

Using a systems genetics approach, Rasmussen et al. identify rare migraine-associated mutations aggregated in modules. The mutations suggest a role in migraine pathophysiology for signalling pathways involving hormones, G-proteins and serotonin receptors.
Keywords: complex trait, gene-gene interaction, genetic network, transcriptomics, migraine
Abstract
Migraine is the most common neurological disorder worldwide and it has been shown to have complex polygenic origins with a heritability of estimated 40–70%. Both common and rare genetic variants are believed to underlie the pathophysiology of the prevalent types of migraine, migraine with typical aura and migraine without aura. However, only common variants have been identified so far. Here we identify for the first time a gene module with rare mutations through a systems genetics approach integrating RNA sequencing data from brain and vascular tissues likely to be involved in migraine pathology in combination with whole genome sequencing of 117 migraine families. We found a gene module in the visual cortex, based on single nuclei RNA sequencing data, that had increased rare mutations in the migraine families and replicated this in a second independent cohort of 1930 patients. This module was mainly expressed by interneurons, pyramidal CA1, and pyramidal SS cells, and pathway analysis showed association with hormonal signalling (thyrotropin-releasing hormone receptor and oxytocin receptor signalling pathways), Alzheimer’s disease pathway, serotonin receptor pathway and general heterotrimeric G-protein signalling pathways. Our results demonstrate that rare functional gene variants are strongly implicated in the pathophysiology of migraine. Furthermore, we anticipate that the results can be used to explain the critical mechanisms behind migraine and potentially improving the treatment regime for migraine patients.
Introduction
Migraine is a common neurovascular disorder that is characterized by a unilateral throbbing head pain, accompanied by sensitivity to light and sound and/or nausea/vomiting. It is often accompanied by neurological disturbances (the aura) (Headache Classification Committee of the International Headache Society (IHS), 2018; Szabó et al., 2018). The prevalent types of migraine, migraine with typical aura (MTA) and migraine without aura are considered complex polygenic disorders with a heritability between 40% and 70% (Sutherland and Griffiths, 2017). Scores of common genetic variants have been associated with migraine explaining a part of the genetic variance (Gormley et al., 2016). It has also been shown that the polygenic risk score is higher in families with MTA and migraine without aura; however, it is believed that rare causal variants could also contribute to the genetic variance of migraine (Hansen et al., 2017; Gormley et al., 2018). Such rare causal variants can be detected in families with mendelian-like segregating pattern of migraine, using e.g. whole genome sequencing (WGS). It is encouraging that family studies have identified multiple rare causal variants for a rare dominant form of migraine, familial hemiplegic migraine (Ophoff et al., 1996; Dichgans et al., 2005; Marini et al., 2012; Barros et al., 2013). However, as the prevalent types of migraine are polygenic and do not follow a complete dominant inheritance pattern, we hypothesize that genes with rare variants of small or medium effect size are aggregating in mechanisms specific for migraine. Such mechanisms can be identified in gene networks identified, for example, by RNA sequencing of migraine-relevant tissues in combination with WGS.
Weighted gene co-expression network analysis (WGCNA) (Zhao et al., 2010) has been useful in identifying such gene networks, which has the potential of pointing towards biologically and clinically relevant disease mechanisms (Eising et al., 2017; Feng and Wang, 2017; Liao et al., 2017; Wang et al., 2017). Gene expression is highly tissue-specific, thus co-expression networks of tissues likely to be involved in migraine could suggest migraine-specific networks. The pathophysiology of migraine is not completely understood but it is widely accepted and shown in several studies that the trigeminal ganglion, the visual cortex and the vasculature are involved in critical mechanisms of migraine (Zhang et al., 2010, 2011; Coppola et al., 2013; Brighina et al., 2015; Gormley et al., 2016; Shelukhina et al., 2017; Gaist et al., 2018). The trigeminal ganglion is a cluster of nerve cells controlling sensory input from the face and, for example, due to release of calcitonin gene-related peptide (CGRP) and expression of serotonin 5-HTd1, a treatment target of migraine (Potrebic et al., 2003; Messlinger and Russo, 2018). The visual cortex is involved in processing of visual information, and cortical spreading depression in visual cortex explains the aura (Marzoli and Criscuoli, 2017; Charles, 2018). The cardiovascular system has, in addition to specific brain tissues, been linked to migraine (Pickrell et al., 2016). This was recently stipulated as aorta, as common genetic variants associated with migraine were significantly higher expressed in aorta (Gormley et al., 2016).
In this study, we used a systems genetics approach integrating rare genetic variants segregating in migraine families with gene expression networks. We first identified gene networks using WGCNA applied to RNA sequencing data from human aorta (tissue), trigeminal ganglion (tissue) and visual cortex (single nuclei). Second, we integrated rare mutations from our WGS data with gene networks to pinpoint networks having rare variants segregating with migraine, suggesting distinct involvement in migraine mechanisms. Third, these results were replicated in an independent cohort of migraineurs without familial incidences of migraine. The complementary results revealed—for the first time—the critical role of rare gene variants in migraine heritability.
Materials and methods
The study design focused on aorta, trigeminal ganglion and visual cortex as they are key tissues in the pathophysiology of migraine (Zhang et al., 2010, 2011; Coppola et al., 2013; Brighina et al., 2015; Gormley et al., 2016; Shelukhina et al., 2017; Gaist et al., 2018), aiming to identify the pathways involved in the migraine mechanisms (Fig. 1). We constructed co-expression networks of aorta, trigeminal ganglion and visual cortex using RNA sequencing data, for identification of tissue-specific modules using WGCNA. We then integrated data on rare functional variants segregating with migraine, derived from WGS data of 874 individuals from 117 families with migraine, to extracted modules associated with migraine. We aimed to replicate the modules in patients with sporadic migraine (n = 1930) and population control subjects (n = 2500). We then performed functional annotation to map the pathways/networks and cell types involved in migraine.
Figure 1.

Study design. Co-expression networks were constructed for the trigeminal ganglion, visual cortex and aorta, to identify tissue-specific modules of highly co-expressed genes. In parallel, the WGS data were analysed to identify mutations. Modules with over-representations of mutations were defined as potential ‘migraine modules’. Similarly, we defined mutations in an independent cohort compared to population controls and replicated the potential migraine modules. Replicated modules were further analysed using functional annotation. RVIS = residual variation intolerance score.
Subjects
We recruited 117 families with Mendelian-like segregation of migraine. These families consisted of 874 subjects, 262 of which were diagnosed with MTA, 213 with migraine without aura, 145 with both MTA and migraine without aura, and 254 familial members with no history of migraine. Initially, a migraine patient, i.e. proband, was recruited from the Danish Headache Center and, subsequently, family members were recruited, enabling segregation analysis. The replication cohort consisted of 1930 sporadic migraine patients, 312 of whom were diagnosed with MTA, 1087 were diagnosed with migraine without aura, and 531 were diagnosed with both MTA and migraine without aura; all were recruited from the Danish Headache Center. All participants were assessed by a trained physician or a senior medical student trained in headache diagnostics using a semi-structured interview. The interview was designed by the previous head of classification committee Prof. Jes Olesen to enable diagnosis according to the International Classification of Headache Disorders second edition (ICHD-3) (Olesen, 2006). The study was approved by the Ethics Committee (H-2-2010-122), reported and approved by the data protection agency, and written informed consent was obtained from all participants.
For population controls, we used sequencing data from the 1000 Genomes project (1000G) (Auton et al., 2015). We used the 1000G phase 3 data (VCF files), which included 2500 individuals who were both WGS and whole exome sequenced (WES). When restricting the 1000G cohort to non-Finish Europeans (NFE), 403 individuals were analysed. The NFE classification was provided by 1000G.
Whole genome sequencing
Preparation of samples
Genomic DNA extraction was prepared from whole blood using Illumina TruSeq DNA kit according to the manufacturers’ instructions. In short, 1 µg genomic DNA was isolated from frozen blood and fragmented into 300–400 bp using a Covaris E220 instrument, followed by end repair. AMPure XP magnetic purification beads were then used for size selection followed by PCR enrichment (10 cycles) using appropriate primers. Subsequently, 3′-adenylation and ligation of sequencing adaptors with a T nucleotide overhang was performed and again followed by AMPure magnetic purification. The sequencing libraries were then analysed with regards to quality and concentration using the LabChip® GX (96 samples) instrument from Perkin Elmer. Sequencing libraries were diluted and stored at −20°C. The optimal cluster densities, library insert size, duplication rates and library diversities were assessed by multiplexing on an Illumina MiSeq instrument. All steps in the workflow were monitored using an in-house laboratory information management system (LIMS) with barcode tracking of all samples and reagents.
Whole genome sequencing
Sequencing libraries were hybridized to the surface of paired-end flow cells using the Illumina cBot. Paired-end sequencing-by-synthesis was performed on an Illumina HiSeq X instrument. Read lengths used were 2 × 150 cycles of incorporation and imaging. Real-time analysis involved conversion of image data to base-calling in real-time.
Reference genome and alignment
The human genome assembly GRCh38 was used as reference sequences map reads. The reference did not include alternate assemblies (GCA_000001405.15_GRCh38_no_alt_analysis_set.fna) in addition to sequences determined to represent common contaminants in the sequencing pipeline. Common contaminants in the sequencing pipeline are the sequences of the bacteriophage PhiX (Enterobacteria_phage_phiX174_sensu_lato_uid14015/NC_001422.fna), the bacteria Ralstonia Pickettii (Ralstonia_pickettii_12D_uid58859) and two sequences from the human microbiome (Coprobacillus_D7_uid32495/NZ_EQ999972.fna and Coprobacillus_D7_uid32495/NZ_EQ999922.fna).
Raw sequences were aligned against the reference with BWA v.0.7.10 (Li and Durbin, 2009). The sequences in the BAM files were realigned around indels with GenomeAnalysisTK- Lite/2.3.9 (McKenna et al., 2010) using a public set of known indels. We marked PCR duplicates with Picard tools v.1.117.
Quality control and variant calling
After alignment to reference genome, BAM files were analysed and removed from the analysis based on following criteria: mean base quality >25, per cent marked duplicate >50, mean n per read >30, per cent mapping quality below 20 > 11, per cent reads unmapped >40, per cent both reads in a pair unmapped >40, per cent first read in a pair unmapped >40, per cent second read in a pair unmapped >40. Following filtering, variants were called, which resulted in 5 017 453 ± 2607 variants per individual and 697 044 ± 3244 variants with minor allele frequency (MAF) < 5% (Supplementary Fig. 1B and C) and a mean sequencing depth of 32.94× ± 0.22× (Supplementary Fig. 1A).
WGS and variant calling were performed in collaboration with deCODE genetics®.
RNA sequencing data
RNA sequencing data of aorta (n = 247 individuals) were downloaded from GTEx using the R-package recount (Collado-Torres et al., 2017). Sequencing and alignment are described at http://gtexportal.org. In short, RNA sequencing was performed using the Illumina TruSeq library construction protocol, and sequenced on the Illumina Hiseq2000 or HiSeq2500, generating 76-bp paired-end reads. Reads were aligned to the human reference genome (GRCh38) using STAR v.2.5.3a based on the GENCODE v.26 annotation. Read counts and TPM values were produced with RNA-SeQC.
RNA sequencing data of human trigeminal ganglia (n = 16 individuals) was downloaded from NCBI’s Sequence Read Archive (SRX3020382). Messenger RNA was isolated from small subregions of 16 human trigeminal ganglia, as described previously (LaPaglia et al., 2018). In short, non-stranded cDNA libraries were prepared using the Illumina TruSeq RNA Library Preparation Kit v2, and then sequenced on the Illumina HiSeq2500, generating 125-bp paired-end reads. The quality of the fastq-files was checked using the NGSQC toolkit and low quality reads were filtered out according to default settings (cut-off read length of 70% and a cut-off quality score of 20). On average, per sample, 85.2% of the reads were of high quality resulting in on average 26.8 million reads [standard deviation (SD) = 1.9 million reads]. Filtered fastq files were aligned to the human reference genome (Release 32, GRCh38.p13) using the pseudo-aligner Kallisto v.0.46.0. Length-scaled transcripts per million were imported into R using tximport v.3.10.
RNA sequencing data of the human primary visual cortex were downloaded from Allen Human Brain Map (n = 8998) and technical details are described in the technical white paper: transcriptomics (Allen Brain Institute, 2018). In short, 8998 single nuclei were isolated from six cortical layers of visual cortex (n = 8 individuals). The Nextera XT kit (Illumina) was used to prepare cDNA libraries. Single nucleus libraries were then sequenced on the Illumina HiSeq2500, generating 50-bp paired-end reads. Reads were aligned to the human reference genome (GRCh38.p2) using STAR v.2.5.3. Final results included quantification of exon and intron counts. We merged exon and intron data by summing exon and intron counts at gene level.
For all tissues, we included only protein-coded genes, using a gene list extracted from BioMart (GRCh38.p18). This resulted in counts of 19 747, 18 299 and 16 665 genes in trigeminal ganglion, visual cortex and aorta, respectively.
Construction of co-expression networks
A co-expression network was constructed of each tissue, i.e. aorta, trigeminal ganglion and visual cortex. Genes for network construction were selected based on their connectivity (sum of correlations of a gene with all other genes); the 5000 most highly connected genes per tissue were retained. A network was constructed based on the default pipeline of WGCNA (Zhang et al., 2005; Langfelder and Horvath, 2008). To ensure scale-free topology, the correlation network was raised to a power (beta) to reach a scale-free topology measure (R2) >0.8. To ensure a stable and highly reliable network, the adjacency matrix and consequently dissimilarity topological overlap matrix (TOM) were calculated 100 times by leaving 10% of the samples out every time. The average dissimilarity TOM was calculated from the 100 networks. Next, a gene dendrogram was created and modules containing at least 30 genes were detected using the dynamic tree cutting algorithm. Modules are defined as clusters of highly co-expressed genes. Modules were validated using a permutation procedure as described by Iancu et al. (2012). For each network, we randomly selected groups of genes with the same size of the detected modules in the network and computed the average topological overlap. All modules in the aorta, trigeminal ganglion and visual cortex networks had a significantly higher topological overlap than the random modules, except for the ‘grey’ module (which includes unassigned genes). This confirmed that we detected modules with statistical and potentially functional relevance for the tissues.
Detection of migraine-associated modules
Detection of mutated genes in migraine families
We used the pedigree variant annotation, analysis and search tool (pVAAST) v.2.2.0 (Hu et al., 2014) to detect genes with rare functional mutations segregating with migraine. We performed a test for each gene in the modules for each of the 117 families.
Genes were identified with pVAAST based on the following criteria: (i) genes were identified based on the penetrance level (0.9–1) in each family; and (ii) genes were identified based on the functionality of variants only giving rise to frameshift deletions, frameshift insertions, non-frameshift deletions, non-frameshift insertions, non-synonymous single nucleotide polymorphisms (SNPs), splice site mutations, stop-gain and stop-loss mutations. We refer to these functional variants as mutations from here on. Third, genes were identified based on mutations having a MAF < 5% in the genome aggregation database (gnomAD) v.2. See Supplementary Fig. 3A–C for MAF distribution of all variants in the three tissues analysed. The rationale for choosing a MAF < 0.05% was based on the prevalence of migraine being 1:5 to 1:6, biallelic, a genetic heterogeneity of 1, several scenarios of allelic heterogeneity (between 0.01 and 0.05) gave rise to MAF up to 5%, thus a MAF cut-off of 5% was chosen. The cut-off was calculated using the allele frequency app https://www.cardiodb.org/allelefrequencyapp/ (Whiffin et al., 2017).
Genes with rare functional mutations are hereafter termed mutated genes. Genes were termed non-mutated when at least one variant could be detected that was not a rare functional variant.
Genic intolerance analysis of modules
The mutation tolerance is gene dependent, thus we calculated the mean residual variation intolerance score (RVIS) (Petrovski et al., 2013) for genes with a rare functional mutation (mutated genes) and for genes without rare functional mutations (non-mutated genes) per module. A one-sided t-test was performed to exclude modules where the mutated genes had a significantly higher mean RVIS than the non-mutated genes (P < 0.05).
Detection of migraine-associated modules in families
For each tissue, modules with more mutated genes than expected by chance were detected by a one-tailed Fisher’s exact test, i.e. the number of mutated genes versus non-mutated genes in a module against number of mutated genes versus non-mutated genes in all other modules. We only included genes in the Fishers’ exact test in which genetic variations were detectable. Therefore, the number of genes included in Fishers’ exact test for familial migraine were: 4894 in aorta, 4652 in trigeminal ganglion, and 4879 in visual cortex. P-values were adjusted using Bonferroni correction for the number of modules (trigeminal ganglion: n = 39, visual cortex: n = 8, aorta: n = 16) and defined as migraine-associated when Padj < 0.05.
Replication of migraine-associated modules in sporadic migraine
Using an independent cohort of 1930 sporadic migraine patients, i.e. with no known history of first-degree relatives with migraine, and using the 1000G control cohort, we assessed the migraine-associated modules found in the migraine family cohort using ANNOVAR (v.2018apr16). ANNOVAR was run using VCF files to annotate genetic variants in mRNAs and to annotate MAF of those genetic variants. We used refSeq and gnomAD v.2 as input databases for ANNOVAR. Module genes with a MAF < 5% were analysed for mutations as defined above. See Supplementary Fig. 3D–F for MAF distribution of mutations. Modules with more mutated genes than expected by chance were determined using one-tailed Fisher’s exact test, i.e. the number of mutated genes versus non-mutated genes in a module against number of mutated genes versus non-mutated genes in all other modules. We only included genes in the Fishers’s exact test in which we could detect genetic variation. Therefore, the number of genes included in the test were for sporadic migraine: 4871 in trigeminal ganglion, 4973 in visual cortex and 4956 in aorta. For 1000G the gene counts were: 3201 in trigeminal ganglion, 2937 in visual cortex and 3251 in aorta. To avoid biases derived from different data processing and batches, the Fisher’s tests were carried out separately for cases and controls. Modules that had significantly more mutated genes in cases but not controls were considered replication.
Functional annotation of migraine-associated modules
Visualization
The Protein-Protein-interaction network was depicted using stringApp v.1.5.0 in Cytoscape v.3.7.2 (Shannon et al., 2003) assessing the protein products and interaction thereof having the threshold for the interaction confidence score > 0.4. Protein interaction data v.11 from www.string-db.org were used as input for Cytoscape.
Pathway analysis
We carried out pathway analysis using PANTHER over-representation test (data version released 20190711) (Mi et al., 2013). The total set of genes used for network construction of the tissue of interest was used as reference. Pathways were tested for over-representation using the binomial test and P-values were adjusted for multiple-testing using FDR correction and defined as significant in case Padj < 0.05.
Enrichment analysis in single cell RNA sequencing data
We used two different approaches to investigate modules with increased mutation burden segregating in families. First, we used single cell RNA sequencing data from mouse cortex and hippocampus for cell-type enrichment testing (Zeisel et al., 2015), allowing for differentiation between six different cell types: astrocytes, interneurons, endothelial cells, microglia, oligodendrocytes, pyramidal CA1 neurons and pyramidal SS neurons. Enrichment analysis was carried out using the Expression Weighted Celltype Enrichment (EWCE) R package (Skene and Grant, 2016) with 10 000 bootstraps for genes in the significant migraine-associated modules.
Data availability
WGS data are considered personal and sensitive data by the scientific ethical committee, thus data can only be shared as part of material agreed upon reasonable request.
Results
We constructed gene co-expression networks for three tissues known to be involved migraine aetiopathology: the aorta, the trigeminal ganglia, and the visual cortex. For each tissue we selected the 5000 most highly connected genes for network construction; 961 genes were present in all three networks (Fig. 2A and Supplementary Table 1). From the derived co-expression networks, we identified 34 modules in the aorta network, 64 modules in trigeminal ganglia and eight modules in visual cortex (Supplementary Fig. 2 and Supplementary Table 1). Though the aorta and trigeminal ganglion networks are based on tissues, the visual cortex network is based on single nuclei data and, therefore, modules may represent subgroups of cell types. Cell type enrichment analysis showed that, for example, the green module was over-represented by genes expressed in interneurons, though other modules (e.g. blue) were covered by different cell types (Fig. 2E).
Figure 2.

Overview of genes and mutations in trigeminal ganglion, visual cortex and aorta. (A and B) Number of genes and mutated genes in the trigeminal ganglia (TG), visual cortex (VC) network and aorta. (C) Module overlap based on the mutated genes in both trigeminal ganglia, visual cortex and aorta. (D) Distribution of mutations in mutated genes in trigeminal ganglion, visual cortex and aorta. (E) Gene-set enrichment analysis using brain single-cell RNA sequencing data. *P < 0.05.
We used WGS data from our cohort of families with Mendelian-like segregation pattern of migraine, to identify migraine-associated modules, with over-representation of mutated genes. We excluded gene modules that had over-representation of highly mutation-tolerant genes, i.e. type I error (Supplementary Fig. 4). From the aorta network, 16 modules remained for analysis, 39 in trigeminal ganglia and eight in visual cortex.
We identified 313 mutated genes in aorta, 875 mutated genes in trigeminal ganglia, and 1243 mutated genes in visual cortex, of which 13 were present in all three networks (Fig. 2A and B). We found no significant overlap of mutated genes between the three networks (Fig. 2C). The rare mutations consisted primarily of non-synonymous SNPs (86.59–90.13%), while 0.54–5.43% are high impact mutations (frameshift mutations, splice site mutations and stop-loss/stop-gain mutations) (Fig. 2D).
Selection of migraine-associated modules
As hypothesized, genes with rare mutations are more likely to aggregate in pathways specific for migraine mechanisms. Such genes are likely to be enriched within our constructed networks, based on the expression profile of migraine-relevant tissue. In the aorta network we found one migraine-associated module (Fig. 3A, darkgreen), in the trigeminal ganglion network we found five migraine-associated modules (yellow, salmon, honeydew1, darkturquoise and darkslateblue), and in the visual cortex network we found three migraine-associated modules (blue, brown, yellow) (Fig. 3A).
Figure 3.

Identification of migraine-associated modules. (A) Over-representation of mutated genes for familial migraine within modules of the trigeminal ganglia, the visual cortex and in the aorta network. *Modules with an over-representation of mutated genes in familial migraine than are assessed for replication analysis. (B) Replication in sporadic migraine (Spo. mig.) compared to 1000G. The odds ratio (OR) is displayed for each module and the error bars show the 95% confidence interval.
We were able to replicate the brown module (visual cortex), showing an increased burden of mutated genes in our sporadic migraine cohort and not in the 1000G cohort. We were not able to replicate any of the other modules, although the yellow module (trigeminal ganglia) was significant in the sporadic migraine it was also significantly increased in the 1000G cohort (Fig. 3B). Restricting the 1000G cohort to non-Finish Europeans did not affect the results (Supplementary Fig. 5A–C).
We analysed the visual cortex brown module and the trigeminal ganglion yellow module on a protein level. The majority (82.5%) of the proteins in the visual cortex brown module were participating in the network, suggesting a strong functional module network (Fig. 4A). The trigeminal ganglion yellow module was divided into three smaller networks covering 5.7–32.1% of the proteins, indicating a less strong functional module network (Fig. 4B).
Figure 4.
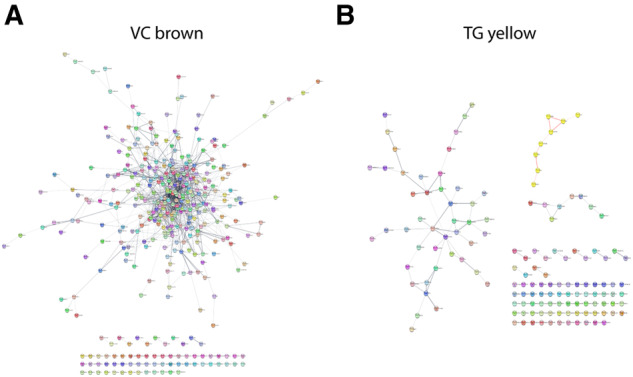
Analysis of modules on protein level. (A) Network of proteins encoded by genes in the brown module (visual cortex, VC). A large fraction of the proteins contribute to the network. (B) Three networks of proteins encoded by genes in the yellow (trigeminal ganglia, TG) module are depicted.
Functional annotation of migraine-associated modules
Cell type enrichment analysis of the migraine-associated module (visual cortex brown) showed that the module was mainly expressed by interneurons, pyramidal CA1, and pyramidal SS cells (Fig. 2E). Furthermore, five pathways were over-represented: the thyrotropin-releasing hormone receptor (TSHR) signalling pathway, oxytocin receptor (OXTR) mediated signalling pathway, Alzheimer’s disease amyloid secretase pathway, serotonin receptor (5HT2R) signalling pathway, and heterotrimeric G-protein signalling pathway-Gq alpha and Go alpha mediated pathway (Table 1). The distribution of mutated genes co-segregating with migraine across families for the module showed that 71 genes have mutations in one family whereas 126 genes harbour functional mutations in two or more families (Fig. 5). Several genes were mutated frequently among families. The top three genes included ATXN1, mutated in 28 families; FAM153B, mutated in 19 families; and CACNA1B, mutated in 16 families.
Table 1.
Pathways detected in the visual cortex brown module that was over-represented for mutations
| Module | Mutated genes | Pathways | P-value |
|---|---|---|---|
| VC brown | 197 | Thyrotropin-releasing hormone receptor signalling pathway | 0.046 |
| Oxytocin receptor mediated signalling pathway | 0.045 | ||
| Alzheimer’s disease-amyloid secretase pathway | 0.040 | ||
| 5HT2 type receptor mediated signalling pathway | 0.040 | ||
| Heterotrimeric G-protein signalling pathway-Gq alpha and Go alpha mediated pathway | 0.017 |
Number of mutated genes in families is listed as well as corrected P-values.
Figure 5.
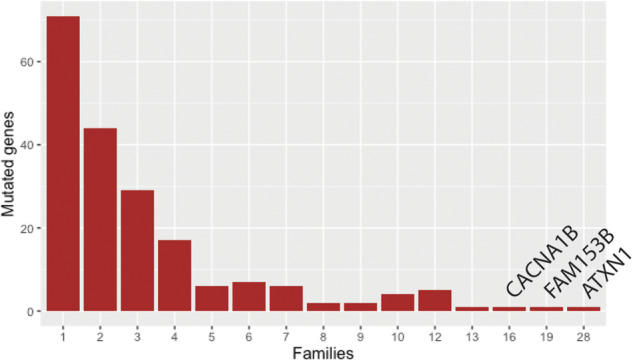
Count of mutated genes in visual cortex brown and in how many families the genes co-segregate with migraine.
Discussion
We present a systems genetics approach integrating WGS data from 117 families with Mendelian-like segregation patterns of migraine with RNA sequencing data from tissues involved in migraine aetiopathology: trigeminal ganglia, visual cortex, and aorta. After replication in an independent cohort of 1930 sporadic cases of migraine, we identified one module with a significant accumulation of mutated genes. The results propose distinct mechanisms affected in migraine, including the TSHR signalling pathway, OXTR signalling pathway, Alzheimer’s disease amyloid secretase pathway, 5HT2R signalling pathway and G-protein signalling pathway combined with activity of interneurons, pyramidal CA1 and SS neurons. Further, we were able to suggest specific genes with mutations to be involved in migraine aetiology.
The co-expression approach has been applied to migraine previously. Focusing on common risk variants and using different brain tissues (Eising et al., 2016). The authors allocated migraine-associated genes to modules across brain tissues, showing that oligodendrocytes and gene transcription regulation may play a causal role in migraine. We have shown a similar approach with two important differences. First, our networks are constructed using human RNA sequencing data from the tissues probably involved in migraine aetiopathology, trigeminal ganglia, visual cortex, and aorta. Second, we integrated the co-expression networks with mutations found segregating in a large family cohort.
WGCNA is a well-established method for studying biological networks and diseases. We used it to identify gene modules in three migraine-relevant tissues. Like WGCNA, WGS is also a well-known resource in disease research. Over recent years, the cost of having a genome sequenced has dropped substantially such that it is now possible to sequence entire cohorts (Hayden, 2014). WGS has an advantage over, for example, genotyping since rare variants and de novo variants are included in the sequencing. We have—for the first time—utilized both methods in an attempt to reveal more of the defective biological mechanisms leading to migraine. Common variants do not explain 100% of the heritability of migraine (Gormley et al., 2016). Integrating RNA sequencing data via WGCNA with WGS, we have closed some of the gap between how much of the phenotype can be explained by common variants and now by rare mutations (MAF < 0.05%).
Serotonin and the serotonin signalling pathway have, for many years, been known for their implications in migraine. Serotonin levels are believed to increase during migraine attacks (Sicuteri et al., 1961) and the family of serotonin receptor agonists—triptans—are the preferred migraine treatment (Dussor, 2014). Serotonin binds to serotonin receptors, which comprise both metabotropic and ionotropic receptors. The two families of metabotropic serotonin receptors: 5HT1 and 5HT2, have both been associated with migraine (Hill and Reynolds, 2007; Ferrari et al., 2010; Nelson et al., 2010; Thompson et al., 2012; Yücel et al., 2016) as well as the serotonin transporter (Ogilvie et al., 1998; Marziniak et al., 2005; Bayerer et al., 2010). We directly associate 5HT2 with migraine in the visual cortex brown module as well as the G-protein-mediated signalling pathway including Go, which signals downstream of the 5HT1 receptor. Moreover, we found an association of the visual cortex brown module gene-set to neurons in our EWCE analysis. Cortical neuronal activity initiates and propagates cortical spreading depression (Leao, 1944), which has been linked to serotonergic activity in functional studies (Supornsilpchai et al., 2006; Guedes et al., 2017). We suggest that the migraine phenotype may be caused by mutated genes in the serotonin signalling pathway as well as the downstream G-protein coupled signalling pathway.
In addition to serotonergic neurons, glutamatergic neurons and synapses have also been linked to migraine through different association studies where SNPs in genes related to glutamate transport or signalling have been detected (Shin et al., 2011; Gormley et al., 2016). In addition, glutamate levels have been increased in migraineurs (Ferrari et al., 1990; Martínez et al., 1993; Campos et al., 2013). Glutamate plays a central role in cortical spreading depression as an activator and propagator and inversely, inhibition of glutamate receptors impairs initiation and propagation of cortical spreading depression (Lauritzen and Hansen, 1992; Peeters et al., 2007; Zhou et al., 2013). In our analysis, we detected three genes (ATXN1, FAM153B and CACNA1B) that were mutated in >15 families. CACNA1B and ATXN1 are both involved in glutamate signalling. CACNA1B encodes a subunit in a voltage-gated calcium channel, which is expressed at the presynapse and involved in release of glutamate from the presynapse (Bunda et al., 2019). ATXN1 encodes a DNA binding protein, which is able to regulate expression of the metabotropic glutamate receptor 1 (mGluR1) at the postsynaptic membrane (Notartomaso et al., 2013). Thereby the glutamatergic signalling is altered on both sides of the synaptic cleft in our migraine patients and may contribute to the migraine pathophysiology. CACNA1B is a member of a family of genes, which includes CACNA1A, that has been linked to a monogenic subtype of MTA, namely familial hemiplegic migraine (FHM) (Ophoff et al., 1996). FHM is a more severe phenotype than MTA and patients with FHM can experience ataxia symptoms. Ataxia is a complex disease and multiple genes have been linked to the disorder including ATXN1. The knowledge regarding FAM153B is limited and no function of the encoded protein has been reported. According to the GTEX data, FAM153B is highly expressed in the brain and likely has a vital function in neurons, which may be affected by mutations (Lonsdale et al., 2013).
Oxytocin and thyrotropin are both hormones that regulate the activity of OXTR and TSHR. Both receptors are G-protein coupled receptors expressed throughout the brain; however, TSHR is expressed in lower amounts than OXTR in the brain according to GTEX data (Lonsdale et al., 2013; Warfvinge et al., 2020). Oxytocin has been the subject of migraine research as it is a strong analgesic in neuroinflammation (Yu et al., 2003; Yang et al., 2007; Engle et al., 2012; Kubo et al., 2017). Moreover, oxytocin is increased during pregnancy, which inversely correlates to the decrease in headaches and migraine of pregnant females (Callaghan, 1968; Kuwabara et al., 1987; Goadsby et al., 2008; Hoshiyama et al., 2012). Oxytocin levels also increase during intercourse and orgasms for both males and females. For the latter, it is reported that 47% experience relief from migraine during sex (Carmichael et al., 1987; Evans and Couch, 2001). Clinical testing of oxytocin in migraine patients led to a long term (24 h) effect on pain after dosing. In the course of 8 weeks, oxytocin had an effect on the migraine frequency (Tzabazis et al., 2017). Thyrotropin is less studied with respect to migraine compared to oxytocin. However, high levels of thyrotropin were associated with lower occurrences of headache (Hagen et al., 2001) and low levels of thyrotropin may be associated with a prolonged and refractory clinical course of migraine (Starikova et al., 2019). Taken together, hormone receptor misregulation seems to play a role in migraine. Our data point to a dysregulation in the hormone receptor pathways, which most likely derives from mutations affecting proteins active downstream from oxytocin and thyrotropin receptors.
Finally, we detected the Alzheimer’s disease amyloid secretase pathway in the visual cortex brown module. It has been suggested that migraine may be associated with mild types of cognitive changes (Araújo et al., 2012) but there is no clear evidence showing whether or not migraine is a risk factor for Alzheimer’s disease (Breteler et al., 1991; Wang et al., 2018; Lee et al., 2019; Morton et al., 2019).
Strengths and limitations
In this study we used a novel approach by combining gene-gene interaction networks based on RNA sequencing data with rare mutations found in families using WGS data. The co-expression networks enable investigation of biological mechanisms in migraine-relevant tissues. The construction of those networks is affected by the origin and quality of the RNA sequencing data, i.e. inter-tissue comparison is not possible becuase of, for example, their origin, bulk versus single cell, and sequencing depth. The modules revealed in the aorta and trigeminal ganglia data are driven by differences in gene expression levels between individuals, while in the visual cortex network, using single nuclei data, modules are driven by differences in cell types. Interpretation of pathways detected in visual cortex, therefore, needs to be done with caution. Similarly, thresholds applied to mutation calling affect results. We have analysed mutations with a MAF < 0.05 in both familial and sporadic migraine. The recent migraine GWAS included variants with a MAF as low as 0.01 (Gormley et al., 2016). Our study may, therefore, overlap with respect to these variants. However, we believe it is a strength to include variants with a MAF up to 0.05 because of the high prevalence of migraine in the population.
The differences in the number of genes having genetic variants in the families was marginal, which is also reflected by the mean coverage of the genome. The difference between the replication cohorts most likely derives from different sample handling and sequencing, hence the separate assessment of the sporadic migraine cohort and 1000G cohort. Thus, a more ideal comparison of having controls from the same population and process together would have been preferred.
Our familial migraine cohort has a large fraction of migraine patients with aura. This is due to the initial recruiting of our patients and may be considered as a limitation. However, our replication cohort of sporadic migraine has a small fraction of MTA patients compared to migraine without aura patients. Despite the fact that there are no differences in clinical presentation between familial migraine and sporadic migraine, our results may be slightly biased towards MTA since we initially analysed our familial cohort (Ravn et al., 2019). If more familial migraine without aura patients were included in future studies resembling our study, it could perhaps result in more gene modules implicated in the migraine biology.
Conclusion
Migraine-associated mechanisms derived from a visual cortex module exist and are likely to contribute to the migraine pathophysiology. Our data support previous findings that the serotonin, glutamate, hormonal and G-protein signalling pathways are involved in migraine. We suggest that a dysfunction in neurotransmitter and hormone signalling is part of migraine pathophysiology.
Supplementary Material
Acknowledgements
We thank all participating patients and the employees at the Danish Headache Center for their help during the recruitment of the patients. We would also like thank Kari Stefansson and Hreinn Stefansson from deCODE Genetics, Iceland Reykjavik for genetic analysis.
Funding
This project was financed by a grant from Candys Foundation “CEHEAD” (J.O.).
Competing interests
The authors report no competing interests.
Glossary
- MAF =
minor allele frequency
- MTA =
migraine with typical aura
- WGCNA =
weighted gene co-expression network analysis
- WGS =
whole genome sequencing
References
- Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2018; 38: 1–211. [DOI] [PubMed] [Google Scholar]
- Allen Brain Institute. Allen Cell Types Database Transcriptomics. 2018: 1–15. http://help.brain-map.org/download/attachments/8323525/CellTypes_Transcriptomics_Overview.pdf (10 April 2018, date last accessed).
- Araújo CD, Barbosa IG, Lemos SMA, Domingues RB, Teixeira AL.. Cognitive impairment in migraine: a systematic review. Dement Neuropsychol 2012; 6: 74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature 2015; 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros J, Damásio J, Tuna A, Alves I, Silveira I, Pereira-Monteiro J, et al. Cerebellar ataxia, hemiplegic migraine, and related phenotypes due to a CACNA1A Missense mutation. JAMA Neurol 2013; 70: 235. [DOI] [PubMed] [Google Scholar]
- Bayerer B, Engelbergs J, Savidou I, Boes T, Küper M, Schorn CF, et al. Single nucleotide polymorphisms of the serotonin transporter gene in migraine-an association study. Headache J Head Face Pain 2010; 50: 319–22. [DOI] [PubMed] [Google Scholar]
- Breteler MM, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Medical history and the risk of Alzheimer’s disease: a collaborative re-analysis of case-control studies Int J Epidemiol 1991; 20: S36-42. [DOI] [PubMed] [Google Scholar]
- Brighina F, Bolognini N, Cosentino G, MacCora S, Paladino P, Baschi R, et al. Visual cortex hyperexcitability in migraine in response to sound-induced flash illusions. Neurology 2015; 84: 2057–61. [DOI] [PubMed] [Google Scholar]
- Bunda A, Lacarubba B, Bertolino M, Akiki M, Bath K, Lopez-Soto J, et al. Cacna1b alternative splicing impacts excitatory neurotransmission and is linked to behavioral responses to aversive stimuli. Mol Brain 2019; 12: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan N. The migraine syndrome in pregnancy. Neurology 1968; 18: 197–201. [DOI] [PubMed] [Google Scholar]
- Campos F, Sobrino T, Pérez-Mato M, Rodríguez-Osorio X, Leira R, Blanco M, et al. Glutamate oxaloacetate transaminase: a new key in the dysregulation of glutamate in migraine patients. Cephalalgia 2013; 33: 1148–54. [DOI] [PubMed] [Google Scholar]
- Carmichael MS, Humbert R, Dixen J, Palmisano G, Greenleaf W, Davidson JM.. Plasma oxytocin increases in the human sexual response. J Clin Endocrinol Metab 1987; 64: 27–31. [DOI] [PubMed] [Google Scholar]
- Charles A. The migraine aura. Continuum (Minneap Minn). 2018; 24: 1009–22. [DOI] [PubMed] [Google Scholar]
- Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, et al. Reproducible RNA-seq analysis using recount2. Nat Biotechnol 2017; 35: 319–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Parisi V, Di Lorenzo C, Serrao M, Magis D, Schoenen J. Lateral inhibition in visual cortex of migraine patients between attacks. J Headache Pain 2013; 14: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005; 366: 371–7. [DOI] [PubMed] [Google Scholar]
- Dussor G. Serotonin, 5HT1 agonists, and migraine: new data, but old questions still not answered. Curr Opin Support Palliat Care 2014; 2: 137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising E, H Huisman SM, Mahfouz A, Vijfhuizen LS, F Lelieveldt BP, J M van den Maagdenberg AM, et al. Gene co-expression analysis identifies brain regions and cell types involved in migraine pathophysiology: a GWAS-based study using the Allen Human Brain Atlas. Hum Genet 2016; 135: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eising E, Pelzer N, Vijfhuizen LS, Vries B, De Ferrari MD, THoen PAC, et al. Identifying a gene expression signature of cluster headache in blood. Sci Rep 2017; 7: 40218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle MP, Ness TJ, Robbins MT. Intrathecal oxytocin inhibits visceromotor reflex and spinal neuronal responses to noxious distention of the rat urinary bladder. Reg Anesth Pain Med 2012; 37: 515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RW, Couch JR. Orgasm and migraine. Headache 2001; 41: 512–4. [DOI] [PubMed] [Google Scholar]
- Feng Y, Wang X.. Systematic analysis of microarray datasets to identify Parkinson’s disease-associated pathways and genes. Mol Med Rep 2017; 15: 1252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari MD, Färkkilä M, Reuter U, Pilgrim A, Davis C, Krauss M, et al. Acute treatment of migraine with the selective 5-HT1F receptor agonist Lasmiditan: a randomised proof-of-concept trial. Cephalalgia 2010; 10: 1170–8. [DOI] [PubMed] [Google Scholar]
- Ferrari MD, Odink J, Bos KD, Malessy M, Bruyn GW.. Neuroexcitatory plasma amino acids are elevated in migraine. Neurology 1990; 40: 1582–6. [DOI] [PubMed] [Google Scholar]
- Gaist D, Hougaard A, Garde E, Reislev NL, Wiwie R, Iversen P, et al. Migraine with visual aura associated with thicker visual cortex. Brain 2018; 141: 776–85. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Goldberg J, Silberstein SD.. Migraine in pregnancy. BMJ 2008; 336: 1502–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 2016; 48: 856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley P, Kurki MI, Hiekkala ME, Veerapen K, Häppölä P, Mitchell AA, et al. Common variant burden contributes to the familial aggregation of migraine in 1,589 families. Neuron 2018; 99: 1098. [DOI] [PubMed] [Google Scholar]
- Guedes RCA, Araújo M. D G D, Verçosa TC, Bion FM, de Sá AL, Pereira A, et al. Evidence of an inverse correlation between serotonergic activity and spreading depression propagation in the rat cortex. Brain Res 2017; 1672: 29–34. [DOI] [PubMed] [Google Scholar]
- Hagen K, Bjøro T, Zwart J. A, Vatten L, Stovner LJ, Bovim G.. Low headache prevalence amongst women with high TSH values. Eur J Neurol 2001; 8: 693–9. [DOI] [PubMed] [Google Scholar]
- Hansen RD, Christensen AF, Olesen J.. Family studies to find rare high risk variants in migraine. J Headache Pain 2017; 18: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden EC. The $1,000 genome. Nature 2014; 507: 294–5. [DOI] [PubMed] [Google Scholar]
- Hill MJ, Reynolds GP.. 5-HT2C receptor gene polymorphisms associated with antipsychotic drug action alter promoter activity. Brain Res 2007; 1149: 14–7. [DOI] [PubMed] [Google Scholar]
- Hoshiyama E, Tatsumoto M, Iwanami H, Saisu A, Watanabe H, Inaba N, et al. Postpartum migraines: a long-term prospective study. Intern Med 2012; 51: 3119–23. [DOI] [PubMed] [Google Scholar]
- Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL, et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol 2014; 32: 663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Kawane S, Bottomly D, Searles R, Hitzemann R, McWeeney S.. Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics 2012; 28: 1592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo A, Shinoda M, Katagiri A, Takeda M, Suzuki T, Asaka J, et al. Oxytocin alleviates orofacial mechanical hypersensitivity associated with infraorbital nerve injury through vasopressin-1A receptors of the rat trigeminal ganglia. Pain 2017; 158: 649–59. [DOI] [PubMed] [Google Scholar]
- Kuwabara Y, Takeda S, Mizuno M, Sakamoto S.. Oxytocin levels in maternal and fetal plasma, amniotic fluid, and neonatal plasma and urine. Arch Gynecol Obstet 1987; 241: 13–23. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPaglia DM, Sapio MR, Burbelo PD, Thierry-Mieg J, Thierry-Mieg D, Raithel SJ, et al. RNA-Seq investigations of human post-mortem trigeminal ganglia. Cephalalgia 2018; 38: 912–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ.. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab 1992; 12: 223–9. [DOI] [PubMed] [Google Scholar]
- Leao A. Spreading depression of activity in the cerebral cortex. J Neurophysiol 1944; 7: 359–90. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lim JS, Oh DJ, Kong IG, Choi HG, Increased risk of neurodegenerative dementia in women with migraines: A nested case-control study using a national sample cohort. Medicine (United States) 2019; 98: e14467. [DOI] [PMC free article] [PubMed]
- Li H, Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Huang K, Huang R, Liu X, Han C, Yu L, et al. Genome-scale analysis to identify prognostic markers in patients with early-stage pancreatic ductal adenocarcinoma after pancreaticoduodenectomy. OncoTagets Ther 2017; 10: 4493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013; 45: 580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini C, Conti V, Mei D, Battaglia D, Lettori D, Losito E, et al. PRRT2 mutations in familial infantile seizures, paroxysmal dyskinesia, and hemiplegic migraine. Neurology 2012; 79: 2109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez F, Castillo J, Rodríguez JR, Leira R, Noya M.. Neuroexcitatory amino acid levels in plasma and cerebrospinal fluid during migraine attacks. Cephalalgia 1993; 13: 89–93. [DOI] [PubMed] [Google Scholar]
- Marziniak M, Mössner R, Schmitt A, Lesch KP, Sommer C.. A functional serotonin transporter gene polymorphism is associated with migraine with aura. Neurology 2005; 64: 157–9. [DOI] [PubMed] [Google Scholar]
- Marzoli SB, Criscuoli A.. The role of visual system in migraine. Neurol Sci 2017; 38: 99–102. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K, Russo AF.. Current understanding of trigeminal ganglion structure and function in headache. Cephalalgia 2018; 39: 1661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD.. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc 2013; 8: 1551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton RE, St. John PD, Tyas SL.. Migraine and the risk of all-cause dementia, Alzheimer’s disease, and vascular dementia: a prospective cohort study in community-dwelling older adults. Int J Geriatr Psychiatry 2019; 34: 1667–76. [DOI] [PubMed] [Google Scholar]
- Nelson DL, Phebus LA, Johnson KW, Wainscott DB, Cohen ML, Calligaro DO, et al. Preclinical pharmacological profile of the selective 5-HT1F receptor agonist lasmiditan. Cephalalgia 2010; 10: 1159–69. [DOI] [PubMed] [Google Scholar]
- Notartomaso S, Zappulla C, Biagioni F, Cannella M, Bucci D, Mascio G, et al. Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol Brain 2013; 6: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie AD, Russell MB, Dhall P, Battersby S, Ulrich V, Smith CAD, et al. Altered allelic distributions of the serotonin transporter gene in migraine without aura and migraine with aura. Cephalalgia 1998; 18: 26. [DOI] [PubMed] [Google Scholar]
- Olesen J. International Classification of Headache Disorders, Second Edition (ICHD-2): current status and future revisions. Cephalalgia 2006; 26: 1409–10. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, Van Eijk R, Oefner PJ, Hoffman SMG, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996; 87: 543–52. [DOI] [PubMed] [Google Scholar]
- Peeters M, Gunthorpe MJ, Strijbos P, Goldsmith P, Upton N, James MF.. Effects of pan- and subtype-selective N-methyl-D-aspartate receptor antagonists on cortical spreading depression in the rat: therapeutic potential for migraine. J Pharmacol Exp Ther 2007; 321: 564–72. [DOI] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB.. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet 2013; 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA, Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet 2016; 48: 709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrebic S, Ahn AH, Skinner K, Fields HL, Basbaum AI. Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci 2003; 23: 10988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravn J, Chalmer MA, Oehrstroem EL, Kogelman LJA, Hansen TF, Characterization of Familial and Sporadic Migraine. Headache J Head Face Pain 2019; 59: 1802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelukhina I, Mikhailov N, Abushik P, Nurullin L, Nikolsky EE, Giniatullin R.. Cholinergic nociceptive mechanisms in rat meninges and trigeminal Ganglia: potential implications for migraine pain. Front Neurol 2017; 27: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HE, Han SJ, Lee KS, Park JW.. Polymorphism of the glutamate transporter protein EAAT2 and migraine transformation into chronic daily headache. J Clin Neurol 2011; 7: 143–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicuteri F, Testi A, Anselmi B.. Biochemical investigations in headache: increase in the hydroxyindoleacetic acid excretion during migraine attacks. Int Arch Allergy Immunol 1961; 19: 55–8. [Google Scholar]
- Skene NG, Grant S.. Identification of vulnerable cell types in major brain disorders using single cell transcriptomes and expression weighted cell type enrichment. Front Neurosci 2016; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starikova NL, Baidina TV, Kalashnikova TP.. Thyrotropin levels and severity of symptoms in migraine patients of tertiary headache center. Cephalalgia 2019; 39: 148–52. [DOI] [PubMed] [Google Scholar]
- Supornsilpchai W, Sanguanrangsirikul S, Maneesri S, Srikiatkhachorn A.. Serotonin depletion, cortical spreading depression, and trigeminal nociception. Headache 2006; 46: 34–9. [DOI] [PubMed] [Google Scholar]
- Sutherland HG, Griffiths LR.. Genetics of migraine: insights into the molecular basis of migraine disorders. Headache J Head Face Pain 2017; 57: 537–69. [DOI] [PubMed] [Google Scholar]
- Szabó N, Faragó P, Király A, Veréb D, Csete G, Tóth E, et al. Evidence for plastic processes in migraine with aura: a diffusion weighted MRI study. Front Neuroanat 2018; 11: 2381–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MD, Noble-Topham S, Percy ME, Andrade DM, Ebers GC.. Chromosome 1p36 in migraine with aura: association study of the 5HT 1D locus. Neuroreport 2012; 23: 45–8. [DOI] [PubMed] [Google Scholar]
- Tzabazis A, Kori S, Mechanic J, Miller J, Pascual C, Manering N, et al. Oxytocin and migraine headache. Headache 2017; 57: 64–75. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu W, Sun S, Yu S, Fan L.. Headache disorder and the risk of dementia: a systematic review and meta-analysis of cohort studies. J Headache Pain 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, He X, Liu X, Liu Y, Zhang W, Huang Q, et al. Weighted gene co-expression network analysis identifies FKBP11 as a key regulator in acute aortic dissection through a NF-kB dependent pathway. Front Physiol 2017; 8: 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfvinge K, Krause D, Edvinsson L.. The distribution of oxytocin and the oxytocin receptor in rat brain: relation to regions active in migraine. J Headache Pain 2020; 21: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med 2017; 19: 1151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Yang Y, Chen JM, Liu WY, Wang CH, Lin BC.. Central oxytocin enhances antinociception in the rat. Peptides 2007. [DOI] [PubMed] [Google Scholar]
- Yu SQ, Lundeberg T, Yu LC, Involvement of oxytocin in spinal antinociception in rats with inflammation. Brain Res 2003; 983: 13–22. [DOI] [PubMed] [Google Scholar]
- Yücel Y, Coskun S, Cengiz B, Özdemir HH, Uzar E, Çim A, et al. Association of Polymorphisms within the serotonin receptor genes 5-HTR1A, 5-HTR1B, 5-HTR2A and 5-HTR2C and migraine susceptibility in a Turkish population. Clin Psychopharmacol Neurosci 2016; 14: 250–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lönnerberg P, Manno GL, Juréus A, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science (80-) 2015; 347: 1138–42. [DOI] [PubMed] [Google Scholar]
- Zhang X, Burstein R, Jakubowski M, Noseda R, Kainz V, Levy D.. Activation of Meningeal Nociceptors by Cortical Spreading Depression: implications for Migraine with Aura. J Neurosci 2010; 30: 8807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S, Carlson M, Dong J, Drake T, Geschwind D, et al. Statistical Applications in Genetics and Molecular Biology A General Framework for Weighted Gene Co-Expression Network Analysis A General Framework for Weighted Gene Co-Expression Network Analysis. 2005. [DOI] [PubMed]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R.. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 2011; 69: 855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Langfelder P, Fuller T, Dong J, Li A, Hovarth S.. Weighted gene coexpression network analysis: state of the art. J Biopharm Stat 2010; 20: 281–300. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang J, Zhang X, Zeng L, Wang L, Jiang W, Effect of metabotropic glutamate 5 receptor antagonists on morphine efficacy and tolerance in rats with neuropathic pain. Eur J Pharmacol 2013; 718: 17–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
WGS data are considered personal and sensitive data by the scientific ethical committee, thus data can only be shared as part of material agreed upon reasonable request.