

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer-related death with a median survival time of 6–12 months. Most patients present with disseminated disease and the majority are offered palliative chemotherapy. With no approved treatment modalities for patients who progress on chemotherapy, we explored the effects of long-term Gemcitabine on the tumor microenvironment in order to identify potential therapeutic options for chemo-refractory PDAC. Using a combination of mouse models, primary cell line-derived xenografts, and established tumor cell lines, we first evaluated chemotherapy-induced alterations in the tumor secretome and immune surface proteins by high throughput proteomic arrays. In addition to enhancing antigen presentation and immune checkpoint expression, Gemcitabine consistently increased the synthesis of CCL/CXCL chemokines and TGFβ-associated signals. These secreted factors altered the composition of the tumor stroma, conferring Gemcitabine resistance to cancer-associated fibroblasts in vitro and further enhancing TGFβ1 biosynthesis. Combined Gemcitabine and anti-PD-1 treatment in transgenic models of murine PDAC failed to alter disease course unless mice also underwent genetic or pharmacologic ablation of TGFβ signaling. In the setting of TGFβ signaling deficiency, Gemcitabine and anti-PD-1 led to a robust CD8+ T-cell response and decrease in tumor burden, markedly enhancing overall survival. These results suggest that Gemcitabine successfully primes PDAC tumors for immune checkpoint inhibition by enhancing antigen presentation only following disruption of the immunosuppressive cytokine barrier. Given the current lack of third-line treatment options, this approach warrants consideration in the clinical management of Gemcitabine-refractory PDAC.
Keywords: Pancreatic Cancer, Tumor Microenvironment, Chemotherapy, Chemoresistance, Immunotherapy
Graphical Abstract
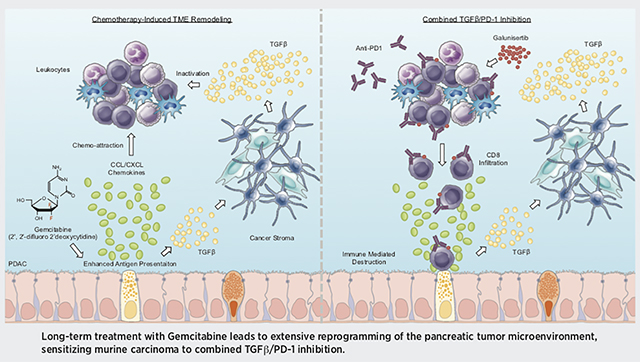
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) often presents at late clinical stages and is associated with poor outcomes. As the majority of patients are diagnosed with metastatic disease and are not eligible for surgery, most are managed through broad-spectrum chemotherapy (1). While aggressive chemotherapy marginally improves survival, nearly all tumors either have or develop some degree of drug resistance. Thus median survival remains a dismal 6–12 months (2). Gemcitabine was first approved for use in metastatic PDAC in 1996 (3–5), and is used first line with albumin conjugated (nab) Paclitaxel (6). Additionally, the FOLFIRINIOX regimen (Folinic Acid, 5-Fluorouracil, Irinotecan, and Oxaliplatin) is also widely used in metastatic PDAC, showing superior efficacy but a higher rate of serious adverse effects (7). While both regimens offer a survival benefit to most patients, most eventually progress on treatment. With no FDA-approved third line medications, chemo-refractory PDAC patients are generally provided only symptomatic or hospice care. As such, there is an urgent clinical need to identify novel therapeutic approaches for patients in the second or third line setting.
While there has been extensive research into the mechanisms that underlie the emergence of chemoresistance in PDAC, none has successfully translated to a new treatment modality. However, recent evidence suggests that in addition to their well-documented effects on nucleotide synthesis, DNA polymerization, and translation, select anti-neoplastic agents may alter a variety of additional cell functions, including the processing and presentation of self-peptide (8–10). Classically, PDAC is poorly immunogenic with diminished antigen presentation (11) and a highly immunosuppressive TME that further impedes the functional activation of cytotoxic T-lymphocytes (CTLs) (12). As such, single agent immune checkpoint inhibitors (ICIs) have yet to show clear efficacy in the PDAC (13–16). As invasive strategies to promote antigen presentation such as irreversible electroporation have helped overcome the innate resistance of PDAC to ICIs (17), should cytotoxic chemotherapy similarly enhance the presentation of tumor antigen, its addition to select immunotherapies may improve drug responses.
We therefore evaluated the effects of prolonged Gemcitabine treatment using both established models of murine PDAC and in vitro models of Gemcitabine resistance. We determined that tumor-bearing mice treated with Gemcitabine for several months developed large-scale alterations in the expression of several immune surface proteins including MHC Class 1, PD-L1, and PD-L2. Gemcitabine similarly altered the composition of the tumor secretome, increasing a variety of CCL/CXCL family cytokines and transforming growth factor β (TGFβ)-associated signals. These secreted factors conferred a Gemcitabine resistant phenotype to tumor stromal cells in vitro, further enhancing stellate cell synthesis of immunomodulating cytokines including TGFβ1.
We next challenged transgenic models of early and advanced PDAC with a combination of Gemcitabine and anti-PD-1, which failed to significantly alter disease course unless mice also had genetic or pharmacologic ablation of TGFβ signaling. In the setting of TGFβ signal deficiency, Gemcitabine and anti-PD-1 led to significant regression of established disease, hallmarked by increased CTL infiltration and activity exceeding that observed with PD-1/TGFβ inhibition alone. Combined, these observations suggest that select anti-neoplastic agents such as Gemcitabine may prime tumor cells for ICIs by enhancing the presentation of self-peptide, but require the added intervention of an immunosuppressive cytokine barrier. Given the current lack of third line treatment options, this combined approach warrants consideration in Gemcitabine-refractory PDAC, potentially offering a new therapeutic option in patients for which there is currently no effective treatment.
MATERIALS & METHODS
Antibodies
All antibodies were purchased from established commercial vendors, and were verified by the manufacturer for the specific species and applications for which they were used. A full list of all antibodies used as well as the vendor, clone, and product numbers can be found in Table S1.
Transgenic Mice
Nongenic B6 (Wild Type), Ptf1a-Cre x LSL-KrasG12D (KC), Tgfbr1+/−, KC/Tgfbr1+/− (KCT), Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H (KPC), and Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H+/+ (KPPC) mice were generated as described in our previous work (18), or purchased from an in house vendor at the University of Illinois at Chicago. At roughly 12 weeks of age, KC and KCT mice were administered an intraperitoneal (IP) injection of either a PBS vehicle or daily Galunisertib (75mg/kg) and a fixed dose of 200μg anti-PD-1 twice per week. Mice were euthanized after four months of treatment and tissues collected for analysis. For studies involving KPC mice, animals were administered IP injection of a PBS vehicle, Gemcitabine (100mg/kg), Galunisertib, anti-PD-1, Galunisertib and Gemcitabine, Galunisertib and anti-PD-1, or triple combination of Galunisertib, Gemcitabine, and anti-PD-1 as described. KPC mice were sacrificed when moribund or showing clear signs of health decline, e.g., fur loss, weight loss, or lethargy, or when they reached 8 months of age in the case of mice with prolonged survival. For euthanasia, animals were anesthetized with isoflurane until unresponsive to toe tap and/or agonal breathing. Thoracotomy served as the primary method of euthanasia and exsanguination the secondary method. For all mouse studies, males and females were randomized at a 50:50 ratio.
Primary Cell Line-Derived Xenografts
The G-68 cell line was established from a non-Hispanic, white female with a T3N1, moderately differentiated, pancreatic ductal adenocarcinoma using methods described previously (19), and cultured in DMEM/F12 media supplemented with 10% heat-inactivated FBS, penicillin (100 U/mL), and streptomycin (100mg/ml). 5×106cells were suspended in 100 μL of a 1:1 DMEM/F-12 with Glautamax media and matrigel (Corning, Corning, NY) and inoculated subcutaneously into the right flank of NSG mice (The Jackson Laboratory, Bar Harbor, ME). Tumor size was measured twice weekly with digital caliper. For treatment, mice were randomized into treatment groups when the tumors reached 100–200 mm3, with an average tumor volume of 145 mm3. Once randomized, animals were treated with either PBS vehicle or 40mg/kg Gemcitabine via IP injection. Mice were euthanized when moribund, when the maximum tumor size allowed per to institutional policy (2 cm), or when tumors became ulcerated. For euthanasia, animals were scarified by CO2 suffocation followed by cervical dislocation, and tumors subsequently harvested and processed as described above.
Study Approval
All experiments involving the use of mice were performed following protocols approved by the IACUC at the University of Illinois at Chicago. All human tissues were obtained from patients providing fully informed, written consent. For histology, tissues were obtained in a de-identified manner from the Northwestern University Pathcore following local IRB approval. Similarly, the G-68 cell line was generated from a fully consenting patient and following local IRB approval/de-identification at the University of Florida.
Statistical Analysis
Data were analyzed by either Student’s T-test, simple linear regression analysis, or ANOVA fit to a general linear model in Minitab express, the validity of which was tested by adherence to the normality assumption and the fitted plot of the residuals. Results were arranged by the Tukey method, and considered significant at p < 0.05 unless otherwise noted. Results are presented as either boxplot showing the median value and all other values arranged into quartiles, or as the mean of individual replicates plus standard deviation.
Additional Methodology
For additional materials and methods, please see the supplemental materials, which also contains the following references (12–14, 18, 20–32).
RESULTS
Long-term Gemcitabine treatment alters the immune landscape of murine PDAC
To evaluate the effects of long-term chemotherapy within the pancreatic TME, we used the well-established Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H (KPC) model of invasive PDAC. This model faithfully recapitulates human PDAC histotypes with several key features including poor immunogenicity and a dense, reactive tumor stroma. KPC animals were initially reported to develop precursor pancreatic intraepithelial neoplasms (PanIN) at 6 weeks, focal PDAC lesions at approximately 15 weeks, and advanced PDAC at 18 weeks (33), though this can vary by individual colony and background strain. In our colony, KPC mice are maintained in full a C57/B6 background and have a mean survival of 124 days (N=30). Therefore, to model the long-term effects of Gemcitabine we elected for an early intervention study in which animals were allowed to develop overt disease for a minimum of 90 days (roughly 13 weeks), at which point mice in our colony generally have a combination of normal glandular tissue, low and high grade PanIN lesions, as well as scant, focal areas of PDAC (Figure S1A–C). Animals were then randomized at a 50:50 male to female ratio into one of two treatment groups (N=4/group). Mice were treated with IP injections of either PBS vehicle or 100mg/kg Gemcitabine twice per week, and sacrificed when showing clear signs of health decline e.g. weight loss, ascites, or lethargy. Consistent with previous reports, Gemcitabine provided a modest survival benefit averaging three weeks (Figure S1C).
After collection, tissues were sectioned and stained either with H&E, or by immunohistochemistry for clinically relevant immune surface proteins. Consistent with improved antigen presentation, Gemcitabine led to significant increases in MHC Class 1 in neoplastic tissues, as well as immune checkpoints PD-L1 and PD-L2 (Figure 1A,B). However, despite the presumptive increase in antigen presentation, Gemcitabine failed to increase the number of tumor infiltrating lymphocytes, which were largely confined to the periphery of tumor specimens in both treatment groups (Figure 1A,B). Based on these observations, we next evaluated Gemcitabine-induced alterations in tumor cytokines by homogenizing tumor tissue and conducting a high throughput array of 111 immunoregulatory proteins (Figure 1C,D and S1D, N=4/group).
Figure 1. Long-term Gemcitabine treatment alters the immune landscape of murine PDAC.

(A) Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H (KPC) mice were generated as a model of advanced PDAC. Starting at 90 days (~13 weeks) of age, mice were administered twice-weekly intraperitoneal injections of either PBS vehicle or 100mg/kg Gemcitabine. Pancreas tissues were collected when the animals were moribund. Tissues from vehicle and Gemcitabine treated mice were then stained with H&E or by immunohistochemistry for MHC Class 1, PD-L1, or PD-L2. (B) Tissue sections were evaluated by three blinded investigators. For IHC images, slides were assigned a score by each investigator from 0–3+ based on staining intensity, and composite values displayed as a box plot. For tumor infiltrating lymphocytes, each investigator quantified four H&E stained 40X fields per animal. These values were averaged and represented by box plot (*p < 0.05, N=4/group). (C) Tissues were homogenized and 200μg of tumor lysate evaluated by a high throughput proteome profiler array (ARY028). Representative blots from each group are displayed above. (D) Pixel density was evaluated using ImageJ, and samples normalized to the mean intensity of the reference spots for each blot minus the background density. Composite normalized values for Gemcitabine treated mice were divided by those for vehicle treated mice, and are presented as fold change (*p < 0.05).
After normalizing to reference samples, we identified consistent alterations to the tumor secretome of Gemcitabine-treated animals, as well as select immune surface proteins. Specifically, Gemcitabine-treated mice had significant increases in a variety of CCL and CXCL-family chemokines, with CCL6 and 17 being the highest expressed (Figure 1D). Gemcitabine-treated mice also displayed increased expression of several interleukins, including IL10 and IL28 (Figure S1D). Gemcitabine similarly enhanced the expression of TNFα and IFNγ, the latter consistent with the increased expression of MHC-Class 1 and PD-L1/PD-L2 (Figure S1D). We also observed Gemcitabine-induced upregulation of a variety of immune surface proteins, including CD26, CD40, CD54, and TGFβ receptor complex member CD105 (Figure S1D), as well as neutrophil associated proteins myeloperoxidase (MPO) and Neutrophil Gelatinase-Associated Lipocalin (NGAL) (Figure S1D). Tumor lysates were also evaluated for expression of TGFβ1 by ELISA, which showed a similar increase in TGFβ1 expression in the pancreas of Gemcitabine-treated mice (Figure S1D).
We next used the KPC-derived PDAC cell line KPC105 and conducted a similar study in vitro. Cells were incubated with 2.5μM Gemcitabine and lysates collected after 48 hours. Cell extracts were then subjected to the same high throughput cytokine screen described above, which showed a similar upregulation of CCL and CXCL-family chemokines in response to a transient course of Gemcitabine, as well as TGFβ1 by ELISA (Figure S1E,F).
Long-term Gemcitabine treatment similarly alters the immune profile of primary tumor cell line-derived xenografts
Given the apparent alterations in the immunogenicity of murine PDAC, we next sought to evaluate the effects of long-term chemotherapy on human tumor cells. We therefore generated a primary pancreatic cancer cell line (G-68) from a non-Hispanic, white female with a T3N1 tumor harboring both KRASG12D and TP53R248W mutations similar to those used in the KPC model. 5×106 cells were subsequently injected into the flank of right flank of NSG mice, and once tumors reached 100–200 mm3 in size, animals were treated with either vehicle or 40mg/kg of Gemcitabine once per week (N=4–5/group). Tumors were initially responsive to Gemcitabine for an average of four weeks, after which tumor growth accelerated to the normal rate. At this point, mice were considered to have developed clinical Gemcitabine resistance. This temporary tumor stasis corresponded to delayed mortality (Figure 2A), though this was not reflected in tumor size or any observable change in histopathology (Figure 2B,C). Similar to results observed in KPC mice, while chemotherapy did not alter the frequency of neoplastic lesions, Gemcitabine treatment led to significant increases in the human MHC Class 1 analog HLA-A,B,C in the neoplastic epithelium, as well as immune checkpoints PD-L1 and PD-L2 (Figure 2D,E).
Figure 2. Long-term Gemcitabine treatment similarly alters the immune profile of primary cell line-derived xenografts.

(A-B) G-68 human cells were injected subcutaneously into NSG mice, and once tumors reached 100–200mm3, animals were treated with either a saline vehicle or 40mg/kg Gemcitabine. Animals were sacrificed when moribund or when tumors ulcerated. Survival in days post enrollment is displayed via the Kaplan-Meier method, and tumor size in diameter (N=4–5/group). (C,D) Tissues from vehicle and Gemcitabine treated mice were then stained with either H&E or by immunohistochemistry for CK19, HLA-A,B,C, PD-L1, or PD-L2. (E) Tissue sections were evaluated by three blinded investigators. For IHC images, CK19+ lesions were quantified per 20X field, or slides were assigned a score by each investigator from 0–3+ based on staining intensity, and composite values displayed as a box plot. (F) Tissues from control and Gemcitabine treated tumors were lysed, and 200μg of total protein was evaluated by a high throughput proteome profiler array (ARY022B). Pixel density was evaluated using ImageJ, and samples normalized to the mean intensity of the reference spots for each blot minus the background density. Composite normalized values for all treated tumors were divided by those for control tumors, and are presented as fold change plus standard deviation (*p < 0.05). Using the same lysates, 20μg of total protein from control and Gemcitabine treated mice was also subjected to TGFβ1 ELISA and are similarly presented as fold change plus standard deviation (*p < 0.05). (G) Using the TCGA genomic databases of pancreatic cancer patients (N=186), the relationship between mRNA expression of individual genes was plotted with that of the cytotoxic surrogate CD8A. All mRNA expression values are plotted in log scale and are displayed with the associated P and Spearmen (S) coefficient values.
As with KPC mice, we next evaluated Gemcitabine-induced alterations in tumor cytokines by homogenizing tumor tissue and conducting a high throughput array of 105 human immunoregulatory proteins (Figure S1G, N=4/group). Again paralleling results in KPC mice, despite the lack of an adaptive immune system, G-68-derived tumors treated with Gemcitabine displayed highly significant increases in a variety of CCL/CXCL family chemokines, including CCL5, CCL20, CXCL8, CXCL10, and CXCL12 (Figure 2F). Gemcitabine treated mice also had increased intratumoral expression of inflammatory cytokines IL1 and IL32, as well as CD54, NGAL, MIF, and the TGFβ family ligand MIC-1, (Figure 2F). Like MIC-1, tumor lysate from Gemcitabine treated mice also had significant upregulation of the immunosuppressive cytokine TGFβ1 as determined by ELISA (Figure 2F, N=3/group). We then repeated this experiment using G-68 tumor cells in vitro, which confirmed the upregulation of several previously identified cytokines in response to Gemcitabine (Figure S2A,B). Interestingly, when evaluating the TCGA genomic databases of pancreatic cancer patients (N=186), we determined that mRNA expression of several of these CCL/CXCL chemokines, as well as genetically and functionally-related family members, had statistically significant associations with mRNA expression of the cytotoxic T-cell marker CD8A (Figure 2G).
Cytotoxic chemotherapy alters tumor cell immunogenicity in vitro
To determine that the observed alterations are indeed a consequence of Gemcitabine resistance in the cancer epithelium, we next sought to evaluate the effects of both transient and long-term chemotherapy on established human tumor cell lines in vitro. We therefore used Panc1 cells and determined the IC50 of the five currently FDA approved first-line chemotherapy agents. These include Gemcitabine (1μM), Paclitaxel (100nM), 5-Fluorouracil (2.5μM), Irinotecan (2.5μM), and Oxaliplatin (2.5μM). After 48 hours, dead cells were excluded by live/dead gating, and live cells were analyzed by flow cytometry for surface expression of the HLA-A,B,C, PD-L1, PD-L2, CTLA-4, and CD86, as well as intracellular expression of TGFβ1.
In response to Gemcitabine or Paclitaxel, Panc1 cells had enhanced surface expression of PD-L1, PD-L2, CTLA-4, HLA-A,B,C, and modestly increased expression of CD86 (Figure 3A,B). While 5-FU reduced cell viability as expected, we observed no significant change in PD-L1 or CD86 expression, and only slight increase in PD-L2, CTLA-4 and HLA-A,B,C levels (Figure 3A,B). Similarly, Irinotecan strongly enhanced expression of PD-L2, and mildly induced CTLA-4, HLA-A,B,C, and CD86 with no observable effect on PD-L2 (Figure 3A,B and S2A). Oxaliplatin was the least immunogenic of the drugs evaluated, causing only slight elevations in PD-L2, CTLA-4, and HLA-A,B,C (Figure 3A,B and S2A). After confirming these results in other cell lines (Figure S3A,B), we incubated Panc1 cells in increasing concentrations of Gemcitabine for several passages until viable in a high concentration of 10μM. These Gemcitabine resistant (GR) cells now had stronger, persistent upregulation of PD-L1, PD-L2, CTLA-4, HLA-A,B,C, and CD86, as well a modest increase in intracellular TGFβ1 (Figure 3A,B).
Figure 3. Cytotoxic chemotherapy alters tumor cell immunogenicity in vitro.

(A) Panc1 cells were incubated with the known IC50 of the first line chemotherapy agents Gemcitabine (Gem, 1μM), Paclitaxel (Pct, IC50 = 100nM), 5-Fluorouracil (5-FU, 2.5μM), Irinotecan (Irin, 2.5μM), and Oxaliplatin (Ox, 2.5μM), after which the expression of both surface and intracellular immmunomodulatory proteins was evaluated by flow cytometry. (B) Using unstained Panc1 cells, we identified the number of cells positive for each antigen in vehicle treated control (C) cells. We then determined the percent of the parent population for Gemcitabine (G), Paclitaxel (P), 5-Fluorouracil (F), Irinotecan (I), and Oxaliplatin (O) treated Panc1 cells, or Panc1-GR (GR) cells with expression above the geometric mean for control cells (*p < 0.05). (C) Panc1 cells were incubated with 0, 1, or 5μM Gemcitabine over a 48-hour period, and Panc1-GR cells were grown in 10μM Gemcitabine for several passages. Cells were incubated with a protein transport inhibitor for one hour, lysed, and 200μg of total cell lysate was evaluated by a high throughput proteome profiler array (ARY022B). Pixel density was evaluated using ImageJ, and samples normalized to the mean intensity of the reference spots for each blot minus the background density. Composite normalized values for all experimental groups were divided by those for untreated Panc1 cells, and are presented as fold change plus standard deviation (*p < 0.05).
After verifying these observations by immunocytochemistry (Figure S3C), we next incubated Panc1 cells with a transient 48-hour course of 0, 1, or 5μM Gemcitabine, or used Panc1-GR cells grown in 10μM Gemcitabine for several passages. After 48 hours, cells were treated with a protein transport inhibitor, and lysates subjected to a high throughput screen of 105 tumor-derived immunoregulatory proteins analogous to that described in KPC mice. This allows for the simultaneous evaluation of 105 cytokines, chemokines, and immunomodulating proteins (Figure 3C and S3D). While transient incubation with Gemcitabine had modest effects on cytokine production, Panc1-GR cells had significant alterations in a wide variety of functionally-related proteins, particularly those involved in inflammation/chemotaxis, angiogenesis, and/or TGFβ signaling. For instance, Panc1-GR cells displayed highly significant increases in the expression of the pro-inflammatory chemokines CCL2, CCL4, CCL20, CXCL1, CXCL5, and CXCL8, closely resembling the phenotype of KPC and xenografted mice (Figure 3C). Panc1-GR cells also had robust expression of the IL6 regulator FGF2, as well as the pro-inflammatory cytokine IL32 and CD56 (Figure 3C). Additionally, GR cells had upregulation of pro-angiogenic factors Angiogenin and the TGFβ family ligand MIC-1 (Figure 3C). Contrasting our observations in mice, Panc1-GR cells had diminished expression of the TGFβ receptor complex member CD105 (Figure 3C).
Gemcitabine-resistant tumor cells confer drug resistance to stromal cells via paracrine TGFβ signaling
Given the apparent aberrations to TGFβ signaling and the established roles for TGFβ in immune evasion in PDAC (12, 18, 34), we next evaluated the concentration of TGFβ1 in the culture media of Panc1 in response to a 72-hour course of 0, 1, or 5μM Gemcitabine, or in Panc1-GR cells by ELISA. While high dose Gemcitabine led to a modest increase in TGFβ1 in the culture media, Panc1-GR cells had a near 3-fold increase in secreted TGFβ1 compared to chemo-naive Panc1 cells (Figure S4A). Given the established roles of TGFβ in expanding the tumor stroma (12), which itself can also contribute to the clinical failure of Gemcitabine by acting as a drug scavenger (35), we next explored the capability of Panc1-GR cells to modify the activity of stromal cells.
Next, an equal number of Panc1 and Panc1-GR cells were grown in serum free DMEM for 24 hours, after which media was collected, filtered, and evaluated for TGFβ1 expression (Figure 4A). This media was then supplemented with 10% FBS and transferred to one of three stromal cell lines: cancer-associated fibroblast cells CAF2 and CAF3 or the human Pancreatic Stellate Cell line hPSC. After 48 hours, media was collected and re-evaluated for TGFβ1 by ELISA. While Panc1 conditioned media increased TGFβ1 secretion by CAF2 and hPSC cells, Panc1-GR conditioned media led to greater increases in TGFβ1 in all cell lines, surpassing the sum of TGFβ1 produced by tumor and stromal cells in isolation (Figure 4B).
Figure 4. Gemcitabine-resistant tumor cells confer drug resistance to stromal cells via paracrine TGFβ signaling.

(A) An equal number of Panc1 cells and Panc1-GR cells were seeded into 6-well plates and grown in serum free media. After 24 hours, culture media was collected, filtered, and subjected to TGFβ1 ELISA (*p < 0.05). (B) Serum free media was conditioned in either Panc1 or Panc1-GR cells for 24 hours as described and sterile filtered. Media was supplemented with 10% FBS, and transferred to an equal number of CAF2, CAF3, or hPSC stromal cell lines. After 48 hours, control (C), Panc1 conditioned (P), and Panc1-GR (GR) conditioned media was collected and re-evaluated for TGFβ by ELISA (*p < 0.05). (C) CAF2, CAF3, and hPSC cells were incubated with increasing concentrations of Gemcitabine delivered in either control media (red), Panc1 conditioned media (blue), or Panc1-GR conditioned media (GR, green). After 72 hours cell viability was evaluated by MTT assay. (D) hPSC cells were grown in either control (C), Panc1 conditioned (P), Panc1-GR (GR) conditioned media, or GR media with 10μM of the TGFBR1-inhbitor Galunisertib. After 48 hours, cells were incubated with a protein transport inhibitor for one hour, lysed, and 200μg of total cell lysate was evaluated by a high throughput proteome profiler array (ARY022B). Pixel density was evaluated using ImageJ, and samples normalized to the mean intensity of the reference spots for each blot minus the background density. Mean normalized values for experimental groups were divided by those for untreated hPSC cells, and are presented as fold change plus standard deviation (*p < 0.05). (E) CAF2, CAF3, and hPSC cells were again incubated with increasing concentrations of Gemcitabine delivered in either Panc1-GR conditioned media (green) or Panc1-GR conditioned media supplemented with 10μM of the TGFBR1-inhbitor Galunisertib (purple). After 72 hours cell viability was evaluated by MTT assay.
Given the pro-survival role TGFβ1 in mesenchymal cells, the experiment was repeated, and cells grown in increasing concentrations of Gemcitabine ranging from 0.5 to 10μM. While Panc1 conditioned media modestly enhanced Gemcitabine resistance in all cell lines, the addition of Panc1-GR conditioned media further enhanced this response, particularly in CAF2 and CAF3 cells (Figure 4C, N=4). Given the known immunosuppressive role of stromal-derived TGFβ in PDAC (12), we next grew hPSC cells in either control media, Panc1 conditioned media, Panc1-GR conditioned media, or Panc1-GR conditioned media supplemented with 10μM of the type 1 TGFβ receptor (TGFBR1) inhibitor Galunisertib, which was not toxic to any cell line at the concentration used (Figure S4B). After 48 hours, we repeated the described high throughput screen of immunoregulatory proteins (Figure 4D and S4C, N=2/group). Despite the difference with respect to Gemcitabine resistance, both Panc1 and Panc1-GR conditioned media had similar effects on the stellate cell secretome, leading to increases in several factors observed in our previous arrays. Both Panc1 and Panc1-GR media enhanced hPSC synthesis of CXCL8, CD56, FGF2, CD54, MIC-1, CD105, and the lymphokine MIF; however, only Panc1-GR media significantly upregulated IL17 (Figure 4D). While Galunisertib had little effect on unstimulated stromal cells, the addition of Galunisertib to Panc1-GR media neutralized the induction of all upregulated immunoregulatory proteins, with the exception of MIF (Figure 4D). Similarly, Galunisertib prevented the Gemcitabine resistance conferred to stromal cells by Panc1-GR media, restoring drug sensitivity to near basal levels (Figure 4E, N=4).
TGFβ functions as a cytokine barrier impeding the efficacy of combined Gemcitabine and anti-PD-1
Though we observed some heterogeneity across model systems, Gemcitabine appears to ubiquitously alter the expression of several tumor-derived cytokines (Figure S4D). Of the most frequently altered signals, TGFβ remains the only such cytokine with an inhibitor currently in clinical trial for PDAC. Further, the contributions of TGFβ signals to immune evasion in PDAC are well established (12, 18), and our data to this point suggest that in the setting of Gemcitabine resistance, TGFβ serves as a master regulator of the tumor microenvironment. Therefore, to determine the potential clinical relevance of our findings, we first evaluated the mRNA expression of clinically actionable immune checkpoints PD-L1 (CD247) and PD-L2 (PCDCLG2), as well as a variety of genes associated with the TGFβ signaling pathway, and leukocyte specific genes including CD3D, CD3E, CD3G, CD4, CD8A, CD8E, and CD45 (Figure S5A and Table S2). Interestingly, though only PD-L1 expression was a significant predictor of poor outcomes, a high TGFβ gene signature positively associated with expression of PD-L1 and PD-L2, as did increased expression of several leukocyte markers including CD8A and CD45 (Figure S5B–G). We therefore explored the relationship between PD-L1 expression (by immunohistochemistry) and lymphocyte infiltration (by H&E staining) in a group pancreatic cancer excisional biopsies collected from Northwestern University, examining both adjacent normal (N=27) and PDAC (N=47) specimens. While the mean lymphocyte infiltrate PDAC exceeded that of adjacent normal sections, this was highly varied with some sections having almost no tumor infiltrating lymphocytes, whereas others had areas of overwhelming lymphocytosis (Figure S5H–J).
Based on these observations, we next set to determine the combined therapeutic efficacy of Gemcitabine and immune checkpoint inhibition in vivo, with and without TGFβ signal inhibition. To address this, we used both the Ptf1-Cre x LSL-KrasG12D (KC) model of neoplastic disease, as well as KC mice with heterozygous deletion of Tgfbr1+/− (KCT) (Figure 5A). Consistent with our previous report (18), these two models had no observable difference in tumor development at six months of age (Figure 5B, N=6/group). While KCT mice had increased expression of PD-L1 in the neoplastic epithelium (Figure S6A), they had no significant difference in MHC Class 1 or PD-L2 expression compared to aged matched KC controls (Figure 5B). Similarly, there was no significant difference between KC and KCT mice with respect to tumor specific localization of the cytotoxic surrogate GranzymeB (Figure 5C).
Figure 5. TGFβ functions as a cytokine barrier impeding the efficacy of combined Gemcitabine and anti-PD-1.

(A-C) Ptf1a-Cre x LSL-KrasG12D (KC) mice were bred to generate a model of conditional expression of oncogenic KRASG12D. KC mice were also crossed to the Tgfbr1 haplo-insufficient animals to generate KC/Tgfbr1+/− (KCT). Tissues were collected at six months and stained with H&E, or via immunohistochemistry for MHC Class 1, PD-L1, or PD-L2 (N=6/group). (C) The anti-tumor immune response of KC and KCT mice was evaluated by dual staining for the duct cell marker CK19 (green) and the cytotoxic surrogate GranzymeB (GrzB, red). (D) KC and KCT mice were allowed to reach 12 weeks of age, and randomized at a 50:50 male to female ratio into one of two groups. Mice were either administered intraperitoneal injection every other day of either a vehicle control (Vehicle, N=3/group) or 200μg of an anti-PD-1 neutralizing antibody (Anti-PD-1) with twice-weekly doses of 100mg/kg Gemcitabine (Gem + Anti-PD-1, N=3–7/group). (E) The pancreas was collected at the conclusion of the study (100 days post enrollment), and gross changes in pancreas gland structure evaluated. Representative images from each group are displayed. (F,G) The pancreas from control and drug treated mice was stained with either H&E or Masson’s Trichrome allowing for evaluation of changes in histopathology and fibrosis respectively. Tissue sections were quantified by three blinded investigators, averaged, and displayed as box plot. Additionally, the pancreas was weighed at the time of tissue collection, normalized to bodyweight, and displayed accordingly. (H,I) Tissue sections were also stained via immunohistochemistry for the duct marker CK19 and amylase, MHC Class 1, PD-L1, PD-L2, and the TGFβ surrogate pSMAD2 (N=3–7/group). Tissue sections were quantified as described and displayed as box plot (*p < 0.05, NS = non-significant where p > 0.05). (J) The pancreas from control and drug-treated mice were dual-stained for the T-cell marker CD3 and the epithelial surrogate E-Cadherin (E-Cad), for the duct cell marker CK19 and the cytotoxic surrogate GranzymeB (GrzB), or for the apoptotic surrogate Cleaved Caspase 3 (Cleaved C3). Tissue sections were quantified as described and displayed as box plot (p < 0.05). (K) Mesenteric lymph nodes from drug treated mice were collected, and evaluated for CD8+CD69+ or CD8+Perforin+ cells.
Mice were then allowed to develop extensive PanIN disease for approximately 3 months, at which time KC and KCT mice were administered either a PBS vehicle, or a twice-weekly 200μg injection of the PD-1 neutralizing antibody RMP1–14 (anti-PD-1) in combination with 100mg/kg Gemcitabine. After four months on treatment, animals were sacrificed and tissues examined (Figure 5D). At the conclusion of the study, vehicle treated KC and KCT mice developed an enlarged, firm, nodular pancreas, as did KC mice administered Gemcitabine + anti-PD-1 (N=3). Contrastingly, 6/7 Gemcitabine + anti-PD-1 treated KCT mice had a smaller, malleable pancreas resembling that of a wild type animal (Figure 5E, N=7). On histologic evaluation, Gemcitabine + anti-PD-1 failed to alter disease course in KC mice, which had no observable change in normalized pancreas weight, lesion frequency, or fibrosis. However, KCT mice administered Gemcitabine + anti-PD-1 had significant reductions in PanIN incidence, fibrosis, and the mass of the pancreas (Figure 5F,G).
Despite the changes in phenotype, both drug treated KC and KCT mice displayed increased expression of PD-L1, PD-L2, and MHC Class 1 on the neoplastic epithelium, (Figure 5H,I). Expectedly, KCT mice had substantial reductions in the phosphorylation of SMAD2, affirming the disruption of TGFβ signaling (Figure 5H,I). These observations were paralleled by changes in cell-mediated immunity, with Gemcitabine + anti-PD-1 treated KCT mice displaying increased tumor infiltrating CD3+ and CD8+ lymphocytes, as well as GranzymeB deposition in remaining neoplastic tissues. This was paralleled by increased activation of the GranzymeB effector and apoptotic surrogate Caspase 3 (Figure 5J and S6B). We then confirmed the apparent increase in CTL activation by flow cytometry of regional lymph nodes, which showed an increase in CD8+Perforin+ and CD8+CD69+ cells in drug-treated KCT mice (Figure 5K).
Gemcitabine potentiates dual-agent immunotherapy in advanced PDAC
While the combination of Gemcitabine and anti-PD-1 had significant efficacy in early PanIN disease with genetic ablation of TGFβ signals, the translational relevance of these findings are limited, as they do not address the individual contributions of each drug. Further, patients seldom present with such early stage disease, and genetic suppression of TGFβ signals does not fully recapitulate the effects of a TGFβ pathway inhibitor. We therefore used the KPC model of advanced PDAC, and randomized 41 mice into one of seven treatment arms. In order to allow adequate time for Gemcitabine-induced remodeling of the TME, mice were again enrolled at 90 days (roughly 12.85 weeks) of age, at which point they develop extensive high-grade PanIN disease and rare, focal areas PDAC. At this time, mice were then administered either a saline vehicle every other day (N=7), 100mg/kg Gemcitabine twice per week (N=7), 75mg/kg of the TGFβ signaling inhibitor Galunisertib every other day (N=4), staggered doses of Gemcitabine and Galunisertib (N=5), a fixed 200μg dose of anti-PD-1 twice-weekly (N=4), staggered doses of Galunisertib and anti-PD-1, or twice-weekly Gemcitabine starting at 90 days (N=6), with the addition of Galunisertib and anti-PD-1 two weeks later (N=8). Tissues were collected either when the animals were moribund or when mice reached 8 months of age (Figure 6A).
Figure 6. Gemcitabine potentiates dual-agent immunotherapy in advanced PDAC.

(A) Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H (KPC) mice were used as a model of aggressive PDAC. At 90 days (~13 weeks) of age, animals were randomized at a 50:50 male to female ratio into one of seven groups. Mice were administered an intraperitoneal injection of either a saline vehicle every other day (N=7), 100mg/kg Gemcitabine twice per week (N=7), 75mg/kg of the TGFβ signaling inhibitor Galunisertib every other day (N=4), staggered doses of Gemcitabine and Galunisertib (N=5), a fixed 200μg dose of anti-PD-1 twice-weekly (N=4), staggered doses of Galunisertib and anti-PD-1, or twice-weekly Gemcitabine starting at 90 days (N=6), with the addition of Galunisertib and anti-PD-1 two weeks later (N=8). The pancreata were then collected either when the animals were moribund or at the conclusion of the study (8 months). (B) Kaplan-Meier curve indicating survival for mice across all six groups in days post enrollment (N=4–8/group). (C,D) After tissue collection, gross changes in pancreas gland structure were evaluated, including gland weight, which was also normalized to each animal’s body weight, and results displayed as box plot. Tissues were also stained with H&E or Masson’s Trichrome, and the relative percentage of normal tissue, number of lesions per high power field, percent area fibrosis, and number of tumor-infiltrating lymphocytes quantified by three blinded investigators and displayed as box plot. Sections were also stained via immunohistochemistry for the TGFβ effector pSMAD2 or the proliferation surrogate PCNA and quantified/displayed as described (*p < 0.05). (E,F) The pancreas from control and drug-treated mice were stained by immunohistochemistry for MHC Class 1, PD-L1, the T-cell marker CD3 and the epithelial surrogate E-Cadherin (E-Cad), CD3 and regulatory T-cell (Treg) marker FoxP3, the duct cell marker CK19 and the cytotoxic surrogate GranzymeB (GrzB), or for the apoptotic surrogate Cleaved Caspase 3 (Cleaved C3). Tissue sections were quantified and displayed as described (*p < 0.05).
Consistent with our previous results (18), neither Galunisertib nor anti-PD-1 monotherapy significantly altered disease course. However, the combination of Galunisertib and anti-PD-1 led to a modest reduction in tumor burden with limited fibrosis in remaining neoplastic tissues. The addition of Gemcitabine further enhanced the efficacy of the combined immunotherapy, leading to improved preservation of normal gland architecture, added reductions in PanIN frequency and fibrosis, as well as comparable reductions in SMAD2 phosphorylation, mitoses, and intratumoral TGFβ1 (Figure 6B–D and S6C–F). Additionally, 7/8 mice receiving all three drugs showed no mortality or obvious signs of disease-associated morbidity at the conclusion of the study, and tissues were collected from healthy mice after 150 days on treatment. One mouse did not meet the study endpoint, and was sacrificed 103 days post enrollment. Contrastingly, significant mortality was observed in all other groups, though this was modestly delayed in Galunisertib/Gemcitabine treated mice, and more significantly delayed in Galunisertib/anti-PD-1 treated mice (Figure 6B).
While areas of residual disease were scarce compared to other groups, lesions in mice administered all three drugs had uniform, overwhelming lymphocytosis exceeding mice given Galunisertib and anti-PD-1 (Figure 6C,D and S6D). In addition to increased expression of MHC Class 1 and PD-L1, these tumor-infiltrating lymphocytes were uniformly positive for the T-cell marker CD3 (Figure 6E,F), negative for regulatory T-cell (Treg) surrogate FoxP3, and largely positive for CD8, contrasting the sparse but FoxP3-dominant T-cell infiltrate of control mice (Figure 6E,F). These findings were paralleled by increased tumor-specific staining for GranzymeB as well as its downstream target cleaved Caspase 3, affirming the increased apoptosis in remaining neoplastic areas (Figure 6E,F).
In addition to the reduction in lesion incidence, Gemcitabine/Galunisertib/anti-PD-1 treated mice had substantial changes with respect to the vasculature in remaining areas of disease. Consistent with previous reports, vehicle treated KPC mice had severe fibrosis and clear vascular dysfunction (36). Namely, we observed scant arteries/arterioles within the tumor as well as diffuse capillaries and small, compressed veins (Figure S6G). Contrastingly, Gemcitabine/Galunisertib/anti-PD-1 treated mice had an increased frequency of large vessels, including patent arteries and distended veins, as well as increased capillarity density (Figure S6G). These changes were confirmed by dual staining for vascular marker CD31 and αSMA, which allowed for more accurate quantification and differentiation of veins and arteries/arterioles. In addition to the increased frequency of intratumoral arteries, Gemcitabine/Galunisertib/anti-PD-1 treated mice had an increased mean arteriolar diameter. Similar changes were observed in the venous system, which were also more frequent and more distended in triple treated mice, with similar results in intratumoral capillary density (Figure S6G).
Combining Gemcitabine, Galunisertib, and anti-PD-1 leads to intratumoral accumulation and activation of cytotoxic T lymphocytes
To better evaluate the restoration of cytotoxic immunity in Gemcitabine/Galunisertib/anti-PD-1 treated mice, we next repeated our in vivo experiment and examined the tumor infiltrating lymphocytes by flow cytometry. The pancreata and spleens from vehicle and Gemcitabine/Galunisertib/anti-PD-1 treated mice (N=3/group) were collected at their respective study endpoints and stained for CD4 and CD8. Consistent with our prior histopathology, both CD4+ and CD8+ T-cells were scarce in the pancreata of vehicle treated mice, whereas those treated with the combination of Gemcitabine/Galunisertib/anti-PD-1 had highly significant increases in both T-cell subsets (Figure 7A,B). However, Gemcitabine/Galunisertib/anti-PD-1 treated mice only displayed modest increases in CD4+ and CD8+ populations in the spleen, suggestive of a largely tumor specific phenotype (Figure 7C–F).
Figure 7. Combining Gemcitabine, Galunisertib, and anti-PD-1 leads to intratumoral accumulation and activation of cytotoxic T lymphocytes.
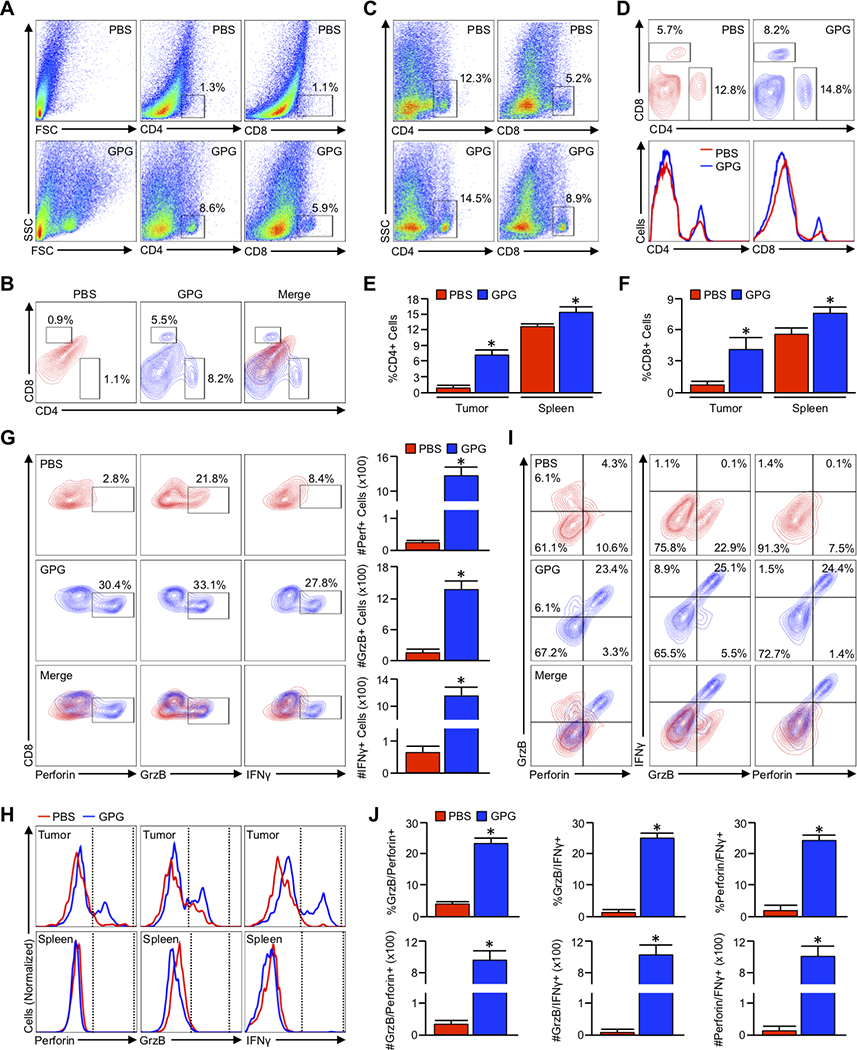
(A,B) Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H (KPC) mice were again used as a model of aggressive PDAC. At 90 days (~13 weeks) of age, mice were administered an intraperitoneal injection every other day of either saline (PBS) or twice-weekly Gemcitabine starting at 90 days with the addition of Galunisertib and anti-PD-1 two weeks later (G/P/G). The pancreas was then collected either when the animals were moribund or at the conclusion of the study (150 days post enrollment) and analyzed by flow cytometry for tumor infiltrating CD4+ and CD8+ T-cells, respectively (N=3/group). (C,D) The spleens from both PBS and GPG-treated mice were collected and analyzed as described for CD4+ and CD8+ T-cells. (E,F) The relative percent of total cells positive for CD4 and CD8 from both the tumor and spleen of PBS and GPG treated mice (*p < 0.05). (G) Tumor infiltrating cells were gated based on the CD8 and CD4 staining shown above, and CD8+ events isolated and analyzed for expression of cytotoxic T-cell activation markers Perforin (Perf), GranzymeB (GrzB), and Interferon γ (IFNγ). The percent of CD8+ cells positive for each activation marker was normalized to the total number of CD8+ T-cells per 100,000 events, and displayed to the right of the flow cytometry plots (*p < 0.05). (H) The modal expression of Perforin, GranzymeB, and IFNγ within both intratumoral and splenic CD8+ T-cells are displayed as a histogram plot. (I) CD8+ cells were gated as previously, and analyzed for the simultaneous expression of the aforementioned T-cell activation markers including GranzymeB and Perforin, GranzymeB, and IFNγ, as well as Perforin and IFNγ. (J) Using the described gating, the relative percent of GranzymeB+Perforin+, and GranzymeB+IFNγ+, and Perforin+IFNγ+ are plotted, as are the absolute number of each per 100,000 events (*p < 0.05).
As the cytotoxic effects of immune checkpoint inhibitors are largely dependent on the activation of CD8+ T-cells, we next gated to CD8+ cells evaluated intracellular expression of three distinct activation markers: GranzymeB, Perforin, and IFNγ. Though CD8+ T-cells were again scarce in the pancreas of vehicle treated mice, those that were present remained refractory from full activation with little to no expression of Perforin and IFNγ, and moderate expression of GranzymeB. Contrastingly, approximately 30% of the more robust CD8+ infiltrate of Gemcitabine/Galunisertib/anti-PD-1 treated mice were positive for all three cytokines, a phenomenon that again was exclusive to intratumoral lymphocytes and not observed in the spleen (Figure 7G,H). CD8+ cells were next evaluated for the simultaneous expression of multiple activation markers, which again confirmed the presence of fully activated CD8+ cells exclusively in the pancreas of Gemcitabine/Galunisertib/anti-PD-1 treated mice (Figure 7I,J and S7A–D).
DISCUSSION
Cytotoxic chemotherapy has been the backbone of the clinical management of PDAC for several decades. Despite the emergence of new multi-drug regimens such as Gemcitabine/nab-Paclitaxel or FOLFIRINOX, five-year survival remains a dismal 9% with nearly all patients eventually progressing on treatment (37). As discussed, there are no standard-of-care medications that can currently be offered to patients who progress on chemotherapy. Here, we sought to address this issue by exploring the biologic consequences of long-term Gemcitabine treatment within the pancreatic TME in order to identify potential treatment strategies in the second or third line setting. Through these experiments, we determined that long-term Gemcitabine leads to extensive reprogramming of the tumor microenvironment, particularly with respect to tumor immunogenicity.
Our results suggest that Gemcitabine enhances the expression of antigen presenting molecules and inflammatory cytokines/chemokines, as well as negative immune checkpoints PD-L1 and PD-L2. This is consistent with prior studies that hint at a potential interplay between chemotherapy and the presentation of self-peptide (38, 39). These collective observations suggest that chemotherapy may prime pancreatic cancers for ICIs, particularly as Gemcitabine has been shown to enhance the cross-presentation of tumor antigens (40). However, ICIs have yet to show substantial efficacy in clinical trials (Table S3). Particularly, the combination of Gemcitabine/nab-Paclitaxel and pembrolizumab provided only a modest benefit compared to Gemcitabine/nab-Paclitaxel in chemotherapy-naïve PDAC patients (30). This was recapitulated by our in vivo experiments, in which the combination of Gemcitabine and anti-PD-1 had a marginal effect on tumor-bearing mice. Interestingly, these observations were contrasted by experiments using immune-competent xenografts. There, Gemcitabine and anti-PD-1 led to complete responses and improved survival (41). However, a likely explanation of this discrepancy is that xenograft models generally lack the desmoplastic stroma inherent to genetically modified mice (42). This may be of particular relevance, as the stroma is itself a source of several cytokines including TGFβ (12, 43), and select stromal cells express negative immune checkpoints including PD-L1 (44).
As demonstrated in our earlier works, TGFβ signals are largely produced by the pancreatic cancer stroma, and impede the functional activation of tumor infiltrating lymphocytes (12). In the current study, we determined that prolonged administration of Gemcitabine enhances tumor cell secretion of TGFβ1, thereby conferring drug resistant phenotypes to neighboring stromal cells and further enhancing the production of inflammatory cytokines/chemokines. Based on these observations, we challenged models of murine tumorigenesis with Gemcitabine with anti-PD-1, first in the setting of genetic ablation of Tgfbr1, followed by pharmacologic inhibition of TGFβ signaling. In the setting of TGFβ signaling inhibition, Gemcitabine and Anti-PD-1 led to near uniform drug responses, CTL-mediated regression of disease, and improved survival.
While encouraging, it is important to note that progress for immunotherapy in PDAC has been difficult, both with ICIs (45) and vaccine-based immunotherapy (46–48). Similarly, combination immunotherapy has also shown less than promising results (25). For instance, the bispecific PD-L1/TGFβ antibody M7824 only produced a partial response in one microsatellite instability-high (MSI-H) patient, with no benefit observed in the other four enrolled (29). Interestingly, in the setting of MSI-H, patients generally have a high mutational burden, and following ICIs, responders undergo rapid expansion of neoantigen-specific T-cell clones (16). This led to the approval of pembrolizumab for MSI-H-PDAC in 2017. Similarly, despite early mortality, murine tumor models with homozygous TP53 loss (KPPC) display increased MHC Class 1 expression and robust T-cell infiltration (Figure S8A–C). However, though high TMB is associated with poor clinical outcomes (Figure S8D,E), MSI-H is rare in PDAC and its predictive value for ICIs is not clear (16, 49–51).
As highlighted in this paper, the immunosuppressive TME, particularly TGFβ signaling, may be an important consideration for pancreatic cancer immunotherapy. We have previously demonstrated that suppression of TGFβ signaling augments PD-1 inhibition in murine PDAC (18). Additionally, Galunisertib is showing clear efficacy in early clinical trial, particularly in combination with Gemcitabine (52). Similarly, the addition of TGFβ signaling inhibition and ICIs is showing significant early efficacy in several cancers (53–57). In PDAC, the combination of Galunisertib and durvalumab has shown early promise in a phase Ib trial (NCT02734160). While these results are no doubt encouraging, more work is needed to explore TGFβ as a potential immune checkpoint in PDAC, particularly in combination with anti-PD-1 in Gemcitabine-refractory disease.
Our data also appears to substantiate the long-standing hypothesis that the relatively scant presentation of self-antigen limits the therapeutic efficacy of ICIs in PDAC. Hence, the use of cytotoxic chemotherapy followed by combined TGFβ inhibitor and anti-PD-1 had marked efficacy in vivo, cooperating to produce durable immune responses in murine PDAC. The combination of chemotherapy and ICIs has been highly effective in many solid tumor malignancies including non-small cell lung cancer (58–60) and triple negative breast cancer (61, 62). In PDAC, however, the combination of Gemcitabine/nab-Paclitaxel and pembrolizumab provided only a modest benefit compared to Gemcitabine/nab-Paclitaxel alone (30). Similar results were observed using the combination of nivolumab with nab-Paclitaxel, or nivolumab with GVAX-based immunotherapy (63). However, as none of these strategies contend with the immunosuppressive TME precipitated by long-term chemotherapy, the addition of Galunisertib or alternate suppressive cytokine inhibitor may be a highly effective means of enhancing the efficacy of either chemo-immunotherapy in the first line, or ICIs and palliative Gemcitabine in the third line.
Finally, though our data supports this potential approach, there are several added factors that must be considered when translating these findings to the bedside. While we showed that single agent Gemcitabine appears to prime the tumor microenvironment for immunotherapy, it remains to be seen whether the same will be true of other cytotoxic regimens such as FOLFIRONOX. Additionally, both Gemcitabine/nab-Paclitaxel and FOLFIRINOX regimens are associated with Grade III hematologic adverse effects (6, 7), and severe neutropenia has also been observed with Gemcitabine monotherapy (64). The resultant paucity of leukocytes may limit the efficacy of immune checkpoint inhibition, thus dose adjustments may be required. However, despite these potential hurdles, our data suggest that Gemcitabine primes the TME for immune evasion, providing an opportunity for intervention in the third line setting. Given the lack of options at this stage, the combination of anti-PD-1, Galunisertib, and maintenance Gemcitabine warrants consideration in patients who begin to progress on cytotoxic chemotherapy.
Supplementary Material
SIGNIFICANCE.
These data suggest that long-term treatment with Gemcitabine leads to extensive reprogramming of the pancreatic tumor microenvironment and that patients who progress on Gemcitabine-based regimens may benefit from multi-drug immunotherapy.
ACKNOWLEDGEMENTS
This work is dedicated to the memory of our friend Tami Bichrt Robson who recently passed away after a long and courageous fight with breast cancer. The authors would like to thank our friend and mentor Dr. Larry Tobacman for his guidance, leadership, and dedication to the Medical Scientist Training Program at the University of Illinois College of Medicine, and we wish him well in his retirement. We would also like to thank Dr. Enrico Benedetti and the Department of Surgery at the University of Illinois at Chicago. This work was supported by Veterans Affairs Merit Award I01BX002703 and Career Scientist Award IK6 BX004855 to A. Rana, by NIH F30CA236031 to D.R Principe, and by NIH R01CA217907 and Veterans Affairs Merit Award I01BX002922 to H.G. Munshi.
Grant Support: This work was supported by Veterans Affairs Merit Award I01BX002703 and Career Scientist Award IK6 BX004855 to A. Rana, by NIH F30CA236031 to D.R Principe, and by NIH R01CA217907 and Veterans Affairs Merit Award I01BX002922 to H.G. Munshi.
Abbreviations:
- PDAC
Pancreatic Ductal Adenocarcinoma
- ICIs
Immune checkpoint inhibitors
- PanIN
Pancreatic Intraepithelial Neoplasm
- IP
Intraperitoneal
- TGFβ
Transforming Growth Factor β
- KC
Ptf1a-Cre x LSL-KrasG12D
- KCT
Ptf1a-Cre x LSL-Kras/Tfgbr1+/−
- KPC
Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H
- KPPC
Pdx1-Cre x LSL-KrasG12D x LSL-TP53R172H+/+
- GR
Gemcitabine resistant
- αPD-1
Anti-PD-1
Footnotes
Conflict of Interest Disclosure: The authors have no conflicts to disclose.
REFERENCES
- 1.McGuigan A, et al. (2018) Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol 24(43):4846–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, et al. (2011) Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol 8(1):27–33. [DOI] [PubMed] [Google Scholar]
- 3.Moore M (1996) Activity of gemcitabine in patients with advanced pancreatic carcinoma. A review. Cancer 78(3 Suppl):633–638. [DOI] [PubMed] [Google Scholar]
- 4.Carmichael J, et al. (1996) Phase II study of gemcitabine in patients with advanced pancreatic cancer. Br J Cancer 73(1):101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casper ES, et al. (1994) Phase II trial of gemcitabine (2,2’-difluorodeoxycytidine) in patients with adenocarcinoma of the pancreas. Invest New Drugs 12(1):29–34. [DOI] [PubMed] [Google Scholar]
- 6.Von Hoff DD, et al. (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369(18):1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conroy T, et al. (2018) FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med 379(25):2395–2406. [DOI] [PubMed] [Google Scholar]
- 8.Emens LA & Middleton G (2015) The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res 3(5):436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gravett AM, Trautwein N, Stevanovic S, Dalgleish AG, & Copier J (2018) Gemcitabine alters the proteasome composition and immunopeptidome of tumour cells. Oncoimmunology 7(6):e1438107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu WM, Fowler DW, Smith P, & Dalgleish AG (2010) Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer 102(1):115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torphy RJ, Zhu Y, & Schulick RD (2018) Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann Gastroenterol Surg 2(4):274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Principe DR, et al. (2016) TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res 76(9):2525–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Royal RE, et al. (2010) Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33(8):828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brahmer JR, et al. (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366(26):2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le DT, et al. (2015) PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372(26):2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le DT, et al. (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349):409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao J, et al. (2019) Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat Commun 10(1):899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Principe DR, et al. (2019) TGFbeta Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol Cancer Ther 18(3):613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pham K, et al. (2016) Isolation of Pancreatic Cancer Cells from a Patient-Derived Xenograft Model Allows for Practical Expansion and Preserved Heterogeneity in Culture. Am J Pathol 186(6):1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aglietta M, et al. (2014) A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol 25(9):1750–1755. [DOI] [PubMed] [Google Scholar]
- 21.Cerami E, et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao J, et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melisi D, et al. (2018) Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer 119(10):1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melisi D, et al. (2019) A phase Ib dose-escalation and cohort-expansion study of safety and activity of the transforming growth factor (TGF) β receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. Journal of Clinical Oncology 37(15_suppl):4124–4124. [Google Scholar]
- 25.Naing A, et al. (2018) Abstract CT177: Epacadostat plus durvalumab in patients with advanced solid tumors: preliminary results of the ongoing, open-label, phase I/II ECHO-203 study. Cancer Research 78(13 Supplement):CT177–CT177. [Google Scholar]
- 26.Principe DR, et al. (2016) PEDF inhibits pancreatic tumorigenesis by attenuating the fibro-inflammatory reaction. Oncotarget 7(19):28218–28234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Principe DR, et al. (2017) Loss of TGFbeta signaling promotes colon cancer progression and tumor-associated inflammation. Oncotarget 8(3):3826–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Principe DR, et al. (2017) TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 36(30):4336–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strauss J, et al. (2018) Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFbeta, in Advanced Solid Tumors. Clin Cancer Res 24(6):1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss GJ, et al. (2018) Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Invest New Drugs 36(1):96–102. [DOI] [PubMed] [Google Scholar]
- 31.Weiss GJ, et al. (2017) A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br J Cancer 117(1):33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winograd R, et al. (2015) Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 3(4):399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hingorani SR, et al. (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7(5):469–483. [DOI] [PubMed] [Google Scholar]
- 34.Principe DR, et al. (2014) TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst 106(2):djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hessmann E, et al. (2018) Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 67(3):497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobetz MA, et al. (2013) Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62(1):112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siegel RL, Miller KD, & Jemal A (2017) Cancer Statistics, 2017. CA Cancer J Clin 67(1):7–30. [DOI] [PubMed] [Google Scholar]
- 38.Ma Y, et al. (2013) Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38(4):729–741. [DOI] [PubMed] [Google Scholar]
- 39.Shurin GV, Tourkova IL, Kaneno R, & Shurin MR (2009) Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol 183(1):137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anyaegbu CC, Lake RA, Heel K, Robinson BW, & Fisher SA (2014) Chemotherapy enhances cross-presentation of nuclear tumor antigens. PLoS One 9(9):e107894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nomi T, et al. (2007) Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res 13(7):2151–2157. [DOI] [PubMed] [Google Scholar]
- 42.Saluja AK & Dudeja V (2013) Relevance of animal models of pancreatic cancer and pancreatitis to human disease. Gastroenterology 144(6):1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vennin C, et al. (2018) Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 154(4):820–838. [DOI] [PubMed] [Google Scholar]
- 44.Ebine K, et al. (2018) Interplay between interferon regulatory factor 1 and BRD4 in the regulation of PD-L1 in pancreatic stellate cells. Sci Rep 8(1):13225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Postow MA, Callahan MK, & Wolchok JD (2015) Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 33(17):1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coveler AL, Rossi GR, Vahanian NN, Link C, & Chiorean EG (2016) Algenpantucel-L immunotherapy in pancreatic adenocarcinoma. Immunotherapy 8(2):117–125. [DOI] [PubMed] [Google Scholar]
- 47.Le DT, et al. (2017) Results from a phase 2b, randomized, multicenter study of GVAX pancreas and CRS-207 compared to chemotherapy in adults with previously-treated metastatic pancreatic adenocarcinoma (ECLIPSE Study). Journal of Clinical Oncology 35(4_suppl):345–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Middleton G, et al. (2014) Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol 15(8):829–840. [DOI] [PubMed] [Google Scholar]
- 49.Hu ZI, et al. (2018) Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin Cancer Res 24(6):1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu C, et al. (2017) The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J Natl Cancer Inst 109(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lutz ER, et al. (2014) Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2(7):616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Melisi D, et al. (2016) A phase II, double-blind study of galunisertib+gemcitabine (GG) vs gemcitabine+placebo (GP) in patients (pts) with unresectable pancreatic cancer (PC). Journal of Clinical Oncology 34(15_suppl):4019–4019. [Google Scholar]
- 53.Chen X, et al. (2018) Dual TGF-beta and PD-1 blockade synergistically enhances MAGE-A3-specific CD8(+) T cell response in esophageal squamous cell carcinoma. Int J Cancer. [DOI] [PubMed] [Google Scholar]
- 54.Holmgaard RB, et al. (2018) Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer 6(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mariathasan S, et al. (2018) TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554(7693):544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tauriello DVF, et al. (2018) TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554(7693):538–543. [DOI] [PubMed] [Google Scholar]
- 57.Vanpouille-Box C & Formenti SC (2018) Dual Transforming Growth Factor-beta and Programmed Death-1 Blockade: A Strategy for Immune-Excluded Tumors? Trends Immunol 39(6):435–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paz-Ares L, et al. (2018) Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med 379(21):2040–2051. [DOI] [PubMed] [Google Scholar]
- 59.Gandhi L, et al. (2018) Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 378(22):2078–2092. [DOI] [PubMed] [Google Scholar]
- 60.Socinski MA, et al. (2018) Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 378(24):2288–2301. [DOI] [PubMed] [Google Scholar]
- 61.Horn L, et al. (2018) First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 379(23):2220–2229. [DOI] [PubMed] [Google Scholar]
- 62.Schmid P, et al. (2018) Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379(22):2108–2121. [DOI] [PubMed] [Google Scholar]
- 63.Feng M, et al. (2017) PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett 407:57–65. [DOI] [PubMed] [Google Scholar]
- 64.Otake A, et al. (2017) Chemotherapy-induced neutropenia as a prognostic factor in patients with metastatic pancreatic cancer treated with gemcitabine. Eur J Clin Pharmacol 73(8):1033–1039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.