

Expression pattern of synthetic lethality genes is a previously unknown factor associated with cancer risk across human tissues.
Abstract
Various characteristics of cancers exhibit tissue specificity, including lifetime cancer risk, onset age, and cancer driver genes. Previously, the large variation in cancer risk across human tissues was found to strongly correlate with the number of stem cell divisions and abnormal DNA methylation levels. Here, we study the role of synthetic lethality in cancer risk. Analyzing normal tissue transcriptomics data in the Genotype-Tissue Expression project, we quantify the extent of co-inactivation of cancer synthetic lethal (cSL) gene pairs and find that normal tissues with more down-regulated cSL gene pairs have lower and delayed cancer risk. Consistently, more cSL gene pairs become up-regulated in cells treated by carcinogens and throughout premalignant stages in vivo. We also show that the tissue specificity of numerous tumor suppressor genes is associated with the expression of their cSL partner genes across normal tissues. Overall, our findings support the possible role of synthetic lethality in tumorigenesis.
INTRODUCTION
Cancers of different human tissues have markedly different molecular, phenotypic, and epidemiological characteristics, known as the tissue specificity in cancer. Various aspects of this intriguing phenomenon include a considerable variation in lifetime cancer risk, cancer onset age, and the genes driving the cancer across tissue types. The variation in lifetime cancer risk is known to span several orders of magnitude (1, 2). Such variation cannot be fully explained by the difference in exposure to carcinogens or hereditary factors and has been shown to strongly correlate with differences in the number of lifetime stem cell divisions (NSCD) estimated across tissues (2, 3). As claimed by Tomasetti and Vogelstein (2), these findings are consistent with the notion that tissue stem cell divisions can propagate mutations caused either by environmental carcinogens or random replication error (4). In addition, the importance of epigenetic factors in carcinogenesis has long been recognized (5), and Klutstein et al. (6) have recently reported that the levels of abnormal CpG island DNA methylation (LADM) across tissues are highly correlated with their cancer risk. Although both global (e.g., smoking and obesity) and various cancer type–specific (e.g., HCV infection for liver cancer) risk factors are well known (7), no factors other than NSCD and LADM have been reported to date to explain the across-tissue variance in lifetime cancer risk.
Besides lifetime cancer risk, cancer onset age, as measured by the median age at diagnosis, also varies among adult cancers (1). Although most cancers typically manifest later in life [more than 40 years old (1, 8)], some such as testicular cancer often have earlier onset (1). Many tumor suppressor genes (TSGs) and oncogenes are also tissue specific (9–11). For example, mutations in the TSG BRCA1 are predominantly known to drive the development of breast and ovarian cancer but rarely other cancer types (12). In general, factors explaining the overall tissue specificity in cancer could be tissue intrinsic (10, 13), and their elucidation can further advance our understanding of the forces driving carcinogenesis.
Synthetic lethality/sickness (SL) is a well-known type of genetic interaction, conceptualized as cell death or reduced cell viability that occurs under the combined inactivation of two genes but not under the inactivation of either gene alone. The phenomenon of SL interactions was first recorded in Drosophila (14) and then in Saccharomyces cerevisiae (15). In recent years, much effort has been made to identify SL interactions specifically in cancer, since targeting these cancer SLs (cSLs) has been recognized as a highly valuable approach for cancer treatment (16–19). The effect of cSL on cancer cell viability has led us to investigate whether it plays an additional role even before tumors manifest, i.e., during carcinogenesis. In this study, we quantify the level of cSL gene pair co-inactivation in normal (noncancerous) human tissue as a measure of resistance to cancer development (termed cSL load, explained in detail below). We show that cSL load can explain a considerable level of the variation in cancer risk and cancer onset age across human tissues, as well as the tissue specificity of some TSGs. Together, these correlative findings support the effect of SL in impeding tumorigenesis across human tissues.
RESULTS
Computing cSL load in normal human tissues
To study the potential effects of cSL in normal, noncancerous tissues, we define a measure called cSL load, which quantifies the level of cSL gene pair co-inactivation based on gene expression of normal human tissues from the Genotype-Tissue Expression (GTEx) dataset (20). Specifically, we used a recently published reference set of genome-wide cSLs that are common to many cancer types, identified from both in vitro and The Cancer Genome Atlas (TCGA) cancer patient data (21) via the identification of clinically relevant synthetic lethality (ISLE) (table S1A) (22, 23). For each GTEx normal tissue sample, we computed the cSL load as the fraction of cSL gene pairs (among all the genome-wide cSLs) that have both genes lowly expressed in that sample (Methods; illustrated in Fig. 1). We further defined tissue cSL load (TCL) as the median cSL load value across all samples of each tissue type in GTEx (Methods and table S2A). We then proceed to test our hypothesis that TCL can be a measure of the level of resistance to cancer development intrinsic to each human tissue (outlined in Fig. 1).
Fig. 1. A schematic diagram providing an overview of this study.
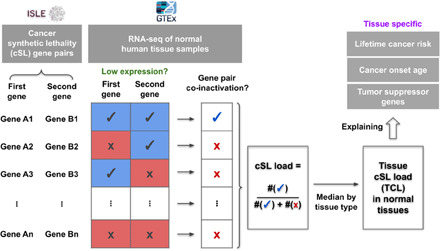
This diagram illustrates the computation of cSL load for each sample and each tissue type (i.e., TCL) and depicts the outline of this study, where we attempted to explain the tissue-specific lifetime cancer risk, cancer onset age, and TSGs using TCL. See main text and Methods for details.
TCL in normal tissues is inversely correlated with their lifetime cancer risk
SL is widely known to be context specific across species, tissue types, and cellular conditions (24). In theory, a cancer-specific cSL gene pair can be co-inactivated in the normal tissue without reducing normal cell fitness, while conferring resistance to the emergence of malignantly transformed cells due to the lethal effect specifically on the cancer cells. Different normal tissues can have varied TCLs (representing the levels of cSL gene pair co-inactivation) as a result of their specific gene expression profiles, and we hypothesized that normal tissues with higher TCLs should have lower cancer risk, as transforming cancerous cells in these tissues will face higher cSL-mediated vulnerability and lethality. To test this hypothesis, we obtained data on the tissue-specific lifetime cancer risk in humans (Methods) and correlated that with the TCL values computed for the different tissue types. We find a strong negative correlation between the TCL (computed from older-aged GTEx samples, age ≥50 years) and lifetime cancer risk across normal tissues (Spearman’s ρ = −0.664, P = 1.59 × 10−4; Fig. 2A and table S2A). This correlation is robust, as comparable results are obtained when this analysis is carried out in various ways (e.g., different cutoffs for low expression of genes, different cSL network sizes, and different cancer type–normal tissue mappings; fig. S1 and note S3). We also showed that this correlation is not confounded by the number of poised genes associated with bivalent chromatin, variation in cancer driver gene expression, and immune cell or fibroblast abundance (notes S11 to S13 and figs. S12 to S14). Notably, the cSL load varies with age due to age-related gene expression changes, and the correlation with lifetime cancer risk is not found when the TCL is computed on samples from the young population (20 ≤ age < 50 years, Spearman’s ρ = −0.0251, P = 0.901; fig. S2A); this is consistent with the observation that lifetime cancer risk is mostly contributed by cancers occurring in older populations (1). We still see a marked negative correlation between TCL and lifetime cancer risk when analyzing samples from all age groups together (Spearman’s ρ = −0.49, P = 0.01; fig. S2B). Repeating these analyses using different control gene pairs including (i) random gene pairs, (ii) shuffled cSL gene pairs, and (iii) degree-preserving randomized cSL network (same size as the actual cSL network; note S4) results in significantly weaker correlations (empirical P < 0.001; fig. S3, A to C, and note S4), confirming that the associations found with cancer risk results from a cSL-specific effect.
Fig. 2. TCL can explain the variance in lifetime cancer risk across human tissues.
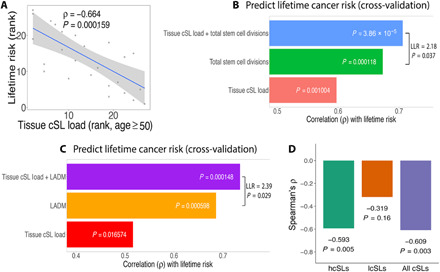
(A) Scatterplot showing Spearman’s correlations between lifetime cancer risk and TCL computed for the older population (age ≥50 years) (ranked values are used as lifetime cancer risk spans several orders of magnitude.) (B) Lifetime cancer risks across tissues were predicted using linear models (under cross-validation) containing different sets of explanatory variables: (i) TCL only, (ii) the number of stem cell divisions (NCSD) only, and (iii) TCL and NSCD (27 data points). The prediction accuracy is measured by Spearman’s ρ, shown by the bar plots. The result of a likelihood ratio test between models (ii) and (iii) is also displayed. (C) A similar bar plot as in (B) comparing the predictive models for cancer risk involving the following variables: (i) TCL only, (ii) the LADM only, and (iii) TCL and LADM combined (21 data points only due to the smaller set of LADM data). A model containing all the three variables does not increase the prediction power (Spearman’s ρ = 0.77 under cross-validation) and is not shown. (D) Bar plot showing the correlations between lifetime cancer risk with TCLs computed (age ≥50 years) using subsets of cSLs: hcSLs, lcSLs, and all cSLs. Spearman’s ρ and P values are shown. The hcSLs and lcSLs are identified using data of matched TCGA cancer types and GTEx normal tissues (Methods), which correspond to only a subset of tissue types. To facilitate comparison, here, the correlation for all cSLs was also computed for the same subset of tissues, and therefore, the resulting correlation coefficient is different from that in (A).
While the randomized cSL networks used in the control tests described above provide significantly weaker correlations with cancer risk than those observed with cSLs, many of these correlations are still significant by themselves (fig. S3, B and C). This suggests that there may be a possible association between the expression of single genes in the cSL network (cSL genes) and cancer risk. To investigate this, we computed the tissue cSL single-gene load (SGL; the fraction of lowly expressed cSL genes) for each tissue (Methods). We do find a significant negative correlation between tissue SGL levels and cancer risk (Spearman’s ρ = −0.49, P = 0.01; fig. S3D and note S5). This correlation vanishes when we use random sets of single genes (fig. S3F). However, after controlling for the single-gene effect, the partial correlation between TCL and cancer risk is still highly significant (Spearman’s ρ = −0.69, P = 6.10 × 10−5; fig. S3G), pointing to the dominant role of the SL genetic interaction effect (note S5).
TCL predicts lifetime cancer risk across tissues in addition to the number of tissue stem cell divisions and abnormal DNA methylation levels
We next compared the predictive power of TCL to those obtained with the previously reported measures of NSCD (2, 3) and LADM (6), using the set of GTEx tissue types investigated here (Methods). We first confirmed the strong correlations of NSCD and LADM with tissue lifetime cancer risk in our specific dataset (Spearman’s ρ = 0.72 and 0.74, P = 2.6 × 10−5 and 1.3 × 10−4, respectively; fig. S4). These correlations are stronger than the one we reported above between TCL and cancer risk. However, adding TCL to either NSCD or LADM in linear regression models leads to enhanced predictive models of cancer risk compared to those obtained with NSCD or LADM alone [log-likelihood ratio (LLR) = 2.18 and 2.39, P = 0.037 and 0.029, respectively]. Furthermore, adding TCL to each of these factors increases their prediction accuracy under cross-validation (Spearman’s ρ’s from 0.67 and 0.69 with NSCD and LADM alone to 0.71 and 0.77, respectively; Fig. 2, B and C). LADM and NSCD are significantly correlated (Spearman’s ρ = 0.66, P = 0.02), while the TCL correlates only in a borderline significant manner with either NSCD (Spearman’s ρ = −0.57, P = 0.06) or LADM (Spearman’s ρ = −0.52, P = 0.08). Together, these observations support the hypothesis that TCL is associated with tissue cancer risk, with a partially independent role from either NSCD or LADM.
cSL pairs that are more specific to cancer are more predictive of cancer risk in normal tissues
We have shown results that support the role of TCL in impeding cancer development, and we reason that such an effect is dependent on the notion that many of the cSLs are specific to cancer while having weaker or no lethal effects in normal tissues. We tested and found that the co-inactivation of cSL gene pairs is under much weaker negative selection in GTEx normal tissues versus matched TCGA cancers [Wilcoxon rank sum test P = 2.93 × 10−6 (fig. S5A), also shown using cross-validation (note S7)]. Moreover, we hypothesize that those cSLs with the highest specificity to cancer (i.e., with the strongest SL effect in cancer and no or the weakest effect on normal cells) should have the strongest effect on cancer development. To test this, we identified the subset of such cSLs (termed “highly specific cSLs” or “hcSLs”) and those with the lowest specificity to cancer (termed “lowly specific cSLs” or “lcSLs”; Methods) and recomputed the TCLs of all normal GTEx tissues using these two cSL subsets, respectively. The TCLs computed from the hcSLs correlate much stronger with cancer lifetime risk than those computed from the lcSLs (Spearman’s ρ = −0.593 versus −0.319; Fig. 2D), testifying that these cSLs with high functional specificity to cancer are more relevant to carcinogenesis. These hcSLs are enriched for cell cycle, DNA damage response, and immune-related genes [false discovery rate (FDR) < 0.05; table S5 and Methods], which are known to play key roles in tumorigenesis.
Higher TCL in the younger population is associated with delayed cancer onset
We have thus established that TCL in the older population is inversely correlated with lifetime cancer risk across tissues. We next hypothesized that higher cSL load in a given normal tissue in the young population may delay cancer onset, which typically occurs later (age >40 years) (1). To test this, we use the median age at cancer diagnosis (1) of a certain tissue as its cancer onset age (table S3 and Methods). We find that the TCL values (for age ≤40 years) are markedly correlated with cancer onset age (Spearman’s ρ = 0.502, P = 0.011; Fig. 3A). This result is again robust to variations in our methods to compute TCL and cancer onset age (fig. S6, table S3, and note S3). We note that the cancer onset age is not significantly correlated with lifetime cancer risk (Spearman’s ρ = 0.279, P = 0.28).
Fig. 3. TCL can explain the variance in cancer onset age across human tissues.
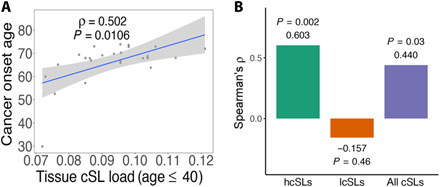
(A) Scatterplot showing Spearman’s correlations between cancer onset age and TCL (age ≤40 years). (B) Bar plot showing the correlations between cancer onset age with TCLs computed (age ≤40 years) using subsets of cSLs: hcSLs, lcSL, and all cSLs. Spearman’s ρ and P values are shown. As in Fig. 2D, this analysis was done for a subset of GTEx normal tissues for which we had matched TCGA cancer types to identify the hcSLs and lcSLs (Methods); therefore, the correlation result for all cSLs is also different from that in (A).
Similar to our earlier analysis, we see that the TCLs computed from the hcSLs correlate much stronger with onset age than those from the lcSLs or all cSLs (Spearman’s ρ = 0.603 versus −0.157; Fig. 3B and fig. S7A) and also stronger than those obtained from control tests performed as before (empirical P < 0.001; fig. S7, B to D). As with the case of cancer risk, the observed correlation is dominated by the SL genetic interaction effects rather than the single-gene effects (fig. S7, E to G, and note S5).
cSL load decreases in cells treated by chemical carcinogens and throughout pre-cancer stages
To further corroborate the relevance of cSL load to carcinogenesis, we next investigated whether carcinogen treatment in normal (noncancer) cell lines and primary cells in vitro can lead to cSL load decrease. First, we analyzed gene expression data from a recent study where human primary hepatocytes, renal tube epithelial cells, and cardiomyocytes were treated with the carcinogen and hepatotoxin thioacetamide-S-oxide (25). We computed the cSL load in each cell type after treatment versus control and found a significant decrease of cSL load only in the hepatocytes (Wilcoxon rank sum test P = 0.014; Fig. 4A), which is consistent with thioacetamide-S-oxide’s role as a hepatotoxin and a carcinogen primarily in the liver. Second, we collected the gene expression signatures of chemotherapy drug treatments in a total of four primary cells and normal cell lines from the Connectivity Map (CMAP) (26). We quantified the drug-induced cSL load changes indirectly from the gene signatures (Methods), comparing the strongly mutagenic DNA-targeting drugs (n = 6) including alkylating agents and DNA topoisomerase inhibitors to the weak/nonmutagenic taxanes and vinca alkaloids (n = 5), which act on the cytoskeleton and not directly on DNA (27). We find that the strong mutagenic chemotherapy drugs lead to a significantly larger decrease in cSL load (Fig. 4B, P = 0.03 from a linear model controlling for cell type; Methods). The strong mutagenicity of alkylating agents and DNA topoisomerase inhibitors is consistent with their mechanisms of actions; they are also World Health Organization class I carcinogens (28), supported by incidence of secondary cancers in patients treated by these drugs for their primary cancers (29). In contrast, taxanes and vinca alkaloids have shown negative or weak/inconclusive results in mutagenic tests (27, 30). These results are not likely affected by cell death, as the cSL decreased specifically only for the two classes among all tested chemotherapy drugs. Although the CMAP dataset used for this analysis does not include cell viability information, the gene expression of the cells does not show an apoptotic signature after the drug treatment.
Fig. 4. Experimental and clinical evidence further supports that cSL load may play a functional role in cancer development.
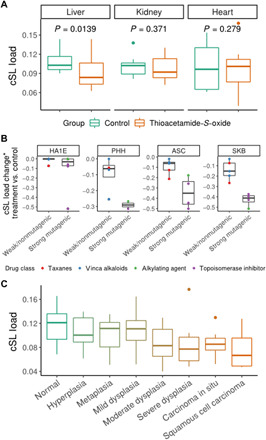
(A) Box plots showing the cSL loads in control versus thioacetamide-S-oxide–treated samples in human primary hepatocytes (“liver”), renal tube epithelial cells (“kidney”), and cardiomyocytes (“heart”), using the data from (25). One-sided Wilcoxon rank-sum test P values are shown. (B) Box plots showing the cSL load changes after treatment by different classes of chemotherapy drugs in four cell types, using the CMAP data (26). Asterisk indicates that the cSL load change is estimated indirectly from the CMAP drug treatment gene expression signatures (Methods). Strongly mutagenic drugs (n = 6), including alkylating agents (green points) and DNA topoisomerase inhibitors (purple points), lead to a significantly larger cSL load decrease compared to weak or nonmutagenic drugs (n = 5), including taxanes (red points) and vinca alkaloids (blue points); P = 0.03 from a linear model controlling for cell type. HA1E is an immortalized kidney cell line; PHH, primary human hepatocyte; ASC, adipose-derived stem cell; SKB, human skeletal myoblast. (C) Box plots showing the cSL load in samples of different stages of premalignant lesions in the lung (including normal tissue and lung squamous cell carcinoma) (28). The cSL load shows an overall decreasing trend from normal to different pre-cancer stages to cancer (one-sided Wilcoxon rank sum test of normal versus cancer P = 4.47 × 10−5; ordinal logistic regression has negative coefficient −28.7, P = 5.89 × 10−7).
Further beyond these in vitro findings, analyzing a recently published lung cancer dataset (31), we find that cSL load decreases progressively as cancers develop from normal tissues throughout the multiple stages of premalignant lesions in vivo (normal versus cancer Wilcoxon rank sum test P = 4.47 × 10−5, ordinal logistic regression P = 5.89 × 10−7 with negative coefficient −28.7; Fig. 4C). These results provide further evidence supporting cSL as a factor that may be involved in cancer development.
The activity state of cSL partners of some TSGs predicts the specific tissues in which they are known to drive cancer
Given the role of cSLs in cancer development, we turned to ask whether cSL may also contribute to the tissue/cancer-type specificity of TSGs (10, 32). Specifically, we reasoned that the loss of function of a gene is unlikely to have cancer-driving effects in tissues where its cSL partner genes are lowly expressed, due to the synthetic lethal effect of such co-inactivation on the emerging cancer cells. In other words, this gene is unlikely to be a TSG in such tissues. To study this hypothesis, we obtained a list of TSGs together with the tissues in which their loss is annotated to have a tumor-driving function from the COSMIC database (table S6A) (11). We further identified the cSL partner genes of each such TSG using ISLE (Methods and table S6B) (22). In total, there are 23 TSGs for which we were able to identify more than one cSL partner gene. Consistent with our hypothesis, we find that in most of the cases, the cSL partner genes of TSGs have higher expression levels in the tissues where the TSGs are known drivers compared to the tissues where they are not established drivers (binomial test for the direction of the effect P = 0.023; Fig. 5A). We identified 10 TSGs whose individual effects are significant (FDR < 0.05) and cSL specific (as shown by the random control test), and all these 10 cases exhibit the expected direction of effect (labeled in Fig. 5A and table S6C; two example TSGs, FAS and BRCA1, are shown in Fig. 5B, details are in fig. S8 and Methods). Reassuringly, these findings disappear under randomized control tests involving random partner genes of the TSGs and shuffled TSG–tissue type mappings (note S9), further consolidating the role of cancer-specific cSLs of normal tissues in cancer risk and development.
Fig. 5. The expression levels of the cSL partner genes of TSGs can explain their tissue specificity.
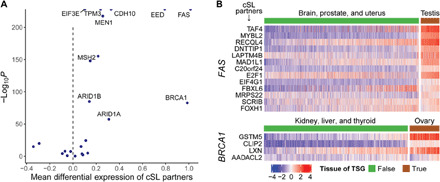
(A) For each tissue-specific TSG gene Gi, the expression levels of its cSL partner genes in the tissue type(s) where gene Gi is a TSG were compared to those where gene Gi is not an established TSG, using GTEx normal tissue expression data. The volcano plot summarizes the result of comparison with linear models. Positive linear model coefficients (x axis) mean that the expression levels of the cSL partner genes are, on average, higher in the tissue(s) where gene Gi is a TSG. Many cases have near-zero P values and are represented by points (half-dots) on the top border line of the plot. Overall, there is a dominant effect of the cSL partner genes of TSGs having higher expression levels in the tissues where the TSGs are known drivers (binomial test P = 0.023). All TSGs with FDR < 0.05 that also passed the random control tests are labeled. (B) Examples of two well-known TSGs, FAS and BRCA1, are given. The heatmaps display the normalized expression levels of their cSL partner genes (rows) in tissues of where these two genes are known to be TSGs [according to the annotation from the COSMIC database (11)] and in tissues where they are not established TSGs (columns), respectively. High and low expressions are represented by red and blue, respectively. For clarity, one typical tissue type where the TSG is a known driver (e.g., testis for FAS) and three other tissue types where the TSG is not an established driver (and the least frequently mutated) are shown.
DISCUSSION
In this work, we show that the cSL load in normal tissues is a strong predictor of tissue-specific lifetime cancer risk and is much stronger than the pertaining predictive power observed on the individual gene level. Consistently, we find that higher cSL load in the normal tissues from young people is associated with later onset of the cancers of that tissue. As far as we know, no other factor has been previously reported to be predictive of cancer onset age across tissues. Furthermore, cSL load decreases upon carcinogen treatment in vitro and during cancer development through stages of precancerous lesions in vivo. Last, we show that the activity status of cSL partners of TSGs can explain their tissue-specific inactivation.
We have shown that the correlation between cSL and cancer risk in normal tissues may be explained by the fact that many of the cSLs are specific to cancer and have weak or no functional lethal effect in the normal tissues (Figs. 2D and 3B and fig. S5); therefore, normal tissues can bear relatively high cSL loads without being detrimentally affected—quite to the contrary, they become more resistant to cancer due to the latent effect of these cSLs on potentially emerging cancer cells. We emphasize that while we quantified the cSL loads using the normal tissue data from GTEx, the set of cSLs we used was derived exclusively in cancer from completely independent cancer datasets (and without using any information regarding lifetime cancer risk, onset, or tumor suppressor tissue specificity), so there is no circularity involved. The cSL load in normal tissues was computed to reflect the summed effects of individual cSL gene pairs. The underlying assumption is that the low expression of each cSL gene pair is synthetic sick (i.e., reducing cell fitness to some extent) and that the effects from different cSL gene pairs are additive, consistent with the ISLE method of cSL identification (22). Many experimental screenings of SL interactions also rely on techniques such as RNA interference that inhibits gene expression rather than completely knocks out a gene (33), and it is evident that most of the resulting SL gene pairs have milder than lethal effects. While these cSLs likely act via a diverse range of biological pathways and thus do not provide pathway-specific mechanisms, the additive cancer-specific lethal effect of such cSL gene pairs, however, could form a negative force impeding cancer development from normal tissues.
Obviously, as we are studying the across-tissue association between cSL load and cancer risk, it is essential to focus on cSLs that are common to many cancer types (i.e., pan-cancer). Therefore, we focused on cSLs identified computationally by ISLE via the analysis of the pan-cancer TCGA patient data (22). In contrast, most experimentally identified cSLs are obtained in specific cancer cell lines and are thus less likely to be pan-cancer [and possibly, less clinically relevant (22)]. However, for completeness, we also compiled a set of experimentally identified cSLs from published studies (22, 34) (note S1 and table S1B). The corresponding TCL values computed using this set of cSLs correlate significantly with lifetime cancer risk but not with cancer onset age; the correlation with cancer risk is also markedly weaker than that obtained from ISLE-derived cSLs [Spearman’s ρ = −0.433, P = 0.024 (fig. S9A), control tests and detailed analysis are explained in note S4]. These experimentally identified cSLs can explain some cases of tissue-specific TSGs including BRCA1 and BRCA2 (fig. S9E) but do not result in overall significant accountability for a large proportion of TSGs present in the analysis (like in Fig. 5A). This corroborates the importance of pan-cancer cSLs and their relevance to cancer risk.
TCL is not likely to be a corollary of NSCD and LADM [while LADM was thought to be closely related to NSCD (6)], as the cSL load is computed by analyzing expression data of bulk tissues, where stem cells occupy only a minor proportion. We have shown that TCL significantly adds to either NSCD or LADM in predicting lifetime cancer risk (Fig. 2, B and C), which also suggests that cSL load is an independent factor correlated with cancer risk with unique underlying mechanisms. Furthermore, NSCD is measured as the product of the rate of tissue stem cell division and the number of stem cells residing in a tissue (2), and we confirmed that TCL is correlated with lifetime cancer risk independent of both of these components (partial Spearman’s ρ = −0.510 and −0.567, P = 0.007 and 0.002, respectively; fig. S10, A and B). We additionally tested and verified that proliferation indices computed for the bulk normal tissues do not correlate with lifetime cancer risk across tissues (Spearman’s ρ = 0.062, P = 0.77; fig. S10C and note S10). Furthermore, we verified that our observed correlations are not confounded by the number of samples from each cancer or tissue type (fig. S11).
Since cSL load can vary with age, one may wonder whether cSL load could be extended to correlate with age-specific cancer risk within a tissue (as opposed to across tissues). However, variations in cancer risk across tissues and across ages can be driven by different factors. We did not find a consistent correlation between cSL load computed by age range and age-specific cancer risk in all tissue types (note S14 and fig. S15). Another extension to our current research question is studying the effect of higher-order genetic interactions on cancer risk, which is plausible but challenging to study due to the limited knowledge available on such complex interactions.
While revealing cSL as a previously unknown factor associated with cancer development, our study has several limitations. First, because of the importance of using pan-cancer cSLs as discussed above, we mainly relied on the cSLs computationally inferred by ISLE (22) as one of the most comprehensive pan-cancer cSL datasets. However, current cSL prediction algorithms are far from perfect and should not be regarded as the gold standard for general cSL identification. Only a minor fraction of the large number of predicted cSLs have been experimentally validated only in specific cell types. The cSLs inferred by ISLE should be best viewed as a set of candidate cSL pairs that emerge from genetic screen data in vitro but with further support from patient and phylogenetic data. Future studies that provide experimentally validated pan-cancer cSLs are needed to consolidate our current findings. Second, we have relied on analyzing the gene expression data of bulk tissues from GTEx and not the expression data of the specific cells of origin of the corresponding cancers. More refined future analysis is desirable using single-cell data across normal human tissues as such data becomes more widely available. Last, our study does not establish a causal relationship between the cSL load and the risk of cancer, as it is challenging to experimentally perturb a large number of cSLs simultaneously. The results shown are descriptive and association based, and the causal role of SLs in carcinogenesis remains to be studied mechanistically.
Together, our findings demonstrate strong associations between SL and cancer risk, onset time, and context specificity of tumor suppressors across human tissues. This suggests that beyond the effect on cancer after it has developed, cSL could also play an important role during the entire course of carcinogenesis, although further studies are needed to establish causality. While SL has been attracting tremendous attention as a way to identify cancer vulnerabilities and target them, this is the first time that its potential role in mediating cancer development is uncovered.
METHODS
cSL interaction networks
The cSL gene pairs computationally identified by the ISLE (identification of clinically relevant SL) pipeline were obtained from (22). We used the cSL network identified with FDR < 0.2 for the main text results, containing 21,534 cSL gene pairs, which is a reasonable size representing only about one cSL partner per gene on average. This also allows us to capture the effects of many weak genetic interactions. Nevertheless, we also used the cSL network with FDR < 0.1 (only 2326 cSLs) to demonstrate the robustness of the results to this parameter (notes S1 and S3). Each gene pair is assigned a significance score [the “SL-pair score” defined in (22)], that a higher score indicates that there is stronger evidence that the gene pair is SL in cancer. Out of these, we used 20,171 cSL gene pairs whose genes are present in the GTEx data (table S1A). The experimentally identified cSL gene pairs were collected from 18 studies [obtained from the supplementary data 1 of Lee et al. (22) except for those from Horlbeck et al. (34)]. Horlbeck et al. (34) provided a gene interaction (GI) score for each gene pair in two leukemia cell lines. Gene pairs with GI scores of <−1 in either cell line were selected as cSLs. A total of 27,975 experimentally identified cSLs were obtained, out of which 27,538 have both their genes present in the GTEx data (table S1B).
GTEx and TCGA data
The V6 release of GTEx (20) RNA sequencing (RNA-seq) data [gene-level reads per kilobase of transcript, per million mapped reads (RPKM) values] was obtained from the GTEx Portal (https://gtexportal.org/home/). The associated sample phenotypic data were downloaded from dbGaP (35) (accession number phs000424.vN.pN). For comparing the level of negative selection to co-inactivation of cSL gene pairs between normal and cancer tissues, the RNA-seq data of TCGA and GTEx as RNA-seq by expectation-maximization (RSEM) values that have been processed together with a consistent pipeline that helps to remove batch effects were downloaded from UCSC Xena (36). The expression data for each tissue type (normal or cancer) was normalized separately (inverse normal transformation across samples and genes) before being used for the downstream analyses. We mapped the GTEx tissue types to the corresponding TCGA cancer types (table S2B), resulting in one-on-many mappings, e.g., the normal lung tissue was mapped to both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC).
Cancer risk data and onset age
Lifetime cancer risk denotes the chance a person has of being diagnosed with cancer during his or her lifetime. Lifetime cancer risk data (table S2A) are from Tomasetti and Vogelstein (2), which are based on the U.S. statistics from the SEER (Surveillance, Epidemiology, and End Results) database (1). We derived the cancer onset age based on the age-specific cancer incidence data from the SEER database with the standard formula (37). Specifically, for each cancer type, SEER provides the incidence rates for 5-year age intervals from birth to 85+ years old. The cumulative incidence (CI) for a specific age range S is computed from the corresponding age-specific incidence rates (IRi, i ∈ S) as CI = 5∑i ∈ S IRi, and the corresponding risk is computed as risk = 1 − exp(−CI). The onset age for each cancer type (table S3) was computed as the age when the CI from birth is 50% of the lifetime CI (i.e., birth to 85+ years old). Usually, the onset age defined as such is between two ages where the actual CI data are available, so the exact onset age was obtained by linear interpolation. Alternative parameters were used to define onset age (note S3) to show the robustness of the correlation between TCL and cancer onset age based on different definitions.
Computing cSL load
For each sample, we computed the number of cancer-derived SL gene pairs that have both genes lowly expressed and divided it by the total number of cSLs available to get the cSL load per sample. In the ISLE method described in (22), low expression was defined as having expression levels below the 33 percentile in each tissue or cell type. Thus, the ISLE-derived cSL gene pairs were shown to exhibit synthetic sickness effects when both genes in the gene pair are expressed at levels below the 33 percentile in each tissue, even though this appears to be a very tolerant cutoff (22). We therefore adopted the same criterion for low expression for the main results, although we also explored other low expression cutoffs to demonstrate the robustness of the results (note S3).
Computing TCL and correlation with lifetime cancer risk and cancer onset age
TCL of each tissue type is the median value of the cSL loads of all the samples (or a subpopulation of samples) in that tissue, with the cSL load of a sample computed as above. For example, TCL for the older population (age ≥50 years) is the median cSL load for the samples of age ≥50 years in each tissue type. For analyzing the correlation between the TCLs computed from GTEx normal tissues and cancer risk, we mapped the GTEx tissue types to the corresponding cancer types for which lifetime risk data are available from Tomasetti and Vogelstein (2), resulting in 16 GTEx types mapped to 27 cancer types (table S2A). Gallbladder nonpapillary adenocarcinoma and osteosarcoma of arms, head, legs, and pelvis are not mapped to GTEx tissues and excluded from our analysis. Similarly for the correlation between TCLs and cancer onset age, we mapped GTEx tissue types to the tissue sites from the SEER database (as given in the data slot “site recode ICD-O-3/WHO 2008”) by their names (table S3).
Computing cSL SGL
To investigate the effect on the single-gene level, we computed the cSL SGL in a paralleling way to the computation of the cSL load. Among all the unique genes constituting the cSL network (i.e., cSL genes), we computed the fraction of lowly expressed cSL genes for each sample as the cSL SGL, where low expression was defined in the same way as the computation of cSL load as elaborated above. Similarly, tissue cSL SGL is the median value of the cSL SGLs of all the samples in a tissue.
Predicting tissue lifetime cancer risk with linear models
The lifetime cancer risks across tissue types were predicted with linear models containing three different sets of explanatory variables: (i) the number of total stem cell divisions (NSCD) alone, (ii) TCL alone, and (iii) NSCD together with TCL. LLR test was used to determine whether model (iii) (the full model) is significantly better than model (i) (the null model) in predicting lifetime cancer risks. The three models were also used to predict the lifetime cancer risks with a leave-one-out cross-validation procedure, and the prediction performances were measured by Spearman correlation coefficient. A similar analysis was performed to predict lifetime cancer risks across tissue types with three linear models involving the level of abnormal DNA methylation levels of the tissues (6): (i) the number of LADM alone, (ii) TCL alone, and (iii) LADM together with TCL.
Identifying and analyzing hcSLs and lcSLs
For each pair of GTEx normal–TCGA cancer of the same tissue type (table S2B), we computed the fraction of samples where a cSL gene pair i has both genes lowly expressed (defined above) among the normal samples (fni) and cancer samples (fci) and computed a specific score as rsi = fni − fci. We selected the hcSLs as those whose specific scores are greater than the 75% percentile of all scores and lcSLs as those with a score below the 25% percentile (table S4, A and B). We compared SL significance scores between the hcSLs and lcSLs in each tissue using a Wilcoxon rank sum test. For each type of the GTEx normal tissues used in this analysis (i.e., those that can be mapped to TCGA cancer types), we also computed the TCL as above but using the hcSLs, lcSLs, or all cSLs, respectively, and analyzed their correlation with lifetime cancer risk or cancer onset age across the tissues.
Pathway enrichment of the hcSLs
We designed an empirical enrichment test as below to account for the fact that each cSL consists of two genes. For the hcSLs in each tissue type and each given pathway from the Reactome database (38), we computed the odds ratio (OR) for the overlap between the genes in hcSLs and the genes within the pathway based on the Fisher’s exact test procedure, with the “background” being all the genes in the ISLE-inferred cSLs. A greater than 1 OR indicates that the hcSLs are positively enriched for the genes of the pathway. To determine the significance of the enrichment, we repeatedly and randomly sampled the same number of cSLs as that of the hcSLs, computed the ORs similarly, and computed the empirical P value as the fraction of cases where the OR from the random cSLs is greater than that from the hcSLs. We corrected for multiple testing across pathways with the Benjamini-Hochberg method.
CMAP data analysis and estimation of drug-induced cSL load change from gene expression signature
The phase I CMAP (26) data were downloaded from the Gene Expression Omnibus database (GSE92742). Level 5 data that represent the consensus perturbation-induced differential expression signature were used. We focused on CMAP data that involve treatment by specific classes of chemotherapy drugs (mutagenic: alkylating agents and DNA topoisomerase inhibitors; nonmutagenic: taxanes and vinca alkaloids) in normal cell lines or primary cells. We identified a total of 11 drugs tested in four cell types. Given the signature (z score) of a drug treatment in a cell, we estimated the drug-induced cSL load change as follows
where S is the set of cSLs, and |S| is the total number of cSL gene pairs. A gene pair is denoted by (i, j), and zi and zj are the z scores of gene i and gene j, respectively. I() is the indicator function. Intuitively, the above formula quantifies the number of cSL gene pairs where both genes are down-regulated with a z score cutoff of −0.5 (i.e., contributing to cSL load increase), minus the number of cSL gene pairs where either gene is up-regulated with a z score cutoff of 0.5 (i.e., contributing to cSL load decrease), normalized by the total number of cSL gene pairs. We then tested whether the mutagenic drugs lead to a larger decrease in cSL load compared to nonmutagenic drugs with a linear model that controls for both cell type and drug.
Analyzing the tissue specificity of TSGs
We obtained the list of TSGs and their associated tissue types from the COSMIC database (11) (https://cancer.sanger.ac.uk/cosmic/download, the “Cancer Gene Census” data; table S6A). For each TSG, their cSL partner genes were identified using the ISLE pipeline (22) with an FDR cutoff of 0.1 (table S6B). Here, the FDR cutoff is more stringent than that used for the pan-cancer genome-wide cSL network (FDR < 0.2 for the main results) since, here, FDR correction was performed for each TSG, corresponding to a much lower number of multiple hypotheses. As a result, the FDR correction has more power, and a relatively more stringent cutoff can give rise to a more reasonable number of cSL partner genes per TSG. We focused our analysis on 23 TSGs for which more than one cSL partner genes were identified (no cSL partner was identified for most of the other TSGs). The expression levels of the cSL partner genes were then compared between tissue type(s) where the TSG is a known driver and the rest of the tissues where the TSG is not an established driver with linear models. Specifically, the expression levels of the cSL partners were modeled with two explanatory variables: (i) driver status of the TSG in the tissue (binary) and (ii) cSL partner gene (categorical, indicating each of the cSL partner genes of a TSG). The coefficient and P value associated with variable (i) were used to analyze the general trend of differential expression among the cSL partner genes. Positive coefficients of variable (i) means that the expression levels of the cSL partner genes are, on average, higher in the tissue(s) where the TSG is a known driver compared to those in the tissues where the TSG is not an established cancer driver.
Supplementary Material
Acknowledgments
We thank S. J. Chanock, B. M. Ryan, T. Misteli, S. Hannenhalli, X. W. Wang, C. C. Harris, A. Schaffer, M. Gertz, K. Wang, S. Patkar, S. Sinha, and F. Schischlik for critical comments and suggestions on our study and the manuscript. Funding: This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, and the Center for Cancer Research. This work used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). The GTEx Project was supported by the Common Fund of the Office of the Director of the NIH and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. K.C. is supported by the NCI-UMD Partnership for Integrative Cancer Research fellowship. Author contributions: K.C., N.U.N., and E.R. formulated the research question and study design. K.C. and N.U.N. carried out the analysis. J.S.L. provided help with the computational cSL prediction pipeline. K.C., N.U.N., and E.R. analyzed the results. K.C., N.U.N., and E.R. wrote the manuscript with inputs from J.S.L. E.R. supervised the research. The manuscript has been read and approved by all the authors. Competing interests: E.R. is a cofounder and scientific consultant of Pangea Therapeutics (www.pangeamedicine.com), which focuses on precision oncology and SL; however, E.R. has divested all shares and receives no salary or financial benefit from this company. The work in this manuscript is not related to the work of this company. The other authors declare that they have no competing interests. Correspondence and requests for materials should be addressed to E.R. at eytan.ruppin@nih.gov. Data and materials availability: The use of publicly available data has been described in Methods. All data needed to evaluate the conclusions in the paper and the R code are available at https://hpc.nih.gov/~chengk6/SL_cancer_risk.zip.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabc2100/DC1
REFERENCES AND NOTES
- 1.Surveillance, Epidemiology, and End Results (SEER) Program, “SEER*Stat Database Incidence - SEER 9 Regs Research Data, Nov 2017 Sub (1973–2015) - Linked To County Attributes - Total U.S., 1969–2016 Counties,” National Cancer Institute, DCCPS, Surveillance Research Program, released April 2018, based on the November 2017 submission (2018); www.seer.cancer.gov.
- 2.Tomasetti C., Vogelstein B., Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomasetti C., Li L., Vogelstein B., Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu S., Powers S., Zhu W., Hannun Y. A., Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feinberg A. P., Ohlsson R., Henikoff S., The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Klutstein M., Moss J., Kaplan T., Cedar H., Contribution of epigenetic mechanisms to variation in cancer risk among tissues. Proc. Natl. Acad. Sci. U.S.A. 114, 2230–2234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sauer A. G., Siegel R. L., Jemal A., Fedewa S. A., Current prevalence of major cancer risk factors and screening test use in the United States: Disparities by education and race/ethnicity. Cancer Epidemiol. Biomark. Prev. 28, 629–642 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Armitage P., Doll R., The age distribution of cancer and a multi-stage theory of carcinogenosis. Int. J. Epidemiol. 33, 1174–1179 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Bailey M. H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M. C., Kim J., Reardon B., Ng P. K.-S., Jeong K. J., Cao S., Wang Z., Gao J., Gao Q., Wang F., Liu E. M., Mularoni L., Rubio-Perez C., Nagarajan N., Cortés-Ciriano I., Zhou D. C., Liang W.-W., Hess J. M., Yellapantula V. D., Tamborero D., Gonzalez-Perez A., Suphavilai C., Ko J. Y., Khurana E., Park P. J., Van Allen E. M., Lian H.; The MC3 Working Group; The Cancer Genome Atlas Research Network, Lawrence M. S., Godzik A., Lopez-Bigas N., Stuart J., Wheeler D., Getz G., Chen K., Lazar A. J., Mills G. B., Karchin R., Ding L., Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haigis K. M., Cichowski K., Elledge S. J., Tissue-specificity in cancer: The rule, not the exception. Science 363, 1150–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Tate J. G., Bamford S., Jubb H. C., Sondka Z., Beare D. M., Bindal N., Boutselakis H., Cole C. G., Creatore C., Dawson E., Fish P., Harsha B., Hathaway C., Jupe S. C., Kok C. Y., Noble K., Ponting L., Ramshaw C. C., Rye C. E., Speedy H. E., Stefancsik R., Thompson S. L., Wang S., Ward S., Campbell P. J., Forbes S. A., COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elledge S. J., Amon A., The BRCA1 suppressor hypothesis: An explanation for the tissue-specific tumor development in BRCA1 patients. Cancer Cell 1, 129–132 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Schneider G., Schmidt-Supprian M., Rad R., Saur D., Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 17, 239–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bridges C. B., The origin of variations in sexual and sex-limited characters. Amer. Nat. 56, 51–63 (1922). [Google Scholar]
- 15.Bender A., Pringle J. R., Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 1295–1305 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartwell L. H., Szankasi P., Roberts C. J., Murray A. W., Friend S. H., Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Lord C. J., Ashworth A., PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Neil N. J., Bailey M. L., Hieter P., Synthetic lethality and cancer. Nat. Rev. Genet. 18, 613–623 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Parameswaran S., Kundapur D., Vizeacoumar F. S., Freywald A., Uppalapati M., Vizeacoumar F. J., A road map to personalizing targeted cancer therapies using synthetic lethality. Trends Cancer. 5, 11–29 (2019). [DOI] [PubMed] [Google Scholar]
- 20.GTEx Consortium , The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R. M., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., Stuart J. M., The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J. S., Das A., Jerby-Arnon L., Arafeh R., Auslander N., Davidson M., McGarry L., James D., Amzallag A., Park S. G., Cheng K., Robinson W., Atias D., Stossel C., Buzhor E., Stein G., Waterfall J. J., Meltzer P. S., Golan T., Hannenhalli S., Gottlieb E., Benes C. H., Samuels Y., Shanks E., Ruppin E., Harnessing synthetic lethality to predict the response to cancer treatment. Nat. Commun. 9, 2546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng X., Arang N., Rigiracciolo D. C., Lee J. S., Yeerna H., Wang Z., Lubrano S., Kishore A., Pachter J. A., König G. M., Maggiolini M., Kostenis E., Schlaepfer D. D., Tamayo P., Chen Q., Ruppin E., Gutkind J. S., A platform of synthetic lethal gene interaction networks reveals that the GNAQ uveal melanoma oncogene controls the Hippo pathway through FAK. Cancer Cell 35, 457–472.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nijman S. M. B., Friend S. H., Cancer. Potential of the synthetic lethality principle. Science 342, 809–811 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Schyman P., Printz R. L., Estes S. K., O’Brien T. P., Shiota M., Wallqvist A., Concordance between thioacetamide-induced liver Injury in rat and human in vitro gene expression data. Int. J. Mol. Sci. 21, 4017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A., Narayan R., Corsello S. M., Peck D. D., Natoli T. E., Lu X., Gould J., Davis J. F., Tubelli A. A., Asiedu J. K., Lahr D. L., Hirschman J. E., Liu Z., Donahue M., Julian B., Khan M., Wadden D., Smith I. C., Lam D., Liberzon A., Toder C., Bagul M., Orzechowski M., Enache O. M., Piccioni F., Johnson S. A., Lyons N. J., Berger A. H., Shamji A. F., Brooks A. N., Vrcic A., Flynn C., Rosains J., Takeda D. Y., Hu R., Davison D., Lamb J., Ardlie K., Hogstrom L., Greenside P., Gray N. S., Clemons P. A., Silver S., Wu X., Zhao W.-N., Read-Button W., Wu X., Haggarty S. J., Ronco L. V., Boehm J. S., Schreiber S. L., Doench J. G., Bittker J. A., Root D. E., Wong B., Golub T. R., A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szikriszt B., Póti Á., Pipek O., Krzystanek M., Kanu N., Molnár J., Ribli D., Szeltner Z., Tusnády G. E., Csabai I., Szallasi Z., Swanton C., Szüts D., A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 17, 99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans , Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 100 (Pt. B), 1–441 (2012). [PMC free article] [PubMed] [Google Scholar]
- 29.Ferguson L. R., Pearson A. E., The clinical use of mutagenic anticancer drugs. Mutat. Res. 355, 1–12 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Gundy S., Baki M., Bodrogi I., Vinblastine, cisplatin and bleomycin (VPB) adjuvant therapy does not induce dose-dependent damage in human chromosomes. Neoplasma 36, 457–464 (1989). [PubMed] [Google Scholar]
- 31.Mascaux C., Angelova M., Vasaturo A., Beane J., Hijazi K., Anthoine G., Buttard B., Rothe F., Willard-Gallo K., Haller A., Ninane V., Burny A., Sculier J.-P., Spira A., Galon J., Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 571, 570–575 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Magen A., Sahu A. D., Lee J. S., Sharmin M., Lugo A., Gutkind J. S., Schäffer A. A., Ruppin E., Hannenhalli S., Beyond synthetic lethality: Charting the landscape of pairwise gene expression states associated with survival in cancer. Cell Rep. 28, 938–948.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullard A., Synthetic lethality screens point the way to new cancer drug targets. Nat. Rev. Drug Discov. 16, 736 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Horlbeck M. A., Xu A., Wang M., Bennett N. K., Park C. Y., Bogdanoff D., Adamson B., Chow E. D., Kampmann M., Peterson T. R., Nakamura K., Fischbach M. A., Weissman J. S., Gilbert L. A., Mapping the Genetic Landscape of Human Cells. Cell 174, 953–967.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mailman M. D., Feolo M., Jin Y., Kimura M., Tryka K., Bagoutdinov R., Hao L., Kiang A., Paschall J., Phan L., Popova N., Pretel S., Ziyabari L., Lee M., Shao Y., Wang Z. Y., Sirotkin K., Ward M., Kholodov M., Zbicz K., Beck J., Kimelman M., Shevelev S., Preuss D., Yaschenko E., Graeff A., Ostell J., Sherry S. T., The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 39, 1181–1186 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldman M., Craft B., Hastie M., Repečka K., Kamath A., McDade F., Rogers D., Brooks A. N., Zhu J., Haussler D., The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv , 326470 (2019). [Google Scholar]
- 37.D. M. Parkin, C. S. Muir, S. J. Whelan, Y. T. Gao, J. Ferlay, J. Powell, Cancer Incidence in Five Continents (IARC, 1992). [Google Scholar]
- 38.Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B., Milacic M., Roca C. D., Rothfels K., Sevilla C., Shamovsky V., Shorser S., Varusai T., Viteri G., Weiser J., Wu G., Stein L., Hermjakob H., D’Eustachio P., The Reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–D655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wendt H. W., Dealing with a common problem in social science: A simplified rank-biserial coefficient of correlation based on the U statistic. Eur. J. Soc. Psychol. 2, 463–465 (1972). [Google Scholar]
- 40.Venet D., Dumont J. E., Detours V., Most random gene expression signatures are significantly associated with breast cancer outcome. PLOS Comput. Biol. 7, e1002240 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P. W., Levine D. A., Carter S. L., Getz G., Stemke-Hale K., Mills G. B., Verhaak R. G., Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G. N., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., Mc Manus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aryee M. J., Jaffe A. E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A. P., Hansen K. D., Irizarry R. A., Minfi: A flexible and comprehensive bioconductor package for the analysis of Infinium DNA Methylation microarrays. Bioinformatics 30, 1363–1369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabc2100/DC1