

Summary
Electrophilic compounds originating from nature or chemical synthesis have profound effects on immune cells. These compounds are thought to act by cysteine modification to alter the functions of immune-relevant proteins; however, our understanding of electrophile-sensitive cysteines in the human immune proteome remains limited. Here, we present a global map of cysteines in primary human T cells that are susceptible to covalent modification by electrophilic small molecules. More than 3000 covalently liganded cysteines were found on functionally and structurally diverse proteins, including many that play fundamental roles in immunology. We further show that electrophilic compounds can impair T-cell activation by distinct mechanisms involving the direct functional perturbation and/or degradation of proteins. Our findings reveal a rich content of ligandable cysteines in human T cells and point to electrophilic small molecules as a fertile source for chemical probes and ultimately therapeutics that modulate immunological processes and their associated disorders.
In Brief:
Integrated chemical proteomics and phenotypic screening furnishes a global portrait of cysteine reactivity and ligandability in primary human T cells and enables the discovery of electrophilic small molecules that suppress T-cell activation and promote the degradation of immunomodulatory proteins.
Graphical Abstract
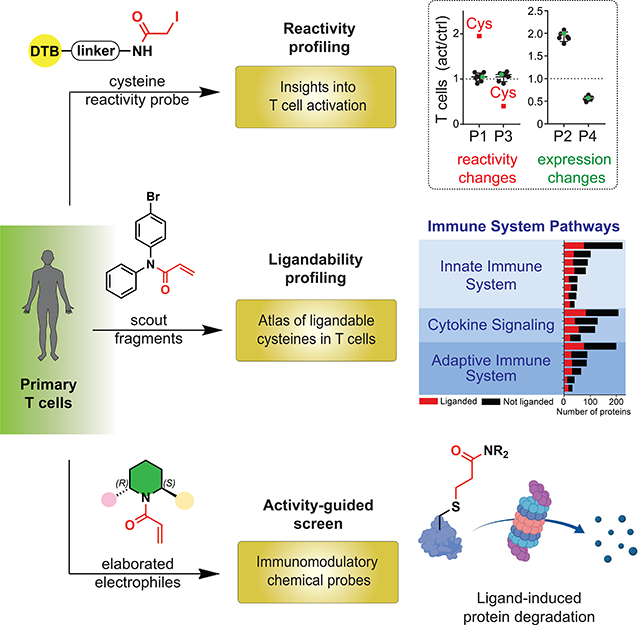
Introduction
Human genetics has emphasized the central role that the immune system plays as both a causal basis for and guardian against a broad range of diseases (Hansen et al., 2018; Lucherini et al., 2018; Sims et al., 2017). This knowledge has been translated into new medicines, including drugs that stimulate immune responses to cancer (Ribas and Wolchok, 2018) and suppress autoimmunity (Telliez et al., 2016). Advanced profiling technologies, such as RNA-sequencing (Papalexi and Satija, 2018) and mass spectrometry (MS)-based proteomics (Rieckmann et al., 2017; Tan et al., 2017), have also improved our understanding of the molecular and cellular composition of the human immune system, leading to the discovery of many genes and proteins with enriched expression in specific subtypes of immune cells.
The aberrant activation of T cells specifically contributes to a number of autoimmune syndromes (Baecher-Allan et al., 2018; Suarez-Fueyo et al., 2016). The molecular pathways that both execute and are influenced by T-cell activation have been studied using gene and protein expression profiling (Hiemer et al., 2019; Rieckmann et al., 2017), as well as phosphoproteomic (Chylek et al., 2014), metabolomic (Hiemer et al., 2019), and protein-protein interaction (Voisinne et al., 2019) methods that compare resting versus activated T cells. Some of the discovered changes in activated T cells occur in general biochemical pathways associated with, for instance, cell proliferation, while others reflect immune-restricted processes. The extent to which these types of activation state-dependent changes in biochemistry might also create a landscape of new targets for chemical probes that regulate T-cell function remains largely unexplored. More generally, most immune-relevant proteins lack chemical probes, which hinders the pharmacological study of immune system function for basic and translational research purposes.
Among the categories of chemical probes, electrophilic small molecules that act by forming covalent bonds with nucleophilic amino acids may offer a particularly attractive class for studying proteins involved in immune responses. Multiple immunomodulatory drugs, either approved (Saidu et al., 2019) or in clinical development (Rip et al., 2018; Telliez et al., 2016), covalently modify cysteine residues in immune-relevant proteins. Cysteines also serve as oxidative stress sensors in signaling pathways, providing a way for the immune system to swiftly respond to diverse environmental insults (Franchina et al., 2018). From a chemical biology perspective, electrophilic compounds, by combining features of molecular recognition and reactivity, can target proteins and sites on proteins that have proven challenging to address with reversibly binding small molecules (Backus et al., 2016; Maurais and Weerapana, 2019; Roberts et al., 2017).
In previous studies, we used the chemical proteomic method activity-based protein profiling (ABPP) to identify cysteines targeted by the immunosuppressive drug dimethyl fumarate (DMF) in primary human T cells, which included cysteines at protein-protein interaction sites in interleukin-1 receptor-associated kinase 4 (IRAK4) and protein kinase C theta (PRKCQ) (Blewett et al., 2016) (Zaro et al., 2019). ABPP has also uncovered changes in cysteine reactivity in genetically defined cancer cells (Bar-Peled et al., 2017), bacteria exposed to oxidative stress (Deng et al., 2013), and C. elegans with perturbations in lifespan regulatory pathways (Martell et al., 2016). Inspired by these initial findings, we set out to perform a series of chemical proteomic experiments that together would furnish a global and quantitative portrait of cysteine reactivity (“reactivity profiling”, Figure 1A and see Glossary in Methods S1 (Weerapana et al., 2010)) and electrophilic small-molecule interactions (“ligandability profiling”, Figure 1A and see Methods S1; (Backus et al., 2016; Wang et al., 2014)) in primary human T cells in resting versus stimulated states. Using broadly reactive electrophilic fragments (“scout” fragments; Figure 1A and see Methods S1), we mapped >3400 liganded cysteines in >2200 proteins, many of which show reactivity and/or expression changes following T-cell stimulation and populate diverse adaptive and innate immune signaling networks. We acquired further knowledge on the pharmacological effects and tractability of small molecule-cysteine interactions by performing an activity-guided screen with a structurally and stereochemically diversified set of “elaborated” electrophiles (Figure 1A and see Methods S1) to uncover immunomodulatory compounds that suppress T-cell activation by diverse mechanisms, including the direct inhibition and/or degradation of proteins.
Figure 1. Chemical proteomic map of cysteine reactivity changes in activated T cells.

(A) Organizational diagram outlining the types of chemical proteomic experiments and electrophilic small molecules used to investigate and modulate the activation of primary human T cells, as well as the major advances enabled by each experiment type. IA-DTB - desthiobiotin polyethyleneoxide iodoacetamide; SAR – structure-activity relationship; MoA – mechanism of action.
(B) Workflow for proteomic experiments measuring cysteine reactivity (TMT-ABPP) and protein expression (TMT-exp) in primary human T cells. See STAR Methods for more details.
(C) Protein expression differences between control and activated T cells. Results represent mean values from four biological replicates.
(D) Representative protein expression differences between control and activated T cells, where results from both TMT-ABPP (black dots) and TMT-exp (green dots) concordantly support expression changes.
(E) Fraction of proteins with cysteine reactivity changes observed for total proteins with the indicated numbers of quantified cysteines in TMT-ABPP experiments. Proteins with only 1–2 quantified cysteines were not interpreted for reactivity changes (gray bars).
(F) Fraction of proteins with human genetics-based immune phenotypes and immune-enriched expression from total proteins showing cysteine reactivity changes in activated T cells (Data S1).
(G) GO-term enrichment analysis for proteins undergoing reactivity (top, 160 total proteins) or expression (bottom, 1106 total proteins) changes in activated T cells. Top-10 enriched biological processes are shown for the expression changes group (Data S1). Red bold font highlights immune-relevant and cell redox homeostasis pathways enriched in expression and reactivity changes groups, respectively.
(H-J) Representative cysteine reactivity changes in activated T cells organized by functional category. Horizontal black lines mark average reactivity value for quantified cysteines from each protein, excluding the reactivity-changing cysteine(s), which are shown in red. (H) Active-site cysteines in redox-related proteins; crystal structure of glutathione reductase (GSR; PDB: 1GRF) with the reactivity-changing C102 shown in blue. (I) Metal-binding cysteines; NMR structure of the EF-hand domain of LCP-1 (PDB: 5JOJ) with the reactivity-changing C42 shown in blue. (J) Cysteines at cofactor/metabolite-binding sites; crystal structure of isocitrate dehydrogenase 1 (IDH1) in complex with NADP (red), isocitrate (orange), and calcium (green ball) (PDB: 1T0L) with the reactivity-changing C269 highlighted in blue.
Results
Chemical proteomic map of cysteine reactivity in activated T cells
Stimulated T cells enter a growth phase that induces changes in, for instance, redox state, cytoskeletal structure (Acuto and Cantrell, 2000), and metabolism (Almeida et al., 2016). We hypothesized that activation would also affect the landscape of reactive cysteines in T cells through direct (e.g., posttranslational modifications) and indirect (e.g., alterations in protein structure and interactions) mechanisms. We mapped cysteine reactivity changes in unstimulated (control) versus stimulated (activated) T cells by performing and integrating quantitative tandem mass tagging (TMT)-based proteomic experiments that measured – i) the extent of labeling of cysteines by an iodoacetamide-desthiobiotin (IA-DTB) probe (TMT-ABPP; see Methods S1); or ii) the expression level of proteins (TMT-exp; see Methods S1) (Figure 1B).
T cells isolated from human blood were activated by exposure to anti-CD3/CD28 antibodies for three days (activated T cells, Figure 1A). Two control populations of T cells were generated – 1) naïve T cells, which were not treated further after negative selection; and 2) expanded T cells, which were obtained by transient activation with anti-CD3/CD28 antibodies for three days followed by growth in IL-2-containing media for another 10–12 days to return the cells to a quiescent state. We first performed TMT-exp experiments comparing naïve, expanded, and activated T cells isolated from four independent donors (4–8 total experimental replicates). Principal component analysis (PCA) of the ~4800 proteins quantified in aggregate confirmed that naïve and expanded T cells were more closely related to each other than to activated T cells based on the main principal component PC1 (68%) (Figure S1A), which was enriched in pathways associated with T-cell activation (Data S1). ~1100 proteins were >2-fold altered in expression in activated T cells (Figure 1C), including many proteins with established immune functions (e.g., IL2RA, TNFAIP3) (Figure 1D). Based on these results, we proceeded to use expanded T cells as the main control group in subsequent proteomic studies, as these cells could be produced on a larger scale than naïve T cells.
We next performed TMT-ABPP experiments on proteomic lysates from expanded versus activated T cells, which quantified 16184 total cysteines, including one or more cysteines on ~80% of the proteins identified in TMT-exp experiments (Data S1). 160 proteins were found to harbor cysteine reactivity changes that differed substantially (>two-fold) from the corresponding expression profiles for these proteins in activated T cells (Figure 1E and Data S1). These cysteine reactivity changes occurred in diverse structural and functional classes of proteins (Figure S1B), including several “immune-relevant” proteins (Figure 1F and Data S1), as defined by immune cell-enriched expression profiles derived from public databases and/or human genetic evidence of causing, upon mutation, immune-related disorders (acquired from the OMIM (Online Mendelian Inheritance in Man) database; see STAR Methods). GO-term enrichment analysis underscored the complementary information provided by TMT-exp and TMT-ABPP experiments. For instance, proteins showing expression changes in activated T cells were enriched, as expected, in processes involved in immune responses (Figure 1G). On the other hand, TMT-ABPP uncovered a distinct set of enriched changes in cellular redox homeostasis (Figure 1G), possibly reflecting the impact of an altered redox environment (Lian et al., 2018; Mak et al., 2017) on the reactivity of cysteines in activated T cells. Notably, several redox proteins showed site-specific reactivity changes in catalytic cysteines in activated T cells (Figure 1H), thus highlighting how ABPP can identify post-translational alterations in protein function that occur in specific cellular states.
Reactivity changes were also found for cysteines: i) in the metal-binding domains of proteins (Figure 1I), including the immune-relevant and calcium-regulated protein L-plastin (LCP-1), which participates in remodeling of the actin cytoskeleton during T-cell activation (Ishida et al., 2017); ii) at sites of protein-protein (Figure S1C) and DNA/RNA-protein (Figure S1D) interactions; and iii) proximal to cofactor- and metabolite-binding sites (Figures 1J and S1E). These cysteine reactivity changes may reflect a landscape of remodeled intermolecular interactions in activated T cells.
Having established quantitative proteomic profiles that uncovered numerous cysteine reactivity and protein expression changes in activated T cells, we next turned our attention to mapping electrophilic small molecule-cysteine interactions in both immunological cell states.
Chemical proteomic map of cysteine ligandability in human T cells
We recently described an ABPP strategy to globally assess the ligandability of cysteines that leverages broadly reactive, electrophilic small-molecule fragments referred to as “scouts” (Backus et al., 2016; Bar-Peled et al., 2017). Two scout fragments bearing an α-chloroacetamide (KB02) or acrylamide (KB05) (Figure 2A, 2B) – reactive groups frequently found in covalent chemical probes and drugs (Baillie, 2016; Honigberg et al., 2010; Ostrem et al., 2013; Xu et al., 2019) – were used to construct in-depth cysteine ligandability maps across human T cells in both control and activated states. We analyzed scout fragment-cysteine interactions using two complementary ABPP methods that provided a balance of confidence in quantitative accuracy (isoTOP-ABPP; see Methods S1) with greater multiplexing capacity (TMT-ABPP) (Figures 2A and S2A, B). Both proteomic methods yielded similar ratio (R) values (DMSO/scout fragment; 500 μM, 1 h) for cysteines in T-cell proteomes, with the MS3-based quantification used in TMT-ABPP resulting in mild ratio compression (Figure S2C, D), which was countered by a substantial increase in the number of quantified cysteines compared to isoTOP-ABPP (Figure S2E). We accordingly designated cysteines as liganded if they showed an R value of ≥ 5 as measured by either isoTOP-ABPP or TMT-ABPP. From a total of 18077 cysteines in 5990 proteins quantified in human T cells, we identified 3466 liganded cysteines in 2283 proteins (Figure 2C, 2D and Data S1). These ligandability events were broadly distributed across cysteines with diverse intrinsic reactivities (Weerapana et al., 2010) (Figure 2E and Data S1), indicating contributions from both the binding and electrophilic groups of scout fragments in conferring strong engagement of cysteines in the T-cell proteome.
Figure 2. Chemical proteomic map of fragment electrophile-cysteine interactions in T cells.

(A) Workflow for chemical proteomic experiments measuring scout fragment engagement of cysteines in primary human T cells. See STAR Methods for more details.
(B) Structures of scout fragments KB02 and KB05. Red color indicates the reactive group for each fragment.
(C, D) Pie chart representations of cysteines (C) and proteins (D) liganded by scout fragments. Results were obtained by combining soluble and particulate proteomic data for KB02 and KB05 treatments (500 μM, 1 h) of both control and activated T cells. R-values within each group were derived from 3–5 independent isoTOP-ABPP and TMT-ABPP experiments.
(E) Total number (left) and percentage (right) of liganded cysteines per total number of cysteines quantified across the indicated intrinsic reactivity ranges, which were determined as described previously (Weerapana et al., 2010).
(F) Total number (left) and percentage (right) of liganded proteins with expression or reactivity changes in activated T cells.
(G) Quantification of cysteines in PDCD1, revealing elevated expression of this protein in activated T cells (increased intensity of DMSO (act) signals for C93 and C241) and scout fragment-sensitivity for C93. Error bars represent SD from 2–4 independent experiments. ****, p < 0.0001 compared to DMSO (act) group.
(H) Overlap of liganded proteins with immune-relevant proteins (Data S1).
(I) Fractions of liganded and quantified proteins from total proteins in OMIM database with immune phenotypes (Data S1, see STAR Methods for details).
(J) Location of liganded C408 (blue) and pathogenic missense mutations (yellow – mutation of H112, which is within 5 Å of C408, red – other mutations) in structure of adenosine deaminase CECR1 (PDB: 3LGD).
Among the liganded cysteines were several targeted by existing covalent probes and drugs (e.g., C909 in JAK3 (Xu et al., 2019); C528 in XPO1 (Haines et al., 2015); Data S1). Liganded cysteines were also well-represented within the subset of proteins showing expression and/or cysteine reactivity changes in activated T cells, where cysteines with altered reactivity showed a greater propensity for liganding by scout fragments (Figure 2F). We identified, for instance, a liganded cysteine (C93) in the programmed cell death protein 1 (PDCD1 or PD-1), which was only observed in activated T cells (Figure 2G), likely reflecting the induced expression of this key immune checkpoint protein following T cell stimulation (Agata et al., 1996).
Nearly a quarter of the proteins harboring liganded cysteines were immune-relevant (Figure 2H and Data S1), of which 118 cause, upon mutation in humans, monogenic diseases with a strong immune phenotype (Figure 2I). Such Mendelian genetic relationships to immune disorders can be used to prioritize proteins for drug development programs aimed at treating autoimmune or autoinflammatory disorders (e.g., JAK3 (Xu et al., 2019)) as well as promoting immune responses to cancer (e.g., CTLA4 (Wei et al., 2018)). Pathogenic missense mutations were found in >75% of the liganded proteins with Mendelian immune phenotypes (Figure S2F), which inspired us to assess the extent to which these mutated residues are in structural proximity to liganded cysteines. For proteins with crystal structures, nearly half of the liganded cysteines were located within 15 Å of mutated residues (Figure S2G, H and Data S1), and, in 13 cases, the liganded cysteine was within 5 Å of a pathogenic mutation site (e.g., CECR1 (Figure 2J) and ZAP70 (Figure S2I). This spatial proximity may designate liganded cysteines that reside in functional pockets on immune-relevant proteins. Integrating our ligandability map with additional genomic and proteomic studies identified liganded cysteines in proteins: 1) found by genome-wide CRISPR screening to regulate T-cell proliferation (Shifrut et al., 2018) (Figure S2J and Data S1); and 2) that are part of immune-enriched modules, including those relevant for T-cell activation (Figure S2K), established by expression-based proteomics (Rieckmann et al., 2017) (Data S1).
Liganded cysteines were well-distributed across the biological processes enriched in activated T cells (Figure S3A, B and Data S1), and pathway analysis further supported the striking breadth of liganded cysteines found in key immune signaling networks (Figure 3A), including those mediating TNF-receptor and T-cell receptor (TCR) signaling and NF-κB activation (Figures 3B and S3C and Data S1). The proteins in these pathways harboring liganded cysteines included enzymes (e.g., DGKA/Z, IKBKB), adaptor proteins (e.g., MYD88) and transcription factors (e.g., NFKB1) (Figure 3B–D). Among the >350 liganded adaptor proteins and transcription factors were several that have Mendelian links to immunological disorders (Figure 3E and Data S1). Some of the liganded cysteines in these proteins reside in proximity to DNA-protein (Figure 3C) and protein-protein (Figure S3E) interfaces. Even for more classically druggable proteins like kinases, liganded cysteines were often found not only within, but also outside of the ATP-binding pocket (Figures 3D and S3D). These findings underscore the potential for covalent ligands to target both functional and non-canonical sites on structurally and functionally diverse protein classes.
Figure 3. Liganded cysteines in immune-relevant proteins.

(A) Ligandability analysis of Reactome pathways within Immune System (left, hierarchical level 2 grouped according to their parent nodes) or Signal Transduction (right, hierarchical level 2) categories. Liganded and quantified proteins are colored red and black, respectively. Also see Data S1.
(B) Diagram of TCR, TNF-alpha and NF-κB pathways marking proteins that possess cysteines liganded by scout fragments (green) or elaborated electrophilic compounds (blue). The rectangular frame around each protein is colored to reflect expression changes (blue, >2 fold, red, <2 fold, and black, unchanged, in activated T cells; yellow not quantified).
(C) Location of a liganded C61 at the DNA-binding interface of NFKB1 (CPDB: 2O61).
(D) Location of liganded active-site (C179) and non-active site (C464) cysteines in IKBKB (PDB: 4E3C).
(E) Fractions of liganded transcription factor and adaptor proteins that are also immune-relevant; Data S1.
Our ABPP experiments with scout fragments established an extensive landscape of ligandable cysteines in immune-relevant proteins, pointing to a broad opportunity to discover covalent chemical probes that modulate T-cell function. We next aimed to establish an experimental workflow that would illuminate the functional effects and tractability of electrophilic small molecule-cysteine interactions, while also preserving the globality and biological integrity afforded by profiling these interactions in primary human T cells.
A functional screen of elaborated electrophilic compounds in T cells
Fragment-based screening offers advantages for discovering compounds that target challenging protein classes; however, progressing fragments to more advanced chemical probes can be confounded by the low-affinity and promiscuity of initial hits and the tractability of fragment-binding sites on proteins (Scott et al., 2012). These problems have been historically addressed by labor-intensive, structure-guided protocols that require purified protein and have limited throughput. We aspired instead to create an in cellulo strategy that integrates phenotypic screening with chemical proteomics to furnish structure-activity relationships (SARs) on many electrophilic small molecule-cysteine interactions in parallel, such that the tractability and potential functional effects of these interactions could be comparatively evaluated.
We performed a multidimensional screen of a focused library of structurally elaborated electrophilic small molecules to identify compounds that suppress T-cell activation at low-μM concentrations without causing cytotoxicity (Figure 4A; ~130 compounds tested at 5–10 μM; average MW = 400 Da; Data S1; Methods S2). The screen identified 19 compounds that substantially suppressed T-cell activation (>65% reduction in IL-2) without causing cytotoxicity (viability >85%), including DMF, which served as a positive control (Figure 4B–D, Figure S4A, and Data S1). Representative active acrylamide (BPK-21, BPK-25, EV-96) and α-chloroacetamide (EV-3, EV-93) compounds were selected for further characterization (Figure 4C–F). Among these active compounds, we note that EV-96 was part of a set of four stereoisomeric electrophiles (Figure 4E) and stereoselectively blocked T-cell activation with an EC50 < 2.5 μM (Figures 4F–H and S4B).
Figure 4. Elaborated electrophilic compounds that suppress T-cell activation.

(A) Workflow for T-cell activation screen. Primary human T cells were treated with compounds (10 μM) or DMSO under TCR-stimulating conditions (96-well plates pre-coated with 5 μg/mL αCD3 and 2 μg/mL αCD28) for 24h. T-cell activation was measured by IL-2 and IFN-γ secretion and surface expression of CD25 and CD69. Compounds were considered as active hits if they reduced IL-2 by >65% with <15% reduction in cell viability compared to DMSO control.
(B) Screening results for elaborated electrophilic compounds.
(C) Structures of active compounds selected for follow-up studies: acrylamides (BPK-21, BPK-25), α-chloroacetamides (EV-3, EV-93), and DMF as a positive control. Red color indicates the reactive group for each elaborated compound.
(D) T-cell activation and cytotoxicity profiles for active compounds. Data are mean values ± SEM; n = 3/group.
(E, F) Structures (E) and activity (F) of four stereoisomeric compounds, where one compound (EV-96) inhibited T-cell activation (F). In (E), the stereoisomeric relationships are shown in blue (diastereomers) and red (enantiomers). Red color in chemical structures indicates the acrylamide reactive group. In (F), T-cell activation (CD25) and cytotoxicity profiles are shown for the stereoisomeric compounds (5 μM, 24 h treatment). Data are mean values ± SD; n = 2–5/group. ***p < 0.001 compared to EV-97.
(G) Concentration-dependent effects of EV-96 and EV-97 on T-cell activation (CD25). Data are mean values ± SEM; n = 4–5/group. **, p < 0.01 compared to 0.5 μM treatment groups.
(H) T-cell activation and cytotoxicity profiles for EV-96. Data are mean values ± SEM; n = 3–5/group.
We next mapped the protein targets of active compounds in T cells by ABPP. In these experiments, we set a slightly lower threshold for liganded cysteines (R ≥ 4 vs R ≥ 5 for scout fragments), as treatments with elaborated electrophilic compounds were performed in situ at much lower concentrations (5–20 μM for BKP-21/25, EV-3/93/96; 50 μM for DMF; 3 h treatment), and we surmised that ≥75% engagement of cysteines may be sufficient to produce pharmacological effects in T cells. Each active compound engaged a limited (0.2–1.0% of ~12,000 total quantified cysteines) and distinct set of cysteines in largely non-overlapping proteins (Figures 5A, B and S4C, D and Data S1) that originated from diverse structural and functional classes (Figure 5C and Data S1), including several immune-relevant proteins (Figure 5B).
Figure 5. Cysteines liganded by active compounds in human T cells.

(A) Heatmap showing liganded cysteines for active compounds in human T cells (treated with the indicated concentrations of compounds (μM) for 3 h followed by ABPP). Cysteines quantified for at least two active compounds with R values ≥4 (DMSO/compound) for at least one of the compounds are shown. Results were obtained by combining isoTOP-ABPP and TMT-ABPP data from 2–6 independent experiments. See STAR Methods for details.
(B) Heatmap showing cysteines liganded by active compounds in immune-relevant proteins.
(C) Distribution of protein classes containing cysteines liganded by active compounds.
(D, E) Comparison of cysteines liganded by active compounds versus scout fragments in human T cells, as displayed in correlation plot (D) and pie chart (E) analyses. In (D), cysteines liganded by both active compounds and scout fragments, only by active compounds, and only by scout fragments are showing in purple, blue, and red, respectively.
(F) Prediction success rate querying for pockets within the indicated distances from cysteines liganded by active compounds.
(G) Modeling of active compound interactions with C203 in MYD88 (PDB 4DOM). Predicted pockets highlighted as green mesh and other cysteines in the structure are colored red. Docking shows preferential liganding by BPK-25 due to predicted hydrogen bonds with E183 and R188 (top), which are not accessible in docked structure of BPK-21 (bottom).
(H) Modeling of active compound interactions with C342 of ERCC3 (PDB 5OF4). Docking shows preferential liganding by BPK-21 (bottom) due to predicted hydrogen bonds with T469 and Q497 and π−π interaction with W493, which are less accessible in the docked structure of BPK-25 (bottom).
The vast majority of cysteines liganded by active compounds (~80%) were also engaged by scout fragments (Figure 5D, E and Data S1), underscoring the potential for fragment profiling to discover tractable sites of ligandability across the human proteome that can also be targeted by more elaborated electrophiles with improved potency (low-μM) and interpretable SARs. In support of this conclusion, molecular modeling revealed predicted binding pockets within 5 Å of cysteines targeted by active compounds in ~60% of the proteins for which structural information was available (Figures 5F and S4E and Data S1). Docking studies on protein targets of the structurally related BPK-21 and BPK-25 compounds, further supported the observed SAR profiles (e.g., selectivity of C203 in MYD88 for BPK-25, C342 in ERCC3 for BPK-21, and a lack of selectivity for C91 in TMEM173; Figure 5G, H and S4F). C91 in TMEM173, or STING, has been shown to be palmitoylated (Mukai et al., 2016) and targeted by other covalent ligands that antagonize inflammatory cytokine production (Haag et al., 2018). Consistent with this past work, we found that BPK-25 inhibited TMEM173 activation by the cyclic dinucleotide ligand cGAMP (Figure S4G–I). More global analysis of our scout fragment data sets revealed additional liganded cysteines subject to palmitoylation (Data S1), suggesting the broader potential to pharmacologically target these dynamic lipid modification sites on proteins with electrophilic compounds.
The markedly distinct cysteine engagement profiles for active compounds (Figure 5A, B) suggested that they may suppress T-cell activation by distinct mechanisms. In support of this hypothesis, we found that active compounds differentially impacted key transcriptional and signaling pathways involved in T-cell activation, with EV-3, BPK-25, and EV-96 suppressing NF-κB activation and only EV-3 and BPK-25 blocking NFAT activation, as measured by > 50% reductions in IκBα phosphorylation (Figure 6A, 6B) and NFATc2 dephosphorylation (Figures S5A, B), respectively. BPK-25 also reduced NFATc2 expression (Figure S5A, B). All of the hit compounds negatively affected mTOR pathway activation, as measured by > 50% reductions in S6K phosphorylation (Figure 6A, B), while none of the active compounds substantially affected ERK phosphorylation (Figure S5C, D). Finally, we excluded GSH reductions as a candidate mechanism because buthionine sulphoximine (BSO), an inhibitor of the GSH biosynthetic enzyme gamma-glutamyl cysteine ligase (GCLC), did not affect T-cell activation despite depleting GSH content (Figure S5E), a result that is consistent with previous studies (Mak et al., 2017).
Figure 6. Mechanistic analysis of active compounds in human T cells.

(A, B) Effects of active compounds on NFκB and mTOR pathways, as determined by western blotting for phosphorylation of IκBα (S32/S36) and S6K (T389), respectively, in stimulated T cells treated with DMSO, active or control (EV-97) compounds for 24 h. (A) Representative western blots. (B) Quantitation of p-IκBα (S32/S36) and p-S6K (T389). Data are mean ± SEM; n = 3–8/group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared to DMSO (αCD3/αCD28) control.
(C) Top: Heat map showing active compounds interactions with cysteines in BIRC2 and BIRC3. Bottom: Domain maps highlighting location of EV-3-sensitive C45 and C28 in BIRC2 and BIRC3, respectively.
(D) Location of C28 in structure of a BIRC3-TRAF2 protein complex (PDB: 3M0A).
(E) Western blots showing reductions in BIRC2 and BIRC3 content in human T cells treated with EV-3 (10 μM), but not other active compounds (DMF (50 μM), BPK-21 (20 μM), and BPK-25 (10 μM)). The BIR3 domain ligand AT406 (1 μM) caused loss of BIRC2, but not BIRC3. Right panels: western blots showing that the proteasome inhibitor MG132 (10 μM) blocks EV-3-induced loss of BIRC2 and BIRC3. All treatments were for 24 h.
(F-G) Impact of cysteine mutagenesis on EV-3-mediated degradation of BIRC2 and BIRC3. FLAG-tagged wild-type (WT) or the indicated cysteine-to-alanine mutants of BIRC2 (C45A) and BIRC3 (C28A) were expressed in primary human T cells. An mCherry-expressing plasmid was used to control for transfection efficiency. Cells were then treated with DMSO, EV-3 (10 μM), or AT-406 (1 μM) for 24 h and analyzed by anti-FLAG blotting (F). (G) Quantitation of data, shown as mean values ± SEM; n = 3/group. *p < 0.05; **p < 0.01 compared to respective DMSO controls.
(H) Effect of genetic disruption of BIRC2 and/or BIRC3 by CRISPR/Cas9 genome editing on T-cell activation. Target disruption was considered to have an effect on T-cell activation if suppression was >33% with a p value < 0.01. Data are mean values ± SEM; n = 6/group. **, p < 0.01 compared to control guides.
(I) Workflow for TMT-exp experiments evaluating protein expression changes caused by active compound treatment in human T cells.
(J) Volcano plot representation of protein expression changes caused by BPK-25 (10 μM, 24 h) with significant decreases in NuRD complex proteins highlighted in red.
(K) Heatmap of top proteins with decreased expression in BPK-25-treated T cells showing that NuRD complex proteins (asterisks) were largely unaltered by other active compounds and blocked in degradation by MG132. Additional NuRD complex proteins also showed evidence of reduced expression (25–50%) in T cells treated with BPK-25.
(L) Representative western blot of time-dependent reductions in NuRD complex proteins in human T cells treated with BPK-25 (10 μM).
(M) T-cell activation and cytotoxicity profile of the pan-HDAC inhibitor vorinostat. Data are mean values ± SD; n = 2–4/group.
Among the active compounds, BPK-21 was unique in that it did not appear to impact the NF-κB or NFAT pathways. As noted above, a specific target of BPK-21, but not other active compounds, was C342 in the helicase ERCC3. This active-site cysteine is also targeted by the electrophilic immunosuppressive natural product triptolide (Titov et al., 2011) (Figure S5F). Like BPK-21, triptolide impairs T cell activation (Chang et al., 2001) (Figure S5G) without blocking NF-κB DNA binding activity (Qiu et al., 1999). We found using CRISPR/Cas9 technology that disruption of the ERCC3 gene (sgERCC3 cells), but not other representative targets of BPK-21, significantly impaired T-cell activation to a similar degree as BPK-21 treatment (Figure S5H–K). Western blotting estimated an ~80% loss of ERCC3 protein in sgERCC3 cells, which also showed only a modest further reduction in activation when treated with BPK-21 (Figure S5J, K). These data, taken together, indicate that BPK-21 likely suppresses T-cell activation through blockade of ERCC3 function, which may in turn act downstream or separately from pathways involved in NFAT and NF-κB activation.
EV-3-mediated degradation of BIRC2 and BIRC3
Within the NF-κB pathway, C28 in BIRC3 was a unique target of EV-3 (Figures 5B and 6C), and the corresponding cysteine (C45) in BIRC2 was also engaged by EV-3, as well as by DMF, but not other active compounds (Figures 6C and S6A). Other quantified cysteines in BIRC2 and BIRC3 were not affected by EV-3 treatment (Figure 6C). These proteins regulate both canonical and non-canonical NF-κB activation (Gyrd-Hansen and Meier, 2010) through ubiquitination of diverse substrates (Figure 3B) (Samuel et al., 2006; Yang et al., 2016). C28 of BIRC3 (and C45 of BIRC2) is located in close proximity to the BIR1 domain, which interacts with TRAF2 (Figure 6C, D) to facilitate recruitment to the TNF receptor. This interaction has been suggested to stabilize BIRC2, preventing its autoubiquitination and subsequent degradation (Csomos et al., 2009), and mutations in the BIR1 domain (Samuel et al., 2006; Zheng et al., 2010) impair interactions with TRAF2, but, to our knowledge, chemical probes targeting this region of BIRC2/3 have not yet been described.
We found that treatment of human T cells with EV-3, but not other active compounds, including DMF, led to the concentration-dependent (Figure S6B, C), time-dependent (Figure S6D), and proteasome-sensitive (Figure 6E) loss of both BIRC2 and BIRC3. This profile differed from the described Smac mimetic inhibitor AT406, which targets the BIR3 domain (Cai et al., 2011) and promoted the loss of BIRC2, but not BIRC3 (Figures 6E and S6B). EV-3 caused minimal changes in mRNA content for BIRC2 or BIRC3 (Figure S6E), supporting a direct effect on protein stability in T cells. Consistent with this conclusion, EV-3 promoted the degradation of wild-type (WT), but not cysteine-to-alanine mutants of BIRC2 (C45A) and BIRC3 (C28A) when recombinantly expressed in T cells (Figure 6F, G). In contrast, AT406 maintained its degradative activity with both WT and C45A variants of BIRC2 (Figure 6F, G). Finally, CRISPR/Cas9-mediated disruption of BIRC2 or BIRC3 impaired T-cell activation, with the combined disruption of both proteins producing a more substantial effect (Figures 6H and S6F, G). Treatment with EV-3 further decreased T-cell activation in sgBIRC2/sgBIRC3 cells, which may reflect an impact on the residual ~30% of BIRC2 and BIRC3 protein still present in these cells (Figure S6H) or on additional targets of EV-3 that contribute to its suppressive effects in T cells. In this regard, we evaluated several additional EV-3 targets by CRISPR/Cas9, but none were found to substantially impair T-cell activation (Figure S6F).
BPK-25 promotes degradation of the NuRD complex
A survey of the cysteines engaged by BPK-25 did not reveal obvious candidate proteins within the immunological pathways affected by this compound (e.g., NF-κB, NFAT; Data S1). Motivated by the finding that EV-3 promoted the degradation of BIRC proteins (Figures 6E and S6B–D), we performed expression-based proteomic (TMT-exp) experiments of human T cells treated with BPK-25 (Figure 6I and Data S1). This study revealed that BPK-25, but not other active compounds, promoted the striking and selective reduction of several proteins in the Nucleosome Remodeling and Deacetylation Complex (NuRD) (Figure 6J–L and Data S1). Only two other proteins across the >3000 quantified proteins in our proteomic experiments showed substantial (>50%) reductions in BPK-25-treated cells (FAM213B and HLA-F) (Figure 6K). BPK-25-mediated reductions in NuRD complex proteins were both concentration- (Figure S6I) and time- (Figure 6L) dependent and blocked by treatment with the proteasome inhibitor MG132 (Figure 6K). The reductions in NuRD complex proteins were not accompanied by corresponding changes in mRNA expression (Figure S6J), and a non-electrophilic propanamide analogue of BPK-25 (BPK-25-ctrl) did not suppress T-cell activation (Figure S6K) or affect NuRD complex proteins in T cells (Figure S6L), supporting that BPK-25 lowers NuRD complex proteins by a post-translational mechanism involving covalent protein engagement. Whether BPK-25 promotes NuRD complex degradation by engaging one or more cysteines in the protein constituents of this complex remains to be determined. Despite this mechanistic uncertainty, we confirmed previous results (Takahashi et al., 1996) that histone deacetylase (HDAC) inhibition blocks T-cell activation (Figure 6M). HDACs can also support NF-κB function (Jung et al., 2009; Kumar et al., 2017; Wagner et al., 2015). These findings indicate that BPK-25-mediated loss of the NuRD complex may be relevant to the T cell-suppressive activity of this compound.
Stereoselective degradation of immune kinases by EV-96
The cysteine engagement profiles of the active compound EV-96 and its stereoisomeric analogues revealed a striking number of stereoselective interactions (Figures 7A and S7A, Data S1), especially for the EV-96 and EV-97 pair of enantiomers. Several of these cysteines were found in immune-relevant proteins (Figure S7A), including the active-site cysteine (C449) of the kinase TEC, which was stereoselectively engaged by EV-96 (Figures 7A and S7B). Western blotting revealed that EV-96, but not EV-97, also promoted the loss of TEC protein in T cells (Figure 7B).
Figure 7. EV-96 stereoselectively engages and degrades immune kinases in T cells.

(A) Heatmap showing cysteines engaged >50% by stereoisomeric compounds (5 μM, 3 h). For inclusion in the map, cysteines were also required to show increased engagement by the relevant stereoisomeric electrophile at 20 μM (see Data S1).
(B) Western blot showing decreased TEC protein in human T cells treated with EV-96, but not EV-97 (5 μM each, 24 h).
(C) TMT-exp experiments comparing protein expression in DMSO-treated αCD3/CD28-stimulated (DMSO-stim)-versus-naïve control (DMSO-ctrl) T cells (y-axis) and EV-97-treated-versus-EV-96-treated stimulated T cells (x-axis). T cells were treated with DMSO or compounds (5 μM each) for 8 h. Red background denotes proteins with: i) > 2-fold expression in stimulated T cells treated with EV-97 versus EV-96; and ii) < 1.5 fold change in expression in DMSO-stim vs DMSO-ctrl T cells. The two proteins in this region are colored green. Proteins showing >2-fold changes in expression in DMSO-stim vs DMSO-ctrl T cells were removed from the analysis.
(D) Protein sequences showing EV-96-liganded cysteine in TEC (C449) and its conservation in ITK (C442).
(E, F) Western blot analysis (E) showing reductions in ITK protein (8 h) and PLCG1 phosphorylation (Y783, 24 h) in αCD3/CD28-stimulated (stim) T cells treated with EV-96, but not EV-97 (5 μM each). (F) Quantitation of data, shown as mean values ± SD; n = 2–5/group. **p < 0.01 compared to EV-97 treatment.
(G) Western blot analysis showing reductions in ITK protein in stimulated, but not control (naïve) T cells treated with EV-96 (5 μM). Co-treatment with the proteasome inhibitor MG132 (10 μM) blocks EV-96-induced reductions in ITK. All treatments were performed for 8 h. See Figure S7D for quantitation of these western blotting data.
(H) Quantitation of TMT-exp data showing effects of EV-96 and EV-97 (5 μM each, 8 h) on ITK protein in naïve control (ctrl) T cells versus αCD3/CD28-treated (stimulated, stim) T cells. Data are mean values ± SEM; n = 4/group. ****p < 0.0001 compared to DMSO-treated stim control.
(I) Western blot showing that pre-treatment with the ITK inhibitor PF-064655469 (1 h, 5 μM) blocks EV-96-induced degradation of ITK, but did not independently alter ITK protein in T cells.
To better understand the global effects of EV-96 on the protein content of T cells, we performed TMT-exp experiments on control versus activated T cells treated with DMSO, EV-96, or EV-97. To account for the immunosuppressive activity of EV-96, which we expected would indirectly block activation-dependent changes in protein expression in T cells, we compared the profiles of DMSO-treated stimulated (DMSO-stim)-versus-control (DMSO-ctrl) T cells to EV-97-treated-versus-EV-96-treated stimulated T cells. From a total of 3750 quantified proteins that displayed < 2-fold expression changes between DMSO-stim and DMSO-ctrl T cells, two proteins were found to be substantially reduced in expression (> two-fold) in EV-96-treated, but not EV-97-treated stimulated T cells – the immune-relevant proteins ITK and CYTIP (Figure 7C and Data S1). Interestingly, ITK is a kinase that shares > 55% identity with TEC, including conservation of the active-site cysteine engaged by EV-96 (C442 in ITK; Figure 7D). While we did not detect TEC kinase in TMT-exp experiments or C442 of ITK in TMT-ABPP experiments, we interpreted the acquired data to indicate that EV-96 may stereoselectively engage a shared active-site cysteine in both kinases, leading to their degradation.
ITK is a tyrosine kinase that plays a major role in T-cell signaling, undergoing recruitment to the plasma membrane following TCR stimulation, where ITK is activated by phosphorylation and in turn phosphorylates PLCG1 to promote downstream signaling (Andreotti et al., 2018). We verified by western blotting that EV-96, but not EV-97, caused the loss of ITK protein in stimulated T cells, and this effect also led to a stereoselective blockade of PLCG1 phosphorylation (Figures 7E, F and S7C). Treatment with the proteasome inhibitor MG132 blocked EV-96-mediated loss of ITK (Figure 7G). Strikingly, we also found that EV-96 only caused the degradation of ITK in stimulated, but not naïve (Figures 7G, H and S7D) or expanded (Figure S7E) control T cells, suggesting that upstream signaling events may be required to convert ITK into a form that is sensitive to EV-96-dependent degradation. Also consistent with this premise, EV-96 did not inhibit purified, recombinant ITK protein (Figure S7F). While we do not yet fully understand the prerequisite events for enabling EV-96 engagement and degradation of ITK, we found that LCK-dependent phosphorylation of the upstream scaffolding protein SLP-76 was not affected by EV-96 (Figure S7G), indicating the maintenance of early events in TCR signaling in cells treated with EV-96. Additionally, we confirmed that a non-electrophilic analogue of EV-96 (EV-96-ctrl) did not suppress T-cell activation (Figure S7H) or induce ITK degradation (Figure S7I) and pre-treatment with the inactive enantiomer EV-97 did not rescue ITK from EV-96-dependent degradation (Figure S7J). Finally, pre-treatment with PF-064655469, a structurally distinct covalent inhibitor that engages C442 of ITK (Wang et al., 2020; Zapf et al., 2012) and blocks ITK enzymatic activity (Figure S7F), rescued ITK from EV-96-induced degradation without affecting ITK stability on its own (Figures 7I and S7K).
Taken together, our studies indicate that EV-96 stereoselectively engages and promotes the degradation of key immune kinases, providing a plausible mechanism for its immunosuppressive effects. That the degradation of ITK was only observed in stimulated T cells further points to a provocative state-dependent activity for EV-96, which should make this compound a distinctly useful chemical probe for studying T-cell signaling in various biological contexts.
Discussion
In this manuscript, we describe a chemical proteomic strategy aimed at addressing a paradoxical challenge at the interface of immunology and chemical biology, namely that, despite major advances in our understanding of the genetic basis of human immune disorders, most immune-relevant proteins lack small-molecule probes to facilitate their functional characterization. By generating and integrating global maps of cysteine reactivity and ligandability, we provide a resource that greatly expands the landscape of electrophilic compound-protein interactions in primary human T cells. We show how cysteine ligandability maps can be further refined by phenotypic screening to illuminate tractable and functional sites of electrophilic compound action on immune-relevant proteins. We also showcase the value of incorporating stereochemistry into electrophilic compound libraries, which enabled the discovery of pairs of physicochemically matched compounds with differential activity for streamlined mechanistic analysis.
Several active compound-cysteine interactions led to protein degradation without requiring a separate E3 ligase-directing ligand, underscoring the potential for covalent modification by small molecules to directly affect protein stability in cells (Jones, 2018; Yang et al., 2019). That EV-96-mediated degradation of ITK was further only observed in stimulated T cells highlights an exciting opportunity to develop chemical probes that target this important immune kinase in a state-dependent manner. The only other protein showing a similar stereoselective decrease in stimulated T cells was CYTIP (Figure 7C), a scaffolding protein that also localizes to the immune synapse (Chen et al., 2006). While we are not aware of a direct protein-protein interaction between ITK and CYTIP, it is possible that EV-96-mediated loss of ITK leads to changes in CYTIP phosphorylation and stability in T cells.
One of the broader goals of this study was to assess the potential of electrophilic fragment screening to globally map sites of druggability in primary human immune cells. Among the >3400 cysteines liganded by scout fragments in human T cells were not only several known sites of electrophilic drug action (Data S1), but also cysteines in diverse protein components of key immune signaling pathways (e.g., NF-κB, TLR, TCR; Figure 3A, B) and in transcription factors involved in immune cell lineage commitment (e.g., RUNX3, EOMES) (Wang and Bosselut, 2009), thymocyte development (e.g., THEMIS) (Lesourne et al., 2009), and Th1 (e.g., STAT1, STAT4) (Oestreich and Weinmann, 2012), Th2 (e.g., IRF4) (Zhu, 2010), Th17 (e.g., IRF4, STAT3) (Ivanov et al., 2007), and Treg (e.g., FOXP3) (Fontenot et al., 2003) differentiation (Data S1). The number of fragment-sensitive cysteines that were also engaged in T cells by more elaborated electrophilic compounds (Figures 5D and 7A) further indicates that the ligandability maps furnished by scout fragments canvas many tractable sites for covalent ligand development in immune-relevant proteins, including those historically considered challenging to target with small molecules.
Future studies may include more in-depth global comparisons of cysteine ligandability across different immune cell types and states, which may discover additional chemical probes that, like EV-96, affect protein stability and/or function in a context-dependent manner. In this regard, we should acknowledge that, while PCA analysis clearly designated the expanded T cells studied herein as being more closely related to naïve rather than activated T cells, some proteins and pathways relevant to T-cell activation may have been altered in the expanded T cells due to the culturing conditions required for their proliferation. When considering ways to optimize the electrophilic compound-protein interactions discovered herein, we call attention to the potential for structure-guided approaches, but also caution that only a minor fraction of the ligandability events mapped in our chemical proteomic experiments (~25%) were found in proteins or domains in proteins for which high-resolution structural information is available (Figure S4E). This outcome emphasizes the large gap still remaining in our understanding of the three-dimensional structures of full-length human proteins. Finally, we should emphasize that some of the cysteine ligandability events mapped herein, including those showing good tractability, may fail to produce direct functional effects on proteins. Such so-called “silent” compounds still have the potential to be converted into heterobifunctional small-molecule degraders of proteins, an approach that has been successfully used with other covalent ligands (Buckley et al., 2015; Burslem et al., 2018; Tovell et al., 2019).
Cysteine is a uniquely nucleophilic amino acid that performs diverse functions in catalysis, redox regulation, metal coordination, and signaling via post-translational modification (Giles et al., 2003; Jacob et al., 2003), each of which may be affected by biochemical changes associated with T-cell activation. The extent to which such state-dependent changes in cysteine reactivity may be exploited for the development of chemical probes and possibly drugs that have a more selective effect on the activity of T cells remains an open and exciting question for future inquiry. T cell-restricted pharmacology may also emerge from targeting proteins selectively expressed in this immune cell type, and we note, in this regard, the rich content of ligandable cysteines in immune-relevant proteins discovered in our studies. The presence of ligandable cysteines in proteins historically considered challenging to target with small molecules, as well as our demonstration that electrophilic compounds engaging these cysteines can directly affect the functions and/or stability of immune-relevant proteins in cells, points to the broad potential for covalent small molecules to serve as probes and future drugs for modulating diverse immunological processes.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benjamin F. Cravatt (cravatt@scripps.edu).
Materials Availability
All chemical probes and other elaborated electrophilic compounds generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Raw proteomic data is available from the Lead Contact upon request. Raw RNA-sequencing data has been deposited to NCBI under GEO: GSE137756. Processed proteomic and RNA-sequencing data is provided in Data S1 and S2, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Isolation of peripheral blood mononuclear cells (PBMC) and T cells
All studies with primary human cells were performed with samples from human volunteers followed by protocols approved by The Scripps Research Institute Institutional Review Board. Blood from random, de-identified healthy donors (age 18 to 65) was obtained after informed donor consent and sex was not recorded. Peripheral blood mononuclear cells (PBMCs) were isolated over Lymphoprep (STEMCELL Technologies) gradient using slightly modified manufacturer’s instructions. Briefly, 25 mL of freshly isolated blood was layered on top of 12.5 mL of Lymphoprep in a 50 mL Falcon tube minimizing mixing of blood with Lymphoprep. The tubes were centrifuged at room temperature (931 g, 20 min, 23 °C) with brake off and the plasma and Lymphoprep layers containing PBMCs were transferred to new 50 mL Falcon tubes with a 2:1 dilution with PBS. The cells were pelleted (524 g, 8 min, 4 °C) and washed with PBS (20 mL) once. T cells were isolated from fresh PBMCs using EasySep Human T Cell Isolation Kit (STEMCELL Technologies, negative selection) according to manufacturer’s instructions.
METHODS DETAILS
T cell activation for mass-spectrometry analysis (activated T cells)
Non-tissue culture treated 6-well plates were pre-coated with αCD3 (5 μg/mL, BioXCell) and αCD28 antibodies (2 μg/mL, BioXCell) in PBS (2 mL/well) and kept at 4 °C overnight. The next day, the plates were transferred to a 37 °C incubator for 1 h and washed with PBS (2 × 5 mL/well). Freshly isolated T cells were resuspended in RPMI media supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 1 × 106 cells/mL, plated into the pre-coated 6-well plates (6–10 mL/well) and kept at 37 °C in a 5% CO2 incubator for 3 days. Following this incubation period, the cells were combined in 50 mL Falcon tubes, pelleted (524 g, 5 min, 4 °C), and washed with PBS (10 mL). The cells were then transferred into Eppendorf tubes in 1 mL of PBS and pelleted. PBS was then aspirated and the cells were either re-suspended in fresh RPMI media for in situ treatments or flash-frozen and kept at −80 °C until further analysis (in vitro treatments).
T cell expansion for mass-spectrometry analysis (control T cells)
A non-tissue culture treated 6-well plate was pre-coated with αCD3 (1.5 μg/mL) antibody in PBS (3 mL/well) and kept at 4 °C overnight. The next day, the plates were transferred to a 37 °C incubator for 1 h and washed with PBS (2 × 5 mL/well). Freshly isolated T cells were re-suspended in RPMI media (10% FBS, L-glutamine (2 mM), penicillin (100 U/mL), streptomycin (100 μg/mL)), containing αCD28 antibody (1 μg/mL) at 1 × 106 cells/mL, plated into the pre-coated 6-well plate (6–10 mL/well) and kept at 37 °C in a 5% CO2 incubator for 3 days. Following this incubation period the cells were combined in 50 mL Falcon tubes, pelleted (524 g, 5 min, 4 °C), and washed with PBS (10 mL). The cells were then re-suspended in RPMI media containing recombinant IL-2 (10 U/mL) and kept at 37 °C in a 5% CO2 incubator for 10–12 days, splitting the cells every 3–4 days to keep cell density below 2 × 106 cells/mL. After this time, the cells were pelleted (524 g, 5 min, 4 °C), washed with PBS (10 mL) and either re-suspended in fresh RPMI media for in situ treatments or flash-frozen and kept at −80 °C until further analysis (in vitro treatments).
Proteomic platforms: Whole proteome TMT (TMT-exp, related to Figures 1, S1, S3, and Data S1)
• Sample preparation
Freshly isolated naive, activated or expanded T cells (1.6 × 10^7 cells, 2 × 106 cells/mL in RPMI media) were pelleted (600 g, 5 min), and washed with PBS (1 × 10 mL). The cells were then transferred to an Eppendorf tube in additional PBS (1 mL), pelleted (600 g, 5 min), flash frozen, and kept at −80 °C until further analysis. Cell pellets were thawed on ice and lysed in lysis buffer (150 μL, 1 tablet of Roche complete, mini, EDTA-free Protease Inhibitor Cocktail dissolved in 10 mL of PBS) using a probe sonicator (2 × 8 pulses). Protein concentration was adjusted to 2.0 mg/mL and the samples (100 μL, 200 μg protein) were transferred to new Eppendorf tubes (1.5 mL) containing urea (48 mg/tube, final urea concentration: 8 M). DTT (5 μL, 200 mM fresh stock in H2O, final DTT concentration: 10 mM) was then added to the tubes and the samples were incubated at 65 °C for 15 min. Following this incubation, iodoacetamide (5 μL, 400 mM fresh stock in H2O, final IA concentration: 20 mM) was added and the samples were incubated in the dark at 37 °C with shaking for 30 min. Ice-cold MeOH (600 μL), CHCl3 (200 μL), and H2O (500 μL) were then added, the mixture was vortexed and centrifuged (10,000 g, 10 min, 4 °C) to afford a protein disc at the interface of CHCl3 and aqueous layers. The top layer was aspirated without perturbing the disk, additional MeOH (600 μL) was added and the proteins were pelleted (10,000 g, 10 min, 4 °C) and used in the next step or stored at −80 °C overnight.
• Whole proteome TMT LysC and trypsin digestion
The resulting protein pellets were resuspended in EPPS buffer (160 μL, 200 mM, pH 8) using probe sonicator (2 × 6 pulses). LysC solution (4 μL/sample, 20 μg in 40 μL of HPLC grade water) was added and the samples were incubated at 37 °C with shaking for 2 h. Trypsin (10 μL, 0.5 μg/μL in trypsin buffer) and CaCl2 (1.8 μL, 100 mM in H2O) were then added and the samples were incubated at 37 °C with shaking overnight.
• Whole proteome TMT labeling with TMT tags
Peptide concentration was determined using the microBCA assay (Thermo Scientific) according to manufacturer’s instructions. For each sample, a volume corresponding to 25 μg of peptides was transferred to a new Eppendorf tube and the total volume was brought up to 35 μL with EPPS buffer (200 mM, pH 8). The samples were diluted with CH3CN (9 μL) and incubated with the corresponding TMT tags (3 μL/channel, 20 μg/μL) at rt for 30 min. Additional TMT tag (3 μL/channel, 20 μg/μL, 30 min) was added and the samples were incubated for another 30 min. Labeling was quenched by the addition of hydroxylamine (6 μL, 5% in H2O). Following a 15 min incubation at rt, formic acid was added (2.5 μL, final FA concentration: 5%) and the samples were stored at −80 °C until further analysis.
• Whole proteome TMT ratio check and high pH fractionation
Small aliquots (2 μL) from each channel were combined in a separate Eppendorf tube and dried using a SpeedVac vacuum concentrator. The residue was re-dissolved in Buffer A (20 μL) and desalted using C18 stage tips (made in-house using 200 μL pipette tips and C18 discs (3M Empore)). Briefly, the stage-tip was activated by passing MeOH (2 × 50 μL) and washed with Buffer B (2 × 50 μL, 5% H2O, 95% CH3CN, 0.1% FA), followed by Buffer A (2 × 50 μL, 5% CH3CN/95% H2O, 0.1% FA). The sample was then loaded and the stage-tip was washed with Buffer A. The sample was eluted into a new Eppendorf tube with Buffer B (2 × 50 μL) and dried using SpeedVac vacuum concentrator. The residue was re-dissolved in Buffer A (10 μL) and analyzed by mass-spectrometry using the following LC-MS gradient: 5% buffer B in buffer A from 0–15 min, 5–15% buffer B from 15–17.5 min, 15–35% buffer B from 17.5–92.5 min, 35–95% buffer B from 92.5–95 min, 95% buffer B from 95–105 min, 95–5% buffer B from 105–107 min, and 5% buffer B from 107–125 min (buffer A: 95% H2O, 5% CH3CN, 0.1% FA; buffer B: 5% H2O, 95% CH3CN, 0.1% FA) and standard MS3-based quantification described below. Ratios were determined from the average peak intensities corresponding to each channel. For a ten-plex experiment, samples (20 μL/channel, final volumes adjusted based on the determined ratios) were combined in a new low binding Eppendorf tube (1.5 mL) and dried using SpeedVac. The residue was subjected to high pH fractionation.
• High pH fractionation
The spin columns for high pH fractionation were pre-equilibrated prior to use. Briefly, the columns were placed in Eppendorf tubes (2 mL), spun down to remove the storage solution (5,000 g, 2 min), and washed with CH3CN (2 × 300 μL, 5,000 g, 2 min) and buffer A (2 × 300 μL, 95% H2O, 5% CH3CN, 0.1% FA, 5,000 g, 2 min). TMT labeled peptides were re-dissolved in buffer A (300 μL, 95% H2O, 5% CH3CN, 0.1% FA) and loaded onto pre-equilibrated spin columns for high pH fractionation. The columns were spun down (2,000 g, 2 min) and the flow through was used to wash the original Eppendorf tube and passed through the spin column again (2,000 g, 2 min). The column was then washed with buffer A (300 μL, 2,000 g, 2 min) and 10 mM aqueous NH4HCO3 containing 5% CH3CN (300 μL, 2,000 g, 2 min), and the flow through was discarded. The peptides were eluted from the spin column into fresh Eppendorf tubes (2.0 mL) with a series of 10 mM NH4HCO3 / CH3CN buffers (2000 g, 2 min). The following buffers were used for peptide elution (% CH3CN): 7.5, 10, 12.5, 15, 17.5, 20, 22.5, 25, 27.5, 30, 32.5, 35, 37.5, 40, 42.5, 45, 47.5, 50, 52.5, 55, 75. Every 7th fraction was combined into a new clean Eppendorf tube (2 mL) and the solvent was removed using SpeedVac vacuum concentrator. The resulting 7 combined fractions were re-suspended in buffer A (24 μL) and analyzed on the Orbitrap Fusion mass-spectrometer (5 μL injection volume).
Alternative desalting and fractionation protocol:
Some of the samples were desalted and fractionated using an alternative protocol. In this protocol, the samples were re-suspended in buffer A (1 mL, 95% H2O, 5% CH3CN, 0.1% FA) and desalted by passing through Sep-Pak C18 cartridges (55–105 μm, Waters). Briefly, the cartridge was conditioned with CH3CN (3 × 1 mL) and equilibrated with buffer A (3 × 1 mL) before the sample was loaded by dropwise addition. The cartridge was then washed with the flow-through from the sample (1 × 1 mL) and desalted by passing buffer A (3 × 1 mL). The peptides were then eluted by adding 1 mL of 80% CH3CN, 20% H2O, 0.1% FA and the eluent was evaporated to dryness using SpeedVac vacuum concentrator. The sample was then re-suspended in buffer A (500 μL) and fractionated into a 96 deep-well plate using HPLC (Agilent), for offline high pH fractionation. The peptides were eluted onto a capillary column (ZORBAX 300Extend-C18, 3.5 μm) and separated at a flow rate of 0.5 mL/min using the following gradient: 100% buffer A from 0–2 min, 0–13% buffer B from 2–3 min, 13–42% buffer B from 3–60 min, 42–100% buffer B from 60–61 min, 100% buffer B from 61–65 min, 100–0% buffer B from 65–66 min, 100% buffer A from 66–75 min, 0–13% buffer B from 75–78 min, 13–80% buffer B from 78–80 min, 80% buffer B from 80–85 min, 100% buffer A from 86–91 min, 0–13% buffer B from 91–94 min, 13–80% buffer B from 94–96 min, 80% buffer B from 96–101 min, and 80–0% buffer B from 101–102 min (buffer A: 95% H2O, 5% acetonitrile, 0.1% FA; buffer B: 10 mM aqueous NH4HCO3). Each well in the 96-well plate contained 20 μL of 20% FA to acidify the eluting peptides. The eluent was evaporated to dryness in the plate using SpeedVac vacuum concentrator. The peptides were re-suspended in 80% CH3CN, 20% H2O, 0.1% FA buffer (100 μL/well) and every 12th fraction was combined into a new clean Eppendorf tube (1.5 mL), following an additional wash with the same buffer (200μL/column, washing wells in each row). The solvent was removed using SpeedVac vacuum concentrator, the resulting 12 combined fractions were re-suspended in buffer A (24 μL) and analyzed on the Orbitrap Fusion mass-spectrometer (5 μL injection volume).
• Whole proteome TMT liquid chromatography-mass-spectrometry (LC-MS) analysis
Samples were analyzed by liquid chromatography tandem mass-spectrometry using an Orbitrap Fusion mass spectrometer (Thermo Scientific) coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex). The peptides were eluted onto a capillary column (75 μm inner diameter fused silica, packed with C18 (Waters, Acquity BEH C18, 1.7 μm, 25 cm) and separated at a flow rate of 0.25 μL/min using the following gradient: 5% buffer B in buffer A from 0–15 min, 5–35% buffer B from 15–155 min, 35–95% buffer B from 155–160 min, 95% buffer B from 160–169 min, 95–5% buffer B from 169–170 min, and 5% buffer B from 170–200 min (buffer A: 95% H2O, 5% acetonitrile, 0.1% FA; buffer B: 5% H2O, 95% CH3CN, 0.1% FA). The voltage applied to the nano-LC electrospray ionization source was 1.9 kV. Data was acquired using an MS3-based TMT method adapted from Wang, Y. et al. (Wang et al., 2019). Briefly, the scan sequence began with an MS1 master scan (Orbitrap analysis, resolution 120,000, 400−1700 m/z, RF lens 60%, automatic gain control [AGC] target 2E5, maximum injection time 50 ms, centroid mode) with dynamic exclusion enabled (repeat count 1, duration 15s). The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of: quadrupole isolation (isolation window 0.7) of precursor ion followed by collision-induced dissociation (CID) in the ion trap (AGC 1.8E4, normalized collision energy 35%, maximum injection time 120 ms). Following the acquisition of each MS2 spectrum, synchronous precursor selection (SPS) enabled the selection of up to 10 MS2 fragment ions for MS3 analysis. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (collision energy 55%, AGC 1.5E5, maximum injection time 120 ms, resolution was 50,000). For MS3 analysis, we used charge state–dependent isolation windows. For charge state z = 2, the MS isolation window was set at 1.2; for z = 3–6, the MS isolation window was set at 0.7. The MS2 and MS3 files were extracted from the raw files using RAW Converter (version 1.1.0.22; available at http://fields.scripps.edu/rawconv/), uploaded to Integrated Proteomics Pipeline (IP2), and searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, non-redundant variant of the Human UniProt database (release-2012_11). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146 Da). N-termini and lysine residues were also searched with a static modification corresponding to the TMT tag (+229.1629 Da). Peptides were required to be at least 6 amino acids long, and to have at least one tryptic terminus. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a peptide false-positive rate below 1%. The MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2).
• Whole proteome protein ratios calculation for state-dependent reactivity dataset
The MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2). At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of non-unique peptides, removal of half-tryptic peptides, removal of peptides with more than one internal missed cleavage sites, removal of peptides with low (<10,000) sum of reporter ion intensities (5 channels/donor), and peptides with high variation between either of the replicate channels for expanded or activated T cells (coefficient of variance >0.5). R values (activated vs. expanded) for each peptide entry were calculated using the average reporter ion intensities of activated and expanded TMT channels. Then the ratios of all quantified peptides for a protein were averaged to obtain the final protein ratio. Proteins were required to have at least two unique quantified peptides in each experiment.
Proteomic platforms: Reactivity profiling (Related to Figures 1, S1, and Data S1)

• Sample preparation and treatment
Activated or expanded (control) primary human T cells were pelleted (524 g, 5 min) and washed with PBS (10 mL). The cells were then transferred to an Eppendorf tube in additional PBS (1 mL), pelleted (600 g, 5 min), flash frozen, and kept at −80 °C until further analysis. Cell pellets were thawed on ice and lysed by sonication (2 × 8 pulses). Soluble and particulate proteomic fractions were separated by ultracentrifugation (100,000 g, 45 min), and protein concentration was normalized to 1.7 mg/mL using a standard DC protein assay (Bio-Rad). The resulting proteomes (500 μL, 1.7 mg/mL) were treated with iodoacetamide polyethyleneoxide desthiobiotin (IA-DTB (Santa Cruz), 5 μL of 10 mM stock in DMSO, final concentration: 100 μM) for 1 h at ambient temperature. Ice-cold MeOH (500 μL) and CHCl3 (200 μL) were then added, the mixture was vortexed and centrifuged (10,000 g, 10 min, 4 °C) to afford a protein disc at the interface of CHCl3 and aqueous layers. Both layers were aspirated without perturbing the disk, which was re-suspended in cold methanol (500 μL) and CHCl3 (200 μL) by sonication. The proteins were pelleted (10,000 g, 10 min, 4 °C), and the resulting pellets were re-suspended in 90 μL of buffer containing 9M urea, 10 mM DTT and 50 mM triethylammonium bicarbonate (1/20 dilution of 1.0 M stock solution, pH 8.5) by thorough pipetting up and down. The resulting mixture was heated at 65 °C for 20 min. Sample was cooled to room temp, iodoacetamide (10 μL, 500 mM solution in H2O) was added, and the samples were incubated at 37 °C for 30 min with shaking.
• Trypsin digestion and streptavidin enrichment
Following the labeling with iodoacetamide, samples were diluted with 305 μL of triethylammonium bicarbonate buffer (50 mM, 1/20 dilution of 1.0 M stock, pH 8.5; Final urea concentration: 2.0 M). Trypsin (4 μL of 0.25 μg/μL trypsin in trypsin buffer, containing 25 mM CaCl2) was then added and the proteins were digested at 37 ˚C overnight. The following day, samples were diluted with wash buffer (400 μL, 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2% NP-40), streptavidin-agarose beads (50% slurry in wash buffer) were added to each sample (40 μL/sample) and the bead mixture was rotated for 2 h at rt. Briefly, for a 10-plex sample, streptavidin-agarose bead slurry (440μL, 50% slurry) was washed (2 × 1 mL, 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% NP-40) and brought up to the initial volume in the wash buffer prior to the addition to the sample. After incubation, the beads were pelleted by centrifugation (2,000 g, 1 min), transferred to a BioSpin column and washed (3 × 1 mL wash buffer, 3 × 1 mL PBS, 3 × 1 mL H2O). Peptides were eluted by the addition of 300 μL of 50% aqueous CH3CN containing 0.1% FA. The eluate was then evaporated to dryness using SpeedVac vacuum concentrator.
• TMT tag labeling
Peptides were re-suspended in 100 μL EPPS buffer (200 mM, pH 8.0) with 30% dry CH3CN, vortexed and spun down (2,000 g, 1 min). TMT tags (3 μL/channel in dry CH3CN, 20 μg/μL) were added to the corresponding tubes and the reaction was allowed to proceed for 75 min. The reaction was quenched by the addition of 5% hydroxylamine (3 μL per sample), vortexed and left at room temperature for 15 min. FA (5 μL) was then added to each tube, the tubes were vortexed, spun down and combined in a low binding 1.5 mL Eppendorf tube. The final combined sample was dried in a SpeedVac vacuum concentrator and kept at −80 °C until the high pH fractionation step.
• High pH fractionation
High pH fractionation was performed according to the procedure described above to yield 7 or 12 fractions which were re-suspended in buffer A (10 μL/sample) and analyzed by liquid chromatography tandem mass-spectrometry.
• TMT ABPP broad ligandability liquid chromatography-mass-spectrometry (LC-MS) analysis
Samples were analyzed by liquid chromatography tandem mass-spectrometry using an Orbitrap Fusion mass spectrometer (Thermo Scientific) coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex). The peptides were eluted onto a capillary column (75 μm inner diameter fused silica, packed with C18 (Waters, Acquity BEH C18, 1.7 μm, 25 cm) and separated at a flow rate of 0.25 μL/min using the following gradient: 5% buffer B in buffer A from 0–15 min, 5–35% buffer B from 15–155 min, 35–95% buffer B from 155–160 min, 95% buffer B from 160–169 min, 95–5% buffer B from 169–170 min, and 5% buffer B from 170–200 min (buffer A: 95% H2O, 5% acetonitrile, 0.1% FA; buffer B: 5% H2O, 95% CH3CN, 0.1% FA). The voltage applied to the nano-LC electrospray ionization source was 1.9 kV. Data was acquired using an MS3-based TMT method adapted from Wang, Y. et al. (Wang et al., 2019) Briefly, the scan sequence began with an MS1 master scan (Orbitrap analysis, resolution 120,000, 400−1700 m/z, RF lens 60%, automatic gain control [AGC] target 2E5, maximum injection time 50 ms, centroid mode) with dynamic exclusion enabled (repeat count 1, duration 15s). The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of: quadrupole isolation (isolation window 0.7) of precursor ion followed by collision-induced dissociation (CID) in the ion trap (AGC 1.8E4, normalized collision energy 35%, maximum injection time 120 ms). Following the acquisition of each MS2 spectrum, synchronous precursor selection (SPS) enabled the selection of up to 10 MS2 fragment ions for MS3 analysis. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (collision energy 55%, AGC 1.5E5, maximum injection time 120 ms, resolution was 50,000). For MS3 analysis, we used charge state–dependent isolation windows. For charge state z = 2, the MS isolation window was set at 1.2; for z = 3–6, the MS isolation window was set at 0.7. The MS2 and MS3 files were extracted from the raw files using RAW Converter (version 1.1.0.22; available at http://fields.scripps.edu/rawconv/), uploaded to Integrated Proteomics Pipeline (IP2), and searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, non-redundant variant of the Human UniProt database (release-2012_11). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146 Da) and up to one differential modification for the desthiobiotin (DTB) tag (+398.2529 Da). N-termini and lysine residues were also searched with a static modification corresponding to the TMT tag (+229.1629 Da). Peptides were required to be at least 6 amino acids long, to have at least one tryptic terminus, and to contain the DTB modification. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a peptide false-positive rate below 1%. The MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2).
• TMT-ABPP R value calculation for cysteine state-dependent reactivity dataset
At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of non-unique peptides, removal of half-tryptic peptides, removal of peptides with more than one internal missed cleavage site, removal of peptides with low (<10,000) sum of reporter ion intensities in both expanded or activated channels, removal of peptides with high variation (coefficient of variance >0.5) between the replicate expanded or activated channels if their sum of reporter ion intensities is greater than 5,000. R values (activated vs. expanded) for each peptide were calculated using the average reporter ion intensities of activated and expanded TMT channels. Once the R values were calculated, two types of grouping were performed to aggregate peptide quantification data: 1) overlapping peptides with the same modified cysteine (e.g., different charge states, high pH fractionation fractions, or tryptic termini) were grouped together, then their R values were averaged, and the shortest unique tryptic peptide was reported; 2) multiple modified cysteines on a tryptic peptide were grouped together, then the averaged R values were reported for further data processing. The median value derived from at least two biological replicates was reported as the final R value for each peptide with a maximum ratio cap of 20.
• Data processing and analysis for IA-DTB reactivity dataset
Proteins must have at least three unique quantified peptides in either particulate or soluble fraction in the TMT-ABPP experiments within the state-dependent dataset to be analyzed. The fraction with the most quantified unique peptides was selected for analysis for each protein. If a protein had an equal number of unique quantified peptides in both fractions, the peptide R ratios (activated vs. expanded) from both fractions were averaged. To account for potential donor variations in protein expression level, proteins were required to have at least one peptide R ratio within 1.5-fold of the protein expression level measured in TMT-exp experiments (if available) and were excluded from the analysis if all peptide R ratios were greater than 2.0 or less than 0.5. For proteins with 5 or more quantified peptides, a cysteine was considered for potential change in reactivity if its peptide R value differed more than two-fold from both the median R value of all quantified cysteines on the same protein and from the protein expression level measured in TMT-exp experiments (if available). For proteins with three or four quantified peptides, a cysteine was considered for potential change in reactivity if its peptide R value differed more than two-fold from the protein expression level measured by TMT-exp data, with an additional requirement that the maximum peptide R ratio differed more than 2-fold from the minimum peptide R ratio. All the cysteines that passed the initial filters described above were manually curated to remove low quality profiles.
Proteomic platforms: Ligandability profiling isoTOP-ABPP (related to Figures 2, S2B, and Data S1)

• Sample preparation
Activated or expanded primary human T cells were pelleted (524 g, 5 min), washed with PBS, and lysed by sonication (2 × 8 pulses). Soluble and particulate proteomic fractions were separated by ultracentrifugation (100,000 g, 45 min), and protein concentration was normalized to 1.7 mg/mL using a standard DC protein assay (Bio-Rad). The resulting proteomes were analyzed by competitive isotopic Tandem Orthogonal Proteolysis Activity-Based Protein Profiling (isoTOP-ABPP) using a protocol from Gao et al. (Gao et al., 2018)
• IA-alkyne labeling and click chemistry
Samples (500 μL, 1.7 mg/mL) were treated with scout fragments (5 μL of 50 mM DMSO stock, final concentration: 500 μM) or DMSO for 1 h, followed by treatment with iodoacetamide alkyne (IA-alkyne, 5 μL of 10 mM stock in DMSO, final concentration: 100 μM) for 1 h at ambient temperature. Modified proteins were then conjugated to isotopically labeled, TEV-cleavable biotin tags (TEV-tags) using copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Reagents for the CuAAC reaction were pre-mixed prior to their addition to the proteome samples. TEV tags (light or heavy, 10 μL of 5 mM stocks in DMSO, final concentration = 100 μM), tris(benzyltriazolylmethyl)amine ligand (TBTA; 30 μL of 1.7 mM stock in DMSO:t-butanol 1:4, final concentration = 100 μM), tris(2-carboxyethyl)phosphine hydrochloride (TCEP; 10 μL of fresh 50 mM stock in water, final concentration = 1 mM), and CuSO4 (10 μL of 50 mM stock in water, final concentration = 1 mM) were combined in an Eppendorf tube, vortexed and added to the proteomes (55 μL/sample). The CuAAC reaction mixture containing “heavy” TEV tag was added to the DMSO-treated control samples and the CuAAC reaction mixture containing “light” TEV tag was added to compound-treated samples. The reaction was allowed to proceed at rt for 1 h, “heavy” and “light” samples were combined pairwise in 15 mL conical Falcon tubes on ice containing 4 mL of cold methanol (pre-chilled at −80 °C), 1 mL CHCl3, and 1 mL H2O. Eppendorf tubes from the reaction mixtures were washed with additional H2O (1mL each) and the washes were added to the same Falcon tube (final ratios MeOH : CHCl3 : H2O = 4 : 1 : 4). Following centrifugation (5,000 g, 10 min, 4 °C), a protein disk formed at the interface of CHCl3 and aqueous layers. Both layers were aspirated without perturbing the disk, which was resuspended in cold MeOH (2 mL) and CHCl3 (1 mL) by vortexing. The proteins were pelleted (5,000 g, 10 min, 4 °C), and the resulting pellets were solubilized in 1.2% SDS in PBS (1 mL) with sonication and heating (95 °C, 5 min).
• isoTOP-ABPP sample streptavidin enrichment
Once solubilized, the samples were diluted with PBS (4 mL) and streptavidin-agarose beads were added for the enrichment (final SDS concentration: 0.2% in PBS). The beads (100 μL of a 50% slurry per sample) were washed with PBS (2 × 10 mL) and resuspended in 1 mL of PBS per sample prior to addition. The final mixture was rotated for 3 h at rt. Following this enrichment step, the beads were pelleted by centrifugation (2,000 g, 2 min) and washed to remove non-specifically binding proteins (2 × 10 mL 0.2% SDS in PBS, 2 × 10 mL PBS, and 2 × 10 mL H2O).
• isoTOP-ABPP sample trypsin and TEV digestion
After the last wash, the beads were transferred to new Eppendorf tubes in water (2 × 0.5 mL), pelleted (4,000 g, 3 min), and resuspended in 6M urea in PBS (0.5 mL). DTT (25 μL of a fresh 200 mM stock in water, final concentration – 10 mM) was added and the beads were incubated at 65 °C for 15 min. Iodoacetamide (25 μL of a 400 mM stock in water, final concentration – 20 mM) was then added and the samples were incubated in the dark at 37 °C with shaking for 30 min. Following this incubation, the mixture was diluted with PBS (900 μL), the beads were pelleted by centrifugation and resuspended in 2M urea in PBS (200 μL). Trypsin (Promega, sequencing grade; 2 μg in 6 μL of trypsin buffer containing 1 mM CaCl2) was added to the mixture and the digestion was allowed to proceed overnight at 37 °C with shaking. The beads were pelleted (2,000 g, 2 min) and the tryptic digest was aspirated. The beads were then washed (3 × 1 mL PBS, 3 × 1 mL H2O), transferred to a new Eppendorf tube in H2O (2 × 0.5 mL), washed with TEV buffer (200 μL, 50 mM Tris, pH 8, 0.5 mM EDTA, 1 mM DTT), and resuspended in TEV buffer (140 μL). TEV protease (4 μL, 80 μM) was then added and the beads were incubated at 30 °C overnight with rotation. Following the overnight digestion, the beads were pelleted by centrifugation (2,000 g, 2 min) and the TEV digest was separated from the beads using Micro Bio-Spin columns (Bio-Rad) with centrifugation (800 g, 0.5 min) and an additional wash (100 μL H2O). The samples were then acidified by the addition of 0.1% FA (14 μL, final concentration: 5% v/v) and stored at −80 °C prior to analysis.
• isoTOP-ABPP liquid-chromatography-mass-spectrometry (LC-MS/MS) analysis
Samples were pressure-loaded onto a 250 μm (inner diameter) fused silica capillary columns packed with C18 resin (Aqua 5 μm, Phenomenex) and analyzed by multidimensional liquid chromatography tandem mass-spectrometry (MudPIT) using an LTQ-Velos Orbitrap mass spectrometer (Thermo Scientific) coupled to an Agilent 1200-series quaternary pump. The peptides were eluted onto a biphasic column with a 5 μm tip (100 μm fused silica, packed with C18 (10 cm) and bulk strong cation exchange resin (3 cm, SCX, Phenomenex) in a 5-step MudPIT experiment, using 0%, 30%, 60%, 90%, and 100% salt bumps of 500 mM aqueous ammonium acetate and a 5%–100% gradient of buffer B in buffer A (buffer A: 95% water, 5% CH3CN, 0.1% FA; buffer B: 5% water, 95% CH3CN, 0.1% FA) as previously described (Weerapana et al., 2007). Data were collected in data-dependent acquisition mode with dynamic exclusion enabled (20 s, repeat of 2). One full MS (MS1) scan (400–1800 m/z) was followed by 30 MS2 scans (ITMS) of the nth most abundant ions.
• isoTOP-ABPP peptide identification
The MS2 spectra data were extracted from the raw file using RAW Converter (version 1.1.0.22; available at http://fields.scripps.edu/rawconv/), uploaded to Integrated Proteomics Pipeline (IP2), and searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, non-redundant variant of the Human UniProt database (release-2012_11). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146) and up to one differential modification for either the light or heavy TEV tags (+464.28595 or +470.29976 respectively). Peptides were required to have at least one tryptic terminus and to contain the TEV modification. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a peptide false-positive rate below 1%.
• isoTOP-ABPP R value calculation and data processing
The heavy/light isoTOP-ABPP ratios (R values) for each unique peptide (DMSO/compound treated) were quantified with in-house CIMAGE software (Weerapana et al., 2010) using default parameters (3 MS1 acquisitions per peak and signal to noise threshold set to 2.5). Site-specific engagement of cysteine residues was assessed by blockade of IA-alkyne probe labeling. A maximal ratio of 20 was assigned for peptides that showed a ≥ 95% reduction in MS1 peak area in the compound treated proteome (light TEV tag) compared to the control DMSO-treated proteome (heavy TEV tag). Ratios for unique peptide sequences were calculated for each experiment; overlapping peptides with the same modified cysteine (e.g., different charge states, elution times or tryptic termini) were grouped together and the median ratio was reported as the final ratio (R). Additionally, ratios for peptide sequences containing multiple cysteines were grouped together. When aggregating data across experimental replicates, the mean of each experimental median R was reported. The peptide ratios reported by CIMAGE were further filtered to ensure the removal or correction of low-quality ratios in each individual dataset. The quality filters applied were the following: removal of half-tryptic peptides, removal of peptides with more than one tryptic miscleavage site, removal of peptides with R = 20 and only a single MS2 event triggered during the elution of the parent ion, removal of non-unique peptides. Further filtering was then performed as described below for each experiment type.
Proteomic platforms: Ligandability profiling TMT-ABPP (Figures 2, S2A)
• Sample preparation and treatment
Broad ligandability TMT-ABPP samples were processed according to the same protocol as for IA-DTB reactivity profiling samples described above (Steps 1–5) with the exception of pre-treatment of the lysates with scout fragments KB02 or KB05 (5 μL of 50 mM DMSO stock, final concentration: 500 μM) for 1 h prior to the IA-DTB treatment step.
• TMT-ABPP R value calculation for broad ligandability datasets
At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of non-unique peptides, removal of half-tryptic peptides, removal of peptides with more than one internal missed cleavage sites, removal of peptides with low (<20,000) sum of reporter ion intensities for either expanded or activated control channels, removal of peptides with high variation between the replicate control channels (coefficient of variance >0.5), and peptides corresponding to the lower average reporter ion intensity control channels (activated vs expanded) if the difference in the average reporter ion intensity between expanded and activated control channels was more than two-fold. R-value (DMSO-treated vs. KB02/KB05-treated) for each peptide entry was calculated using the reporter ion intensities of DMSO and KB02/KB05 treated TMT channels for each treatment group with a maximum ratio cap of 20. Once the R values were calculated, two types of grouping were performed to aggregate peptide quantification data: 1) overlapping peptides with the same modified cysteine (e.g., different charge states, high pH fractionation fractions, or tryptic termini) were grouped together, then their R values were averaged, and the shortest unique tryptic peptide was reported; 2) multiple modified cysteines on a tryptic peptide were grouped together, then the averaged R values were used for further data processing. Peptides with high donor variation (R>5 for one donor, while R<2 for the other donor) were discarded (<1%), then the R values of replicate channels of the same condition were averaged to obtain the final reported data. A cysteine was required to be quantified in at least two TMT channels for each proteomic fraction to be reported.
• Combining data across experimental groups
To combine data across replicates from different experiment groups (e. g., broad ligandability and elaborated fragment data or hyper-reactivity) or different experiment types (e. g., TMT and isoTOP), we used identifiers consisting of the Uniprot accession concatenated with the tryptic sequence associated with the particular peptide. Peptides that contained the same modified cysteine or where multiple cysteines were modified on that peptide were combined. When data from an experiment group associated with a miscleaved peptide sequence was combined with data from another group which contained a non miscleaved variant of the same peptide, all data was reported under the fully tryptic identifier, unless the non miscleaved variant introduced an additional cysteine, in which case the data was not merged.
• Filtering of broad ligandability data
All peptides with R = 20 were manually reviewed. Peptides with R = 20 were discarded if the ratio set contained a single 20, and the minimum ratio in the set was less than 4. If the ratio set contained two or more 20 values and the minimum ratio in the set was less than 2, these 20 values were also discarded. This filter was applied on R values within a single experiment and when aggregating data from replicate experiments.
When aggregating data from replicate experiments, for peptides that had standard deviations greater than 60% of the mean, the lowest ratio of that set was reported, unless the minimum ratio of the set was ≥ 4, in which case the average ratio was reported. Individual peptide sequences were required to have been quantified (R ≠ 0) in at least two replicates per condition. Peptides were considered liganded if they had a final value of R ≥ 5.
Proteomic platforms: Elaborated compound TMT-ABPP (related to Figure 5 and Data S1)

• In situ labeling with cysteine-reactive electrophiles
Activated or expanded (control) T cells were re-suspended in RPMI media supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 3 × 106 cells/mL. The compounds were added to cells as 1000x DMSO stocks and mixed well with the media by pipetting up and down after addition. The cells were kept at 37 °C in 5% CO2 containing incubators for 3h, then pelleted by centrifugation (524 g, 5 min, 4 °C), washed with cold PBS (10 mL) and transferred to Eppendorf tubes (1 mL PBS). The cells were pelleted again (524 g, 5 min, 4 °C), flash-frozen, and kept at −80 °C until further analysis.
• TMT-ABPP
TMT-ABPP samples were processed according to the same protocol as for IA-DTB reactivity profiling samples described above (Steps 1–5).
• TMT-ABPP R value calculation for elaborated compounds dataset
At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of non-unique peptides; removal of half-tryptic peptides, removal of peptides with more than one internal missed cleavage site, removal of peptides with low (<10,000) sum of reporter ion intensities for control channels, and peptides with high variation between the replicate control channels (coefficient of variance >0.5). R values (compound-treated vs. DMSO-treated) for each peptide entry were calculated using the reporter ion intensities of DMSO and compound treated TMT channels for each treatment group with a maximum ratio cap of 20. Once the R values for each peptide entry were calculated, two types of grouping were performed to aggregate peptide quantification data: 1) overlapping peptides with the same modified cysteine (e.g., different charge states, high pH fractionation fractions, or tryptic termini) were grouped together, then their R values were averaged, and the shortest unique tryptic peptide was reported; 2) multiple modified cysteines on a tryptic peptide were grouped together, then the averaged R values were used for further data processing. The R values of replicate channels of the same condition were averaged to obtain the final reported data with the requirement that all included peptides have been quantified in at least two individual experiments.
• Filtering of elaborated compound data
All peptides with R = 20 were manually reviewed. Within individual replicates, peptides with R = 20 were discarded if the ratio set contained a single 20, and the minimum ratio in the set was less than 4. When aggregating ratios across replicates, peptides with R = 20 were discarded if the ratio set contained a single 20, and the minimum ratio in the set was less than 3. Individual peptide sequences were required to have been quantified (R ≠ 0) in at least two replicates per condition, unless they had R ≥ 4 in both particulate and soluble conditions for a given compound. During manual review of the data, some peptides were exempted from specific filters due to additional evidence of their validity. BIRC2 (C45) R = 20 and CASP2 (C366, C370) R = 20 values derived from DMF datasets values derived from DMF datasets were exempted from applied 20-filters as the same residues were convincingly liganded in the TMT datasets. Peptides were considered liganded if they had a final value of R ≥ 4.
Proteomic platforms: Hyper-reactivity profiling (related to Figure 2E and Data S1)

• Sample preparation
Activated or expanded (control) primary human T cells were pelleted (524 g, 5 min), washed with PBS, and lysed by sonication (2 × 8 pulses). Soluble and particulate proteomic fractions were separated by ultracentrifugation (100,000 g, 45 min), and protein concentration was normalized to 1.7 mg/mL using a standard DC protein assay (Bio-Rad).
• IA-alkyne labeling
Samples (500 μL, 1.7 mg/mL) were treated with iodoacetamide alkyne (IA-alkyne, 5 μL of 10 mM stock in DMSO and 5 μL of 1 mM stock in DMSO, final concentrations: 100 μM and 10 μM) for 1 h at ambient temperature. Hyper-reactivity samples were then processed according to the same protocol as isoTOP-ABPP broad ligandability profiling samples described above. The CuAAC reaction mixture containing “heavy” TEV tag was added to the 100 μM-treated samples and the CuAAC reaction mixture containing “light” TEV tag was added to 10 μM-treated samples.
• Filtering and processing of hyper-reactivity data
Peptides with R = 20 were discarded if the ratio set contained a single 20, and the minimum ratio in the set was less than 4. This filter was applied on R values within a single experiment and when aggregating data from replicate experiments. Data from these experiments was separated according to activation state and the minimal ratio between soluble and particulate fractions for each state was reported for each peptide.
Proteomic platforms: Whole proteome TMT (TMT-exp) for elaborated compounds dataset (related to Figures 6H–6J, 7C and Data S1)
• In situ labeling with elaborated cysteine-reactive electrophiles (DMF, EV3, BPK-21, BPK-25)
Freshly isolated T cells were re-suspended in RPMI media supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 2 × 106 cells/mL. The compounds were added to cells (1.6 × 107 cells/treatment, 2 × 106 cells/mL) as 1000x DMSO stocks and mixed well with the media by pipetting up and down after addition. The cells were kept at 37 °C in 5% CO2 containing incubators for 24h, then pelleted by centrifugation (524 g, 5 min, 4 °C), washed with cold PBS (10 mL) and transferred to Eppendorf tubes (1 mL PBS). The cells were pelleted again (524 g, 5 min, 4 °C), flash-frozen, and kept at −80 °C until further analysis.
• In situ labeling with elaborated enantiomeric probes EV-96 and EV-97
Non-tissue culture treated 6-well plates were pre-coated with either PBS or αCD3 (5 μg/mL) and αCD28 (2 μg/mL) antibodies (2 mL/well) and stored at 4 °C overnight. The next day, the plates were kept at 37 °C in 5% CO2 containing incubators for 2h and washed with PBS (2 × 5 mL/well). Expanded T cells were re-suspended in RPMI media supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 2 × 106 cells/mL. DMSO and compounds were added to cells as 1000x DMSO stocks and mixed well with the media by pipetting up and down after addition. The cells (1.6 × 107 cells/treatment, 2 × 106 cells/mL) were then plated onto pre-coated 6-well plates (8 mL/well) and kept at 37 °C in 5% CO2 containing incubators for 8h. The treatment groups were as follows: DMSO (no activation), EV-96 (no activation), DMSO (αCD3, αCD28), EV-96 (αCD3, αCD28), and EV-97 (αCD3, αCD28). Following the treatment, the cells were transferred to falcon tubes and pelleted by centrifugation (524 g, 5 min, 4 °C), washed with cold PBS (10 mL) and transferred to Eppendorf tubes (1 mL PBS). The cells were pelleted again (524 g, 5 min, 4 °C), flash-frozen, and kept at −80 °C until further analysis.
• Mass-spectrometry sample preparation
Samples were processed according to the same protocol as TMT-exp samples described above (Steps 2–6).
• Whole proteome protein ratio calculation for elaborated electrophiles DMF, EV3, BPK-21, BPK-25 (related to Figures 6I, 6J)
At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of non-unique peptides, removal of half-tryptic peptides, removal of peptides with more than one internal missed cleavage site, removal of cysteine-containing peptides, removal of peptides with low (<10,000) sum of reporter ion intensities for control channels, and peptides with high variation between the replicate control channels (coefficient of variance >0.5). R values (compound-treated vs. DMSO-treated) were calculated using the reporter ion intensities of compound and DMSO treated TMT channels for each treatment group. Then the ratios of all peptides of a protein were averaged to be reported as the final protein ratio. Proteins were required to have at least two unique quantified peptides in each experiment and were quantified in at least two independent experiments.
• Whole proteome protein ratio calculation for enantiomeric probes EV-96 and EV-97 (related to Figure 7C)
At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of non-unique peptides, removal of half-tryptic peptides, removal of peptides with more than one internal missed cleavage site, removal of peptides with low (<5,000) average of reporter ion intensities for control channels, and peptides with high variation between the replicate control channels (coefficient of variance >0.5). R values (compound-treated vs. DMSO-treated) were calculated using the reporter ion intensities of compound and DMSO treated TMT channels for each treatment group. Then the ratios of all peptides of a protein were averaged to be reported as the final protein ratio. Proteins were required to have at least two unique quantified peptides in each experiment and were quantified in all the independent experiments. The following additional quality filters were applied to generate the plot in Figure 7C: (1) proteins that show ≥ 2-fold changes in DMSO-treated naïve and DMSO-treated activated T cells were removed; (2) keratins were removed.
Principal Component Analysis of TMT-exp data (related to Figures 1 and S1)
Principal component analysis (PCA) based on singular value decomposition was performed using scikit-learn (Pedregosa et al., 2011) library (version 0.22) for Python 3.7.5. IA reactivity (activated vs. expanded) values were log2 transformed, and, since PCA cannot operate on sparse matrices, only data for proteins that were quantified in all experiments was used (3806 out of a total of 4824 after log2 transform). Enrichment of principal components was performed using Perseus 1.6.10.43 (Tyanova et al., 2016) using the latest annotations available on the Perseus website (tagged 2019_01). Note that the scikit-learn and Perseus produced identical PCA and loadings. Enriched p-values were corrected using the Benjamini-Hochberg FDR (Benjamini and Hochberg, 1995) method and a significance cutoff of 0.05 was used.
Gene Ontology (GO) Enrichment Analysis (related to Figure 1)
GO Enrichment Analysis was performed using the GOATOOLS (Klopfenstein et al., 2018) (v0.9.9) library for Python 3.7.5 using a gene association file downloaded from Uniprot (Bateman et al., 2019) (goa_human.gaf, version 2.1) and the latest ontology file downloaded from the GO Consortium (Ashburner et al., 2000) (go-basic.obo, version 1.2). To identify enriched GO terms associated with changes in cell state, enrichments were carried out using the list of 1106 proteins identified to be changing in expression (defined as having a two-fold decrease or increase in expression, see Data S1), against the background list of all 4824 proteins characterized in the TMT-exp experiments. Fisher’s exact test was used to compute p-values for each category, and these values were corrected for multiple hypothesis testing using the Benjamini-Hochberg FDR (Benjamini and Hochberg, 1995) method and a significance threshold of 0.05 was applied. Only GO terms from the Biological Process namespace were included in downstream analyses. Enriched terms were then passed through REVIGO (Supek et al., 2011), which identifies redundant terms and chooses representative terms for each group. We further reduced redundancies by removing terms that were subsets of other terms. The following settings were used with REVIGO: allowed similarity of 0.4, searched against the human Uniprot database, using the SimRel (Schlicker et al., 2006) semantic similarity measure. For the plots in Figure 1G and S3A, we plotted the top 20 non-redundant enriched terms.
To identify enriched GO terms associated with changes in cysteine reactivity following T-cell activation, enrichments were carried out as described above using the list of 160 proteins that were identified to change in reactivity (Data S1 against a background of all 4347 proteins characterized in the 4 experiments. Since only the “cell-redox homeostasis term” was enriched, it was not necessary to remove redundant terms using REVIGO or other means.
Reactome Pathways Liganded by Scout Fragments (related to Figure 3)
Reactome pathways were downloaded from the Reactome website (version 72) (Jassal et al., 2020), and for each pathway proteins that were (a) liganded (R ≥ 5) or (b) quantified (R > 0 for at least 1 peptide in TMT-ABPP experiments), but not liganded by scout fragments were listed. For Figure 3A, only pathways belonging to the third and second hierarchical levels of the Immune System and Signal Transduction top-level pathways, respectively, were highlighted, as these pathways were deemed most relevant in the context of the paper. Further, for inclusion in Figure 3A, pathways were required to contain at least 25 proteins that were liganded. Data S1 lists all the Reactome pathways (at all hierarchy levels) with pathways plotted in Figure 3A listed at the top.
Activity-guided profiling: multidimensional screen for inhibition of T cell activation (related to Figure 4)
Non-tissue culture treated 96-well plates were pre-coated with αCD3 (5 μg/mL) and αCD28 antibodies (2 μg/mL) in PBS (100 μL/well) and left at 4 °C overnight. Freshly isolated T cells were re-suspended in RPMI media supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 2 × 106 cells/mL. Compound stocks (200x) in DMSO were diluted to 2x stocks in the working RPMI media in another 96-well plate. The pre-coated 96-well treatment plates were washed with PBS (2 × 200μL), T cells (100 μL/well, 2 × 105 cells/well) were then added to the wells, followed by the addition of 2x compound stocks in RPMI media (100 μL). The outer wells of the plates were filled with media without cells to avoid the edge effect in the assay. The plates were then incubated at 37 °C in a 5% CO2 containing incubator for 24 h. Following the treatment, the cells were transferred to a U-bottom 96-well plate and harvested by centrifugation (600 g, 3 min, 4 °C). The supernatants were kept and stored at −80 °C for further cytokine analysis, while the cells were washed with PBS (2 × 150 μL) prior to staining for flow cytometry analysis.
Flow cytometry analysis (related to Figures 4 and 6)
Following the PBS washes, the cells were stained with fixable near-IR LIVE/DEAD cell stain (Invitrogen) according to manufacturer’s instructions. Briefly, one vial of near-IR LIVE/DEAD dye was resuspended in DMSO (50 μL) and diluted with PBS (1:1000). The diluted stain was added to each well (200 μL) and the cells were incubated for 30 min at room temperature in the dark. After this time, the cells were pelleted (600 g, 3 min, 4 °C), washed once with PBS (200 μL/well) and incubated with a freshly made cocktail of antibodies for the appropriate cell surface markers diluted in PBS containing 2% FBS (1:400 antibody dilution). The corresponding data in Figures 4D, 4G, 4H, and 6L is presented as the mean percentage of DMSO treated control ± SEM, n = 3/group (Figures 4D, 4H, and 6L); n = 4–5/group (Figure 4G). The corresponding data in Figure 4F is presented as the mean percentage of DMSO treated control ± SD, n = 2–5/group.
Duoset ELISA quantification of secreted cytokines (IL-2, IFN-γ) (related to Figures 4 and 6)
The levels of secreted IL-2, IFN-γ, and TNFα after incubating T cells in the presence of DMSO or electrophilic compounds under TCR-stimulating conditions were measured using DuoSet ELISA cytokine kits (R&D Systems) in clear microplates (R&D Systems) according to manufacturer’s instructions and read using a CLARIOstar (BMG Labtech) plate reader (450 nm). All cytokine concentrations were calculated according to the standard curve generated for each experiment. The corresponding data in Figures 4D, 4H, and 6A is presented as the mean percentage of DMSO-treated control ± SEM, n = 3/group.
ISRE-luciferase and CellTiter Glo assays (related to Figure S4G)
THP1-Lucia ISG cells were resuspended in low-serum growth media (2% FBS) at a density of 5 × 105 cells/mL and treated with BPK-25 or vehicle (DMSO) in the presence of viral dsDNA (2 μg/mL). 50 μL of cells/well were seeded into each well of a 384-well white Greiner plates and incubated for 24 h. To evaluate expression of the luciferase reporter, 30 μL of Quanti-luc (Invivogen) detection reagent was added to each well and luminescence was read using an Envision plate reader (Perkin Elmer) set with an integration time of 0.1 seconds. To evaluate cell viability, 30 μL of CellTiter-Glo (Promega) reagent was added to each well and each plate was read using the same instrument settings utilized for the luciferase assay. For each cell type and assay, luminescence signals for test article samples were normalized to vehicle-treated samples and reported as relative light units (RLU). The corresponding data in Figure S4G is presented as the mean percentage of DMSO-treated control ± SEM, n = 3/group.
Bio-Plex quantification of secreted cytokines (related to Figure S4I)
Freshly isolated PBMCs (4 × 106 cells/mL, 1 mL/well), were treated with BPK-25 (10 μM) or vehicle (DMSO) for 6 h in a 24-well plate, after which cGAMP (10 μM) was added to the wells and the cells were incubated for additional 20 h. Following this treatment, the cells were transferred to 1.5 mL Eppendorf tubes and harvested by centrifugation (600 g, 8 min, 4 °C). The supernatants were saved (−80 °C) and used for further cytokine analysis using Bio-Plex Pro Human Cytokine assay (Bio-Rad) according to manufacturer’s instructions. Bio-Plex Assay is a multiplex flow immunoassay that simultaneously detects and identifies cytokines based on fluorescent dye-labeled 6.5 μm magnetic beads in a single reaction. When run on the Bioplex 200 system, 50 μL of supernatant was mixed with 50 μL of beads and quantified against human cytokines standard curves. The corresponding data in Figure S4I is presented as the mean percentage of DMSO-treated control ± SEM, n = 3/group.
ELISA quantification of secreted IFN-β (related to Figure S4I)
Concentrations of IFN-β were determined with VeriKine-HS human IFN-β serum ELISA kit (PBL Assay Science) according to manufacturer’s instructions. All concentrations of IFN-β were calculated according to the standard curve generated for each experiment. The corresponding data in Figure S4I is presented as the mean percentage of DMSO-treated control ± SD, n = 2/group.
Measurement of intracellular glutathione levels (related to Figure S5A)
Intracellular glutathione levels were determined using GSH-Glo glutathione assay (Promega Corporation) according to manufacturer’s instructions. Briefly, freshly isolated T cells were treated with compounds or DMSO for 24 h under TCR-stimulating conditions (96-well plate, 1 × 105 cells/well) at 37 °C in 5% CO2 containing incubator, then transferred to a U-shape bottom 96-well plate and pelleted (600 g, 3 min, 4 °C). The supernatants were kept and stored at −80 °C for cytokine analysis. The cells were washed with PBS (2 × 150 μL) and resuspended in 50 μL of PBS. An aliquot of treated cells (25 μL) was then added to an equal volume of 2x GSH reaction buffer containing Glutathione S-transferase and Luciferin-NT substrate (1:50 dilution in GSH-Glo Reaction Buffer). The reaction was incubated for 30 min at rt, after which Luciferin Detection Reagent (in reconstitution buffer with esterase, 25 μL/well) was added, and the plate was incubated for an additional 15 min and luminescence was read using a CLARIOstar (BMG Labtech) plate reader. The corresponding data in Figure S5A is presented as the mean percentage of DMSO-treated control ± SEM, n = 3/group.
Western blot analysis (related to Figures 6 and 7)
Western blot analysis was performed on freshly isolated or expanded T cells. For Western blot protein degradation analysis, primary human T cells (2 × 107 cells/treatment) were re-suspended in RPMI media at 2 × 106 cells/mL and treated with the compounds or DMSO at 37 °C in a CO2 containing incubator for 24 h (or otherwise indicated times). Following this incubation period, the cells were pelleted (600 g, 5 min, 4 °C), washed with PBS (10 mL), transferred to 1.5 mL Eppendorf tubes, flash-frozen, and stored at −80 °C until further analysis. On the day of the analysis, the cell pellets were thawed on ice, re-suspended in cold PBS and lysed by sonication with probe sonicator (2 × 8 pulses). Protein concentrations for all the samples were adjusted to 1 mg/mL, 4x loading buffer was added (10 μL to 30 μL of proteome), and the samples were heated at 95 °C for 5 min. The proteins were resolved using SDS-PAGE (10% acrylamide gel) and transferred to 0.45 μM nitrocellulose membranes (GE Healthcare). The membrane was blocked with 5% milk in Tris-buffered saline (20 mM Tris-HCl 7.6, 150 mM NaCl) with 0.1% tween 20 (TBST) buffer at rt for 1 h (or at 4 °C overnight), washed 3 times with TBST, and incubated with primary antibodies in 5% BSA in TBST at 4 °C overnight. Following another TBST wash (3 times), the membrane was incubated with secondary antibody (1:5000 in 5% milk in TBST) at 4 °C overnight. The membrane was washed with TBST (3 times), developed with ECL western blotting detection reagent kit (Thermo Scientific) and recorded on CL-XPosure film (Thermo Scientific). Relative band intensities were quantified using ImageJ software.(Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018.)
Western blot analysis of chromatin-bound proteins (related to Figures 6K, S6I, S6L)
BPK-25 (10 μM) treated expanded T cells were washed with PBS before permeabilization by rotation at 4 °C for 10 min with cytoplasm lysis buffer (10 mM sodium phosphate pH 7.4, 25 mM KCl, 1.5 mM MgCl2, 10% glycerol, and 0.025% NP-40 supplemented with 1x HALT protease inhibitor cocktail (Thermo Scientific)). Nuclei were pelleted (500 g, 5 min), and washed with cytoplasm lysis buffer without detergent, before being lysed by gentle sonication (Branson Sonifier 250) in cell lysis buffer (10 mM sodium phosphate pH 7.4, 25 mM KCl, 1.5 mM MgCl2, 10% glycerol, and 1% NP-40, 0.1% SDS supplemented with 1x HALT, and 1x Benzoase (Pierce)) and rotated for 2 h at 4 °C. Insoluble material was precipitated by centrifugation (12,000 g, 10 min) and the protein concentration of nuclear extracts was measured using standard BCA assay (Thermo Scientific) and normalized. Electrophoretic separation was performed on Novex 4–20% Tris-Glycine Mini Gels (Invitrogen) using the Novex Wedgewell system, and transferred to 0.45 μM Nitrocellulose membranes (GE Healthcare). Primary antibodies were applied overnight at 4 °C in 5% BSA/TBST. Secondary antibodies were applied for 1 h at rt in 5% milk/TBST. Blots were imaged using fluorescence-labeled secondary antibodies (LI-COR) on the Odyssey CLx Imager. Relative band intensities were quantified using ImageJ software.
For dose response and BPK-25-control treatment (Figures S6I, S6L), the above procedure was followed as written with the omission of nuclei isolation by permeabilization and washing steps (proceeding instead directly to sonication in cell lysis buffer (10 mM sodium phosphate pH 7.4, 25 mM KCl, 1.5 mM MgCl2, and 1% NP40 0.1% SDS supplemented with 1x HALT, and 1x Benzoase (Pierce)).
Gene expression (qPCR) analysis (related to Figures S4H, S6E)
Total RNA from compound or DMSO treated T cells (1.5 × 107 cells/group) was isolated using RNeasy Mini Kit (Qiagen) according to manufacturer’s protocol. RNA concentration was determined using NanoDrop and adjusted to 1 μg RNA in 15 μL RNAse free water for the reverse transcription reaction. cDNA amplification was done using iScript Reverse Transcription Supermix kit (BioRad) according to manufacturer’s instructions. The following PCR settings were used for the reverse transcription reaction: 5 min at 25 °C (priming), 20 min at 46 °C (Reverse transcription), 1 min at 95 °C (RT inactivation), hold at 4 °C. qPCR analysis was performed on ABI Real Time PCR System (Applied Biosystems) with the SYBR green Mastermix (Applied Biosystems). Relative gene expression was normalized to actin. qPCR primers used (5’ to 3’): actin-fwd (AGAGCTACGAGCTGCCTGAC), actin-rev (AGCACTGTGTTGGCGTACAG), BIRC2-fwd (AGCACGATCTTGTCAGATTGG), BIRC2-rev (GGCGGGGAAAGTTGAATATGTA), BIRC3-fwd (AAGCTACCTCTCAGCCTACTTT), BIRC3-rev (CCACTGTTTTCTGTACCCGGA), IL6-fwd (AATTCGGTACATCCTCGACGG), IL6-rev (GGTTGTTTTCTGCCAGTGCC), IL1-beta-fwd (ACAGATGAAGTGCTCCTTCCA), IL1-beta-rev (GTCGGAGATTCGTAGCTGGAT), CXCL10-fwd (CCAGAATCGAAGGCCATCAA), CXCL10-rev (CATTTCCTTGCTAACTGCTTTCAG).
RNA sequencing (related to Figure S6J and Data S2)
Total RNA from compound or DMSO treated T cells (1.5 × 107 cells/group) was isolated using RNeasy Mini Kit (Qiagen) using RNAse free DNAse set (Qiagen) for on column DNA digestion according to manufacturer’s protocol and stored at −80 °C until further analysis. RNA quality was assessed using TapeStation 4200 and RNA-Seq libraries were prepared using the TruSeq stranded mRNA Sample Preparation Kit v2 according to Illumina protocols. Multiplexed libraries were validated using TapeStation 4200, normalized, pooled and quantified by qPCR for sequencing. High-throughput sequencing was performed on the NextSeq 500 system (Illumina). Image analysis and base calling were done with Illumina CASAVA-1.8.2. Sequenced reads were quality-tested using FASTQC (Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the hg19 human genome using the STAR (Dobin et al., 2013) version 2.5.3a. Mapping was carried out using default parameters (up to 10 mismatches per read, and up to 9 multi-mapping locations per read). The genome index was constructed using the gene annotation supplied with the hg19 Illumina iGenomes (iGenomes online. Illumina. 2015. http://support.illumina.com/sequencing/sequencing_software/igenome.html) collection and overhang value of 100. Gene expression counts were quantified with HOMER (Heinz et al., 2010) v4.10.4 analyzeRepeats.pl using the parameters -raw -count exons -condenseGenes -strand +. Normalized gene expression counts were calculated using HOMER v4.10.4 analyzeRepeats.pl using the parameters -fpkm -count exons -condenseGenes -strand + -normMatrix 1000000. Differential expression was carried out with HOMER v4.10.4 getDiffExpression.pl using the default DESeq2 v1.14.1 subroutine on the raw count table.
ITK enzyme activity assay (related to Figure S7F)
ITK Kinase Enzyme System (V3191, Promega) was used for ITK in vitro assay. Briefly, ITK (139 nM) was pre-incubated with 25, 50 or 100 μM of EV-96 or 100 μM of PF-06465469 in 25 μL of reaction buffer (50 μM DTT, 2 mM MnCl2 and 1X reaction buffer A) for 1 h at room temperature. 50 μM of ATP was then added and reaction proceeded for additional 30 min at room temperature. ADP-Glo™ Kinase Assay (V6930, Promega) was used to measure ADP generated from the reaction following manufacturer’s instructions.
CRISPR/Cas9 experiments (related to Figures 6, S5 and S6)
• Generation of in vitro transcribed sgRNAs
DNA templates consisting of a T7 RNA Polymerase promoter, the ~20 nucleotide target-specific sequence, and a 20 nucleotide overlap to the generic sgRNA template were generated for each desired target by overlapping PCR using Q5 High Fidelity Master Mix (New England Biolabs) under the following conditions: 98 °C for 2 min; 50 °C for 10 min; 72 °C for 10 min. Guide RNA templates were used to transcribe guide RNAs using the HiScribe T7 High Yield RNA Synthesis kit (New England Biolabs) according to the manufacturer’s instructions. Following in vitro transcription, guides were purified using Monarch RNA Cleanup Kit (New England Biolabs) following manufacturer’s instructions.
• Oligos for Generation of T7 sgRNA production
The generic template design is as follows: 5′-TAATACGACTCACTATA-N(19–21)-GTTTTAGAGCTAGAAATAGC-3′. For a list of Oligos for T7 sgRNA production, see Methods S3. To generate the 80-nt chimeric sgRNA core sequence (5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTA TTTCTAGCTCTAAAAC-3′), an ultramer was ordered from IDT.
• Cas9 Ribonucleoprotein (RNP) Assembly and Electroporation
The Cas9 RNPs were assembled before transfection using the ArciTect™ Cas9-eGFP Nuclease (StemCell) with the T7 transcribed RNAs at a molar ratio of 1:3 in Buffer T. For each target of interest, the genome was tiled with 3 unique guide RNAs. Before the transfection, primary T cells were preactivated on αCD3/αCD28-precoated plates in complete RPMI medium supplemented with 100 U/mL IL-2 for 48 h. The T cells were then washed with PBS and resuspended in Buffer T (10 × 106 cells/mL) and the Cas9 RNP transfections were performed using the Neon Transfection system (ThermoFisher). Following the Cas9 RNP transfection, T cells were cultured in RPMI supplemented with 50 U/mL IL-2 for 7 days.
• FACS Analysis of T cell Activation (related to Figures 6G, S5H–S5J, S6F, S6G)
Seven days post-transfection, the cells were stimulated for a second time using αCD3/αCD28-precoated plates in the presence of IL-2 (100 IU/mL) for 24 h. Cell surface staining for T cell activation was performed using αCD25-PE (Biolegend) and αCD69-APC (Biolegend) antibodies for 1 h at 4 °C. Viable cells gating was performed using eBioscience™ Fixable Viability Dye eFluor™ 780 (ThermoFisher).
• Analysis of ERCC3, BIRC2 and BIRC3 protein expression levels after CRISPR/Cas9 Genome Editing (related to Figures S5K, S6H)
Using the previously described protocol, the Cas9-GFP protein was incubated with guide RNA transcripts to generate the Cas9:sgRNA RNP complex. The Cas9 RNP complexes were Neon transfected into αCD3/αCD28 activated T-cells using the aforementioned experimental procedures. Following 72 h after transfection, GFP-positive T-cells were FACS sorted and subsequently lysed on ice for 45 min using 1X radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology) combined with 1x Halt Protease and Phosphatase Single-Use Inhibitor Cocktail (Thermo Scientific). The lysates were centrifuged to remove cellular debris, the lysates were collected, and samples were stored at −80°C until time of analysis.
• Western blot analysis for CRISPR/Cas9 gene editing experiments
On the day of the analysis, cell lysates were thawed on ice and then combined with 2x Laemmli loading Buffer (BioRad) with 0.05% BME. The samples were heat denatured for 10 min at 95 °C and loaded into a Bolt™ 4 to 12% (vol/vol), Bis-Tris precast polyacrylamide gels (Invitrogen by Thermo Fisher). Gels were run at 120V in 1x MOPS Buffer and were then transferred to a 0.2 μm PVDF nitrocellulose membrane (Trans-Blot Turbo Transfer Pack, Bio-Rad) using the Trans-Blot Turbo Transfer System (13A, 25V, 7 min) (BioRad). The membrane was blocked in 5% BSA for 1 h followed by overnight incubation at 4 °C with primary antibodies (1:1000 anti-mCherry [Cell Signaling Technology] or 1:1000 anti-DYKDDDDK [Cell Signaling Technology]). Membranes were washed with TBST (3x, 5 min) and incubated with secondary anti-rabbit HRP antibody for 1 h at room temperature followed by a subsequent washing with TBST (3x, 5 min). The membrane was developed with ECL western blotting detection reagent kit (Thermo Scientific) and imaged on ChemiDoc XRS+ with Image Lab Software (Bio-Rad).
• Impact of cysteine mutagenesis on EV-3-mediated decay of BIRC2 and BIRC3 (related to Figures 6E, 6F).
BIRC2 and BIRC3 Open Reading Frames were cloned into the pcDNA3.1(+)_Flag expression plasmid (Thermo Fisher Scientific) from Jurkat cell (ATCC) mRNA. RNA was isolated from cells using the RNA-Easy Kit (Qiagen) per manufacturer’s instructions and cDNA was generated via reverse transcription the Quantitect Reverse Transcription Kit (Qiagen). Cloning into the pcDNA expression plasmid was performed using the NEBuilder® HiFi DNA Assembly Master Mix (New England BioLabs). The cysteine-to-alanine mutations of interest were generated using Q5 Site-Directed Mutagenesis Kit (New England BioLabs). For a list of primers used for site-directed mutagenesis, see Methods S3.
Primary human T cells (2 × 107 cells) that had been previously activated with αCD3/αCD28 as described above were co-transfected in triplicate with 5 μg of the BIRC expressing plasmid and 1 μg of an mCherry-expressing plasmid pcDNA-mCherry to control for transfection efficiency using the Neon Transfection System at a cell density of 2 × 107 cells/mL. Transfected cells were cultured in complete RPMI medium supplemented with 50 IU/mL of IL-2 for 24 h, at which point the transfected cells were treated with either DMSO, EV-3 (10 μM), or AT406 (1 μM) for 24 h, pelleted, washed with PBS, and lysed on ice for 45 min using 1X radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology) combined with 1x Halt Protease and Phosphatase Single-Use Inhibitor Cocktail (Thermo Scientific). The lysates were centrifuged, the supernatants were collected, and samples were stored at −80 °C until time of analysis. Western blot analysis of the samples was performed according to the same protocol as described above for the CRISPR/Cas9 Gene knock out experiments.
Molecular modeling (related to Figures 5 and S4)
• Description of the methods
In order to gain structural insights on the systems considered, we applied two different docking methods based on the software Autodock (Morris et al., 2009): the reactive docking and the flexible side chain covalent docking. The reactive docking is a predictive method that allows to identify the residues most likely to be modified by covalent binding. This is accomplished in two steps: first, it performs a scan of all solvent accessible residues of a given type (cysteines, in this case), then it applies conventional, untethered docking with a special potential to simulate the incipient reaction to identify the most likely ones to be modified by the ligands. Reactive docking was successfully applied in previous studies, where it was used to model electrophile reactions with cysteine (Backus et al., 2016), tyrosine and lysine (Mortenson et al., 2018), and serine (Qinheng et al., 2019) residues. For the flexible side chain covalent method, ligands are modeled attached to the alkylated residue via covalent bond, then processed following the covalent docking protocol (Bianco et al., 2016) (available online at http://autodock.scripps.edu/resources/covalentdocking), where both ligand and residue are modeled as flexible during the docking. This method is used to analyze the non-covalent interactions of the bound ligands and target residues constituting the binding site.
The crystal structures of the proteins were retrieved from the Protein Data Bank: TIR domain of MYD88 (PDB 4DOM), ERCC3 (PDB 5OF4), and TMEM173 (PDB 6NT5). Hydrogens were added with Reduce (Word et al., 1999), then were prepared using AutoDockTools (Morris et al., 2009) following the standard AutoDock protocol (Forli et al., 2016). Reactive docking was performed following the protocol reported previously (Backus et al., 2016) with a grid box defined for each cysteine: C168, C192, C216, C203, C247 and C280 of MYD88 (size x: 60, y: 60, z: 60 points). All dockings were performed using AutoDock 4.2.6 (Morris et al., 2009), generating 100 poses using the default LGA parameters. Poses with the best energy score were selected and analyzed. Figures were generated using Pymol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.).
• Pocket analysis with AutoSite
AutoSite (Ravindranath and Sanner, 2016) was used to analyze ligand-binding pockets located in proximity to the liganded cysteine sites identified as targets of elaborated compounds for which X-ray structures were available in the PDB. Briefly, AutoSite is a pocket prediction method based on the AutoDock maps, which provides an energy-based analysis used for identifying and characterizing small molecule binding sites on biological macromolecules. 74 cysteine sites in 68 structures have been considered for the pocket prediction (see Data S1) and the results have been analyzed by measuring the distance between the thiol sulfur of the labeled cysteine and the closest point of the pocket using a custom Python script.
By taking into account the length of acrylamide or chloroacetamide warheads and the optimal placement of ligands for establishing a covalent bond with the cysteine thiol, the distance of 6 Å was defined as the minimum distance cut-off between the thiol sulfur and the closest point of the predicted pocket in order to consider a pocket as adjacent to the modified cysteine. Using this cut-off, the software identified pockets for 68 % of the alkylated cysteines, suggesting that not all the modified residues lie in close proximity of a pocket that can be predicted using this approach. It must be noted that this approach does not address the possibility that transient pockets might be formed when the proteins under consideration interact with other proteins in larger assemblies or crowded cellular environments. Success rates for the distances of 5, 4 and 3 Å have been calculated as well. All the analyzed structures are derived from X-ray crystallography experiments except for ERCC3 and TMEM173, whose structures were solved using cryo-EM experiments.
• Reactive docking and flexible side chain covalent docking on MYD88
isoTOP-ABPP and TMT-ABPP experiments showed that the TIR domain of MYD88 is covalently modified with different potency by BPK-25 and BPK-21 at C203, and by KB02 and KB05 at either C274 or C280 within the tryptic peptide (270–282). Consequently, we applied two different docking techniques to rationalize the different potencies of the first compounds on C203, and to attempt resolving the ambiguity between C274 and C280 modification. In the first approach, we used the reactive docking method to sort the ambiguity between the labeling of the C274 and C280, then the flexible side chain covalent docking was used to generate putative binding modes of all the compounds and provide structural insight for their different activities.
We performed reactive docking simulations on the entire domain (PDB 4DOM) with ligands KB02 and KB05 and in addition, with BPK-25 and BPK-21 as a proof of concept, since experimental studies show direct labeling of C203 with BPK-25, but not BPK-21. Reactive docking analysis on BPK-25 and BPK-21 confirmed that the most favorable residue is C203, while C274 is the predicted residue for the covalent binding of KB02 and KB05. These results show that the position of C280 on the protein surface is less likely to be modified because it is largely solvent exposed, while on the contrary, C274 is located inside a cleft of the domain. Flexible side chain covalent docking was then used to refine the binding mode of compounds BPK-25 and BPK-21 (Figure 5G). In particular, the predicted binding mode of BPK-25 shows that it could bind by establishing two hydrogen bonds with R188 and E183 via the amide moiety (Figure 5G, top right), which is missing in BPK-21 (Figure 5G, bottom right). The lack of these interactions justifies the lower efficacy reported for BPK-21 in modifying C203.
• Flexible side chain covalent docking on ERCC3
isoTOP-ABPP and TMT-ABPP demonstrated that compounds BPK-25 and BPK-21 bind to ERCC3 by alkylating C342. Flexible side chain docking simulations were performed to rationalize the higher efficacy of BPK-21 with respect to BPK-25, by modeling the two ligands bound to C342 on a low resolution Cryo-EM structure of the protein (PDB 5OF4, 4.4 Å resolution). Results showed that BPK-21 can form two hydrogen bonds with T469 and Q497 side chains, while its central aromatic ring establishes a π−π interaction with W493 (Figure 5H, bottom right). None of these interactions are possible for BPK-25 (Figure 5H, top right), which is reflected in a lower docking score.
• Flexible side chain covalent docking on TMEM173
In order to provide a structural insight of the direct labeling of C91 of the protein TMEM173 with ligands BPK-21 and BPK-25, we applied flexible side chain covalent docking method on a low-resolution Cryo-EM structure (PDB 6NT5, res. 4.1 Å). Results show that both BPK-21 and BPK-25 engage the pocket by establishing a hydrogen bond with the carbonyl oxygen of the amide group and R86 (Figure S4I), while the rest of the molecule is involved in hydrophobic interactions. Despite their structural differences, both ligands occupy the pocket by establishing mostly hydrophobic interactions in a very similar manner, in agreement with the comparable reported efficacy.
Generation of reference protein tables (related to Data S1)
Immune-enriched genes were identified by analyzing microarray data from BioGPS (U133A and MOE430 datasets for human and mouse, respectively) and RNASeq data from GTex (release V7). Briefly, data were first filtered to restrict analyses to microarray signals above 150 and median RPKM values above 10. Samples from each transcriptomic dataset were grouped to identify immune related cells and tissues. Within each group the highest-expressing sample was chosen and group-level values were converted to Z-scores to identify genes showing immune enrichment within each dataset. Immune-enriched Z-scores above 3 or 4 (for RNA-Seq or microarray data, respectively) were summed across all probes and datasets and the summed Z-score was used to rank-order all genes. There were ~2004 genes with a summed Z-score above 11.0 and these were defined as “immune-enriched” as these represented the approximately 10% most-immune-enriched genes in the genome.
Genes with immune-related phenotypes were identified by parsing data in the Online Mendelian Inheritance of Man (OMIM) database (https://www.omim.org). OMIM associations were extracted from the human UniProt database downloaded in February 2019. From the 3925 genes for which human phenotypic associations could be identified, 655 genes with immune-related phenotypes were selected by querying phenotype titles and descriptions for immune-related substrings (‘*immun*’, ‘*inflam*’, ‘*rheum*’, ‘*psoria*’, etc).
Immune-relevant genes were defined as those genes that were immune-enriched and/or associated with immune-related phenotypes, as described above. In total there were 2476 genes that met this criteria (Data S1).
• T cell proliferation gene list (SLICE) (Shifrut et al., 2018)
Hits (genes with FDR < 0.2 and |Z score| > 2, authors’ criteria) were taken from the manuscript by Shifrut et al.(Shifrut et al., 2018) and cross-referenced with cysteine ligandability profiling data (Data S1) based on UniProt accessions.
• Immune module lists (Rieckmann et al., 2017)
Genes corresponding to immune modules were taken from Data S1 from the manuscript by Rieckmann et al.(Rieckmann et al., 2017) and cross-referenced with cysteine ligandability profiling data (Data S1) based on UniProt accessions.
• Transcription Factors
The list of putative transcription factors was adapted from the GSEA website (http://software.broadinstitute.org/gsea/msigdb/gene_families.jsp). (Messina et al., 2004)
• Adaptors and Scaffolding Proteins
To generate a list of putative adaptor and scaffolding proteins we combined data from several different sources including GO (Ashburner et al., 2000; The Gene Ontology, 2019), UniProt (UniProt, 2019), the scaffold protein database ScaPD (Han et al., 2017), manual literature review, and a reagent list from R&D Biosystems (Adaptor Proteins Research Areas: R&D Systems https://www.rndsystems.com/research-area/adaptor-proteins (accessed Sep 4, 2019)). Proteins associated with following GO terms were included: GO:0035591 (signaling adaptor activity), GO:0060090 (molecular adaptor activity), GO:0008093 (cytoskeletal adaptor activity), GO:0035615 (clathrin adaptor activity). Lists of proteins for these GO terms were downloaded from the Gene Ontology project website using the AmiGO tool (http://amigo.geneontology.org/amigo/; version 2.5.12)(Carbon et al., 2009) with filters requiring that entries were of the type “protein” belonging to the “Homo sapiens” organism. Additionally, a keyword search was performed on a downloaded copy of SwissProt human data from Uniprot. Data was queried using BioPython (Cock et al., 2009) and the following search terms were used: “adapter”, “adaptor”, and “scaffold” and the search was performed against the following columns: comments prefixed with “FUNCTION”, associated GO term cross-references, keywords, and entry descriptions. The data that was used from the ScaPD database consisted of experimentally verified scaffold proteins.
The resulting list was then cross-referenced with cysteine ligandability profiling data (Data S1), and the categorization of every target protein was reviewed manually.
• SwissPalm list
SwissPalm proteins and sites (Release 3 (06/28/2019))(Blanc et al., 2015) were downloaded and cross-referenced with the cysteine ligandability profiling data (Data S1) based on UniProt accessions. Sites were deemed a match if any of the cysteine residues in the detected tryptic peptide matched the SwissPalm reference (Data S1). These lists are likely an underrepresentation of the full extent of palmitoylated proteins (especially in immune cells) and this remains an active area of research.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was preformed using GraphPad Prism version 7.03 for Windows (GraphPad Software, La Jolla California USA, www.graphad.com). Statistical values including the n and statistical significance are also reported in the Figure Legends. Statistical significance of western Blotting, ELISA, and FACS quantification data was defined as p < 0.05 and determined by two-tailed unpaired t test with Welch’s correction (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001) compared to respective control treatment groups.
Supplementary Material
Figure S1. Chemical proteomic map of cysteine reactivity changes in activated T cells. Related to Figure 1.
(A) Principal component analysis (PCA) of protein expression profiles in naïve, expanded, and activated T cells. Different cell states are indicated by colors in the legend, with each independent replicate shown separately (4-8 replicates from 4 independent donors per group). Reactome pathways that were enriched (Benjamini-Hochberg corrected p-values ≤ 0.05) for each principal component as determined by Perseus are listed in Data S1.
(B) Bar graph representation of proteins with cysteine reactivity changes organized by molecular function GO terms. OR – oxidoreductase.
(C-E) Representative cysteine reactivity changes in activated human T cells organized by functional categories. Dashed line marks unchanged R value for act vs cntrl cells, and horizontal black lines for each protein measurement mark average R value for the quantified peptides (excluding the reactivity-changing cysteine(s)) from that protein. Green dots represent act/ctrl expression value for proteins in TMT-exp experiments. (C) cysteines at protein-protein interaction (PPI) surfaces. (D) Top: cysteines at DNA/RNA-binding sites; bottom: cryo-EM structure of human ribonuclease P RPP30 (PDB: 6AHU) bound to mature tRNA with the reactivity-changing cysteine C225 highlighted in blue. (E) Cysteines in cofactor/metabolite-binding sites; x-ray crystal structure of the active form of human origin recognition complex subunit 1 (ORC1) in complex with ATP and Mg2+ (PDB: 5UJ7) with the reactivity-changing cysteine C506 highlighted in blue (top left image); x-ray crystal structure of human 3’-phosphoadenosine-5’-phosphosulfate synthetase 1 (PAPSS1) in complex with ADP (PDB: 1X6V) with the reactivity-changing cysteine C165 highlighted in blue (top right image); x-ray crystal structure of human glucose-6-phosphate dehydrogenase (G6PD) in complex with NADP+ (PDB: 2BH9) with the reactivity-changing cysteine C385 highlighted in blue (bottom image).
Figure S2. Chemical proteomic map of fragment electrophile-cysteine interactions in human T cells. Related to Figure 2.
(A) Extended experimental workflow for chemical proteomic experiments measuring scout fragment-cysteine interactions in primary human T cells using isobaric tandem mass tags for mass differentiation and MS3-based quantification (TMT-ABPP).
(B) Extended experimental workflow for chemical proteomic experiments measuring scout fragment electophile effects on cysteine reactivity in primary human T cells using clickable, TEV protease-sensitive, isotopically labeled tags for mass differentiation and MS1-based quantification (isoTOP-ABPP).
(C, D) Comparison of R-values from isoTOP-ABPP and TMT-ABPP experiments, as displayed in correlation plot (C) and bar graph (D) analyses. Results represent mean R-values derived from 3-5 independent isoTOP-ABPP experiments and 4 independent TMT-ABPP experiments (6 TMT channels per experiment) for each compound treatment and proteomic fraction (2-5 biological donors for each method). A cysteine was required to be quantified in at least two experiments for each compound treatment or proteomic fraction group. KB02-treated soluble proteome samples are used as an example for the correlation plot. For (C) and (D), data points with R(TMT-ABPP) ≥5 are colored in blue with the exception of rare outlier cases that also exhibited R(isoTOP-ABPP) <5 and R(isoTOP-ABPP)<1/2R(TMT), which are colored in red.
(E) Bar graphs showing the total number of quantified peptides in isoTOP-ABPP and TMT-ABPP experiments for soluble and particulate proteomic fractions of primary human T cells. Results represent a combination of data from KB02 and KB05 experiments (500 μM, 1 h) with both control and activated T cells. R-values within each experimental treatment group were derived from 3-5 independent isoTOP-ABPP experiments and 4 independent TMT-ABPP experiments. A cysteine was required to be quantified in at least two experiments for each compound treatment or proteomic fraction group to be reported.
(F) Bar graph showing types of pathogenic mutations in liganded proteins with immune phenotypes (OMIM).
(G) Fraction of proteins with pathogenic missense mutations and immune phenotypes (OMIM) for which crystal structures are available. Crystal structures that contain at least one liganded cysteine and pathogenic missense mutation are shown in green. MM – missense mutations.
(H) Bar graph showing distance of pathogenic missense mutations from liganded cysteines. For cases with distances of <15 Å, the number of liganded cysteines is cumulatively as distance increases.
(I) Location of the liganded cysteine C346 (blue) in the immune-relevant kinase ZAP70 and pathogenic missense mutations (yellow – mutation within 5 Å of C346; red – other mutations). PDB: 4K2R.
(J) Fraction of liganded proteins from total proteins encoded by T cell proliferation genes (Shifrut et al., 2018).
(K) Fraction of liganded proteins from total proteins found in previously described immune-enriched modules (Rieckmann et al., 2017).
Figure S3. Analysis of cysteine ligandability in immune signaling pathways. Related to Figure 3.
(A) Cysteine ligandability analysis of the enriched GO terms for proteins undergoing reactivity (top) or expression (bottom) changes in activated T cells. Enriched terms were passed through REVIGO, which identifies redundant terms and chooses representative terms for each group (See STAR Methods for details). Red sub-bars on right graphs represent both percentage and total number of proteins in each GO-term category having one or more liganded cysteines. Top-10 enriched biological processes are shown for the expression changes group.
(B) Venn diagram showing overlap between proteins quantified by TMT-exp and proteins liganded with scout fragments.
(C) Cysteine quantification events in experiments with scout fragments and elaborated compounds with the following colors used to mark proteins with no (grey), one (yellow), two (brown), or > two (orange) quantified cysteines.
(D) Location of liganded cysteines C59 (active site) and C406 (non-active site) in the immune-relevant kinase CHUK (PDB: 5EBZ).
(E) Location of the liganded cysteine C313 in IRF9 (mouse orthologous residue to human C319) at the site of its protein-protein interaction with STAT2 (PDB: 5OEN).
Figure S4. Analysis of active compound effects in human T cells. Related to Figures 4 and 5.
(A) Flow cytometry analysis of T cell activation. T cell activation was monitored by measuring CD25 expression levels after overnight stimulation with αCD3 and αCD28 antibodies in the presence of active compounds. The following FACS gating strategy was used: 1) Doublet discrimination was achieved by plotting forward scatter area (x-axis) versus side scatter width (y-axis) and selecting the area containing singlets for further analysis; 2) Live cells were selected by plotting forward scatter area (x-axis) versus Live/Dead Near-IR channel (y-axis, area, logarithmic scale) and gating on the negative cell population; 3) CD4+ cells were differentiated from CD8+ cells by plotting Pacific Blue channel (x-axis, area, logarithmic scale, PB-CD4 antibody) versus PerCP-Cy5.5 channel (y-axis, area, logarithmic scale, PerCP-Cy5.5-CD8a antibody). CD4+ positive, CD8+ negative cells were further analyzed for CD25 expression levels by measuring mean fluorescence intensity of the PE-channel (PE-CD25). Similar results were obtained by gating on the CD8+ positive, CD4+ negative cell population. BPK-25-treated cells are used as an example for the gating strategy. Representative histogram showing CD25 levels in unstimulated T cells (red), stimulated T cells in the presence of DMSO (light blue), and stimulated T cells in the presence of active compounds (orange) is also presented. Data is representative of three biological replicates used in Figure 4D.
(B) Concentration-dependent effects of EV-96 and EV-97 on T-cell activation parameters (left graphs: IL-2 and IFN-γ) and cytotoxicity (right graph) (24 h treatment). Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 4-5/group. *, p < 0.05; **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to EV-97 (0.5 μM) treatment group.
(C) Bar graph showing the total number of quantified (black) and liganded (R≥4, red) cysteines in cells treated with active compounds. Results are obtained by combining isoTOP-ABPP and TMT-ABPP data for both soluble and particulate proteomic fractions. R-values within each experimental treatment group were derived from 3-6 independent isoTOP-ABPP experiments and 2-3 independent TMT-ABPP experiments (4-6 TMT channels). A cysteine was required to be quantified in at least two experiments for each proteomic fraction to be reported.
(D) Representative bar graph showing the total number of liganded proteins as relates to the corresponding number of quantified (blue) and liganded (R≥4, red) cysteines per protein for BPK-25 treatments. Results are obtained by combining isoTOP-ABPP and TMT-ABPP data for both soluble and particulate proteomic fractions for each compound treatment. A cysteine was required to be quantified in at least two experiments for each proteomic fraction to be reported.
(E) Fraction of protein targets of active compounds with available crystal structures containing the corresponding liganded cysteine residues.
(F) Modeling of BPK-21 and BPK-25 interactions with C91 of stimulator of interferon genes protein (STING, or TMEM173).
(G) IRF response of THP1-Lucia™ ISG cells. THP-1 Lucia ISG cells were treated with DMSO or BPK-25 in the presence of viral dsDNA (2 μg/mL) for 24h and the levels of IRF-induced Lucia luciferase were determined using QUANTI-Luc™. Cell viability was measured using CellTiter-Glo™ assay. Luminescence signals for test samples were normalized to DMSO-treated samples and reported as relative light units (RLU) ± SEM; n = 3/group.
(H) Effect of BPK-25 treatment on gene expression related to TMEM173/STING pathway activation in PBMCs. PBMCs were treated with DMSO or BPK-25 (10 μM) for 5 h and stimulated with cGAMP (2 μM) for 2 h. Relative expression of IL-6, IL-1β, and IP10 (CXCL10) genes was measured by qPCR and normalized to actin.
(I) Effect of BPK-25 treatment on secretion of cytokines related to TMEM173/STING pathway activation in PBMCs. PBMCs were treated with DMSO or BPK-25 (10 μM) for 5 h and stimulated with cGAMP (10 μM) for 20 h. Cytokine levels (IFN-β, IP-10 (CXCL10), IL-6, MCP-1, TNFα) were measured using standard ELISA and Bio-Plex protocols (see STAR Methods for details). Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group. *, p < 0.05; **, p < 0.01 by two-tailed unpaired t test with Welch’s correction between indicated groups.
Figure S5. Functional analysis of protein targets of active compounds in human T cells. Related to Figure 6.
(A-B) Effects of active compounds on NFAT pathway activation. (A) Western blot analysis of NFATc2 content and phosphorylation in T cells treated with active (EV-3 (10 μM), BPK-21 (20 μM), BPK-25 (10 μM) (EV-96 (5 μM)) and control (EV-97 (5 μM)) compounds for 4 h in stimulated T cells. The anti-NFATc2 antibody recognizes both phosphorylated (upper band) and dephosphorylated (lower band) NFATc2. (B) Bar graph representation of western blot data showing compound effects on p-NFATc2 (green) and NFATc2 (red) content. Protein band intensity calculations were made using ImageJ software and are presented as the band intensity ± SEM; n = 3-4/group. *, p < 0.05 by two-tailed unpaired t test with Welch’s correction compared to NFATc2. p-NFATc2 levels were normalized using a mean (p-NFATc2/NFATc2) ratio for DMSO-treated controls to account for the differences between p-NFATc2 and NFATc2 in DMSO (stim) control. Dashed line shows 50% of mean signal intensities for dephosphorylated NFATc2 in DMSO (stim) control.
(C-D) Effects of active compounds on MAPK pathway. (C) Western blot data showing compound effects on p-ERK1/2 (T202/Y204) content. (D) Bar graph representation of western blot data showing the compound effects on p-ERK1/2 (T202/Y204) content. All treatments were done for 24 h. Protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO treated control cells ± SD; n = 3-5/group.
(E) Effects of active compounds versus the GCLC inhibitor BSO on glutathione (GSH) content and T cell activation parameters. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3-5/group. *, p < 0.05; **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to BSO (10 μM) treatment (IL-2 and IFN-γ) or unstimulated control (GSH).
(F) Structure of immunosuppressive natural product triptolide (left) and MS1 signal intensities (right) for ERCC3_C342 in isoTOP-ABPP experiments of expanded T cells treated with BPK-21 (20 μM, 3 h), BPK-25 (10 μM, 3h), or triptolide (0.2 μM, 3 h).
(G) T cell activation and cytotoxicity profile of the immunosuppressive natural product triptolide. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group.
(H) Flow cytometry analysis of T cell activation following ERCC3 gene disruption by CRISPR/Cas9 genome editing. T cells were activated for 2 days prior to Cas9 RNP transfection and were then cultured in IL-2 containing RPMI media to return the cells to a quiescent state. Seven days post-transfection, T cells were stimulated overnight with αCD3 and αCD28 antibodies in the presence of DMSO or BPK-21 with the activation monitored by measuring CD25 and CD69 expression levels. The following FACS gating strategy was used: 1) Lymphocyte gating; 2-3) Doublet discrimination by plotting forward scatter height (x-axis) versus forward scatter width (y-axis) and side scatter height (x-axis) versus side scatter width (y-axis); 4) Live cells were selected by plotting APC-Cy7 channel (x-axis, area, logarithmic scale, eBioscience™ Fixable Viability Dye eFluor™ 780) versus side scatter area (y-axis) and gating on the negative cell population; 5) Transfected cells were selected by gating on FITC-positive cell population (GFP-positive cells). GFP-positive cells were further analyzed for CD25 and CD69 expression levels by measuring mean fluorescence intensity of the PE- (PE-CD25) and APC-channels (APC-CD69). Representative histograms showing CD25 levels in stimulated T cells in the presence of scrambled sgRNA control (light blue) or ERCC3 guide RNAs (green) in the presence of DMSO or BPK-21 (orange) are presented. Data is representative of a total of six replicate treatments used in Figures S5I and S5J.
(I) Effect of genetic disruption of representative targets of active compound BPK-21 by CRISPR/Cas9 genome editing on T cell activation. Target disruption was considered to have an effect if T cell activation was suppressed >33% with a p value < 0.01. Data are presented as the mean percentage of control guide sgRNA-treated control cells ± SEM; n = 6/group. **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to control guides.
(J) Genetic disruption of ERCC3 and BPK-21 treatment produce similar degrees of blockade of T cell activation. Data are presented as the mean percentage of control sgRNA-treated control cells ± SEM; n = 6/group. **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to control guides.
(K) Efficiency of CRISPR/Cas9 genome editing. Left: western blot analysis showing ERCC3 protein content after CRISPR/Cas9 genome editing using control or ERCC3 sgRNA and sorting for GFP-positive T cells. Right: Quantitation of western blot data, where protein band intensity calculations were made using ImageJ software and represent the mean percentage of control guide sgRNA-treated cells ± SD; n = 2/group.
Figure S6. Functional analysis of protein targets of active compounds in human T cells. Related to Figure 6.
(A) MS1 signal intensities for BIRC3_C28, BIRC3_C164, and BIRC2_C45 in isoTOP-ABPP experiments of expanded T cells treated with EV-3 (10 μM, 3 h) or DMF (50 μM, 3 h).
(B) Bar graph representation of western blot data shown in Figure 6E, where protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO treated control ± SEM; n = 2-5 independent experiments per group. **p < 0.01; ****, p < 0.0001 by two-tailed unpaired t test with Welch’s correction compared to EV-3 (3h).
(C, D) EV-3 causes loss of BIRC2 and BIRC3 in human T cells. (C) Full western blot from Figure 6E showing reductions in BIRC2 and BIRC3 content in human T cells treated with EV-3 (1-10 μM, 24 h) and the blockade of EV-3-induced loss of BIRC2 and BIRC3 by co-treatment with the proteasome inhibitor MG132 (10 μM). (D) Western blot showing time-dependent reductions in BIRC2 and BIRC3 content in human T cells treated with EV-3 (10 μM).
(E) mRNA content for BIRC2 and BIRC3 quantified by RNA-sequencing of T cells treated with EV-3 (10 μM), DMF (50 μM), AT-406 (1 μM), BPK-25 (10 μM), or BPK-21 (20 μM). All treatments were done with T cells for 24 h. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group.
(F) Effect of genetic disruption of representative targets of active compound EV-3 by CRISPR/Cas9 genome editing on T cell activation. Target disruption was considered to have an effect if T cell activation was suppressed >33% with a p value < 0.01. Data are presented as the mean percentage of control guide sgRNA-treated control cells ± SEM; n = 3/group. **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to control guides.
(G) Flow cytometry analysis of T cell activation following BIRC2, BIRC3, or BIRC2 and BIRC3 gene disruption by CRISPR/Cas9 genome editing. T cells were activated for 2 days prior to Cas9 RNP transfection and were then cultured in IL-2 containing RPMI media to return the cells to a quiescent state. Seven days post-transfection, T cells were stimulated overnight with αCD3 and αCD28 antibodies in the presence of DMSO or BPK-21 with the activation monitored by measuring CD25 and CD69 expression levels. The same FACS gating strategy as in Figure S5H was used. Representative histograms showing CD25 levels in stimulated T cells in the presence of scrambled sgRNA control (light blue) or BIRC2/BIRC3/BIRC2+BIRC3 guide RNAs (green) in the presence of DMSO or EV-3 (orange) are presented. Data is representative of a total of six replicate treatments used in Figure 6H and S6F.
(H) Efficiency of CRISPR/Cas9 genome editing. Left: western blot analysis of BIRC2 and BIRC3 protein content using control, BIRC2, or BIRC3 sgRNA and sorting for GFP-positive T cells. Right: Quantitation of western blot data, where protein band intensity calculations were made using ImageJ software and represent the mean percentage of control guide sgRNA-treated cells ± SD; n = 2/group.
(I) Quantification of BPK-25 concentration-dependent changes in protein expression. Western blot protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO treated control ± SD; n = 2.
(J) Active compound treatment did not, in general, reduce the mRNA content for NuRD complex proteins. mRNA quantities were measured by RNAseq. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group.
(K) BPK-25-ctrl, a non-electrophilic propanamide analogue of BPK-25, does not inhibit T-cell activation. Left: Structure of BPK-25. Middle: Structure of BPK-25-ctrl. Right: T-cell activation (CD25) and cytotoxicity profiles for BPK-25 and BPK-25-ctrl (10 μM of each compound, 24 h). Data are presented as the mean percentage of DMSO-treated control cells ± SD; n = 2/group.
(L) Western blot data showing that BPK-25-ctrl, does not alter protein content for NuRD complex members.
Figure S7. Characterization of protein targets of EV-96 in human T cells. Related to Figure 7.
(A) Left: Bar graph showing fraction of total targets that show enantioselective (ES) engagement with one of the stereoisomeric compounds EV-96, EV-97, EV-98, and EV-99 at either 5 μM or 20 μM test concentrations in T cells (3 h). A target was considered enantioselective if it was engaged by one of the stereoisomers with an R value ≥4 and showed a reduction in engagement by at least 50% with the corresponding enantiomeric compound. Middle: Bar graph showing the percentage of enantioselective targets that were also liganded by scout fragments. Data for enantioselective (ES) and non-enantioselective (other) targets are shown separately. Right: Fraction of enantioselective targets that are immune-relevant (immune-enriched (blue) and/or have human genetics-based immune phenotypes (green)).
(B) Enantioselective engagement of C449 of TEC kinase by EV-96. Bar graphs showing percent reductions in IA-DTB labeling of C449 of TEC in T cells treated with the indicated concentrations of stereoisomeric electrophiles (3 h treatment; left: 5 μM, right: 20 μM), as quantified by TMT-ABPP experiments. Results represent mean values ± SD; n = 2-4/group.
(C) Western blot results showing enantioselective reductions in ITK protein and PLCG1 phosphorylation (Y783) in stimulated T cells treated with EV-96 (5 μM) compared to DMSO- or EV-97 (5 μM), EV-98 (5 μM), and EV-99 (5 μM)-treated T cells, measured at the indicated time points. Results are from one experiment representative of 2-5 independent experiments.
(D) Quantitation of western blot data showing effects of EV-96 and EV-97 on ITK protein content in naïve control (ctrl) versus αCD3/CD28-treated (stimulated, stim) T cells and the blockade of EV-96-induced loss of ITK by co-treatment with the proteasome inhibitor MG132 (10 μM). Cells were treated with compounds (5 μM each) or DMSO control for 8 h. Protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO-treated control ± SD; n = 2-5 independent experiments per group. **p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to DMSO-treated stim control.
(E) Western blot showing time-dependent decrease in ITK protein content in EV-96-treated αCD3/CD28-stimulated (Stim), but not expanded control (Ctrl) T cells. Cells treated with DMSO or EV-96 (5 μM) for 1–8 h. Results are from a single experiment representative of 2-4 independent experiments.
(F) EV-96 does not block the kinase activity of recombinant, purified ITK protein, as measured by the ADP-Glo™ Kinase Assay (Promega) following manufacturer’s instructions. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group. ***p < 0.001 by two-tailed unpaired t test with Welch’s correction compared to EV-96(100 μM)-treated control. *PF – PF-06465469, an irreversible ITK inhibitor that reacts with C442.
(G) Western blot results showing that EV-96 (5 μM, 2-8 h) does not impair the phosphorylation of SLP-76 (S376) in stimulated (Stim) T cells.
(H) EV-96-ctrl, a non-electrophilic propanamide analogue of EV-96, does not inhibit T-cell activation. Left: Structure of EV-96-ctrl. Right: T-cell activation and cytotoxicity profiles for EV-96 and EV-96-ctrl (5 μM, 24 h). Data are presented as the mean percentage of DMSO-treated control cells ± SD; n = 2-5/group.
(I) Western blot results showing that EV-96-ctrl does not alter ITK protein content in stimulated T cells. Cells were treated with compounds (5 μM each) or DMSO for 8 h. Results are from one experiment representative of 2 independent experiments.
(J) Western blot results showing that pre-treatment (1 h) with EV-97 (5 μM) does not block EV-96-dependent decreases in ITK protein content in stimulated T cells. After 1 h pre-treatment, cells were treated with EV-96 (5 μM) or DMSO for 8 h.
(K) Structure of PF-06465469, an irreversible ITK inhibitor that reacts with C442 (left) and the corresponding crystal structure of a PF-06465469-ITK complex showing covalent modification of C442 (red).
Methods S1 (related to STAR Methods)
Glossary of Key Terms used in the manuscript (TMT-ABPP, isoTOP-ABPP, TMT-exp, scout fragment electrophiles, elaborated electrophiles, stereoisomeric electrophiles).
Methods S2 (Related to Figure 4)
Synthetic procedures and characterization of the elaborated small-molecule electrophiles tested in the inhibition of T cell activation screen.
Methods S3 (related to STAR Methods)
List of oligos used for T7 sgRNA production and for BIRC2_C45A and BIRC3_C28A mutagenesis studies.
Data S1 (related to Figures 1–7)
Tab 1. Table of contents for Data S1.
Tab 2 (Table S1, related to Figure 1 and S1A). Reactome pathways that were enriched (Benjamini-Hochberg corrected p-values ≤ 0.05) for each principal component as determined by Perseus. The size column refers to the size of the pathway in Reactome. Pathways colored in red are enriched in both principal components (albeit with different p-values), whereas uncolored pathways are enriched selectively in one or the other component.
Tab 3 (Table S2, related to Figure 1). TMT-exp proteomic data showing changes in protein expression in control vs activated primary human T cells.
Tab 4 (Table S3, related to Figure 1). TMT-ABPP proteomic data showing changes in cysteine reactivity in control vs activated primary human T cells.
Tab 5 (Table S4, related to Figures 1, 2, 3, and 5). List of proteins with identified cysteine reactivity changes, along with designations of immune-relevance and GO-terms: “RNA-binding”, “Metal ion binding”, “DNA binding”, “disulfide oxidoreductase activity”, and “ATP binding”. The summary of GO-term analysis is provided in Figure S1B.
Tab 6 (Table S5, related to Figure 1). Resource table for determination of immune-relevant genes. Multiple Z-scores were calculated from several transcriptomic tissue profiles, and these Z-scores were summed to generate the value in column H. Genes with summed Z-scores in the top ~10% were considered “immune-enriched”, which corresponded to Z scores > 11. Annotation of immune phenotype (column T) was performed by parsing data in the Online Mendelian Inheritance of Man (OMIM) database and querying phenotype titles and descriptions for immune-related substrings (‘*immun*’, ‘*inflam*’, ‘*rheum*’, ‘*psoria*’, etc), as described in the STAR Methods. In total, there were 2004 immune-enriched genes and 655 genes associated with immune phenotypes which, when combined, result in 2746 total immune-relevant genes (column U).
Tab 7 (Table S6, related to Figures 2 and 5). Master table containing total cysteine reactivity proteomic data from combined isoTOP-ABPP and TMT-ABPP experiments with scout fragments (“broad ligandability”, KB02 and KB05), active compounds (DMF, EV-3, EV-93, BPK-21, BPK-25, EV-96), and inactive stereoisomers (EV-97, EV-98, EV-99), as well as isoTOP-ABPP hyperreactivity experiments. Values for each treatment group are final average values resulting from at least 2 replicate experiments described in STAR Methods. Maximum values from all treatment groups (Columns J and P) for active compounds and scout fragments are used to determine “ligandability” of a select residue (related to Figure 2).
Tabs 8-9 (Tables S7, S8, related to Figure 2). Comparison of isoTOP-ABPP and isoTOP-ABPP hyperreactivity R values for activated and control T cells (related to Figure 2E).
Tab 10 (Table S9, related to Figure 2). List of liganded cysteines in immune-relevant proteins that are also targeted by other electrophile compounds.
Tab 11 (Table S10, related to Figures 2 and S2). Analysis of missense pathogenic mutations and their distance from liganded cysteines in proteins with immune phenotype (OMIM). Only proteins containing crystal structures with a liganded cysteine and at least one pathogenic missense mutation are included.
Tab 12 (Table S11, related to Figures 2 and S2). Analysis of proteins liganded with scout fragments (Table S6) for assignment to T cell proliferation genes (related to Figure S2K) (Shifrut et al., 2018).
Tab 13 (Table S12, related to Figures 2 and S2). Analysis of liganded and quantified proteins for assignment to previously described immune-enriched modules (modules 10, 17, 19, 23, 27, related to Figure S2L). (Rieckmann et al., 2017).
Tab 14 (Table S13, related to Figures 3). Ligandability analysis of Reactome pathways. All proteins for a specified pathway with at least one liganded cysteine are listed in column H, proteins with at least one quantified cysteine, but without liganded cysteines are listed in column I. All members for each pathway are listed in column J.
Tab 15 (Table S14, related to Figures 3). List of immune-enriched transcription factors and adaptor proteins liganded with scout fragments (related to Figure 3E). See STAR Methods for details.
Tab 16 (Table S15, related to Figure 4). Chemical structures of compounds used in the inhibition of T cell activation screen.
Tab 17 (Table S16, related to Figure 4). Summary of the results of the phenotypic screen for inhibition of T cell activation.
Tab 18 (Table S17, related to Figure 5). isoTOP-ABPP and TMT-ABPP proteomic data for active compounds (DMF, EV-3, EV-93, BPK-21, BPK-25, EV-96 and inactive stereoisomers EV-97, EV-98, EV-99). Values for each treatment group are final average values resulting from at least 2 replicate experiments described in STAR Methods. For each compound, maximum values across both methods and proteomic fractions are reported in the Master table (Tab 6, Table S6).
Tab 19 (Table S18, related to Figure 5). Distribution of protein classes containing cysteines liganded by active compounds (Figure 5C).
Tab 20 (Table S19, related to Figure 5). Comparison of isoTOP-ABPP and TMT-ABPP R values for cysteines liganded by active compounds versus scout fragments in human T cells, as displayed in correlation plot (Figure 5D) and pie chart (Figure 5E) analyses.
Tab 21 (Table S20, related to Figure 5). Prediction of pocket volumes within the indicated distances from cysteines liganded by active compounds (related to Figure 5F).
Tab 22 (Table S21, related to Figures 2 and 5). Liganded cysteine residues with scout fragments at known sites of palmitoylation (obtained by cross-referencing with SwissPalm proteins and sites).
Tab 23 (Table S22, related to Figure 6). Protein expression changes after BPK-25 (10 μM, 24 h) treatment as determined by TMT-exp (10 replicates from 5 donors, related to Figure 6J).
Tab 24 (Table S23, related to Figure 6). Summary of protein expression changes after DMF (50 μM, 24 h), EV-3 (10 μM, 24 h), BPK-21 (20 μM, 24 h), BPK-25 (10 μM, 24 h), and BPK-25 (10 μM, 24h) + MG132 (10 μM, 24 h) treatment as determined by TMT-exp (average values from 2-5 donors are reported, related to Figure 6K).
Tab 25 (Table S24, related to Figure 7). Summary of protein expression changes in human T cells treated with DMSO, EV-96, or EV-97 with or without αCD3/αCD28 stimulation as determined by TMT-exp (average values from 6 replicate treatments performed with cells from two donors (4 and 2 for each donor) are reported, related to Figure 7C).
Data S2 (Related to Figure 6)
RNA-sequencing data for primary human T cells treated with DMF (50 μM, 24 h), EV-3 (10 μM, 24 h), AT406 (1 μM, 24 h), BPK-21 (20 μM, 24 h), and BPK-25 (10 μM, 24 h).
Highlights:
Chemical proteomics identifies cysteine reactivity changes in activated T cells
Chemical proteomics maps ligandable cysteines in diverse immune-relevant proteins
Cysteine-directed electrophilic compounds suppress T cells by diverse mechanisms
Electrophile-cysteine interactions promote the degradation of immune proteins
ACKNOWLEDGEMENTS
We thank D. Tallman for technical assistance, B. Lu for help with Table S4, and M. Niphakis for helpful discussions. This work was supported by NIH CA231991 (B.F.C.), NIH-NCI K99CA248715 (X.Z.), CA212467 (V.M.C.), CA211526 (M.M.D.), GM069832 (S.F.), a Clinical Translational Science Award (UL1 TR001114), Damon Runyon Cancer Research Foundation DRG-2341-18 (X.Z.), the Life Sciences Research Foundation (E.V.V.), and Vividion Therapeutics. M.N.S. was supported by funding from NIH-NCI CCSG: P30 014195, R01 GM102491-07, and the Helmsley Trust. D.B.K. received funding from the European Union’s Framework Programme for Research and Innovation Horizon 2020 (2014-2020).
Footnotes
DECLARATION OF INTERESTS
Dr. Cravatt is a founder and scientific advisor to Vividion Therapeutics. Drs. Cravatt, Crowley, Melillo, Remillard, Schafroth, Vinogradova, Zhang, and Mr. Yokoyama are co-inventors on a patent application related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Acuto O, and Cantrell D (2000). T cell activation and the cytoskeleton. Annu Rev Immunol 18, 165–184. [DOI] [PubMed] [Google Scholar]
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, and Honjo T (1996). Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. International immunology 8, 765–772. [DOI] [PubMed] [Google Scholar]
- Almeida L, Lochner M, Berod L, and Sparwasser T (2016). Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol 28, 514–524. [DOI] [PubMed] [Google Scholar]
- Andreotti AH, Joseph RE, Conley JM, Iwasa J, and Berg LJ (2018). Multidomain Control Over TEC Kinase Activation State Tunes the T Cell Response. Annu Rev Immunol 36, 549–578. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baecher-Allan C, Kaskow BJ, and Weiner HL (2018). Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 97, 742–768. [DOI] [PubMed] [Google Scholar]
- Baillie TA (2016). Targeted Covalent Inhibitors for Drug Design. Angew Chem Int Ed Engl 55, 13408–13421. [DOI] [PubMed] [Google Scholar]
- Bar-Peled L, Kemper EK, Suciu RM, Vinogradova EV, Backus KM, Horning BD, Paul TA, Ichu TA, Svensson RU, Olucha J, et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 171, 696–709 e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, Orchard S, Magrane M, Alpi E, Bely B, Bingley M, Britto R, Bursteinas B, Busiello G, et al. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 57, 289–300. [Google Scholar]
- Bianco G, Forli S, Goodsell DS, and Olson AJ (2016). Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci 25, 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, David F, Abrami L, Migliozzi D, Armand F, Burgi J, and van der Goot FG (2015). SwissPalm: Protein Palmitoylation database. F1000Res 4, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, and Cravatt BF (2016). Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci Signal 9, rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, and Crews CM (2015). HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol 10, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, et al. (2018). The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol 25, 67–-77 e63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Sun H, Peng Y, Lu J, Nikolovska-Coleska Z, McEachern D, Liu L, Qiu S, Yang CY, Miller R, et al. (2011). A potent and orally active antagonist (SM-406/AT-406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem 54, 2714–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, Ami GOH, and Web Presence Working G (2009). AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WT, Kang JJ, Lee KY, Wei K, Anderson E, Gotmare S, Ross JA, and Rosen GD (2001). Triptolide and chemotherapy cooperate in tumor cell apoptosis. A role for the p53 pathway. J Biol Chem 276, 2221–2227. [DOI] [PubMed] [Google Scholar]
- Chen Q, Coffey A, Bourgoin SG, and Gadina M (2006). Cytohesin binder and regulator augments T cell receptor-induced nuclear factor of activated T Cells.AP-1 activation through regulation of the JNK pathway. J Biol Chem 281, 19985–19994. [DOI] [PubMed] [Google Scholar]
- Chylek LA, Akimov V, Dengjel J, Rigbolt KT, Hu B, Hlavacek WS, and Blagoev B (2014). Phosphorylation site dynamics of early T-cell receptor signaling. PLoS One 9, e104240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csomos RA, Brady GF, and Duckett CS (2009). Enhanced cytoprotective effects of the inhibitor of apoptosis protein cellular IAP1 through stabilization with TRAF2. J Biol Chem 284, 20531–20539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Weerapana E, Ulanovskaya O, Sun F, Liang H, Ji Q, Ye Y, Fu Y, Zhou L, Li J, et al. (2013). Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe 13, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, and Rudensky AY (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4, 330–336. [DOI] [PubMed] [Google Scholar]
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, and Olson AJ (2016). Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchina DG, Dostert C, and Brenner D (2018). Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol 39, 489–502. [DOI] [PubMed] [Google Scholar]
- Gao DW, Vinogradova EV, Nimmagadda SK, Medina JM, Xiao Y, Suciu RM, Cravatt BF, and Engle KM (2018). Direct Access to Versatile Electrophiles via Catalytic Oxidative Cyanation of Alkenes. J Am Chem Soc 140, 8069–8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles NM, Watts AB, Giles GI, Fry FH, Littlechild JA, and Jacob C (2003). Metal and redox modulation of cysteine protein function. Chem Biol 10, 677–693. [DOI] [PubMed] [Google Scholar]
- Gyrd-Hansen M, and Meier P (2010). IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer 10, 561–574. [DOI] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, et al. (2018). Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273. [DOI] [PubMed] [Google Scholar]
- Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, Fung HY, Albrecht S, Alexandropoulos K, McCauley D, et al. (2015). Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci 18, 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang J, Wang J, Liu S, Hu J, Zhu H, and Qian J (2017). ScaPD: a database for human scaffold proteins. BMC Bioinformatics 18, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J Cell Biol 217, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemer S, Jatav S, Jussif J, Alley J, Lathwal S, Piotrowski M, Janiszewski J, Kibbey R, Alves T, Dumlao D, et al. (2019). Integrated Metabolomic and Transcriptomic Profiling Reveals Novel Activation-Induced Metabolic Networks in Human T cells. bioRxiv, 635789. [Google Scholar]
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al. (2010). The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A 107, 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida H, Jensen KV, Woodman AG, Hyndman ME, and Vogel HJ (2017). The Calcium-Dependent Switch Helix of L-Plastin Regulates Actin Bundling. Sci Rep 7, 40662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Zhou L, and Littman DR (2007). Transcriptional regulation of Th17 cell differentiation. Semin Immunol 19, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C, Giles GI, Giles NM, and Sies H (2003). Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl 42, 4742–4758. [DOI] [PubMed] [Google Scholar]
- Jassal B, Matthews L, Viteri G, Gong CQ, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Research 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LH (2018). Small-Molecule Kinase Downregulators. Cell Chem Biol 25, 30–35. [DOI] [PubMed] [Google Scholar]
- Jung ID, Lee JS, Jeong YI, Lee CM, Chang JH, Jeong SK, Chun SH, Park WS, Han J, Shin YK, et al. (2009). Apicidin, the histone deacetylase inhibitor, suppresses Th1 polarization of murine bone marrow-derived dendritic cells. Int J Immunopathol Pharmacol 22, 501–515. [DOI] [PubMed] [Google Scholar]
- Klopfenstein DV, Zhang LS, Pedersen BS, Ramirez F, Vesztrocy AW, Naldi A, Mungall CJ, Yunes JM, Botvinnik O, Weigel M, et al. (2018). GOATOOLS: A Python library for Gene Ontology analyses. Sci Rep-Uk 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Gogulamudi VR, Periasamy R, Raghavaraju G, Subramanian U, and Pandey KN (2017). Inhibition of HDAC enhances STAT acetylation, blocks NF-kappaB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am J Physiol Renal Physiol 313, F781–F795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesourne R, Uehara S, Lee J, Song KD, Li L, Pinkhasov J, Zhang Y, Weng NP, Wildt KF, Wang L, et al. (2009). Themis, a T cell-specific protein important for late thymocyte development. Nat Immunol 10, 840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian G, Gnanaprakasam JR, Wang T, Wu R, Chen X, Liu L, Shen Y, Yang M, Yang J, Chen Y, et al. (2018). Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucherini OM, Rigante D, Sota J, Fabiani C, Obici L, Cattalini M, Gattorno M, and Cantarini L (2018). Updated overview of molecular pathways involved in the most common monogenic autoinflammatory diseases. Clin Exp Rheumatol 36 Suppl 110, 3–9. [PubMed] [Google Scholar]
- Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, Binsfeld C, Hao Z, Brustle A, Itsumi M, et al. (2017). Glutathione Primes T Cell Metabolism for Inflammation. Immunity 46, 675–689. [DOI] [PubMed] [Google Scholar]
- Martell J, Seo Y, Bak DW, Kingsley SF, Tissenbaum HA, and Weerapana E (2016). Global Cysteine-Reactivity Profiling during Impaired Insulin/IGF-1 Signaling in C. elegans Identifies Uncharacterized Mediators of Longevity. Cell Chem Biol 23, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurais AJ, and Weerapana E (2019). Reactive-cysteine profiling for drug discovery. Curr Opin Chem Biol 50, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina DN, Glasscock J, Gish W, and Lovett M (2004). An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res 14, 2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortenson DE, Brighty GJ, Plate L, Bare G, Chen W, Li S, Wang H, Cravatt BF, Forli S, Powers ET, et al. (2018). “Inverse Drug Discovery” Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J Am Chem Soc 140, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, and Taguchi T (2016). Activation of STING requires palmitoylation at the Golgi. Nat Commun 7, 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreich KJ, and Weinmann AS (2012). Transcriptional mechanisms that regulate T helper 1 cell differentiation. Curr Opin Immunol 24, 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papalexi E, and Satija R (2018). Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 18, 35–45. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J Mach Learn Res 12, 2825–2830. [Google Scholar]
- Qinheng Z, Jordan L, W., Seiya K, Diogo S-M, Christopher J, S., Gencheng L, Stefano F, John E, M., Dennis W, W., and K. Barry S (2019). “Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery of Selective Covalent Inhibitors of Human Neutrophil Elastase.
- Qiu D, Zhao G, Aoki Y, Shi L, Uyei A, Nazarian S, Ng JC, and Kao PN (1999). Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-kappaB transcriptional activation. J Biol Chem 274, 13443–13450. [DOI] [PubMed] [Google Scholar]
- Ravindranath PA, and Sanner MF (2016). AutoSite: an automated approach for pseudo-ligands prediction-from ligand-binding sites identification to predicting key ligand atoms. Bioinformatics 32, 3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieckmann JC, Geiger R, Hornburg D, Wolf T, Kveler K, Jarrossay D, Sallusto F, Shen-Orr SS, Lanzavecchia A, Mann M, et al. (2017). Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol 18, 583–593. [DOI] [PubMed] [Google Scholar]
- Rip J, Van Der Ploeg EK, Hendriks RW, and Corneth OBJ (2018). The Role of Bruton’s Tyrosine Kinase in Immune Cell Signaling and Systemic Autoimmunity. Crit Rev Immunol 38, 17–62. [DOI] [PubMed] [Google Scholar]
- Roberts AM, Ward CC, and Nomura DK (2017). Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr Opin Biotechnol 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F, and Alexandre J (2019). Dimethyl fumarate, a two-edged drug: Current status and future directions. Med Res Rev 39, 1923–1952. [DOI] [PubMed] [Google Scholar]
- Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, and Reed JC (2006). Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem 281, 1080–1090. [DOI] [PubMed] [Google Scholar]
- Schlicker A, Domingues FS, Rahnenfuhrer J, and Lengauer T (2006). A new measure for functional similarity of gene products based on Gene Ontology. Bmc Bioinformatics 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DE, Coyne AG, Hudson SA, and Abell C (2012). Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971 e1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Fueyo A, Bradley SJ, and Tsokos GC (2016). T cells in Systemic Lupus Erythematosus. Curr Opin Immunol 43, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, and Smuc T (2011). REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. Plos One 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi I, Miyaji H, Yoshida T, Sato S, and Mizukami T (1996). Selective inhibition of IL-2 gene expression by trichostatin A, a potent inhibitor of mammalian histone deacetylase. J Antibiot (Tokyo) 49, 453–457. [DOI] [PubMed] [Google Scholar]
- Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, Wang X, Cho JH, Wang H, Rankin S, et al. (2017). Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 46, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, Moy E, Balbo P, Li W, Zhao Y, et al. (2016). Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem Biol 11, 3442–3451. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology C (2019). The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, et al. (2011). XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol 7, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovell H, Testa A, Maniaci C, Zhou H, Prescott AR, Macartney T, Ciulli A, and Alessi DR (2019). Rapid and Reversible Knockdown of Endogenously Tagged Endosomal Proteins via an Optimized HaloPROTAC Degrader. ACS Chem Biol 14, 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- UniProt C (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisinne G, Kersse K, Chaoui K, Lu L, Chaix J, Zhang L, Goncalves Menoita M, Girard L, Ounoughene Y, Wang H, et al. (2019). Quantitative interactomics in primary T cells unveils TCR signal diversification extent and dynamics. Nat Immunol 20, 1530–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Kiweler N, Wolff K, Knauer SK, Brandl A, Hemmerich P, Dannenberg JH, Heinzel T, Schneider G, and Kramer OH (2015). Sumoylation of HDAC2 promotes NF-kappaB-dependent gene expression. Oncotarget 6, 7123–7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Weerapana E, Blewett MM, and Cravatt BF (2014). A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, and Bosselut R (2009). CD4-CD8 lineage differentiation: Thpok-ing into the nucleus. J Immunol 183, 2903–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xue G, and Pan Z (2020). Design, synthesis and structure-activity relationship of indolylindazoles as potent and selective covalent inhibitors of interleukin-2 inducible T-cell kinase (ITK). Eur J Med Chem 187, 111918. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dix M, Remsberg J, Lee H. y., Kalocsay M, Gygi S, Vite G, Lawrence M, Parker C, and Cravatt B (2019). Expedited Mapping of the Ligandable Proteome Using Fully Functionalized Enantiomeric Probe Pairs. 7764638.v7764631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Speers AE, and Cravatt BF (2007). Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)--a general method for mapping sites of probe modification in proteomes. Nat Protoc 2, 1414–1425. [DOI] [PubMed] [Google Scholar]
- Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SC, Duffy CR, and Allison JP (2018). Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- Word JM, Lovell SC, Richardson JS, and Richardson DC (1999). Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 285, 1735–1747. [DOI] [PubMed] [Google Scholar]
- Xu H, Jesson MI, Seneviratne UI, Lin TH, Sharif MN, Xue L, Nguyen C, Everley RA, Trujillo JI, Johnson DS, et al. (2019). PF-06651600, a Dual JAK3/TEC Family Kinase Inhibitor. ACS Chem Biol 14, 1235–1242. [DOI] [PubMed] [Google Scholar]
- Yang J, Li Y, Yan W, Li W, Qiu Q, Ye H, and Chen L (2019). Covalent modification of Cys-239 in beta-tubulin by small molecules as a strategy to promote tubulin heterodimer degradation. J Biol Chem 294, 8161–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kelly P, Shaffer AL 3rd, Schmitz R, Yoo HM, Liu X, Huang DW, Webster D, Young RM, Nakagawa M, et al. (2016). Targeting Non-proteolytic Protein Ubiquitination for the Treatment of Diffuse Large B Cell Lymphoma. Cancer Cell 29, 494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapf CW, Gerstenberger BS, Xing L, Limburg DC, Anderson DR, Caspers N, Han S, Aulabaugh A, Kurumbail R, Shakya S, et al. (2012). Covalent inhibitors of interleukin-2 inducible T cell kinase (itk) with nanomolar potency in a whole-blood assay. J Med Chem 55, 10047–10063. [DOI] [PubMed] [Google Scholar]
- Zaro BW, Vinogradova EV, Lazar DC, Blewett MM, Suciu RM, Takaya J, Studer S, de la Torre JC, Casanova JL, Cravatt BF, et al. (2019). Dimethyl Fumarate Disrupts Human Innate Immune Signaling by Targeting the IRAK4-MyD88 Complex. J Immunol 202, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Kabaleeswaran V, Wang Y, Cheng G, and Wu H (2010). Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell 38, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J (2010). Transcriptional regulation of Th2 cell differentiation. Immunol Cell Biol 88, 244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chemical proteomic map of cysteine reactivity changes in activated T cells. Related to Figure 1.
(A) Principal component analysis (PCA) of protein expression profiles in naïve, expanded, and activated T cells. Different cell states are indicated by colors in the legend, with each independent replicate shown separately (4-8 replicates from 4 independent donors per group). Reactome pathways that were enriched (Benjamini-Hochberg corrected p-values ≤ 0.05) for each principal component as determined by Perseus are listed in Data S1.
(B) Bar graph representation of proteins with cysteine reactivity changes organized by molecular function GO terms. OR – oxidoreductase.
(C-E) Representative cysteine reactivity changes in activated human T cells organized by functional categories. Dashed line marks unchanged R value for act vs cntrl cells, and horizontal black lines for each protein measurement mark average R value for the quantified peptides (excluding the reactivity-changing cysteine(s)) from that protein. Green dots represent act/ctrl expression value for proteins in TMT-exp experiments. (C) cysteines at protein-protein interaction (PPI) surfaces. (D) Top: cysteines at DNA/RNA-binding sites; bottom: cryo-EM structure of human ribonuclease P RPP30 (PDB: 6AHU) bound to mature tRNA with the reactivity-changing cysteine C225 highlighted in blue. (E) Cysteines in cofactor/metabolite-binding sites; x-ray crystal structure of the active form of human origin recognition complex subunit 1 (ORC1) in complex with ATP and Mg2+ (PDB: 5UJ7) with the reactivity-changing cysteine C506 highlighted in blue (top left image); x-ray crystal structure of human 3’-phosphoadenosine-5’-phosphosulfate synthetase 1 (PAPSS1) in complex with ADP (PDB: 1X6V) with the reactivity-changing cysteine C165 highlighted in blue (top right image); x-ray crystal structure of human glucose-6-phosphate dehydrogenase (G6PD) in complex with NADP+ (PDB: 2BH9) with the reactivity-changing cysteine C385 highlighted in blue (bottom image).
Figure S2. Chemical proteomic map of fragment electrophile-cysteine interactions in human T cells. Related to Figure 2.
(A) Extended experimental workflow for chemical proteomic experiments measuring scout fragment-cysteine interactions in primary human T cells using isobaric tandem mass tags for mass differentiation and MS3-based quantification (TMT-ABPP).
(B) Extended experimental workflow for chemical proteomic experiments measuring scout fragment electophile effects on cysteine reactivity in primary human T cells using clickable, TEV protease-sensitive, isotopically labeled tags for mass differentiation and MS1-based quantification (isoTOP-ABPP).
(C, D) Comparison of R-values from isoTOP-ABPP and TMT-ABPP experiments, as displayed in correlation plot (C) and bar graph (D) analyses. Results represent mean R-values derived from 3-5 independent isoTOP-ABPP experiments and 4 independent TMT-ABPP experiments (6 TMT channels per experiment) for each compound treatment and proteomic fraction (2-5 biological donors for each method). A cysteine was required to be quantified in at least two experiments for each compound treatment or proteomic fraction group. KB02-treated soluble proteome samples are used as an example for the correlation plot. For (C) and (D), data points with R(TMT-ABPP) ≥5 are colored in blue with the exception of rare outlier cases that also exhibited R(isoTOP-ABPP) <5 and R(isoTOP-ABPP)<1/2R(TMT), which are colored in red.
(E) Bar graphs showing the total number of quantified peptides in isoTOP-ABPP and TMT-ABPP experiments for soluble and particulate proteomic fractions of primary human T cells. Results represent a combination of data from KB02 and KB05 experiments (500 μM, 1 h) with both control and activated T cells. R-values within each experimental treatment group were derived from 3-5 independent isoTOP-ABPP experiments and 4 independent TMT-ABPP experiments. A cysteine was required to be quantified in at least two experiments for each compound treatment or proteomic fraction group to be reported.
(F) Bar graph showing types of pathogenic mutations in liganded proteins with immune phenotypes (OMIM).
(G) Fraction of proteins with pathogenic missense mutations and immune phenotypes (OMIM) for which crystal structures are available. Crystal structures that contain at least one liganded cysteine and pathogenic missense mutation are shown in green. MM – missense mutations.
(H) Bar graph showing distance of pathogenic missense mutations from liganded cysteines. For cases with distances of <15 Å, the number of liganded cysteines is cumulatively as distance increases.
(I) Location of the liganded cysteine C346 (blue) in the immune-relevant kinase ZAP70 and pathogenic missense mutations (yellow – mutation within 5 Å of C346; red – other mutations). PDB: 4K2R.
(J) Fraction of liganded proteins from total proteins encoded by T cell proliferation genes (Shifrut et al., 2018).
(K) Fraction of liganded proteins from total proteins found in previously described immune-enriched modules (Rieckmann et al., 2017).
Figure S3. Analysis of cysteine ligandability in immune signaling pathways. Related to Figure 3.
(A) Cysteine ligandability analysis of the enriched GO terms for proteins undergoing reactivity (top) or expression (bottom) changes in activated T cells. Enriched terms were passed through REVIGO, which identifies redundant terms and chooses representative terms for each group (See STAR Methods for details). Red sub-bars on right graphs represent both percentage and total number of proteins in each GO-term category having one or more liganded cysteines. Top-10 enriched biological processes are shown for the expression changes group.
(B) Venn diagram showing overlap between proteins quantified by TMT-exp and proteins liganded with scout fragments.
(C) Cysteine quantification events in experiments with scout fragments and elaborated compounds with the following colors used to mark proteins with no (grey), one (yellow), two (brown), or > two (orange) quantified cysteines.
(D) Location of liganded cysteines C59 (active site) and C406 (non-active site) in the immune-relevant kinase CHUK (PDB: 5EBZ).
(E) Location of the liganded cysteine C313 in IRF9 (mouse orthologous residue to human C319) at the site of its protein-protein interaction with STAT2 (PDB: 5OEN).
Figure S4. Analysis of active compound effects in human T cells. Related to Figures 4 and 5.
(A) Flow cytometry analysis of T cell activation. T cell activation was monitored by measuring CD25 expression levels after overnight stimulation with αCD3 and αCD28 antibodies in the presence of active compounds. The following FACS gating strategy was used: 1) Doublet discrimination was achieved by plotting forward scatter area (x-axis) versus side scatter width (y-axis) and selecting the area containing singlets for further analysis; 2) Live cells were selected by plotting forward scatter area (x-axis) versus Live/Dead Near-IR channel (y-axis, area, logarithmic scale) and gating on the negative cell population; 3) CD4+ cells were differentiated from CD8+ cells by plotting Pacific Blue channel (x-axis, area, logarithmic scale, PB-CD4 antibody) versus PerCP-Cy5.5 channel (y-axis, area, logarithmic scale, PerCP-Cy5.5-CD8a antibody). CD4+ positive, CD8+ negative cells were further analyzed for CD25 expression levels by measuring mean fluorescence intensity of the PE-channel (PE-CD25). Similar results were obtained by gating on the CD8+ positive, CD4+ negative cell population. BPK-25-treated cells are used as an example for the gating strategy. Representative histogram showing CD25 levels in unstimulated T cells (red), stimulated T cells in the presence of DMSO (light blue), and stimulated T cells in the presence of active compounds (orange) is also presented. Data is representative of three biological replicates used in Figure 4D.
(B) Concentration-dependent effects of EV-96 and EV-97 on T-cell activation parameters (left graphs: IL-2 and IFN-γ) and cytotoxicity (right graph) (24 h treatment). Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 4-5/group. *, p < 0.05; **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to EV-97 (0.5 μM) treatment group.
(C) Bar graph showing the total number of quantified (black) and liganded (R≥4, red) cysteines in cells treated with active compounds. Results are obtained by combining isoTOP-ABPP and TMT-ABPP data for both soluble and particulate proteomic fractions. R-values within each experimental treatment group were derived from 3-6 independent isoTOP-ABPP experiments and 2-3 independent TMT-ABPP experiments (4-6 TMT channels). A cysteine was required to be quantified in at least two experiments for each proteomic fraction to be reported.
(D) Representative bar graph showing the total number of liganded proteins as relates to the corresponding number of quantified (blue) and liganded (R≥4, red) cysteines per protein for BPK-25 treatments. Results are obtained by combining isoTOP-ABPP and TMT-ABPP data for both soluble and particulate proteomic fractions for each compound treatment. A cysteine was required to be quantified in at least two experiments for each proteomic fraction to be reported.
(E) Fraction of protein targets of active compounds with available crystal structures containing the corresponding liganded cysteine residues.
(F) Modeling of BPK-21 and BPK-25 interactions with C91 of stimulator of interferon genes protein (STING, or TMEM173).
(G) IRF response of THP1-Lucia™ ISG cells. THP-1 Lucia ISG cells were treated with DMSO or BPK-25 in the presence of viral dsDNA (2 μg/mL) for 24h and the levels of IRF-induced Lucia luciferase were determined using QUANTI-Luc™. Cell viability was measured using CellTiter-Glo™ assay. Luminescence signals for test samples were normalized to DMSO-treated samples and reported as relative light units (RLU) ± SEM; n = 3/group.
(H) Effect of BPK-25 treatment on gene expression related to TMEM173/STING pathway activation in PBMCs. PBMCs were treated with DMSO or BPK-25 (10 μM) for 5 h and stimulated with cGAMP (2 μM) for 2 h. Relative expression of IL-6, IL-1β, and IP10 (CXCL10) genes was measured by qPCR and normalized to actin.
(I) Effect of BPK-25 treatment on secretion of cytokines related to TMEM173/STING pathway activation in PBMCs. PBMCs were treated with DMSO or BPK-25 (10 μM) for 5 h and stimulated with cGAMP (10 μM) for 20 h. Cytokine levels (IFN-β, IP-10 (CXCL10), IL-6, MCP-1, TNFα) were measured using standard ELISA and Bio-Plex protocols (see STAR Methods for details). Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group. *, p < 0.05; **, p < 0.01 by two-tailed unpaired t test with Welch’s correction between indicated groups.
Figure S5. Functional analysis of protein targets of active compounds in human T cells. Related to Figure 6.
(A-B) Effects of active compounds on NFAT pathway activation. (A) Western blot analysis of NFATc2 content and phosphorylation in T cells treated with active (EV-3 (10 μM), BPK-21 (20 μM), BPK-25 (10 μM) (EV-96 (5 μM)) and control (EV-97 (5 μM)) compounds for 4 h in stimulated T cells. The anti-NFATc2 antibody recognizes both phosphorylated (upper band) and dephosphorylated (lower band) NFATc2. (B) Bar graph representation of western blot data showing compound effects on p-NFATc2 (green) and NFATc2 (red) content. Protein band intensity calculations were made using ImageJ software and are presented as the band intensity ± SEM; n = 3-4/group. *, p < 0.05 by two-tailed unpaired t test with Welch’s correction compared to NFATc2. p-NFATc2 levels were normalized using a mean (p-NFATc2/NFATc2) ratio for DMSO-treated controls to account for the differences between p-NFATc2 and NFATc2 in DMSO (stim) control. Dashed line shows 50% of mean signal intensities for dephosphorylated NFATc2 in DMSO (stim) control.
(C-D) Effects of active compounds on MAPK pathway. (C) Western blot data showing compound effects on p-ERK1/2 (T202/Y204) content. (D) Bar graph representation of western blot data showing the compound effects on p-ERK1/2 (T202/Y204) content. All treatments were done for 24 h. Protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO treated control cells ± SD; n = 3-5/group.
(E) Effects of active compounds versus the GCLC inhibitor BSO on glutathione (GSH) content and T cell activation parameters. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3-5/group. *, p < 0.05; **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to BSO (10 μM) treatment (IL-2 and IFN-γ) or unstimulated control (GSH).
(F) Structure of immunosuppressive natural product triptolide (left) and MS1 signal intensities (right) for ERCC3_C342 in isoTOP-ABPP experiments of expanded T cells treated with BPK-21 (20 μM, 3 h), BPK-25 (10 μM, 3h), or triptolide (0.2 μM, 3 h).
(G) T cell activation and cytotoxicity profile of the immunosuppressive natural product triptolide. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group.
(H) Flow cytometry analysis of T cell activation following ERCC3 gene disruption by CRISPR/Cas9 genome editing. T cells were activated for 2 days prior to Cas9 RNP transfection and were then cultured in IL-2 containing RPMI media to return the cells to a quiescent state. Seven days post-transfection, T cells were stimulated overnight with αCD3 and αCD28 antibodies in the presence of DMSO or BPK-21 with the activation monitored by measuring CD25 and CD69 expression levels. The following FACS gating strategy was used: 1) Lymphocyte gating; 2-3) Doublet discrimination by plotting forward scatter height (x-axis) versus forward scatter width (y-axis) and side scatter height (x-axis) versus side scatter width (y-axis); 4) Live cells were selected by plotting APC-Cy7 channel (x-axis, area, logarithmic scale, eBioscience™ Fixable Viability Dye eFluor™ 780) versus side scatter area (y-axis) and gating on the negative cell population; 5) Transfected cells were selected by gating on FITC-positive cell population (GFP-positive cells). GFP-positive cells were further analyzed for CD25 and CD69 expression levels by measuring mean fluorescence intensity of the PE- (PE-CD25) and APC-channels (APC-CD69). Representative histograms showing CD25 levels in stimulated T cells in the presence of scrambled sgRNA control (light blue) or ERCC3 guide RNAs (green) in the presence of DMSO or BPK-21 (orange) are presented. Data is representative of a total of six replicate treatments used in Figures S5I and S5J.
(I) Effect of genetic disruption of representative targets of active compound BPK-21 by CRISPR/Cas9 genome editing on T cell activation. Target disruption was considered to have an effect if T cell activation was suppressed >33% with a p value < 0.01. Data are presented as the mean percentage of control guide sgRNA-treated control cells ± SEM; n = 6/group. **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to control guides.
(J) Genetic disruption of ERCC3 and BPK-21 treatment produce similar degrees of blockade of T cell activation. Data are presented as the mean percentage of control sgRNA-treated control cells ± SEM; n = 6/group. **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to control guides.
(K) Efficiency of CRISPR/Cas9 genome editing. Left: western blot analysis showing ERCC3 protein content after CRISPR/Cas9 genome editing using control or ERCC3 sgRNA and sorting for GFP-positive T cells. Right: Quantitation of western blot data, where protein band intensity calculations were made using ImageJ software and represent the mean percentage of control guide sgRNA-treated cells ± SD; n = 2/group.
Figure S6. Functional analysis of protein targets of active compounds in human T cells. Related to Figure 6.
(A) MS1 signal intensities for BIRC3_C28, BIRC3_C164, and BIRC2_C45 in isoTOP-ABPP experiments of expanded T cells treated with EV-3 (10 μM, 3 h) or DMF (50 μM, 3 h).
(B) Bar graph representation of western blot data shown in Figure 6E, where protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO treated control ± SEM; n = 2-5 independent experiments per group. **p < 0.01; ****, p < 0.0001 by two-tailed unpaired t test with Welch’s correction compared to EV-3 (3h).
(C, D) EV-3 causes loss of BIRC2 and BIRC3 in human T cells. (C) Full western blot from Figure 6E showing reductions in BIRC2 and BIRC3 content in human T cells treated with EV-3 (1-10 μM, 24 h) and the blockade of EV-3-induced loss of BIRC2 and BIRC3 by co-treatment with the proteasome inhibitor MG132 (10 μM). (D) Western blot showing time-dependent reductions in BIRC2 and BIRC3 content in human T cells treated with EV-3 (10 μM).
(E) mRNA content for BIRC2 and BIRC3 quantified by RNA-sequencing of T cells treated with EV-3 (10 μM), DMF (50 μM), AT-406 (1 μM), BPK-25 (10 μM), or BPK-21 (20 μM). All treatments were done with T cells for 24 h. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group.
(F) Effect of genetic disruption of representative targets of active compound EV-3 by CRISPR/Cas9 genome editing on T cell activation. Target disruption was considered to have an effect if T cell activation was suppressed >33% with a p value < 0.01. Data are presented as the mean percentage of control guide sgRNA-treated control cells ± SEM; n = 3/group. **, p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to control guides.
(G) Flow cytometry analysis of T cell activation following BIRC2, BIRC3, or BIRC2 and BIRC3 gene disruption by CRISPR/Cas9 genome editing. T cells were activated for 2 days prior to Cas9 RNP transfection and were then cultured in IL-2 containing RPMI media to return the cells to a quiescent state. Seven days post-transfection, T cells were stimulated overnight with αCD3 and αCD28 antibodies in the presence of DMSO or BPK-21 with the activation monitored by measuring CD25 and CD69 expression levels. The same FACS gating strategy as in Figure S5H was used. Representative histograms showing CD25 levels in stimulated T cells in the presence of scrambled sgRNA control (light blue) or BIRC2/BIRC3/BIRC2+BIRC3 guide RNAs (green) in the presence of DMSO or EV-3 (orange) are presented. Data is representative of a total of six replicate treatments used in Figure 6H and S6F.
(H) Efficiency of CRISPR/Cas9 genome editing. Left: western blot analysis of BIRC2 and BIRC3 protein content using control, BIRC2, or BIRC3 sgRNA and sorting for GFP-positive T cells. Right: Quantitation of western blot data, where protein band intensity calculations were made using ImageJ software and represent the mean percentage of control guide sgRNA-treated cells ± SD; n = 2/group.
(I) Quantification of BPK-25 concentration-dependent changes in protein expression. Western blot protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO treated control ± SD; n = 2.
(J) Active compound treatment did not, in general, reduce the mRNA content for NuRD complex proteins. mRNA quantities were measured by RNAseq. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group.
(K) BPK-25-ctrl, a non-electrophilic propanamide analogue of BPK-25, does not inhibit T-cell activation. Left: Structure of BPK-25. Middle: Structure of BPK-25-ctrl. Right: T-cell activation (CD25) and cytotoxicity profiles for BPK-25 and BPK-25-ctrl (10 μM of each compound, 24 h). Data are presented as the mean percentage of DMSO-treated control cells ± SD; n = 2/group.
(L) Western blot data showing that BPK-25-ctrl, does not alter protein content for NuRD complex members.
Figure S7. Characterization of protein targets of EV-96 in human T cells. Related to Figure 7.
(A) Left: Bar graph showing fraction of total targets that show enantioselective (ES) engagement with one of the stereoisomeric compounds EV-96, EV-97, EV-98, and EV-99 at either 5 μM or 20 μM test concentrations in T cells (3 h). A target was considered enantioselective if it was engaged by one of the stereoisomers with an R value ≥4 and showed a reduction in engagement by at least 50% with the corresponding enantiomeric compound. Middle: Bar graph showing the percentage of enantioselective targets that were also liganded by scout fragments. Data for enantioselective (ES) and non-enantioselective (other) targets are shown separately. Right: Fraction of enantioselective targets that are immune-relevant (immune-enriched (blue) and/or have human genetics-based immune phenotypes (green)).
(B) Enantioselective engagement of C449 of TEC kinase by EV-96. Bar graphs showing percent reductions in IA-DTB labeling of C449 of TEC in T cells treated with the indicated concentrations of stereoisomeric electrophiles (3 h treatment; left: 5 μM, right: 20 μM), as quantified by TMT-ABPP experiments. Results represent mean values ± SD; n = 2-4/group.
(C) Western blot results showing enantioselective reductions in ITK protein and PLCG1 phosphorylation (Y783) in stimulated T cells treated with EV-96 (5 μM) compared to DMSO- or EV-97 (5 μM), EV-98 (5 μM), and EV-99 (5 μM)-treated T cells, measured at the indicated time points. Results are from one experiment representative of 2-5 independent experiments.
(D) Quantitation of western blot data showing effects of EV-96 and EV-97 on ITK protein content in naïve control (ctrl) versus αCD3/CD28-treated (stimulated, stim) T cells and the blockade of EV-96-induced loss of ITK by co-treatment with the proteasome inhibitor MG132 (10 μM). Cells were treated with compounds (5 μM each) or DMSO control for 8 h. Protein band intensity calculations were made using ImageJ software and represent the mean percentage of DMSO-treated control ± SD; n = 2-5 independent experiments per group. **p < 0.01 by two-tailed unpaired t test with Welch’s correction compared to DMSO-treated stim control.
(E) Western blot showing time-dependent decrease in ITK protein content in EV-96-treated αCD3/CD28-stimulated (Stim), but not expanded control (Ctrl) T cells. Cells treated with DMSO or EV-96 (5 μM) for 1–8 h. Results are from a single experiment representative of 2-4 independent experiments.
(F) EV-96 does not block the kinase activity of recombinant, purified ITK protein, as measured by the ADP-Glo™ Kinase Assay (Promega) following manufacturer’s instructions. Data are presented as the mean percentage of DMSO-treated control cells ± SEM; n = 3/group. ***p < 0.001 by two-tailed unpaired t test with Welch’s correction compared to EV-96(100 μM)-treated control. *PF – PF-06465469, an irreversible ITK inhibitor that reacts with C442.
(G) Western blot results showing that EV-96 (5 μM, 2-8 h) does not impair the phosphorylation of SLP-76 (S376) in stimulated (Stim) T cells.
(H) EV-96-ctrl, a non-electrophilic propanamide analogue of EV-96, does not inhibit T-cell activation. Left: Structure of EV-96-ctrl. Right: T-cell activation and cytotoxicity profiles for EV-96 and EV-96-ctrl (5 μM, 24 h). Data are presented as the mean percentage of DMSO-treated control cells ± SD; n = 2-5/group.
(I) Western blot results showing that EV-96-ctrl does not alter ITK protein content in stimulated T cells. Cells were treated with compounds (5 μM each) or DMSO for 8 h. Results are from one experiment representative of 2 independent experiments.
(J) Western blot results showing that pre-treatment (1 h) with EV-97 (5 μM) does not block EV-96-dependent decreases in ITK protein content in stimulated T cells. After 1 h pre-treatment, cells were treated with EV-96 (5 μM) or DMSO for 8 h.
(K) Structure of PF-06465469, an irreversible ITK inhibitor that reacts with C442 (left) and the corresponding crystal structure of a PF-06465469-ITK complex showing covalent modification of C442 (red).
Methods S1 (related to STAR Methods)
Glossary of Key Terms used in the manuscript (TMT-ABPP, isoTOP-ABPP, TMT-exp, scout fragment electrophiles, elaborated electrophiles, stereoisomeric electrophiles).
Methods S2 (Related to Figure 4)
Synthetic procedures and characterization of the elaborated small-molecule electrophiles tested in the inhibition of T cell activation screen.
Methods S3 (related to STAR Methods)
List of oligos used for T7 sgRNA production and for BIRC2_C45A and BIRC3_C28A mutagenesis studies.
Data S1 (related to Figures 1–7)
Tab 1. Table of contents for Data S1.
Tab 2 (Table S1, related to Figure 1 and S1A). Reactome pathways that were enriched (Benjamini-Hochberg corrected p-values ≤ 0.05) for each principal component as determined by Perseus. The size column refers to the size of the pathway in Reactome. Pathways colored in red are enriched in both principal components (albeit with different p-values), whereas uncolored pathways are enriched selectively in one or the other component.
Tab 3 (Table S2, related to Figure 1). TMT-exp proteomic data showing changes in protein expression in control vs activated primary human T cells.
Tab 4 (Table S3, related to Figure 1). TMT-ABPP proteomic data showing changes in cysteine reactivity in control vs activated primary human T cells.
Tab 5 (Table S4, related to Figures 1, 2, 3, and 5). List of proteins with identified cysteine reactivity changes, along with designations of immune-relevance and GO-terms: “RNA-binding”, “Metal ion binding”, “DNA binding”, “disulfide oxidoreductase activity”, and “ATP binding”. The summary of GO-term analysis is provided in Figure S1B.
Tab 6 (Table S5, related to Figure 1). Resource table for determination of immune-relevant genes. Multiple Z-scores were calculated from several transcriptomic tissue profiles, and these Z-scores were summed to generate the value in column H. Genes with summed Z-scores in the top ~10% were considered “immune-enriched”, which corresponded to Z scores > 11. Annotation of immune phenotype (column T) was performed by parsing data in the Online Mendelian Inheritance of Man (OMIM) database and querying phenotype titles and descriptions for immune-related substrings (‘*immun*’, ‘*inflam*’, ‘*rheum*’, ‘*psoria*’, etc), as described in the STAR Methods. In total, there were 2004 immune-enriched genes and 655 genes associated with immune phenotypes which, when combined, result in 2746 total immune-relevant genes (column U).
Tab 7 (Table S6, related to Figures 2 and 5). Master table containing total cysteine reactivity proteomic data from combined isoTOP-ABPP and TMT-ABPP experiments with scout fragments (“broad ligandability”, KB02 and KB05), active compounds (DMF, EV-3, EV-93, BPK-21, BPK-25, EV-96), and inactive stereoisomers (EV-97, EV-98, EV-99), as well as isoTOP-ABPP hyperreactivity experiments. Values for each treatment group are final average values resulting from at least 2 replicate experiments described in STAR Methods. Maximum values from all treatment groups (Columns J and P) for active compounds and scout fragments are used to determine “ligandability” of a select residue (related to Figure 2).
Tabs 8-9 (Tables S7, S8, related to Figure 2). Comparison of isoTOP-ABPP and isoTOP-ABPP hyperreactivity R values for activated and control T cells (related to Figure 2E).
Tab 10 (Table S9, related to Figure 2). List of liganded cysteines in immune-relevant proteins that are also targeted by other electrophile compounds.
Tab 11 (Table S10, related to Figures 2 and S2). Analysis of missense pathogenic mutations and their distance from liganded cysteines in proteins with immune phenotype (OMIM). Only proteins containing crystal structures with a liganded cysteine and at least one pathogenic missense mutation are included.
Tab 12 (Table S11, related to Figures 2 and S2). Analysis of proteins liganded with scout fragments (Table S6) for assignment to T cell proliferation genes (related to Figure S2K) (Shifrut et al., 2018).
Tab 13 (Table S12, related to Figures 2 and S2). Analysis of liganded and quantified proteins for assignment to previously described immune-enriched modules (modules 10, 17, 19, 23, 27, related to Figure S2L). (Rieckmann et al., 2017).
Tab 14 (Table S13, related to Figures 3). Ligandability analysis of Reactome pathways. All proteins for a specified pathway with at least one liganded cysteine are listed in column H, proteins with at least one quantified cysteine, but without liganded cysteines are listed in column I. All members for each pathway are listed in column J.
Tab 15 (Table S14, related to Figures 3). List of immune-enriched transcription factors and adaptor proteins liganded with scout fragments (related to Figure 3E). See STAR Methods for details.
Tab 16 (Table S15, related to Figure 4). Chemical structures of compounds used in the inhibition of T cell activation screen.
Tab 17 (Table S16, related to Figure 4). Summary of the results of the phenotypic screen for inhibition of T cell activation.
Tab 18 (Table S17, related to Figure 5). isoTOP-ABPP and TMT-ABPP proteomic data for active compounds (DMF, EV-3, EV-93, BPK-21, BPK-25, EV-96 and inactive stereoisomers EV-97, EV-98, EV-99). Values for each treatment group are final average values resulting from at least 2 replicate experiments described in STAR Methods. For each compound, maximum values across both methods and proteomic fractions are reported in the Master table (Tab 6, Table S6).
Tab 19 (Table S18, related to Figure 5). Distribution of protein classes containing cysteines liganded by active compounds (Figure 5C).
Tab 20 (Table S19, related to Figure 5). Comparison of isoTOP-ABPP and TMT-ABPP R values for cysteines liganded by active compounds versus scout fragments in human T cells, as displayed in correlation plot (Figure 5D) and pie chart (Figure 5E) analyses.
Tab 21 (Table S20, related to Figure 5). Prediction of pocket volumes within the indicated distances from cysteines liganded by active compounds (related to Figure 5F).
Tab 22 (Table S21, related to Figures 2 and 5). Liganded cysteine residues with scout fragments at known sites of palmitoylation (obtained by cross-referencing with SwissPalm proteins and sites).
Tab 23 (Table S22, related to Figure 6). Protein expression changes after BPK-25 (10 μM, 24 h) treatment as determined by TMT-exp (10 replicates from 5 donors, related to Figure 6J).
Tab 24 (Table S23, related to Figure 6). Summary of protein expression changes after DMF (50 μM, 24 h), EV-3 (10 μM, 24 h), BPK-21 (20 μM, 24 h), BPK-25 (10 μM, 24 h), and BPK-25 (10 μM, 24h) + MG132 (10 μM, 24 h) treatment as determined by TMT-exp (average values from 2-5 donors are reported, related to Figure 6K).
Tab 25 (Table S24, related to Figure 7). Summary of protein expression changes in human T cells treated with DMSO, EV-96, or EV-97 with or without αCD3/αCD28 stimulation as determined by TMT-exp (average values from 6 replicate treatments performed with cells from two donors (4 and 2 for each donor) are reported, related to Figure 7C).
Data S2 (Related to Figure 6)
RNA-sequencing data for primary human T cells treated with DMF (50 μM, 24 h), EV-3 (10 μM, 24 h), AT406 (1 μM, 24 h), BPK-21 (20 μM, 24 h), and BPK-25 (10 μM, 24 h).
Data Availability Statement
Raw proteomic data is available from the Lead Contact upon request. Raw RNA-sequencing data has been deposited to NCBI under GEO: GSE137756. Processed proteomic and RNA-sequencing data is provided in Data S1 and S2, respectively.