

Abstract
The 43 kDa subunit of the chloroplast signal recognition particle, cpSRP43, is an ATP-independent chaperone essential for the biogenesis of the light harvesting chlorophyll-binding proteins (LHCP), the most abundant membrane protein family on earth. cpSRP43 is activated by a stromal factor, cpSRP54, to more effectively capture and solubilize LHCPs. The molecular mechanism underlying this chaperone activation is unclear. Here, a combination of hydrogen-deuterium exchange, electron paramagnetic resonance, and NMR spectroscopy experiments reveal that a disorder-to-order transition of the ankyrin repeat motifs in the substrate binding domain of cpSRP43 drives its activation. An analogous coil-to-helix transition in the bridging helix, which connects the ankyrin repeat motifs to the cpSRP54 binding site in the second chromodomain, mediates long-range allosteric communication of cpSRP43 with its activating binding partner. Our results provide a molecular model to explain how the conformational dynamics of cpSRP43 enables regulation of its chaperone activity and suggest a general mechanism by which ATP-independent chaperones with cooperatively folding domains can be regulated.
Keywords: chaperone, membrane protein biogenesis, protein dynamics, ankyrin repeat proteins, NMR spectroscopy
Introduction
Cells employ a diverse network of chaperones to assist in a variety of processes needed to maintain protein homeostasis, including de novo protein folding, sequestration and remodeling of misfolded proteins, and localization to target cellular compartments. To precisely time the capture, folding, and release of their substrates, chaperones undergo controlled structural rearrangements to complete their functional cycle. Many chaperones, such as members of the ubiquitous Hsp70, Hsp90, and Hsp100 families, use regulated ATPase cycles to alternate between conformations that promote client capture or release and even to remodel client conformations during this cycle.1–3 Many ATP-independent small heat shock proteins undergo changes in oligomeric state and/or unfolding transitions upon activation under stress conditions.4–7
A distinct group of chaperones are dedicated to the post-translational targeting of integral membrane proteins, and their chaperone cycle needs to be spatially regulated. Membrane proteins contain hydrophobic transmembrane domains (TMDs) that are highly prone to misfolding and aggregation in the aqueous environments where they are initially synthesized. Therefore, nascent membrane proteins require effective molecular chaperones to protect them from aggregation, maintain them in an insertion-competent state, and guide them to the appropriate cellular membrane. Many examples illustrate the intimate link between chaperone function and membrane protein biogenesis: SecB, Skp and SurA protect bacterial outer membrane proteins,8–14 and components of the GET and SND pathways deliver membrane proteins harboring late TMDs.15,16 A conserved feature of membrane protein chaperones is that they need to turn ‘on’ in the aqueous phase to tightly capture and protect membrane protein clients, and to turn ‘off’ at the target membrane to facilely release clients for membrane insertion. Failure to properly protect membrane proteins leads to the formation of toxic aggregates and is implicated in a variety of neurodegenerative disease pathologies.17 Despite their importance, only a few membrane protein chaperone systems have been studied in detail, and the regulatory mechanism of most membrane protein chaperones remain not well understood.
The light harvesting chlorophyll-binding proteins (LHCPs) in green plants comprise ~50% of the proteins in the thylakoid membrane and are the most abundant family of membrane proteins on earth.18 Like the vast majority (>95%) of organellar proteins, LHCPs are translated in the cytosol and imported into the chloroplast stroma, where they form a soluble “transit complex” with the chloroplast signal recognition particle (cpSRP) comprised of cpSRP43 and cpSRP54 subunits.19–22 Via the interaction of cpSRP54 with the membrane-bound receptor, cpFtsY, the transit complex is recruited to the thylakoid membrane and hands off LHCP to the Alb3 translocase.23–29 Alb3 mediates the insertion of LHCPs into the membrane, where they assemble into the light harvesting complex that forms the antenna of the photosynthetic reaction center.18,30 The abundance of LHCPs and their crucial roles in energy generation of green plants necessitate highly effective chaperones to assist in their biogenesis, making it a robust system to understand the function and mechanism of membrane protein chaperones.
The cpSRP43 subunit in cpSRP is responsible for chaperoning LHCPs during their targeted delivery to the thylakoid membrane.31,32 cpSRP43 contains a substrate binding domain (SBD) consisting of four ankyrin repeat motifs capped by an N-terminal chromodomain (CD1) and a C-terminal bridging helix (BH).32–34 The ankyrin repeat motifs in the SBD recognize a conserved L18 motif between TMD2 and TMD3 of LHCP.22,33–36 The SBD also forms extensive contacts with all three TMDs in LHCP, potentially via hydrophobic surfaces on the ankyrin repeat motifs and the BH, to protect them from aggregation.37 The chaperone activity of the cpSRP43 SBD is regulated by interaction partners that bind at two additional chromodomains C-terminal to the SBD. Wildtype cpSRP43 displays lower chaperone activity than the SBD, but is activated upon binding of a motif from the C terminus of the cpSRP54 M-domain (54 M) to CD2.38–40 In contrast, the stromal domain of the Alb3 translocase binds to CD3 and triggers the selective release of the LHCP TMDs from cpSRP43.34 These regulations in chaperone activity strongly correlated with changes in the HSQC spectra of cpSRP43. At least two component peaks were observed for multiple residues in CD1 in apo cpSRP43. One of the component peaks became dominant under activating conditions, such as in the isolated SBD or upon addition of the 54 M or L18 peptide, whereas the opposite component peak became dominant in the presence of the Alb3 stromal domain.34 These observations led to a model in which free cpSRP43 samples both a chaperone-inactive, open state and a chaperone-active, closed state. cpSRP54 drives cpSRP43 to the closed state to enable effective capture of LHCPs in the stroma, whereas the Alb3 translocase drives its transition to the open state to effect the facile release of LHCPs at the site of membrane insertion.
Despite this conceptual framework, the nature of the conformational changes underlying cpSRP43 activation remains poorly understood. Due to crowding of the HSQC spectra, component cross peaks for both the open and closed states of cpSRP43 could be assigned for only 12 residues in CD1, whereas only the peaks for the closed state were assigned for the majority of residues in the SBD.34 Crystal structures are available for the SBD and SBD-CD2 fragments bound to the 54 M peptide, which primarily capture the closed conformation.38,41 A complete picture of cpSRP43′s conformational landscape throughout its chaperone cycle is therefore lacking. To address this gap in knowledge, we used a combination of biophysical approaches, including hydrogen-deuterium exchange (HDX), electron paramagnetic resonance (EPR) and NMR spectroscopy, to probe the structural changes of cpSRP43 during its activation. Our results show that the ankyrin repeat motifs and the BH of cpSRP43 undergo a disorder-to-order transition, whereas CD1 shifts between two folded structures during the open-to-closed rearrangement. We propose a model in which a coil-to-helix transition in the BH is propagated across the ankyrin repeat domain of cpSRP43 and allows long-range communication of the activating ligand to drive folding of the sites required for substrate binding.
Results
The cpSRP43 SBD is better solvent-protected in the activated state
To probe for potential structural changes in the cpSRP43-SBD during its activation, we compared the amide hydrogen/deuterium exchange (HDX) efficiencies between free cpSRP43 and cpSRP43 bound to a peptide containing its recognition motif on LHCP (L18) or on the M-domain of SRP54 (54M peptide; Figure 1). HDX coupled to mass spectrometry (MS) measures the local solvent accessibility of amide protons, which vary the most at sites of protein-protein interactions or sites undergoing largescale conformational changes.42 Over 160 peptides were observed and analyzed from each MS spectrum of cpSRP43, covering >90% of its protein sequence across all domains (Figures S1–S4). Many regions of cpSRP43 (CD1, ankyrin repeat motifs, BH, CD3, and the C terminus of CD2) were identified by multiple peptides, providing redundant data that ensure reliable and accurate quantification.
Figure 1.
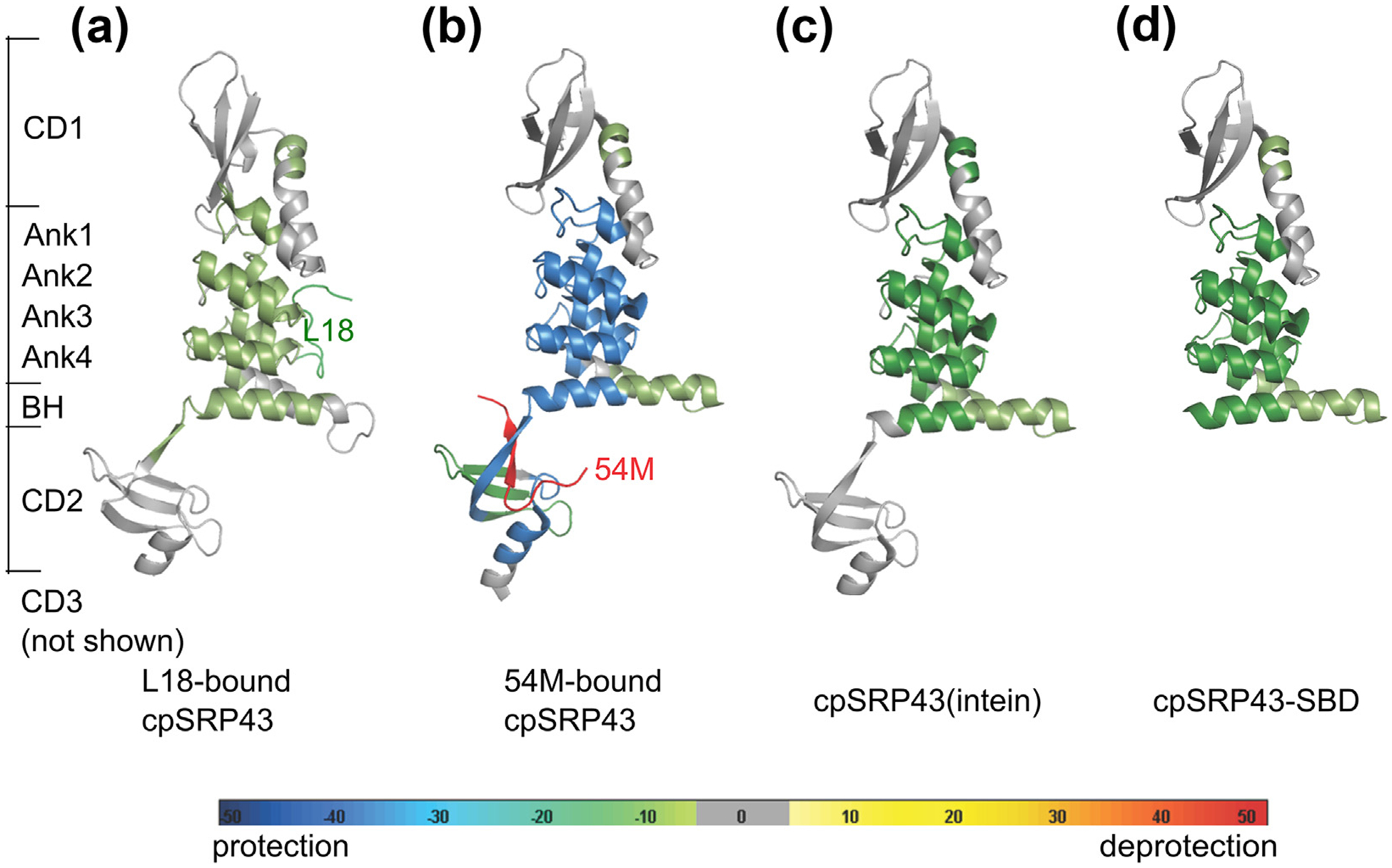
Substrate, activating ligand, and activating mutations lead to increased global protection of the cpSRP43 SBD from solvent exchange as determined by HDX mass spectrometry. The differences in HDX incorporation (Figures S1–S4) relative to wildtype, apo-cpSRP43 are mapped onto the structure for the following samples: L18-bound cpSRP43 (a); 54 M-bound cpSRP43 (b); superactive cpSRP43(intein) (c); and cpSRP43-SBD (d). Protection (reduced solvent exchange) or deprotection (increased solvent exchange) are denoted by pseudo-colors as indicated, and gray denotes no change.
We found that both the L18 and 54 M peptides induced slower H/D exchange of backbone amides in ankyrin repeat motifs 1–4 and the BH in the SBD of cpSRP43 (Figures 1(a), (b) and S1–S2). In general, the 54 M peptide had a larger effect than L18 in reducing the rate of H/D exchange. Additional sites in CD2 also showed significantly lower HDX in the presence of 54 M (Figures 1(b) and S2), presumably due to shielding of the 54 M binding site from solvent and possibly 54 M-induced local rearrangements in CD2. Although ligand-induced protection from H/D exchange could arise from either shielding of the ligand binding site or from induced conformational changes, the reduction in solvent accessibility was observed across almost all the helices in the SBD and includes regions that are remote from the immediate L18 or 54 M binding sites. These results suggest that binding of the substrate (L18) and activator (54 M) induce global changes in the structure or dynamics of cpSRP43-SBD that render this domain better solvent protected.
To directly test this model, we analyzed the H/D exchange profile of two previously identified superactive cpSRP43 mutants that are pre-organized into the active conformation: cpSRP43 (intein), in which the short Gln-Val linker between the BH and CD2 was replaced by a longer and more flexible sequence; and cpSRP43-SBD, a construct containing only the SBD but lacking CD2 and CD3.34 Both mutants exhibit higher chaperone activity than wildtype cpSRP43, are predominantly in the active state based on HSQC NMR data, and largely bypass activation by cpSRP54.34 HDX analyses showed that, compared to wildtype cpSRP43, significant reductions in backbone solvent accessibility across the entire SBD, including the ankyrin repeat motifs and the BH (Figures 1 (c), (d), S3 and S4), were observed with both mutants. In contrast, few changes in HDX efficiency were observed in CD1, CD2 and CD3 between these mutants and wildtype cpSRP43 (Figures 1 and S1–S4). The protection patterns in the SBD observed with the superactive chaperone mutants were similar to those induced by 54 M and L18, although 54 M induced the strongest protections. Together, these results indicate that the SBD of cpSRP43, specifically the ankyrin repeats and BH, becomes better solvent protected in the activated state.
Substrate and activator rigidify the cpSRP43 SBD
To gain higher resolution information into the substrate and 54 M-induced structural changes in cpSRP43-SBD, we used site-directed spin labeling and electron paramagnetic resonance (EPR) spectroscopy, which provides a sensitive probe for the local backbone mobility at sites of interest. The nitroxide probe (1-oxy-2,2,5,5-tetrame thyl-3-pyrrolinyl-3-methyl) methanethiosulfonate (MTSSL) is small with a short spacer, which can be placed throughout a protein without significantly affecting structure.43 The mobility of MTSSL is highly dependent on the local secondary or tertiary structure,44 which can be used to distinguish loops from helices and sites on the surface from those buried in the protein interior.45,46 Using a previously characterized cysteineless cpSRP43 mutant that retains chaperone activity within 2–3 fold of wildtype cpSRP43 and is strongly activated by 54 M (37 and Figure S5), we introduced single cysteine mutations at selected residues in the SBD where the mutation affected chaperone activity less than three-fold (Figure 2(a) & (b) and37). The native cysteine (C175) and the engineered single cysteines allowed site-specific labeling with MTSSL. We verified that the MTSSL-labeled cpSRP43 could still bind the 54 M peptide with affinities that are within 3-fold of WT cpSRP43 (Figure S5(a) and38) and that 54 M binding restored full chaperone activity of MTSSL-labeled cpSRP43 variants towards LHCP (Figure S5(b) & (c)). We measured the effects of 54 M and the substrate protein, LHCP, on the EPR spectra of spin-labeled cpSRP43. The changes in the EPR spectra were characterized by the linewidth of the central resonance (Figure 2(c) and (d), ΔH0) and the overall breadth of the EPR spectra, especially the intensity of hyperfine splitting that arises from highly immobile populations of spin probes relative to the mobile population (Figure 2(c)).47–49
Figure 2.
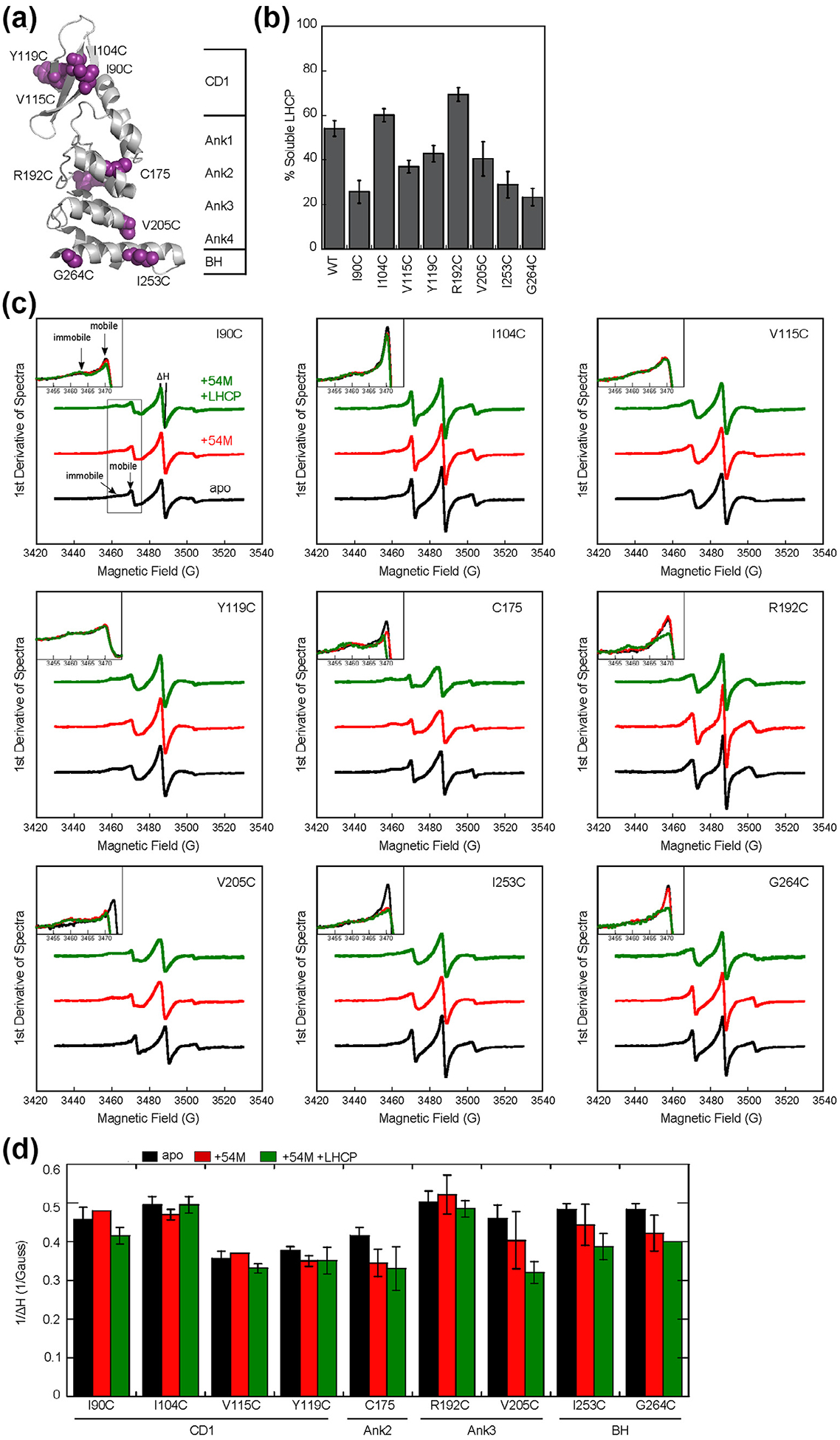
The cpSRP54 M-domain and substrate induce immobilization of spin probes across the ankyrin repeat domain and the BH of cpSRP43. (a) Structure of the cpSRP43 SBD highlighting residues used for SDSL-EPR analyses. (b) The chaperone activities of single cysteine cpSRP43 mutants were analyzed using the turbidity assay. Reactions used 1 μM LHCP and 2.5 μM WT or mutant cpSRP43. All comparisons were relative to cysteineless cpSRP43(C175A, C297S), which is denoted as WT. (c) EPR spectra for spin labels at indicated positions in apo-cpSRP43 (black), cpSRP43 bound to 54 M (red), and cpSRP43 bound with both 54 M and LHCP (green). The insets show a zoom-in of the spectra at 3450–3475 G, which contain the peaks reporting on the mobile and immobile populations of the spin probe. (D) Summary of the effects of 54 M and LHCP on the central line width (ΔH0) of the spectra for spin labels at various positions in the SBD, quantified from the data in part C. All values are reported as mean ± S.D., with n ≥ 2.
All five spin labels in the ankyrin repeat motifs and the BH (C175, R192C, V205C, I253C, G264C) underwent significant EPR spectral changes upon binding the 54 M peptide and/or LHCP (Figure 2(c) and (d), black vs. red and green). Four of the spin probes at these positions become less mobile, as reflected by an increase in the central linewidth and the reductions in the mobile fraction in the EPR spectra (Figure 2(c) and (d)). For the remaining residue, R192C, changes in probe mobility were not obvious from the central linewidth but were readily detectable from changes in EPR spectral shape upon the addition of both 54 M and LHCP, which showed a higher relative population of immobile species (Figure 2(c)). As 54 M binds outside the SBD (Figure 1), the observed immobilization of multiple spin probes indicates long-range allosteric communications propagated throughout the ankyrin repeat domain. On the other hand, none of the four spin probes in CD1 underwent significant spectral changes upon binding of 54 M or LHCP (Figure 2(c) and (d)). Collectively, the EPR data show that the 54 M- and substrate-induced conformational changes in the cpSRP43-SBD lead to increased backbone rigidity of the ankyrin repeat domain and the BH, but these changes did not propagate to CD1.
NMR spectroscopy reveals disorder-to-order transitions in the SBD
To obtain dynamic information on the changes in the cpSRP43-SBD during its activation, we employed site specific fluorine-labeling and NMR spectroscopy. We attached the 3-bromo-1,1,1-tri fluoropropan-2-one (BTFA) probe to single cysteines engineered at surface-exposed residues throughout cysteineless cpSRP43 and collected 1D 19F NMR spectra (Figure 3(a)). To test whether cysteine mutation and BTFA-labeling disrupted the activity or activation of cpSRP43, we measured the ability of each BTFA-labeled cpSRP43 variant to prevent LHCP aggregation with and without 54 M present using light scattering assays (Figure S6). Only the variants that retained chaperone activity and could be activated by 54 M were subjected to further NMR analysis.
Figure 3.
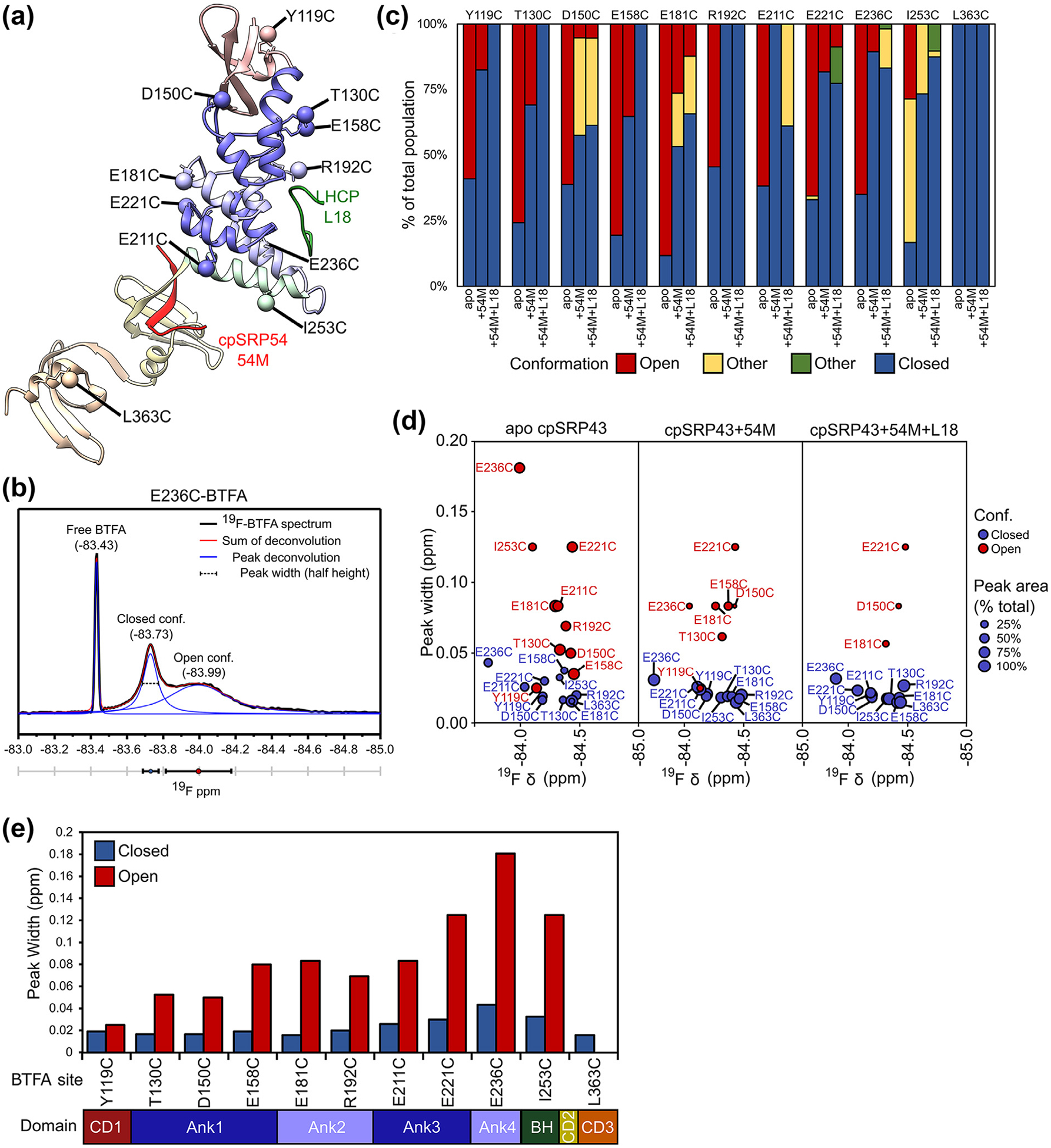
Analysis of the19F NMR spectra of BTFA-labeled cpSRP43. (a) Composite structural model of cpSRP43 based on the coordinates for SBD-CD2 (PDB: 3UI2) and CD2-CD3 (PDB: 5E4W). BTFA terminal carbons are shown as labeled spheres. The individual domains are colored to match the diagram in C. Interacting peptides from binding partners (L18, 54 M) are indicated. (b) Example of a deconvolution of the 19F spectrum for E236C-BTFA, with the experimental data in black, individual component peaks in blue, and the sum of the component peaks in red. Three parameters can be extracted: the chemical shift of the peak center (ppm), the peak width (ppm), and the peak area. (c) Summary of the relative abundance of component cross peaks for the indicated BTFA probe sites in apo cpSRP43, cpSRP43 + 54 M, and cpSRP43 + 54 M + L18, calculated from deconvolution of the 19F NMR spectra for BTFA shown in (b). (d) Comparison of the chemical shift and peak width for component cross peaks for the closed (blue circles) and open (red circles) conformations in apo cpSRP43 (left), cpSRP43 bound to 54 M peptide (middle), and cpSRP43 bound to both the 54 M and L18 peptides. The size of the circle indicates the relative abundance of the component peak. (e) Summary of the line widths of the 19F resonances in the closed (blue) and open (red) conformations of apo cpSRP43 for the indicated BTFA-labeled sites. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Most cpSRP43 variants labeled with a single BTFA probe produced spectra with two or more peaks. The spectra were deconvoluted into individual component peaks, which allowed us to extract three fitting parameters for each peak: the chemical shift, peak width, and peak area (Figures 3(b) and S7). With the exception of residue L363C (in CD3), addition of the activating peptides, 54 M or L18, changed the relative abundance of the component peaks at most BTFA labeled sites (Figure 3(c) and (d)), whereas the chemical shifts of each component peak remained largely unchanged (Figure S7). This supports the model that interaction partners stabilize specific conformational states that are already sampled by apo cpSRP43.34 We assigned the component peaks that increased in relative abundance in response to L18 or 54 M to the chaperone-active closed conformation, and those that decreased in abundance upon L18 and 54 M binding to the chaperone-inactive open conformation (Figure 4(a)). In contrast to most probes in the SBD, we detected only one peak for the BTFA probe in CD3 (L363C) that was unchanged upon ligand binding; this is consistent with previous observations from amide HSQC data that CD3 does not sense or respond to 54 M or L18 binding.34 Finally, 19F Diffusion Ordered Spectroscopy (DOSY) measurements on apo cpSRP43 D150C-BTFA showed nearly identical diffusion coefficients for the two component peaks (7 × 10−7 cm2 s−1, Figure S8), excluding the possibility that one of the peaks arises from oligomerization.50 The ability to observe both conformations of cpSRP43 in 19F NMR allowed us to directly extract information about the structural changes during its activation.
Figure 4.
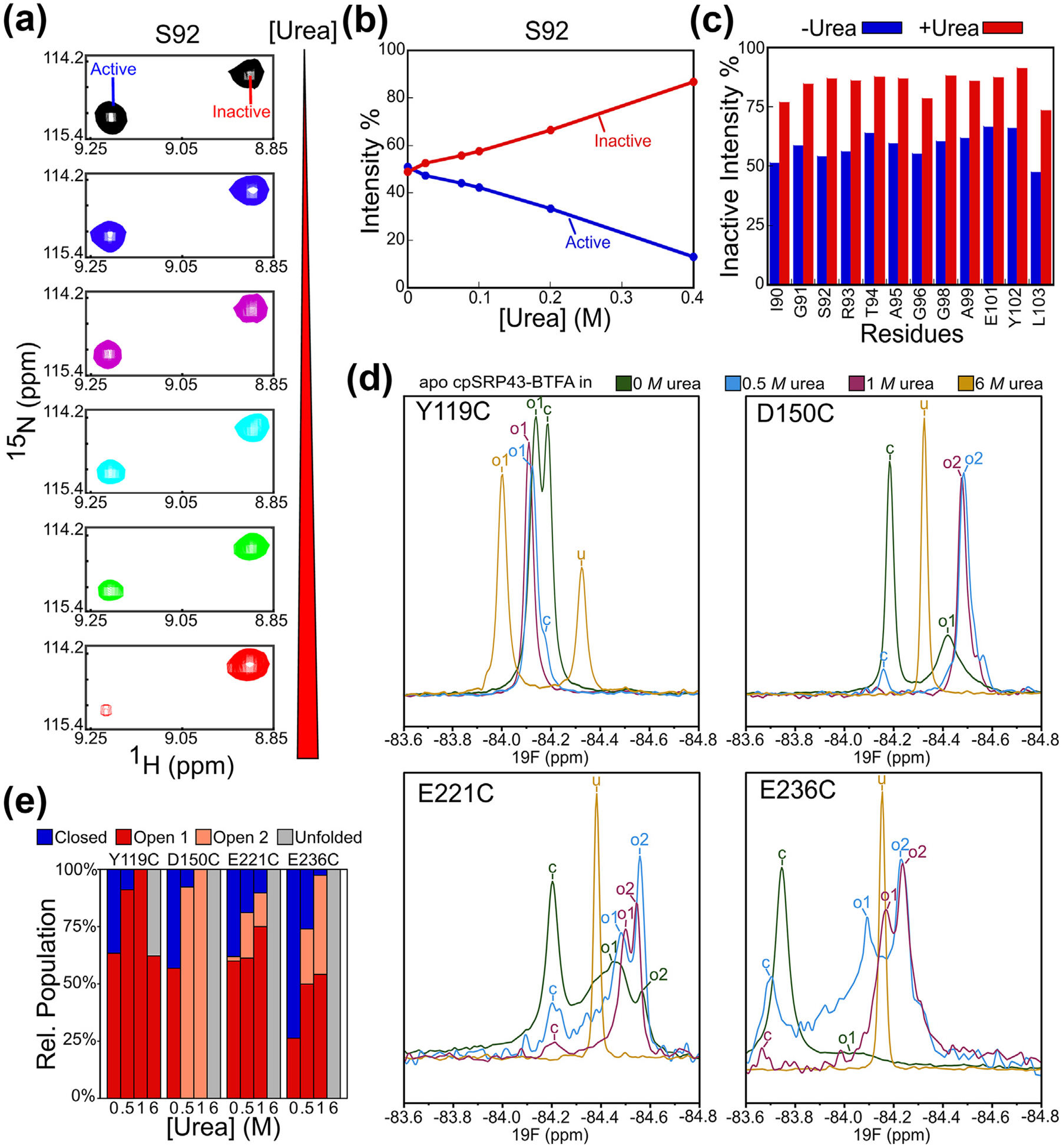
Low doses of urea drive cpSRP43 into the open state. (A) Effect of urea on the relative intensity of the component cross peaks for Ser92 in the TROSY spectra of 2H, 15N-labeled apo cpSRP43. (B) Quantification of the data in part A. (C) Summary of the effects of urea on the relative intensities of the open conformation cross peaks at the indicated residues. Relative intensities were determined from cross peak heights. (D) 19F 1D NMR spectra on BTFA-labeled apo cpSRP43 at 0 M (green), 0.5 M (blue), 1 M (purple), and 6 M (yellow) urea. The assigned peaks are labeled by conformation: closed (c), open 1 (o1), open 2 (o2), and unfolded (u). (E) Relative populations of closed (blue), open 1 (red), and open 2 (pink), and unfolded (gray) peaks at the indicated urea concentrations. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We first compared the chemical shifts of 19F-BTFA labeled cpSRP43 between the closed and open conformations, which provide information on the local environment of the BTFA probes. The 19F peaks for probes in Ank1, Ank3 and Ank4 (D150C, E211C, E221C, and E236C) shifted significantly upfield in the open conformation compared to the closed conformation (Figures S7 and S9), suggesting that these sites are more solvent exposed in the open conformation.51,52 The 19F peaks for the probe in the BH (I253C) showed the opposite change; however, the complexity of the 19F spectra for I253C-BTFA suggests the presence of additional interactions or conformational states that complicate interpretation of its chemical shift values. Importantly, the component peaks for the closed conformation are well dispersed across all the BTFA probes in the ankyrin repeat motifs and the BH (Figures S7 and 3(d), blue circles). In contrast, the open conformation peaks for probes in the first three ankyrin repeats are narrowly dispersed around −84.4 ppm (Figures S7 and 3(d), red circles). This strongly suggests that, in contrast to the well-folded, closed conformation of cpSRP43 in which each BTFA probe is in a unique chemical environment, many sites in the ankyrin repeat motifs and the BH are partially disordered in the open conformation.
To identify changes in protein dynamics upon ligand-induced activation of cpSRP43, we compared the peak widths of the BTFA probes between the open and closed conformations. In the closed state, the 19F linewidths are relatively uniform for the apo cpSRP43 and its complexes with 54 M and L18 (Figures S7, 4(b) and (c)). For all BTFA probes in the ankyrin repeat motifs and the BH, the 19F peaks corresponding to the open conformation are significantly broader than those for the closed conformation (Figure 3(d) and (e)). This suggests conformational fluctuations in the open state on the μs-ms time scale53 and is consistent with the evidence from HDX and EPR that the SBD in apo cpSRP43 is flexible and becomes more rigid upon binding of L18 or 54 M. The greatest broadening was observed for BTFA probes in Ank3, Ank4, and the BH (E221C, E236C, and I253C; Figure 4(c)), suggesting that the C-terminal half of the SBD is particularly flexible in the open conformation. These observations of structural disorder are analogous to the findings with IKBa,54,55 Notch,56,57 and p19(INK4d),58, and appear to be a conserved feature among ankyrin repeat domain proteins.59 In contrast, the component peaks for both the open and closed conformations have similarly narrow peak widths for BTFA probes in CD1 (Y119C) (Figure 3(d) and (e)), suggesting that CD1 is well-folded in both states.
To obtain additional evidence for a disorder-to-order transition during the open-to-closed rearrangement in cpSRP43, we tested the effect of urea on the 1H, 15N-HSQC TROSY spectra of apo cpSRP43. If the open state is partially disordered and the closed state is fully folded, denaturant should shift the equilibrium towards the open state. Previous HSQC NMR studies of cpSRP43 identified 12 residues in CD1 that show two component cross peaks; the change in their relative abundance upon addition of L18 and 54 M peptide further allowed the assignment of the peaks to the open or closed conformation.34 Intriguingly, titration of low doses of urea (up to 0.4 M) significantly reduced the component cross peaks for the closed conformation, with a corresponding increase in the intensity of the component cross peak for the open conformation (example for S92 shown in Figure 4(a) and (b)). The same reduction was observed for all the residues in CD1 for which component cross peaks could be confidently assigned (Figure 4(c)). These results show that denaturant such as urea promotes the open conformation in cpSRP43 and moreover, that the closed conformation of cpSRP43 is only marginally more stable than the open conformation and can therefore be substantially destabilized by low doses of denaturant.
To further probe the conformational changes in the ankyrin repeat motifs, whose open conformation cross peaks could not be assigned in the 1H, 15N-HSQC TROSY spectra, we collected 19F NMR spectra on BTFA-labeled apo cpSRP43 at increasing urea concentrations. Addition of 0.5–1 M urea reduced the component peak for the closed conformation, with a corresponding increase in the open conformation peaks for the BTFA probes in CD1 (Y119C) and Ank2–4 (D150C, E221C, E236C) (Figure 4(d) and (e)), consistent with the effect of urea on the HSQC spectra (Figure 4(a) and (b)). As described earlier, the open state peaks for BTFA probes in Ank3 (E221C) and Ank4 (E236C) were broad (Figure 4(c)) and suggested the presence of multiple conformations in intermediate exchange. In support of this model, multiple component peaks with narrower widths were observed for the open state in the presence of 0.5–1 M urea (Figure 4(d) and (e); ‘o1’ and ‘o2’), suggesting that urea accelerated the exchange between multiple conformations with different degrees of solvent exposure in the open state. For BTFA probes in the ankyrin repeat motifs, the component peaks for the open state also shifted upfield at 0.5–1 M urea (Figure 4(d)), in contrast to the solvent effect of urea that shifts the 19F peaks downfield in general (see Fig. S10 for the effect of urea on a BTFA-labeled peptide, and Figure 4(d) for the effect of urea on the closed state peaks), suggesting that urea enhanced the solvent exposure of the open conformations at these sites.60,61 Finally, while the BTFA probes in the Ankyrin repeat domain were completely unfolded in 6 M urea, only 35% of the probe in CD1 (Y119C) was unfolded in 6 M urea (Figure 5 (d) and (e)), indicating that CD1 is stably folded in the open state. Together, these results provide additional evidence that the ankyrin repeat motifs exchange between multiple partially unfolded conformations in the open state.
Figure 5.
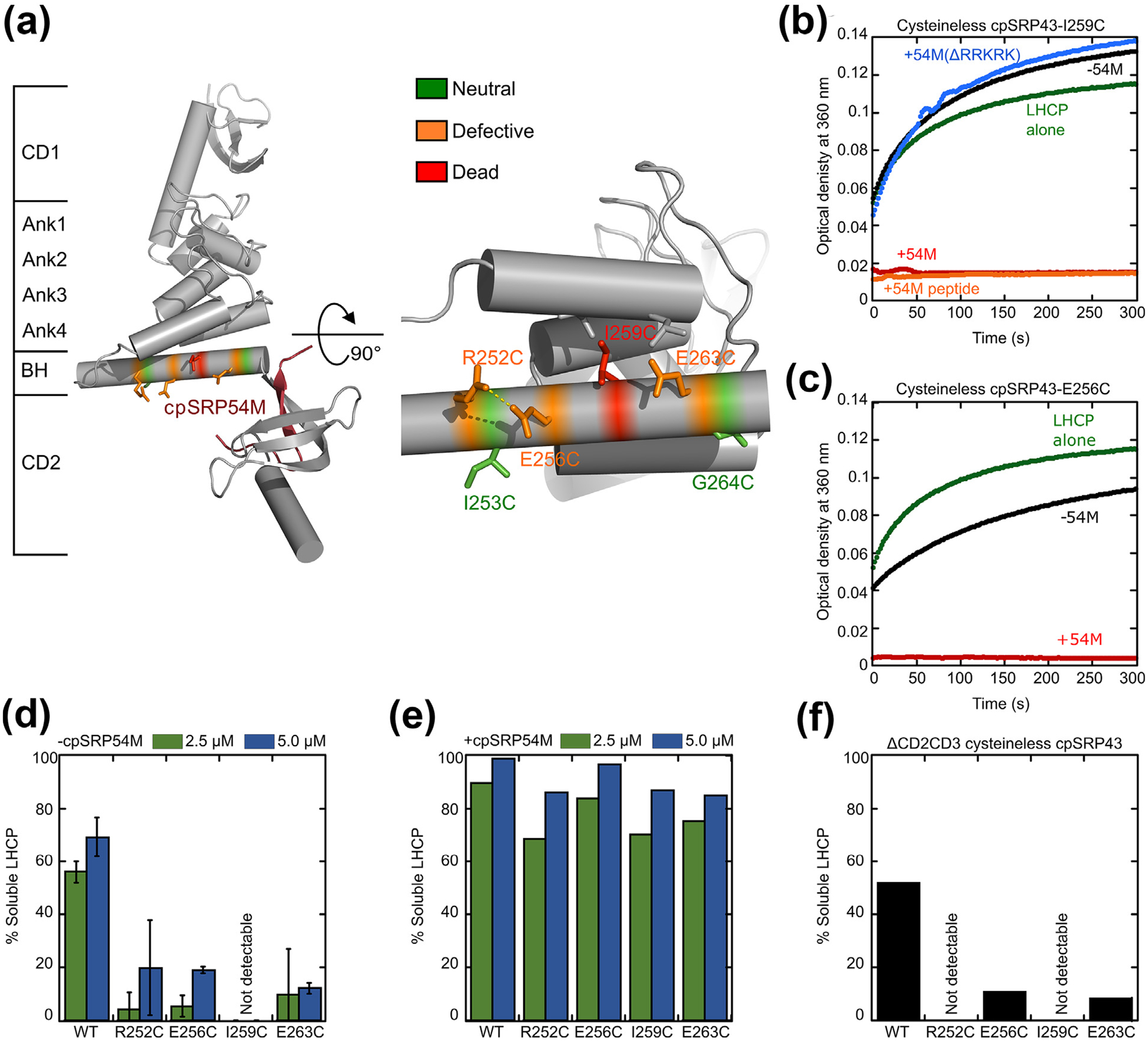
Disruption of intra- and inter-helical contacts in the bridging helix compromises chaperone activity and renders cpSRP43 more dependent on cpSRP54. (a) Structure of the SBD-CD2 fragment of cpSRP43 highlighting the residues in the BH subject to mutational analyses. Colors indicate different mutational effects on the chaperone activity of cpSRP43. (b) Time courses for the aggregation of LHCP, monitored by turbidity at 360 nm, upon dilution into buffer (green) or into solutions containing mutant cpSRP43(I259C) in the absence (black) and presence of the indicated regulatory factors: 54M (red); 54M peptide (orange); mutant 54M lacking the basic C-terminal residues that bind cpSRP43 (54M(ΔRRKRK); blue). (c) Time courses for aggregation of LHCP upon dilution into buffer (green), or into solutions containing mutant cpSRP43(E256C) in the absence (black) and presence (red) of the cpSRP54 M-domain. (d, e) Summary of the chaperone activities of indicated BH mutants in the absence (d) and presence (e) of the cpSRP54 M-domain, quantified from the degree of solubilization of LHCP at the indicated chaperone concentrations based on turbidity measurements. (f) Mutations in the BH disrupt chaperone activity even in the absence of CD2 and CD3. All mutations were constructed in cysteinless cpSRP43 (C175A, C297S), which is denoted as WT in this figure.
In summary, 19F NMR allowed us to directly observe both the open and closed conformations in cpSRP43 and detect widespread disorder and flexibility in the ankyrin repeat motifs and the BH in the open conformation compared to the well-structured closed state. These results support findings from the H/D exchange and EPR data and provide direct evidence that the open-to-closed rearrangement of cpSRP43 involves widespread changes across the SBD, with the ankyrin repeat motifs and BH undergoing a disorder-to-order transition and CD1 rearranging from one well-folded structure to another.
A folded bridging helix is essential for chaperone activity
To test if rearrangement to a well-folded ankyrin repeat domain is required for chaperone activity, we re-examined the effects of several mutations in the BH, which serves as a C-terminal capping element for the ankyrin repeat domain of cpSRP43 and bridges the ankyrin repeat motifs to the cpSRP54 binding site in CD2 (Figure 5(a)). The BH is essential for maintaining the closed state of cpSRP43, and its deletion abolishes the chaperone activity of cpSRP43-SBD towards LHCP,34 indicating its critical role in cpSRP43 function. Multiple mutations in the BH have been introduced that disrupt either intra- or inter-helical contacts. The side chains of R252 and E256 form an (i, i + 4) salt bridge that stabilizes helix formation in the BH. I259 and E262 engage in close contacts with residues from the neighboring helix in Ank4 that stabilize inter-helical packing (Figure 5(a)). Mutation of any of these residues severely disrupted cpSRP43′s chaperone activity (Figure 5(b–d), Supplementary Table 1; see also37). In contrast, mutations of I253 and G264, which extend out towards the solvent in the closed conformation structure, did not substantially affect chaperone activity (Figure S6 and37). The R252C, E256C, I259C and E262C mutations caused large deleterious effects on chaperone activity even with cpSRP43-SBD, in which CD2 and CD3 have been deleted (Figure 5(f)), excluding models in which the mutational defects are caused by disruption of interactions between the BH and the nearby chromodomains. Thus, a properly folded BH is essential to arrange the cpSRP43-SBD into an active conformation, and any disruption of intra- or inter-helical interactions in the BH have strong deleterious effects on its chaperone activity.
To provide additional evidence for this model, we tested the effect of 54 M on these chaperone-deficient BH mutants. In the absence of 54 M, none of them effectively suppressed the aggregation of LHCP, whereas LHCP was close to completely solubilized by these chaperone mutants in the presence of 54 M (Figure 5(b) and (c), summarized in (d) and (e)). Deletion of the conserved C-terminal basic residues in 54 M, which disrupts the binding of 54 M with cpSRP43,62 abolished the rescue of chaperone activity (Figure 5(b), blue line), supporting the specificity of this effect. Further, 54 M peptide was sufficient to produce the same rescue as the intact 54 M domain (Figure 5(b), red vs. orange lines), excluding the possibility that the 54 M domain contributed additional interaction sites for binding and solubilization of LHCP as previously proposed.19 Thus, 54 M was able to completely rescue deleterious mutations that severely disrupt helix formation in the BH and the chaperone activity of cpSRP43. The more extensive rescue of the loss-of-function BH mutants compared to wildtype cpSRP43 is consistent with a model in which helix-destabilizing mutations shifted the conformational equilibrium of cpSRP43 further towards the open conformation, and are therefore more dependent on cpSRP54 for attaining the chaperone-active, closed conformation.
Discussion
cpSRP43 is a small, ATP-independent chaperone that captures LHCPs imported into the chloroplast, protects them during their transit through the stroma, and delivers them to the translocation machinery for insertion into the thylakoid membrane.20 Its small size, robust activity, well-characterized cellular role, and amenability to structural methods make it an excellent system to study the structure-dynamics-function relationship of a membrane protein chaperone. Previous work demonstrated that the chaperone activity of the cpSRP43 SBD is extensively regulated by binding of cpSRP54 in the stroma and the Alb3 translocase at the thylakoid membrane to effect spatial regulation of its coupled chaperone and protein targeting cycle34. In this work, we employed EPR, HDX, and NMR spectroscopy to define the structural changes in cpSRP43 upon its activation. Our results reveal extensive dynamics in the open conformation of cpSRP43 and strongly suggest that a disorder-to-order transition of the ankyrin repeat domain is responsible for its activation. Such a transition provides an attractive mechanism for long-range molecular communication of the SBD of cpSRP43 with binding partners that bind in its CD2 and CD3 domains.
The results of multiple biophysical methods support a disorder-to-order transition during the activation of cpSRP43. Under conditions where cpSRP43 is chaperone-active, either in gain-of-function variants pre-organized into the closed state or when it is bound to the 54 M or L18 peptides, we observed substantially reduced solvent exchange across the entire ankyrin repeat domain that could not be explained by ligand-induced shielding, whereas no such changes were observed in the chromodomains. These data suggest a global restructuring of the ankyrin repeat domain during its activation that renders it better protected from solvent. A clue to the nature of this structural change was provided by EPR studies, which showed that paramagnetic probes across the ankyrin repeat motifs and BH displayed reduced local backbone mobility upon substrate or activator binding, whereas those in CD1 did not change. These results suggest that the ankyrin repeat domain is structurally more flexible in the open conformation.
NMR experiments provided direct evidence for this model. With BTFA probes placed at sites throughout the SBD, we observed 19F peaks for both the open and closed conformations of cpSRP43, showing that these conformers interconvert slowly on the NMR time scale. For 19F-BTFA probes in the ankyrin repeat domain, the peaks for the closed state have well dispersed chemical shift distributions and narrow peak widths, suggesting a defined three-dimensional fold consistent with the existing crystal structures of the cpSRP43 SBD. In contrast, the open state peaks are poorly dispersed and become increasingly broad towards the C terminus of the ankyrin repeat domain, suggesting a high degree of disorder and dynamics that explain the HDX and EPR data. This model is further supported by the observation that low doses of urea shifts the conformational equilibrium towards the open conformation. In contrast to the ankyrin repeat motifs, both the open and closed state peaks for 19F-BTFA probes in CD1 are well dispersed and have narrow peak widths. We could further observe CD1 during the open-to-closed rearrangement using our assignments of both the open and closed state peaks for 12 residues in CD1.34 Despite the chemical shift differences in the amide HSQC cross peaks of these residues between the two states,34 the Cα and Cβ chemical shifts of both states are identical, suggesting little change in the secondary structure of CD1 during the open-to-closed rearrangement. As these residues cluster in a pair of β-strands that pack against Ank1 in the crystal structure of the closed state, their observed amide chemical shift differences between the two states likely reflect changes in the packing of Ank1 against CD1 in the open state.33 Together, these results indicate that CD1 remains well-folded in both the open and closed conformations, in agreement with the results from HDX and EPR analyses. The different effects of urea on CD1 and the ankyrin repeats in the open state further support a model where CD1 adopts a stable fold in both the closed and open states, whereas the ankyrin repeat domain samples many partially unfolded conformations with relatively low stability but is driven to a single, well-folded closed conformation upon activation. Collectively, all three biophysical methods show that CD1 remains folded, while the ankyrin repeat domain undergoes a disorder-to-order transition during the open-to-closed rearrangement that activates cpSRP43.
A growing body of work points to the transition between folded and disordered structures in ATP-independent chaperones as a general mechanism to regulate chaperone activity.63 Many of these chaperones are activated by order-to-disorder transitions. For example, small heat shock proteins deoligomerize and become partially disordered upon heat stress, which activates them to prevent misfolded proteins from aggregation.4,7,64–66 Other stresses, such as acidic pH or oxidation, trigger unfolding transitions that activate chaperones such as HdeA, Hsp33, and Hsp26.5,6,67,68 Our work shows that the opposite transition is used to activate cpSRP43, analogous to the Spy13 and Skp11 chaperones that protect nascent membrane proteins during their transit across the bacterial periplasm. The use of a disorder-to-order transition by cpSRP43 may arise in part from the folding property of the ankyrin repeat domain, in which the helices from individual repeats stack together to form a conserved, concave-shaped scaffold that presents binding surfaces for target proteins.69 As interactions within each repeat are less stable than the interface between repeat motifs, ankyrin repeat domain proteins fold and unfold cooperatively despite the lack of long-range tertiary contacts.59 Disorder-to-order transitions have been observed in IκBα, in which two of the ankyrin repeat motifs are disordered in the apo protein but becomes structured upon binding to NFκB,54,55 with the notch intracellular domain upon binding the CSL transcription factor,56,57 and with p19(INK4d).58 Importantly, a disorder-to-order transition of the ankyrin repeat domain provides an attractive molecular mechanism to explain the long-range molecular communication of cpSRP43 with cpSRP54, whose binding in CD2 induces structural changes that propagate across the entire SBD.
We propose a model in which a disorder-to-order transition in the ankyrin repeat domain of cpSRP43 reshapes the surface of the SBD to favor tight substrate binding (Figure 6). Apo cpSRP43 extensively samples a chaperone-inactive, open conformation in which the ankyrin repeat domain is partially disordered and highly dynamic, especially towards the C-terminal repeat motifs and the BH. Binding of cpSRP54 in CD2, adjacent to the BH, induces a coil-to-helix transition in the BH, enabling it to initiate inter-repeat interactions that propagate across the ankyrin repeat domain. This generates a well-folded, closed conformation in the SBD that presents both the L18 binding site and contiguous hydrophobic surfaces needed for efficient capture of the LHCP TMDs, whereas these contact surfaces would be disrupted in the partially unfolded open conformation. We speculate that other allosteric regulators, such as the Alb3 translocase, could act by inducing additional conformational transitions in cpSRP43 when the targeting complex is delivered to the thylakoid membrane and thus favor selective release of LHCPs at the site of their membrane insertion.
Figure 6.
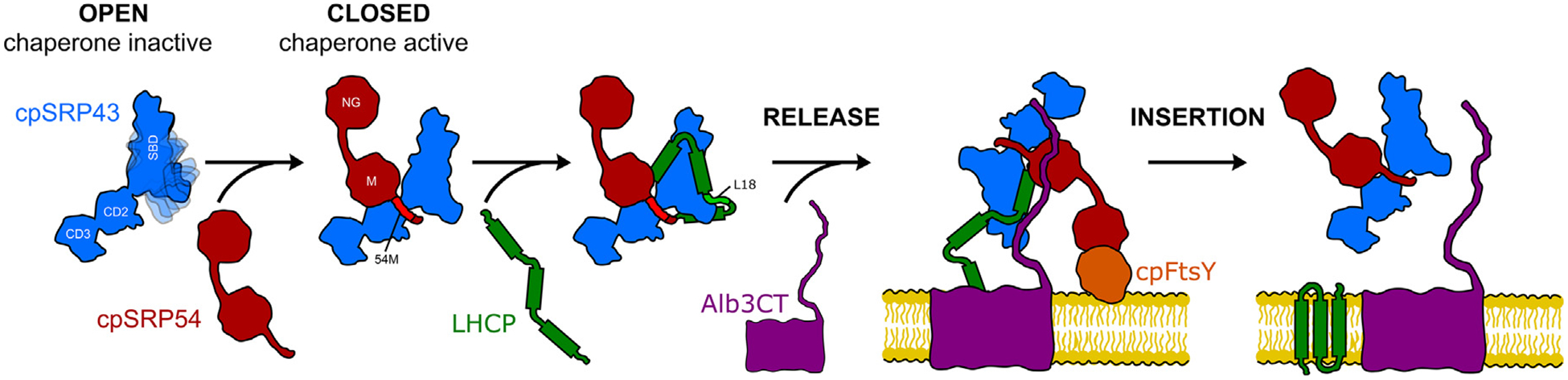
Model for allosteric regulation of cpSRP43’s chaperone activity. In the open conformation, the SBD’s ankyrin repeat domain and BH are flexible and disordered, resulting in an inactive chaperone. The interaction of cpSRP43 with the cpSRP54 M-domain or with the L18 motif on the LHCP substrate stabilizes the closed conformation, in which the Ankyrin repeat domain is more stably folded. This rearrangement enables the formation of contiguous contact surfaces on the cpSRP43-SBD for the substrate TMDs and thus high chaperone activity. The interaction of cpSRP54 with cpFtsY delivers the transit complex to the Alb3 translocase at the thylakoid membrane, where Alb3 triggers cpSRP43 to release LHCP and mediates its insertion into the membrane.
Materials and methods
Protein expression and purification
Single cysteine mutants of Lhcb5 and cpSRP43 were constructed using the QuikChange Mutagenesis procedure (Stratagene) according to manufacturer’s instructions. WT and mutant cpSRP43, LHCP, and wildtype and mutant Lhcb5 were overexpressed and purified as previously described.31
Chaperone activity of cpSRP43
The ability of cpSRP43 to prevent LHCP aggregation was measured as described previously.32,36 Aggregates were removed via ultra-centrifugation in a TLA-100 rotor (Beckman Coulter) at 100,000 rpm for 30 min at 4 °C prior to the experiment. Light scattering experiments were performed by addition of 3 μL of 50 μM LHCP denatured 8 M urea to 150 μL buffer D (50 mM KHEPES, pH 7.5 and 200 mM NaCl) or 2.5 μM cpSRP43 in buffer D. Light scattering was monitored at 360 nm on a UV-Vis spectrometer (Beckman Coulter) over time until equilibrium was reached. The percentage of soluble LHCP (% soluble) at equilibrium was plotted for each single-cysteine cpSRP43 mutant.
Measurement of cpSRP54M binding
Experiments were carried out as previously described.34 Binding of cpSRP43 to the cpSRP54M peptide (QKQKAPPGTARRKRKAC) was detected by changes in fluorescence anisotropy of Fluorescein labeled on the C-terminal cysteine. Anisotropy measurements were performed in 50 mM HEPES (pH 7.5), 200 mM NaCl at 25 °C on FluoroLog 3–22 (Yobin Yvon), using 10 nM fluorescein-labeled 54 M peptide and indicated concentrations of MTSSL-labeled cpSRP43. The samples were excited at 500 nm, and the fluorescence anisotropy was recorded at 527 nm. To obtain the equilibrium dissociation constant between cpSRP43 and 54 M, the data were fit to:
| (1) |
in which Aobs is the observed anisotropy value, A0 is the anisotropy value of 54 M-fluorescein alone, ΔA is the change in anisotropy at saturating concentrations of cpSRP43, and Kd is the equilibrium dissociation constant for the interaction between MTSSL-labeled cpSRP43 and 54 M-fluorescein.
Site-directed spin labeling and EPR measurements
Spin labeling reactions were performed in 50 mM HEPES (pH 7.5), 200 mM NaCl. Reduced and degassed single cysteine mutants of cpSRP43 were labeled with a five- to tenfold molar excess of MTSSL (Toronto Research Chemicals) at room temperature in the dark for 2–3 h. Excess MTSSL was removed by gel filtration chromatography. The concentrations of spin-labeled samples were 30–100 μM cpSRP43, at least 10-fold higher than the apparent Kd of cysless cpSRP43 for 54 M (0.39 μM)38 and LHCP (2–3 μM).34 After formation of the cpSRP43-LHCP complex, samples were ultracentrifuged at 100,000 rpm for 30 minutes to remove aggregated proteins. EPR spectra were acquired using a 9.4-GHz (X-band) EMX EPR spectrometer (Bruker) equipped with an ER 4119HS cavity at 20–23 °C. 5–30 scans were accumulated and averaged using microwave power of 5 mW with modulation amplitude set at 1 gauss and a magnetic field sweep width of 100 gauss. The central linewidth of EPR spectra was the same at microwave powers of 0.2 – 5 mW. Less than 2% background labeling was observed; background subtraction was therefore not necessary.
HDX detected by mass spectrometry
Solution-phase amide HDX experiments were carried out with a fully automated system (CTC HTS PAL, LEAP Technologies, Carrboro, NC; housed inside a 4 °C cabinet) as described previously with slight modifications.70 HDX of full-length cpSRP43, cpSRP43(intein), and cpSRP43-SBD was performed at 4 °C in 50 mM KHEPES (pH 8.0) and 200 mM NaCl using an automated system described previously.70 All samples were run at 10 μM. L18 and 54 M peptides were added at a 1:1 molar ratio to cpSRP43. Briefly, protein was incubated at 4 °C in a D2O buffer for a range of exchange times (0 s, 10 s, 30 s, 60 s, 900 s, and 3600 s) before quenching the exchange reaction with an acidic quench solution (pH 2.4) containing 3 M urea and 1% TFA. All mixing and pepsin digestions were carried out on a LEAP Technologies Twin HTS PAL liquid handling robot housed inside a temperature controlled fridge. Protein digestion was performed in-line with chromatography using an immobilized pepsin column. Mass spectra were acquired on a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo-Fisher Scientific).
Peptides were identified using tandem MS/MS experiments performed with QExactive (Thermo Fisher Scientific, San Jose, CA) over a 70 min linear gradient. Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis per scan event. The MS/MS*.raw data files were converted to *.mgf files and then submitted to MASCOT (version 2.3 Matrix Science, London, UK) for peptide identification.71 The maximum number of missed cleavages was set at 4 with the mass tolerance for precursor ions ±0.6 Da and for fragment ions ±8 ppm. Oxidation to methionine was selected for variable modification. Pepsin was used for digestion and no specific enzyme was selected in the MASCOT during the search. Peptides included in the peptide set used for HDX detection had a MASCOT score of 20 or greater. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence and false positives were ruled out if they did not pass a 1% false discovery rate. The MS/MS spectra of all the peptide ions from the MASCOT search were further manually inspected, and only the unique charged ions with the highest MASCOT score were included in HDX peptide set.
HDX analyses were performed in triplicate, with single preparations of each purified protein/complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation. This is accomplished determining the observed averages of the undeuterated and fully deuterated spectra and using the conventional formula described elsewhere.72 Statistical significance for the differential HDX data is determined by an unpaired t-test for each time point, and is integrated into the HDX Workbench software.70,73
BTFA labeling and 19F NMR
Labeling reactions were performed in 50 mM HEPES (pH 7.5), 200 mM NaCl. 200 μM concentrations of reduced and degassed single cysteine mutants of cpSRP43 were labeled with a 2 mM (tenfold molar excess) of BTFA (Sigma Aldrich) at room temperature for 2 hours. Excess BTFA was removed by two rounds of buffer exchange into 50 mM sodium phosphate (pH 6.5), 150 mM NaCl then supplemented with 10% D2O (NMR buffer). The final concentrations of BTFA-labeled samples were 25–100 μM cpSRP43. For urea titrations, samples were buffer exchanged into NMR buffer supplemented with either 0.5, 1, or 6 M urea. 1D 19F spectra were acquired at 17 °C on a Bruker Avance 600 spectrometer equipped with a 5 mm QCI 1H/19F/13C/15N quadruple resonance cryoprobe with a single axis Z-gradient. 19F-DOSY measurements were performed and used to estimate the diffusion constant as described previously.52 NMR data were processed with NMRPipe.74 Peaks were analyzed in MestReNova with peak deconvolutions fit to a generalized Lorentzian using simulated annealing.75
1H-15N NMR spectroscopy
Multidimensional NMR spectra were recorded on a Bruker Avance 800 spectrometer using a 5 mm cryogenic TCI probe with a single Z-axis gradient, and on an Avance 900 spectrometer using a 5 mm room temperature TXI probe with a triple axis gradient. NMR spectra were acquired at 17 °C using 2H, 15N-labeled protein (~0.2 mM) in NMR buffer containing 10% (vol/vol) D2O at pH 6.5 or with low doses (0–0.4 M) of urea. NMR data were processed with NMRPipe74 and analyzed with NMRView Java.76
Supplementary Material
Acknowledgement
We thank members of the Shan group for helpful comments on the manuscript and Dr. Peter Qin for helpful discussions in interpretation of the EPR data. This work was supported by NIH Training Grant 2T32 GM 7616-36 to Paul Sternberg, NIH R01 GM114390, NIH R35 GM136321, and DOE DE.SC0020661 to Shu-ou Shan, and the Skaggs Institute for Chemical Biology to Peter Wright.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2020.11.007.
References
- 1.Zhuravleva A, Gierasch LM, (2011). Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc. Natl. Acad. Sci. USA, 108, 6987–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH, (2006). Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature, 440, 1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schirmer EC, Glover JR, Singer MA, Lindquist S, (1996). HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem. Sci, 21, 289–296. [PubMed] [Google Scholar]
- 4.Bepperling A, Alte F, Kriehuber T, Braun N, Weinkauf S, Groll M, Haslbeck M, Buchner J, (2012). Alternative bacterial two-component small heat shock protein systems. Proc. Natl. Acad. Sci. USA, 109, 20407–20412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bardwell JCA, Jakob U, (2012). Conditional disorder in chaperone action. Trends Biochem. Sci, 37, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foit L, George JS, Zhang BW, Brooks CL, Bardwell JCA, (2013). Chaperone activation by unfolding. Proc. Natl. Acad. Sci. USA, 110, E1254–E1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baughman HER, Pham T-H-T, Adams CS, Nath A, Klevit RE, (2020). Release of a disordered domain enhances HspB1 chaperone activity toward tau. Proc. Natl. Acad. Sci. USA, 117, 2923–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang C, Rossi P, Saio T, Kalodimos CG, (2016). Structural basis for the antifolding activity of a molecular chaperone. Nature, 537, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walton TA, Sandoval CM, Fowler CA, Pardi A, Sousa MC, (2009). The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc. Natl. Acad. Sci. USA, 106, 1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randall LL, Hardy SJS, (2002). SecB, one small chaperone in the complex milieu of the cell. Cell. Mol. Life Sci, 59, 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burmann BM, Wang C, Hiller S, (2013). Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat. Struct. Mol. Biol, 20, 1265–1272. [DOI] [PubMed] [Google Scholar]
- 12.Thoma J, Burmann BM, Hiller S, Müller DJ, (2015). Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nat. Struct. Mol. Biol, 22, 795–802. [DOI] [PubMed] [Google Scholar]
- 13.He L, Sharpe T, Mazur A, Hiller S, (2016). A molecular mechanism of chaperone-client recognition. Sci. Adv, 2, e1601625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hennecke G, Nolte J, Volkmer-Engert R, Schneider-Mergener J, Behrens S, (2005). The periplasmic chaperone SurA exploits two features characteristic of integral outer membrane proteins for selective substrate recognition. J. Biol. Chem, 280, 23540–23548. [DOI] [PubMed] [Google Scholar]
- 15.Hegde RS, Keenan RJ, (2011). Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat. Rev. Mol Cell. Biol, 12, 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aviram N, Ast T, Costa EA, Arakel EC, Chuartzman SG, Jan CH, Haßdenteufel S, Dudek J, et al. , (2016). The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature, 540, 134–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartl FU, (2017). Protein misfolding diseases. Annu. Rev. Biochem, 86, 21–26. [DOI] [PubMed] [Google Scholar]
- 18.Schünemann D, (2004). Structure and function of the chloroplast signal recognition particle. Curr. Genet, 44, 295–304. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Henry R, Yuan J, Cline K, Hoffman NE, (1995). A chloroplast homologue of the signal recognition particle subunit SRP54 is involved in the posttranslational integration of a protein into thylakoid membranes. Proc. Natl. Acad. Sci. USA, 92, 3789–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuenemann D, Gupta S, Persello-Cartieaux F, Klimyuk VI, Jones JDG, Nussaume L, Hoffman NE, (1998). A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc. Natl. Acad. Sci. USA, 95, 10312–10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klimyuk VI, Persello-Cartieaux F, Havaux M, Contard-David P, Schuenemann D, Meiherhoff K, Gouet P, Jones JD, et al. , (1999). A chromodomain protein encoded by the arabidopsis CAO gene is a plant-specific component of the chloroplast signal recognition particle pathway that is involved in LHCP targeting. Plant Cell, 11, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tu CJ, Peterson EC, Henry R, Hoffman NE, (2000). The L18 domain of light-harvesting chlorophyll proteins binds to chloroplast signal recognition particle 43. J. Biol. Chem, 275, 13187–13190. [DOI] [PubMed] [Google Scholar]
- 23.Tu C-J, Schuenemann D, Hoffman NE, (1999). Chloroplast FtsY, chloroplast signal recognition particle, and GTP are required to reconstitute the soluble phase of light-harvesting chlorophyll protein transport into thylakoid membranes. J. Biol. Chem, 274, 27219–27224. [DOI] [PubMed] [Google Scholar]
- 24.Moore M, Harrison MS, Peterson EC, Henry R, (2000). Chloroplast oxa1p homolog Albino3 is required for post-translational integration of the light harvesting chlorophyll-binding protein into thylakoid membranes. J. Biol. Chem, 275, 1529–1532. [DOI] [PubMed] [Google Scholar]
- 25.Eichacker LA, Henry R, (2001). Function of a chloroplast SRP in thylakoid protein export. BBA-Mol. Cell Res, 1541, 120–134. [DOI] [PubMed] [Google Scholar]
- 26.Kuhn A, Stuart R, Henry R, Dalbey RE, (2003). The Alb3/Oxa1/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion?. Trends Cell Biol, 13, 510–516. [DOI] [PubMed] [Google Scholar]
- 27.Lewis NE, Marty NJ, Kathir KM, Rajalingam D, Kight AD, Daily A, Kumar TKS, Henry RL, et al. , (2010). A dynamic cpSRP43-Albino3 interaction mediates translocase regulation of chloroplast signal recognition particle (cpSRP)-targeting components. J. Biol. Chem, 285, 34220–34230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falk S, Ravaud S, Koch J, Sinning I, (2010). The C terminus of the Alb3 membrane insertase recruits cpSRP43 to the thylakoid membrane. J. Biol. Chem, 285, 5954–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dünschede B, Bals T, Funke S, Schünemann D, (2011). Interaction studies between the chloroplast signal recognition particle subunit cpSRP43 and the full-length translocase Alb3 reveal a membrane-embedded binding region in Alb3 protein. J. Biol. Chem, 286, 35187–35195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghanotakis DF, Demetriou DM, Yocum CF, (1987). Isolation and characterization of an oxygen-evolving Photosystem II reaction center core preparation and a 28 kDa Chl-a-binding protein. BBA-Bioenergetics, 891, 15–21. [Google Scholar]
- 31.Jaru-Ampornpan P, Chandrasekar S, Shan S, (2007). Efficient interaction between two GTPases allows the chloroplast SRP pathway to bypass the requirement for an SRP RNA. Mol. Biol. Cell, 18, 2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaru-Ampornpan P, Shen K, Lam VQ, Ali M, Doniach S, Jia TZ, Shan S, (2010). ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat. Struct. Mol. Biol, 17, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stengel KF, Holdermann I, Cain P, Robinson C, Wild K, Sinning I, (2008). Structural basis for specific substrate recognition by the chloroplast signal recognition particle protein cpSRP43. Science, 321, 253–256. [DOI] [PubMed] [Google Scholar]
- 34.Liang F-C, Kroon G, McAvoy CZ, Chi C, Wright PE, Shan S, (2016). Conformational dynamics of a membrane protein chaperone enables spatially regulated substrate capture and release. Proc. Natl. Acad. Sci. USA, 113, E1615–E1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cain P, Holdermann I, Sinning I, Johnson AE, Robinson C, (2011). Binding of chloroplast signal recognition particle to a thylakoid membrane protein substrate in aqueous solution and delineation of the cpSRP43-substrate interaction domain. Biochem. J, 437, 149–155. [DOI] [PubMed] [Google Scholar]
- 36.Jaru-Ampornpan P, Liang F-C, Nisthal A, Nguyen TX, Wang P, Shen K, Mayo SL, Shan S, (2013). Mechanism of an ATP-independent protein disaggregase: II. Distinct molecular interactions drive multiple steps during aggregate assembly. J. Biol. Chem, 288, 13431–13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McAvoy CZ, Siegel A, Piszkiewicz S, Miaou E, Yu M, Nguyen T, Moradian A, Sweredoski MJ, et al. , (2018). Two distinct sites of client protein interaction with the chaperone cpSRP43. J. Biol. Chem, 293, 8861–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holdermann I, Meyer NH, Round A, Wild K, Sattler M, Sinning I, (2012). Chromodomains read the arginine code of post-translational targeting. Nat. Struct. Mol. Biol, 19, 260–263. [DOI] [PubMed] [Google Scholar]
- 39.Gao F, Kight AD, Henderson R, Jayanthi S, Patel P, Murchison M, Sharma P, Goforth RL, et al. , (2015). Regulation of structural dynamics within a signal recognition particle promotes binding of protein targeting substrates. J. Biol. Chem, 290, 15462–15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kathir KM, Rajalingam D, Sivaraja V, Kight A, Goforth RL, Yu C, Henry R, Kumar TKS, (2008). Assembly of chloroplast signal recognition particle involves structural rearrangement in cpSRP43. J. Mol. Biol, 381, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horn A, Hennig J, Ahmed YL, Stier G, Wild K, Sattler M, Sinning I, (2015). Structural basis for cpSRP43 chromodomain selectivity and dynamics in Alb3 insertase interaction. Nat. Commun, 6, 8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engen JR, (2009). Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem, 81, 7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Islam SM, Stein RA, Mchaourab HS, Roux B, (2013). Structural refinement from restrained-ensemble simulations based on EPR/DEER data: Application to T4 lysozyme. J. Phys. Chem, 117, 4740–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.López CJ, Oga S, Hubbell WL, (2012). Mapping molecular flexibility of proteins with site-directed spin labeling: A case study of myoglobin. Biochemistry-US, 51, 6568–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van SP, Birrell GB, Griffith OH, (1974). Rapid anisotropic motion of spin labels. models for motion averaging of the ESR parameters. J. Magn. Reson, 15, 444–459. [Google Scholar]
- 46.Timofeev VP, Nikolsky DO, (2003). The role of the fast motion of the spin label in the interpretation of EPR spectra for spin-labeled macromolecules. J. Biomol. Struct. Dyn, 21, 367–378. [DOI] [PubMed] [Google Scholar]
- 47.Altenbach C, Froncisz W, Hyde JS, Hubbell WL, (1989). Conformation of spin-labeled melittin at membrane surfaces investigated by pulse saturation recovery and continuous wave power saturation electron paramagnetic resonance. Biophys. J, 56, 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altenbach C, Flitsch SL, Khorana HG, Hubbell WL, (1989). Structural studies on transmembrane proteins. 2. Spin labeling of bacteriorhodopsin mutants at unique cysteines. Biochemistry-US, 28, 7806–7812. [DOI] [PubMed] [Google Scholar]
- 49.Hubbell WL, Gross A, Langen R, Lietzow MA, (1998). Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struc. Biol, 8, 649–656. [DOI] [PubMed] [Google Scholar]
- 50.Altieri AS, Hinton DP, Byrd RA, (1995). Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J. Am. Chem. Soc, 117, 7566–7567. [Google Scholar]
- 51.Sun X, Dyson HJ, Wright PE, (2017). Fluorotryptophan incorporation modulates the structure and stability of transthyretin in a site-specific manner. Biochemistry-US, 56, 5570–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun X, Dyson HJ, Wright PE, (2018). Kinetic analysis of the multistep aggregation pathway of human transthyretin. Proc. Natl. Acad. Sci. USA, 115, E6201–E6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mittermaier AK, Kay LE, (2009). Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci, 34, 601–611. [DOI] [PubMed] [Google Scholar]
- 54.Sue S-C, Cervantes C, Komives EA, Dyson HJ, (2008). Transfer of flexibility between ankyrin repeats in IκBα upon formation of the NF-κB complex. J. Mol. Biol, 380, 917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamboy JA, Kim H, Dembinski H, Ha T, Komives EA, (2013). Single-molecule FRET reveals the native-state dynamics of the IκBα ankyrin repeat domain. J. Mol. Biol, 425, 2578–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nam Y, Sliz P, Song L, Aster JC, Blacklow SC, (2006). Structural basis for cooperativity in recruitment of MAML coactivators to notch transcription complexes. Cell, 124, 973–983. [DOI] [PubMed] [Google Scholar]
- 57.Wilson JJ, Kovall RA, (2006). Crystal structure of the CSL-notch-mastermind ternary complex bound to DNA. Cell, 124, 985–996. [DOI] [PubMed] [Google Scholar]
- 58.Löw C, Weininger U, Zeeb M, Zhang W, Laue ED, Schmid FX, Balbach J, (2007). Folding mechanism of an ankyrin repeat protein: Scaffold and active site formation of human CDK inhibitor p19INK4d. J. Mol. Biol, 373, 219–231. [DOI] [PubMed] [Google Scholar]
- 59.Barrick D, Ferreiro DU, Komives EA, (2008). Folding landscapes of ankyrin repeat proteins: experiments meet theory. Curr. Opin. Struc. Biol, 18, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abbott GL, Blouse GE, Perron MJ, Shore JD, Luck LA, Szabo AG, (2004). 19F NMR studies of plasminogen activator inhibitor-1. Biochemistry-US, 43, 1507–1519. [DOI] [PubMed] [Google Scholar]
- 61.Robertson DE, Kroon PA, Ho C, (1977). Nuclear magnetic resonance and fluorescence studies of substrate-induced conformational changes of histidine-binding protein J of Salmonella typhimurium. Biochemistry-US, 16, 1443–1451. [DOI] [PubMed] [Google Scholar]
- 62.Funke S, Knechten T, Ollesch J, Schünemann D, (2005). A unique sequence motif in the 54-kDa subunit of the chloroplast signal recognition particle mediates binding to the 43-kDa subunit. J. Biol. Chem, 280, 8912–8917. [DOI] [PubMed] [Google Scholar]
- 63.Hiller S, (2019). Chaperone-bound clients: The importance of being dynamic. Trends Biochem. Sci, 44, 517–527. [DOI] [PubMed] [Google Scholar]
- 64.Alderson TR, Roche J, Gastall HY, Dias DM, Pritišanac I, Ying J, Bax A, Benesch JLP, et al. , (2019). Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun, 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alderson TR, Ying J, Bax A, Benesch JLP, Baldwin AJ, (2020). Conditional disorder in small heat-shock proteins. J. Mol. Biol, 432, 3033–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jovcevski B, Kelly MA, Rote AP, Berg T, Gastall HY, Benesch JLP, Aquilina JA, Ecroyd H, (2015). Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol, 22, 186–195. [DOI] [PubMed] [Google Scholar]
- 67.Tapley TL, Körner JL, Barge MT, Hupfeld J, Schauerte JA, Gafni A, Jakob U, Bardwell JCA, (2009). Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc. Natl. Acad. Sci. USA, 106, 5557–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knoefler D, Thamsen M, Koniczek M, Niemuth NJ, Diederich A-K, Jakob U, (2012). Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol. Cell, 47, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sedgwick SG, Smerdon SJ, (1999). The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem. Sci, 24, 311–316. [DOI] [PubMed] [Google Scholar]
- 70.Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, Griffin PR, (2006). Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem, 78, 1005–1014. [DOI] [PubMed] [Google Scholar]
- 71.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS, (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis, 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Z, Smith DL, (1993). Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci, 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, et al. , (2012). HDX Workbench: Software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectr, 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, (1995). NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 75.Willcott MR, (2009). MestRe Nova. J. Am. Chem. Soc, 131 13180–13180. [Google Scholar]
- 76.Johnson BA, Blevins RA, (1994). NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR, 4, 603–614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.