

ABSTRACT
Background
Aberrations in the regulation of cell proliferation perturb cellular homeostasis and lead to malignancies in which dysregulation of the cell cycle and suppressed apoptosis are 2 common phenomena. Combinatorial nutritional approaches could be efficacious in ameliorating these aberrations.
Objectives
We sought to investigate the effect of dietary broccoli sprouts (BSp) and green tea polyphenol (GTP) administration on cell cycle progression and apoptosis in mammary tumors.
Methods
Forty female HER2/neu transgenic mice were randomly divided into 4 groups and treated with control, 26% BSp (wt:wt) in food, 0.5% GTPs (wt:vol) in drinking water, or combined BSp and GTPs from dams’ conception until their pups were killed at 29 wk of age. Pups’ tumor growth was monitored weekly for 27 wk. Tumor cell cycle– and apoptosis-related protein expression was measured. Data were analyzed with 2-factor or 3-factor (repeated-measures) ANOVA.
Results
Compared with the control group, BSp and/or GTPs decreased tumor incidence (P < 0.05) and combined BSp and GTPs synergistically [combination index (CIn) < 1] reduced tumor volume over time (P-time < 0.01). BSp and/or GTPs upregulated the expression of phosphatase and tension homolog, P16, and P53 (P < 0.05) and downregulated myelocytomatosis oncogene, Bmi1 polycomb ring finger oncogene, and telomerase reverse transcriptase (P < 0.05) compared with the control group. Combined BSp and GTPs synergistically (CIn < 1) downregulated the expression of cyclin B1, D1, and E1 and cyclin-dependent kinase 1, 2, and 4 (P < 0.05) compared with the control group. Moreover, combined BSp and GTPs induced apoptosis by regulating Bcl-2-associated X protein and B-cell lymphoma 2 (P < 0.05). BSp and/or GTPs also reduced the expression of DNA methyltransferase 1, 3A, and 3B and histone deacetylase 1 compared with the control group (P < 0.05).
Conclusions
Collectively, lifelong BSp and GTP administration can prevent estrogen receptor–negative mammary tumorigenesis through cell cycle arrest and inducing apoptosis in HER2/neu mice.
Keywords: breast cancer, broccoli sprouts, green tea polyphenols, cell cycle arrest, apoptosis
Introduction
Mammary tumor initiation and progression are chronic courses involving both genetic mutation and posttranslational dysregulation, resulting from sophisticated etiological disorders that are normally triggered by an imbalance between environmental impacts (endogenous, exogenous, and pathogenic) and the genetically susceptible host. An increasing body of evidence shows that a class of bioactive dietary compounds referred to as “epigenetic diets,” such as genistein in soybean products, resveratrol in grapes and berries, ascorbic acid in fruits, sulforaphane (SFN) in broccoli sprouts (BSp), and epigallocatechin-3-gallate (EGCG) in green tea, may act as environmental regulators that can neutralize genetic/epigenetic abnormalities during carcinogenesis, leading to breast cancer prevention (1). Studies have reported that several mechanisms may contribute to the preventive effect of these bioactive phytochemicals on breast cancer, including, but not limited to, induction of cell cycle arrest (2), promotion of apoptosis (3), regulation of phase 2 detoxification enzymes (4), and suppression of key regulators involved in the inflammatory response.
Conventional cancer prevention studies have primarily focused on singly administered dietary compounds that may not be sufficiently efficacious and there is a paucity of investigations targeting the collaborative effects of certain compounds on the regulation of molecular mechanisms involved in breast cancer prevention. This is a major gap in our knowledge because the human diet commonly consists of a multitude of nutritional ingredients. Recent advances have been made by studying the interactions between dietary compounds and combinatorial strategies in breast cancer prevention, among which the synergistic effect of SFN and EGCG shows promising efficacy owing to their similar favorable biological effects and potent anticancer properties despite their different mechanistic properties (5). SFN is a naturally occurring isothiocyanate often found in cruciferous vegetables, which have been of increasing interest owing to the rich presence of various bioactive compounds that show promising preventive or therapeutic effects on numerous cancers (6, 7). EGCG is the most abundant catechin which accounts for >50% of the total polyphenols in green tea, which is consumed extensively by humans. Green tea polyphenols (GTPs) have been shown to prevent breast cancer through inhibition of tumor angiogenesis and metastasis as well as induction of cell cycle arrest and apoptosis (5, 8). Previously, we found that combinatorial treatment with SFN and EGCG synergistically resensitized tamoxifen treatment in estrogen receptor (ER)-negative breast cancer via reactivation of ERα expression (9). In vivo studies further revealed that the combination of BSp and GTP diets significantly inhibited mammary tumor growth in ER-negative mouse xenografts (9). However, their efficacy on mammary tumor suppression in a spontaneous transgenic mouse model has not yet been proven and their mechanisms are not fully understood.
Accumulating studies propose that exposure windows are crucial to nutritional intervention against chronic diseases, including cancers. The Fetal Basis of Adult Disease hypothesis put forward that prenatal/maternal interventions such as nutrition can affect developmental programming, leading to changed disease susceptibility in later life (10). Early life, including embryogenesis, fetal development, and early postnatal life, is susceptible to external environmental cues that can disrupt individual development. This key window with considerable developmental plasticity also provides the possibility for parental nutritional intervention to maintain or reasonably moderate the genetic/epigenetic background in the offspring, leading to the health of descendants that may last for a lifetime. An interesting study revealed that early-life consumption of cabbage, a cruciferous vegetable, showed a lower incidence of breast cancer in the Polish (11). Maternal dietary supplementation of indole-3-carbinol, another bioactive compound derived from cruciferous vegetables, protected the fetus from transplacental carcinogenesis (12). Our previous studies also indicated that prenatal/maternal BSp dietary treatment exhibited maximal preventive effects in ER-negative mammary tumor development compared with adult BSp treatments through affecting epigenetic profiles (13). Thus, early life, especially maternal, exposure to key phytochemicals shows promising chemopreventive effects on breast cancer.
It has generally been assumed that because tumorigenesis carries a seemingly endless combination of genetic and epigenetic alterations resulting in hugely disparate types of cancers, cancer interventions must be diverse as well. Fortunately, the development of etiology offers an increasing number of therapeutic and preventive targets of various cancers, among which deregulated cell proliferation and the obligate compensatory suppression of apoptosis are 2 of a limited number of “mission critical” events that underlie the complexity and idiopathy of every cancer and can propel tumor cells into uncontrolled neoplastic progression (14). Because cancer is not a single disease that could be treated with a single cure, developing prevention and therapeutic strategies for the full gamut of cancers is almost impossible. However, it will be a promising direction if we focus on the molecular aberrations of proliferation and apoptosis that are intimately coupled in tumor progression.
It is known that cancer can be classified as an epigenetic disease, because epigenome dysregulation is involved in neoplastic processes of many cancers, and the epigenetic impact of bioactive dietary compounds on cancer attracts considerable attention. As an epigenetic diet, green tea–derived EGCG is a well-documented DNA methyltransferase (DNMT) inhibitor which can correct excessive hypermethylation in cancer cells (15, 16). EGCG also exhibits the most potent histone modification–inhibiting properties compared with other GTPs (17). Interest in SFN-mediated chemoprevention has recently surged partly owing to its proven activity against key modulators of histone modification, histone deacetylases (HDACs), which lead to increased acetylation globally and in the specific gene locus (18–20). SFN has also been shown to inhibit DNMT in cancer cells (21, 22). Interestingly, studies have suggested the involvement of DNMT inhibitors and HDAC inhibitors in the activation of the cell cycle (23, 24).
Herein, we hypothesized that the maternal/prenatal stage of development could be a potential critical window for BSp and/or GTP dietary administration against mammary tumor progression in a spontaneous breast cancer mouse model. In addition, the present study focuses on exploring the underlying mechanisms with respect to cell cycle arrest and apoptosis induction as well as epigenetic modification. Our findings indicate that combinatorial BSp and GTPs synergistically prevented ER-negative mammary tumors through arresting the cell cycle in most phases and checkpoints. Apoptosis and epigenetic regulation may also play a significant role in this process.
Methods
Animal experiments
Mouse model
We used a wild-type FVB/N-Tg (MMTVneu)202NK1/J (HER2/neu) mouse model which can spontaneously begin to develop ER-negative breast cancer tumors at ∼20 wk (13). The female mice of this model overexpress an activated rat c-neu oncogene Erbb2 which drives the development of ER-negative mammary tumors in mice (25). Because the HER2/neu gene is often overexpressed in human malignant breast tumors, the HER2/neu transgenic mouse model can closely approximate human breast cancer (13). Breeder Her2/neu mice (4 wk of age) were obtained from Jackson Laboratory. Genotyping of each generation was performed at week 3 to verify the Tag genotypes according to the genotyping protocol from Jackson Laboratory. Mice were housed in the Animal Resource Facility of the University of Alabama at Birmingham (UAB) and were maintained under the following conditions: 12-h dark/light cycle, at a temperature of 24 ± 2°C with 40%–60% humidity. Sample sizes were determined by 2-proportion comparison using a power calculator (http://powerandsamplesize.com/).
Dietary treatment
SFN-rich BSp powder (Natural Sprout Co.) was added at 26% (wt:wt) into modified AIN-93G diet pellets (26) (TestDiet). The details of the dietary ingredients have been previously published (13, 27) and are rearranged and provided in Supplemental Table 1. Orally fed GTP Sunphenon 90D (SP90D; Taiyo International, Inc.) was added at 0.5% (wt:vol) in drinking water. SP90D, which contains polyphenols at >90%, catechins at >80%, EGCG at >45%, and caffeine at <1%, is a decaffeinated extract of green tea containing purified polyphenols rich in green tea catechins. Supplemental Table 2 provides detailed specification values and the nutrient composition of SP90D.
In addition, the BSp and GTP diets used in this study are equivalent to daily intakes of ∼234 g BSp and ∼5.7 g GTPs for an adult (60-kg) human, respectively (28), which are considered pharmacologically achievable and have translational potential (10, 22). Our preliminary data revealed that dietary BSp and GTPs had no effects on the HER2/neu transgenic gene, transoncoprotein, or its phosphorylation state. These 2 dietary botanicals also showed no negative effects on maternal and offspring health, mammary gland development, and other tissues, indicating its safe application in this study.
Experimental design
Consistent with our power calculations, 40 Her2/neu female mice were randomly divided into 4 groups (10 mice/group) and were fed 1 of 4 different dietary regimens upon conception (determined upon expulsion of a vaginal plug): 1) control diet: mice were fed AIN-93G basal mix diet pellets; 2) BSp diet: mice were fed BSp at 26% (wt:wt, modified AIN-93G supplemented with 26% BSp; TestDiet) in food; 3) GTP diet: GTPs were administered in drinking water at 0.5% (wt:vol, 5 mg/mL); or 4) combination diet: mice were fed both the BSp and GTP diets. Treatments were first continued throughout the gestation and lactation periods of the dams. Twenty female pups were then randomly selected from each group upon weaning at 3 wk of age, and fed the same diet as their mother until they were killed at 29 wk of age, when all mice in the control group developed tumors with a mean diameter >1 cm (Figure 1).
FIGURE 1.

Schematic representation of study design. Female HER2/neu transgenic mice were treated with control, BSp, GTPs, or BSp + GTPs from dams’ conception through pups’ age of 29 wk. Pups’ mammary tumor growth was monitored weekly from weaning through the end of the experiment. Dams were mated at 10 wk of age; pups were weaned at 3 wk of age; the experiment was terminated with sampling at 29 wk of age. BSp, broccoli sprouts; GTP, green tea polyphenol.
Growth performance, tumor observation, and sampling
The body weight of pups was recorded biweekly from 4 wk to 28 wk of age. Food and water intakes of pups were measured at 4, 12, 20, and 28 wk of age, respectively.
Tumor volume and tumor incidence of pups were measured and recorded weekly. Tumor volumes were calculated using the formula: tumor volume (cm3) = 0.523 × [length (cm) × width2 (cm2)] (29). The experiment was terminated at 29 wk of age when all mice in the control group developed tumors with a mean diameter > 1 cm. At the end of the experiment, carbon dioxide was used to kill mice and the mammary tumors were excised, weighed, and snap-frozen in liquid nitrogen for further analysis. Animal procedures were reviewed and approved by the UAB Institutional Animal Care and Use Committee (IACUC; Animal Project Numbers: 10088 and 20653). All animal-related experiments and procedures were performed in accordance with the guidelines of the IACUC at UAB.
Western blotting analysis
Western blotting analysis was conducted as described previously (27). β-actin was used as the loading control in this study. Supplemental Table 3 lists detailed information on the primary antibodies used in the Western blotting analysis. Immunoreactive bands were visualized by ChemiDoc™ Imaging Systems (Bio-Rad). Protein expression was quantified by the densitometry function in the ImageJ (NIH) software.
Statistical analyses
Power calculations were conducted using an online calculator (http://powerandsamplesize.com/). Sample size was calculated by 1-sided 2-proportion comparison.
Statistical analysis was performed by SPSS (IBM) version 24.0. Tumor incidence was analyzed with the chi-square test and binary logistic regression (for time effect). Significant differences in tumor volume between treatments were determined using a 2 (BSp) × 2 (GTPs) × 11 (Time) mixed factorial design with repeated measures on the time factor. A 3-factor ANOVA was performed for food and water intakes (2 × 2 × 4) and body weight (2 × 2 × 13) with BSp, GTPs, and Time as the main factors. Tumor weight, latency, and protein expression were analyzed for main effects of BSp and GTPs, and their interactions using a 2-factor ANOVA (2 × 2). For post hoc analyses, significant differences between groups were inspected with Duncan's new multiple range test. Levene's test was performed to check the homogeneity of group variances. In the case of heterogeneity, the Welsh test was used for testing equality of group means, followed by the Games-Howell post hoc test. Obtained data were presented as means ± SEMs. Differences between groups were considered significant at P < 0.05.
The CompuSyn software (http://www.combosyn.com/; ComboSyn, Inc.) was used to determine combination index (CIn) values for tumor growth and protein expression. CIn < 1 indicates synergism, CIn = 1 indicates an additive effect, and CIn > 1 indicates antagonism (30, 31).
Results
Dietary BSp and/or GTP administration had no effect on the growth performance of HER2/neu mice
With the exception that the GTP group showed higher body weight than the control or BSp group at 8, 12, and 16 wk of age, lifelong BSp and/or GTP administration, overall, had no effect on body weight (Supplemental Figure 1, Supplemental Table 4) and daily food and water intakes (Supplemental Figure 1, Supplemental Table 5) of HER2/neu mice at the same point in time. Based on mouse daily food and water intake, daily BSp intakes of the BSp and combined groups were 1.24 g and 1.23 g, respectively; daily GTP intakes of the GTP and combined groups were 28.2 mg and 28.1 mg, respectively, after the mammary tumor formation in HER2/neu mice.
Lifelong BSp and/or GTP administration prevented ER-negative mammary tumorigenesis in HER2/neu mice
As Figure 2 depicts, dietary BSp or GTP administration notably suppressed tumor growth and the combined group showed a stronger inhibiting effect than individually administered BSp or GTPs. Both the single-intervention and combined groups prominently decreased tumor incidence in comparison with the control group (P < 0.05) (Figure 2A) and tumor incidence was significantly affected by time (P-Time < 0.01; OR: 1.94; 95% CI: 1.75, 2.16) (Figure 2A). For all 3 treatments, the weight of tumors was significantly decreased (P < 0.05) (Figure 2B) and the combined treatment exhibited a synergistic effect (CIn = 0.59) (Supplemental Table 6). In addition, dietary BSp and/or GTP supplementation were also shown to significantly reduce tumor volume (P < 0.05) (Figure 2C) and combined BSp and GTPs showed synergistic effects at 23 (CIn = 0.21), 25 (CIn = 0.87), 26 (CIn = 0.69), 28 (CIn = 0.63), and 29 (CIn = 0.72) wk (Supplemental Table 6). Importantly, mouse tumor volume of all treatment groups was significantly affected by time (P-Time < 0.01) (Figure 2C). Moreover, the BSp and GTP diets showed significant interaction in reducing tumor volume (P-interaction < 0.05) (Supplemental Table 6) and the combined group had a higher effect than either of the BSp or GTP groups. Strikingly, the mice in the control group started to form mammary tumors from 18 wk of age, whereas the dietary BSp, GTP, and combined groups started to have tumors from 21, 22, and 23 wk, respectively (Figure 2A). Compared with the control group (tumor latency = 23.2 wk), the BSp group (tumor latency = 27.3 wk), GTP group (tumor latency = 26.5 wk), and combined group (tumor latency = 28.7 wk) significantly extended tumor latency with ratios of 17.7%, 14.1%, and 23.5%, respectively (Supplemental Table 6). Taken together, the combined group was more effective than BSp or GTPs administered alone in suppressing tumor development, as shown by its lower tumor incidence, lighter tumor weight, and smaller tumor size (Figure 2), and showed, to some extent, synergistic effects. In addition, the BSp treatment group was more efficacious than the GTP treatment group in terms of inhibiting tumor growth.
FIGURE 2.

Tumors of control female HER2/neu mice and those exposed to BSp, GTPs, or both from conception through 29 wk of age. (A) Tumor incidence, (B) weight, and (C) volume. (D) Representative images of the excised tumors recorded under a light microscope. Values are means ± SEMs, n = 15–20. Labeled means, or means at a time, without a common letter differ, P < 0.05. BSp, broccoli sprouts; GTP, green tea polyphenol.
Dietary BSp and/or GTP administration increased the protein expression of tumor suppressors in HER2/neu mice
To unravel the molecular mechanisms of the aforementioned cancer prevention effect, we first analyzed the protein expression amount of several tumor suppressor proteins in mammary tumor samples from all 3 treatments. The BSp treatment significantly increased the expression of P16 and P53 (P < 0.05) (Figure 3) and tended to increase the expression of phosphatase and tension homolog (PTEN) (P = 0.10) (Figure 3). The GTP treatment significantly increased protein expression of PTEN and P53. The combinatorial treatment with BSp and GTPs significantly increased the expression of P16 and P53 (P < 0.05) (Figure 3) and showed a stronger effect than BSp and GTPs administered singly. The results indicated that all 3 dietary treatments had profound effects on increasing the expression of tumor suppressor proteins and contributed to cancer prevention. Notably, the combination of BSp and GTPs showed synergistic effects on increasing the expression of P16 (CIn = 0.39) and P53 (CIn = 0.06) (Supplemental Table 7) and their expression was significantly affected by BSp (main effect: P < 0.05) or GTP (main effect: P < 0.05) administration singly (Supplemental Table 7).
FIGURE 3.
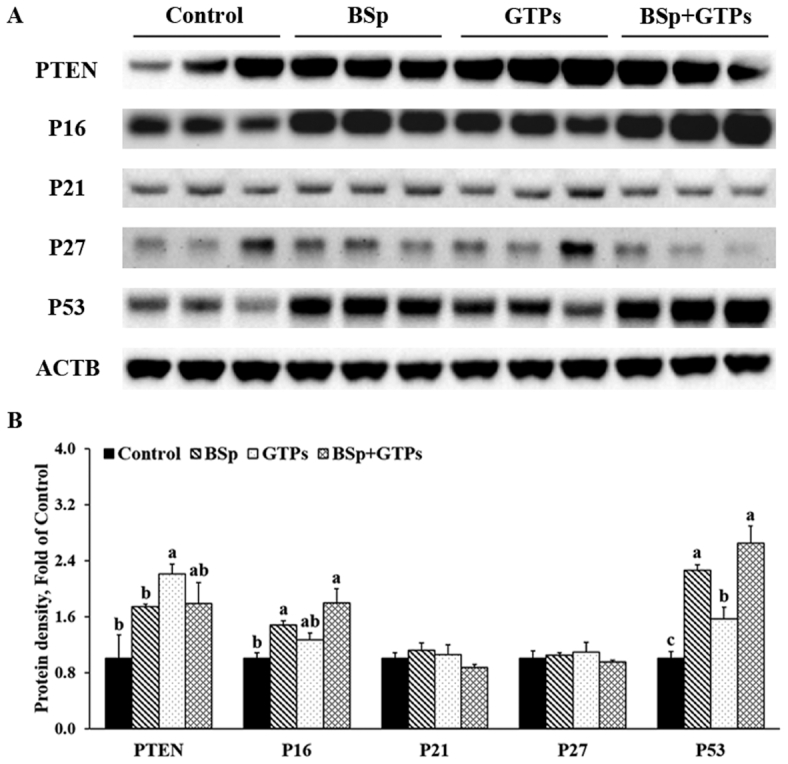
Western blot bands (A) and quantification (B) for PTEN, P16, P21, P27, and P53 in the tumors of female HER2/neu mice treated with control, BSp, GTPs, or BSp + GTPs. ACTB was used as a loading control. Values are means ± SEMs, n = 3. Labeled means without a common letter differ, P < 0.05. ACTB, β-actin; BSp, broccoli sprouts; GTP, green tea polyphenol; PTEN, phosphatase and tension homolog.
Dietary BSp and/or GTP administration induced cell cycle arrest by regulating cyclins and cyclin-dependent kinases in HER2/neu mice
We then evaluated the effect of BSp and/or GTPs on the expression of cyclin (CCN) and cyclin-dependent kinase (CDK) in mammary tumors of offspring. As Figures 4 and 5 show, the combined BSp and GTP diet significantly reduced the expression of CCNB1 and CDK1 (P < 0.05) and the G2/M phase CCN–CDK complex (Figures 4, 5). Further, G1/S phase CDK2 and CCNE1, and CDK4 and CCND1 also significantly decreased in the combined group (Figures 4, 5). Strikingly, we found that the expression of retinoblastoma protein (RB) significantly increased (P < 0.05), whereas the phosphorylation of RB (pRB) showed a decrease although the difference was not significant in the combined group. In addition, S phase CDKA2 also decreased but not at a significant level. Interestingly, the expression of CDK6 significantly increased in this study. Moreover, lifelong combinatorial administration of BSp and GTPs exhibited synergistic effects on inhibiting the protein expression amounts of CDK1 (CIn = 0.11), CDK4 (CIn = 0.08), CDK6 (CIn = 0.03), CCNA2 (CIn = 0.29), and CCNB1 (CIn = 0.17) and their expression amounts were more likely to be affected by BSp administration (main effect: P < 0.05) (Supplemental Table 7). These results indicated that dietary BSp and GTPs induce cell cycle arrest during the G1 phase and slow the G1–S phase and G2–M phase transitions.
FIGURE 4.
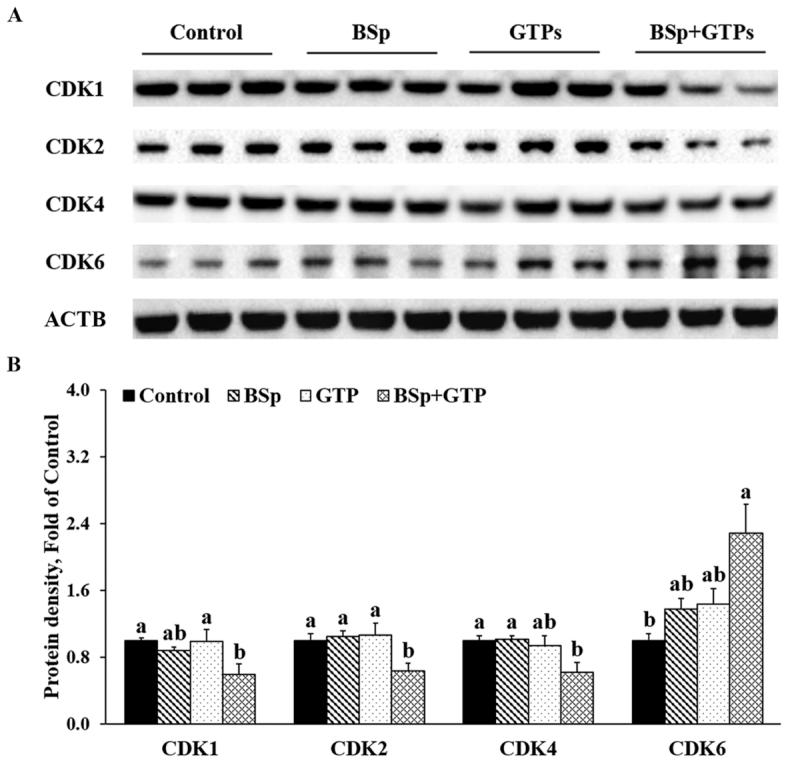
Western blot bands (A) and quantification (B) for CDK1, 2, 4, and 6 in the tumors of female HER2/neu mice treated with control, BSp, GTPs, or BSp + GTPs. ACTB was used as a loading control. Values are means ± SEMs, n = 3. Labeled means without a common letter differ, P < 0.05. ACTB, β-actin; BSp, broccoli sprouts; CDK, cyclin-dependent kinase; GTP, green tea polyphenol.
FIGURE 5.
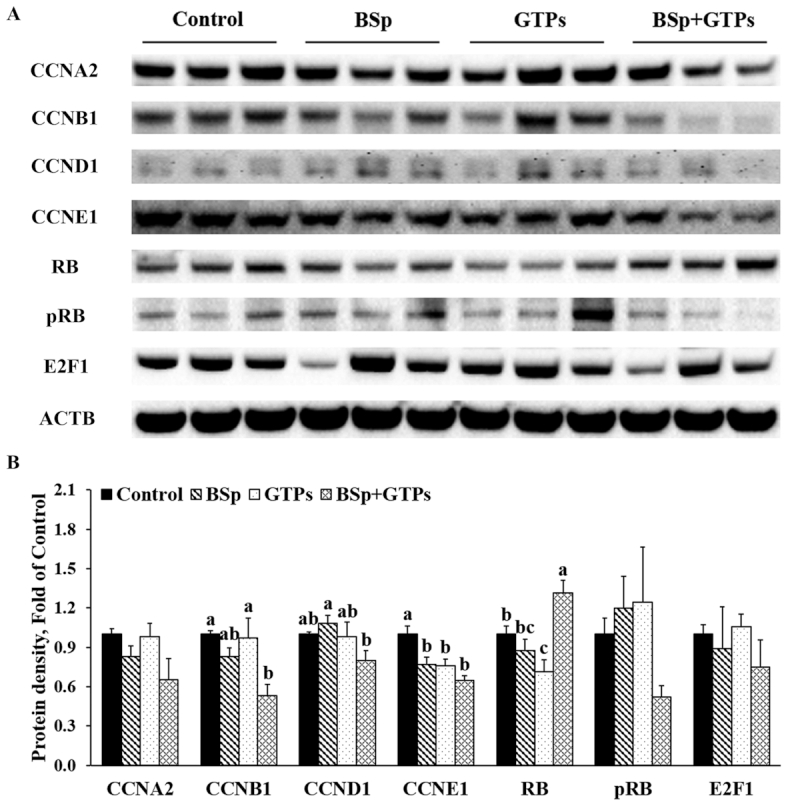
Western blot bands (A) and quantification (B) for CCNA2, CCNB1, CCND1, CCNE1, RB, pRB, and E2F1 in the tumors of female HER2/neu mice treated with control, BSp, GTPs, or BSp + GTPs. ACTB was used as a loading control. Values are means ± SEMs, n = 3. Labeled means without a common letter differ, P < 0.05. ACTB, β-actin; BSp, broccoli sprouts; CCN, cyclin; GTP, green tea polyphenol; pRB, phosphorylation of retinoblastoma protein; RB, retinoblastoma protein.
Dietary BSp and/or GTPs promoted apoptosis in the mammary tumors of HER2/neu mice
We also measured the expression of 2 important members of the Bcl-2 family, Bcl-2-associated X protein (BAX) and B-cell lymphoma 2 (BCL2), that are involved in the evolutionary pathway for programmed cell death or apoptosis. As Figure 6 shows, BSp or GTP diets administered alone and their combination significantly increased the expression of BAX and decreased the expression of BCL2 (P < 0.05) (Figure 6). The ratio of BAX to BCL2 also showed a remarkable increase in all 3 treatment groups. In addition, we found that combined BSp and GTPs can synergistically inhibit the protein expression of BCL2 (CIn = 0.88) and at the same time showed significant interaction on the expression of BAX (P-interaction < 0.05) (Supplemental Table 7). These results suggest that BSp and GTPs, treated singly or in combination, induce apoptosis through upregulating BAX and downregulating BCL2.
FIGURE 6.
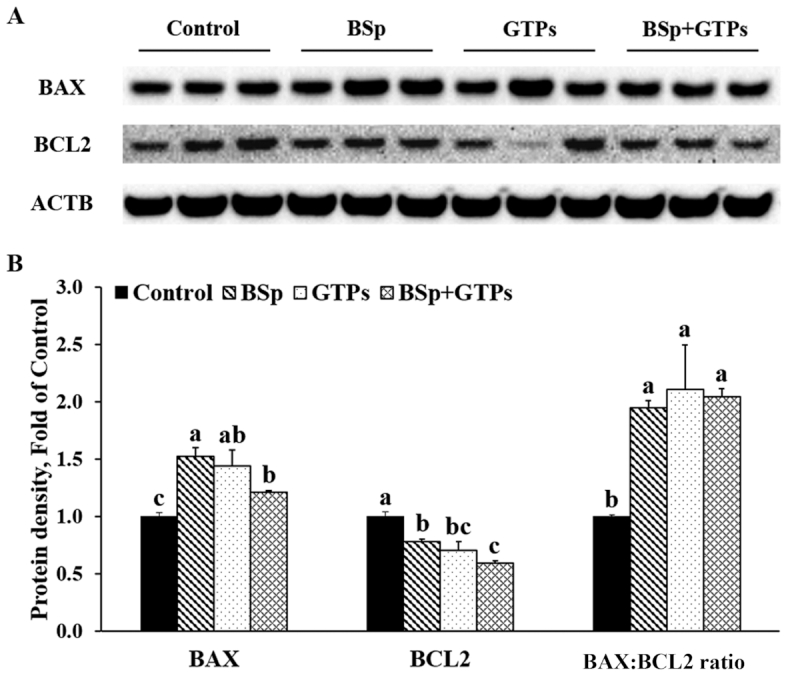
Western blot bands (A) and quantification (B) for BAX and BCL2 in the tumors of female HER2/neu mice treated with control, BSp, GTPs, or BSp + GTPs. ACTB was used as a loading control. Values are means ± SEMs, n = 3. Labeled means without a common letter differ, P < 0.05. ACTB, β-actin; BAX, Bcl-2-associated X protein, BCL2, B-cell lymphoma 2; BSp, broccoli sprouts; GTP, green tea polyphenol.
Dietary BSp and/or GTP administration led to decreased expression of tumor-promoting proteins in HER2/neu mice
To explore the inhibiting role of BSp and GTPs in growth-promoting pathways, we moved forward to detect the expression changes of myelocytomatosis oncogene (MYC), telomerase reverse transcriptase (TERT), and Bmi1 polycomb ring finger oncogene (BMI1). Results indicated that BSp, GTPs, and the combinatorial treatments significantly decreased the expression of the tumor-promoting proteins MYC, TERT, and BMI1 (P < 0.05) (Figure 7). The BSp and GTP combinatorial treatment had a stronger effect on inhibiting the protein amounts of TERT and BMI1 and showed a significant interaction on the expression of TERT (P-interaction < 0.01) (Supplemental Table 7). These results suggested that BSp and GTPs together can inhibit tumor progression and then drive the tumor cells toward cell cycle arrest and/or apoptosis.
FIGURE 7.
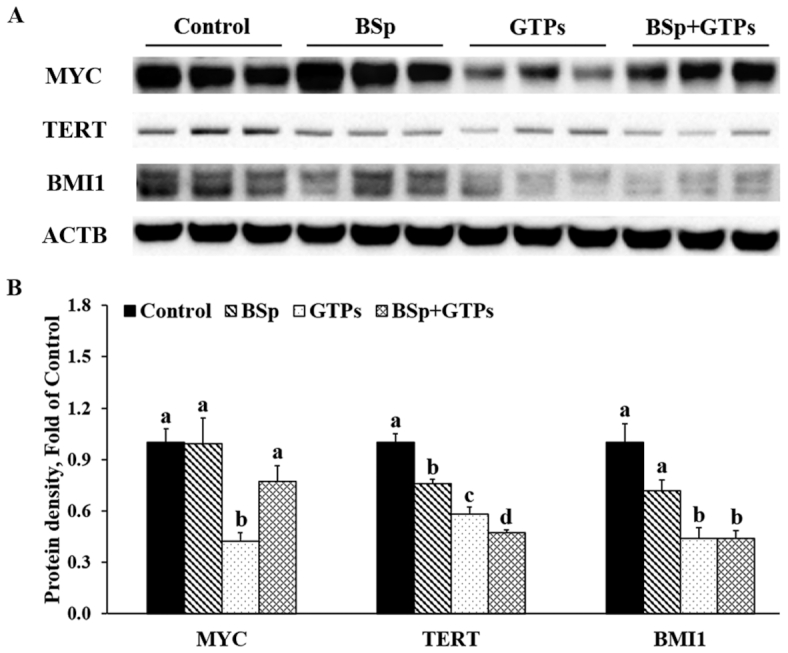
Western blot bands (A) and quantification (B) for MYC, TERT, and BMI1 in the tumors of female HER2/neu mice treated with control, BSp, GTPs, or BSp + GTPs. ACTB was used as a loading control. Values are means ± SEMs, n = 3. Labeled means without a common letter differ, P < 0.05. ACTB, β-actin; BSp, broccoli sprouts; BMI1, Bmi1 polycomb ring finger oncogene; GTP, green tea polyphenol; MYC, myelocytomatosis oncogene; TERT, telomerase reverse transcriptase.
BSp and/or GTP administration induced expression changes of DNMTs and HDACs in HER2/neu mice
Because SFN is known as an HDAC inhibitor (32) and GTPs inhibit DNMTs (15), we further investigated the epigenetic influence of BSp and GTPs in cancer prevention. As Figure 8 shows, both BSp and GTPs administered alone significantly decreased the expression of DNMT1 and DNMT3B (P < 0.05) (Figure 8). The combined treatment with BSp and GTPs had significant synergistic effects on decreasing the protein expression of DNMT3A (CIn = 0.70) and HDAC1 (CIn = 0.54) (P < 0.05) but showed no significant effect on DNMT1 (Figure 8). The expression of DNMTs was shown to be affected by BSp (main effect: P < 0.01) and/or GTP (main effect: P < 0.05) administration (P-interaction < 0.01) (Supplemental Table 7). Together, these results suggested that the combinatorial treatment with BSp and GTPs greatly inhibited the expression of epigenetics-related enzymatic modulators, including DNMT3A, DNMT3B, and HDAC1.
FIGURE 8.

Western blot bands (A) and quantification (B) for DNMT1, 3A, and 3B and HDAC1 in the tumors of female HER2/neu mice treated with control, BSp, GTPs, or BSp + GTPs. ACTB was used as a loading control. Values are means ± SEMs, n = 3. Labeled means without a common letter differ, P < 0.05. ACTB, β-actin; BSp, broccoli sprouts; DNMT, DNA methyltransferase; GTP, green tea polyphenol; HDAC, histone deacetylase.
Discussion
Our previous studies of the efficacy of combinatorial SFN and EGCG revealed a strong inhibition of the cell viability of 2 ER-negative breast cancer cell lines, MDA-MB-157 and MDA-MB-231, as well as 2 ovarian cancer cell lines, paclitaxel-sensitive (SKOV3-ip 1) and -resistant (SKOV3TR-ip 2) (9, 33). We found that both SFN and EGCG were capable of suppressing breast cancer cell growth and that combined SFN and EGCG treatment can increase apoptosis by downregulating hTERT and Bcl-2, and arrest cell proliferation in both the G2/M and S phases in ovarian cancer cells (33). Further in vivo studies revealed that combinatorial diets of BSp, a known source of SFN, and GTPs, in which EGCG accounts for >50% of catechins, significantly inhibited mammary tumor growth in breast cancer mouse xenografts (9, 34), which led us to hypothesize that BSp and GTPs may inhibit the growth of mammary tumors in mouse through cell cycle arrest and inducing apoptosis. Furthermore, our recent findings suggested that both maternal (pregestational or perinatal) and early-life (from weaning to adulthood) BSp singly administrated can profoundly reduce mammary tumorigenesis through affecting epigenetic profiles and tumor-related gene expression in the nontreated mouse offspring, whereas an adulthood-administered BSp diet only showed slight suppressive effects on mammary tumors, suggesting that time window of the intervention could be a key factor in bioactive compound administration for coping with mammary cancer (13, 27). We therefore conducted an in vivo study using a spontaneous transgenic breast cancer mouse model (HER2/neu) with a lifelong treatment with BSp and/or GTPs from the conception of dams until the termination of their pups and focused our analysis on cell proliferation– and apoptosis related-pathways.
As expected, lifelong BSp and/or GTP administration can prominently decrease tumor incidence, reduce tumor volume, and delay tumor latency, among which combined BSp and GTPs was more effective in inhibiting the tumor growth than BSp or GTPs administered alone. The dramatic tumor prevention effects, especially reduced tumor size, observed in this study could be a mixed effect of tumorigenesis prevention before the stage of full-blown tumors and tumor shrinking (cancer cell killing) after they are formed. The significantly prolonged tumor latency of the BSp and/or GTP treatment groups indicated that dietary compounds can delay spontaneous tumorigenesis in HER2/neu mice. On the other hand, the dietary compounds can also induce cell cycle arrest and apoptosis in mammary cancer, which will cause tumor cell death, after tumor formation. Our previous results showed that both maternal treatment (gestation and lactation periods) and postnatal early-life (from weaning to adulthood) BSp administration can significantly suppress mammary tumor progression in both SV40 and HER2/neu mouse models, indicating that the tumor inhibition effects observed in this study were a combined effect of maternal and postnatal treatments (13).
Because it is known that P21 and P27 are 2 critical members from the Cip/Kip family, which is 1 of the 2 classes of Cdk-inhibitory subunits (CKIs) and predominantly inhibits the G1–S phase transition (35), we expected to observe an increase in these 2 proteins in the mammary tumors that we obtained from treated mice. Although BSp or GTPs administered singly and their combination did not significantly upregulate P21 and P27, we did observe a remarkable increase in P16 in BSp- and/or GTP-treated mice. In mammalian cells, the Cip/Kip family and another class of CKIs, the Ink4 family, provide a tissue-specific mechanism by which CDK activity is mediated and cell cycle progression can be restrained in response to various cellular signals (35). Because P16 is an important member of the Ink4 (inhibitors of CDK4) family (35), this suggests that BSp and GTPs may arrest the G1 phase through promoting the expression of an Ink4 family member rather than Cip/Kip subunits. A study carried out on vascular smooth muscle cells showed that HDAC inhibitors can upregulate the transcript amount of p21, whereas this induction did not translate to increased protein amounts, owing to unrecognized translational or posttranslational regulation of the P21 protein (24). We also detected the protein expression amounts of P53, which is an important cell growth regulator whose function is lost in approximately half of all human cancers and is proven to be involved in several different aspects of cell proliferation, apoptosis, and DNA repair (36). It was shown that activated P53 induced the expression of P21, which can further inhibit CCND/CDKs, resulting in the arrest of the G1 phase (37). Overexpression of P53 can also induce G2-M growth arrest probably by suppressing CCNB1 transcription and synthesis in a cell-type-specific manner (38, 39). Moreover, studies demonstrate that P53 can downregulate CDK2 via the regulation of CDK-activating kinase activity in the S phase (37). In the present study, we found that dietary BSp and GTPs significantly increased the expression of P53 but had no effect on P21, demonstrating that this dietary regimen may mainly participate in cell cycle arrest in mammary tumors through targeting the S and G2 phases (Supplemental Figure 2). In addition, we observed an increased expression of PTEN in GTP-treated mice. A large number of findings have shown that PTEN acts as a brake in 2 major transitions during the cell cycle, the G1–S and G2–M phases, preventing controlled proliferation and chromosome instability. PTEN has a nuclear function of downregulating CCND, resulting in G1 arrest. PTEN can also form a complex with CCNB1 and CDK1, preventing bypass of the G2 checkpoint. Although we did not observe significant expression increases of PTEN from the BSp and the combination treatment groups, we did observe noticeable rises in these 2 groups, indicating dietary BSp and GTPs may reasonably control G1 progression and the G2–M transition through upregulating the protein expression of PTEN. MYC is another tumor progression–related protein that was investigated in this study. The activation of MYC is associated with mammary tumorigenesis and it is considered as a promising therapeutic target against breast cancer (40). We found that long-term dietary GTPs significantly decreased the expression of MYC, indicating that GTPs participate in mammary tumor suppression by downregulating MYC. It is worth noting that MYC also has a key role in normal cell cycle progression. In particular, P53-dependent transcriptional repression of MYC is required for G1 cell cycle arrest (41). Like MYC, TERT is also a cancer-promoting protein and underwent highly significant decreases in all 3 of our treatment groups. Celeghin et al. (42) conducted a study indicating that the inhibition of TERT impairs cell cycle progression in TERT-positive cells, suggesting the possible role of TERT on proliferation of mammary tumors in this study.
Progression of the 4 phases in the cell cycle of the eukaryotic cell is mediated by sequential activation and inactivation of CDKs as well as their cognate CCN partners. The progression of the G1 phase is mainly driven by CDK2 and CDK4/6, after binding to CCND and CCNE. Our study shows decreases of CDK2, CDK4, and CCNE1, as well as a decreasing tendency of CCND1, at the protein level in BSp- and GTP-treated mammary tumors. A recent study showed that combinatorial SFN and withaferin A, a bioactive compound found in the Indian winter cherry, downregulated the amounts of CCND1, CDK4, and pRB; conversely, the expression amounts of E2F and P21 were increased in breast cancer cells (2). Interestingly, we report a dramatic increase of the protein amount of CDK6, which is typically associated with positively regulating the cell cycle from the G1 to the S phase (43). In recent years, new evidence has suggested that the role of CDK6 for proliferation might be less important in certain cell types, where CDK4 or CDK2 can compensate for its role (44, 45). In addition, CDKs and CCNs form activated complexes before function, and the decreasing of CCND may also counteract the increased expression of CDK6, but further studies are needed to reveal potential modes of action. RB is a tumor suppressor and is negatively associated with cell cycle progression. The phosphorylation of RB, which is normally driven by the active kinase CCND-CDK4/6, causes dissociation of the transcription factor E2F1 as well as HDAC1. The release of E2F1 and HDAC1 allows transcriptional activation of CCNE which will activate CDK2 to further phosphorylate RB. As Supplemental Figure 2 shows, dietary BSp and GTPs may inhibit the phosphorylation of RB through downregulating the protein amounts of CCND1 and CCNE1, along with their binding partners, CDK4 and CDK2, resulting in lower release of E2F1 and HDAC1, which further arrests the progression of the G1 phase and inhibits the transition from G1 to S. We also report downregulated expression of 3 CDK complexes, CCNB1/CDK1, CCNA2/CDK2, and CCNA2/CDK1, in the combined treatment with BSp and GTPs (Supplemental Figure 2). CCNA can bind to CDK2 in the S phase or promote the transition from G2 to M when it binds to CDK1. The CCNB/CDK1 complex also functions during the G2–M transition. Our previous study showed that SFN can arrest the progression from G2 to M phase in the proliferation of ovarian cancer cells, and the inhibitory effect can extend to the S phase when it is combined with EGCG (33). In the present study, major components of GTPs (EGCG) and BSp (SFN) may, to a large extent, also synergistically inhibit cell proliferation through arresting the S phase and G2–M transition in mammary tumors.
The convergence of deregulated cell proliferation and suppressed cell death constitutes the common platform for neoplastic evolution and is required for the development of all cancers, although tumors are extremely heterogeneous and diverse (14). Moreover, proliferation and apoptosis are intimately coupled (46). We therefore detected several key proteins involved in apoptosis regulation as well (Supplemental Figure 3). BCL2 is an oncogene-derived protein that negatively controls cellular death. As a BCL2-homologous protein, BAX can promote cell death by competing with BCL2. As suspected, we reported significant increases of BAX at the protein level in BSp- and/or GTP-treated mice; inversely, decreases of BCL2 were found in the same treatment group, indicating that these dietary regimens suppress mammary tumor development via inducing apoptosis. More solid evidence in favor of the link between cell cycle progression and apoptosis is based on some studies in which apoptosis was regulated by those genes that are also involved in cell proliferation. As aforementioned, P53 can induce cell cycle arrest, but in some other circumstances it triggers apoptosis. Activated P53 has been shown to repress the transcription of BCL2 to induce apoptosis (47). In some systems, the proapoptotic protein, BAX, is also a transcriptional target of P53 (48). Another study showed that cell death caused by the aberrant expression of P53 can be reverted by pRB in HeLa cells with the presence of human papillomavirus (49). pRB not only can bind with E2F1 forming stable complexes throughout the cell cycle, but also has a protective function against apoptosis. In addition, results show that pRB may also inhibit apoptosis by regulating the activation of E2F1, indicating E2F1 is involved in apoptosis pathways as well (50). Interestingly, a study conducted by Alajez et al. (51) using the C666-1 nasopharyngeal cancer model demonstrated that targeted depletion of BMI1 can sensitize cancer cells to P53-mediated apoptosis. Overexpression of BMI1 is a common characteristic during cancer development and progression. We observed a greater reduction of the protein expression of BMI1 associated with suppression by the combination of BSp and GTPs in this study. Moreover, BMI1 was shown to collaborate with MYC in tumorigenesis via inhibiting MYC-induced apoptosis (52). Thus, apoptosis partly induced by the downregulation of BMI1 and MYC may be also involved in the mammary tumor suppression observed in our study. Our previous studies showed that the combination of SFN and EGCG can increase apoptosis by downregulating hTERT and BCL2 in ovarian cancer cells (33), suggesting that TERT may be also involved in mammary tumor apoptosis in the present study.
It is known that SFN is an HDAC inhibitor and EGCG acts as a potent DNMT inhibitor, and they both have been also shown to have properties involved in DNMT inhibition or HDAC inhibition (9, 15). As epigenetic regulators, HDACs and DNMTs are both critical in the regulation of cell cycle progression as well (2). As aforementioned, the phosphorylation of RB causes dissociation of HDAC1, which further allows the transcriptional activation of CCNE. Our previous results showed the downregulation of HDAC1, 2, and 3 induced by SFN and/or WA was responsible for the expression changes of P21 and P53 (2). In smooth muscle cells, the phosphorylation of RB was shown to be entirely abolished by HDAC inhibition through transcriptional repression of CCND1, resulting in downregulation of downstream E2F target genes (24). The present study also found that a combinatorial BSp and GTPs diet is capable of significantly decreasing the protein expression of HDAC1 in mammary tumors, indicating the role of this dietary regimen in cell cycle arrest via HDAC inhibition. Moreover, we reported notably decreased expression amounts of DNMT1, 3A, and 3B in this study. A recent study revealed that DNMT1 was involved in the activation of cell cycle progression in diffuse large B-cell lymphoma cells (23). Gu et al. also reported that microRNA-129-5p can induce cell cycle arrest in the G1 phase by directly targeting DNMT3A (53). These add supporting evidence to our study that downregulation of DNMTs is linked to cell cycle arrest in mammary tumor cells. It is worth noting that the significantly reduced expression of DNMTs and HDACs in this study may also indicate that the dietary compounds of BSp and/or GTPs specifically target mammary tumor cells because normal tissues do not have excess amounts of DNMTs and HDACs (54, 55), and our preliminary data also revealed that dietary BSp and/or GTPs showed no negative effects on benign mammary gland development or other tissues.
In summary, lifelong BSp and/or GTP administration can profoundly suppress ER-negative mammary tumorigenesis through arresting the cell cycle and inducing apoptosis. Importantly, combined BSp and GTPs showed synergistic effects on mammary tumor suppression in HER2/neu mice, suggesting that the combined addition of bioactive botanicals could be a more promising approach to protect against tumor initiation and progression. This study may also provide mechanistic understanding with respect to appropriate administration of key dietaries for coping with breast cancer development in humans.
Supplementary Material
Acknowledgments
We acknowledge the Tollefsbol laboratory members for their advice and support in this study. The authors’ responsibilities were as follows—SL and TOT: designed the research; SL and HW: conducted the research and wrote the first draft; SL: analyzed the data; TOT: had primary responsibility for the final content; and all authors: read and approved the final manuscript.
Notes
Supported by NIH grants R01CA178441 (to TOT) and R01CA204346 (to TOT).
Author disclosures: the authors report no conflicts of interest.
Supplemental Figures 1–3 and Supplemental Tables 1–7 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
SL and HW contributed equally to this work.
Abbreviations used: BAX, Bcl-2-associated X protein; BCL2, B-cell lymphoma 2; BMI1, Bmi1 polycomb ring finger oncogene; BSp, broccoli sprouts; CCN, cyclin; CDK, cyclin-dependent kinase; CIn, combination index; CKI, cyclin-dependent kinase inhibitory subunit; DNMT, DNA methyltransferase; EGCG, epigallocatechin-3-gallate; ER, estrogen receptor; GTP, green tea polyphenol; HDAC, histone deacetylase; IACUC, Institutional Animal Care and Use Committee; MYC, myelocytomatosis oncogene; pRB, phosphorylation of retinoblastoma protein; PTEN, phosphatase and tension homolog; RB, retinoblastoma protein; SFN, sulforaphane; SP90D, Sunphenon 90D; TERT, telomerase reverse transcriptase; UAB, University of Alabama at Birmingham.
Contributor Information
Shizhao Li, Department of Biology, University of Alabama at Birmingham, Birmingham, AL, USA.
Huixin Wu, Department of Biology, University of Alabama at Birmingham, Birmingham, AL, USA.
Trygve O Tollefsbol, Department of Biology, University of Alabama at Birmingham, Birmingham, AL, USA; Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, AL, USA; Nutrition Obesity Research Center, University of Alabama at Birmingham, Birmingham, AL, USA; Comprehensive Center for Healthy Aging, University of Alabama at Birmingham, Birmingham, AL, USA; Comprehensive Diabetes Center, University of Alabama at Birmingham, Birmingham, AL, USA.
References
- 1. Li S, Chen M, Li Y, Tollefsbol TO. Prenatal epigenetics diets play protective roles against environmental pollution. Clin Epigenet. 2019;11(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Royston KJ, Paul B, Nozell S, Rajbhandari R, Tollefsbol TO. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp Cell Res. 2018;368(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Royston KJ, Udayakumar N, Lewis K, Tollefsbol TO. A novel combination of withaferin A and sulforaphane inhibits epigenetic machinery, cellular viability and induces apoptosis of breast cancer cells. Int J Mol Sci. 2017;18(5):1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci U S A. 1997;94(19):10367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meeran SM, Patel SN, Li Y, Shukla S, Tollefsbol TO. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS One. 2012;7(5):e37748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gasparello J, Gambari L, Papi C, Rozzi A, Manicardi A, Corradini R, Gambari R, Finotti A. High levels of apoptosis are induced in the human colon cancer HT-29 cell line by co-administration of sulforaphane and a peptide nucleic acid targeting miR-15b-5p. Nucleic Acid Ther. 2020;30(3):164–74. [DOI] [PubMed] [Google Scholar]
- 7. Shankar S, Ganapathy S, Srivastava RK. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin Cancer Res. 2008;14(21):6855–66. [DOI] [PubMed] [Google Scholar]
- 8. Steed KL, Jordan HR, Tollefsbol TO. SAHA and EGCG promote apoptosis in triple-negative breast cancer cells, possibly through the modulation of cIAP2. Anticancer Res. 2020;40(1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Meeran SM, Tollefsbol TO. Combinatorial bioactive botanicals re-sensitize tamoxifen treatment in ER-negative breast cancer via epigenetic reactivation of ERα expression. Sci Rep. 2017;7(1):9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Law C, Barker D, Osmond C, Fall C, Simmonds S. Early growth and abdominal fatness in adult life. J Epidemiol Community Health. 1992;46(3):184–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nelson NJ. Migrant studies aid the search for factors linked to breast cancer risk. J Natl Cancer Inst. 2006;98(7):436–8. [DOI] [PubMed] [Google Scholar]
- 12. Yu Z, Mahadevan B, Löhr CV, Fischer KA, Louderback MA, Krueger SK, Pereira CB, Albershardt DJ, Baird WM, Bailey GS. Indole-3-carbinol in the maternal diet provides chemoprotection for the fetus against transplacental carcinogenesis by the polycyclic aromatic hydrocarbon dibenzo[a,l]pyrene. Carcinogenesis. 2006;27(10):2116–23. [DOI] [PubMed] [Google Scholar]
- 13. Li Y, Buckhaults P, Li S, Tollefsbol T. Temporal efficacy of a sulforaphane-based broccoli sprout diet in prevention of breast cancer through modulation of epigenetic mechanisms. Cancer Prev Res. 2018;11(8):451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–8. [DOI] [PubMed] [Google Scholar]
- 15. Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63(22):7563–70. [PubMed] [Google Scholar]
- 16. Lee WJ, Shim J-Y, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68(4):1018–30. [DOI] [PubMed] [Google Scholar]
- 17. Choi K-C, Jung MG, Lee Y-H, Yoon JC, Kwon SH, Kang H-B, Kim M-J, Cha J-H, Kim YJ, Jun WJ. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009;69(2):583–92. [DOI] [PubMed] [Google Scholar]
- 18. Paul B, Li Y, Tollefsbol TO. The effects of combinatorial genistein and sulforaphane in breast tumor inhibition: role in epigenetic regulation. Int J Mol Sci. 2018;19(6):1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin SL, Kala R, Tollefsbol TO. Mechanisms for the inhibition of colon cancer cells by sulforaphane through epigenetic modulation of microRNA-21 and human telomerase reverse transcriptase (hTERT) down-regulation. Curr Cancer Drug Targets. 2018;18(1):97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang L-L, Zhou S-J, Zhang X-M, Chen H-Q, Liu W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed Pharmacother. 2016;78:74–80. [DOI] [PubMed] [Google Scholar]
- 21. Gao L, Cheng D, Yang J, Wu R, Li W, Kong A-N. Sulforaphane epigenetically demethylates the CpG sites of the miR-9-3 promoter and reactivates miR-9-3 expression in human lung cancer A549 cells. J Nutr Biochem. 2018;56:109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan H, Zhang R, Tesfaye D, Tholen E, Looft C, Hölker M, Schellander K, Cinar MU. Sulforaphane causes a major epigenetic repression of myostatin in porcine satellite cells. Epigenetics. 2012;7(12):1379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loo SK, Hamid SSA, Musa M, Wong KK. DNMT1 is associated with cell cycle and DNA replication gene sets in diffuse large B-cell lymphoma. Pathol Res Pract. 2018;214(1):134–43. [DOI] [PubMed] [Google Scholar]
- 24. Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011;31(4):851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54(1):105–15. [DOI] [PubMed] [Google Scholar]
- 26. Reeves PG, Nielsen FH, Fahey GC Jr. AIN-93 Purified Diets for Laboratory Rodents: Final Report of the American Institute of Nutrition ad hoc Writing Committee on the Reformulation of the AIN-76A Rodent Diet. Am J Clin Nutr. 1993;123(11):1939–51. [DOI] [PubMed] [Google Scholar]
- 27. Li S, Chen M, Wu H, Li Y, Tollefsbol TO. Maternal epigenetic regulation contributes to prevention of estrogen receptor–negative mammary cancer with broccoli sprout consumption. Cancer Prev Res. 2020;13(5):449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen G, Khor TO, Hu R, Yu S, Nair S, Ho C-T, Reddy BS, Huang M-T, Newmark HL, Kong A-NT. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007;67(20):9937–44. [DOI] [PubMed] [Google Scholar]
- 29. Zhou J-R, Gugger ET, Tanaka T, Guo Y, Blackburn GL, Clinton SK. Soybean phytochemicals inhibit the growth of transplantable human prostate carcinoma and tumor angiogenesis in mice. J Nutr. 1999;129(9):1628–35. [DOI] [PubMed] [Google Scholar]
- 30. Gao Y, Tollefsbol TO. Combinational proanthocyanidins and resveratrol synergistically inhibit human breast cancer cells and impact epigenetic–mediating machinery. Int J Mol Sci. 2018;19(8):2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chou T-C. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008;49(11):2059–80. [DOI] [PubMed] [Google Scholar]
- 32. Myzak MC, Karplus PA, Chung F-L, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64(16):5767–74. [DOI] [PubMed] [Google Scholar]
- 33. Chen H, Landen CN, Li Y, Alvarez RD, Tollefsbol TO. Epigallocatechin gallate and sulforaphane combination treatment induce apoptosis in paclitaxel-resistant ovarian cancer cells through hTERT and Bcl-2 down-regulation. Exp Cell Res. 2013;319(5):697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Y, Buckhaults P, Cui X, Tollefsbol TO. Combinatorial epigenetic mechanisms and efficacy of early breast cancer inhibition by nutritive botanicals. Epigenomics. 2016;8(8):1019–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev. 1996;6(1):56–64. [DOI] [PubMed] [Google Scholar]
- 36. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–31. [DOI] [PubMed] [Google Scholar]
- 37. Schneider E, Montenarh M, Wagner P. Regulation of CAK kinase activity by p53. Oncogene. 1998;17(21):2733–41. [DOI] [PubMed] [Google Scholar]
- 38. Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. The p53 network. J Biol Chem. 1998;273(1):1–4. [DOI] [PubMed] [Google Scholar]
- 39. Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci U S A. 1995;92(18):8493–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. D'Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, Cox JD, Ha SI, Belka GK, Golant A. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7(2):235–9. [DOI] [PubMed] [Google Scholar]
- 41. Ho JSL, Ma W, Mao DYL, Benchimol S. p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol. 2005;25(17):7423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Celeghin A, Giunco S, Freguja R, Zangrossi M, Nalio S, Dolcetti R, De Rossi A. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016;7(12):e2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14(3):2077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kozar K, Sicinski P. Cell cycle progression without cyclin D-CDK4 and cyclin D-CDK6 complexes. Cell Cycle. 2005;4(3):388–91. [DOI] [PubMed] [Google Scholar]
- 45. Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118(4):493–504. [DOI] [PubMed] [Google Scholar]
- 46. Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif. 2003;36(3):165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 1994;54(12):3131–5. [PubMed] [Google Scholar]
- 48. Pucci B, Kasten M, Giordano A. Cell cycle and apoptosis. Neoplasia. 2000;2(4):291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haupt Y, Rowan S, Shaulian E, Vousden K, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev. 1995;9(17):2170–83. [DOI] [PubMed] [Google Scholar]
- 50. Fan G, Ma X, Kren BT, Steer CJ. The retinoblastoma gene product inhibits TGF-beta1 induced apoptosis in primary rat hepatocytes and human HuH-7 hepatoma cells. Oncogene. 1996;12(9):1909–19. [PubMed] [Google Scholar]
- 51. Alajez N, Shi W, Hui A, Yue S, Ng R, Lo K, Bastianutto C, O'Sullivan B, Gullane P, Liu F. Targeted depletion of BMI1 sensitizes tumor cells to P53-mediated apoptosis in response to radiation therapy. Cell Death Differ. 2009;16(11):1469–79. [DOI] [PubMed] [Google Scholar]
- 52. Jacobs JJ, Scheijen B, Voncken J-W, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13(20):2678–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gu X, Gong H, Shen L, Gu Q.. MicroRNA-129-5p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting DNMT3A. Am J Transl Res. 2018;10(9):2834–47. [PMC free article] [PubMed] [Google Scholar]
- 54. Pechalrieu D, Etievant C, Arimondo PB. DNA methyltransferase inhibitors in cancer: from pharmacology to translational studies. Biochem Pharmacol. 2017;129:1–13. [DOI] [PubMed] [Google Scholar]
- 55. Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280(2):168–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.