reads with longer stretches of soft-clipped bases required 10 A’s to be removed. Finally, only reads overlapping intron-containing genes were considered for analyses presented here, with a required 50 bp minimum overlap. For Figure 3C, reads were filtered to start within 100 bp of the TSS (thereby excluding intrusive transcripts). For all other analyses, reads were filtered for start positions no more than 100 bp downstream of the annotated TSS. Reads were classified into 3 groups to encapsulate their readthrough status. "readthrough transcripts" begin near the annotated TSS but terminate > 150 bp downstream of the annotated PAS, indicating that readthrough occurred downstream. "Intrusive transcripts" overlap the gene of interest, but begin > 100 bp upstream of the TSS, indicating that transcription from an upstream gene failed to terminate. If a read met both of these conditions (as would be the case for a read which covers multiple gene bodies), it was assigned as an "intrusive transcript" relative to that gene. All other reads fall into the "no readthrough" category. All data were visualized in IGV (Robinson et al., 2011) and exported to produce genome browser figures.
RT-PCR
Nascent RNA was purified from chromatin as described above and depleted of poly(A)+ RNA. Samples were reverse transcribed with SuperScript III and random hexamers (Roche). For validation of splicing levels, intron-spanning primers amplified spliced and unspliced products which were visualized on an agarose gel. For validation of readthrough transcription, a forward primer in the gene body was paired with reverse primers either in the gene body (control) or in the region downstream of the PAS (downstream readthrough). Products were visualized on agarose gel using GelStar stain.
QUANTIFICATION AND STATISTICAL ANALYSIS
The Mann Whitney U test was applied to distributions of ΔSMIT parameter in Figure 2B as is appropriate for determining significance between small datasets (n = 53 genes). Detailed information about the clustering and modeling techniques in Figure 1 can be found in the Clustering of features and Machine learning – Lasso regression sections of the STAR Methods.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rapamycin | Calbiochem | 553211 |
| G418 | Thermofisher | 11811023 |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S |
| Advantage 2 PCR kit | Clontech | 639201 |
| SuperScript III Reverse Transcriptase | Invitrogen | 18080051 |
| SMARTer PCR cDNA synthesis kit | Clontech | 634925 |
| Random Hexamer Primers | ThermoFisher | SO142 |
| Terminator 5′-Phosphate-dependent Exonuclease | Lucigen | TER51020 |
| T4 RNA ligase II (truncated K227Q) | NEB | M0351 |
| TurboDNase | Invitrogen | AM2238 |
| Blunt TA/Ligase Master Mix | NEB | M0367 |
| NEBNext Ultra II repair/dA-tailing module | NEB | E7546 |
| Critical Commercial Assays | ||
| AMPure XP beads | Agencourt | A63880 |
| Zirconia beads | Biosepc | 11079110 |
| Dynabeads mRNA DIRIECT Micro Purification Kit | ThermoFisher | 61021 |
| RNA Clean & Concentrator kit | Zymo Research | R1017 |
| MinElute PCR purification kit | QIAGEN | 28004 |
| PCR barcoding kit | Oxford Nanopore Technologies | SQK-PBK004 |
| Deposited Data | ||
| Raw image data | This study, Mendeley data | http://dx.doi.org/10.17632/dddf2vhjyg.1 |
| Raw and processed SMIT data | This study | GSE156133 |
| Raw and processed nanopore data | This study | GSE156133 |
| Nab2 PAR-CLIP data | Baejen et al., 2014 | GSM1442550 |
| SMIT data | Oesterreich et al., 2016 | GSE70907 |
| Machine learning features, see Table S3 | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| Nab2-AA and Control-AA | Schmid et al., 2015 | N/A |
| S. cerevisiae: Strain background: BY4741 | N/A | |
| S. cerevisiae: ORF deletions | Saccharomyces Genome Deletion Project | N/A |
| Oligonucleotides | ||
| Primers for cloning deletion strains, see Table S4 | This study | N/A |
| Primers for SMIT library amplification, see Table S4 | This study | N/A |
| Nascent RNA 3′ end adaptor | Oesterreich et al., 2016 | N/A |
| Primers for RT-PCR validation of Nab2 SMIT, see Table S4 | This study | N/A |
| Software and Algorithms | ||
| R version 3.6.1 | R foundation for Statistical Computing | https://www.r-project.org/; RRID: SCR_001905 |
| Prinseq-lite | Schmieder and Edwards, 2011 | http://prinseq.sourceforge.net/ |
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Hisat2 | (Kim etal., 2019) | https://github.com/DaehwanKimLab/hisat2 |
| Bedtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Samtools | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| Qcat | Oxford Nanopore Technologies | https://github.com/nanoporetech/qcat |
| Guppy (v3.3.0) | Oxford Nanopore Technologies | N/A |
| Minimap2 | Li, 2018 | https://github.com/lh3/minimap2 |
| Fastx Toolkit | N/A | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Ggplot2 | https://ggplot2.tidyverse.org/ | RRID:SCR_014601 |
| R stats | N/A | https://www.rdocumentation.org/packages/stats/versions/3.6.2 |
| DescTools | N/A | https://www.rdocumentation.org/packages/DescTools/versions/0.99.37 |
| Caret | Kuhn, 2008 | http://caret.r-forge.r-project.org/ |
| Plyr | N/A | https://www.rdocumentation.org/packages/plyr/versions/1.8.6 |
| Reshape2 | N/A | https://www.rdocumentation.org/packages/reshape2/versions/1.4.4 |
| IGV | Robinson et al., 2011 | RRID:SCR_011793 |
| Other | ||
| MinION Flow Cell | Oxford Nanopore Technologies | FLO-MIN106 |
Highlights.
Machine learning predicts Nab2 is a regulator of co-transcriptional splicing
Loss of Nab2 causes transcriptional readthrough, and chimeric RNAs do not splice
Therefore, the role of Nab2 in co-transcriptional splicing is indirect
Organization of neighboring genes affects RNA processing events
ACKNOWLEDGMENTS
We thank L. Herzel for help with designing the multiplexed SMIT protocol, L. Schärfen for writing a script for the Nanopore sequencing data analysis, M. Hochstrasser for sharing deletion strains, and T.H. Jensen for sharing the Nab2 Anchor-Away strain. We thank M. Ares Jr. and A. Stark for feedback and discussions about data analysis. We are grateful to members of the Neugebauer lab, especially K. Reimer and D. Phizicky, for helpful discussions and comments on the manuscript. This work was supported by the National Institutes of Health (CMB TG T32GM007223 to T.A. and NIH R01 GM112766 from the NIGMS to K.M.N.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108324.
REFERENCES
- Aibara S, Gordon JMB, Riesterer AS, McLaughlin SH, and Stewart M (2017). Structural basis for the dimerization of Nab2 generated by RNA binding provides insight into its contribution to both poly(A) tail length determination and transcript compaction in Saccharomyces cerevisiae. Nucleic Acids Res. 45, 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert T, Herzel L, and Neugebauer KM (2017). Perfect timing: splicing and transcription rates in living cells. Wiley Interdiscip. Rev. RNA 8, wrna.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares M Jr., Grate L, and Pauling MH (1999). A handful of intron-containing genes produces the lion’s share of yeast mRNA. RNA 5, 1138–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baejen C, Torkler P, Gressel S, Essig K, Söding J, and Cramer P. (2014). Transcriptome maps of mRNP biogenesis factors define pre-mRNA recognition. Mol. Cell 55, 745–757. [DOI] [PubMed] [Google Scholar]
- Baejen C, Andreani J, Torkler P, Battaglia S, Schwalb B, Lidschreiber M, Maier KC, Boltendahl A, Rus P, Esslinger S, et al. (2017). Genome-wide Analysis of RNA Polymerase II Termination at Protein-Coding Genes. Mol. Cell 66, 38–49.e6. [DOI] [PubMed] [Google Scholar]
- Batisse J, Batisse C, Budd A, Böttcher B, and Hurt E (2009). Purification of nuclear poly(A)-binding protein Nab2 reveals association with the yeast transcriptome and a messenger ribonucleoprotein core structure. J. Biol. Chem. 284, 34911–34917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieberstein NI, Carrillo Oesterreich F, Straube K, and Neugebauer KM (2012). First exon length controls active chromatin signatures and transcription. Cell Rep. 2, 62–68. [DOI] [PubMed] [Google Scholar]
- Carrillo Oesterreich F, Preibisch S, and Neugebauer KM (2010). Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell 40, 571–581. [DOI] [PubMed] [Google Scholar]
- Carrocci TJ, and Neugebauer KM (2019). Pre-mRNA Splicing in the Nuclear Landscape. Cold Spring Harb. Symp. Quant. Biol. 84, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, and Ares M (2002). Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296, 907–910. [DOI] [PubMed] [Google Scholar]
- Cooke C, Hans H, and Alwine JC (1999). Utilization of splicing elements and polyadenylation signal elements in the coupling of polyadenylation and last-intron removal. Mol. Cell. Biol. 19, 4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, and West S (2013). Splicing-coupled 3′ end formation requires a terminal splice acceptor site, but not intron excision. Nucleic Acids Res. 41, 7101–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HL, Choquet K, and Churchman LS (2020). Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nano-pores. Mol. Cell 77, 985–998.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, and Bentley DL (2001). Capping, splicing, and 3′ processing are independently stimulated by RNA polymerase II: different functions for different segments of the CTD. Genes Dev. 15, 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. [DOI] [PubMed] [Google Scholar]
- Görnemann J, Kotovic KM, Hujer K, and Neugebauer KM (2005). Co-transcriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell 19, 53–63. [DOI] [PubMed] [Google Scholar]
- Green DM, Marfatia KA, Crafton EB, Zhang X, Cheng X, and Corbett AH (2002). Nab2p is required for poly(A) RNA export in Saccharomyces cerevisiae and is regulated by arginine methylation via Hmt1p. J. Biol. Chem. 277, 7752–7760. [DOI] [PubMed] [Google Scholar]
- Grosso AR, Leite AP, Carvalho S, Matos MR, Martins FB, Vitor AC, Desterro JMP, Carmo-Fonseca M, and de Almeida SF (2015). Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. eLife 4, e09214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackmann A, Wu H, Schneider UM, Meyer K, Jung K, and Krebber H (2014). Quality control of spliced mRNAs requires the shuttling SR proteins Gbp2 and Hrb1. Nat. Commun. 5, 3123. [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, and Laemmli UK (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 31, 925–932. [DOI] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, and Neugebauer KM (2017). Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 18, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzel L, Straube K, and Neugebauer KM (2018). Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Res. 28, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Claw-son H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. (2006). The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34, D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irniger S, Egli CM, and Braus GH (1991). Different classes of polyadenylation sites in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 3060–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Guisbert K, Duncan K, Li H, and Guthrie C (2005). Functional specificity of shuttling hnRNPs revealed by genome-wide analysis of their RNA binding profiles. RNA 11, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotovic KM, Lockshon D, Boric L, and Neugebauer KM (2003). Cotranscriptional recruitment of the U1 snRNP to intron-containing genes in yeast. Mol. Cell. Biol. 23, 5768–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TL, Krogan NJ, and Guthrie C (2008). A single SR-like protein, Npl3, promotes pre-mRNA splicing in budding yeast. Mol. Cell 32, 727–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M (2008). Building predictive models in R using the caret package. J. Stat. Softw. 28.
- Lacadie SA, and Rosbash M (2005). Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5’ss base pairing in yeast. Mol. Cell 19, 65–75. [DOI] [PubMed] [Google Scholar]
- Lacadie SA, Tardiff DF, Kadener S, and Rosbash M (2006). In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev. 20, 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SW, Apponi LH, Cornejo OE, Kitchen CM, Valentini SR, Pav-lath GK, Dunham CM, and Corbett AH (2009). Splice variants of the human ZC3H14 gene generate multiple isoforms of a zinc finger polyadenosine RNA binding protein. Gene 439, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Izaurralde E, Jarmolowski A, McGuigan C, and Mattaj IW (1996). A nuclear cap-binding complex facilitates association of U1 snRNP with the cap-proximal 50 splice site. Genes Dev. 10, 1683–1698. [DOI] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen Z-Y, Wang W, Baker CC, and Krug RM (2001). The 3′-end-processing factor CPSF is required for the splicing of single-intron pre-mRNAs in vivo. RNA 7, 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map Format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Geiger G, Minet M, Schroeder S, Cilli K, McNeil JB, and Bentley DL (2002). Functional interaction of yeast pre-mRNA 3′ end processing factors with RNA polymerase II. Mol. Cell 9, 1101–1111. [DOI] [PubMed] [Google Scholar]
- Manning KS, and Cooper TA (2017). The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell Biol. 18, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. [Google Scholar]
- Martins SB, Rino J, Carvalho T, Carvalho C, Yoshida M, Klose JM, de Almeida SF, and Carmo-Fonseca M (2011). Spliceosome assembly is coupled to RNA polymerase II dynamics at the 3′ end of human genes. Nat. Struct. Mol. Biol. 18, 1115–1123. [DOI] [PubMed] [Google Scholar]
- Moore MJ, and Proudfoot NJ (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136, 688–700. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, and Snyder M (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves LT, Douglass S, Spreafico R, Venkataramanan S, Kress TL, and Johnson TL (2017). The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae. Genes Dev. 31, 702–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen KE, Homer CM, Ryan CJ, Shales M, Krogan NJ, Patrick KL, and Guthrie C (2017). The histone variant H2A.Z promotes splicing of weak introns. Genes Dev. 31, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, and Berget SM (1991). Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes Dev. 5, 2086–2095. [DOI] [PubMed] [Google Scholar]
- Oesterreich FC, Herzel L, Straube K, Hujer K, Howard J, and Neugebauer KM (2016). Splicing of Nascent RNA Coincides with Intron Exit from RNA Polymerase II. Cell 165, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak C, Garshasbi M, Kahrizi K, Gross C, Apponi LH, Noto JJ, Kelly SM, Leung SW, Tzschach A, Behjati F, et al. (2011). Mutation of the conserved polyadenosine RNA binding protein, ZC3H14/dNab2, impairs neural function in Drosophila and humans. Proc. Natl. Acad. Sci. USA 108, 12390–12395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, and Guthrie C (2007). Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 5, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan Aaron R., and Hall Ira M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed]
- Rigo F, and Martinson HG (2008). Functional coupling of last-intron splicing and 3′-end processing to transcription in vitro: the poly(A) signal couples to splicing before committing to cleavage. Mol. Cell. Biol. 28, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan DP, Herschlag D, and Brown PO (2011). Identification of RNA recognition elements in the Saccharomyces cerevisiae transcriptome. Nucleic Acids Res. 39, 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldi T, Cortazar MA, Sheridan RM, and Bentley DL (2016). Coupling of RNA Polymerase II Transcription Elongation with Pre-mRNA Splicing. J. Mol. Biol. 428, 2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Olszewski P, Pelechano V, Gupta I, Steinmetz LM, and Jensen TH (2015). The Nuclear PolyA-Binding Protein Nab2p Is Essential for mRNA Production. Cell Rep. 12, 128–139. [DOI] [PubMed] [Google Scholar]
- Schmieder R, and Edwards R (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek S, Zeng Y, Bellur DL, Bergkessel M, Morris KJ, Deng Q, Du-ong D, Seyfried NT, Guthrie C, Staley JP, et al. (2016). The Evolutionarily-conserved Polyadenosine RNA Binding Protein, Nab2, Cooperates with Splicing Machinery to Regulate the Fate of pre-mRNA. Mol. Cell. Biol. 36, 2697–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff DF, and Rosbash M (2006). Arrested yeast splicing complexes indicate stepwise snRNP recruitment during in vivo spliceosome assembly. RNA 12, 968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier M, Maudlin I, and Murphy S (2020). Transcription and splicing: A two-way street. WIREs RNA 11, e1593. [DOI] [PubMed] [Google Scholar]
- Teng X, Dayhoff-Brannigan M, Cheng WC, Gilbert CE, Sing CN, Diny NL, Wheelan SJ, Dunham MJ, Boeke JD, Pineda FJ, and Hardwick JM (2013). Genome-wide consequences of deleting any single gene. Mol. Cell 52, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R (1994). Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. B Methodol. 58, 267–288. [Google Scholar]
- Tilgner H, Jahanbani F, Gupta I, Collier P, Wei E, Rasmussen M, and Snyder M (2018). Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome. Genome Res. 28, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck AC, and Tollervey D (2013). A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell 154, 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudek A, Schmid M, Makaras M, Barrass JD, Beggs JD, and Jensen TH (2018). A Nuclear Export Block Triggers the Decay of Newly Synthesized Polyadenylated RNA. Cell Rep. 24, 2457–2467.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viphakone N, Voisinet-Hakil F, and Minvielle-Sebastia L (2008). Molecular dissection of mRNA poly(A) tail length control in yeast. Nucleic Acids Res. 36, 2418–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windgassen M, Sturm D, Cajigas IJ, González CI, Seedorf M, Bastians H, and Krebber H (2004). Yeast shuttling SR proteins Npl3p, Gbp2p, and Hrb1p are part of the translating mRNPs, and Npl3p can function as a translational repressor. Mol. Cell. Biol. 24, 10479–10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- Yeo INK, and Johnson RA (2000). A new family of power transformations to improve normality or symmetry. Biometrika 87, 954–959. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SMIT processing code is available on GitHub: https://github.com/carrillo/SMITproject. All other code including machine learning modeling is available at https://github.com/NeugebauerLab/Alpert2020_Nab2. The accession number for the SMIT and long-read sequencing data reported in this paper is GEO:GSE156133. Original data have been deposited to to Mendeley Data: http://dx.doi.org/10.17632/dddf2vhjyg.1.