

Abstract
Neurons are frequently classified into distinct types on the basis of structural, physiological, or genetic attributes. To better constrain the definition of neuronal cell types, we characterized the transcriptomes and intrinsic physiological properties of over 4,200 mouse visual cortical GABAergic interneurons and reconstructed the local morphologies of 517 of those neurons. We find that most transcriptomic types (t-types) occupy specific laminar positions within visual cortex, and, for most types, the cells mapping to a t-type exhibit consistent electrophysiological and morphological properties. These properties display both discrete and continuous variation among t-types. Through multimodal integrated analysis we define 28 met-types that have congruent morphological, electrophysiological, and transcriptomic properties and robust mutual predictability. We identify layer-specific axon innervation pattern as a defining feature distinguishing different met-types. These met-types represent a unified definition of cortical GABAergic interneuron types, providing a systematic framework to capture existing knowledge and bridge future analyses across different modalities.
Graphical Abstract
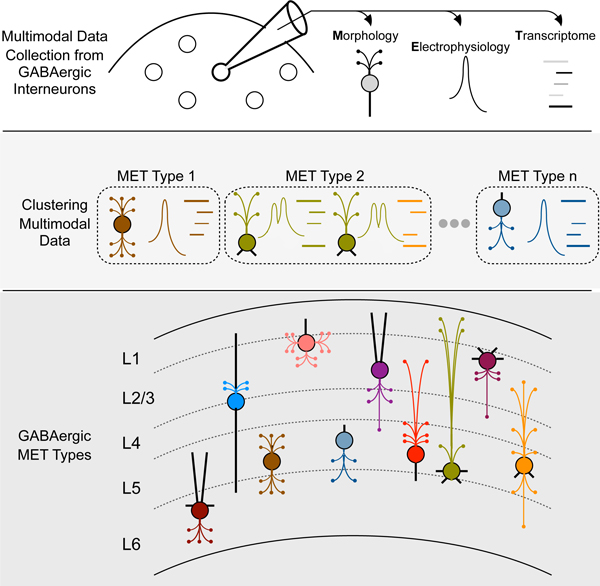
ETOC:
GABAergic cortical interneurons of the mouse visual cortex can be defined into 28 types based on their morphological, electrophysiological and transcriptomic properties and are distinguished by their layer-specific axon innervation patterns.
Introduction
The mammalian brain contains many millions, and in some cases billions, of neurons. No two neurons are identical, yet neurons are regularly categorized into classes and types based on a variety of criteria. Cell type categorization provides a logistical and conceptual framework for understanding how cells and circuits enable brain function.
Early efforts to define and distinguish groups of neurons focused on the brain areas in which they reside, their precise locations within a given brain area, and/or features of their dendritic, somatic, and/or axonal compartments. Electrophysiological, biochemical, immunohistochemical, and genetic methods have subsequently helped define and distinguish increasingly more specific sets of neurons (reviewed recently in (Klausberger and Somogyi, 2008; Huang and Paul, 2019; Tremblay et al., 2016; Zeng and Sanes, 2017; Cembrowski and Spruston, 2019)). Despite the diversity and amount of data collected and analyzed to this point, the field has not readily converged on a consensus number, or even definition, of neuronal cell types; indeed, efforts to determine a consensus number of cell types have been limited by well-documented challenges (Ascoli et al., 2008; DeFelipe et al., 2013; Tebaykin et al., 2018).
Recent advances in the collection and analysis of single cell RNA-sequencing (scRNAseq) data provide a promising new way to define cell types and how they relate to one another (Saunders et al., 2018; Tasic et al., 2018; Zeisel et al., 2018; Zeisel et al., 2015; Tasic et al., 2016; Shekhar et al., 2016). This relatively new, transcriptomic approach to neuronal classification is powerful due to its high dimensionality — quantitative expression data is obtained for thousands of genes from each neuron — and high scalability — scRNAseq data can be collected from thousands to millions of individual cells. Moreover, new methods such as the Patch-seq recording technique (Fuzik et al., 2016; Cadwell et al., 2016; Scala et al., 2019; Földy et al., 2016) enable one to directly relate the transcriptomic features of a given neuron to other identifying characteristics of the same neuron - e.g., the neuron’s precise location, morphology, and/or intrinsic electrophysiological properties.
Our previous efforts have systematically defined neuronal cell types in mouse visual cortex based on either transcriptomic characteristics (Tasic et al., 2018; Tasic et al., 2016) or electrophysiological and morphological characteristics (Gouwens et al., 2019). In this study we endeavored to classify neuronal types in a more integrated manner by capturing the transcriptomic, electrophysiological and morphological properties of individual neurons via the Patch-seq technique. We chose to focus our initial efforts on cortical GABAergic interneurons for several reasons. First, the morphology of GABAergic interneurons is better preserved in ~350 um thick in vitro slice preparations than that of glutamatergic pyramidal neurons with known long-range projections. Second, transcriptomically-defined types of GABAergic interneurons are more readily conserved across cortical brain areas (Tasic et al., 2018). Third, the number of GABAergic cell types estimated from our scRNAseq data (Tasic et al., 2018) and morpho-electric data (Gouwens et al., 2019) from the mouse primary visual cortex (VISp) - i.e., the number of VISp GABAergic “t-types” (60) and “me-types” (26) - differs by more than a factor of two. Such discrepancies are not unique to VISp; studies applying anatomical, physiological, and neurochemical approaches in other cortical areas have also arrived at varying numbers of GABAergic neuronal types (Tremblay et al., 2016; Jiang et al., 2015; Markram et al., 2015). Resolving these discrepancies and deriving a unified set of GABAergic cell types based on congruence among different modalities will provide an essential foundation for achieving a common understanding of the function of these cell types across the field.
Evidence collected to date suggests the presence of four major subclasses of cortical GABAergic neurons: parvalbumin (Pvalb)-positive, somatostatin (Sst)-positive, vasoactive intestinal peptide (Vip)-positive cells, and cells that express 5-hydroxytryptamine receptor 3A but lack Vip (Htr3a+/Vip-) (Tremblay et al., 2016). Each of these subclasses can be subdivided into several types, largely defined by their axonal morphologies. For example, the Pvalb subclass can be split into basket (and related) cells and chandelier cells; the Sst subclass into Martinotti cells and non-Martinotti cells; the Vip subclass into bipolar, bitufted and multipolar cells; and the Htr3a+/Vip- subclass into neurogliaform cells and single bouquet cells. Our recent single-cell transcriptomic study of two mouse cortical areas (Tasic et al., 2018), VISp and anterolateral motor cortex (ALM), confirmed these four major subclasses and revealed an additional subclass (Sncg) that is related to the Vip and Htr3a+/Vip-(renamed to Lamp5) subclasses.
At the finest (terminal) level of our transcriptomic cell type taxonomy, we identified 60 GABAergic transcriptomic types altogether, each belonging to one of the five subclasses. Furthermore, using highly specific transgenic driver lines, we were able to correlate some transcriptomic types (t-types) with some well-known or newly defined morpho-electric types (me-types) (Tasic et al., 2018; Gouwens et al., 2019); for example, the Pvalb Vipr2 and Sst Chodl t-types correspond to chandelier cells and Nos1+ long-projecting Sst neurons, respectively. The morpho-electric profiles of most t-types, however, are largely unknown and their correspondence to historically defined GABAergic types has not been established.
Here we report results obtained from the analysis of over 4,200 standardized, quality-controlled Patch-seq recordings from GABAergic interneurons in visual cortex of the adult mouse. The dataset, freely available to the public via https://portal.brain-map.org, facilitates answers to a variety of critical questions, including: (1) to what extent do neurons categorized into the same transcriptomic type (t-type) on the basis of scRNAseq data exhibit similar morphological and/or electrophysiological features; (2) to what extent do neurons belonging to different t-types exhibit distinct electrophysiological and/or morphological features? Answering these and other related questions will facilitate the interpretation of high-throughput in situ molecular surveys of cell types in the brain currently being undertaken (Lee et al., 2014; Chen et al., 2015; Coskun and Cai, 2016; Kebschull et al., 2016; Codeluppi et al., 2018; Zhang et al., 2020). The answers will also help define neuronal cell types and features that differ between them, reveal the nature of cellular diversity and its underlying rules, and ultimately improve our understanding of cellular and circuit functions. In addition, the increased inhibitory neuron diversity accessed through transcriptomic types may reveal new insights about structural and electrophysiological organization of mouse visual cortex.
Results
To establish direct correspondence between the electrical, morphological, and transcriptomic features of mouse cortical neurons, we built a standardized pipeline to extract high quality data from all three modalities from single cells (see Figure 1A, Figure S1, and Methods). In particular, using a modified version of the Patch-seq technique (Cadwell et al., 2016; Fuzik et al., 2016; Földy et al., 2016), we characterized electrophysiological responses to a series of hyperpolarizing and depolarizing current injections, extracted and reverse transcribed nuclear and cytosolic mRNA, sequenced the resulting cDNA using the same scRNAseq method, SMART-Seq v4, as in our transcriptomic study (Tasic et al., 2018), and mapped each cell’s soma location and cortical depth into the Allen Mouse Common Coordinate Framework (CCF) (Kuan et al., 2015; Wang et al., 2020), a 3D anatomical reference space for the mouse brain. Both the transcriptomic and electrophysiological data from 4,270 cortical GABAergic interneurons passed our QC criteria (see Methods and Figure S1A); the 2,341 cells that exhibited adequate biocytin filling were imaged at 63X resolution, and the dendritic and axonal morphologies of 517 of these cells were reconstructed.
Figure 1: Transcriptomic analysis of scRNAseq data obtained from Patch-seq recordings.
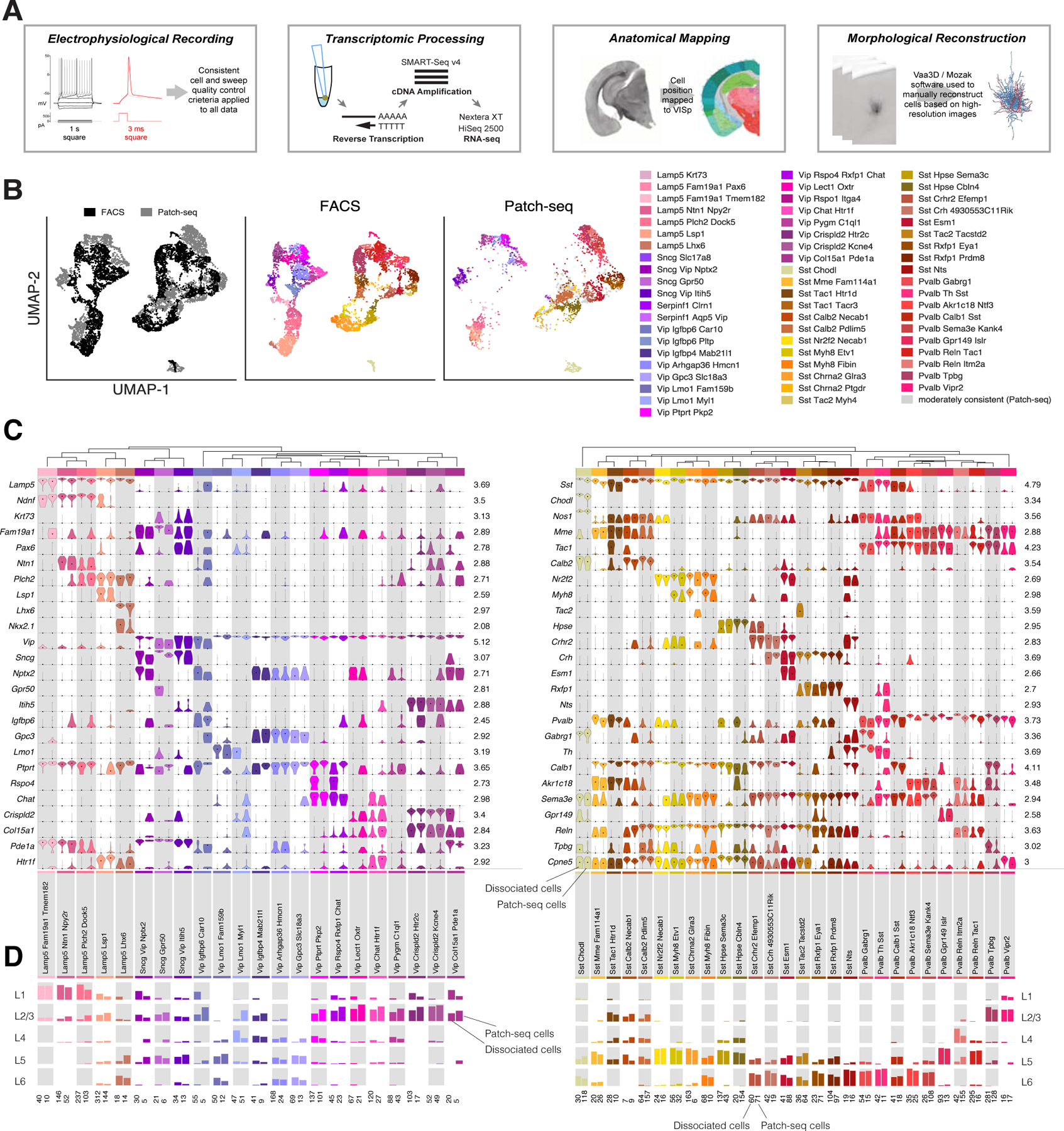
(A) Summary of the Patch-seq data collection and processing pipeline. (B) Transcriptomic UMAP plots based on principal components of gene expression (Methods; left: n=6,080 dissociated cells, black, and n=3,855 cells from Patch-seq recordings, gray). Colors in middle and right plots indicate t-type. (C) Marker gene expression distributions within each t-type are represented by pairs of violin plots corresponding to Patch-seq recordings and dissociated cells from (Tasic et al., 2018) (right and left in each column for each type, respectively). Rows are genes, black dots are medians. Values within each row are normalized between 0 and maximum detected (shown on the y axis), displayed on a log10 scale. (D) Layer distribution of cells for each t-type in both datasets. In each column the dissociated cells and Patch-seq recordings are shown on the left and right, respectively. Total number of cells from Patch-seq recordings and dissociated cells in each type is shown below each column on right and left, respectively. For (C) and (D), only cells from transgenic lines common to both data sets (see Table S1) and only types with at least 5 cells were used (n=4,651 dissociated cells and n=2,260 Patch-seq recordings). For part (D) only cells with specific layer assignment are shown (n=3,767 dissociated cells and n=2,260 Patch-seq recordings). See also Figure S2 and Table S1.
Relating neurons assayed via Patch-seq recordings to transcriptomic types
The scRNAseq transcriptomes from cDNA obtained from Patch-seq recordings of individual GABAergic interneurons exhibited a similar number of total genes as the scRNAseq transcriptomes obtained from individual dissociated neurons in our previous transcriptomic study (Figure S2A). Moreover, the two transcriptomic datasets had similar structure when visualized together via a Uniform Manifold Approximation and Projection (UMAP, (Becht et al., 2018)) method (Figure 1B). However, we (Figure S2B) and others (Tripathy et al., 2018) found that the mRNA collected from one neuron via a Patch-seq recording often includes contaminating mRNA from adjacent neurons and/or non-neuronal cells. In addition, compared to those of equivalent dissociated cells, the transcriptomes of cells obtained by Patch-seq recordings more frequently exhibit “dropout” — a technical failure to detect some mRNA species that were actually present (Figure S2B).
To leverage the quality and large size of transcriptomic datasets obtained from dissociated neurons, we mapped the transcriptomes obtained via Patch-seq recordings to transcriptomic types (t-types) derived from dissociated cells (Tasic et al., 2018). Our mapping procedure for GABAergic neurons was designed to minimize the contribution of contamination from nearby non-neuronal cells (such as glia) and glutamatergic neurons. The mapping method was based on comparing a cell’s marker genes expression at each branch point of the reference hierarchical transcriptomic tree with the marker genes expression of the reference cell types (Methods, Figure S1D); robustness was assessed by a bootstrap approach (Methods).
We applied additional filters after the initial mapping (see Figure S1 and Methods). One particularly important filter focused on the confidence with which Patch-seq transcriptome mapped to one or more reference t-types. Some closely related t-types in the reference taxonomy have many cells with an “intermediate” identity between those t-types (Tasic et al., 2018). However, due to contamination and dropout, some Patch-seq transcriptomes unexpectedly have ambiguous identities between presumably distinct types. To distinguish between “expected” and “unexpected” ambiguity in mapping between t-types, we computed the reference mapping probability matrix between t-types (defined as the probability of a cell type being confused with other cell types in the reference dissociated cell data set). We then used Kullback-Leibler (KL) divergence between the mapping probability distributions and the reference mapping probability distribution as a measure of discrepancy; a larger KL divergence value indicates that the mapping distribution differs from the reference distribution. Cell mappings were categorized as “highly consistent,” “moderately consistent,” and “inconsistent” based on the KL divergence, gene expression correlation with reference cells, and the frequency of mapping to a single type (Methods, Figure S1G) . We found that 3,855 of the 4,270 cells had consistent t-type mapping (2,955 “highly consistent” and 900 “moderately consistent”, Figures S1G and S2C). When we performed analyses by t-type, we restricted the data set to the 2,955 cells with highly-consistent mapping to limit the effect of mapping ambiguities due to technical issues on our results (unless otherwise noted).
Several results suggested that this mapping approach is meaningful. First, an independent approach (Figure S1E, Methods) that did not rely on the hierarchical structure of the transcriptomic tree assigned cells to similar t-types (79% of highly-consistent cells were assigned to the same t-type by both methods). Second, cells obtained from transgenic driver mice that label subclasses or specific types of GABAergic interneurons map to similar t-types regardless of whether the examined transcriptomes originated from dissociated cells or Patch-seq recordings (Table S1). Third, the relative expression level of key marker genes was similar in cells from the two data sets assigned to the same t-type (Figure 1C), and cells of the same t-type from each method were near each other in the UMAP projection (Figure 1B). Fourth, the layer distribution of cells from Patch-seq recordings that mapped with high confidence to a given t-type closely resembled the layer distribution of dissociated cells mapping to the same t-type (Figure 1D).
Alignment of brain slices used for our Patch-seq recordings to the CCF 3D reference space (Wang et al., 2020) enabled detailed investigation into the cortical areas and layers from which scRNAseq data were obtained (Figure 2A). While data obtained from dissociated cells indicated a laminar preference for multiple t-types (see Figure 1D), the additional anatomical precision gained from targeting individual cells with recording electrodes in brain slices revealed further layer specificity (Figure 2B). For example, we found that some Sst t-types in L5 were preferentially located in the upper part (e.g., Sst Calb2 Pdlim5, Sst Hpse Cbln4, Sst Hpse Sema3c), while others occupied the lower part of L5 (e.g., Sst Esm1, Sst Rxfp1 Eya1, Sst Crhr2 Efemp1). Similarly, some t-types were found right at the L1-L2/3 border (Vip Col15a1 Pde1a, Pvalb Vipr2), while others were distributed across L2/3 (Vip Chat Htr1f, Pvalb Tpbg) (Figure 2B). A smaller number of t-types were notably found across multiple layers (e.g., Lamp5 Lsp1, Sncg Vip Itih5, and Sst Calb2 Pdlim5). The specific laminar (or sub-laminar) locations of many GABAergic t-types across subclasses provides a strong anatomical correlate of the cell groupings defined based on transcriptomic data alone.
Figure 2: Positions of cells from different t-types in a common reference space.
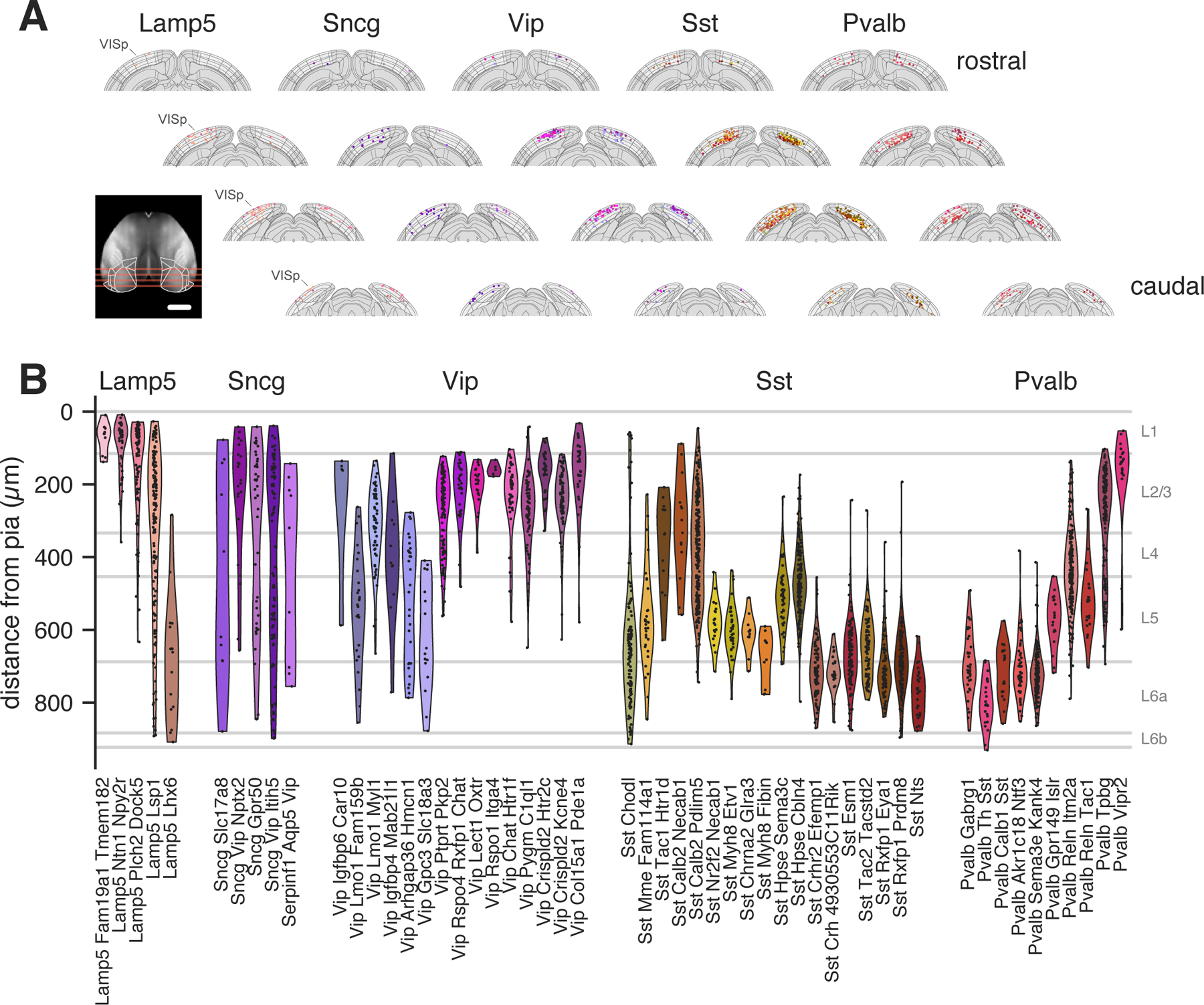
(A) Coronal views of positions of recorded cells (n=2,930 cells) organized by transcriptomic subclass and colored by t-type. Visual areas are indicated by a lighter background. Each t-type is visualized on a single cortical hemisphere for clarity. Inset shows the top-down positions of the virtual coronal slices. Inset scale bar: 2 mm. (B) Distance from pia by t-type (n=2,930 cells) where positions are aligned to average cortical layer thicknesses. Only t-types with at least 5 highly consistently mapped cells are shown in (A) and (B).
Characterizing the intrinsic electrophysiological features of transcriptomic types
How homogenous are the electrophysiological properties of neurons within a given t-type? How distinguishable are the electrophysiological properties of cells mapped to different t-types? To begin answering these questions, we analyzed all 4,270 neurons’ electrophysiological responses to a standardized current-clamp protocol composed of square-pulse current injections (Figure 3A, Figure S3A). Based on these measurements, we calculated 12 “feature vectors,” capturing properties such as action potential shape, the normalized response to hyperpolarizing current indicative of sag potential, and others. Applying a sparse principal components analysis (sPCA) to each feature vector revealed the most informative sparse principal components (sPCs, see Methods, (Gouwens et al., 2019)), which were aggregated to form a set of 44 sPCs that characterized the electrophysiological properties of the cells in the data set (Methods). Projecting the sPCs onto two dimensions using the UMAP method provided a useful visualization of electrophysiological feature gradients across all 4,270 cells (Figure 3B). Many of the sPCs were strongly related to traditional electrophysiological features; for example, the “subthreshold 2” sPC was correlated with input resistance (Pearson’s r = 0.90), and the “subthreshold (normalized) 1” sPC reflected the amount of sag induced by hyperpolarizing current steps (lower values of the sPC corresponding with more sag).
Figure 3: Electrophysiological characterization of transcriptomic types.
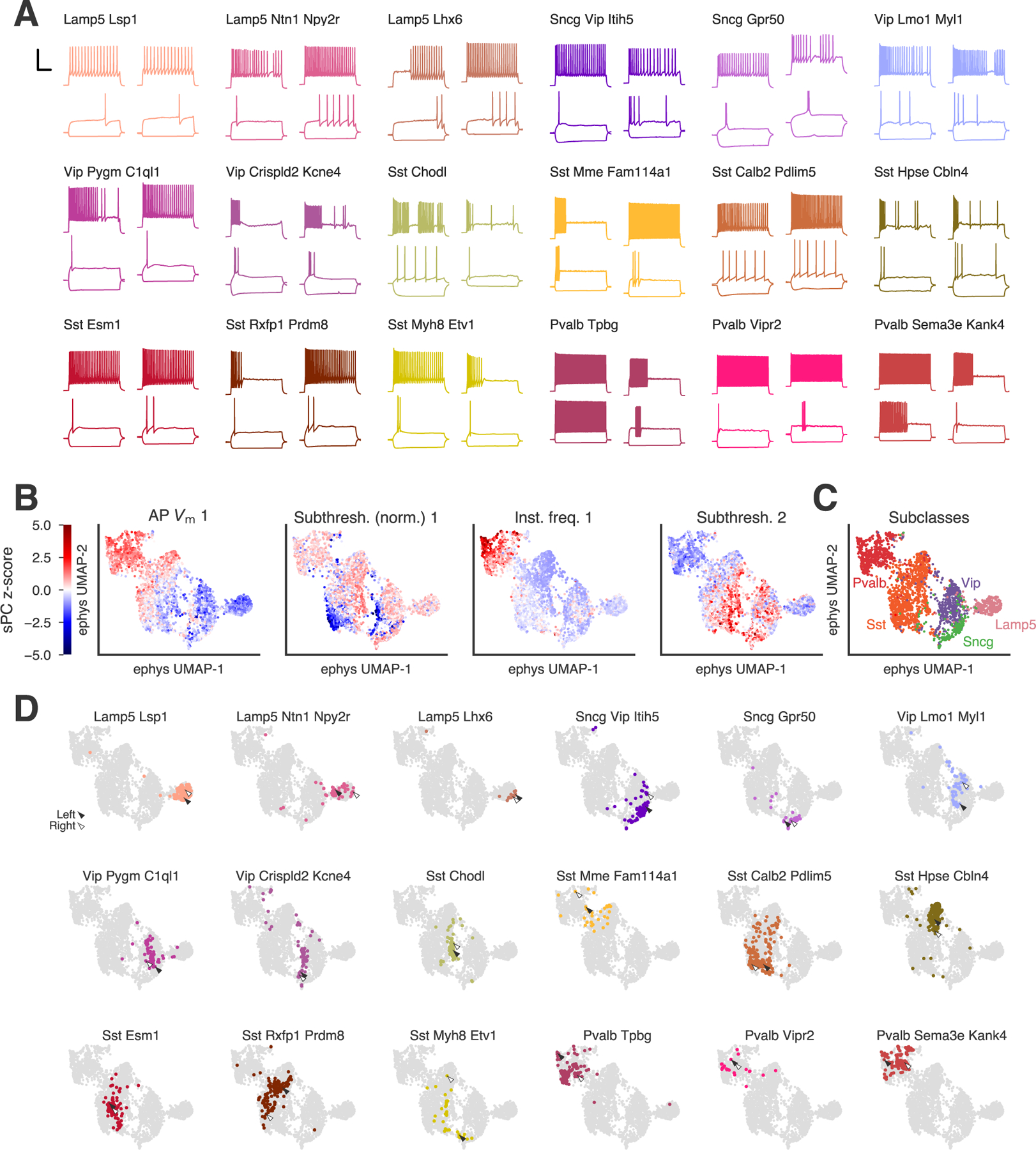
(A) Example responses from different t-types to 1 second-long current steps with stimulus amplitudes equal to −70 pA and rheobase for that cell (lower traces) and rheobase + 80 pA (upper trace). Two randomly-chosen examples shown for each t-type. Scale bar: vertical 50 mV, horizontal 250 ms. (B) UMAP plots based on a projection of the z-scored electrophysiology sparse principal components (sPCs). The values of four example sPCs are shown for all cells (n=4,270). (C) Electrophysiology UMAP plots with 2,955 cells (“highly consistent cells,” see Figure S1G) labeled by transcriptomic subclass. (D) Electrophysiology UMAP plots with individual t-types highlighted. Arrowheads indicate the locations of the examples (filled: left, hollow: right) shown in (A). See also Figures S3, S4, and S5.
Examining the location of the 2,955 cells with “highly consistent” t-type mapping demonstrated that, at the subclass level, most GABAergic interneurons occupied one of 4 large clouds in the electrophysiology UMAP projection, corresponding to the Lamp5, Vip, Sst, and Pvalb transcriptomic subclasses (Figure 3C). Cells of the Sncg subclass fell into a smaller region adjacent to the Vip cells. Cells mapping to individual t-types were typically found in a similar location (Figure 3D, Figure S3B), suggesting some degree of consistency in their electrophysiological properties. However, this consistency varied across t-types, with some having tight distributions (e.g., Lamp5 Lhx6) and others (e.g., Sst Mme Fam114a1) being spread across spaces occupied by different transcriptomic subclasses.
Pairs of t-types that occupy nearby or overlapping locations in electrophysiological space may have distributions of electrophysiological features that were essentially the same as each other. To test whether this was the case, we fit statistical models of pairs of t-types with either two components (one for each t-type) or a single component describing the cells from both t-types combined (Methods). We assessed the difference in fit between the models by calculating the log-likelihood ratio (LLR) (Figure S4A). Most pairs were significantly better fit by separate distributions for each t-type rather than by a single distribution describing all cells from the pair, indicating that most t-types differed from each other in their electrophysiological properties. Non-significant differences were typically found between closely related pairs of t-types.
The correspondence between electrophysiological and transcriptomic properties naturally raises the question of whether electrophysiological features can be quantitatively predicted from scRNAseq gene expression values. There are several challenges to doing this, including technical noise in gene expression measurements and potential differences between transcript levels and protein levels in a neuron. However, as a test case, we examined whether we could predict the median electrophysiology sPC values of t-types from the median ion channel gene expression values (Figure S5). The ability to predict the values with a sparse regression varied across the sPCs, but several had relatively high scores, suggesting that genes could be used to predict certain aspects of the electrophysiological responses.
Characterizing the morphological features of transcriptomic types
For a subset (517 cells) of transcriptomically and electrophysiologically characterized neurons from the Sst, Pvalb, Vip, Sncg, and Lamp5 subclasses, we also performed morphological analyses. Example morphologies from selected t-types are shown aligned to an average cortical template (Methods), followed by soma depth distributions and averaged dendrite and axon depth distributions (Figure 4A; all reconstructions can be found in Figures S6 and S7). Example neurons were chosen by calculating a pairwise similarity score for axonal branch structure and location (NBLAST score) (Costa et al., 2016; Kohl et al., 2013) for all reconstructed neurons in a t-type. The five neurons with the highest NBLAST scores are shown left to right in descending order, with the leftmost morphology being most characteristic for that t-type.
Figure 4: Morphological characterization of transcriptomic types.
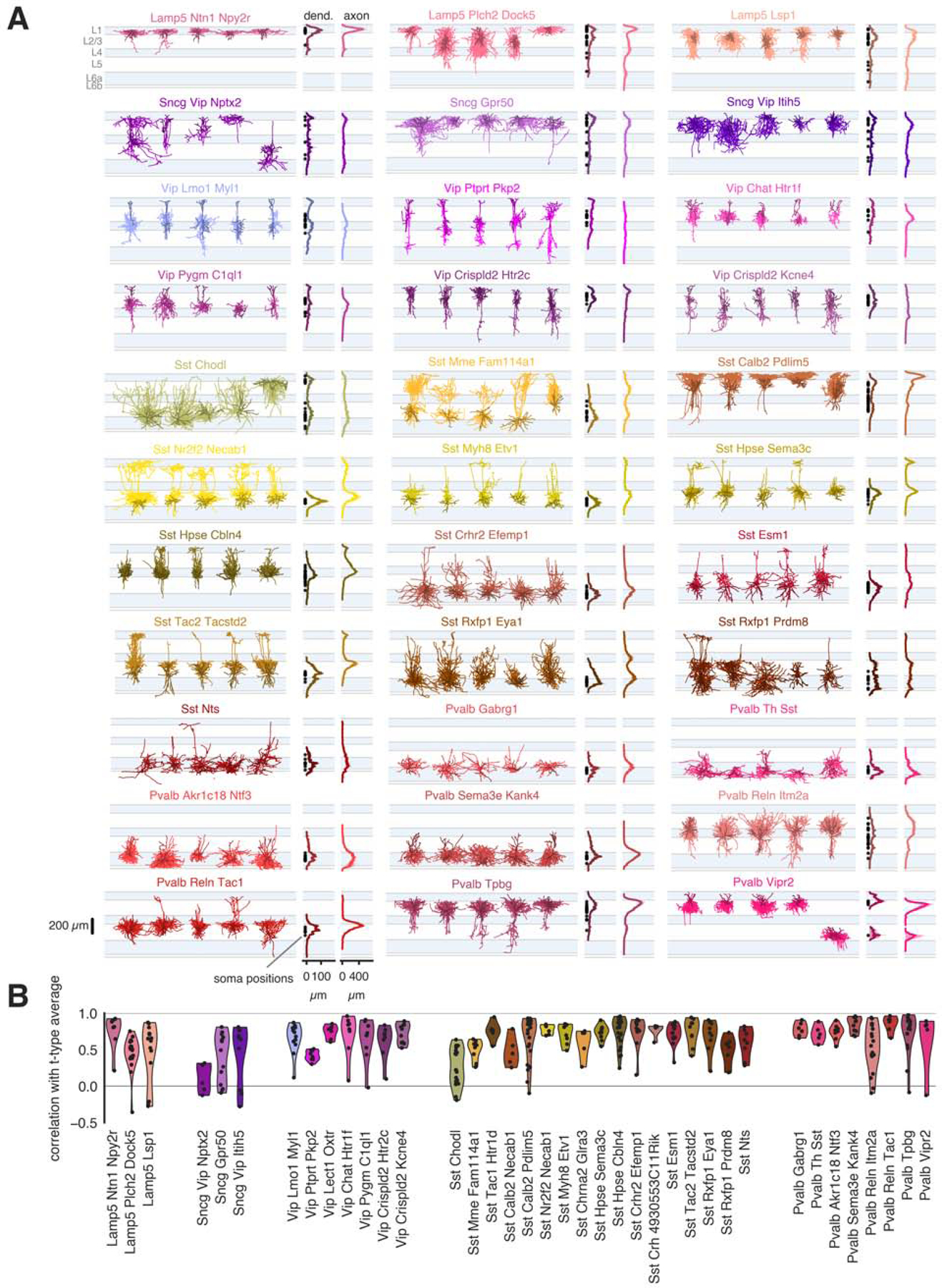
(A) Representative morphological reconstructions from t-types (selected by NBLAST similarity scores, Methods). Dendrite and axon depth histograms calculated from all reconstructions of the t-type are shown to the right, along with soma depth positions (black dots). Dendrites are in darker colors, axon in lighter colors. Histograms are shown as mean (lines) ± SEM (shaded regions). (B) Correlations between individual cell axon depth histograms and the average histogram of its t-type (excluding itself). Only t-types with at least 5 highly-consistent mapped cells are shown in (B). See also Figures S4, S6, and S7.
Across subclasses, we observed t-types with distinct dendrite and axon distribution patterns. Specifically, many t-types were distinguished by a layer-selective axonal projection. For example, in the Sst subclass, the Sst Calb2 Pdlim5 type had a L1-dominant (Martinotti-type) axon (DeFelipe et al., 2013), while Sst Tac1 Htr1d had a L2/3-dominant axon, Sst Hpse Cbln4 had a L4-dominant axon, and Sst Rxfp1 Prdm8 had a L5/L6-dominant axon. Other Sst t-types sent axon into L1 (Martinotti-like), but either had axon innervation that evenly split across two layers (Mme Fam114a1, Hpse Sema3c) or L5-dominant axon innervation (Myh8 Etv1 and Tac2 Tacstd2). Similarly, the Pvalb subclass had t-types that predominantly targeted L2/3 (Pvalb Tpbg), L2/3 and L4 (Pvalb Reln Itm2a), and L5 and L6 (Pvalb Sema3e Kank4, Pvalb Gabrg1). In contrast, t-types in the Vip subclass exhibited dendrites and axons that were relatively evenly distributed across multiple layers, while some t-types in the Lamp5 subclass (Lamp5 Plch2 Dock5, Lamp5 Lsp1) were found in multiple layers and had more variable axon distribution. Interestingly, layer-selective projection patterns often corresponded to the soma locations of each t-type, though not in all cases. Sst Calb2 Pdlim5 and Sst Tac1 Htrd1 cells each exhibited a reliable axon pattern despite their somas being distributed across L2/3, L4, and L5.
To evaluate the consistency of morphologies within a t-type, we measured the correlation between an individual cell’s laminar axon distribution pattern and the t-type average (Figure 4B). Most cells had a pattern that was positively correlated with the average, indicating similar axonal innervation patterns. We also examined differences in morphological features among pairs of t-types by LLR analysis as described above for electrophysiological properties (Figure S4A). Though more comparisons fell below the significance levels using morphological properties (possibly due to less statistical power because of fewer samples with morphologies), the Sst and Pvalb t-types had particularly strong correspondence with morphology.
Integrated classification of GABAergic neurons by correspondence between transcriptomic and morpho-electric types
The above analyses revealed that in many cases the cells mapping to a given t-type had consistent electrophysiological and morphological features. However, some t-types had overlapping morpho-electric features, while others exhibited electrophysiological (e.g., Sst Mme Fam114a1, Sst Tac1 Htr1d) or morphological (e.g., Sst Calb2 Pdlim5, Pvalb Reln Itm2a) heterogeneity. In light of the continuous variation observed between some t-types (Tasic et al., 2018), we investigated whether we could group cells from similar t-types that also shared similar electrophysiological and morphological properties and define morphological/electrophysiological/transcriptomic types (met-types) that exhibit a high degree of congruence across the three modalities assessed here.
To first identify sets of cells with similar morphological and electrophysiological properties, we performed an unsupervised me-clustering analysis (Gouwens et al., 2019) on the 517 cells with Patch-seq recordings for which we had electrophysiological and morphological data. We then compared the me-type assignments to the t-types (Figure 5A). Here, we examined cells with either highly-consistent or moderately-consistent t-type mapping (n=503) so that we would not potentially exclude cells that appeared as intermediate between t-types for true biological reasons.
Figure 5: Definition of met-types and type prediction accuracy.
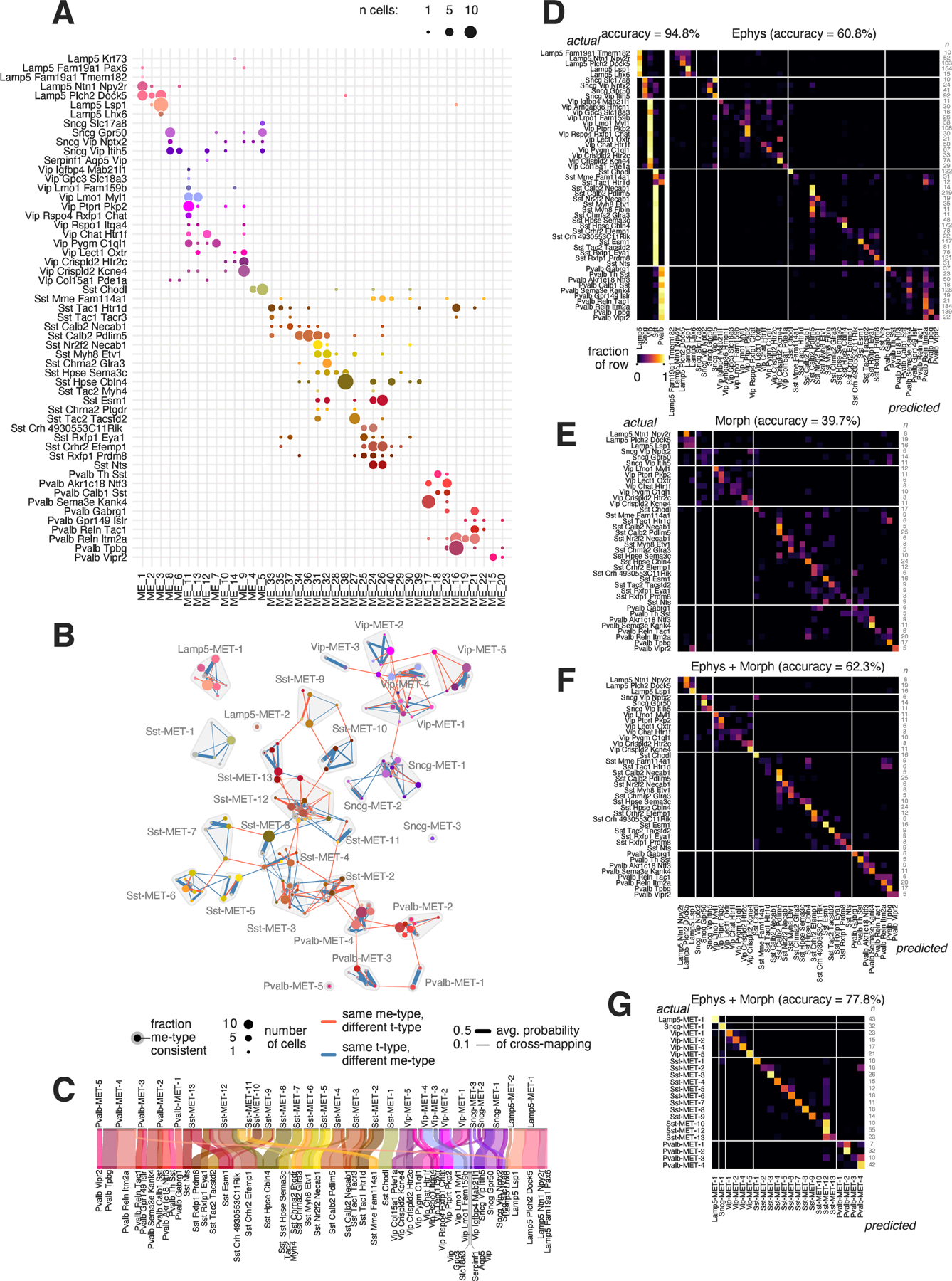
(A) Comparison of t-types with unsupervised joint electrophysiological and morphological clustering results (me-types). Both highly-consistent and moderately-consistent cells are shown (n=503). (B) Graph visualization of cross-mapping between me-type/t-type combinations. The nodes represent cells with a particular me-type/t-type combination as in (A), and the size of the node indicates the number of cells. Edges represent the presence of cells in a given me-type/t-type combination that have some probability of mapping to another t-type (orange lines) or me-type (blue lines); thicker lines indicate a higher probability. Nodes with only a single cell and no connections are not shown. Outlined groups indicate well-connected me-type/t-type combinations forming 28 met-types (Methods). (C) River plot showing the relationships between t-types (bottom) and met-types (top). (D) Out-of-bag confusion matrices of a random forest classifier trained to predict t-subclasses (left) and t-types (right) from electrophysiological features. Only t-types with at least 10 highly consistent mapped cells are used. (E–F) Same as (D) but for predicting t-types from morphological features alone (E) and from morphological and electrophysiological features together (F). Only t-types with at least 5 highly consistent mapped cells are used. (G) Same as (D) but for predicting met-types from morphological and electrophysiological features. Only met-types with at least 5 cells are used. For (D–G), values are normalized to the row sums. See also Figures S4 and S8.
Overall, we observed that transcriptomic similarity was closely related to similarity in the other modalities, leading to a roughly diagonal correspondence matrix. At the same time, straightforward one-to-one correspondence was rarely seen. As expected, cells that mapped to different transcriptomic subclasses mapped to mostly non-overlapping sets of me-types. The exceptions included types like Sst Mme Fam114a1 and Sst Tac1 Htr1d which, as mentioned above, contained cells that showed more Pvalb-like electrophysiological properties. The Sst Tac1 Htr1d t-type has previously been shown to contain cells with gene expression similar to some Pvalb t-types (Tasic et al., 2018). Certain t-types, such as Sst Chodl, Sst Hpse Cbln4, and Pvalb Sema3e Kank4, were each found mainly in a single respective me-type. For most t-types, however, cells were distributed across a number of me-types. The reverse was also true; most me-types comprised multiple t-types. This was also the case when cells were mapped to a different set of t-types based on a larger dissociated cell data set from all of cortex and the hippocampus (Figure S8, (Yao et al., 2020)).
To identify sets of cells that could be grouped into met-types, we made use of the fact that our mapping procedure estimated the probability of assigning a given cell to t-types other than its best match (Figure S2C). We computed a similar estimate for alternative me-type assignments by using a random-forest classifier and subsampling procedure (Methods). We used these t-type and me-type cross-mapping probabilities to construct a graph where the nodes were t-type/me-type combinations (i.e., the non-zero elements of the matrix in Figure 5A). Figure 5B shows the resulting graph after adding edges that connect nodes with the same me-type but different t-types (orange lines) or the same t-type but different me-types (blue lines); edges were weighted by the average cross-mapping probability. We then used a community detection algorithm (Traag et al., 2019) to identify 28 met-types from the graph. Most t-types were strongly associated with a single met-type, and most met-types were composed of several t-types (Figure 5C).
We next examined how well t-types and met-types could be predicted from electrophysiological and morphological features. As discussed above, the assignment of an individual cell to a t-type based on electrophysiological and/or morphological features may be difficult if features vary continuously between t-types. On the other hand, we expect met-types to be more reliably predicted using these modalities. To test this, we trained supervised classifiers to predict t-types and met-types based on electrophysiological and morphological data modalities.
We trained random forest classifiers on all 44 electrophysiology sPCs to predict either transcriptomic subclass or t-type (Figure 5D). The subclass classifier performed at >90% accuracy, indicating that GABAergic transcriptomic subclasses have distinctive electrophysiological features. However, the t-type classifier had a prediction accuracy of ~61%, and prediction error rates varied considerably across t-types. A t-type classifier trained with morphological features only had an accuracy of ~40% (Figure 5E), and the pattern of confused types differed from the electrophysiology classifier. The overall lower accuracy could be in part due to the smaller available number of cells with morphologies. However, classifier accuracy improved to 62% when electrophysiological features were used together with morphological features (Figure 5F). Prediction of met-types using combined electrophysiological and morphological features did have a higher accuracy (~78%) than t-type prediction (Figure 5G), although we note that this is likely due in part to also having fewer categories to predict (22 met-types with at least 5 cells versus 38 t-types).
In addition to met-type prediction by a supervised classifier, we also quantified the degree to which met-types were distinct or overlapping using methods similar to those developed to assess the degree of distinctiveness versus continuity in transcriptomic properties (Harris et al., 2018). For each category of electrophysiological and morphological feature and each pair of met-types, we calculated a cross-validated likelihood ratio statistic (comparing a given held-out cell to either its own class or the other of the pair), then summarized the separation of those likelihood ratios by a d′ statistic (Figure S4B; higher values indicate more separation). As expected, pairs of met-types that were confused by the supervised classifier, such as Sst-MET-10 and Sst-MET-12, exhibited a high degree of overlap in most feature categories. This analysis also enables understanding of how met-types relate to one another; for example, the Pvalb met-types had a large degree of overlap in electrophysiological features but had more morphological differences in part due to their different laminar locations (see Figure 2B).
Characteristics of met-types
We identified thirteen met-types within the Sst subclass (Figure 6A). These met-types were fairly cohesive in low-dimensional projections of transcriptomic and electrophysiological space (Figure 6B) and could be identified by particular combinations of marker genes (Figure 6C). Sst-MET-1 mapped to the Sst Chodl t-type, generally occupied deeper layers, and corresponded to non-Martinotti, long-range projecting inhibitory neurons (Tasic et al., 2018; Paul et al., 2017; He et al., 2016). Sst-MET-2 was composed mostly of cells mapping to Sst Tac1 Htr1d and had electrophysiological properties similar to Pvalb interneurons. These cells had APs with lower upstroke/downstroke ratios and less axonal innervation of L1. A previous study also observed that cells of this t-type had fast-spiking-like firing properties (Scala et al., 2019). Sst-MET-3 and Sst-MET-4 contained mostly cells mapping to Sst Calb2 Pdlim5, which were found in either cortical layers L2/3 (for Sst-MET-3) or L5 (for Sst-MET-4); both met-types exhibited the most exuberant axonal elaborations in L1 among Sst cells (Figure 6D). As mentioned, this met-type reflects the well-described Martinotti-type neurons (Wang et al., 2004) and includes cells that resemble the recently described L2/3 Martinotti and L5/6 fanning-out Martinotti cells (Nigro et al., 2018; Munõz et al., 2017). Note that the dominant layer of innervation for the fanning-out type may differ between visual and somatosensory cortex, while overall axon shape remains the same.
Figure 6: Summary of identified Sst and Pvalb inhibitory met-types.
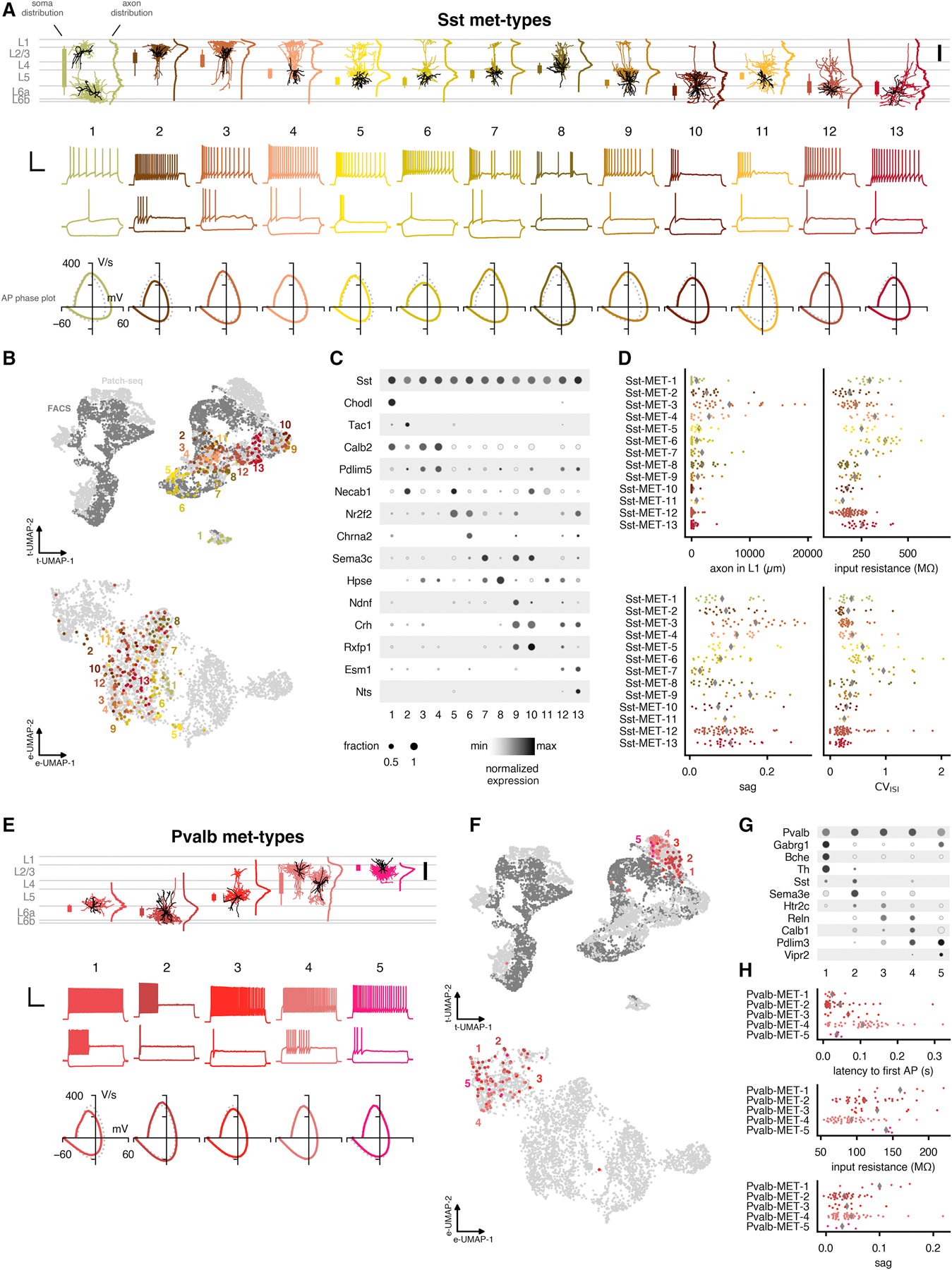
(A) Example morphologies and electrophysiological responses for each Sst met-type. In the morphology plot, soma distributions are indicated by bars to the left (thicker: 25% to 75% range, thinner: 5% to 95% range). Average axon depth histograms are shown to the right (normalized to their maximum values). Scale bar: 200 µm. Electrophysiology examples include responses evoked by a hyperpolarizing current step (−70 or −90 pA, lower), the response at rheobase (lower), and the response evoked by a rheobase + 40 pA stimulus (upper). Scale bar: vertical 50 mV, horizontal 250 ms. Phase plots of the average first spike of the rheobase trace are shown below the example traces; the subclass-wide average is the dotted gray line. (B) UMAPs based on gene expression (upper, see Figure 1B) and electrophysiology features (lower, see Figure 3B–C) with Sst met-types shown in colors. On the top, Patch-seq cells are shown in light gray and dissociated cells are shown in dark gray. (C) Dot plots of marker genes that are differentially expressed across the Sst met-types. Expression values (shading) are log-transformed and normalized to the maximum; size of dot indicates fraction of cells expressing the gene within the t-type. (D) Selected morphological and electrophysiological features by Sst met-type. Averages shown as gray diamonds. CVISI (coefficient of variation of the interspike interval) was measured for the rheobase + 40 pA amplitude response. (E–H) Same as (A–D) but for Pvalb met-types. In (D), the latency to first AP was measured at rheobase. See also Figures S6 and S7 and Tables S2 and S3.
Sst-MET-5 contained cells (many mapping to Sst Nr2f2 Necab1) that possessed burst-like or very strongly adapting firing patterns; their axons predominantly innervated the region near their somas in L5, with a less pronounced projection to L2/3 and superficial L1. This met-type also resembles the L5/6 fanning-out neurons described in those same papers. Sst-MET-6 (containing cells mapping to Sst Myh8 Etv1 and Sst Chrna2 Glra3) was similar to Sst-MET-5 but with more variable L1 axon and its appearance more closely resembles T-shaped (Munõz et al., 2017) and alpha-2 Martinotti cells (Hilscher et al., 2017). Compared with Sst-MET-5, Sst-MET-6 cells expressed more Chrna2 and less Necab1 (Figure 6C) and had a higher input resistance (Figure 6D). Sst-MET-7 contains cells mapping to Sst Hpse Sema3c that often have a T-shaped Martinotti-like appearance. Sst-MET-7 cells differed in their morphology from Sst-MET-6 cells in that they are located more superficially in L5, and their axon does not branch until it enters L1, unlike Sst-MET-6 cells, which tend to be in deep L5 and branch before entering L1. Their electrophysiological properties were also more distinctive compared with Sst-MET-6, having more irregular firing (higher CVISI) and less hyperpolarization-evoked sag (Figure 6D). Sst-MET-8 (containing primarily cells mapped to Sst Hpse Cbln4) was found in L4 and upper L5; it strongly innervated L4 (with a less dominant projection to L1) and exhibited distinct electrophysiological properties including lower AP thresholds (Figure 6A), little hyperpolarization-evoked sag (Figure 6D), and relatively low input resistance (Figure 6D). This met-type aligns with L5 Non-Martinotti cells in somatosensory cortex (Ma et al., 2006; Naka et al., 2019) that have distinct connectivity profiles compared to Martinotti neurons, though in visual cortex these neurons have more L1-innervating axon.
Sst-MET-9 was found in lower L5 and primarily innervated that layer with some projection to L1. Most of its cells belonged to the Sst Tac2 Tacstd2 t-type, and many of them expressed Ndnf as a marker (Figure 6C). Sst-MET-10 was similar to Sst-MET-9 but found deeper in cortex with more consistently sparse or absent innervation of L2/3 and L4 and more pronounced innervation of L6; it also exhibited much less sag than Sst-MET-9 (Figure 6D). Sst-MET-10 contained many cells of the Sst Rxfp Prdm8 t-type. Sst-MET-11 only contained three cells in our analysis, so it may be a less stable or robustly identified type. However, it was notable for containing L5 cells with a low upstroke/downstroke ratio and low input resistances. Sst-MET-12 contained L5/L6 cells from several t-types, including Sst Crhr2 Efemp1, Sst Rxfp1 Eya1, and Sst Crh 4930553C11Rik. These cells primarily innervated the deeper layers with relatively few axonal projections to L1. Sst-MET-13 contained cells in L5 and L6 and expressed the markers Esm1 and Nts (Figure 6C); these cells had few projections toward upper layers, as well. Sst neurons with these dominantly deep-layer axon innervation patterns have not been previously described, but do fit into the general category of non-Martinotti cells.
Five met-types were identified in the Pvalb subclass and mostly reflected different laminar positions (Figure 6E–H). AP shapes were quite consistent across the Pvalb met-types and were typical of a fast-spiking phenotype. The first two met-types (Pvalb-MET-1 and Pvalb-MET-2) were found predominantly in L5 and L6 and had basket-like morphologies with axon and dendrites restricted to the deep part of L5 and L6. Pvalb-MET-1 contained cells mapped to the transcriptomically-distinct Pvalb Gabrg1 t-type and also exhibited more hyperpolarization-induced sag than the other Pvalb met-types (Figure 6H); Pvalb-MET-2 was composed of four Pvalb t-types and contained fast-spiking cells resembling locally-projecting L6 cells (Bortone et al., 2014) and deep basket cells (Markram et al., 2015; Jiang et al., 2015). Marker genes also could distinguish these deep Pvalb met-types, as Pvalb-MET-1 cells expressed Gabrg1, Bche, and Th, while Pvalb-MET-2 cells expressed Sema3e (Figure 6G). Pvalb-MET-3 had cells that mapped to Pvalb Reln Tac1 and contained mostly L5 fast-spiking cells with L5-dominant axon innervation and a few collaterals extending into superficial layers. Pvalb-MET-4 contained cells that mapped to both Pvalb Reln Itm2a and Pvalb Tpbg. These consisted of mainly L4 and L2/3 fast-spiking cells (but included some deeper cells as well) with a L2/3-dominant axonal projection and sparse projections to deeper layers. These interlaminar or translaminar projections resemble those described previously for fast-spiking neurons in L6 (Frandolig et al., 2019; Bortone et al., 2014). Among these first four Pvalb met-types, there appeared to be depth-dependent delays in the onset of AP generation at rheobase that tended to decrease with increasing depth from pia (Figure 6H). Pvalb-MET-5 (containing Pvalb Vipr2 cells) represented chandelier cells as previously identified (Woodruff and Yuste, 2008; Taniguchi et al., 2013; Inan and Anderson, 2014; Tasic et al., 2018; Gouwens et al., 2019) and had higher input resistances compared to Pvalb-MET-4 (Figure 6H).
We identified two met-types among the Lamp5 cells (Figure 7A–C). However, we had relatively few cells from the Lamp5 Krt73, Lamp5 Fam19a1 Pax6, and Lamp5 Fam19a1 Tmem182 t-types; those cells may form other distinct met-types that we were not able to characterize here. Most of the other Lamp5 cells were in the Lamp5-MET-1 type, except for the Lamp5 Lhx6 cells which formed Lamp5-MET-2. Lamp5-MET-1 had APs with high upstroke/downstroke ratios and a neurogliaform-like morphology (Jiang et al., 2013; Jiang et al., 2015; Schuman et al., 2019); these cells were found primarily in L1 and L2/3 but also appeared in deeper cortical layers. This met-type was fairly heterogenous, with variation in gene expression (as cells came from the Lamp5 Lsp1, Lamp5 Plch2 Dock5, and Lamp5 Ntn1 Npy2r t-types), morphology (ranging from L1 cells with horizontally-extending axon to deeper cells with round, dense axon), and electrophysiology (such as differences in the late-spiking phenotype, as measured by the latency to the first action potential at rheobase). Lamp5-MET-2 was distinguishable by the presence or absence of several marker genes (Figure 7B) and was found in deep layers; the electrophysiological properties resembled Lamp5-MET-1, with high-upstroke downstroke ratios and late-spiking.
Figure 7: Summary of identified Lamp5, Sncg, and Vip inhibitory met-types.
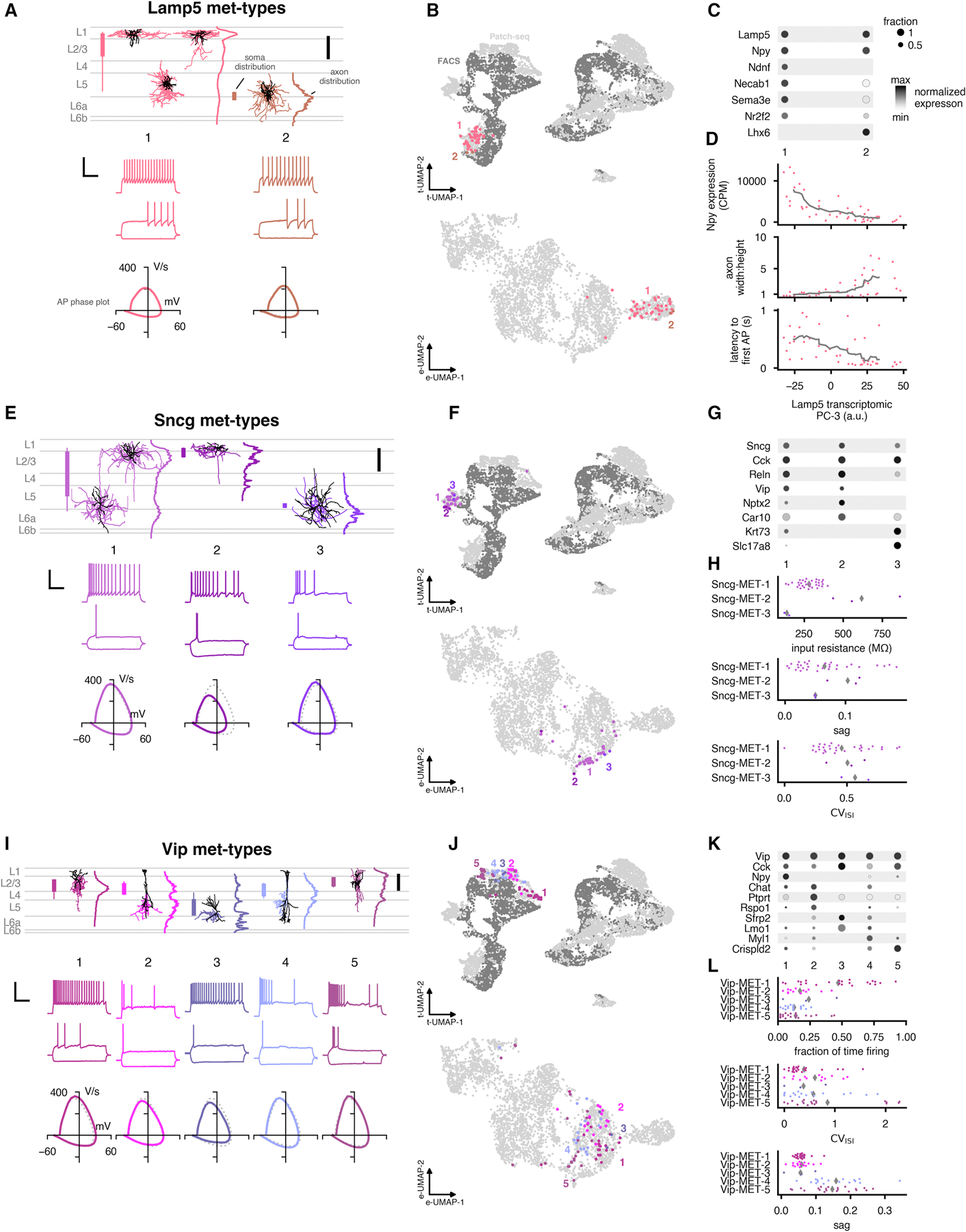
(A) Example morphologies and electrophysiological responses for each Lamp5 met-type. In the morphology plot, soma distributions are indicated by bars to the left (thicker: 25% to 75% range, thinner: 5% to 95% range). Average axon depth histograms are shown to the right (normalized to their maximum values). Scale bar: 200 µm. Electrophysiology examples include responses evoked by a hyperpolarizing current step (−70 or −90 pA, lower), the response at rheobase (lower), and the response evoked by a rheobase + 40 pA stimulus (upper) . Scale bar: vertical 50 mV, horizontal 250 ms. Phase plots of the average first spike of the rheobase trace are shown below the example traces; the subclass-wide average is the dotted gray line. (B) UMAPs based on gene expression (upper, see Figure 1B) and electrophysiology features (lower, see Figure 3B–C) with Lamp5 met-types shown in colors. On the top, Patch-seq cells are shown in light gray and dissociated cells are shown in dark gray. (C) Dot plots of marker genes that are differentially expressed across the Lamp5 met-types. Expression values (shading) are log-transformed and normalized to the maximum; size of dot indicates fraction of cells expressing the gene within the t-type. (D) Variation in Npy expression, axon width:height ratio, and latency to first AP on the rheobase sweep for cells of the Lamp5-MET-1 met-type. Values are plotted against the third transcriptomic principal component (PC-3) calculated from 4,020 differentially expressed genes from all Lamp5 Patch-seq cells. Gray line is rolling mean (window of 10 cells). (E–G) Same as (A–C) but for Sncg met-types. (H) Electrophysiology feature differences between Sncg met-types. Averages shown as gray diamonds. CVISI (coefficient of variation of the interspike interval) was measured for the rheobase + 40 pA amplitude response. (I–K) Same as (A–C) but for Vip met-types. (L) Same as (H) but for Vip met-types. Fraction of time firing was measured for the rheobase + 40 pA amplitude response. See also Figure S7 and Table S4.
We attempted to characterize the heterogeneity of Lamp5-MET-1 by performing principal component analysis (with 20 components; see Figure 1B) on the gene expression data for the Lamp5 Patch-seq cells in our data set. The third principal component (PC-3) varied systematically across the t-types found in Lamp5-MET-1. Interestingly, several other properties of these cells also varied along this dimension (Figure 7D). One end of the spectrum defined by PC-3 had cells with low Npy expression, wide axons, and fewer cells with a late-spiking phenotype, while the other end had cells expressing more Npy with rounder axons that were consistently late-spiking. Therefore, this met-type exhibits continuous variation in multiple data modalities in a concerted way. We noted that each end of the spectrum resembled populations of L1 interneurons described in a recent study in barrel cortex (Schuman et al., 2019): the cells here with higher Npy expression have electrophysiology and morphology consistent with Ndnf+/Npy+ neurogliaform cells, and those at the other end have properties more similar to the “canopy cells” described in that study.
Three met-types were identified in the Sncg subclass (Figure 7E–H). Sncg-MET-1 was found in most cortical layers. These cells had bitufted or multipolar dendrites with wide, translaminar axon that mostly avoided L1. They exhibited regular, adapting firing patterns. Based on gene expression, Sncg neurons have been linked to Vip+/Cck + basket cells (Tasic et al., 2018). The other two Sncg met-types had few cells (n=3 and n=2) but fairly distinctive properties. Sncg-MET-2 cells expressed Car10 and Nptx2 (Figure 7G), had very high input resistances (Figure 7H), and were found at the L1-L2/3 border (Figure 7E). Sncg-MET-3 cells were deep and belonged to the Sncg Slc17a8 t-type.
We identified five Vip met-types (Figure 7I–L), all resembling bipolar or bitufted cells (Peters and Kara, 1985; Prönneke et al., 2015). Vip-MET-1 had cells primarily in L2/3 and L4, with bitufted dendrites extending into L1, and a fairly concentrated axon innervation of lower L2/3. Vip-MET-2 had a similar laminar position to Vip-MET-1 but with longer dendrites and a narrower profile with wider axonal branching only in deep layers. Vip-MET-2 also exhibited more distributed axon innervation, spanning L2/3 to L6, but mostly avoiding L4. Vip-MET-1 and −2 cells also differed in their gene expression. Vip-MET-1 cells expressed Npy while Vip-MET-2 cells expressed Ptprt and Rspo1 (Figure 7K). Vip-MET-1 also typically had regular, sustained firing while Vip-MET-2 firing was usually irregular (Figure 7L). Vip-MET-3 was found in L5 (Figure 7I) and had short, bitfuted dendrites and complex axon elaboration at layer borders. Vip-MET-3 expressed the marker genes Sfrp2 and Lmo1 (Figure 7K). Vip-MET-4 was found in lower L2/3 and innervated L2/3 through L6 similar to Vip-MET-2. However, these cells had enriched L4 axonal innervation; they also exhibited irregular and/or transient firing and had more hyperpolarization-evoked sag than the first three Vip met-types (Figure 7L). The final Vip met-type, Vip-MET-5 (containing cells from the t-types Vip Crispld2 Htr2c and Vip Crispld2 Kcne4) innervated all layers, including L1 (Figure 7I) and had more hyperpolarization-induced sag, similar to Vip-MET-4 (Figure 7L). These cells also frequently fired bursts near rheobase.
The anatomical distribution of the marker genes selective for specific t- or met-types have been characterized in a recent MERFISH study in the mouse primary motor cortex (MOp) (Zhang et al., 2020); they may be similarly distributed in the visual cortex since GABAergic t-types are shared across cortical areas (Tasic et al., 2018). Summaries of the morphological, electrophysiological, and transcriptomic properties of each met-type are provided in Tables S2, S3, and S4.
Discussion
Groups, classes, or types of neurons have often been distinguished on the basis of structural, physiological, biochemical, or genetic data. The (sometimes dramatically) different number of distinguishable neuronal cell types inferred from one data modality versus another, and the challenge of relating groups of neurons distinguished on the basis of different cellular attributes highlights the power and utility of a more integrated experimental approach. Here, the high-throughput, standardized application of the Patch-seq technique enabled the intrinsic electrophysiological, morphological, and transcriptomic characteristics of individual neurons to be assessed simultaneously. This dataset, publicly available at https://portal.brain-map.org/, will allow researchers familiar with collecting and interpreting one type of data (e.g., electrophysiological data) to better interpret it in the context of other studies.
Major findings from our study include: 1) Morpho-electrical features of GABAergic interneurons largely co-vary with transcriptomic types, with discrete and continuous variations co-existing along the three modalities. 2) Correspondence between the independently derived t- and me-types is mostly consistent but not strictly one-to-one. 3) Integrated analysis results in a classification of 28 met-types of cortical interneurons with congruent transcriptomic and morpho-electrical characteristics. 4) The 28 met-types are labeled by salient gene markers and provide a systematic framework to encapsulate historical knowledge of cortical interneuron types. 5) Unexpectedly, we found that nearly all t- and met-types in each subclass are arranged along the cortical depth; the soma locations of each type are often centered in a layer or sublayer, with continuous variations both within and between types. 6) The axons of many types also target specific layers, and this is a hallmark distinction between met-types; in particular, differential axon targeting specificity redefines the traditional concept of Sst interneuron types. These findings reveal patterns and some underlying rules of the complex landscape of cortical GABAergic cellular diversity, which may be generalizable to other brain cell types.
The inherent variations displayed in our data – sometimes consistent and sometimes differential across the modalities – and the resulting lower granularity of met-types compared to the original t-types (28 vs 60) may be due to both technical and biological reasons. The limited number of stimuli in our standardized patch clamp protocol, the relatively small number of morphological reconstructions, and the much lower dimensionality of electrophysiological and morphological data compared to transcriptomic data may all contribute to this difference. Contamination and dropout in the Patch-seq transcriptomic data can also introduce ambiguity in t-type mapping. Also, we note cases in which scRNAseq data from Patch-seq recordings mapped more frequently to one t-type (Sst Hpse Cbln4, Pvalb Reln Itm2a) while scRNAseq data from dissociated cells mapped more frequently to another, related t-type (Sst Hpse Sema3c, Pvalb Reln Tac1, respectively). Such discrepancies may in part reflect gene expression differences in slice versus dissociated cell preparations.
However, some of the ambiguity in types due to the existence of continuous and sometimes differential variations along the three modalities could also reflect the biological reality. Consistent with our results, (Scala et al., 2020) describe a continuum of variability in the morphology and electrophysiology of glutamatergic and GABAergic neurons in primary motor cortex of the mouse — similar transcriptomic cell types frequently exhibit similar morpho-electric features, often without clear boundaries between them. Also, (Que et al., 2020) report that neurons mapping to distinct Pvalb t-types in the mouse hippocampus exhibit qualitatively similar morphological characteristics. Even in transcriptomic data from isolated cells, continuous variations were frequently observed (Stanley et al., 2020; Harris et al., 2018; Tasic et al., 2018). How such continuous variation is subdivided into separate t-types depends on the specific statistical criterion chosen, which can be arbitrary in the absence of other supporting evidence. Thus, currently described t-types, which are defined by unsupervised clustering of transcriptomic data alone, need to be validated by other modalities of data.
Altogether, our results support the presence of at least 28 GABAergic interneuron met-types that have congruent transcriptomic, electrophysiological and morphological properties (Figures 6 and 7). These met-types represent a version of a unified definition of cortical GABAergic interneuron types established with multimodal experimental data and enabling cross-modal feature prediction. The number of distinguishable met-types could change in the future with further anatomical, connectional, and functional studies. There may be other intrinsic electrophysiological differences between cells that could be revealed by stimuli not used in our standardized recording protocol; these differences could potentially expand the number of met-types. Studies of local and long-range connections could also reveal further cell type-specific differences. A recent study showed that long-range connections from visual cortex layer 2/3 neurons do not correlate with transcriptomic types but rather with within-type gene expression differences (Kim et al., 2020). This intriguing result suggests that transcriptomic types may be revised based on the combination with another modality (e.g., connectivity) via approaches such as supervised clustering to achieve better consistency.
A major insight gained from our study is that the somas of many GABAergic t- or met-types are restricted to a single cortical layer or sublayer (Figure 2) and, in the case of Sst interneurons, elaborate their axons in a dominant cortical layer (Figure 4). We identify 13 Sst met-types (Figure 6), which is substantially more than described in previous morphological and/or electrophysiological studies (Kawaguchi and Kubota, 1996; McGarry et al., 2010; Markram et al., 2015; Jiang et al., 2015; Munõz et al., 2017). Our data show that two met-types corresponding to t-type Sst Calb2 Pdlim5, Sst-MET-3 (in L2/3) and Sst-MET-4 (in L5, the fanning-out Martinotti cells), exhibit extensive axon elaborations in L1 relative to other layers as described for classically-defined Martinotti cells. The axons of neurons mapping to all other Sst met-types, by comparison, preferentially arborize outside of L1. However, all these Sst met-types contain at least one cell that had some degree of L1 projection and could therefore technically be considered a Martinotti cell (usually the T-shaped) as well. We are not aware of an absolute definition of a Martinotti cell in terms of total amount of axon in L1, though it is generally described as “significant” (Tremblay et al., 2016; Zhou et al., 2020). In any case, we find that these met-types consistently target one or two other layers to a greater extent than L1. For example, Sst-MET-2 densely innervates L2/3 and largely avoids L1. The axons of Sst-MET-8 neurons (mapping to the Sst Hpse Cbln4 t-type) — one of two major Sst met-types found in L4 — extensively innervate L4 and innervate L1 to a lesser degree. A dominant L4 projection appears to be a common feature of Sst Hpse Cbln4-type neurons across cortical areas (Naka et al., 2019), while the L1 projection is not always present. Sst met-types found mainly in the deeper part of L5 and in L6 have predominantly L5 and L6 axonal projections. These results reveal that Sst neurons have clear laminar innervation preferences that vary across met-types; consequently, their dominant laminar targets may be a more reliable distinguishing feature than the presence or absence of axon in layer 1. Recent evidence also suggests that different morphological phenotypes correspond to differential synaptic connectivity profiles (Nigro et al., 2018; Naka et al., 2019; Scala et al., 2019).
As we have shown here, integrating multiple modalities is a powerful approach for classifying neurons into types. We expect that additional information about cellular and circuit properties will continue to improve the identification of archetypal interneuron groups. This could include further refinement of the transcriptomic taxonomy profiled under different states, more detailed electrophysiological and morphological characterization, the addition of proteomic information, local and long-range synaptic connectivity, and in vivo functional characterization. With the availability of large-scale electromicrograph images, we will soon be able to map morphological phenotypes across datasets to understand the relationship between transcriptomic, morphological and electrophysiological phenotypes and connectivity profiles. In addition, multiplex FISH is already being used to measure the proportions of transcriptomic types within a given brain tissue (Hodge et al., 2019; Moffitt et al., 2018; Zhang et al., 2020). Combined, these methods will facilitate the development of high-resolution cell type-specific circuit maps.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Requests for further information should be directed to and will be fulfilled by the Lead Contact, Gabe J. Murphy (gabem@alleninstitute.org).
Materials availability
Two new transgenic mouse lines, Sncg-IRES-Cre and Gpr139-IRES2-FlpO-WPRE-neo, were used in this study. Both lines have been deposited with and will be available from The Jackson Laboratory; moreover, characteristics of both lines will be described in a forthcoming publication (Daigle et al., in preparation).
Data and code availability
Transcriptomic data supporting the findings of this study are available at the NeMO archive. Electrophysiological data for this study are available at the DANDI archive. Morphological reconstructions from this study are available at the Brain Image Library (BIL) archive. Information for accessing these data are in the Key Resources Table and summarized at https://portal.brain-map.org/explore/classes/multimodal-characterization (“Neurons in Mouse Primary Visual Cortex”).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Takara | 634894 |
| Nextera XT Index Kit V2 Set A-D | Nextera | FC-131– 2001,2002,2003,2004 |
| Deposited Data | ||
| Transcriptomic data – fastq files | This paper; NeMO | http://data.nemoarchive.org/other/grant/AIBS_patchseq/transcriptome/scell/SMARTseq/raw/20200611/ |
| Transcriptomic data – BAM files | This paper; NeMO | http://data.nemoarchive.org/other/grant/AIBS_patchseq/transcriptome/scell/SMARTseq/processed/align/20200611/ |
| Transcriptomic data – processed count files | This paper; NeMO | http://data.nemoarchive.org/other/grant/AIBS_patchseq/transcriptome/scell/SMARTseq/processed/analysis/20200611/ |
| Raw electrophysiology data | This paper; DANDI | DANDI: 000020 https://dandiarchive.org/dandiset/000020 |
| Morphological reconstructions | This paper; Brain Image Library | ftp://download.brainlib.org:8811/biccn/zeng/pseq/morph/200526/ |
| Specimen metadata | This paper; Allen Institute for Brain Science | https://brainmapportal-live-4cc80a57cd6e400d854-f7fdcae.divio-media.net/filer_public/5e/2a/5e2a5936-61da-4e09-b6da-74ab97ce1b02/20200711_patchseq_metadata_mouse.csv |
| Experimental Models: Organisms/Strains | ||
| Mouse: Calb1-IRES2-Cre | Daigle et al., Cell, 2018; The Jackson Laboratory | RRID:IMSR_JAX:028532 |
| Mouse: Calb2-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:010774 |
| Mouse: Cck-IRES0Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:012706 |
| Mouse: Chat-IRES-Cre | Rossi et al., Cell Metab. 2011; The Jackson Laboratory | RRID:IMSR_JAX:006410 |
| Mouse: Chrna2-Cre_OE25 | Gong, et al., J Neurosci 2007; MMRRC | RRID:MMRRC_036502-UCD |
| Mouse: Crh-IRES-Cre_ZJH | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:012704 |
| Mouse: Ctgf-T2A-dgCre | Tasic et al., Nat. Neurosci 2016; The Jackson Laboratory | RRID:IMSR_JAX:028535 |
| Mouse: Esr2-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:030158 |
| Mouse: Etv1-CreERT2 | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:013048 |
| Mouse: Gad2-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:010802 |
| Mouse: Glt25d2-Cre_NF107 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_036504-UCD |
| Mouse: Gpr139-IRES2-FlpO-WPRE-neo | Daigle et al. (in preparation) | N/A |
| Mouse: Htr3a-Cre_NO152 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_036680-UCD |
| Mouse: Ndnf-IRES2-dgCre | Tasic et al., Nat. Neurosci 2016; The Jackson Laboratory | RRID:IMSR_JAX:028536 |
| Mouse: Nkx2.1-CreERT2 | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:014552 |
| Mouse: Nos1-CreERT2 | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:014541 |
| Mouse: Npr3-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:031333 |
| Mouse: Npy-IRES2-FlpO | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:030211 |
| Mouse: Nr5a1-Cre | Dhillon et al., Neuron 2006; The Jackson Laboratory | RRID:IMSR_JAX:006364 |
| Mouse: Ntsr1-Cre_GN220 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_030648-UCD |
| Mouse: Oxtr-T2A-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:031303 |
| Mouse: Pvalb-T2A-Dre | Madisen et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:021190 |
| Mouse: Pvalb-T2A-FlpO | Madisen, et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:022730 |
| Mouse: Pvalb-IRES-Cre | Hippenmeyer et al., PLoS Biol 2005; The Jackson Laboratory | RRID:IMSR_JAX:008069 |
| Mouse: Rbp4-Cre_KL100 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_031125-UCD |
| Mouse: Rorb-IRES2-Cre | Harris et al., Front. Neural Circuits, 2014; The Jackson Laboratory | RRID:IMSR_JAX:023526 |
| Mouse: Slc17a8-iCre | Grimes et al., Vis Neurosci 2011; The Jackson Laboratory | RRID:IMSR_JAX:018147 |
| Mouse: Slc17a8-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:028534 |
| Mouse: Slc32a1-IRES-Cre | Tong et al., Nat. Neu. 2008; The Jackson Laboratory | RRID:IMSR_JAX:012897 |
| Mouse: Slc32a1-T2A-FlpO | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:029591 |
| Mouse: Sncg-IRES2-FlpO-neo | Daigle et al. (in preparation) | N/A |
| Mouse: Sst-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:013044 |
| Mouse: Sst-IRES-FlpO | He et al., Neuron, 2016; The Jackson Laboratory | RRID:IMSR_JAX:028579 |
| Mouse: Tac1-IRES2-Cre | Harris et al., Front. Neural Circuits, 2014; The Jackson Laboratory | RRID:IMSR_JAX:021877 |
| Mouse: Th-Cre_FI172 | Gong et al., J Neurosci 2007; The Jackson Laboratory | RRID:MMRRC_031029-UCD |
| Mouse: Th-P2A-FlpO | Poulin et al., Nat. Neurosci 2018 | N/A |
| Mouse: Tlx3-Cre_PL56 | Gerfen et al., Neuron 2013; The Jackson Laboratory | RRID:MMRRC_036547-UCD |
| Mouse: Vip-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:010908 |
| Mouse: Vip-IRES-FlpO | He et al., Neuron, 2016; The Jackson Laboratory | RRID:IMSR_JAX:028578 |
| Mouse: Vipr2-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:031332 |
| Mouse: Ai14(RCL-tdT) | Madisen et al., Nat. Neu. 2010; The Jackson Laboratory | RRID:IMSR_JAX:007914 |
| Mouse: Ai65(RCFL-tdT) | Madisen et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:021875 |
| Mouse: Ai66(RCRL-tdT) | Madisen et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:021876 |
| Mouse: Gad67-GFP_X94 | Ma et al., J Neurosci 2006; The Jackson Laboratory | RRID:IMSR_JAX:006334 |
| Software and Algorithms | ||
| Igor Pro | WaveMetrics | https://www.wavemetrics.com/products/igorpro |
| MIES | Allen Institute for Brain Science | https://github.com/alleninstitute/mies |
| Vaa3D | Peng et al., 2010 | http://www.vaa3d.org |
| Allen SDK | Allen Institute for Brain Science | https://github.com/alleninstitute/allensdk |
| IPFX | Allen Institute for Brain Science | https://github.com/alleninstitute/ipfx |
| DRCME | Gouwens et al., 2019; Allen Institute for Brain Science | https://github.com/alleninstitute/drcme |
The custom electrophysiology data acquisition software (MIES) is available at https://github.com/alleninstitute/mies. The Vaa3D morphological reconstruction software, including the Mozak extension, is freely available at http://www.vaa3d.org and its code is available at https://github.com/Vaa3D. The code for electrophysiological and morphological feature analysis and clustering is available as part of the open-source Allen SDK repository (https://github.com/AllenInstitute/AllenSDK), IPFX repository (https://github.com/alleninstitute/ipfx), and DRCME repository (https://github.com/alleninstitute/drcme).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse breeding and husbandry
Mice (male and female) were between the ages of P45 and P70 at the time of euthanasia; all procedures were carried out in accordance with the Institutional Animal Care and Use Committee at the Allen Institute for Brain Science. Animals (≤5 mice per cage) were provided food and water ad libitum and were maintained on a regular 12-h light–dark cycle. Animals were maintained on the C57BL/6J background, and newly received or generated transgenic lines were backcrossed to C57BL/6J. Experimental animals were heterozygous for the recombinase transgenes and the reporter transgenes. Transgenic lines used in this study are summarized in the Key Resources Table. Standard tamoxifen treatment for CreER lines included a single dose of tamoxifen (40 μL of 50 mg mL−1) dissolved in corn oil and administered via oral gavage at P10–P14. Tamoxifen treatment for Nkx2.1-CreERT2;Ai14 was performed at embryonic day (E)17 (oral gavage of the dam at 1 mg per 10 g of body weight), pups were delivered by cesarean section at E19 and then fostered. Ndnf-IRES2-dgCre animals did not receive trimethoprim induction, since the baseline dgCre activity (without trimethoprim) was sufficient to label the cells with the Ai14 reporter.
METHOD DETAILS
Tissue processing
Mice were anesthetized with 5% isoflurane and intracardially perfused with 25 or 50 mL of ice-cold slicing artificial cerebral spinal fluid (ACSF; 0.5 mM calcium chloride (dehydrate), 25 mM D-glucose, 20 mM HEPES buffer, 10 mM magnesium sulfate, 1.25 mM sodium phosphate monobasic monohydrate, 3 mM myo-inositol, 12 mM N-acetyl-L-cysteine, 96 mM N-methyl-D-glucamine chloride (NMDG-Cl), 2.5 mM potassium chloride, 25 mM sodium bicarbonate, 5 mM sodium L-ascorbate, 3 mM sodium pyruvate, 0.01 mM taurine, and 2 mM thiourea (pH 7.3), continuously bubbled with 95% O2/5% CO2). Slices (350 μm) were generated (Compresstome VF-300 vibrating microtome, Precisionary Instruments or VT1200S Vibratome, Leica Biosystems), with a block-face image acquired (Mako G125B PoE camera with custom integrated software) before each section to aid in registration to the common mouse reference atlas. Brains were mounted for slicing either in the coronal plane or at a 17° angle from the coronal plane to preserve the intactness of neuronal processes in the visual cortex.
Slices were transferred to an oxygenated and warmed (34 °C) slicing ACSF for 10 min, then transferred to room temperature holding ACSF (2 mM calcium chloride (dehydrate), 25 mM D-glucose, 20 mM HEPES buffer, 2 mM magnesium sulfate, 1.25 mM sodium phosphate monobasic monohydrate, 3 mM myo-inositol, 12.3 mM N-acetyl-L-cysteine, 84 mM sodium chloride, 2.5 mM potassium chloride, 25 mM sodium bicarbonate, 5 mM sodium L-ascorbate, 3 mM sodium pyruvate, 0.01 mM taurine, and 2 mM thiourea (pH 7.3), continuously bubbled with 95% O2/5% CO2) for the remainder of the day until transferred for patch-clamp recordings.
Patch-clamp recording
Slices were bathed in warm (34 °C) recording ACSF (2 mM calcium chloride (dehydrate), 12.5 mM D-glucose, 1 mM magnesium sulfate, 1.25 mM sodium phosphate monobasic monohydrate, 2.5 mM potassium chloride, 26 mM sodium bicarbonate, and 126 mM sodium chloride (pH 7.3), continuously bubbled with 95% O2/5% CO2). The bath solution contained blockers of fast glutamatergic (1 mM kynurenic acid) and GABAergic synaptic transmission (0.1 mM picrotoxin). Thick-walled borosilicate glass (Warner Instruments, G150F-3) electrodes were manufactured (Narishige PC-10) with a resistance of 4–5 MΩ. Before recording, the electrodes were filled with ~1.0–1.5 µL of internal solution with biocytin (110 mM potassium gluconate, 10.0 mM HEPES, 0.2 mM ethylene glycol-bis (2-aminoethylether)-N,N,N’,N’-tetraacetic acid, 4 mM potassium chloride, 0.3 mM guanosine 5’-triphosphate sodium salt hydrate, 10 mM phosphocreatine disodium salt hydrate, 1 mM adenosine 5’-triphosphate magnesium salt, 20 µg/mL glycogen, 0.5U/µL RNAse inhibitor (Takara, 2313A) and 0.5% biocytin (Sigma B4261), pH 7.3). The pipette was mounted on a Multiclamp 700B amplifier headstage (Molecular Devices) fixed to a micromanipulator (PatchStar, Scientifica).
The composition of bath and internal solution as well as preparation methods were made to maximize the tissue quality of slices from adult mice, to align with solution compositions typically used in the field (to maximize the chance of comparison to previous studies), modified to reduce RNAse activity and ensure maximal gain of mRNA content.
Electrophysiology signals were recorded using an ITC-18 Data Acquisition Interface (HEKA). Commands were generated, signals processed, and amplifier metadata were acquired using MIES (https://github.com/AllenInstitute/MIES/), written in Igor Pro (Wavemetrics). Data were filtered (Bessel) at 10 kHz and digitized at 50 kHz. Data were reported uncorrected for the measured (Neher, 1992) –14 mV liquid junction potential between the electrode and bath solutions.
Prior to data collection, all surfaces, equipment and materials were thoroughly cleaned in the following manner: a wipe down with DNA away (Thermo Scientific), RNAse Zap (Sigma-Aldrich), and finally with nuclease-free water.
After formation of a stable seal and break-in, the resting membrane potential of the neuron was recorded (typically within the first minute). A bias current was injected, either manually or automatically using algorithms within the MIES data acquisition package, for the remainder of the experiment to maintain that initial resting membrane potential. Bias currents remained stable for a minimum of 1 s before each stimulus current injection.
To be included in the analysis, a cell needed to have a >1 GΩ seal recorded before break-in and an initial access resistance <20 MΩ and <15% of the Rinput. To stay below this access resistance cut-off, cells with a low input resistance were successfully targeted with larger electrodes. For an individual sweep to be included, the following criteria were applied: (1) the bridge balance was <20 MΩ and <15% of the Rinput; (2) bias (leak) current 0 ± 100 pA; and (3) root mean square noise measurements in a short window (1.5 ms, to gauge high frequency noise) and longer window (500 ms, to measure patch instability) <0.07 mV and 0.5 mV, respectively.
Extracting the nucleus at the conclusion of the electrophysiology experiment led to a substantial increase in transcriptomic data quality. After electrophysiological recording, the pipette was centered on the soma or placed near the nucleus (if visible). A small amount of negative pressure was applied (~ –30 mbar) to begin cytosol extraction and to attract the nucleus to the tip of pipette. After approximately one minute, the soma had visibly shrunk and/or the nucleus was near the tip of the pipette. While maintaining negative pressure, the pipette was slowly retracted; slow, continuous movement was maintained while monitoring the pipette seal. Once the pipette seal reached >1GΩ and the nucleus was visible on the tip of the pipette, the speed was increased to remove the pipette from the slice. The pipette containing internal solution, cytosol, and the nucleus was removed from pipette holder, and its contents were expelled into a PCR tube containing the lysis buffer (Takara, 634894).
cDNA amplification and library construction
We used the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara, 634894) to reverse transcribe poly(A) RNA and amplify full-length cDNA according to the manufacturer’s instructions. We performed reverse transcription and cDNA amplification for 20 PCR cycles in 0.65 mL tubes, in sets of 88 tubes at a time. At least 1 control 8-strip was used per amplification set, which contained 4 wells without cells and 4 wells with 10 pg control RNA. Control RNA was either Mouse Whole Brain Total RNA (Zyagen, MR-201) or control RNA provided in the SMART-Seq v4 kit. All samples proceeded through Nextera XT DNA Library Preparation (Illumina FC-131–1096) using either Nextera XT Index Kit V2 Set A-D (FC-131–2001,2002,2003,2004) or custom dual-indexes provided by IDT (Integrated DNA Technologies). Nextera XT DNA Library prep was performed according to manufacturer’s instructions except that the volumes of all reagents including cDNA input were decreased to 0.2×— by volume. Each sample was sequenced to approximately 1 million reads.
Sequencing data processing
Fifty-base-pair paired-end reads were aligned to GRCm38 (mm10) using a RefSeq annotation gff file retrieved from NCBI on 18 January 2016 (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/all/). Sequence alignment was performed using STAR v2.5.3 (Dobin et al., 2013) in two pass Mode. PCR duplicates were masked and removed using STAR option “bamRemoveDuplicates.” Only uniquely aligned reads were used for gene quantification. Gene counts were computed using the R GenomicAlignments package (Lawrence et al., 2013) summarizeOverlaps function using “IntersectionNotEmpty” mode for exonic and intronic regions separately. Exonic and intronic reads were added together to calculate total gene counts; this was done for both the reference dissociated cell data set and the Patch-seq data set of this study. Data were analyzed as counts per million reads (CPM).
Identifying transcriptomic types
For the reasons explained above, we used transcriptomes of dissociated cells from (Tasic et al., 2018) as reference dataset and mapped Patch-seq transcriptomes to the reference data to identify their cell types. The details are explained below and schematized in Figure S1.
Preparation of reference cells and taxonomy tree
24,411 dissociated cells from VISp and ALM regions and their corresponding cell types and a list of 4,020 genes (the top 50 DE genes in each direction for all pairwise cluster comparisons) were adopted from (Tasic et al., 2018). Only neuronal cells and their corresponding cell types from VISp region were selected from the above dataset (in total 13,464 cells and 93 cell types) as the reference dataset for mapping Patch-seq transcriptomes (Figure S1B). Two different mapping algorithms are explained in the following.
Mapping on the reference taxonomy tree
The Patch-seq transcriptomes were mapped to the reference taxonomy tree in the top down manner, trying to resolve broad classes first, followed by subclasses and types (Figure S1C and D). For each single cell transcriptome, starting from the root and at each branch point, we computed its correlation with the reference cell types using the markers associated with the given branch point (i.e., the genes that best distinguished groups at each branch of the tree) and chose the most correlated branch. The process was repeated until reaching the leaves. To determine the confidence of mapping, we applied the 100 bootstrapped iterations at each branch point, and in each iteration, 70% of the reference cells and 70% of markers were randomly sampled for mapping. This was done to evaluate the degree to which mappings were robust to the effects of individual genes or reference cells. The percentage of times a cell was mapped to a given leaf or branch point in the reference taxonomy was defined as the corresponding mapping probability. For each cell, the mapping probability was sorted and the cell type to which that cell was mapped with highest probability was assigned as the corresponding cell type of that cell.
Neural-network-based mapping
We built a feedforward neural network classifier which receives gene expression data as input to predict the cell type for each individual cell. To train the classifier, we used the reference cells and their corresponding types. The reference cells were divided into two groups of training (80%) and test (20%) sets. The input was the log2(CPM+1) gene expression data and the output was a probability vector which predicted the type of each cell (see Figure S1E). After the model was trained, it was used to predict the cell type for transcriptomes obtained by Patch-seq. The training on the reference dataset and the subsequent prediction of the label of each cell (with Patch-seq recording) was performed 100 times (each time the training involved a new set of randomly selected 80% of reference cells). At the end, the percentage of times a cell was mapped to a given leaf or branch point in the reference taxonomy is defined as the corresponding mapping probability.
Assessing mapping quality
Due to presence of more significant contamination and dropout in the Patch-seq transcriptomes (see Figure S2), we occasionally observed ambiguous mapping of a cell to highly distinct cell types. To quantify the quality of mapping by considering what is “expected” and “unexpected” ambiguity of mapping between cell types we further categorized cells with Patch-seq recordings into three main groups: 1) highly consistent cells, 2) moderately consistent cells and 3) inconsistent cells. To do so, we first mapped the reference dissociated cells to the reference taxonomy to obtain the original or “expected” ambiguity which was present in the reference data set. By aggregating the mapping results and the clustering results (original cell types of the reference cells), a reference mapping probability matrix was constructed (Figure S1F). Then we examined whether the ambiguity in the mapping of Patch-seq transcriptomes is similar to this reference cells ambiguity by computing the Kullback-Leibler (KL) divergence (Berger et al., 2009) between the mapping probability distributions of cells with Patch-seq recordings and the reference mapping probability distribution. The KL divergence quantifies how different a probability distribution is from another reference distribution. Cells with a KL divergence greater than 2 were categorized as “inconsistent” cells. If the KL divergence was less than 2, then the correlation of the gene expression between the cell and the reference cells from the same type was computed. If the correlation was less than 0.5, the cell was given an “inconsistent” label. Cells with “inconsistent” mapping were not used in analyses that incorporated t-type assignments. The quality of the remaining cells were further quantified by the probability of the mapping to one or more than one cell types. First, the mapping probabilities of each cell to each of the 93 cell types were sorted. If the sum of the first two highest mapping probabilities for a cell was more than 70% and the ratio of the mapping probability of the first type to the next highest was more than 2, then the cell was labeled as “highly consistent.” Otherwise the cell was labeled “moderately consistent” (Figure S1G).
Visualization of reference cells and Patch-seq cells
Transcriptomic UMAP plots of both reference dissociated cells and Patch-seq cells were based on 20 principal components derived from the log2(CPM + 1) values of the 4,020 most differentially expressed genes across neuron types in the reference data set. Neurons from both data sets were combined for the principal component analysis; two PCs that exhibited high correlation with collection method (Pearson’s r = 0.94 and 0.22) were removed to reduce the effects of technical bias in the plots.
Electrophysiology feature analysis
Electrophysiological features were measured from responses elicited by short (3 ms) current pulses and long (1 s) current steps as previously described (Gouwens et al., 2019). Briefly, APs were detected by first identifying locations where the smoothed derivative of the membrane potential (dV/dt) exceeded 20 mV/ms, then refining based on several criteria including threshold-to-peak voltage and time differences and absolute peak height. After refinement, AP thresholds were re-calculated by finding the point for each AP where the dV/dt was 5% of the average maximal dV/dt across all APs. For each AP, threshold, peak, fast trough, and width (at half-height) were calculated, along with the upstroke/downstroke ratio (i.e., ratio of the peak upstroke dV/dt to the peak downstroke dV/dt).
Waveforms of the first APs elicited by the lowest-amplitude current pulses and steps were concatenated together for sparse principal component analysis (see below); the derivatives of these waveforms were also analyzed in this way. The voltage trajectory of the ISI was characterized by extracting the trace between the fast trough of the initial AP and the threshold of the following AP, normalizing their durations, and averaging together. AP features across the responses to long current steps were compared by binning APs into 20 ms intervals and averaging within a bin. If no APs fell within a bin, the value was interpolated from neighboring bins that had APs. This was done for stimulus amplitudes starting at a given cell’s rheobase, as well +40 pA and +80 pA above rheobase. If a sweep of an expected amplitude was unavailable (for example, if it failed one of the QC criteria), the missing values were interpolated from neighboring QC-passing sweeps. The instantaneous firing frequency (defined as the inverse of the ISI) was also binned and interpolated with 20 ms bins. The adaption of the instantaneous firing frequency was also analyzed by normalizing to the maximum rate observed during the step. Subthreshold responses to hyperpolarizing current steps were analyzed using downsampled (to 10 ms bins) membrane potential traces that were concatenated together. Responses from –10 pA to –90 pA steps (at a –40 pA interval) were used, and 200 ms of the time before and after the step were included as well. In addition, the largest amplitude hyperpolarizing step response was analyzed by normalizing to the minimum membrane potential reached and the baseline membrane potential.
Data sets were built by accumulating the feature vectors in each category (AP waveform, each AP feature across long steps, subthreshold response waveforms, etc). Sparse principal component analysis was performed separately on each data set. Principal components with an adjusted explained variance exceeding 1% were kept (typically 1 to 8 components from a given data set). Analysis of the data set yielded 44 total components. The components were z-scored and combined to form a reduced dimension feature matrix. The electrophysiological feature matrix used was visualized with a two-dimensional projection using Uniform Manifold Approximation and Projection (UMAP, (Becht et al., 2018)).
Sparse regression of gene expression and electrophysiology features
Sparse regression was performed on ion channel genes with non-zero expression to predict electrophysiology sparse principal components using the LassoLarsCV function of scikit-learn (Pedregosa et al., 2011) with ten-fold cross validation. The regression was trained on half the data set and used to predict the other half. Median gene expression values of a t-type were used as the features of the regression, and median sparse principal component value of t-types were predicted.
Anatomical and Morphological Annotation
Biocytin histology
A horseradish peroxidase (HRP) enzyme reaction using diaminobenzidine (DAB) as the chromogen was used to visualize the filled cells after electrophysiological recording, and 4,6-diamidino-2-phenylindole (DAPI) stain was used identify cortical layers as described previously (Gouwens et al., 2019).
Imaging
Mounted sections were imaged as described previously (Gouwens et al., 2019). Briefly, operators captured images on an upright AxioImager Z2 microscope (Zeiss, Germany) equipped with an Axiocam 506 monochrome camera and 0.63x optivar. Two-dimensional tiled overview images were captured with a 20X objective lens (Zeiss Plan-NEOFLUAR 20X/0.5) in brightfield transmission and fluorescence channels. Tiled image stacks of individual cells were acquired at higher resolution in the transmission channel only for the purpose of automated and manual reconstruction. Light was transmitted using an oil-immersion condenser (1.4 NA). High-resolution stacks were captured with a 63X objective lens (Zeiss Plan-Apochromat 63x/1.4 Oil or Zeiss LD LCI Plan-Apochromat 63x/1.2 Imm Corr) at an interval of 0.28 µm (1.4 NA objective) or 0.44 µm (1.2 NA objective) along the Z axis. Tiled images were stitched in ZEN software and exported as single-plane TIFF files.
Anatomical location
To characterize the position of biocytin-labeled cells in the mouse brain, a 20x brightfield and/or fluorescent image of DAPI (4’,6-diamidino-2-phenylindole) stained tissue was captured and analyzed to determine layer position and region. Soma position of reconstructed neurons was annotated and used to calculate soma depth relative to drawings of the pia and/or white matter. Individual cells were also manually placed in the appropriate cortical region and layer within the Allen Mouse Common Coordinate Framework (CCF) by matching the 20x image of the slice with a “virtual” slice at an appropriate location and orientation within the CCF. Laminar locations (see Figure 2) were calculated by finding the path connecting pia and white matter that passed through the cell’s coordinate, identifying its distance to pia and white matter as well as position within its layer, then aligning those values to an average set of layer thicknesses. Using the DAPI image, laminar borders were also drawn for all reconstructed neurons.
Morphological reconstruction
Reconstructions of the dendrites and the full axon were generated for a subset of neurons with good quality transcriptomics, electrophysiology and biocytin fill. Reconstructions were generated based on a 3D image stack that was run through a Vaa3D-based image processing and reconstruction pipeline (Peng et al., 2010). Images were used to generate an automated reconstruction of the neuron using TReMAP (Zhou et al., 2016). Alternatively, intial reconstructions were created manually using the reconstruction software PyKNOSSOS (Ariadne-service) or the citizen neuroscience game Mozak (Mozak.science) (Roskams and Popović, 2016). Automated or manually-initiated reconstructions were then extensively manually corrected and curated using a range of tools (e.g., virtual finger, polyline) in the Mozak extension (Zoran Popović, Center for Game Science, University of Washington) of Terafly tools (Peng et al., 2014; Bria et al., 2016) in Vaa3D. Every attempt was made to generate a completely connected neuronal structure while remaining faithful to image data. If axonal processes could not be traced back to the main structure of the neuron, they were left unconnected.
Before morphological feature analysis, reconstructed neuronal morphologies were expanded in the dimension perpendicular to the cut surface to correct for shrinkage (Deitcher et al., 2017; Egger et al., 2008) after tissue processing. The amount of shrinkage was calculated by comparing the distance of the soma to the cut surface during recording and after fixation and reconstruction. A tilt angle correction was also performed based on the estimated difference (via CCF registration) between the slicing angle and the direct pia-white matter direction at the cell’s location (Gouwens et al., 2019).
Morphology feature analysis
Calculating morphological features
Morphological features were calculated as previously described (Gouwens et al., 2019). Briefly, feature definitions were collected from prior studies (Scorcioni et al., 2008; Markram et al., 2015). Reconstructed neurons were aligned in the direction perpendicular to pia and white matter. Additional features, such as the laminar distribution of axon, were calculated from the aligned morphologies. While slice angle tilt and shrinkage correction were performed (see above), features predominantly determined by differences in the z-dimension were not analyzed to minimize technical artifacts due to z-compression of the slice after processing.
NBLAST scores
To assess the similarity of axonal morphologies directly, we used the NBLAST method of the nat.nblast R package (Costa et al., 2016; Kohl et al., 2013). This method compares a pair of morphologies by analyzing the distance and local direction of nearby points between the two structures. NBLAST similarity scores, which range from 0 (completely dissimilar) to 1 (completely identical), were calculated using version 1 of the algorithm with a standard deviation parameter of 10 microns. Morphologies were aligned on their somas in the z-direction (perpendicular to the cut surface of the slice) before NBLAST scoring. Scores were symmetrized by averaging the scores in each direction between a given pair. Representative morphologies for a t-type were chosen by selecting cells with high average NBLAST scores versus other cells of the same t-type.
Feature correspondence analysis
We used several methods to determine how the different data modalities under investigation varied with respect to each other. The log-likelihood ratio (LLR) allows us to test whether data from each of a pair of types actually appears to come from two separate populations, or whether it is likely that both data sets came from the same population. We used this to evaluate whether pairs of t-types were systematically different from one another. Even if multiple populations are different, they may heavily overlap, making it difficult to tell where a given cell came from; we used supervised classifiers to estimate how reliable predictions about type could be using different modalities. Along similar lines, discriminant analysis is another measure of how well-separated the distributions of different features are between pairs of types. Finally, we used clustering analyses with different modalities to evaluate how well different type assignments match with each other and to find groupings that maximize the degree of correspondence.
Likelihood ratio analysis
The distributions of electrophysiological and morphological features between pairs of t-types were compared by a log-likelihood ratio method. The data from a pair of t-types were fit with either a one-component model (combining all cells) or a two-component model (separate for each t-type). The dimensionality of the data sets was reduced by performing principal components analysis (PCA) on the set of elecrophysiological or morphological features and keeping the top three principal components. Multivariate Gaussian fits with a diagonal covariance matrix were fit to these data sets, and the log-likelihood ratio (LLR) of the data under the two-component versus one-component model were calculated. Since the two-component model would produce a higher likelihood due to overfitting even if the underlying distributions were the same, the statistical significance of the observed LLR was assessed by parametric boostrap. Random samples of the same size as the actual data set were drawn 2,000 times from the one-component model, and LLRs were calculated in the same way as with the actual data. These bootstrapped LLRs were used to estimate the p values of the observed LLRs.
Supervised classification
Supervised classification was performed by training a random-forest classifier with 500 trees using balanced class weights, implemented with the scikit-learn Python package (Pedregosa et al., 2011). Out-of-bag confusion matrices and accuracies were reported.
Discriminant analysis
To assess the degree to which pairs of t-types exhibited discrete or continuous variation in electrophysiological and morphological feature space, we used a cross-validated discriminant analysis method based on one developed to address a similar question in transcriptomic feature space (Harris et al., 2018). For each pair of t-types, multivariate Guassians were fit to “training” cells from each t-type to describe a group of electrophysiological or morphological features (typically with one to six dimensions in each feature group). Then, the likelihood ratio was calculated for each held-out “test” cell between its own assigned t-type versus the other t-type. This produced a distribution of likelihood ratios for each t-type, and the separation of these distributions was summarized by a d′ statistic, , where μi and σi are the mean and standard deviations of the likelihood ratios for cells in t-type i. Larger values of d′ indicate more separation between t-types.
ME-clustering
To identify types that had both consistent electrophysiological and morphological characteristics, we employed the same strategy developed for our previous work (Gouwens et al., 2019). Briefly, this method includes generating multiple combined electrophysiological and morphology feature matrices with different electrophysiological feature weights, then using multiple unsupervised clustering algorithms with each combined feature matrix. A cell-wise co-clustering matrix was calculated from these variants, and consensus clusters were determined by iterative hierarchical clustering. One difference from the prior work was that clusters with lower stability (as assessed by repeating the entire procedure on 90% subsamples and calculating the average Jaccard similarity) were not merged with other clusters closest-matching cluster. Since cells in different me-types could be merged as part of the met-type identification analysis (see below), we kept the me-types at a more granular level in this study.
MET-type identification
To identify sets of cells that could be grouped into met-types, we made use of the fact that our mapping procedure estimated the probability of assigning a given cell to t-types other than its best match (see “Mapping on the reference taxonomy tree” above and Figure S2C). We computed a similar estimate for alternative me-type assignments by using random-forest classifiers trained on 90% of the data to assign labels for the other 10%; this procedure was repeated with different subsamples to obtain 100 predicted assignments for each cell. We used these t-type and me-type cross-mapping probabilities to construct a graph where the nodes were t-type/me-type combinations. Edges were then added to connect nodes with the same me-type but different t-types or the same t-type but different me-types; the edges were weighted by the average cross-mapping probability. Nodes that contained only a single cell with no connections to other nodes in the graph were removed. Finally, we used the Leiden community detection algorithm (Traag et al., 2019) to identified nodes that could be grouped together due to strong connections between them.
Quantification and statistical analysis
No statistical methods were used to predetermine sample sizes, but the sample sizes here are similar to those reported in previous publications. No randomization was used during data collection as there was a single experimental condition for all acquired data. The different stimulus protocols were not presented in a randomized order. Data collection and analyses were not performed blind to the conditions of the experiments as there was a single experimental condition for all acquired data.
Supplementary Material
(A) Each cell underwent standardized collection of electrophysiology data, followed by a series of processing and quality control steps in parallel for each modality. Transcriptomic analysis included extraction and reverse transcription of the cell’s nuclear and cytosolic mRNA, followed by cDNA amplification, sequencing and evaluation of data quality based on normalized summed expression of “on”-type marker genes (NMS) adapted from the single-cell quality control measures in (Tripathy et al., 2018). Electrophysiological analysis included cell and sweep-level quality control gates, and automated extraction of feature vectors. Electrophysiology slices were stained, mounted onto coverslips, imaged at 20x and mapped to the Allen Mouse Common Coordinate Framework (CCF). QC-Qualified cells had an NMS > 0.4, passed electrophysiology quality control and feature extraction and had a confirmed soma location in primary visual cortex (VISp). A subset of cells with high-quality cell fills were further processed for morphological reconstruction. (B) Schematic of reference data set and mapping tree. (C) Assignment of t-types to cells assayed via Patch-seq recordings. QC-qualified cells from (A) were mapped on the reference dataset obtained in (B) using two different mapping algorithms (see (D) and (E)). (D) Schematic illustrating mapping procedure using the reference taxonomy tree. (E) Schematic illustrating mapping using a neural network classifier. (F) Calculation of reference mapping confusion matrix. The reference dissociated cells from (Tasic et al., 2018) were mapped to the reference taxonomy tree using the same method in (D), and the mapping probability matrix of those cells was computed. This probability matrix and the clustering results from (Tasic et al., 2018) of those dissociated cells were used as inputs to compute the reference confusion matrix. (G) Schematic illustrating the determination of mapping quality to each cell assayed via Patch-seq recordings.
(A) Comparing transcriptomic data quality of dissociated cells from (Tasic et al., 2018) and cells assayed via Patch-seq recordings. Distribution of total number of genes detected in reference dissociated cells (dark grey) vs cells assayed via Patch-seq recordings (light grey) are presented by violin plots. The black dots are the medians. (B) Expression of selected excitatory marker genes (Slc17a7, Nrn1, Fam19a1), Glia marker genes (Aqp4, Plp1, Olig1 and Trf) and inhibitory marker genes (Gad1, Gad2, Slc32a1, Dlx2, Dlx5 and Dlx1) for each subclasses (Lamp5, Sncg, Vip, Sst, Pvalb) are represented by violin plots. For each gene, three violin plots are shown. The one on the left is the gene expression of Glutamatergic dissociated cells and are repeated in all subclasses for comparison purposes. The violin plot in the middle is the gene expression of the GABAergic dissociated cells (n=6,080) and the one on the right is the gene expression of all GABAergic interneurons assayed via Patch-seq recordings that passed transcriptomic QC criteria (n=4,270) . (C) Mapping probability for three groups of cells described in Figure S1G (highly consistent, moderately consistent and inconsistent cells) using the reference tree mapping method (Figure S1D and Methods). Horizontal dashed lines divide mapping categories; vertical dashed lines divide transcriptomic subclasses.
Example responses and UMAP plots for t-types not shown in Figure 3. (A) Responses to 1 second-long current steps with stimulus amplitudes equal to −70 pA and rheobase for that cell (lower traces) and rheobase + 80 pA (upper trace). Two randomly-chosen examples shown for each t-type. Scale bar: vertical 50 mV, horizontal 250 ms. (B) UMAP plots generated using the 44 z-scored sparse principal component values collected from electrophysiology feature vectors. Cells from a specific t-type are shown as colored points. Arrowheads indicate the locations of the examples (filled: left, hollow: right) shown in (A). In (A) and (B), t-types with a minimum of 10 cells with highly-consistent mapping are shown.
(A) Heatmap of electrophysiology-based (lower half) and morphology-based (upper half) log-likelihood ratios (LLRs), indicating the improvement gained by fitting each of a pair of t-types separately vs together (lighter colors indicate greater improvement). The statistical significance of the improvement was assessed by parametric bootstrap; pairs that do not reach statistical significance levels of less than p=0.05 and p=0.001 are indicated by crosses and slashes, respectively. Only t-types with at least 10 highly-consistent mapped cells are shown for electrophysiology (lower), and only t-types with at least 5 highly-consistent mapped cells are shown for morphology (upper; missing values indicate t-types with fewer than 5 morphologies). Note that the generally lower LLRs for morphology are likely due in part to the smaller size of the morphology data set. (B) Cross-validated d′ values between pairs of met-types by electrophysiological (green) and morphological (purple) feature groups. Larger d′ values indicate greater discriminability between pairs of t-types. Only met-types with at least 10 cells are shown. Radial tick interval: 1.
(A) Prediction of the AP Vm sparse principal component 1 (AP Vm sPC-1) for half the data set using a sparse linear regression (lasso) over ion channel gene expression values trained on the other half of the data set. Median values of log-normalized gene expression electrophysiology sparse principal components were calculated and used for the regression. Median actual and predicted values are shown (each point is a t-type). Higher values of this sPC are associated with shorter, narrower APs. (B) The non-zero coefficient weights of the trained regression. Note that these weights can be interpreted to some extent. For example, the gene Kcnc3 (aka Kv3.3) has a high weight and is known to contribute to narrow APs in fast-spiking cells. (C) Regression scores (above) and ion channel gene coefficient weights (below) for all the electrophysiology sPCs. All 44 sPCs (columns) are grouped by category (AP Vm, AP dV/dt, etc.) and numbered within each category (bottom labels). The example in (A) and (B) is in the first column. Note that in some cases all coefficients were zero, indicating that the regression was unable to predict that feature. Only voltage-gated ion channel genes that had non-zero median expression in at least one t-type are shown.
All morphological reconstructions from Sst met-types (colors), aligned by layer to an average cortical template and ordered by distance from pia within met-type. Dendrites are in darker colors, axon in lighter colors. Scale bar = 250 µm.
All morphological reconstructions from Pvalb, Lamp5, Sncg and Vip met-types (colors), aligned by layer to an average cortical template and ordered by distance from pia within met-type. Dendrites are in darker colors, axon in lighter colors. Scale bar = 250 µm.
As in Figure 5, me-types are compared to t-types, but here the t-types are from a new study by (Yao et al., 2020) analyzing data from the whole mouse cortex and hippocampus. (A) Comparison of t-types with unsupervised joint electrophysiological and morphological clustering results (me-types). Both highly-consistent and moderately-consistent cells are shown (n=491). (B) Graph visualization of cross-mapping between me-type/t-type combinations. The nodes represent cells with a particular me-type/t-type combination as in (A), and the size of the node indicates the number of cells. Edges represent the presence of cells in a given me-type/t-type combination that have some probability of mapping to another t-type (orange lines) or me-type (blue lines). Thicker lines indicate a higher probability of mapping to another type. Nodes that had only a single cell and no connections to other nodes are not shown. The Leiden community detection algorithm was used to group sets of well-connected me-type/t-type combinations into 28 met-types (outlined groups) . (C) River plot showing the relationships between t-types (left) and met-types (right) . (D) River plot showing the close correspondence between met-types defined using the whole-cortex and hippocampus taxonomy (CTX-HPF, left) of (Yao et al., 2020) and the met-types defined from the visual cortex taxonomy (VISp, right) of (Tasic et al., 2018) that are defined in Figure 5.
Highlights.
Patch-seq data obtained from >4,200 GABAergic interneurons with >500 morphologies
Comprehensive characterization of morpho-electric features of transcriptomic types
28 interneuron met-types with congruent properties across data modalities
Different Sst met-types preferentially innervate different cortical layers
Acknowledgments
We are grateful to Cathryn Cadwell, Federico Scala, and Andreas Tolias for sharing their Patch-seq experience with us in the early stage of our pipeline development. We appreciate feedback on the manuscript provided by Scott Owen, Jeremy Miller, and Brian Kalmbach. We thank Adrian Wanner for providing reconstruction services through Ariadne and Roy Szeto for facilitating the reconstruction work contributed by Mozak.science. Finally, we thank the Mozak citizen-scientists for their valuable contribution. The research was partially supported by several grant awards from institutes under the National Institutes of Health (NIH), including award numbers R01EY023173 from The National Eye Institute and U01MH105982 from the National Institute of Mental Health and Eunice Kennedy Shriver National Institute of Child Health & Human Development to H.Z. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH and its subsidiary institutes. This work was funded by the Allen Institute for Brain Science. We thank Sil Coulter, John Philips, and Allan Jones for leadership and guidance, and dedicate this paper to the vision, encouragement, and long-term support of our founder, Paul G. Allen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsáki G, Cauli B, Defelipe J, Fairén A, et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Berger JO, Bernardo JM, and Sun D (2009). The Formal Definition of Reference Priors. The Annals of Statistics 37, 905–938. [Google Scholar]
- Bortone DS, Olsen SR, and Scanziani M (2014). Translaminar inhibitory cells recruited by layer 6 corticothalamic neurons suppress visual cortex. Neuron 82, 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bria A, Iannello G, Onofri L, and Peng H (2016). TeraFly: real-time three-dimensional visualization and annotation of terabytes of multidimensional volumetric images. Nat Methods 13, 192–194. [DOI] [PubMed] [Google Scholar]
- Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, Reimer J, Shen S, Bethge M, Tolias KF, et al. (2016). Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat Biotechnol 34, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, and Spruston N (2019). Heterogeneity within classical cell types is the rule: lessons from hippocampal pyramidal neurons. Nat Rev Neurosci 20, 193–204. [DOI] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X (2015). RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codeluppi S, Borm LE, Zeisel A, La MG, van LJA, Svensson CI, and Linnarsson S (2018). Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 15, 932–935. [DOI] [PubMed] [Google Scholar]
- Coskun AF, and Cai L (2016). Dense transcript profiling in single cells by image correlation decoding. Nat Methods 13, 657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Manton JD, Ostrovsky AD, Prohaska S, and Jefferis GS (2016). NBLAST: Rapid, Sensitive Comparison of Neuronal Structure and Construction of Neuron Family Databases. Neuron 91, 293–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018) . A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, López-Cruz PL, Benavides-Piccione R, Bielza C, Larrañaga P, Anderson S, Burkhalter A, Cauli B, Fairén A, Feldmeyer D, et al. (2013). New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci 14, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher Y, Eyal G, Kanari L, Verhoog MB, Atenekeng KGA, Mansvelder HD, de Koch CPJ, and Segev I (2017). Comprehensive Morpho-Electrotonic Analysis Shows 2 Distinct Classes of L2 and L3 Pyramidal Neurons in Human Temporal Cortex. Cereb Cortex 27, 5398–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, et al. (2006). Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Nevian T, and Bruno RM (2008). Subcolumnar dendritic and axonal organization of spiny stellate and star pyramid neurons within a barrel in rat somatosensory cortex. Cereb Cortex 18, 876–889. [DOI] [PubMed] [Google Scholar]
- Frandolig JE, Matney CJ, Lee K, Kim J, Chevée M, Kim SJ, Bickert AA, and Brown SP (2019). The Synaptic Organization of Layer 6 Circuits Reveals Inhibition as a Major Output of a Neocortical Sublamina. Cell Rep 28, 3131–3143.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuzik J, Zeisel A, Máté Z, Calvigioni D, Yanagawa Y, Szabó G, Linnarsson S, and Harkany T (2016). Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat Biotechnol 34, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Darmanis S, Aoto J, Malenka RC, Quake SR, and Südhof TC (2016). Single-cell RNAseq reveals cell adhesion molecule profiles in electrophysiologically defined neurons. Proc Natl Acad Sci U S A 113, E5222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, and Heintz N (2013). GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, and Gerfen CR (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27, 9817–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Berg J, Lee C, Jarsky T, Ting J, Sunkin SM, Feng D, Anastassiou CA, Barkan E, et al. (2019). Classification of electrophysiological and morphological neuron types in the mouse visual cortex. Nat Neurosci 22, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes WN, Seal RP, Oesch N, Edwards RH, and Diamond JS (2011). Genetic targeting and physiological features of VGLUT3+ amacrine cells. Vis Neurosci 28, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, et al. (2014). Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front Neural Circuits 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Hochgerner H, Skene NG, Magno L, Katona L, Bengtsson GC, Somogyi P, Kessaris N, Linnarsson S, and Hjerling-Leffler J (2018). Classes and continua of hippocampal CA1 inhibitory neurons revealed by single-cell transcriptomics. PLoS Biol 16, e2006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, et al. (2016). Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron 92, 555. [DOI] [PubMed] [Google Scholar]
- Hilscher MM, Leão RN, Edwards SJ, Leão KE, and Kullander K (2017). Chrna2-Martinotti Cells Synchronize Layer 5 Type A Pyramidal Cells via Rebound Excitation.. PLoS Biol 15, e2001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, and Arber S (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol 3, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, and Paul A (2019). The diversity of GABAergic neurons and neural communication elements. Nat Rev Neurosci 20, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan M, and Anderson SA (2014). The chandelier cell, form and function.. Curr Opin Neurobiol 26, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shen S, Cadwell CR, Berens P, Sinz F, Ecker AS, Patel S, and Tolias AS (2015). Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462–aac9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang G, Lee AJ, Stornetta RL, and Zhu JJ (2013). The organization of two new cortical interneuronal circuits.. Nat Neurosci 16, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, and Kubota Y (1996). Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci 16, 2701–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull JM, Garcia da S.P., Reid AP, Peikon ID, Albeanu DF, and Zador AM (2016). High-Throughput Mapping of Single-Neuron Projections by Sequencing of Barcoded RNA. Neuron 91, 975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Zhang Z, Huang L, Ito-Cole T, Jacobs MW, Juavinett AL, Senturk G, Hu M, Ku M, Ecker JR, et al. (2020). Extraction of Distinct Neuronal Cell Types from within a Genetically Continuous Population.. Neuron. [DOI] [PMC free article] [PubMed]
- Klausberger T, and Somogyi P (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl J, Ostrovsky AD, Frechter S, and Jefferis GS (2013). A bidirectional circuit switch reroutes pheromone signals in male and female brains. Cell 155, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan L, Li Y, Lau C, Feng D, Bernard A, Sunkin SM, Zeng H, Dang C, Hawrylycz M, and Ng L (2015). Neuroinformatics of the Allen Mouse Brain Connectivity Atlas. Methods 73, 4–17. [DOI] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, et al. (2014). Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hu H, Berrebi AS, Mathers PH, and Agmon A (2006). Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice.. J Neurosci 26, 5069–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der B.A., Niino Y, Egolf L, Monetti C, et al. (2015) . Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Muller E, Ramaswamy S, Reimann MW, Abdellah M, Sanchez CA, Ailamaki A, Alonso-Nanclares L, Antille N, Arsever S, et al. (2015). Reconstruction and Simulation of Neocortical Microcircuitry. Cell 163, 456–492. [DOI] [PubMed] [Google Scholar]
- McGarry LM, Packer AM, Fino E, Nikolenko V, Sippy T, and Yuste R (2010). Quantitative classification of somatostatin-positive neocortical interneurons identifies three interneuron subtypes. Front Neural Circuits 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, Rubinstein ND, Hao J, Regev A, Dulac C, et al. (2018). Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz W, Tremblay R, Levenstein D, and Rudy B (2017). Layer-specific modulation of neocortical dendritic inhibition during active wakefulness. Science 355, 954–959. [DOI] [PubMed] [Google Scholar]
- Naka A, Veit J, Shababo B, Chance RK, Risso D, Stafford D, Snyder B, Egladyous A, Chu D, Sridharan S, et al. (2019). Complementary networks of cortical somatostatin interneurons enforce layer specific control. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (1992). Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207, 123–131. [DOI] [PubMed] [Google Scholar]
- Nigro MJ, Hashikawa-Yamasaki Y, and Rudy B (2018). Diversity and Connectivity of Layer 5 Somatostatin-Expressing Interneurons in the Mouse Barrel Cortex. J Neurosci 38, 1622–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A, Crow M, Raudales R, He M, Gillis J, and Huang ZJ (2017). Transcriptional Architecture of Synaptic Communication Delineates GABAergic Neuron Identity. Cell 171, 522–539.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830. [Google Scholar]
- Peng H, Bria A, Zhou Z, Iannello G, and Long F (2014). Extensible visualization and analysis for multidimensional images using Vaa3D. Nat Protoc 9, 193–208. [DOI] [PubMed] [Google Scholar]
- Peng H, Ruan Z, Long F, Simpson JH, and Myers EW (2010). V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat Biotechnol 28, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, and Kara DA (1985). The neuronal composition of area 17 of rat visual cortex. II. The nonpyramidal cells.. J Comp Neurol 234, 242–263. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, Chan CS, Dombeck DA, Deisseroth K, and Awatramani R (2018). Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci 21, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prönneke A, Scheuer B, Wagener RJ, Möck M, Witte M, and Staiger JF (2015). Characterizing VIP Neurons in the Barrel Cortex of VIPcre/tdTomato Mice Reveals Layer-Specific Differences. Cereb Cortex 25, 4854–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que L, Lukacsovich D, and Földy C (2020). Transcriptomic homogeneity and an age-dependent onset of hemoglobin expression characterize morphological PV types. bioRxiv doi: 10.1101/2020.01.21.913103. [DOI]
- Roskams J, and Popović Z (2016). Power to the People: Addressing Big Data Challenges in Neuroscience by Creating a New Cadre of Citizen Neuroscientists. Neuron 92, 658–664. [DOI] [PubMed] [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, and Elmquist JK (2011). Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de RH, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scala F, Kobak D, Bernabucci M, Bernaerts Y, Cadwell CR, Castro JR, Hartmanis L, Jiang X, Laturnus S, Miranda E, et al. (2020) . Phenotypic variation within and across transcriptomic cell types in mouse motor cortex. bioRxiv doi: 10.1101/2020.02.03.929158. [DOI] [PMC free article] [PubMed]
- Scala F, Kobak D, Shan S, Bernaerts Y, Laturnus S, Cadwell CR, Hartmanis L, Froudarakis E, Castro JR, Tan ZH, et al. (2019). Layer 4 of mouse neocortex differs in cell types and circuit organization between sensory areas. Nat Commun 10, 4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman B, Machold RP, Hashikawa Y, Fuzik J, Fishell GJ, and Rudy B (2019). Four Unique Interneuron Populations Reside in Neocortical Layer 1.. J Neurosci 39, 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorcioni R, Polavaram S, and Ascoli GA (2008). L-Measure: a web-accessible tool for the analysis, comparison and search of digital reconstructions of neuronal morphologies. Nat Protoc 3, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016) . Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley G, Gokce O, Malenka RC, Südhof TC, and Quake SR (2020). Continuous and Discrete Neuron Types of the Adult Murine Striatum.. Neuron 105, 688–699.e8. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, Lu J, and Huang ZJ (2013). The spatial and temporal origin of chandelier cells in mouse neocortex.. Science 339, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebaykin D, Tripathy SJ, Binnion N, Li B, Gerkin RC, and Pavlidis P (2018) . Modeling sources of interlaboratory variability in electrophysiological properties of mammalian neurons. J Neurophysiol 119, 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, and Lowell BB (2008). Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities.. Sci Rep 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy SJ, Toker L, Bomkamp C, Mancarci BO, Belmadani M, and Pavlidis P (2018). Assessing Transcriptome Quality in Patch-Seq Datasets. Front Mol Neurosci 11, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua SJ, and Lowell BB (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Ding SL, Li Y, Royall J, Feng D, Lesnar P, Graddis N, Naeemi M, Facer B, Ho A, et al. (2020). The Allen Mouse Brain Common Coordinate Framework: A 3D Reference Atlas. Cell 181, 936–953.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Toledo-Rodriguez M, Gupta A, Wu C, Silberberg G, Luo J, and Markram H (2004). Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J Physiol 561, 65–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff A, and Yuste R (2008). Of mice and men, and chandeliers.. PLoS Biol 6, e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Nguyen TN, Velthoven C.T.J. van, Goldy J, Sedeno-Cortes AE, Baftizadeh F, Bertagnolli D, Casper T, Crichton K, Ding S-L, et al. (2020). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. bioRxiv doi: 10.1101/2020.03.30.015214. [DOI] [PMC free article] [PubMed]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, Zwan J. van der, Häring M, Braun E, Borm LE, Manno GL, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, Manno GL, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zeng H, and Sanes JR (2017) . Neuronal cell-type classification: challenges, opportunities and the path forward. Nat Rev Neurosci 18, 530–546. [DOI] [PubMed] [Google Scholar]
- Zhang M, Eichhorn SW, Zingg B, Yao Z, Zeng H, Dong H, and Zhuang X (2020). Molecular spatial and projection diversity of neurons in primary motor cortex revealed by in situ single-cell transcriptomics. bioRxiv doi: 10.1101/2020.06.04.105700. [DOI]
- Zhou X, Mansori I, Fischer T, Witte M, and Staiger JF (2020). Characterizing the morphology of somatostatin-expressing interneurons and their synaptic innervation pattern in the barrel cortex of the GFP-expressing inhibitory neurons mouse. J Comp Neurol 528, 244–260. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Liu X, Long B, and Peng H (2016). TReMAP: Automatic 3D Neuron Reconstruction Based on Tracing, Reverse Mapping and Assembling of 2D Projections. Neuroinformatics 14, 41–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Each cell underwent standardized collection of electrophysiology data, followed by a series of processing and quality control steps in parallel for each modality. Transcriptomic analysis included extraction and reverse transcription of the cell’s nuclear and cytosolic mRNA, followed by cDNA amplification, sequencing and evaluation of data quality based on normalized summed expression of “on”-type marker genes (NMS) adapted from the single-cell quality control measures in (Tripathy et al., 2018). Electrophysiological analysis included cell and sweep-level quality control gates, and automated extraction of feature vectors. Electrophysiology slices were stained, mounted onto coverslips, imaged at 20x and mapped to the Allen Mouse Common Coordinate Framework (CCF). QC-Qualified cells had an NMS > 0.4, passed electrophysiology quality control and feature extraction and had a confirmed soma location in primary visual cortex (VISp). A subset of cells with high-quality cell fills were further processed for morphological reconstruction. (B) Schematic of reference data set and mapping tree. (C) Assignment of t-types to cells assayed via Patch-seq recordings. QC-qualified cells from (A) were mapped on the reference dataset obtained in (B) using two different mapping algorithms (see (D) and (E)). (D) Schematic illustrating mapping procedure using the reference taxonomy tree. (E) Schematic illustrating mapping using a neural network classifier. (F) Calculation of reference mapping confusion matrix. The reference dissociated cells from (Tasic et al., 2018) were mapped to the reference taxonomy tree using the same method in (D), and the mapping probability matrix of those cells was computed. This probability matrix and the clustering results from (Tasic et al., 2018) of those dissociated cells were used as inputs to compute the reference confusion matrix. (G) Schematic illustrating the determination of mapping quality to each cell assayed via Patch-seq recordings.
(A) Comparing transcriptomic data quality of dissociated cells from (Tasic et al., 2018) and cells assayed via Patch-seq recordings. Distribution of total number of genes detected in reference dissociated cells (dark grey) vs cells assayed via Patch-seq recordings (light grey) are presented by violin plots. The black dots are the medians. (B) Expression of selected excitatory marker genes (Slc17a7, Nrn1, Fam19a1), Glia marker genes (Aqp4, Plp1, Olig1 and Trf) and inhibitory marker genes (Gad1, Gad2, Slc32a1, Dlx2, Dlx5 and Dlx1) for each subclasses (Lamp5, Sncg, Vip, Sst, Pvalb) are represented by violin plots. For each gene, three violin plots are shown. The one on the left is the gene expression of Glutamatergic dissociated cells and are repeated in all subclasses for comparison purposes. The violin plot in the middle is the gene expression of the GABAergic dissociated cells (n=6,080) and the one on the right is the gene expression of all GABAergic interneurons assayed via Patch-seq recordings that passed transcriptomic QC criteria (n=4,270) . (C) Mapping probability for three groups of cells described in Figure S1G (highly consistent, moderately consistent and inconsistent cells) using the reference tree mapping method (Figure S1D and Methods). Horizontal dashed lines divide mapping categories; vertical dashed lines divide transcriptomic subclasses.
Example responses and UMAP plots for t-types not shown in Figure 3. (A) Responses to 1 second-long current steps with stimulus amplitudes equal to −70 pA and rheobase for that cell (lower traces) and rheobase + 80 pA (upper trace). Two randomly-chosen examples shown for each t-type. Scale bar: vertical 50 mV, horizontal 250 ms. (B) UMAP plots generated using the 44 z-scored sparse principal component values collected from electrophysiology feature vectors. Cells from a specific t-type are shown as colored points. Arrowheads indicate the locations of the examples (filled: left, hollow: right) shown in (A). In (A) and (B), t-types with a minimum of 10 cells with highly-consistent mapping are shown.
(A) Heatmap of electrophysiology-based (lower half) and morphology-based (upper half) log-likelihood ratios (LLRs), indicating the improvement gained by fitting each of a pair of t-types separately vs together (lighter colors indicate greater improvement). The statistical significance of the improvement was assessed by parametric bootstrap; pairs that do not reach statistical significance levels of less than p=0.05 and p=0.001 are indicated by crosses and slashes, respectively. Only t-types with at least 10 highly-consistent mapped cells are shown for electrophysiology (lower), and only t-types with at least 5 highly-consistent mapped cells are shown for morphology (upper; missing values indicate t-types with fewer than 5 morphologies). Note that the generally lower LLRs for morphology are likely due in part to the smaller size of the morphology data set. (B) Cross-validated d′ values between pairs of met-types by electrophysiological (green) and morphological (purple) feature groups. Larger d′ values indicate greater discriminability between pairs of t-types. Only met-types with at least 10 cells are shown. Radial tick interval: 1.
(A) Prediction of the AP Vm sparse principal component 1 (AP Vm sPC-1) for half the data set using a sparse linear regression (lasso) over ion channel gene expression values trained on the other half of the data set. Median values of log-normalized gene expression electrophysiology sparse principal components were calculated and used for the regression. Median actual and predicted values are shown (each point is a t-type). Higher values of this sPC are associated with shorter, narrower APs. (B) The non-zero coefficient weights of the trained regression. Note that these weights can be interpreted to some extent. For example, the gene Kcnc3 (aka Kv3.3) has a high weight and is known to contribute to narrow APs in fast-spiking cells. (C) Regression scores (above) and ion channel gene coefficient weights (below) for all the electrophysiology sPCs. All 44 sPCs (columns) are grouped by category (AP Vm, AP dV/dt, etc.) and numbered within each category (bottom labels). The example in (A) and (B) is in the first column. Note that in some cases all coefficients were zero, indicating that the regression was unable to predict that feature. Only voltage-gated ion channel genes that had non-zero median expression in at least one t-type are shown.
All morphological reconstructions from Sst met-types (colors), aligned by layer to an average cortical template and ordered by distance from pia within met-type. Dendrites are in darker colors, axon in lighter colors. Scale bar = 250 µm.
All morphological reconstructions from Pvalb, Lamp5, Sncg and Vip met-types (colors), aligned by layer to an average cortical template and ordered by distance from pia within met-type. Dendrites are in darker colors, axon in lighter colors. Scale bar = 250 µm.
As in Figure 5, me-types are compared to t-types, but here the t-types are from a new study by (Yao et al., 2020) analyzing data from the whole mouse cortex and hippocampus. (A) Comparison of t-types with unsupervised joint electrophysiological and morphological clustering results (me-types). Both highly-consistent and moderately-consistent cells are shown (n=491). (B) Graph visualization of cross-mapping between me-type/t-type combinations. The nodes represent cells with a particular me-type/t-type combination as in (A), and the size of the node indicates the number of cells. Edges represent the presence of cells in a given me-type/t-type combination that have some probability of mapping to another t-type (orange lines) or me-type (blue lines). Thicker lines indicate a higher probability of mapping to another type. Nodes that had only a single cell and no connections to other nodes are not shown. The Leiden community detection algorithm was used to group sets of well-connected me-type/t-type combinations into 28 met-types (outlined groups) . (C) River plot showing the relationships between t-types (left) and met-types (right) . (D) River plot showing the close correspondence between met-types defined using the whole-cortex and hippocampus taxonomy (CTX-HPF, left) of (Yao et al., 2020) and the met-types defined from the visual cortex taxonomy (VISp, right) of (Tasic et al., 2018) that are defined in Figure 5.
Data Availability Statement
Transcriptomic data supporting the findings of this study are available at the NeMO archive. Electrophysiological data for this study are available at the DANDI archive. Morphological reconstructions from this study are available at the Brain Image Library (BIL) archive. Information for accessing these data are in the Key Resources Table and summarized at https://portal.brain-map.org/explore/classes/multimodal-characterization (“Neurons in Mouse Primary Visual Cortex”).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Takara | 634894 |
| Nextera XT Index Kit V2 Set A-D | Nextera | FC-131– 2001,2002,2003,2004 |
| Deposited Data | ||
| Transcriptomic data – fastq files | This paper; NeMO | http://data.nemoarchive.org/other/grant/AIBS_patchseq/transcriptome/scell/SMARTseq/raw/20200611/ |
| Transcriptomic data – BAM files | This paper; NeMO | http://data.nemoarchive.org/other/grant/AIBS_patchseq/transcriptome/scell/SMARTseq/processed/align/20200611/ |
| Transcriptomic data – processed count files | This paper; NeMO | http://data.nemoarchive.org/other/grant/AIBS_patchseq/transcriptome/scell/SMARTseq/processed/analysis/20200611/ |
| Raw electrophysiology data | This paper; DANDI | DANDI: 000020 https://dandiarchive.org/dandiset/000020 |
| Morphological reconstructions | This paper; Brain Image Library | ftp://download.brainlib.org:8811/biccn/zeng/pseq/morph/200526/ |
| Specimen metadata | This paper; Allen Institute for Brain Science | https://brainmapportal-live-4cc80a57cd6e400d854-f7fdcae.divio-media.net/filer_public/5e/2a/5e2a5936-61da-4e09-b6da-74ab97ce1b02/20200711_patchseq_metadata_mouse.csv |
| Experimental Models: Organisms/Strains | ||
| Mouse: Calb1-IRES2-Cre | Daigle et al., Cell, 2018; The Jackson Laboratory | RRID:IMSR_JAX:028532 |
| Mouse: Calb2-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:010774 |
| Mouse: Cck-IRES0Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:012706 |
| Mouse: Chat-IRES-Cre | Rossi et al., Cell Metab. 2011; The Jackson Laboratory | RRID:IMSR_JAX:006410 |
| Mouse: Chrna2-Cre_OE25 | Gong, et al., J Neurosci 2007; MMRRC | RRID:MMRRC_036502-UCD |
| Mouse: Crh-IRES-Cre_ZJH | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:012704 |
| Mouse: Ctgf-T2A-dgCre | Tasic et al., Nat. Neurosci 2016; The Jackson Laboratory | RRID:IMSR_JAX:028535 |
| Mouse: Esr2-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:030158 |
| Mouse: Etv1-CreERT2 | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:013048 |
| Mouse: Gad2-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:010802 |
| Mouse: Glt25d2-Cre_NF107 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_036504-UCD |
| Mouse: Gpr139-IRES2-FlpO-WPRE-neo | Daigle et al. (in preparation) | N/A |
| Mouse: Htr3a-Cre_NO152 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_036680-UCD |
| Mouse: Ndnf-IRES2-dgCre | Tasic et al., Nat. Neurosci 2016; The Jackson Laboratory | RRID:IMSR_JAX:028536 |
| Mouse: Nkx2.1-CreERT2 | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:014552 |
| Mouse: Nos1-CreERT2 | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:014541 |
| Mouse: Npr3-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:031333 |
| Mouse: Npy-IRES2-FlpO | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:030211 |
| Mouse: Nr5a1-Cre | Dhillon et al., Neuron 2006; The Jackson Laboratory | RRID:IMSR_JAX:006364 |
| Mouse: Ntsr1-Cre_GN220 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_030648-UCD |
| Mouse: Oxtr-T2A-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:031303 |
| Mouse: Pvalb-T2A-Dre | Madisen et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:021190 |
| Mouse: Pvalb-T2A-FlpO | Madisen, et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:022730 |
| Mouse: Pvalb-IRES-Cre | Hippenmeyer et al., PLoS Biol 2005; The Jackson Laboratory | RRID:IMSR_JAX:008069 |
| Mouse: Rbp4-Cre_KL100 | Gerfen et al., Neuron 2013; MMRRC | RRID:MMRRC_031125-UCD |
| Mouse: Rorb-IRES2-Cre | Harris et al., Front. Neural Circuits, 2014; The Jackson Laboratory | RRID:IMSR_JAX:023526 |
| Mouse: Slc17a8-iCre | Grimes et al., Vis Neurosci 2011; The Jackson Laboratory | RRID:IMSR_JAX:018147 |
| Mouse: Slc17a8-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:028534 |
| Mouse: Slc32a1-IRES-Cre | Tong et al., Nat. Neu. 2008; The Jackson Laboratory | RRID:IMSR_JAX:012897 |
| Mouse: Slc32a1-T2A-FlpO | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:029591 |
| Mouse: Sncg-IRES2-FlpO-neo | Daigle et al. (in preparation) | N/A |
| Mouse: Sst-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:013044 |
| Mouse: Sst-IRES-FlpO | He et al., Neuron, 2016; The Jackson Laboratory | RRID:IMSR_JAX:028579 |
| Mouse: Tac1-IRES2-Cre | Harris et al., Front. Neural Circuits, 2014; The Jackson Laboratory | RRID:IMSR_JAX:021877 |
| Mouse: Th-Cre_FI172 | Gong et al., J Neurosci 2007; The Jackson Laboratory | RRID:MMRRC_031029-UCD |
| Mouse: Th-P2A-FlpO | Poulin et al., Nat. Neurosci 2018 | N/A |
| Mouse: Tlx3-Cre_PL56 | Gerfen et al., Neuron 2013; The Jackson Laboratory | RRID:MMRRC_036547-UCD |
| Mouse: Vip-IRES-Cre | Taniguchi et al., Neuron 2011; The Jackson Laboratory | RRID:IMSR_JAX:010908 |
| Mouse: Vip-IRES-FlpO | He et al., Neuron, 2016; The Jackson Laboratory | RRID:IMSR_JAX:028578 |
| Mouse: Vipr2-IRES2-Cre | Daigle et al., Cell 2018; The Jackson Laboratory | RRID:IMSR_JAX:031332 |
| Mouse: Ai14(RCL-tdT) | Madisen et al., Nat. Neu. 2010; The Jackson Laboratory | RRID:IMSR_JAX:007914 |
| Mouse: Ai65(RCFL-tdT) | Madisen et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:021875 |
| Mouse: Ai66(RCRL-tdT) | Madisen et al., Neuron 2015; The Jackson Laboratory | RRID:IMSR_JAX:021876 |
| Mouse: Gad67-GFP_X94 | Ma et al., J Neurosci 2006; The Jackson Laboratory | RRID:IMSR_JAX:006334 |
| Software and Algorithms | ||
| Igor Pro | WaveMetrics | https://www.wavemetrics.com/products/igorpro |
| MIES | Allen Institute for Brain Science | https://github.com/alleninstitute/mies |
| Vaa3D | Peng et al., 2010 | http://www.vaa3d.org |
| Allen SDK | Allen Institute for Brain Science | https://github.com/alleninstitute/allensdk |
| IPFX | Allen Institute for Brain Science | https://github.com/alleninstitute/ipfx |
| DRCME | Gouwens et al., 2019; Allen Institute for Brain Science | https://github.com/alleninstitute/drcme |
The custom electrophysiology data acquisition software (MIES) is available at https://github.com/alleninstitute/mies. The Vaa3D morphological reconstruction software, including the Mozak extension, is freely available at http://www.vaa3d.org and its code is available at https://github.com/Vaa3D. The code for electrophysiological and morphological feature analysis and clustering is available as part of the open-source Allen SDK repository (https://github.com/AllenInstitute/AllenSDK), IPFX repository (https://github.com/alleninstitute/ipfx), and DRCME repository (https://github.com/alleninstitute/drcme).