

Abstract
Protein-DNA interactions are essential to establish cell type-specific chromatin architecture and gene expression. We recently developed scDam&T-seq, a multi-omics method that can simultaneously quantify protein-DNA interactions and the transcriptome in single cells. The method effectively combines two existing methods: DamID and CEL-Seq2. DamID works through the tethering of a protein of interest (POI) to the Escherichia coli DNA adenine methyltransferase (Dam). Upon expression of this fusion protein, DNA in proximity of the POI is methylated by Dam and can be selectively digested and amplified. CEL-Seq2, on the other hand, makes use of poly-dT primers to reverse transcribe mRNA, followed by linear amplification through in vitro transcription (IVT). scDam&T-seq is the first technique capable of providing a combined readout of protein-DNA contact and transcription from single-cell samples. Once suitable cell lines have been established, the protocol can be completed in 5 days, with a throughput of hundreds to thousands of cells. The processing of raw sequencing data takes an additional 1–2 days. Our method can be used to understand the transcriptional changes a cell undergoes upon the DNA binding of a protein of interest. It can be performed in any laboratory with access to FACS, robotic and high-throughput sequencing facilities.
Introduction
A myriad of proteins cooperate to establish cell type-specific chromatin architecture and gene expression through their contact with DNA. Such proteins range from post-translationally modified histones to transcription factors, from nuclear lamina constituents to the transcriptional machinery. Methods to measure protein-DNA interactions (ChIP-seq1, DamID2) or their effect on chromatin organisation (DNase-seq3, Hi-C4) have provided valuable insight into the link between epigenetic regulation and transcriptional output. However, these methods originally required thousands to millions of cells and the resulting population-averaged data prohibited the study of diversity and heterogeneity within the sample. Recent technological advances have resulted in single-cell implementations of several methods to study genome architecture5–8, chromatin accessibility9–11, DNA modifications12–17 and protein-DNA interactions18–21. The data generated by these single-cell techniques have revealed that there is heterogeneity between the epigenetic states of individual cells. Moreover, single-cell multi-omics methods combining accessibility or DNA methylation readouts with a transcriptional readout have been able to make a direct connection between epigenetic and transcriptional heterogeneity22–24. However, until recently, a single-cell multi-omics method to study protein-DNA interactions in conjunction with transcription had been lacking.
Here we describe scDam&T-seq, a method we recently developed to measure protein-DNA interactions and transcripts from the same single cell25. scDam&T-seq essentially combines two single-cell methods: scDamID6 to measure protein-DNA interactions and CEL-Seq226, 27 to determine the transcriptional state (Fig. 1). The DamID technique relies on the tethering of a protein-of-interest (POI) to the Escherichia coli DNA adenine methyltransferase (Dam), an enzyme that exclusively methylates adenines in a GATC motif. Expression of the fusion-protein in cells results in methylation of genomic regions where the POI is present. Methylated DNA is then specifically digested and amplified. Through the incorporation of a barcode and a T7 promoter in both the CEL-Seq2 primer and the DamID adapter, DamID and CEL-Seq2 products can be simultaneously amplified and separation of material is not necessary. Once cell lines expressing the Dam-POI fusion have been established, the processing of single cells and library preparation can be completed in 5 days. Data processing takes 1–2 days. The protocol can be performed in any laboratory with access to FACS, robotic and high-throughput sequencing facilities.
Fig 1 |. Overview of the method.
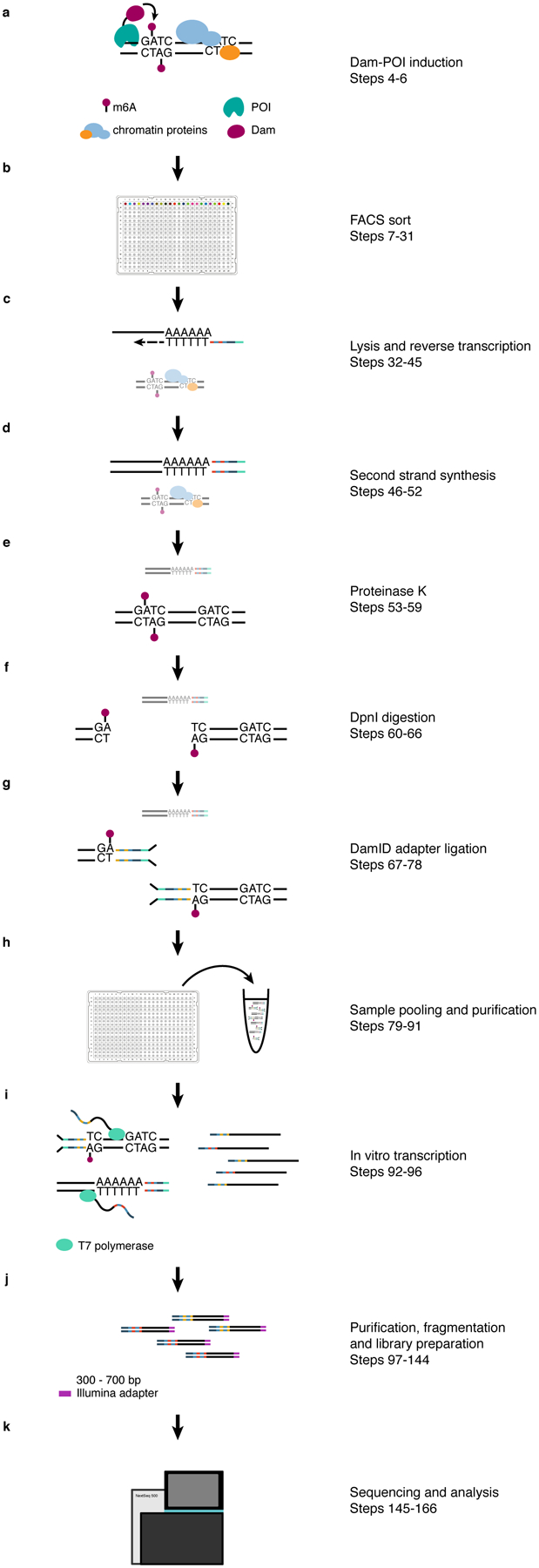
Panels a–k describe the main parts of the protocol (Overview of the Method). The indicated steps refer to the relevant sections of the experimental procedure. In c–g, both transcript and gDNA-derived molecules are shown, with the relevant molecule in each step shown in the foreground.
The dual readout of scDam&T-seq provides the possibility to link transcriptional and epigenetic state in a way that is impossible when these measurements are performed separately. One possible application of the scDam&T-seq data is to compare the transcriptional output of loci in their bound and unbound state. Another possibility is to use the transcriptional information to assign a cell type or state to each cell and subsequently study how the underlying epigenetics differ between these different populations. We have successfully applied both strategies in order to study how contact of chromatin with the nuclear lamina (NL) impacts transcription in mouse embryonic stem cells (mESCs) and how Ring1B is progressively enriched on the inactive × chromosome25.
Overview of the protocol
Prior to the implementation of the protocol, a cell line (or other biological system) capable of expressing the Dam-POI protein needs to be established (Experimental Design: Selecting stable clones). Importantly, a cell line expressing the untethered Dam protein needs to be established as well. The expression of the untethered Dam protein will result in the methylation of the accessible regions in the genome and can therefore be used as a control for the non-targeted GATC methylation by the Dam-POI in the experiment. The expression of the Dam-POI is induced before harvesting the cells to allow for the accumulation of GATC methylation. The majority of the Dam-POI constructs in our lab are controlled by the auxin system but any other induction system can be used28. With the auxin system the Dam-POI construct is continuously degraded in the presence of auxin in the medium. For the stabilization of the protein, auxin-containing medium needs to be removed from the cells and replaced with fresh medium without auxin. Cells express the Dam-POI a certain amount of time, before they are harvested and prepared for FACS sorting (Fig. 1a, b). This is done by live-staining the cells for DNA content quantification with Hoechst before adding propidium iodide to stain and later exclude dead cells. Cells are then sorted as single cells or small populations in 384-well plates containing CEL-Seq2 primers and mineral oil and can be either frozen or used immediately for scDam&T-seq. CEL-Seq2 primers contain a T7 promoter, a P5 Illumina adapter, a Unique Molecular Identifier (UMI), a sample barcode and a poly-dT tail (Fig. 2a, Reagents and Experimental Design: Design and concentration of DamID adapters and CEL-Seq2 primers). Once the cell is lysed, the poly-A tail of the mRNA molecule is annealed by the CEL-Seq2 primer and is reverse transcribed (Fig. 1c, 2a). This is followed by second strand synthesis (Fig. 1d), before proteinase K is added in order to remove all chromatin-associated proteins from the genomic DNA (Fig. 1e). In the next step, the restriction enzyme DpnI is added to specifically digest methylated GATC sites, generating blunt ends and leaving non-methylated GATC sites intact (Fig. 1f). The cell-specific DamID adapters are then added to the reaction and ligated to the digested genomic DNA (Fig. 1g). The DamID adapters contain a forked sequence which prevents adapter concatemer formation, a T7 promoter, a P5 Illumina adapter, a UMI and a sample barcode (Fig. 2b, Reagents). Once the ligation has taken place, genomic DNA of each cell will contain unique barcodes which do not overlap with the mRNA barcodes, and this allows for the samples to be pooled together and cleaned through bead purification (Fig. 1h). The pooled single-cell material is subsequently linearly amplified by the T7 polymerase in an in vitro transcription reaction, generating multiple copies of each molecule (Fig. 1i). The amplified RNA (aRNA) is then bead purified, fragmented by salt buffer at a high temperature and purified again before CEL-Seq2 library preparation with minor adjustments27(Fig. 1j). The concentration and the quality of the finished libraries is assessed before paired-end sequencing on a NextSeq 500 machine (Fig. 1k). The resulting sequencing reads are derived from the DamID and CEL-Seq2 products. The R1 reads of the DamID product contain a DamID barcode, a UMI and the genomic sequence, while the R2 reads solely contain the genomic sequence on the other side of the fragment. The R1 reads of the CEL-Seq2 product contain the sequence of the mRNA molecule, while the R2 reads contain the CEL-Seq2 barcode, a UMI, and part of the poly-A tail. It is thus necessary to sequence paired-end in order to get the barcode and UMI information of the transcript-derived reads. The data is analyzed following the pipeline provided in this protocol. For a step-by-step detailed overview of the molecule structure throughout the protocol please refer to the Supplementary Manual.
Fig 2 |. CEL-Seq2 primer and DamID adapter structure.
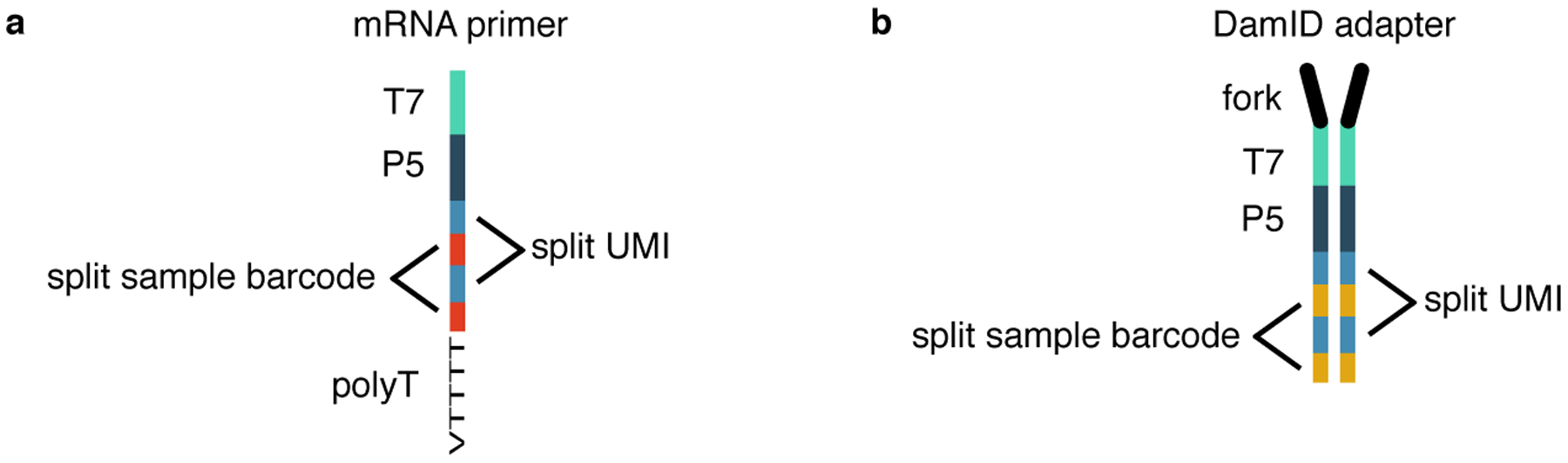
a) CEL-Seq2 primer. b) DamID adapter (Overview of the Method and Experimental Design: Design and concentration of DamID adapters and CEL-Seq2 primers).
Applications of the method
We developed scDam&T-seq as a method to simultaneously detect protein-DNA interactions and poly-adenylated transcripts. However, there are several ways in which these applications may be extended. In the original publication, we showed that untethered Dam itself may be used as a chromatin accessibility readout in addition to its role as a control25. Consequently, the untethered Dam experiment provides extra insight into the system under study and can be used to link accessibility and transcriptional output. In addition, we applied the scDam&T-seq protocol using the methylation-insensitive restriction enzyme AluI instead of DpnI to digest all the genomic AluI motif occurrences and effectively obtain a reduced-representation whole-genome sequencing25. From the resulting data, regions that are enriched or depleted in signal, indicating duplicated and deleted regions, respectively, can be identified across the genome. Such an extension may be of particular interest in systems where frequent large-scale duplications and deletions are known to occur. Moreover, the successful application of AluI opens the door to experimentation with other restriction enzymes, for example those sensitive to DNA modifications such as AbaSI for 5-hydroxymethylcytosine or LpnPI and MspJI for 5-methylcytocine15, 29, 30. Finally, in the original protocol, we employ barcoded poly-dT primers in order to selectively amplify polyadenylated transcripts. Conceivably, an unbiased sampling of the complete RNA pool could be obtained by substituting the poly-dT primers for random hexamer primers.
Next to these extensions of the protocol, we find that limiting the protocol to the DamID-specific steps results in improved results compared to the original scDamID protocol6, 31. This adaptation leaves out all mRNA-processing steps and requires only a few minor adaptations of the DamID steps (Supplementary Methods: scDamID2). In addition, we have extended the DamID-only protocol to population samples (Supplementary Methods: DamID2 in bulk). These adaptations are especially helpful when the experimental question requires no transcriptional information or when performing trial experiments, for example to select a clone with ideal Dam-POI expression levels. Excluding the transcription-specific steps greatly reduces sample processing time, reagent costs and sequencing cost. The reduction in sequencing costs is twofold, since there is less material to sequence and the library can be sequenced single-end.
Comparison with other methods
To our knowledge, scDam&T-seq is the first method capable of simultaneously assaying protein-DNA interactions and transcription. However, there are several technologies that probe transcription and/or epigenetic state in single cells. The most obvious comparison is to scDamID, the method from which scDam&T-seq was derived2, 31. In the original scDamID protocol, methylated DNA is enriched through DpnI digestion followed by the ligation of adapters and PCR amplification. Since a sample-specific barcode is introduced during the PCR via the primer, samples can only be pooled after amplification. In scDam&T-seq, on the other hand, the adapters contain both a sample barcode and a UMI. Therefore, the samples can be pooled prior to amplification, enabling the multiplexing of high numbers of cells, and amplification biases can be minimized by means of the UMIs. In addition, the exponential PCR amplification is replaced by an IVT reaction, which amplifies the material linearly and should result in fewer amplification biases32. It is worth noting that in both scDam&T-seq and scDamID the obtained depth and resolution is much lower than for classic DamID protocols performed on millions of cells. Whereas bulk DamID may give a resolution of individual GATC fragments (< ~1 kb), single-cell based methods typically reach a resolution of 50 – 100 kb.
At the moment, several other single-cell methods are available to probe protein-binding to the DNA18–21, 33, 34. However, these techniques suffer from low coverage due to precipitation steps18 or low sample throughput19. Recently, a ChIP-based method was published in which the nucleosomes of single cells are barcoded in droplets before being pooled together for immuno-precipitation33. Such innovations could potentially improve the efficiency of single-cell ChIP methods. Another promising class of techniques are those based on chromatin immunocleavage (ChIC) based methods35, lately adapted for sequencing as CUT&RUN36. In these methods, nuclei are isolated, permeabilised, and incubated with with antibodies against the POI. Subsequently protein A (pA) fused to micrococcal nuclease37 is added. The pA-MN fusion localizes to the bound antibody and, upon calcium addition, cleaves proximal DNA. The resulting DNA fragments are isolated and processed for sequencing. Using this approach, high quality data can be obtained from low-input (~1000 cells) samples and even single cells20, 21, 34. With some further development, ChIC-based methods could provide a powerful tool for studying protein-DNA interactions in single cells. However, due to the required isolation of nuclei, these methods currently cannot be combined with transcriptome measurements.
Advantages of the method
One of the main advantages of the scDam&T-seq protocol is that it has been designed to minimize the loss of material and technical biases. Due to the addition of a T7 promoter in the DamID adapters and CEL-Seq2 primers, it is possible to amplify DNA and RNA simultaneously and there is no need to perform a separation step in which material may be lost. In general, DamID-based methods preserve material well, since no pull-down is required. Meanwhile, the use of linear amplification through IVT and the presence of UMIs in the adapters minimizes the impact of amplification biases. The early ligation of the barcoded adapters allows many samples to be pooled together at an early stage of the protocol, which minimizes technical variation between samples and enables the processing of hundreds of samples simultaneously. As a result, it is possible to process thousands of single cells in as little as 5 days. Finally, the technique does not rely on the availability of high-quality antibodies, which often require extensive testing and optimization of buffers.
In addition to these technical features, scDam&T-seq has a number of inherent advantages. The fact that methylation is accumulated over time in vivo means that even transient interactions can be recorded, which could be missed with techniques capturing protein-DNA interactions as a snapshot during sample collection. Since the methylation mark is laid down prior to sample collection, this also means that biases in the DamID signal due to cell stress are limited. Moreover, the cumulative nature of the DamID mark provides the possibility to track the history of protein-DNA contacts during the course of one cell cycle.
Limitations of the method
In most instances, the biggest limitation of scDam&T-seq is the fact that the Dam-POI protein needs to be expressed in the system of interest. This requires the design of a suitable construct, the cloning of the construct into a vector, and integration into a cell line or other system of interest. Subsequently, different clones need to be screened to find one showing the best results, as the specificity of the methylation is dependent on the expression level of the Dam-POI. Consequently, scDam&T-seq is applied less readily in samples that are not easily cultured, such as in vivo settings and in clinically derived samples.
Another limitation of scDam&T-seq is its limited applicability to proteins that bind the DNA in very narrow domains, such as transcription factors (TFs). Although DamID has been applied successfully to TFs in low-input samples (1000 cells38), the resolution obtained for single-cell samples is typically too low to study their localization in a meaningful manner. This problem is exacerbated for proteins that bind accessible chromatin, since regions of accessibility tend to accumulate unspecific methylation. Although untethered Dam can be used to control for the accessibility signature, the contributions of true Dam-POI localization and accessibility will be difficult to disentangle at the low resolution obtained for single cells.
Finally, there is an inherent limitation to the resolution that can be obtained with DamID-based technologies, since signal can only be recorded at GATC motifs. On average, GATC motifs occur at 256 bp intervals, which represents the theoretical upper limit of the resolution. However, in practice, the sparsity of the single-cell data is the true limitation of the resolution in a single-cell sample, with a typical bin size of 50 – 100 kb.
Experimental design
Necessary controls
During expression of the Dam-POI construct, the fusion protein will sometimes methylate regions of the DNA without proper localization of the POI, resulting in background signal. Such background methylation preferentially occurs at accessible regions of the chromatin. For that reason, a control experiment should always be performed in which untethered Dam is expressed. The extent to which an accessibility signature is present in the data depends strongly on the nature of the POI. Proteins such as LMNB1 are localized to the inaccessible nuclear lamina, resulting in little to no accessibility signal. On the other hand, proteins that can freely diffuse throughout the nucleoplasm will have stronger accessibility signatures. The accessibility signature obtained from the unthethered Dam experiment can be used for direct normalization of the Dam-POI data or as a negative control. In which way the control data is used depends on the experiment and the specific research question. In general, we find that a normalization works well for population samples or averages of single-cell samples, while treating the untethered Dam as a negative control is more suitable to single-cell data.
In addition to an untethered Dam control, there are a number of technical controls that can be used to optimize the experiments. For instance, leaving a few empty wells in the plate is useful in assaying what the leakage of adapters between samples is. A few wild-type samples not expressing a Dam protein can be included in a library to determine how much of the DamID signal is the result of random genomic DNA (gDNA) breaks that ligate to DamID adapters. Finally, we recommend including up to 4 wells of small populations of 20 or 100 cells that can be used when performing the protocol on a new Dam-POI experiment, to optimize conditions if the single-cell data do not show anticipated results.
Construct design
The design of the construct depends on many factors that are specific to the POI and the biological system. Factors to consider include whether the Dam protein should be fused to the N- or C-terminus of the POI, what kind of induction system will be used and whether Dam will be inserted in a targeted manner into the endogenous POI locus or will be randomly integrated.
In our experience, some biological systems are more sensitive to POI expression levels and duration than others. The DpnI restriction enzyme does not recognize hemi-methylated GATCs and consequently will not digest DNA that has been replicated, because the newly synthesized DNA will only contain unmethylated GATC sites. Therefore, rapidly cycling cells are relatively insensitive to continuous Dam-POI expression, while the genome of senescent or slowly cycling cells may become entirely methylated if Dam-POI expression is not restricted by a proper induction method. In addition, the choice of generating a knock-in of Dam versus an exogenous Dam-POI integration may depend on the expression dynamics of the POI in the biological system.
We recommend users to consider these factors carefully during the design of their constructs and try multiple strategies if necessary. However, not all constructs or Dam-POI fusion proteins work as anticipated. In some cases, the tethering of Dam to the POI affects protein stability, function or localization. Since the generation of a stable expression system can be time-consuming, we suggest that the constructs be first tested using DamID on populations of cells. This can be achieved by transducing or transfecting cells with the construct and performing DamID2 in bulk on these heterogeneous samples (Supplementary Methods: DamID2 in bulk).
Selecting stable clones
Once the construct has been introduced into a cell line (or other system31) via knock-in or random integration, multiple clones should be screened to select the clone with optimal expression levels. Random integration results in much more diversity than knock-in strategies and may require more clones to be screened in order to find one that performs well. There are many ways in which the clones can be compared, but we recommend performing at least the following three steps.
First, the expression levels of Dam-POI can be tested by performing a methyl-PCR on the clones2. In our experience, clones showing a smear of PCR product on a 1 % (wt/vol) agarose gel at 14–24 cycles with 250 ng input material typically have suitable expression levels for single-cell experiments. A subset of clones with varying expression levels can then be selected for further testing. In addition to expression levels, this experiment is ideal to test the inducibility of the different clones. Samples can be collected for each clone prior to induction and at different times post induction to see whether clones are inducible and what their induction dynamics are.
Once several clones have been selected, an initial DamID2 experiment can be performed on bulk samples (Supplementary Methods: DamID2 in bulk). The main purpose of this experiment is to determine whether methylation enrichment is observed at expected regions or in domains of the expected size. Which analyses are most helpful in answering these questions depends entirely on the POI. In the case of very broad domains, such as observed with LMNB1, one suitable measure is the autocorrelation function (ACF), which measures the correlation of a signal with a shifted copy of itself. Since the expected domains are broad, the signal should show a higher correlation with itself over longer distances than in the case for untethered Dam. Another way to validate the signal is to evaluate its enrichment over genomic regions where the POI is known to bind. This could be for example on the promoters of active or inactive genes or over domains determined for the same mark by ChIP-seq, if data is available.
Although the analyses on bulk samples can give insight on whether or not a clone has successful Dam-POI binding, we caution against picking a clone solely on results obtained from population samples. The ideal Dam-POI expression levels for bulk samples tend to be too low for single-cell samples, where it is important that the majority of cells has sufficient signal. As a final step, we therefore suggest to do an experiment comparing single-cell samples of the different clones. In the case of a cell line, we find that 50–100 single-cell samples per clone is typically enough to perform the comparison. Since the transcriptional readout is not relevant for clone selection, the samples can be processed following the scDamID2 protocol (Supplementary Methods: scDamID2).
Induction of Dam-POI expression
The method of induction can differ between biological systems. We make use of the auxin-based degron system, which results in specific and fast degradation of the Dam-POI construct28. In the absence of auxin, degradation stops and the protein is stabilized. In cultured cells, this is achieved by auxin washout. In order to obtain a cell line expressing the Dam-POI in an auxin-inducible manner, first a vector containing the TIR1 sequence as well as a selection marker has to be introduced in the genome. This is achieved either by random integration or in a targeted locus-specific manner. Once a clonal cell line has been selected based on TIR1 protein levels, the Dam-POI sequence has to be introduced, C-terminally tagged with the Auxin-Inducible Degron box (AID). The ideal clonal cell line will contain both the TIR1 sequence and the AID-Dam-POI sequence. Finally, the optimal concentration of auxin needs to be determined, so that the Dam-POI construct is sufficiently degraded without the auxin being inhibitory for cell functions.
The timing of induction (auxin washout) of the Dam-POI construct depends on the activity of the promoter and cell cycle duration. First, time course series are necessary to determine the ideal induction time by checking expression levels of the construct by qPCR or bulk/single cell DamID2. Second, the optimal time window for construct expression needs to be determined. For fast cycling cells in which the m6A mark is lost upon passege through DNA replication, we recommend induction times that allow for the GATC methylation mark to be re-established after S-phase and sorting of the cells in G2/M. In mESCs, for instance, we recommend induction of Dam-POI constructs for 6 to 12 hours. For slow cycling or post-mitotic cells we recommend titration of the induction timings to avoid the accumulation of background methylation.
Sample collection
Depending on the induction time, the research question, and the nature of the POI studied, cells can be either collected in the same cell cycle phase or not. For the correct estimation of the cell cycle phase we live-stain cells with Hoechst dye, which binds to the DNA and allows quantification of the cells’ DNA content. Hoechst staining of the DNA has to be optimized per cell type. We recommend testing different concentrations and times of incubation. As an example, we stain 1 × 106 mESCs at a final concentration of 30 μg/ml with a 45-minute incubation at 37 °C. Finally, when sorting single cells, we recommend saving the index information of each sorted cell (if this option is available), in order to be able to link the DamID and transcriptome information of a cell with its cell cycle phase.
Sample pooling
Once the plate has been processed, samples have to be pooled for IVT amplification. For a successful IVT reaction we recommend pooling a minimum number of 48 single cells. The maximum number of single cells we have successfully pooled for IVT is 384. In order to avoid library preparation biases, pool as many samples without overlapping barcodes as possible in one library. In our experience, single-cell samples and up to 4 small population samples (n”20) can be pooled in the same library. This has the advantage of eliminating library preparation bias between single cells and populations. On the other hand, by doing so one loses the ability of choosing the amount of sequencing reads that will be assigned to either the populations or the single cells (sequencing weight). When the experiment contains multiple conditions, it is important to combine these conditions in the same library. In this way, batch effects can be properly assayed and corrected. Therefore, it is recommended to sort samples from different conditions in the same plate and pool samples together without overlapping barcodes, for both DamID and CEL-Seq2. If this is not possible, different conditions can be sorted in different plates and then pooled together, as long as the barcodes do not overlap.
Library preparation and sequencing
The aRNA product of each sample pool is used for the production of one Illumina library, by following the CEL-Seq2 library preparation protocol27. This way, each sample pool constitutes a library, barcoded with a unique Illumina index (P7). The indices will be used in downstream bioinformatic analysis, to assign reads to each library. We recommend submitting no less than 8 libraries with unique (non-overlapping) Illumina indices per sequencing run, to ensure run complexity and successful cluster formation.
Design and concentration of DamID adapters and CEL-Seq2 primers
The DamID adapters contain a 6 nucleotide (nt) fork, a T7 promoter, a P5 Illumina adapter, an 8 nt sample barcode and a 6 nt UMI. The barcode and the UMI sequences are split and alternate in the sequence (Fig. 2b, Reagents). The CEL-Seq2 primers contain a T7 promoter, a P5 Illumina adapter, an 8 nt sample barcode, a 6 nt UMI and a poly-dT tail to anneal to the poly-A tail of the transcripts. Again, the sample barcode and the UMI sequences are split and alternate in the sequence (Fig. 2a, Reagents).
The design of split barcode and UMI sequences was chosen primarily with the DamID adapters in mind. For the preparation of the double-stranded DamID adapters, the top and bottom oligo strands of each barcode-specific adapter are combined for annealing. During this procedure, the top and bottom strands with different UMI sequences might not anneal. This would result in the formation of “bubbles” of non-annealed UMI sequences, which could interfere with the adapter ligation to the gDNA, since the UMI sequence is close to the 3-prime end of the adapter. Therefore, the split barcode and UMI design minimizes the formation of such “bubbles”. The DamID and CEL-Seq2 barcode sequences were designed according to the following four criteria: i) GC-content is between 35 – 65 % in the barcode sequences; ii) there are no homopolymers of 3 or more nucleotides in the barcode sequences and no homopolymers of 2 or more nucleotides in barcode sequences bordering UMI sequences; iii) the Hamming distance to all other DamID (CEL-Seq2) barcodes is at least 3; iv) the Hamming distance to all CEL-Seq2 (DamID) barcodes is at least 2. The first two criteria ensure that no barcodes of low complexity are generated. The third criterion ensures that each DamID (CEL-Seq2) barcode can be distinguished from all other DamID (CEL-Seq2) barcodes with high confidence. Similarly, the fourth criterion ensures that there is no overlap between DamID and CEL-Seq2 barcodes and reads originating from the two different techniques can be distinguished. As a result, the DamID and the CEL-Seq2 sample barcodes are non-overlapping and can safely be combined in one experiment. Supplementary Table 1 and 2 contain 384 CEL-Seq2 primer sequences and 384 DamID adapter sequences, respectively, as they are currently used in our experiments. These primers and adapters can be used to process 384 samples simultaneously within one sequencing library. CEL-Seq2 primers and DamID adapters can be matched in any combination.
The concentration of the DamID adapters can influence the number of obtained DamID reads. Depending on the nature of the POI binding (narrow or broad domains) and the expected intensity of the DamID readout (Experimental Design: Selecting stable clones), the ideal adapter concentration can vary. In order to avoid excessive adapter in the samples, we recommend a range of 1.25 nM to 100 nM in the reaction. In our experience, increasing concentrations of the DamID adapters does not interfere with the CEL-Seq2 product while increasing DamID output. However, at an equal sequencing depth, an increase in DamID material may result in lower coverage of the CEL-Seq2 material.
Bioinformatic analysis
Raw sequencing data is demultiplexed on CEL-Seq2 and DamID barcodes. After demultiplexing, the reads are aligned and processed according to data type. In general, a high percentage of reads (~ 95 %) can be attributed to a unique CEL-Seq2 or DamID barcode, with the majority of reads being derived from the DamID product (~ 90 %) and a smaller fraction from CEL-Seq2 product (~ 5 %). The final number of unique DamID and CEL-Seq2 reads depends on the complexity of the libraries. For a high-quality library containing 96 single-cell samples, we expect 20 – 40 % of the reads to be unique. The expected output of a scDam&T-seq experiment is discussed in greater detail in the Anticipated Results section at the end of this paper. We have established a pipeline for the processing of the raw sequencing data (Fig. 3). The scripts necessary for the analyses are available on GitHub (https://github.com/KindLab/scDamAndTools) and the main functionalities are described in Table 1.
Fig 3 |. Bioinformatics workflow.
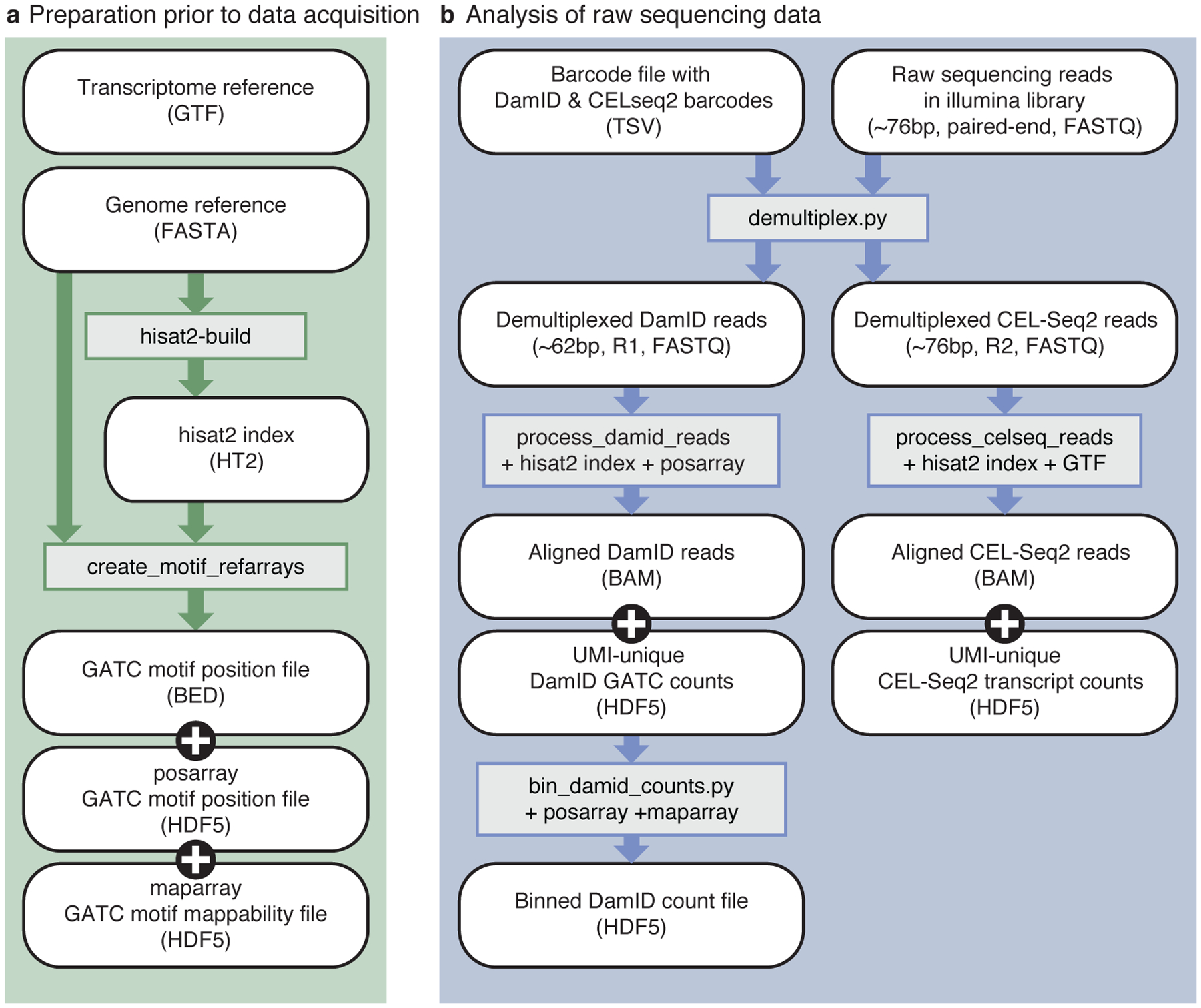
a) Preparation of reference files, which only needs to be performed once per reference genome. The genome reference (FASTA) file is used as input to generate the HISAT2 index, as well as the motif arrays. b) Processing of raw sequencing data to tables of unique DamID and CEL-Seq2 counts. White, rounded boxes show (intermediate) files; grey, rectangular boxes show programs and necessary reference files. Arrows indicate which files are used as input for subsequent programs.
Table 1:
Description of important functions in the scDamAndT package
| Name | Important parameters | Output |
|---|---|---|
| bin_damid_counts.py |
|
An HDF5 file containing a dataset for each chromosome. Each dataset is a vector with the observed UMI-unique GATC counts per genomic bin. The bins all have an equal size. The first bin starts at chromosomal position 0. |
| create_motif_arrays |
|
A BED file containing the strand-specific positions of the GATC motif throughout the genome; a HDF5 file containing the positions of all GATC motifs in the genome (position array); a HDF5 file containing the strand-specific mappability status of all motif occurrences in the genome (mappability array). |
| demultiplex.py |
[input] FASTQ files of the R1 and R2 reads of the library. |
A FASTQ file for each DamID and CEL-Seq2 barcode present in the library with reads that had the corresponding barcode. Barcode sequences are removed during demultiplexing. |
| process_celseq_reads |
|
A BAM file containing all alignments to the reference genome; a HDF5 file containing the number of observed UMI-unique counts per gene, ordered by ENSEMBL ID. |
| process_damid_reads |
|
A BAM file containing all alignments to the reference genome; a HDF5 file containing the number of observed UMI-unique DamID for all GATC occurrences in the genome. |
Expertise needed to implement the protocol
This protocol requires a FACS sorting facility for the single-cell sorting in 384-well plates. Furthermore, this protocol requires a robot facility or knowledge of robotic operation. In case users do not have access to the robotic systems Mosquito HTS and Nanodrop II used in this protocol (Equipment), other robotic machines can be used. These should be able to dispense master mix volumes ranging between 100 and 1920 nl. To avoid contaminations from previous dispensions, the tubing systems and needles should be able to be flushed or changed. The option for transferring volumes from a 96 or 384-well plate to a 384-well plate should be available as well, for CEL-Seq2 primer and DamID adapter dispension. If the user does not have access to a robotic facility, we recommend upscaling the reactions to the point that the volumes can be handled by hand pipetting. However, we lack experience in this and therefore cannot guarantee fail-proof execution of the protocol. Finally, for the successful sequencing of the libraries, a dedicated sequencing facility is needed.
In addition to these experimental facilities, a high-performance computing system with a Linux operating system is necessary for the analysis of the data. The bioinformatic procedures described in step 146–159 give an example of the processing of a single sample. A good understanding of command line usage is necessary in order to perform the processing of multiple samples in parallel. Finally, knowledge of a programming language such as Python or R is necessary for further downstream analyses and data interpretation.
Materials
Biological materials
Cell lines. For Figures 3 and 5 of this protocol we used F1 mouse embryonic stem cells with a hybrid genetic background of 129/Sv and Cast/EiJ RRID CVCL_XY6339. We have also applied the protocol on the haploid human myeloid leukemia cell line KBM76. Cells were negative for mycoplasma contamination and were not systematically authenticated. ! CAUTION Cell lines should be regularly checked for authenticity and mycoplasma contamination.
Fig 5 |. Examples of aRNA bioanalyzer plots.
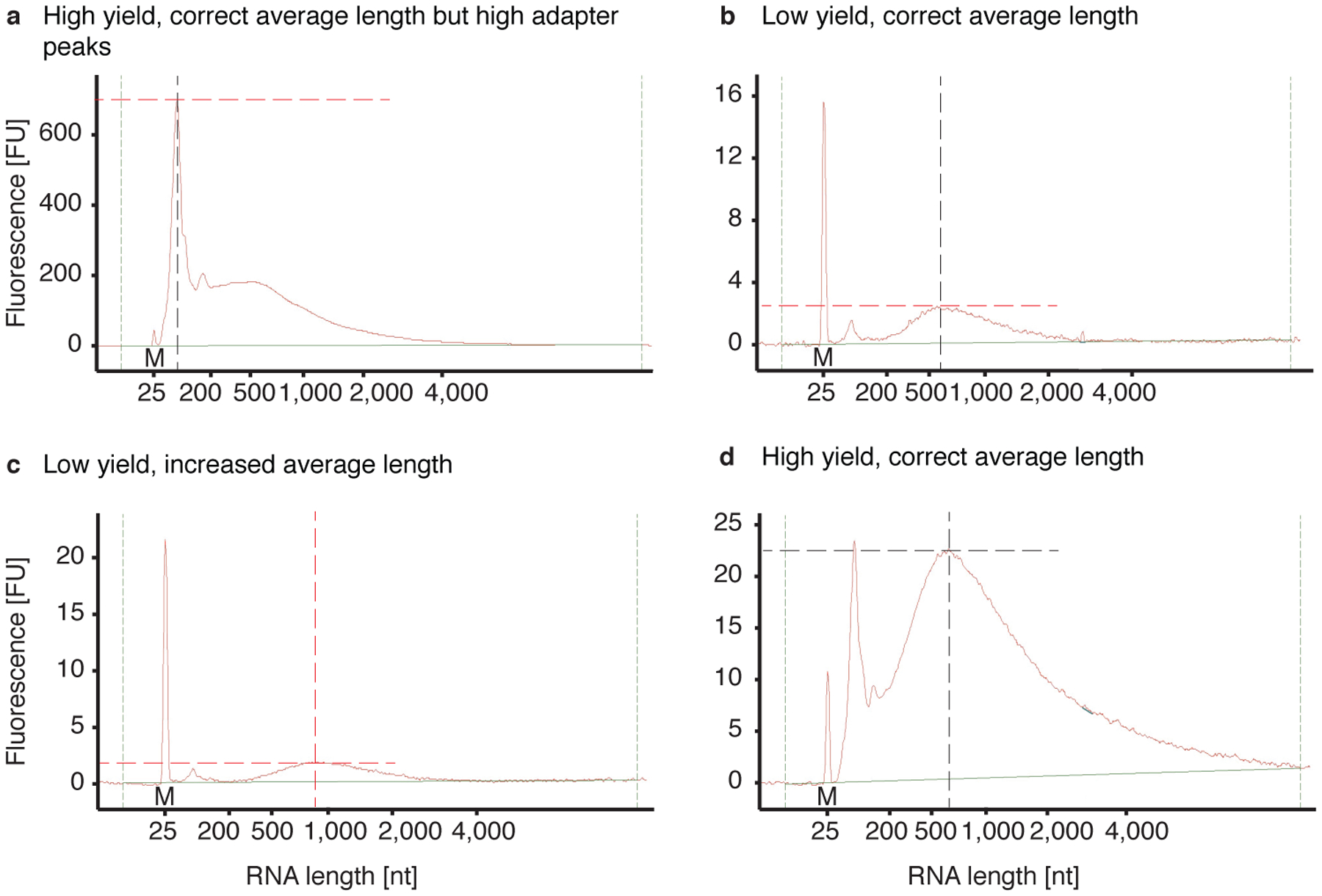
Bioanalyzer results after IVT, bead purification, aRNA fragmentation and another bead purification: a) The aRNA has the correct size distribution of 300 – 700 nt but the adapter peak is extremely high (>600 FU), which can inhibit the library preparation. Extra rounds of aRNA bead purifications are recommended. b) The aRNA has the correct size distribution but the yield is low (<4 FU), possibly due to loss during bead purification. c) The aRNA has an increased size distribution of 500 – 2,000 nt indicating that fragmentation was not complete. In addition, the yield is low (<2.5 FU) indicating loss during bead purification. d) The aRNA has the correct size distribution and good yield (>20 FU) and can be used for library preparation.
Reagents
Wizard genomic DNA purification kit (Promega cat. no. A1120)
DNAZap (Invitrogen cat. no. AM9890) ! CAUTION Chronically toxic for the aquatic systems.
RNase ZAP (Invitrogen cat. no. AM9780) ! CAUTION Aerosols or vapor can cause irritation to lungs and mucous membranes. Work in a fume hood.
Micro-90 concentrated cleaning solution (Sigma-Aldrich cat. no. Z281506)
Mineral oil (Sigma cat. no. M8410)
Glasgow’s MEM (Gibco cat. no. 21710025)
MEM non-essential amino acids solution (100 X; Gibco cat. no. 11140035)
Pen/Strep (10,000 U/ml; Gibco cat. no. 15140122)
Sodium Pyruvate (100 mM; Gibco cat. no. 11360039)
GlutaMAX supplement (100 X; Gibco cat. no. 35050038)
Fetal Bovine Serum (FBS; Sigma cat. no. F7524)
ESGRO mLIF Medium Supplement (10,000,000 U/ml; Milipore cat. no. ESG1107)
B-mercaptoethanol (1M; Sigma cat. no. M3148) ! CAUTION Acutely toxic for humans and aquatic systems. Use hand, eye, nose and mouth protection and do not pour in drain. Indole-3-acetic acid sodium salt (IAA, auxin; Sigma-Aldrich cat. no. I5148)
PBS pH 7.4 (Ambion cat. no. 10010001)
Trypan Blue solution (Sigma-Aldrich cat. no. T8154) ! CAUTION Can cause cancer. Avoid contact with eyes and skin and do not pour in drain.
Hoechst 34580 (Sigma cat. no. 63493)
Propidium iodide (Sigma cat. no. P4864)
Nuclease-free water (Invitrogen cat. no. 1097035)
Magnesium acetate solution (1 M; Sigma-Aldrich cat. no. 63052)
Potassium acetate solution (5 M; Sigma-Aldric cat. no. 95843)
Tween 20 (Sigma-Aldrich cat. no. P1379)
ERCC RNA Spike-In mix 1 (Ambion cat. no. 4456740)
Igepal (Sigma cat. no. I8896) ! CAUTION Skin, mouth and eye irritant. Use with care and wear gloves and mouth mask in case of insufficient ventilation. Chronically toxic for the aquatic systems. Do not pour in drain.
Recombinant ribonuclease inhibitor (Clontech cat. no. 2313A)
dNTPs set (10mM each; Invitrogen cat. no. 10297018)
5x First strand buffer provided with Superscript II package (Thermo Fisher Scientific cat. no. 18064014)
DTT 0.1 M provided with Superscript II package (Thermo Fisher Scientific cat. no. 18064014) ! CAUTION Work with DTT in a ventilated hood. Wear gloves and a coat, and do not pour it into the drain.
RNase OUT (Invitrogen cat. no. 10777019)
SuperScript II (Thermo Fisher Scientific cat. no. 18064014)
5x Second strand buffer (Thermo Fisher Scientific cat. no. 10812014)
E. coli DNA ligase (Invitrogen cat. no. 18052019)
DNA polymerase I (Thermo Fisher Scientific cat. no. 18010025)
RibonucleaseH (Thermo Fisher Scientific cat. no. 18021071)
10x CutSmart buffer (NEB cat. no. B7204S)
Proteinase K (Roche cat. no. 3115879001)
DpnI (NEB cat. no. R0176L)
Tris pH 7.5 (1 M; Roche cat. no. 10708976001)
NaCl (5 M; Sigma-Aldrich cat. no. S5150)
EDTA pH 8 (0.5 M; Invitrogen cat. no. 15575020)
10x T4 ligation buffer provided with T4 ligase (Roche cat. no. 1102430292001)
T4 ligase 5 U/μl (Roche cat. no. 10799009001)
AMPure XP beads (Beckman cat. no. A63881)
PEG8000 (Merck cat. no 1546605)
NaCl (2.5 M; Sigma-Aldrich cat. no. S7653)
Tris-HCl (1M; Roche cat. no. 10812846001)
Ethanol absolute (Scharlau cat. no. ET00052500) ! CAUTION Highly flammable; keep away from heat, hot surfaces, sparks and open flames. Avoid contact with eyes and skin, and wear protective gloves.
MEGAscript T7 Transcription Kit (Thermo Fisher Scientific cat. no. 1334)
Trizma acetate powder (Sigma-Aldrich cat. no. 93337)
Phusion High-Fidelity PCR Master Mix with HF Buffer (NEB cat. no. M0531S)
Agilent RNA 6000 Pico Kit (Agilent cat. no. 50671513)
Agilent High Sensitivity DNA Kit (Agilent cat. no. 50674627)
Qubit 1x dsDNA HS Assay Kit (Invitrogen cat. no. Q33230)
PhiX control V3 (Illumina cat. no. FC-110–3001)
NextSeq 500/550 High Output Kit v2.5 (150 Cycles) (Illumina cat. no. 20024907)
CEL-Seq2 primer
CRITICAL Dissolve primers or pre-order them diluted in nuclease-free water to 500 μM and store at −80 °C indefinitely. Order primers as standard desalted.
CRITICAL Dilute primers in nuclease-free water to a working concentration of 500 nM in a 384-well plate and store at −20 °C indefinitely. Use this as a source plate for the preparation of primer plates (Box 1).
Box 1 | Mosquito robot handling.
Procedure
Primer plate preparation - Timing 20 min for one plate (30 min for 4 max plates)
Turn on the computer workstation and the robot and initialize.
-
Use the “Humidify” function to humidify plate deck chamber to 80 % (wt/vol).
Remove seal and insert the source CEL-Seq2 primer plate (500 nM) in position 1 with corner A1 facing the upper left corner of the magnetic holder.
Remove seals and insert the destination plate(s) containing 5 μl mineral oil in position(s) 2, 3, 4 and 5 with corner A1 facing the upper left corner of the magnetic holder.
Copy the source plate by pipetting 100 nl of CEL-Seq2 primer into the destination plates. Change needles after copying a column to avoid contamination across wells.
DamID adapter dispension - Timing 20 min for one plate (30 min for 4 max plates)
Turn on the computer workstation and the robot and initialize.
Use the “Humidify” function to humidify plate deck chamber to 80 % (wt/vol).
Remove seal and insert the source DamID adapter plate in position 1 with corner A1 facing the upper left corner of the magnetic holder.
Insert the destination plate(s) in position(s) 2, 3, 4 and 5 with corner A1 facing the upper left corner of the magnetic holder. Keep on ice whenever not in the robot.
Copy the source plate by pipetting 50 nl of DamID adapter into the destination plates. Change needles after copying a column and between plates to avoid contamination across samples.
Example of one CEL-Seq2 primer (5’ → 3’): GCCGGTAATACGACTCACTATAGGGAGTTCTACAGTCCGACGATCNNNGATGNNNTCATTT TTTTTTTTTTTTTTTTTTTTTTV
CRITICAL This primer anneals to the poly-A tail of mRNA transcripts with its 3’ poly-dT tail. The 3’ end contains a “V” base, which is G, C or A. This degenerate base prevents the polymerase from slipping over the poly-A sequence and locks the annealing of the primer immediately upstream of the poly-A tail. The primer contains an 8 nt unique barcode (here: GATGTCAT) that labels a single cell. It also contains a 6 nt unique molecular identifier (UMI) that labels a transcript molecule uniquely (here: NNNNNN, where “N” is G, C, A or T). The barcode is split in 2 × 4 nt and alternates in the primer sequence with the UMI which is split in 2 × 3 nt as follows: NNN-GATG-NNN-TCAT (Experimental Design: Design and concentration of DamID adapters and CEL-Seq2 primers). CEL-Seq2 primer sequences 1–384 can be found in Supplementary Table 1.
DamID adapter
CRITICAL Dissolve the adapters or pre-order them diluted in nuclease-free water to 500 μM and store at −80 °C indefinitely. Order adapters as standard desalted. CRITICAL Dilute DamID adapters in annealing buffer to the desired concentration in separate 384-well plates for top and bottom oligos and store at −20 °C indefinitely. Use these plates for the adapter annealing.
CRITICAL Anneal the DamID adapters by combining the complementary top and bottom sequences in one 384-well plate by hand-pipetting or with a robotic system at an equal molar ratio and resuspend. Immerse the plate in a container with water heated up to 100 °C, in a way that the wells are in contact with the water, and the seal stays dry. Leave in container till room temperature (20 – 22 °C) is reached. Vortex plate for 5 sec to resuspend adapters and pulse-spin at room temperature for 10 sec. Store adapters at −20 °C indefinitely. Use this plate for adapter dispensing.
-
Example of one DamID adapter (5’ → 3’): Top oligo GGTGATCCGGTAATACGACTCACTATAGGGGTTCAGAGTTCTACAGTCCGACGATCNNNTGCA NNNTATGGA
Bottom oligo /5Phos/TCCATANNNTGCANNNGATCGTCGGACTGTAGAACTCTGAACCCCTATAGTGAGTCGTA TTACCGGGAGCTT
CRITICAL The 5’ end of the DamID adapter contains a 6 nt non-complementary sequence forming a fork to prevent adapter concatemer formation. The DamID adapter contains an 8 nt unique barcode (here: TGCATATG) that labels a single cell. It contains a 6 nt unique molecular identifier (UMI) that labels a cut GATC site uniquely (here: NNNNNN, where “N” is G, C, A or T). The barcode is split in 2 × 4 nt and alternates in the primer sequence with the UMI which is split in 2 × 3 nt as follows: NNN-GATG-NNN-TCAT (Experimental Design: Design and concentration of DamID adapters and CEL-Seq2 primers). DamID adapter sequences 1–384 can be found in Supplementary Table 2.
Library primers
randomhexRT primer: GCCTTGGCACCCGAGAATTCCANNNNNN, where “N” is G, C, A or T. CRITICAL The N bases should be ordered as hand-mixed whenever this option is available.
RNA PCR Index Primers should follow the Illumina TruSeq Small RNA library prep guidelines (https://www.illumina.com). Example of a RPi index primer (5’ → 3’):
CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATT CC*A
CRITICAL The “*” indicates a phosphorothioate bond which protects the DNA from endo- and exonuclease activity therefore increasing the stability of the oligo.
RNA PCR Primer 1 (RP1) primer, according to the Illumina Truseq Small RNA library prep guidelines (https://www.illumina.com) (5’ → 3’): AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCG*A
CRITICAL The “*” indicates a phosphorothioate bond which protects the DNA from endo- and exonuclease activity therefore increasing the stability of the oligo.
Equipment
Hardshell 384-well PCR plates (Bio-Rad cat. no. HSP3805)
Silverseal sealer, aluminium (Greiner Bio cat. no. 676090)
Cell culture incubator, set at 37 °C and 5 % CO2 (Panasonic cat. no. MCO-170AIC)
Mosquito HTS robot (TTP Labtech)
Vortex (we use the VWR Analog Vortex Mixer VM 3000)
PCR workstation (WVR cat. no. 7322542)
Tabletop centrifuge (we use the Eppendorf cat. no. 5810R)
Burker-Turk hemocytometer (LO-Laboroptik)
Microscope (we use the Nikon Eclipse TS100)
Cell strainer caps (Corning cat. no. 352235)
Falcon Round-bottom polypropylene tubes (Corning cat. no. 352063)
FACS sorter (we sort on the BD Biosciences BD FACSJazz)
Low-retention filter tips (we use the Greiner Bio Sapphire low retention pipette tips)
Nanodrop II robot (BioNex)
Microcentrifuge MiniStar blueline (Vwr cat. no. 5212320)
384-well plate-compatible thermocycler (we use the Eppendorf Mastercycler Pro Thermal Cycler 384 cat. no. 950030030)
Thermocycler with a 96-well holder (we use the Bio-Rad T100 Thermal Cycler cat. no. 1861096)
Magnetic rack (we use DynaMag-2 from Life Technologies cat. no. 12321D)
Heat block (we use the Eppendorf Thermomixer F1.5 cat. no. 5384000012)
Nanodrop 2000 Spectrophotometer (Thermo Scientific cat. no. ND2000)
2100 Bioanalyzer instrument (Agilent cat. no. G2939BA)
Qubit 3.0 Fluorometer (Life Technologies cat. no. Q33216)
Software
Unix/Linux operating system (used version: Ubuntu 16.04.6 LTS)
Bash Shell (used version: bash == 4.2.46(2)-release)
HISAT2 (https://ccb.jhu.edu/software/hisat2/index.shtml, used version: v2.1.0)40
Python3 (https://www.python.org, used version: v3.6.3)
Samtools (http://www.htslib.org/, used version: v1.6)41
scDam&T-seq scripts (https://github.com/KindLab/scDamAndTools), functions are explained in Table 1.
Reagent setup
Auxin42 solution (250 mM)
Weigh 492.93 mg of IAA and dissolve in 10 ml sterile water. Filter-sterilize and aliquot. Keep at −20 °C. Protect from light. When moved to 4 °C it can stored up to a week.
mESC complete culture media without β-mercaptoethanol and ESGROmLIF
In 430 ml of Glasgow’s MEM, add 50 ml FBS, 5 ml Pen/Strep, 5 ml GlutaMAX 100 X, 5 ml non-essential amino acids 100 X, 5 ml sodium pyruvate 100 mM. The final concentrations in the solution are FBS 10 % (vol/vol), Pen/Strep 100 U/ml, GlutaMAX 1 X, non-essential amino acids 1 X, sodium pyruvate 1mM. Store at 4 °C up to 1 month.
mESC complete culture media with β-mercaptoethanol and ESGROmLIF
In 50 ml of mESC complete culture media without β-mercaptoethanol and ESGROmLIF add 5 μl β-mercaptoethanol 1M and 5 μl ESGROmLIF 10,000,000 U/ml. The final concentrations in the solution are β-mercaptoethanol 0.1 mM and ESGRO mLIF 1,000 U/ml. Store at 4 °C up to 1 week.
Hoechst (1 mg/ml)
Add 5 ml of sterile MiliQ water to 5 mg of Hoechst 34580 to make a dilution of 1 mg/ml. Store at −20 °C for up to 6 months. Protect from light.
Propidium iodide (1 mg/ml)
Dissolve 1 mg of propidium iodide in 1 ml of sterile water and filter-sterilize. Store at 4 °C up to 1 year.
2 % (vol/vol) cleaning solution for Nanodrop II robot
To 49 ml nuclease-free water add 1 ml of Micro-90 concentrated cleaning solution. Store at room temperature indefinitely.
ERCC RNA Spike-In (1:50,000)
Add 9990 μl of nuclease-free water to 10 μl of ERCC RNA Spike-In mix 1 to make a dilution 1:1,000 and make aliquots of 20 μl. To make the 1:50,000 working stock add 98 μl of nuclease-free water to 2 μl of 1:1,000 diluted ERCC RNA Spike-Ins. Store at −20 °C up to the date of expiry indicated and do not freeze-thaw the ERCC RNA Spike-Ins more than 8 cycles.
Proteinase K solution 20 mg/ml
Dissolve 100 mg of Proteinase K in 5 ml nuclease-free water. Store at −20 °C up to the date of expiry indicated by the manufacturer.
Tris pH 7.5 (1 M)
Dissolve 6.05 g of Tris base in 30 ml of nuclease-free water. Adjust the pH to 7.5 with HCl and fill up to 50 ml with nuclease-free water and filter-sterilize. Keep at room temperature.
Annealing buffer 5 x
Add 89 ml nuclease-free water to a sterile glass bottle and add 5 ml of Tris 1 M pH 7.5, 5ml of NaCl 5 M and 1 ml EDTA 0.5 M. Final concentrations in the solution are Tris 10 mM, NaCl 50 mM and EDTA 1 mM. Store at room temperature. Prepare new buffer when precipitates start to form.
Igepal 1 % (vol/vol)
Add 49.5 ml nuclease-free water to a 50 ml tube. With a cut tip, take 0.5 ml Igepal and slowly add it to the water. Resuspend till tip is empty. Close tube, invert a few times and leave on rotor until homogenous. Store at room temperature for up to 1 year.
dNTP mix (10 mM)
Add 60 μl of nuclease-free water to a tube. Add 10 μl of dCTP 10 mM, 10 μl of dGTP 10 mM, 10 μl of dATP 10 mM and 10 μl of dTTP 10 mM. Make aliquots of 20 μl and store at −20 °C up to a year. Avoid multiple freeze-thaw cycles.
Bead buffer
Dissolve 20 g of PEG 8000 with 48.75 ml nuclease-free water. Add 1 ml Tris-HCl 1 M, 0.2 ml EDTA 0.5 M, 50 ml NaCl 5 M and 50 μl Tween 20 pH 8.0. Final concentrations in the solution are PEG 8000 20 % (wt/vol), Tris-HCL 10 mM, EDTA 1 mM, NaCl 2.5 M and Tween 20 0.05 % (vol/vol). Store at room temperature for up to 1 year.
Diluted AMPure XP beads
To make a 1:8 bead dilution, add 700 μl of bead buffer to a tube and add 100 μl of AMPure XP beads. Resuspend and vortex till homogenous. Store at 4 °C until indicated expiration date of AMPure XP beads.
Ethanol (80 % vol/vol)
CRITICAL Measure volumes by pipette and not by “adding up” EtOH to 10 ml.
Add 2 ml of nuclease-free water to 15 ml tube and add 8 ml of ethanol 100 % (vol/vol, absolute). Store at room temperature up to one day. Make fresh for each bead purification.
Tris acetate pH 8.1 (1 M)
Weigh 3.623 g of Trizma acetate powder and dissolve in 10 ml of nuclease-free water. Adjust the pH to 8.1 with a base such as NaOH and fill up to 20 ml with nuclease-free water and filter-sterilize. Keep at room temperature up to 1 year.
Fragmentation buffer
Add 5 ml nuclease-free water in a tube and add 2 ml Potassium acetate 5 M, 3 ml Magnesium acetate 1 M and 10 ml Tris-acetate 1 M. Final concentrations in the solution are Potassium acetate 500 mM, Magnesium acetate 150 mM and Tris-acetate 200 mM. Keep at room temperature up to 1 year.
Stop buffer
Use EDTA pH 8 0.5 M. Keep at room temperature up to 1 year.
Lysis buffer 1 × pH 6–6.5 for scDamID2
In a clean bottle add 96.66 ml nuclease-free water, 1 ml Tris acetate 1 M pH 8.1, 1 ml Magnesium acetate 1 M, 1 ml Potassium acetate 5 M, 0.67 ml Igepal and 0.67 ml Tween 20. Final concentrations in the solution are Tris acetate 10 mM, Magnesium acetate 10 mM, Potassium acetate 50 mM, Igepal 0.67 % (vol/vol) and Tween 20 0.67 % (vol/vol). Keep at room temperature up to 1 year.
Procedure
(Day 1) Primer plate preparation - Timing 30 min for one plate
CRITICAL It is crucial that the working area is sufficiently clean when working with single-cell material. DNAZap and RNAseZAP treatment is required in steps 1–3 and 21–78. RNAseZAP treatment is sufficient for steps 79–128. Ethanol 80 % (vol/vol) treatment is sufficient for steps 129–145.
CRITICAL We recommend preparing primer plates, adapter plates and master mixes before single-cell material amplification by IVT in a PCR workstation, in order to avoid contamination and degradation of the single-cell material.
-
1
Pipet 5 μl of mineral oil in each well of a 384-well plate and seal the plate with an aluminium seal.
-
2
Thaw the CEL-Seq2 primer source plate at 4 °C and keep on ice.
CRITICAL STEP Verify seal is covering all wells of the source plate, vortex plate for 5 sec to resuspend primers and pulse-spin at 4 ° C for 10 sec.
-
3
Copy CEL-Seq2 primers from source plate to the mineral oil plate (Box 1). Seal the plate.
PAUSE POINT The primer plates can be kept at −20 °C indefinitely.
Dam-POI induction - Timing 45 min performed in a cell culture hood
CRITICAL The timing of induction can differ depending on the Dam-POI construct, cell line and other parameters (Experimental design). For the data shown in Fig. 4, mESCs expressing Dam-LmnB1 were cultured for 2 days on feeder cells till 80 % colony confluency and the Dam-LmnB1 construct was expressed for 6 hr before collection.
Fig 4 |. Technical statistics of a scDam&T-seq run.
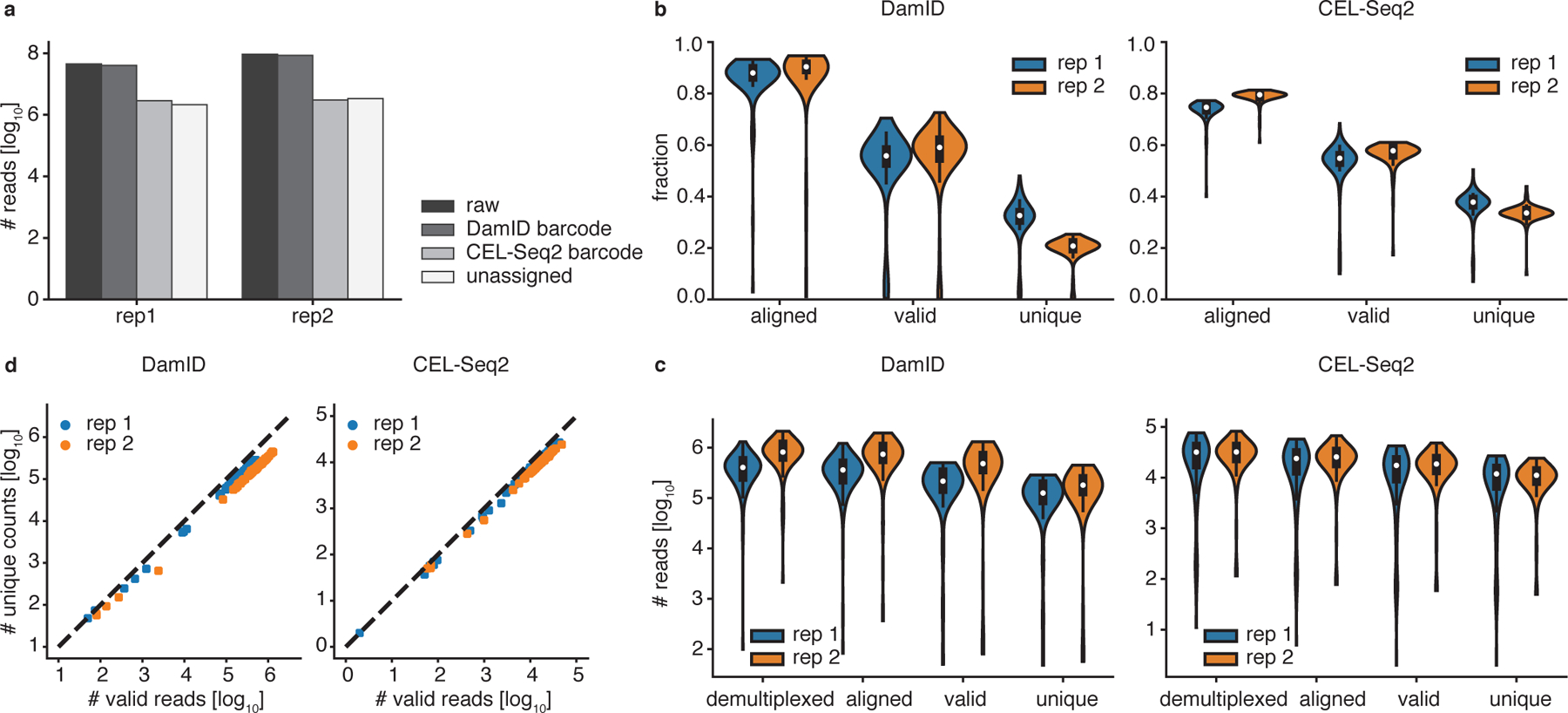
a) Barplot showing the number of raw sequencing reads and the number of DamID, CEL-Seq2 and unassigned reads after demultiplexing. b, c) Overview of progressive read loss during analysis of DamID (left) and CEL-Seq2 (right) data as a fraction of the number of demultiplexed reads per sample (b) and in absolute numbers (c). The black boxplots show the median (white dot), interquartile range (black box), and range of the data within 1.5 IQR of the median (black lines). d) Complexity plot of the DamID (left) and CEL-Seq2 (right) data. Each replicate in plot b–d shows data of one library of 96 single-cell samples, i.e. 96 data points. F1 mESCs were with a hybrid genetic background of 129/Sv and Cast/EiJ were used, RRID CVCL_XY6339. Cells were negative for mycoplasma contamination.
-
4
Pre-warm cell culture medium and PBS at 37 °C for 30 min.
-
5
Remove indole-3-acetic acid 42-containing medium from cells and wash 3 × with warm PBS.
CRITICAL STEP Cells that are easily detached might need to be washed with medium instead of PBS.
-
6
Add cell culture medium without IAA and place cells in 37 °C incubator until harvest.
(Day 2) Prepare cells for FACS sorting by Hoechst staining - Timing 1 hr 30 min
-
7
Harvest cells × hr after induction and prepare a single-cell suspension. For the mESCs presented in Fig. 4 induction was 6 hr prior to harvest.
-
8
Clean hemocytometer with a tissue sprayed with 80 % (vol/vol) ethanol and secure the coverslip.
-
9
Take 100 μl of cell suspension and put in a tube.
-
10
Add 400 μl of Trypan-blue to the tube and mix by pipetting up and down 3–4 times.
-
11
Pipet carefully 100 μl of the Trypan blue-cell suspension mix into the cavity between the hemocytometer and the coverslip. Capillary forces will draw the liquid inside.
-
12
Place the hemocytometer under the microscope set at a 10 × objective and focus the microscope on the grid lines of the hemocytometer.
-
13
Count the live cells (unstained) at the upper left square containing 16 smaller squares. Once done, move to the other 16-corner set square until all the 4 squares are counted.
-
14
To calculate the number of viable cells/ml of cell suspension, divide the number on the tally counter by 4 to obtain the average number per corner square. Multiply the average by 10,000 and then by 5 to correct for the 1:5 dilution of the cell suspension with the Trypan Blue.
-
15
Dilute the cell suspension to 1 × 106 cells/ml. Pipet a minimum of 600 μl of cell suspension in a 15 ml falcon tube.
-
16
Thaw the Hoechst solution on ice and avoid light exposure.
-
17
Add Hoechst to the cell suspension to stain the DNA. For the mESCs presented in Fig. 4 induction cells were stained with 30 μg/ml Hoechst.
CRITICAL STEP The final concentration of the Hoechst needs to be optimized for the cell type used (Experimental design: Sample collection).
-
18
Incubate the cells at 37 °C for 45 min in a cell culture incubator, avoiding light exposure.
-
19
Pass the cells through a falcon cell-strainer cap and in a polypropylene round-bottom tube to exclude cell clumps by gently pipetting the cells on the cap and leting them pass through the filter without force from the pipet tip.
-
20
Keep cells on ice till FACS sort. Do not keep cells on ice for longer than 3 hr.
FACS sorting - Timing 1 hr for 3 plates
-
21
Thaw the primer plate from Step 3 on ice.
-
22
Centrifuge plate at 2,000 g for 1 min at 4 °C for primer droplets to fall at the bottom of each well.
-
23
Transport plate and cells on ice to the FACS sorter.
-
24
Add propidium iodide to a final concentration of 1 μg/ml to the cell suspension for live/dead cell gating.
-
25
Remove the plate seal and load both plate and tube with cell suspension onto machine.
-
26
Exclude debris based on side scatter-forward scatter. See Supplementary Fig. S1 for gating strategy.
-
27
Exclude dead cells based on propidium iodide intensity.
-
28
Create a histogram plot for event counts and Hoechst intensity, to visualize the DNA content. Cells in G2/M should show Hoechst intensity that is twice that of cells in G1.
TROUBLESHOOTING
-
29
On the DNA histogram create a gate for preferred cell cycle phase.
-
30
Sort cells that are alived and in preferred cell cycle phase either as single cells or as small populations of 10, 20 or 100 cells per well (Experimental Design: Sample Collection).
-
31
Seal the plate and spin at 2,000 g for 1 min at 4 °C and store at −80 °C immediately to keep the RNA intact.
PAUSE POINT Sorted plates can be kept at −80 °C for several months.
Lysis - Timing 45 min
-
32
Thaw the RNA ERCC spike-in dilution and the dNTPs on ice. Keep the recombinant ribonuclease inhibitor on an ice block at all times.
Prepare the lysis mix according to the table below. Keep the mix on ice at all times. Incubation at 65 °C (at Step 37) with Igepal will permeabilize/lyse the cellsReagent Amount for 1 well (nl) Amount for 1 plate (μl) Final concentration in mix Nuclease-free water 41 38.8 ERCC RNA Spike-in (1:50,000) 20 18.1 1:250,000 Igepal 1 % (vol/vol) 15 14.2 0.15 % (vol/vol) Recombinant ribonuclease inhibitor (40 U/μl) 4 3.7 1.6 U/μl dNTP mix (10 mM) 20 18.1 2 mM each Total volume 100 92.9 -
33
In an 8-well PCR strip, aliquot 11.1 μl of mix per well of the strip. Spin for 3–5 sec on tabletop spinner. Keep on ice at all times.
CRITICAL STEP It is important to perform steps 34–52 as fast as possible. Keep the plate cold at all times (unless dispension is taking place) to avoid RNA degradation.
-
34
Remove sorted plate (Step 31) from −80 °C, remove seal and dispense the lysis mix immediately.
-
35
Dispense 100 nl per well with the Nanodrop II robot (Box 2). The cumulative reaction volume is 200 nl.
-
36
Seal the plate and centrifuge at 2,000 g for 1 min at 4 °C
-
37
Heat the plate in a thermocycler heated at 65 °C for 5 min with the lid at 100 °C and then place on ice immediately for 1–2 min to cool down.
-
38
Centrifuge at 2,000 g for 1 min at 4 °C.
Box 2 | Nanodrop II robot handling.
Procedure
Master mix dispension - Timing 20 min for one plate (30 min for 4 max plates)
Perform a “Daily clean” program with 2 % (vol/vol) cleaning solution.
Prepare a “mock” 8-well PCR strip containing nuclease-free water at the same volume as the master mix. .
Insert the mock PCR strip and a sealed mock 384-well plate in corresponding positions.
Dispense the desired volume of the particular step in the protocol, to check if robot aspirates the correct volume and that the seal at positions of all wells contains the desired amount of water.
If water check confirms that robot dispenses correctly, repeat steps 3–4 with the actual master mix and destination plate. Remove the seal of the destination plate before dispension.
After the Proteinase K dispension (steps 53–56) perform a “Daily clean” program with 2 % (vol/vol) cleaning solution to remove excess Proteinase K from the tubing systems to avoid contamination in next dispensing steps.
Reverse transcription - Timing 1 hr 45 min
-
39
Thaw the 5 × First strand buffer and DTT on ice. Keep the RNase OUT and Superscript II on an ice block at all times.
-
40Prepare the reverse transcription mix according to the table below. Keep the mix on ice at all times. Reverse transcription will generate a DNA molecule complementary to the transcript sequence (cDNA) by using the barcoded CEL-Seq2 primers added in step 3.
Reagent Amount for 1 well (nl) Amount for 1 plate (μl) Final concentration in mix Nuclease-free water 10 8.6 5x First strand buffer 70 60.5 2.3 x DTT (0.1 M) 35 30.2 0.023 M RNaseOUT (40 U/μl) 17.5 15.1 4.6 U/μl Superscript II (200 U/μl) 17.5 15.1 23.33 U/μl Total volume 150 129.5 -
41
In an 8-well PCR strip, aliquot 15.5 μl of mix per well of the strip. Spin for 3–5 sec on tabletop spinner. Keep on ice at all times.
-
42
Dispense 150 nl per well with the Nanodrop II robot (Box 2). The cumulative reaction volume is 350 nl.
-
43
Seal the plate and centrifuge at 2,000 g for 1 min at 4 °C.
-
44
Put the plate in a thermocycler at °C for 1 hr, 4 °C for 5 min and 70 °C for 10 min, with the lid at 100 °C and then place on ice for 1–2 min to cool down.
-
45
Centrifuge at 2,000 g for 1 min at 4 °C.
Second strand synthesis - Timing 2 h 30 min
-
46
Thaw the 5 × Second strand buffer and dNTPs on ice. Keep the E. coli ligase, the DNA polymerase and the Ribonuclase H on an ice block at all times.
-
47Prepare the second strand mix according to the table below. Keep the mix on ice at all times. Second strand synthesis will generate a second strand of DNA complementary to the cDNA molecule generated during reverse transcription. This is done by using the transcript fragments cleaved by Ribonuclease H, as primers.
Reagent Amount for 1 well (nl) Amount for 1 plate (μl) Final concentration in mix Nuclease-free water 1,347.5 569.3 5x Second strand buffer 437.5 184.8 1.1 x dNTP mix (10 mM) 43.7 18.5 0.22 mM each E. coli ligase (10 U/μl) 15.7 6.6 0.08 U/μl DNA polymerase I (10 U/μl) 61.2 25.9 3.18 U/μl Ribonuclease H (2 U/μl) 15.7 6.6 0.016 U/μl Total volume 1,920.8 811.8 -
48
In an 8-well PCR strip, aliquot 101 μl of mix per well of the strip. Spin for 3–5 sec on tabletop spinner. Keep on ice at all times.
-
49
Dispense 1,920 nl per well with the Nanodrop II robot (Box 2). The cumulative reaction volume is 2270 nl.
-
50
Seal the plate and centrifuge at 2,000 g for 1 min at 4 °C.
-
51
5Put the plate in a thermocycler at 16 °C for 2 hr with the lid at 100 °C and let the thermocycler go to 4 °C at the end of the program.
-
52
Centrifuge at 2,000 g for 1 min at 4 °C.
Proteinase K treatment - Timing 11 hr overnight reaction
-
53
Thaw the Proteinase K and 10 × CutSmart buffer at room temperature and put on ice.
-
54Prepare the proteinase K mix according to the table below. Keep the mix on ice at all times. Proteinase K will cleave all proteins in the cell, including DNA-bound proteins and nucleases.
Reagent Amount for 1 well (nl) Amount for 1 plate (μl) Final concentration in mix Nuclease-free water 84.5 41.5 10x CutSmart buffer 277 136.3 5.5 x Proteinase K (20 mg/ml) 138.5 68.1 5.54 mg/μl Total volume 500 245.9 -
55
In an 8-well PCR strip, aliquot 30.3 μl of mix per well of the strip. Spin for 3–5 sec on tabletop spinner. Keep on ice at all times.
-
56
Dispense 500 nl per well with the Nanodrop II robot (Box 2). The cumulative reaction volume is 2770 nl.
CRITICAL STEP It is important to perform a “Daily clean” on the Nanodrop II robot after dispensing Proteinase K, to remove traces of the proteinase and avoid subsequent reaction contaminations (Box 2).
-
57
Seal the plate and centrifuge at 2,000 g for 1 min at 4 °C.
-
58
Put the plate in a thermocycler at 50 °C for 10 hr, 80 °C for 20 min with the lid at 100 °C and let the machine go to 4 °C at the end of the program.
-
59
Centrifuge at 2,000 g for 1 min at 4 °C.
(Day 3) DpnI digestion - Timing 7 hr
-
60
Thaw the 10x CutSmart buffer at room temperature and put on ice. Keep the DpnI enzyme on an ice block at all times.
-
61Prepare the DpnI mix according to the table below. Keep the mix on ice at all times. DpnI will digest all methylated GATC sites in the genome, leaving blunt free ends.
Reagent Amount for 1 well (nl) Amount for 1 plate (μl) Final concentration in mix Nuclease-free water 177 108.5 10x CutSmart buffer 23 14.1 1 x Dpnl (20 U/μl) 30 18.4 0.4 U/μl Total volume 230 141 -
62
In an 8-well PCR strip, aliquot 17.4 μl of mix per well of the strip. Spin for 3–5 sec on tabletop spinner. Keep on ice at all times.
-
63
Dispense 230 nl per well with the Nanodrop II robot (Box 2). The cumulative reaction volume is 3000 nl.
-
64
Seal the plate and centrifuge at 2,000 g for 1 min at 4 °C
-
65
Put the plate in a thermocycler at 37 °C for 6 hr, then at 80 °C for 20 min with the lid at 100 °C and let the thermocycler go to 4 °C at the end of the program.
-
66
Centrifuge at 2,000 g for 1 min at 4 °C.
Adapter dispension - Timing 45 min
-
67
Thaw the adapter plate at 4 °C during the DpnI digestion.
-
68
Prepare the Mosquito robot (Box 1).
-
69
Dispense 50 nl of DamID adapter per well. The cumulative reaction volume is 3050 nl.
CRITICAL STEP The final concentration of DamID adapter needs to be optimized for the expression of Dam-POI construct of choice. We recommend the final concentration falls within the 1.25 – 100 nM range.
-
70
Remove the plate and the adapter plate from the robot and seal.
-
71
Centrifuge at 2,000 g for 1 min at 4 °C.
Adapter ligation - Timing 12 h 45 min overnight reaction
-
72
Thaw the 10x Ligase buffer on ice. Keep the T4 ligase on an ice block at all times.
-
73Prepare the ligation mix according to the table below. Keep the mix on ice at all times.
Reagent Amount for 1 well (nl) Amount for 1 plate (μl) Final concentration in mix 10x Ligase buffer 350 176.2 7.7 x T4 Ligase (5 U/μI) 100 50.3 0.12 U/μl Total volume 450 226.5 -
74
In an 8-well PCR strip, aliquot 28 μl of mix per well of the strip. Spin for 3–5 sec on tabletop spinner. Keep on ice at all times.
-
75
Dispense 450 nl per well with the Nanodrop II robot (Box 2). The cumulative reaction volume is 3500 nl.
-
76
Seal the plate and centrifuge at 2,000 g for 1 min at 4 °C.
-
77
Put the plate in a thermocycler at 16 °C for 12 hr, then 65 °C for 10 min with the lid at 100 °C. Let the thermocycler go to 4 °C at the end of the program.
-
78
Centrifuge at 2,000 g for 1 min at 4 °C.
PAUSE POINT The processed plate can be kept at −20 °C up to a month.
(Day 4) Pool cells - Timing 1 hr
-
79
If frozen, thaw the plate from step 78 on ice.
CRITICAL STEP Depending on the number of plates, pooling can take longer. We indicate 1 hr approximately for 1 plate.
CRITICAL STEP Depending on the number of adapters used and the distribution of barcodes in the plate, pooling of non-overlapping barcoded material can be done manually or by inversion of the plate onto a collection container such as a clean lid of a tip box and centrifugation at 200 g for 1 min.
-
80
Pool cell lysates in a way that the barcodes do not overlap in 5 ml tubes.
CRITICAL STEP The higher the number of pooled wells, the higher the pool volume. The final reaction volume in each well is 3.5 μl. As an example, when pooling 384 wells, the expected cumulative volume is 384 × 3.5 μl = 1,344 μl. In this case, we recommend splitting the volume over three clean tubes, resulting in a volume of 448 μl per tube.
-
81
Separate oil from aqueous phase by pulse spin and collect the aqueous solution which is the bottom phase, containing the barcoded material. Keep on ice.
-
82
Repeat step 81 and transfer aqueous phase to new tube to remove mineral oil completely.
Purification of barcoded material - Timing 1 hr
CRITICAL Depending on the number of pools, bead purifications can be a bottleneck. We therefore do not recommend cleaning more than 8 reactions simultaneously.
CRITICAL In the following steps bead cleanups are needed to remove byproducts of the previous reactions and for size selection. For pool volumes higher than 30 μl, we recommend using AMPure XP beads that have been diluted with bead binding buffer (Reagent setup). By doing so, the volume of AMPure XP beads is reduced while the size selection is not affected. This enables proper elution of the AMPure XP beads in the small volume of 6 μl in step 91. Taking the example of 448 μl pool volume mentioned above, we recommend using a bead dilution of 1:8, meaning 1 part AMPure XP beads and 7 parts bead binding buffer (Reagent setup). For smaller pool volumes we recommend smaller bead dilutions. Keep an approximate of 30 μl of AMPure XP beads in the final mix, in order to enable the water elution of step 91.
-
83
Equilibrate diluted AMPure XP beads to room temperature for 30 min. Vortex till bead-buffer mix is homogenous.
-
84
Add 0.8 volume diluted AMPure XP beads to 1 volume of pool (Step 82). Allow material to bind to the AMPure XP beads for 10 min at room temperature. Steps 84–95 need to be carried out at room temperature.
-
85
Put the tube on a magnetic rack and allow AMPure XP beads to accumulate. Keep samples on magnetic rack until step 90.
-
86
Remove the aqueous phase carefully without disturbing AMPure XP beads.
-
87
Add 500 μl of fresh 80% (vol/vol) ethanol and leave for 30 sec.
-
88
Remove the ethanol carefully without disturbing AMPure XP beads.
-
89
Repeat steps 87–88. Pulse-spin tube and place in magnetic rack to remove excess ethanol.
TROUBLESHOOTING
-
90
Let AMPure XP beads to air-dry for 5 min or until they appear “matte”.
CRITICAL STEP Do not let AMPure XP beads overdry. Elute in water before cracks start appearing in the bead pellet.
-
91
Add 6 μl of nuclease-free water to the AMPure XP beads to elute the material and resuspend until beads and water form a homogenous mix. Place tube on ice.
Amplification by in vitro transcription - Timing 14 h 15 min
-
92
Thaw the Megascript T7 10x buffer at room temperature. Vortex thoroughly to dissolve precipitates and keep at room temperature.
-
93
Thaw the Megascript T7 NTPs on ice and keep on ice. Keep the enzyme mix on an ice block at all times.
-
94Prepare the Megascript T7 mix as indicated in the table below
Reagent Amount (μl) Final concentration in mix 10x T7 buffer 1.5 9.3 x ATP (75 mM) 1.5 7 mM UTP (75 mM) 1.5 7 mM GTP (75 mM) 1.5 7 mM CTP (75 mM) 1.5 7 mM T7 enzyme mix 1.5 Total volume 9 -
95
Add 9 μl of the Megascript T7 mix to the eluted material from Step 91 and resuspend. Cumulative volume is 16 μl.
-
96
Incubate the mix in a thermocycler at 37 °C for 14 h with the lid heated to 70 °C. Let thermocycler go to 4 °C after the program is finished.
CRITICAL STEP Do not let reaction stay in the thermocycler for more than a few hours after the programs is finished, to keep RNA integrity.
PAUSE POINT The reaction can be kept at −20 °C up to a day.
(Day 5) Purification of aRNA - Timing 1 hr
CRITICAL Depending on the number of samples, bead purifications can be a bottleneck. We therefore do not recommend cleaning too many samples simultaneously.
-
97
Thaw or put the IVT reaction from Step 96 in ice.
-
98
Place reaction on a magnet and transfer the reaction without the AMPure XP beads to a new tube.
-
99
Equilibrate fresh undiluted AMPure XP beads to room temperature for 30 min. Vortex till bead-buffer mix is homogenous.
-
100
Add 0.8 volume undiluted AMPure XP beads to the reaction and allow the material to bind to the AMPure XP beads for 10 min. Cummulative volume is 28.8 μl. Steps 100–108 need to be carried out at room temperature.
-
101
Put the tube on a magnetic rack and allow AMPure XP beads to accumulate. Keep samples on magnetic rack until step 106.
-
102
Remove the liquid carefully without disturbing AMPure XP beads. 103. Add 500 μl of fresh 80 % (vol/vol) ethanol and leave for 30 sec. 104. Remove the ethanol carefully without disturbing AMPure XP beads.
-
103
Repeat steps 103–104. Pulse-spin tube and place in magnetic rack to remove excess ethanol.
-
104
Let AMPure XP beads to air-dry for 5 min or until they appear “matte”.
CRITICAL STEP Do not let AMPure XP beads overdry. Elute in water before cracks start appearing in the bead pellet.
-
105
Add 23 μl of nuclease-free water to the AMPure XP beads and resuspend until beads and water form a homogenous mix. Remove tube from magnetic rack and allow the material to elute for 5 min.
-
106
Place tube in magnetic rack and without disturbing the AMPure XP beads carefully transfer 22 μl solution to a clean tube and place on ice.
aRNA fragmentation - Timing 5 min
-
107
Bring a heat block to 94 °C.
-
108
Add 0.2 volumes of fragmentation buffer to amplified material from Step 108 while on ice and resuspend. Cumulative volume is 26.4 μl.
-
109
Quickly transfer tube to 94 °C for 2 min.
-
110
Remove tube and quickly put on ice.
-
111
Add 0.1 volume of fragmentation STOP buffer to tube as fast as possible and resuspend. Keep on ice. Cumulative volume is 29.04 μl.
TROUBLESHOOTING
Purification and quantification of fragmented aRNA - Timing 1 hr 45 min
CRITICAL Depending on the number of samples, bead purifications can be a bottleneck. We therefore do not recommend cleaning too many samples simultaneously.
CRITICAL Depending on the number of pooled cell lysates, extra rounds of bead purifications can increase the library prep efficiency because of adapter depletion. For 384 pooled cells we recommend at least 2 rounds of bead purification at this stage of the protocol. For 96 pooled cells we recommend 1 round of bead purification, as stated in steps 114–124.
-
112
Equilibrate undiluted AMPure XP beads to room temperature for 30 min. Vortex till bead-buffer mix is homogenous.
-
113
Add 0.8 volume undiluted AMPure XP beads to the reaction and allow the material to bind to the AMPure XP beads for 10 min. Cumulative volume is 52.3 μl. Steps 115–124 need to be carried out at room temperature.
-
114
Put the tube on a magnetic rack and allow AMPure XP beads to accumulate. Keep samples on magnetic rack until step 122.
-
115
Remove the aqueous phase carefully without disturbing AMPure XP beads.
-
116
Add 500 μl of fresh 80 % (vol/vol) ethanol and leave for 30 sec.
-
117
Remove the ethanol carefully without disturbing AMPure XP beads.
-
118
Repeat steps 118–119.
-
119
Pulse-spin tube and place in magnetic rack to remove excess ethanol.
-
120
Let AMPure XP beads to air-dry for 5 min or until they appear “matte”.
CRITICAL STEP Do not let AMPure XP beads overdry. Elute in water before cracks start appearing in the bead pellet.
-
121
Add 13 μl of nuclease-free water to the AMPure XP beads and resuspend until beads and water form a homogenous mix. Remove tube from magnetic rack and allow the material to elute for 5 min.
-
122
Place tube in magnetic rack and without disturbing the AMPure XP beads carefully transfer 12 μl solution to a clean tube and place on ice.
PAUSE POINT The sample can be stored at −80 °C up to 6 months.
-
123
Measure 1 μl of aRNA with a Bioanalyzer RNA pico chip by following the kit manual. Examples of successful and less successful IVT reactions and aRNA fragmentation and bead cleanup for library preparation are shown in Fig. 5.
TROUBLESHOOTING
Reverse transcription - Timing 1 hr 30 min
-
124Prepare the randomhexRT mix as indicated in the table below and heat in thermocycler at 65 °C for 5 min with lid at 100 °C. Immediately put on ice. Reverse transcription will generate a DNA molecule complementary to the aRNA molecule. This is done by using poly-N primers containing the P7 Illumina adapter in their overhang.
Reagent Amount (μl) Final concentration in mix aRNA (step 125) 5 randomhexRT primer (20 μM) 1 13.33 mM dNTP mix (10 mM) 0.5 3.33 mM Total volume of reaction 6.5 -
125Prepare the RT mix as indicated in the table below. Keep mix on ice and enzymes in ice block at all times. Heat in thermocycler at 25 °C for 10 min, then 42 °C for 1 hr with the cycler lid at 50 °C.
Reagent Amount (μl) Final concentration in mix hexRT mix with aRNA (step 126) 6.5 5x First strand buffer 2 2.5 x DTT (0.1 M) 1 25 mM RNase OUT (40 U/μI) 0.5 5 Superscript II (200 U/μI) 0.5 25 Total volume of reaction 10.5
PCR indexing - Timing 30 min
CRITICAL Do not overamplify the material. We recommend always a minimum of 8 cycles of PCR for the generation of enough Illumina indexed molecules. We recommend 10 PCR cycles or more for aRNA product between 1 and 10 Fluorescent Units (FU) and 8–9 cycles for aRNA product higher than 10 FU (Fig. 5). Prepare the indexing mix as indicated in the table below. Keep mix on ice. Index each sample with a unique RPi primer for multiplexing. This step completes the P5 and P7 ends of the molecules by indexing the libraries.
| Reagent | Amount (μl) | Final concentration in mix |
|---|---|---|
| Reverse transcribed aRNA (step 127) | 10.5 | |
| Nuclease-free water | 10.5 | |
| 2x NEBNext High-Fidelity PCR Master Mix | 25 | 1.26 x |
| RNA PCR primer RP1 (10 μM) | 2 | 0.5 μM |
| RNA PCR index primer RPi (10 μM) | 2 | 0.5 μM |
| Total volume of reaction | 50 |
-
126Run PCR program in thermocycler with the lid heated at 105 °C as indicated in the table below.
Cycle number Denature Anneal Extend 1 98°C, 30 s 2–12 98°C, 10s 60°C, 30 s 72°C, 30 s 13 72°C, 10 min
Library purification - Timing 1 hr 30 min
CRITICAL Depending on the number of samples, bead purifications can be a bottleneck. We therefore do not recommend cleaning too many samples simultaneously.
-
127
Equilibrate undiluted AMPure XP beads to room temperature for 30 min. Vortex till bead-buffer mix is homogenous.
-
128
Add 0.8 volume undiluted AMPure XP beads to the reaction and allow the material to bind to the AMPure XP beads for 10 min. Cumulative volume is 90 μl. Steps 130–143 need to be carried out at room temperature.
-
129
Put the tube on a magnetic rack and allow AMPure XP beads to accumulate. Keep samples on magnetic rack until step 138.
-
130
Remove the aqueous phase carefully without disturbing AMPure XP beads.
-
131
Add 500 μl of fresh 80 % (vol/vol) ethanol and leave for 30 sec.
-
132
Remove the ethanol carefully without disturbing AMPure XP beads.
-
133
Repeat steps 133–134. Pulse-spin tube and place in magnetic rack to remove excess ethanol.
-
134
Let AMPure XP beads to air-dry for 5 min or until they appear “matte”.
CRITICAL STEP Do not let AMPure XP beads overdry. Elute in water before cracks start appearing in the bead pellet.
-
135
Add 26 μl of nuclease-free water to the AMPure XP beads and resuspend until beads and water form a homogenous mix. Remove tube from magnetic rack and allow the material to elute for 5 min.
-
136
Place tube in magnetic rack and without disturbing the AMPure XP beads carefully transfer 25 μl elution to a clean tube and place on ice.
-
137
Repeat steps 130–137 to re-clean the eluted material of step 138.
-
138
Add 16 μl of nuclease-free water to the AMPure XP beads and resuspend until beads and water form a homogenous mix. Remove tube from magnetic rack and allow the material to elute for 5 min.
-
139
Place tube in magnetic rack and without disturbing the AMPure XP beads carefully transfer 15 μl solution to a clean tube and place on ice.
PAUSE POINT The finished libraries can be kept at −20 °C indefinitely.
Library quantification and sequencing - Timing 19 hr
-
140
Measure the concentration of the sample with the Qubit dsDNA HS Assay kit by following the kit manual.
-
141
Measure 1 μl of sample with a Bioanalyzer HS DNA chip by following the kit manual. Examples of successful and less successful library prep reactions and purifications are shown in Fig. 6.
TROUBLESHOOTING
-
142
Dilute samples to appropriate molarity for sequencing and pool according to the desired number of reads per sample. We dilute to 4 nM and in case of low concentration, samples can be diluted to 2 nM.
-
143
Submit samples to the sequencing facility. To ensure cluster formation, avoid sequencing fewer than 8 different indexed libraries in one run. We sequence samples on the NextSeq 500, using 75 bp for both Read 1 and Read 2 (paired-end) and spike-in 20 % PhiX. This percentage of PhiX control is specific for the samples generated with this protocol and ensures cluster formation by enriching the complexity of the sequencing pool.
Fig 6 |. Examples of DNA bioanalyzer plots.
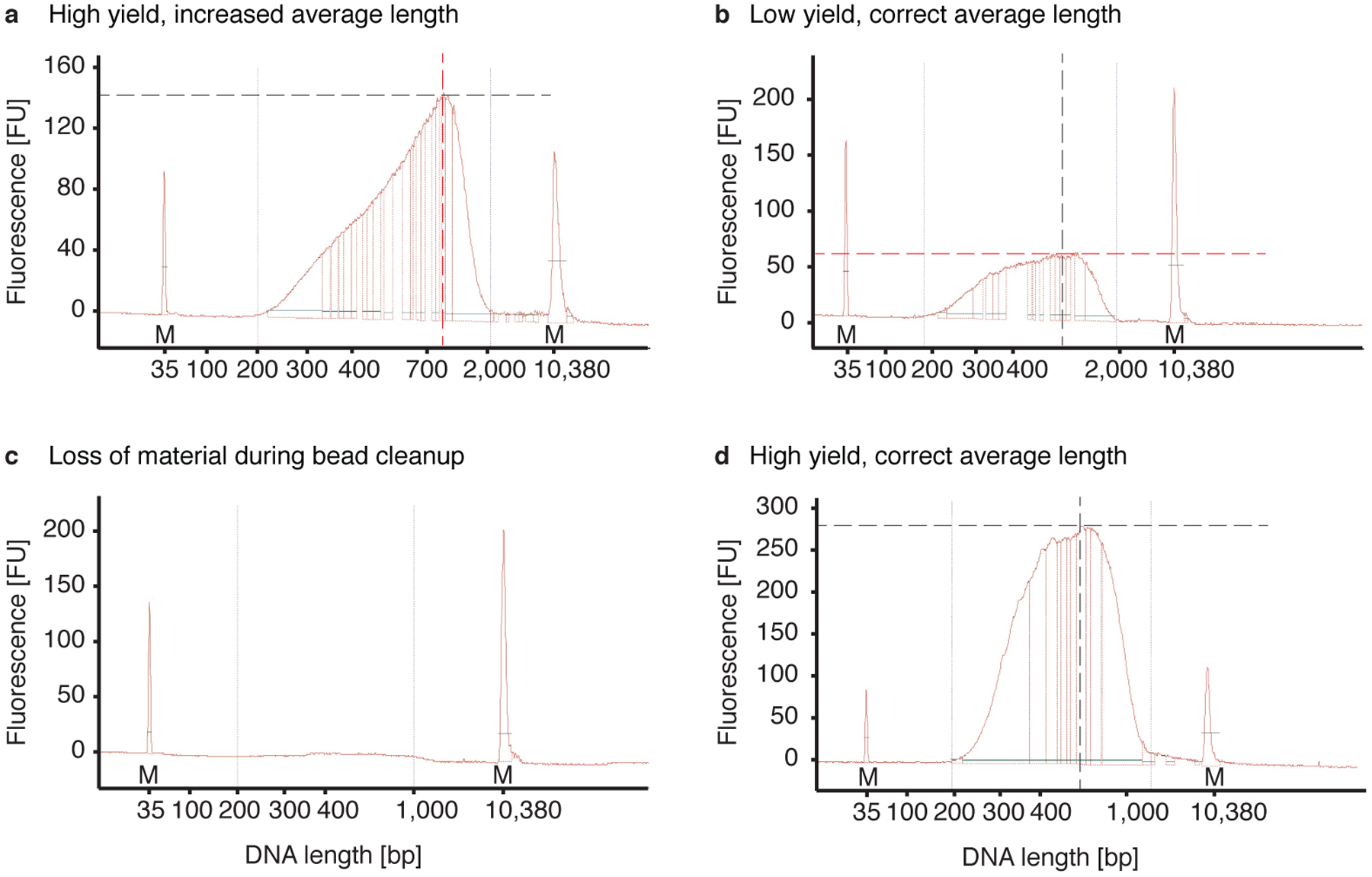
Bioanalyzer results after library construction: a) The library shows good yield (>150 FU) but size distribution is slightly increased (peaking at 1,000 – 1,500 nt), probably due to increased fragment length of aRNA. b) The library shows correct length distribution (250 – 900 nt) but low yield. If library was constructed from aRNA similar to example 4 from 3a, material was probably lost during bead purification. Repeat library preparation with leftover aRNA. c) Lack of both the adapter and the PCR product indicate complete loss during bead purification or failed library preparation. d) The library shows high yield >250 FU and correct size distribution of 250 – 700 nt. FU, fluorescence units. nt, nucleotides. Peaks marked with an “M” indicate the reference markers; black and red dashed lines indicate the relevant optimal and suboptimal features, respectively.
Downloading genome reference files and generating HISAT2 index - Timing 1 hr 30 min
CRITICAL For the purpose of this protocol, the FASTA file, GTF file and HISAT2 index will all be placed in a folder named “references”. For that reason, generate this folder in your own working directory, or replace all mentions of the “references” directory with the path of your own choice:
mkdir ./references
CRITICAL Note that this command and all future commands are executed from the terminal.
-
144
Download the reference sequence of the relevant species, for example from http://www.ensembl.org/info/data/ftp/index.html. Click the “DNA (FASTA)” link and download the file ending with ‘dna.primary_assembly.fa.gz’. Unzip the downloaded file and place it in the “references” directory.
-
145
Download the GTF file for the relevant species, for example from http://www.ensembl.org/info/data/ftp/index.html. Unzip downloaded file and place it in the “references” directory.
-
146
If using ERCC spike-ins, their sequences should be added to the genome FASTA file and the GTF file. For clarity, we add “with_ERCC” to the FASTA and GTF filename in this walkthrough.
-
147Generate a HISAT2 index:
HISAT2_INDEX=“./references/Mus_musculus.GRCm38.dna.primary_assembly.with_ERCC” FASTAFN=“./references/Mus_musculus.GRCm38.dna.primary_assembly.with_ERCC.fa” hisat2-build $FASTAFN $HISAT2_INDEX
Installing scDam&T-seq scripts - Timing 10 min
CRITICAL: The analysis steps in this and subsequent sections demonstrate how scDam&T-seq data can be analysed using the provided software package (scDamAndTools). File names and genome references are chosen to match the test data available as part of the GitHub repository (in the “tutorial” folder). The included data represents five single cells from a mESC Dam-LaminB1 experiment. Care should be taken to modify file names when applying the analysis on other data. A more detailed explanation of all steps and functions can be found in Table 1 and on the GitHub page (https://github.com/KindLab/scDamAndTools), where we have also included the expected results from processing the test data.
-
148Generate a Python3 virtual environment and activate the virtual environment:
python3 -m venv $HOME/.venvs/tutorial source ~/.venvs/tutorial/bin/activate
PAUSE POINT To deactivate the virtual environment:deactivate
-
149Install prerequisite modules:
pip install --upgrade pip wheel setuptools pip install cython
-
150Install scDam&T-seq package:
pip install git+https://github.com/KindLab/scDamAndTools.git
Generate GATC reference arrays - Timing 1 hr 30 min
-
151To efficiently match obtained DamID reads to specific instances of the GATC motif in the genome, we generate two reference arrays. The first array (“position array” or “posarray”) contains the positions of all GATC positions in the genome. The second array (“mappability array” or “maparray”) indicates whether it is possible to uniquely align a read derived from a particular (strand-specific) GATC instance. The mappability array is used to filter out ambiguously aligning GATCs and can serve as an indicator of the (mappable) GATC density along the chromosomes. During the generation of the mappability array, in silico reads are generated for each GATC instance and are subsequently mapped back to the reference genome. The length of the reads should be chosen to be the same as the length of the reads obtained in the experiment (excluding the UMI and barcode):
IN_SILICO_READLENGTH=62 ARRAY_PREFIX=”./refarrays/Mus_musculus.GRCm38.dna.primary_assembly” create_motif_refarrays \ -m “GATC” \ -o $ARRAY_PREFIX \ -r $IN_SILICO_READLENGTH \ -x $HISAT2_INDEX \ $FASTAFN
The script generates three files ending in “.positions.bed.gz” (all occurrences of the GATC motif in BED format), “.posarray.hdf5” (all occurrences of the GATC motif as a HDF5 array), and “.maparray.hdf5” (the mappability of all GATC motifs as a HDF5 array), respectively.
Demultiplex raw data - Timing 1 hr
-
152
If not already done so by the sequencing facility, demultiplex the raw data based on the used Illumina indices.
-
153In a text editor or Microsoft Excel, create a tab-delimited text file43 that describes the barcodes that were used in the library. The file should have two columns, listing the adapter names and sequences respectively. The location and length of UMIs should be indicated with numbers and dashes. Using DamID and CEL-Seq2 barcodes as specified in Experimental Design (Desgin and concentration of DamID adapters and CEL-Seq2 primers), the barcode file of a library with two samples should look as follows:
DamID_BC_001 3-TGCT-3-GAGAGA DamID_BC_002 3-ATTG-3-GAACGA CELseq_BC_001 3-ACAG-3-AGGC CELseq_BC_002 3-GTCT-3-GCCA
Example data and relevant barcode file are included in the scDamAndTools package in the folder “tutorial”.
CRITICAL STEP: There may be multiple raw sequencing files pertaining to the same samples, for example from the different sequencing lanes. These files should be concatenated prior to the processing of DamID and CEL-Seq2 reads (step 157 and 159, respectively).
-
154For each Illumina library, demultiplex the reads based on the used adapters. Make sure the output file format contains the fields “{name}” and “{readname}”, where the barcode name and paired-end read name will be inserted:
OUTFMT=“./data/demultiplexed/index01.{name}.{readname}.fastq.gz” INFOFN=“./data/demultiplexed/index01.demultiplex_info.txt” demultiplex.py \ -vvv \ --mismatches 0 \ --outfmt $OUTFMT \ --infofile $INFOFN \ ./metadata/index01.barcodes.tsv \ ./data/raw/index01_R1_001.fastq.gz \ ./data/raw/index01_R2_001.fastq.gzThe demultiplex script generates a separate FASTQ file for each barcode provided in the barcode information file (see step 155) that contains all reads matching this barcode. In addition, a text file (“index01.demultiplex_info.txt”) is generated that details the number of reads matched to each barcode.
Process DamID demultiplexed files - Timing 2 hr
CRITICAL: The subsequent steps (157–158) need to be performed on all DamID demultiplexed files. It is highly recommended that this process be parallelized on a high-performance computing cluster. The amount of time necessary for these steps depends entirely on the number of libraries, samples per library and available computing cores.
-
155Process the DamID reads to arrays of (UMI-unique) GATC counts. The script aligns the DamID reads to the genome and subsequently matches them to positions as indicated in the position array (see step 153). Since the GATC motif is cleaved in half by DpnI, the prefix “GA” is added to all reads prior to alignment. PCR-duplicates are filtered out based on the available UMI information. For this step, only the R1 reads are used since these contain the genomic sequence aligning to the GATC motif:
OUTPREFIX=“./data/damid/index01.DamID_BC_001”; POSARRAY=“./refarrays/Mus_musculus.GRCm38.dna.primary_assembly.GA TC.posarray.hdf5”; process_damid_reads \ -o $OUTPREFIX \ -m “GA” \ -p $POSARRAY \ -x $HISAT2_INDEX \ -u \ ./data/demultiplexed/index01.DamID_BC_001.R1.fastq.gz
The script generates an alignment file ending in “.sorted.bam”, a GATC count file ending in “.counts.hdf5” and an information file ending in “.counts.stats.tsv”.
-
156Bin the GATC count files into genomically equal-sized bins. The resulting HDF5 file contains for each chromosome the number of observed UMI-unique counts for each bin:
MAPARRAY=“./refarrays/Mus_musculus.GRCm38.dna.primary_assembly.GATC.readlength_62.maparray.hdf5” OUTFN=“./data/damid/index01.DamID_BC_001.counts.binsize_100000.hdf5” bin_damid_counts.py \ -vvv \ --mapfile $MAPARRAY \ --posfile $POSARRAY \ --binsize 100000 \ --outfile $OUTFN \ ./data/damid/index01.DamID_BC_001.counts.hdf5
The output of this step is a single HDF5 file ending in “.binsize_100000.hdf5” that contains the number of unique counts observed in all 100kb bins in the genome.
Process CEL-Seq2 demultiplexed files - Timing 4 hr
CRITICAL: The subsequent step (159) needs to be performed on all CEL-Seq2 demultiplexed files. It is highly recommended that this process be parallelized on a high-performance computing cluster. The amount of time necessary for these steps depends entirely on the number of libraries, samples per library and available computing cores.
-
157Process the CEL-Seq2 reads to an array of UMI-unique counts per gene. For this step, the R2 reads are used, since these contain the genomic sequence:
OUTPREFIX=“./data/celseq/index01.CELseq_BC_001” GTF=“./references/Mus_musculus.GRCm38.98.with_ERCC.gtf” process_celseq_reads \ -o $OUTPREFIX \ -g $GTF \ -x $HISAT2_INDEX \ ./data/demultiplexed/index01.CELseq_BC_001.R2.fastq.gz
The script generates an alignment file ending in “.bam” and a count file ending in “.counts.hdf5”. The count file contains the number of observed UMI-unique transcripts per gene, sorted by their Ensembl gene ID as provided in the GTF file.
Timing
Steps 1 – 6, preparation of primer plates, induction of Dam-POI: 1 hr 15 min
Steps 7 – 31, cell harvest, Hoecht staining, sorting: 2 hr 30 min
Steps 32 – 59, lysis, reverse transcription, second strand synthesis, proteinase K: 16 hr
Steps 60 – 78, DpnI digestion, adapter dispension, adapter ligation: 21 hr
Steps 79 – 96, pooling, bead cleanups, in vitro transcription: 16 hr 15 min
Steps 97 – 125, bead cleanups, fragmentation, bead cleanups, RNA quantification: 3 hr
Steps 126 – 145, library prep, DNA quantification, library pooling, sequencing: 23 hr
Steps 146 – 159, analysis: 10 hr
Anticipated results
Figure 4 shows statistics of two example libraries containing 96 single-cell samples of a Dam-LMNB1 mESC line. The two libraries are biological replicates that were collected, processed and sequenced at different times. We obtained ~45 and ~92 million reads for replicate 1 and 2 respectively, which we consider a high sequencing depth for these samples. Nearly all these reads (>95 %) can be successfully assigned to a DamID or CEL-Seq2 barcode (Fig. 4a). The majority of the demultiplexed reads successfully align (Fig. 4b), after which invalid reads are filtered out. CEL-Seq2 reads are considered to be invalid if they do not align properly to a gene or when a read’s mapping score is lower than a set threshold. DamID reads are considered to be invalid when they do not align to a GATC position or when their mapping quality is too low. In a successful experiment, approximately 60 % of the demultiplexed reads are valid and 20 – 40 % are unique (Fig. 4b).
Although the majority of reads is demultiplexed to a DamID barcode (~90 %) and a much smaller fraction to a CEL-Seq2 barcode (~5 %), the resulting CEL-Seq2 data still provides a median of 11,000 – 12,000 unique transcripts (Fig. 4c) and 3,700 – 3,900 unique genes per cell, which is more than sufficient to perform typical single-cell transcriptome analyses. The DamID data, on the other hand, contains a median of ~125,000 – 175,000 unique GATC counts per sample. We typically exclude samples from our analyses that contain fewer than 10,000 unique counts, but the appropriate threshold depends on the POI. The reason why more DamID material than CEL-Seq2 material is obtained is not entirely clear, but likely has to do with the efficiency with which transcripts and gDNA anneal to the primer and adapter, respectively. We also find that the ratio between DamID and CEL-Seq2 reads varies depending on the POI and methylation level in the cell, with a higher fraction of CEL-Seq2 reads for POIs that methylate a smaller fraction of the genome. If the depth and quality are satisfactory, the resulting DamID data can be used for downstream analysis. We typically work with binned data at a resolution of 50 – 100 kb for single-cell samples.
In general, the final number of unique GATCs is strongly influenced by the number of samples in the library, the number of PCR cycles used during library preparation and the sequencing depth. However, loss of DamID reads can also occur when something goes wrong during the experiment or when the expression of Dam-POI is too low, which results in a high fraction of invalid reads. For that reason, we look at the fraction of valid DamID reads, which should be above 50 % for most samples (Fig. 4b). To establish the complexity of the samples, we compare the number of valid reads to the final number of unique counts (Fig 4d). The libraries shown here have a DamID complexity of 58 % and 35 % and a CEL-Seq2 complexity of 68 % and 58 % for replicate 1 and 2, respectively. If the data is too sparse to execute the desired analyses (e.g. for LMNB1, a median of <10,000 unique counts per cell), libraries with a complexity of more than 30 % may be considered for resequencing. Possible reasons for poor data quality and potential solutions are discussed in Table 2 (Troubleshooting).
Table 2:
Troubleshooting
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 28 | Unexpected cell-cycle profile |
|
Prepare a fresh Hoechst solution. |
| 89, 125 | Low or no aRNA product on Bioanalyzer (Fig. 3a) |
|
Remove all ethanol before elution. |
| 113 | Increased aRNA product distribution (Fig. 3a) |
|
Resuspend fragmentation buffer and make sure whole tube makes contact with the heat block during fragmentation at 94 °C. |
| 143 | Low or no library product on Bioanalyzer (Fig. 3b) |
|
Remove all ethanol before elution. Repeat library prep with leftover aRNA. Increase PCR cycles. |
| 143 | Low product (Fig. 3b) |
|
Bead purify the aRNA 1–2 times extra Repeat library prep. |
| Post data processing | Low complexity |
|
If possible, increase the number of samples in a library and decrease the number of PCR cycles. Make sure the amount of material included in a sequencing run is proportional to the expected output. |
| Post data processing | A uniform DamID signal over the whole genome; signal is very similar to the mappable GATC density |
|
Select a clone with lower expression levels or modify the Dam-POI construct. Optimize the time of induction of the selected clone. Ensure there is no expression of Dam-POI prior to induction. |
| Post data processing | Little DamID signal, despite good signal in positive control |
|
Select a clone with a higher expression level or modify the Dam-POI construct. Induce expression for a longer time. |
| Post data processing | Sparse CEL-Seq2 data compared to DamID data |
|
Include positive controls to make sure single-cell transcription data can be obtained from the material (e.g. WT control). Renew CEL-Seq2 primer plate. Optimize CEL-Seq2 primer and DamID adapter concentrations, e.g. reduce DamID adapter concentration. |
| Post data processing | Low fraction of valid DamID reads |
|
Select clone with optimal expression levels. Include negative control (e.g. WT control). Reduce adapter concentrations or perform additional bead purifications. |
Supplementary Material
Supplementary Methods | scDamID2 and bulk scDamID2 protocols
Supplementary table 1 | CEL-Seq2 primers
Supplementary table 2 | scDamID double-stranded adapters
Supplementary Manual | Step-by-step molecule structure
Supplementary Figure 1 | Gating strategy for FACS
Acknowledgements
We would like to thank the members of the Kind lab for their comments on the manuscript. S.S.D. acknowledges support from the Center for Scientific Computing at UCSB: an NSF MRSEC (DMR-1720256) and NSF CNS-1725797. S.S.D. was also supported by the NIH grant R01HG011013. This work was funded by a European Research Council Starting grant (ERC-StG 678423-EpiID), a Nederlandse Organisatie voor Wetenschappelijk Onderzoek37 Open (824.15.019) and ALW/VENI grant (016.181.013). The Oncode Institute is supported by KWF Dutch Cancer Society.
Footnotes
Competing interests
The authors declare that they have no competing financial interests.
Data availability
A test dataset is available from GitHub (https://github.com/KindLab/scDamAndTools).
Code availability
All codes are available from GitHub (https://github.com/KindLab/scDamAndTools). The code in this manuscript has been peer-reviewed.
References
- 1.Johnson DS, Mortazavi A, Myers RM & Wold B Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Vogel MJ, Peric-Hupkes D & van Steensel B Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc 2, 1467–1478 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Crawford GE et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16, 123–131 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kind J et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flyamer IM et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens TJ et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin W et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo H et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 23, 2126–2135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smallwood SA et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11, 817–820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farlik M et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep 10, 1386–1397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mooijman D, Dey SS, Boisset JC, Crosetto N & van Oudenaarden A Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat Biotechnol 34, 852–856 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Inoue A, Suzuki T & Zhang Y Simultaneous mapping of active DNA demethylation and sister chromatid exchange in single cells. Genes Dev 31, 511–523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu C et al. Single-Cell 5-Formylcytosine Landscapes of Mammalian Early Embryos and ESCs at Single-Base Resolution. Cell Stem Cell 20, 720–731 e725 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Rotem A et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harada A et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol 21, 287–296 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Hainer SJ, Boskovic A, McCannell KN, Rando OJ & Fazzio TG Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 177, 1319–1329 e1311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ku WL et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat Methods 16, 323–325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angermueller C et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 13, 229–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou Y et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 26, 304–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun 9, 781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rooijers K et al. Simultaneous quantification of protein-DNA contacts and transcriptomes in single cells. Nat Biotechnol 37, 766–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimshony T, Wagner F, Sher N & Yanai I CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2, 666–673 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Hashimshony T et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol 17, 77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T & Kanemaki M An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Boers R et al. Genome-wide DNA methylation profiling using the methylation-dependent restriction enzyme LpnPI. Genome Res 28, 88–99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen M et al. Strand-specific single-cell methylomics reveals distinct modes of DNA demethylation dynamics during early mammalian development. bioRxiv, 804526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borsos M et al. Genome-lamina interactions are established de novo in the early mouse embryo. Nature 569, 729–733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu CL, Schreiber SL & Bernstein BE Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics 4, 19 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grosselin K et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet 51, 1060–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmid M, Durussel T & Laemmli UK ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 16, 147–157 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sirunyan AM et al. Search for rare decays of Z and Higgs bosons to J / psi and a photon in proton-proton collisions at s = 13 TeV. Eur Phys J C Part Fields 79, 94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tosti L et al. Mapping transcription factor occupancy using minimal numbers of cells in vitro and in vivo. Genome Res 28, 592–605 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monkhorst K, Jonkers I, Rentmeester E, Grosveld F & Gribnau J X inactivation counting and choice is a stochastic process: evidence for involvement of an X-linked activator. Cell 132, 410–421 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ditzel M et al. Biologic meshes are not superior to synthetic meshes in ventral hernia repair: an experimental study with long-term follow-up evaluation. Surg Endosc 27, 3654–3662 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Aad G et al. Observation of associated near-side and away-side long-range correlations in sqrt[s(NN)]=5.02 TeV proton-lead collisions with the ATLAS detector. Phys Rev Lett 110, 182302 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods | scDamID2 and bulk scDamID2 protocols
Supplementary table 1 | CEL-Seq2 primers
Supplementary table 2 | scDamID double-stranded adapters
Supplementary Manual | Step-by-step molecule structure
Supplementary Figure 1 | Gating strategy for FACS