

Abstract
The relevance of circulating tumor DNA (ctDNA) analysis as a liquid biopsy and minimal residual disease tool in the management of classical Hodgkin lymphoma (cHL) patients was demonstrated in retrospective settings and remains to be confirmed in a prospective setting. We developed a targeted Next-Generation sequencing (NGS) panel for fast analysis (AmpliSeq® technology) of nine commonly mutated genes in biopies and ctDNA of cHL patients. We then conducted a prospective trial to assess ctDNA follow-up at diagnosis and after two cycles (C2) of chemotherapy. Sixty cHL patients treated by first line conventional chemotherapy (BEACOPPescalated [21.3%], ABVD/ABVD-like [73.5%] and other regimens [5.2%, for elderly patients]) were assessed in this noninterventional study. The median age of the patients was 33.5 years (range: 20-86). Variants were identified in 42 (70%) patients. Mutations of NFKBIE, TNFAIP3, STAT6, PTPN1, B2M, XPO1, ITPKB, GNA13 and SOCS1 were found in 13.3%, 31.7%, 23.3%, 5%, 33.3%, 10%, 23.3%, 13.3% and 50% of patients, respectively. ctDNA concentration and genotype were correlated with clinical characteristics and presentation. Regarding early therapeutic response, 45 patients (83%, not available [NA] =6) had a negative positron emission tomography (PET) after C2 (Deauville Score 1-3). The mean of DeltaSUVmax after C2 was -78.8%. ctDNA after C2 was analysed in 54 patients (90%). ctDNA became rapidly undetectable in all cases after C2. Variant detection in ctDNA is suitable to depict the genetic features of cHL at diagnosis and may help to assess early treatment response, in association with PET. Clinical Trial reference: NCT02815137.
Introduction
Classical Hodgkin lymphoma (cHL) patients have greatly benefited from multiagent chemotherapy and improved radiation techniques and 65-90% of patients can achieve disease-free survival after 5 years, depending on stage and clinical risk factors.1 Those with a rapid response to initial treatment have the best outcomes and may benefit from truncated, less-toxic treatment regimens.2 Nevertheless, approximately 20-25% of patients will ultimately experience either primary refractoriness to chemotherapy (within 3 months of doxorubicin-based chemotherapy), early disease relapse (within 12 months after the end of first-line treatment) or late disease relapse,3 underlying the need to understand the mechanisms involved and to identify predictive biomarkers.
Until recent years, the genetic overview of cHL was poorly described because of the scarcity of Hodgkin and Reed-Sternberg (HRS) tumor cells, which usually account for only about 0.1-3% of cells in the tissue,4 complicating biological analyzes that lacked sensitivity. Due to the scarcity of HRS cells in cHL patient biopsies, finding recurrent mutations could be easier in the plasma circulating tumor DNA (ctDNA) of these patients, with potentially fewer heterogeneity issues than tumor tissue testing. The concept of "liquid biopsy" has recently been highlighted in a series of cHL patients,6 for whom high-throughput sequencing (HTS) of a panel of target genes was performed, with successful detection of soma-tic variants both in the tumor and in the plasma. In a previous retrospective study, we investigated the prevalence and clinical relevance of XPO1 E571K in cHL cases7 and demonstrated that this recurrent mutation was detectable in both tumor and plasma, indicating that XPO1 mutations represent a new genetic biomarker, useful both at the time of diagnosis and as a minimal residual disease (MRD) marker.
In order to confirm the results of this previous work, we have developed a prospective observational study (NCT02815137) with a focused and limited array of genes to implement liquid biopsy at different times throughout the standard initial cHL management and to identify in this larger number of patients at least one molecular abnormality with a role in oncogenesis or of prognostic or therapeutic impact that could be used as a biomarker of follow-up during treatment. Our Next-Generation sequencing (NGS) and digital droplet PCR (ddPCR) panel was designed after analysis of literature data on recurrent somatic mutations described in cHL and the validity of this panel was established on a series of 24 patients with cHL in a previous publication. 8 A comparison with previous results recently published by Spina et al.6 showed very similar rates of mutations detected in ctDNA targeting STAT6, B2M, XPO1, NFKBIE and TNFAIP3.
The goal of the present work was to establish the feasibility and validity of prospective ctDNA monitoring, in order to propose new strategies for both tailored diagnosis and treatment based on the simple detection and quantification of acquired mutations in the plasma of cHL patients.
Methods
Patients
For exploratory and feasibility purposes, we prospectively included in this biological study adult patients treated by adriamycin (doxorubicin), bleomycin, vinblastine, dacarbazine (ABVD) or BEACOPPescalated for newly diagnosed stage I-IV cHL at the Henri Becquerel Center (Rouen, France) between 2016 and 2018. According to these inclusion criteria, 64 patients were initially included but four were then excluded (two histological diagnostic errors, one underage patient, one loss to follow-up just after inclusion) leading to a final number of 60 patients analyzed. The exclusion criteria were: patients who had i) already started any treatment (including steroids) before signing the informed consent, ii) contraindication to positron emission tomography (PET), iii) positive HIV, hepatitis B or C serology, and iv) pregnant women. As this was an observational biological study, physicians were blinded to the results of the molecular analysis and these did not alter the routine care of the patients. All experiments were performed in accordance with the Helsinki Declaration and the study was approved by an institutional review board and ethics committee (N° ID-RCB: 2016- A00202-49). All patients gave informed consent for specimen collection, clinical data collection and biomarker analysis. Patients were treated according to routine local recommendations for the management of patients with a diagnosis of de novo cHL.
Blood specimens
Blood samples were obtained by blood draws at diagnosis and after two cycles (C2) of chemotherapy, and another blood sample was obtained in case of disease relapse. Eighteen mL of blood samples were collected in EDTA tubes at the scheduled timepoints during patient management. Samples were centrifuged within 2 hours for 10 minutes at 2,600 g (3,500 rpm) at 4°C to isolate the plasma, which was then stored at -80°C.
NGS and statistical analysis
NGS of targeted genes was performed on an Ion Torrent Personal Genome Machine™ (PGM, Thermo Fisher Scientific). Our panel targets the four basepair hotspot deletion in NFKBIE (exon 1) and the coding regions of the following genes: ITPKB (exons 2 to 8), PTPN1 (exons 1 to 10), TNFAIP3 (exons 2 to 9), SOCS1 (exon 2), STAT6 (exons 12 and 14), B2M (exons 1 to 3), XPO1 (exons 15 to 18) and GNA13 (exons 1 to 4). Based on a theoretical sensitivity of 1%, and on a minimum number of mutated reads equal to 50, the minimum depth of desired sequencing was set at 5,000X. The circulating ctDNA concentrations were expressed in haploid genome equivalents (hGE) per mL of plasma (hGE/mL) and calculated by multiplying the mean variant allelic frequency (VAF) for all mutations used for detection calling by the concentration of cell-free DNA (cfDNA) (pg/mL of plasma) and dividing by 3.3, using the assumption that each haploid genomic equivalent weighs 3.3 pg, as previously described by Scherer et al.9 Taking into account previous experiences in our laboratory,8,10 we retained a threshold of 0.5% of VAF as the lower detection limit for the present study. Statistics were performed with R software v3.3.2 (see the Online Supplementary Materials and Methods).
Results
Somatic mutations in cHL at the time of initial diagnosis
The main clinical features, disease characteristics, and first line treatments of the 60 patients are summarized in Table 1. Briefly, the median age was 33.5 years old, 91.4% of the patients had an excellent performance status (ECOG 0-1) and the majority of patients (55%) displayed B symptoms. The predominance of the scleronodular subtype was confirmed (70%) and we observed a predominance of localized stage I-II disease (51.7%). Regarding disease extension, the bone marrow biopsy, when performed, was normal in the majority of cases (30 of 37 patients, 81.1%). The presence of a bulky mass greater than 10 centimeters (cm) concerned 15% of the patients. Table 2 presents the somatic variants detected by HTS sequencing of biopsy and plasma ctDNA. Patients were considered mutated if they had a mutation in their biopsy-extracted genomic DNA and/or plasma ctDNA. Variants were found in 42 (70%) patients: 21 of 30 (71%) and 41 of 60 (68.8%) of available biopsy and ctDNA samples respectively. In all, 145 different variants were identified: (i) 127 variants in plasma samples, of which were 66 not found in the corresponding biopsies; (ii) 79 in the biopsy samples, of which 18 variants were not found in the corresponding plasma samples. The median VAF were superior in ctDNA than in biopsy (1.99% vs. 1.6%, P=0.024).
Concordance between genetic profiles of biopsy and ctDNA (in terms of double-positivity or double-negativity of samples) was acceptable for 25 of 30 patients (83.3%, Cohen’s k =0.56 [range: 0.23-0.89]). This indicates that 20 patients with a positive genetic biopsy profile also presented the same genetic ctDNA profile, and five patients with a negative genetic biopsy profile also presented no mutations in ctDNA, leading to 25 patients with concordance. More precisely, similarity at the level of the variants in paired biopsy and plasma samples was found for 61 variants (variant identified both in the plasma and in the biopsy of the same patient).
Significant higher plasma ctDNA concentration levels at diagnosis was associated with the following unfavorable clinical characteristics: sedimentation rate ≥50 mm, albumin ≤40 g/L, advanced stage disease, lymphopenia <0.6 G/L, presence of B symptoms; Hasenclever International Prognostic Score ≥3, ≥ 4 involved nodal areas, presence of anemia (hemoglobin level <10.5 g/dL) and elevated lactate dehydrogenase (LDH) (>480 UI/L) (Table 3). Mutations of NFKBIE, TNFAIP3, STAT6, PTPN1, B2M, XPO1, ITPKB, GNA13 and SOCS1 were respectively found in the following proportions of patients (mean number of variants by patient [range]): 13.3% (1.1 [1-2]), 31.7% (1.1 [1-2]), 23.3% (1.4 [1-2]), 5% (1.3 [1-2]), 33.3% (1.3 [1-3]), 10% (1 [1-1]), 23.3% (1.5 [1-4]), 13.3% (1.1 [1-2]) and 50% (1.9 [1-5]).
As a median value, four somatic variants per mutated patient (range: 1-12) were identified. The entire coding sequence of SOCS1was sequenced and was found to be the most frequently mutated gene in this study with 55 somatic variants identified in gDNA and/or ctDNA of 30 patients (50%), some thus presenting two (or more) somatic variants but there is no recurrent variant identified for this gene. Figure 1A shows the number and types of somatic variants identified by gene and patient both in biopsy and ctDNA. In exons 1 to 3 of the B2M gene, 23 somatic variants were found in 20 patients (33.3% of patients). STAT6 is frequently mutated with 10 somatic variants identified in 14 patients (23.3% of patients). These variants are not mutually exclusive and can present on the same allele when the patient is double-mutated (five double-mutated patients). Among these mutations, the N417Y mutation is the most frequent, present in six patients (10%). The coding sequence of the TNFAIP3 gene was sequenced in its entirety, which allowed the identification of 21 somatic variants in 19 patients (31.7% of patients). Two different variants were identified on exons 15 to 18 of the XPO1 gene, in six patients (10%). Unsupervised hierarchical clustering was performed among the nine genes to represent the association of alterations (Figure 1B).
For 8 of the 20 patients presenting similar mutations in both biopsy and plasma samples, the variants identified were strictly identical. For the 12 others, there were discrepancies (somatic variants common between the two tissues, with additional variants in one or both of the tissues). For these 20 patients, the median VAF in the biopsy and the matched plasma were equal to 1.61% and 2.3% respectively (P=0.04).
Mean baseline ctDNA concentration was 323.3 hGE/mL (range: 0-2684.9) at diagnosis. Figure 3A shows the distribution of ctDNA median VAF for the 41 mutated patients with “positive plasma” at diagnosis.
PET measurements, therapeutic response and survival analysis
There was a moderate positive Spearman’s correlation between metabolic tumor volume (MTV) and ctDNA concentration (r=0.47, P<0.001) and between MTV and ctDNA median VAF (r=0.57, P<0.001) for the 41 “positive” plasma samples at diagnosis. Fifty-four interim PET exams were performed after C2: 45 patients displayed a complete metabolic response and nine patients displayed a partial metabolic response (Deauville Scale [DS]).4 Among these nine patients, eight were in complete remission at end of treatment and one elderly patient (universal patient identification number [UPN] 39), who had no detectable mutations either at diagnosis or after C2, relapsed and died from disease progression 1 year after diagnosis. Patient UPN43 had no blood collection after C2 (organizational issue) and was in complete remission at end of treatment. Patient UPN44 had no interim PET scan performed after C2 because the patient was scheduled to receive C8 of ABVD and PET was performed after C4 (metabolic complete response) but we do not have any blood collection after C4.
Table 1.
Patient characteristics.
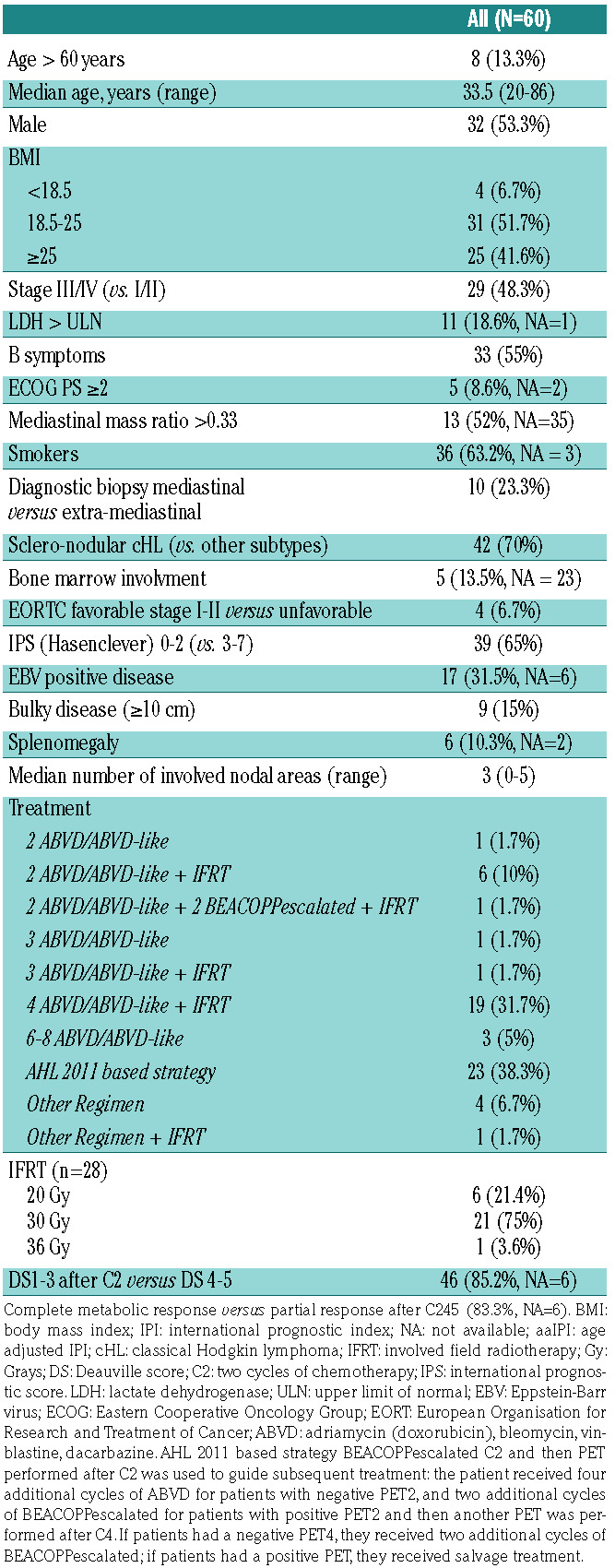
Figure 1.

Number and types of somatic variants identified by gene and patient both in biopsy and circulating tumor DNA. (A) Heatmap representing somatic variants detected in circulating tumor DNA (ctDNA) in the mutated patients (the "Burden" track represents the mutational load [“burden”] of a patient based on the maximum/minimum number of mutations found in the complete cohort). (B) Unsupervised hierarchical clustering performed among the nine genes to represent the association of alterations (numbers within the clustering represent the number of observed associations between two genes).
Table 2.
Synthesis of mutational profiles at diagnosis of the patient cohort.
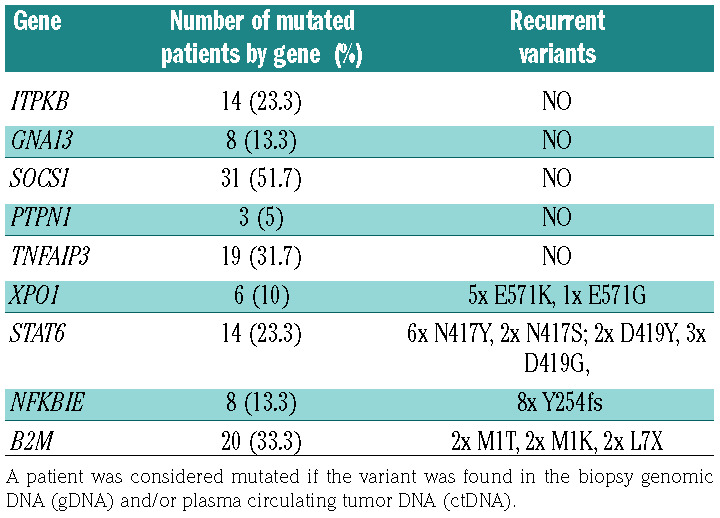
Figure 2.

Prevalence of somatic mutations detected in DNA extracted from the 31 available biopsies and 60 plasma samples of the patients at time of diagnosis. The "genomic DNA (gDNA) and/or circulating tumor DNA (ctDNA) " column corresponds to the patients considered mutated in the biopsy and/or the plasma samples.
The metabolic complete remission rate (DS 1-3) after C2 of chemotherapy and at the end of treatment were 85.2% (not done [ND] =6) and 86% [ND=17]), respectively. The mean of DeltaSUVmax after C2 was -78.8%. Two patients (UPN21 and UPN58) died after the C1 of chemotherapy and two patients experienced an early relapse. The median follow- up of patients was 22.7 months (range: 11.4-38.9 months). Finally, 1-year overall survival and progression-free survival rates were 94.9% (range: 89.3-100%) and 89.8 % (range: 82.4-97.9%) respectively.
ctDNA as a biomarker for therapeutic response assessment in addition to PET
In this ongoing trial, plasma ctDNA after C2 of chemotherapy has only been analyzed for 55 of 60 patients, of which 18 presented no somatic mutations at diagnosis in plasma samples and were therefore not informative for therapeutic molecular response assessment after C2. The remaining five non-analyzed patients had either no available plasma sample after C2 (two patients died before C2, one patient with loss of follow-up and two patients without performed blood collection).
Figure 4 schematically shows the somatic mutation VAF variation in the plasma ctDNA of the 41 mutated evaluable patients between diagnosis and after C2 of chemotherapy. No somatic variant was identified in the blood draw performed concomitantly to interim PET after C2 of chemotherapy in any patient, including patient UPN17 who displayed negative PET after C2 (DS 2) but who was then refractory at end of first line treatment (DS 4). In this patient, the plasma ctDNA was positive for ITPKB p.K232FS mutation at diagnosis, negative after C2 and again positive at relapse at end of first line treatment (2x BEACOPPescalated, 4x ABVD) with VAF of 10.6%, 0% and 2.58%, respectively. The kinetics of ctDNA results were concordant with the PET results for this patient. In the whole cohort, we found no statistically significant difference between the concentration of cfDNA (ng/mL of plasma) after C2 among DS 1-3 patients (35 patients, median 35 ng/mL [range: 20.4-260]) versus DS 4-5 patients (seven patients, median 36.2 ng/mL [range: 21.8-80], P=0.79), which suggests a possible nontumor DNA plasma release by inflammatory mechanisms, regardless of the persistence of an active tumor disease.
In addition, Figure 5 illustrates the case of another patient (UPN38) presenting a positive PET after C2 of ABVD with a residual fixation (DS 5) of a left supraclavi-cular lymph node which was surgically removed and corresponded to a reactive lymph node. Ten identical somatic variants had been identified in this patient at diagnosis in both biopsy and plasma samples. For this patient, we were unable to analyze the second biopsy that was performed after C2 of ABVD due to insufficient materiel. However, this patient no longer had a detectable somatic mutation in the plasma after C2 with a clearance of all baseline mutations that were present at diagnosis. The amount of ctDNA was 1434.85 hGE/mL at diagnosis and after C2, the level of ctDNA was measured at 42 ng/mL of plasma. Taken together, these results argue for a false posi-tive result of PET after C2 with a consistent negativity of concomitant ctDNA sample. The patient received irradiation of the left supraclavicular lymph node and is conside-red in complete remission.
Discussion
Herein we confirm the feasibility of non-invasive somatic mutation analysis in a prospective and indepen-dent setting in a large cohort of patients with cHL, as identified by NGS experiments even when using a limited number of target genes. In this particular and potentially curable disease, highly sensitive techniques like targeted NGS are crucial to highlight low frequency mutations. Low-coverage whole genome sequencing,11 sequencing of circulating cfDNA to detect genomic imbalances in HRS cells12 or targeted exome sequencing of isolated HRS cells13 have recently contributed to investigate genetic lesions underlying cHL but sample sizes were very low. In the present work, we found recurrent mutations in biopsies and plasma ctDNA of a cohort of consecutive 60 patients with cHL. This observation is consistent with previously published data regarding liquid biopsy in cHL with a different set of targeted genes6 and could add new information on driver events and tumorigenesis in this disease. As previously described in the literature, cHL displays a singular mutational signature, close to the primary mediastinal large B-cell lymphoma (PMBL) profile, compared to diffuse large B-cell lymphoma (DLBCL). The mutated genes point to the molecular deregulation of specific programs in cHL: NF-kB signature, STAT6 and cytokine signaling pathway (SOCS1). In our patients, mutations of SOCS1 are frequent and are probably involved with STAT6 in the oncogenesis of cHL. This observation is consistent with the literature in PMBL and cHL populations15–18 except for the recent study by Spina et al.6 that did not cover SOCS1.6
Table 3.
Baseline plasma circulating tumor DNA concentration according to patients’ characteristics at diagnosis.

In total, 70% of our patients with cHL harbored detectable somatic mutations. It is remarkable that 52% (127 variants detected in plasma samples, 66 of which were not detected in the tumors) of all somatic variants were only found in the plasma but not in the tumor because of the well-known tumor cell sparsity in Hodgkin lymphoma (HL). This confirms that ctDNA assessment might play an important role to define somatic mutations in this disease at the time of diagnosis. The detection of somatic alterations in the patients’ ctDNA samples represents a major technological advance and a step towards routine liquid biopsies in cHL. Our ctDNA data are fairly consistent with the results of the landmark study by Spina et al.6 despite several methodological differences and different gene panels. We have highlighted in this study seve-ral associations between median concentration of ctDNA at diagnosis, and disease stage, albumin level, sedimentation rate, LDH level and International Prognostic Score (IPS) which shows a correlation between ctDNA level and tumor mass and the inflammation it generates.
The clearance of the mutations in plasma ctDNA was described as a new prognostic marker for mutated patients in other works in DLBCL patients with undetectable ctDNA after C2 who show a superior PFS compared to patients with positive ctDNA.9 Nevertheless, in our study, the finding that all patients clear ctDNA after C2 of therapy needs to be validated with more sensitive approaches that have a limit of detection lower than 0.5% VAF for disease monitoring. It is unlikely that those patients with cleared ctDNA after C2 have all been cured with only C2 of chemotherapy but future studies will probably assess various therapeutic strategies for these patients including the number of chemotherapy cycles needed to ensure the cure of the disease with real-time and dynamic ctDNA analysis. Outcome of patients in our study was excellent and therefore it is not possible to definitely examine associations between ctDNA levels and events. No somatic variant was identified after C2 of chemotherapy suggesting a possible non tumoral source of cfDNA after chemotherapy or an inflammatory process. Unfortunately, we did not have complete blood cell count data to evaluate the inflammatory syndrome after C2 of chemotherapy. In our previously published ctDNA study in DLBCL patients, we had been able to detect somatic variants after C2 of chemotherapy.17 We can hypothesize that our blood sampling process after C2 of chemotherapy may have been too late compared to the kinetics of clearance of ctDNA after treatment in cHL patients, and that it may have been necessary to perform blood sampling after 15 days or after only C1 of chemotherapy. Future studies will probably address this timing concern.
Figure 3.

Distribution of circulating tumor DNA median variant allelic frequency for the 41 mutated patients with positive plasma at diagnosis. ctDNA: circulating tumor DNA; VAF: variant allelic frequency
Figure 4.

Longitudinal assessment of mutation abundance in plasma circulating tumor DNA upon treatment. Evolution of median circulating tumor DNA (ctDNA) variant allelic frequency (VAF) for each patient (with detectable ctDNA mutation at diagnosis) throughout treatment (at diagnosis [“diag”] n=41) and after two cycles (C2) of chemotherapy (“post C2”, n=31). UPN: universal patient identification number.
One of the limitations of our study, in addition to its noninterventional nature with plasma samples analyzed retrospectively in an unblinded fashion with a freeze-thaw step, is the absence of detectable mutations at time of diagnosis for 18 (30%) patients which potentially led to underestimating the prevalence of somatic mutations in this prospective cohort. We also missed some blood collections and so, in our study, ctDNA may be used as a biomarker for therapeutic response assessment in 31 of 60 (51.7%) of patients.
This might be due to insufficient technological sensitivity, along with the examination of a restricted group of genes. We might possibly not have been able to identify ctDNA in patients with a low tumor burden, which limits its utilization as a MRD tool. Determining the technical limit of detection is a complex equation, as it depends on every sample, every variant and on many factors such as sequencer error, total DNA input, sample nature and qua-lity, coverage depth and sequencing quality. In order to determine whether or not we had missed variants in the initial tumor biopsy of the patient, a microdissection approach of HRS cells followed by a specific allele PCR could be performed but unfortunately, we were unable to do this due to insufficient material. Although additional mutations were found in the plasma compared to the tumor (which remains the gold standard), the false negative rate of the ctDNA analysis remains moderate with 20 of 89 (22.5%) of somatic variants missed in the plasma samples at diagnosis. Nevertheless, the variants detected in the plasma might better reflect the entire genetic panorama of the tumor. The few discrepancies between the detection of the mutation in the plasma and the tumor can be explained partly by the poor quality of some of our biopsies with tumor cell scarcity, potentially rendering the mutation detection in biopsy-extracted DNA impossible, and secondly by the absence of tumor DNA release in plasma by certain tumors and the short half-life (10-15 minutes) of circulating DNA in plasma.18
Our panel of genes analyzed is possibly too targeted and insufficiently sensitive to detect certain subclonal mutations of low allelic frequency, close to the noise threshold of the NGS sequencer (VAF 0.1%). In addition, our panel is not sufficient enough to detect somatic alterations in all patients due to the limited amount of included genes. The knowledge of cHL biology is booming and it will be ne-cessary to include other genes in future NGS panels such as ATM, KMT2D, TP53, ARID1A and CIITA, as suggested by results of the study by Spina et al.,6 in order to approach a more clinically relevant degree of informativeness (90%). However, this panel of genes is simple, reliable and inexpensive and seems to offer a good compromise between cost, sensitivity and easy applicability. In our opinion, the principle of a focused panel analysis for all cHL patients is an appropriate method to depict the genetic features of cHL at diagnosis and relapse. Another limitation of our technique is the absence of coverage of the immunoglobulin heavy chain locus for clonotypic immunoglobulin-gene rearrangement detection in ctDNA.19
Considering the baseline PET, ctDNA was significantly correlated to the MTV. Moreover, for the 41 “positive” plasma at diagnosis, the baseline MTV and the median plasmatic VAF were also significantly correlated. Such a correlation has been found in other lymphomas such as diffuse large cell B lymphomas17 and follicular lymphomas.19 Although significant, the correlations remained moderate (maximal =0.57 between the baseline MTV and the median plasmatic VAF). Several factors could explain these modest correlations, such as the scarcity of tumor cells, the short half-life of circulating DNA in plasma18 and the fact that some mutations may not have been explored. However, it is important to note the moderate value of these correlations.
Regarding other methods of MRD in cHL, it has been demonstrated that pretreatment levels of plasma Epstein Barr virus DNA (EBV-DNA), as determined by quantitative real-time PCR (qRT-PCR), are correlated with unfavorable outcomes among a large prospective cohort of 274 patients with previously untreated, advanced-stage HL a.21 Moreover, patients with plasma EBV(+) at month 6 after treatment also had an inferior outcome compared with plasma EBV(-) patients. In order to improve the clinical relevance of liquid biopsy in cHL, an increase in the sensitivity of somatic variant detection appears to be crucial. Sensitivity of somatic mutation analysis might be increased by (i) implementing new targets like EBV viral load; (ii) supplementary targeted genes or non-coding regions commonly altered by mutations identified in future whole genome analysis; and (iii) technological progress with greater read depth and more advanced bioinformatic pipelines.
Figure 5.

Example of possible application of liquid biopsy analysis. Patient (UPN38) presenting a positive positron emission tomography (PET) scan with residual fixation of a left supraclavicular lymph node (Deauville Scale 5) after two cycles (C2) of adriamycin (doxorubicin), bleomycin, vinblastine, dacarbazine (ABVD): the biopsy showed a reactive lymph node, indicating a false positive PET scan. Plasma circulating tumor DNA (ctDNA) sample was negative at time of the PET post C2 of ABVD with a clearance of all baseline mutations that were present at diagnosis. UPN: universal patient identification number.
Finally, our data point out that ctDNA is an easy method to genotype cHL and might be valuable for the clinical management of the patients. However, our panel is not sufficient to monitor MRD in all patients and could not replace a larger panel of genes in order to precisely evaluate MRD. Our limited gene panel may not perform well enough and should be further improved to better assess its clinical utility in cHL.
Future works with upgraded cfDNA detection, using interventional designs and larger patient cohorts will help to affirm that ctDNA analysis can be decisive for immediate non-invasive detection of patients with primary refractory disease and in post-therapeutic follow-up. We may speculate that ctDNA will soon allow convenient whole exome sequencing, which would enable the identification of somatic variants likely to respond to targeted therapy in relapsing patients.
Conclusion
The assessment of somatic mutations in plasma ctDNA by NGS is suitable as a biomarker in cHL for both diagnostic aid and therapeutic response evaluation, in association with PET. ctDNA concentration levels and genotypes are correlated with clinical characteristics and disease presentation. These data have to be confirmed in a large dedicated interventional study based on ctDNA MRD detection compared to PET.
Supplementary Material
Acknowledgments
The authors would like to thank all the patients that provided samples for the study. The authors would like to thank Mrs Sorina-Dana Mihailescu for statistical analysis support.
Funding Statement
Funding: This work was supported by grants from the Centre Henri Becquerel and the Canceropole nord ouest (CNO).
References
- 1.Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339(21):1506-1514. [DOI] [PubMed] [Google Scholar]
- 2.Hutchings M, Loft A, Hansen M, et al. FDGPET after two cycles of chemotherapy predicts treatment failure and progression-free survival in Hodgkin lymphoma. Blood. 2006;107(1):52-59. [DOI] [PubMed] [Google Scholar]
- 3.Canellos GP, Rosenberg SA, Friedberg JW, Lister TA, Devita VT. Treatment of Hodgkin lymphoma: a 50-year perspective. J Clin Oncol. 2014;32(3):163-168. [DOI] [PubMed] [Google Scholar]
- 4.Schmitz R, Stanelle J, Hansmann M-L, Küppers R. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma. Annu Rev Pathol. 2009;4151-174. [DOI] [PubMed] [Google Scholar]
- 5.Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spina V, Bruscaggin A, Cuccaro A, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018; 131(22):2413-2425. [DOI] [PubMed] [Google Scholar]
- 7.Camus V, Stamatoullas A, Mareschal S, et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica. 2016; 101(9):1094-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bessi L, Viailly P-J, Bohers E, et al. Somatic mutations of cell-free circulating DNA detected by targeted next-generation sequencing and digital droplet PCR in classical Hodgkin lymphoma. Leuk Lymphoma. 2019;60(2):498-502. [DOI] [PubMed] [Google Scholar]
- 9.Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8(364):364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bohers E, Viailly P-J, Becker S, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: analysis of a prospective cohort. Blood Cancer J. 2018; 8(8):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salipante SJ, Adey A, Thomas A, et al. Recurrent somatic loss of TNFRSF14 in classical Hodgkin lymphoma. Genes Chromosomes Cancer. 2016;55(3):278-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandenberghe P, Wlodarska I, Tousseyn T, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin’s lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol. 2015;2(2):e55-e65. [DOI] [PubMed] [Google Scholar]
- 13.Reichel J, Chadburn A, Rubinstein PG, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood. 2015; 125(7):1061-1072. [DOI] [PubMed] [Google Scholar]
- 14.Mottok A, Renne C, Willenbrock K, Hansmann M-L, Brauninger A. Somatic hypermutation of SOCS1 in lymphocytepredominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood. 2007;110(9):3387-3390. [DOI] [PubMed] [Google Scholar]
- 15.Ritz O, Guiter C, Dorsch K, et al. STAT6 activity is regulated by SOCS-1 and modulates BCL-XL expression in primary mediastinal B-cell lymphoma. Leukemia. 2008; 22(11):2106-2110. [DOI] [PubMed] [Google Scholar]
- 16.Weniger MA, Melzner I, Menz CK, et al. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho- STAT5 accumulation. Oncogene. 2006; 25(18):2679-2684. [DOI] [PubMed] [Google Scholar]
- 17.Bohers E, Viailly P-J, Becker S, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: analysis of a prospective cohort. Blood Cancer J. 2018; 8(8):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elshimali Y, Khaddour H, Sarkissyan M, Wu Y, Vadgama J. The clinical utilization of circulating cell free DNA (CCFDNA) in blood of cancer patients. Int J Mol Sci. 2013; 14(9):18925-18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delfau-Larue M-H, van der Gucht A, Dupuis J, et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: distinct prognostic value in follicular lymphoma. Blood Adv. 2018;2(7):807-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanakry JA, Li H, Gellert LL, et al. Plasma Epstein-Barr virus DNA predicts outcome in advanced Hodgkin lymphoma: correlative analysis from a large North American cooperative group trial. Blood. 2013; 121(18): 3547-3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.