

ABSTRACT
Despite the recent success of CAR T cells targeting CD19 and CD22 in hematological malignancies, the production of CAR T cells still requires an extensive manufacturing process. The well-established NK-92 cell line provides a promising alternative to produce CAR-modified effector cells in a GMP-compliant, cost-effective way. NK-92 can be redirected against a variety of surface antigens by our adapter CAR (AdCAR) system utilizing biotinylated antibodies (bAb) as adapter molecules. Selected bAb were capable of inducing significant AdCAR NK-92-mediated lysis of non-Hodgkin lymphoma (NHL) and mantle-cell lymphoma (MCL) cell lines as well as primary MCL and chronic lymphocytic leukemia (CLL) cells. AdCAR specificity was proven using a JeKo-1 CD19/CD20 knockout antigen-loss model. Moreover, through combinations of bAb, AdCAR NK-92 cells are capable of combatting tumor antigen evasion mechanisms. In conclusion, we successfully generated the AdCAR NK-92 cell line which can be manufactured as an “off-the-shelf, on-demand” product allowing universal and tunable tumor targeting.
KEYWORDS: NK-92, chimeric antigen receptors, universal tumor targeting, adapter
Introduction
First-generation chimeric antigen receptors (CARs) are synthetically designed receptors that connect the antigen-binding region of a monoclonal antibody (mAb), typically a single-chain variable fragment (scFv), with an intracellular CD3ζ signaling domain through a T cell receptor transmembrane domain.1–5 Second- and third-generation CARs consist of additional intracellular co-stimulatory domains, such as 4–1BB (CD137), CD28 or OX40.6 When a CAR binds to its respective target antigen, T cell activation is initiated leading to immune effector cell proliferation, cytokine secretion and target cell lysis in an MHC-independent manner.2 Currently, all FDA-approved CAR products are second-generation CAR T cells used to treat a variety of high-grade or refractory hematological malignancies.4,7–9 Despite the recent clinical success of CAR T cell therapies, several obstacles and challenges remain.
The manufacturing process of CAR T cell products is very time-consuming and costly. Furthermore, the quality of patient PBMC can vary greatly and the amount required for production cannot always be met.10,11 Especially for pediatric or heavily pretreated patients, it is often difficult to collect enough lymphocytes with a single round of leukapheresis.12–15 Long-term persistence of CAR T cells can lead to severe neurotoxicity and cytokine release syndrome (CRS).16,17 It was recently reported that NK cells as an allogeneic product with a better safety profile could prove to be a potent alternative effector cell source for CAR therapy.18,19 CAR modification of peripheral blood as well as umbilical cord NK cells has been extensively studied recently with promising results for the targeting of hematological malignancies, first and foremost using CD19 as target structure.20–23
However, similar to primary T cells, expansion capacity varies greatly for NK cells from peripheral blood, and generally, transfection efficiency is especially low which makes CAR NK production challenging.24,25 Although it was shown that retroviral modification of primary NK cells to express genes that encode interleukin-15 increased in vivo expansion as well as persistence, Jeff Miller and colleagues demonstrated that persistence of adoptively transferred NK cells, which is usually very low, is strongly dependent on the conditioning regiment that the patients received.19,26,27
In our study, we used a well-established NK cell line, NK-92, for modification with a CAR. Continuously expandable NK-92 cells can be easily manufactured in a GMP-compliant manner. Irradiated NK-92 cells were shown to be safe in clinical trials even at high intravenous doses of up to 1010 cells.28 Additionally, the efficacy of CAR-engineered NK-92 cells in various pre-clinical studies has been previously shown, and, to date, only few active clinical trials for the treatment of cancer involve CAR NK-92 cells.20,28–30 Although in vivo persistence due to irradiation safety requirements is low, the superb ex vivo expansion potential to high cell numbers makes NK-92 an ideal platform for the generation of CAR-engineered immune cells.26,31
The majority of current CAR therapy strategies target a single-specific tumor antigen which restricts CAR T or CAR NK cells as a potential therapy option for heterogeneous tumor entities and also leaves them vulnerable to classic tumor antigen evasion mechanisms.32,33 Patients with B cell malignancies, for example, being treated with anti-CD19 CAR T cells show high initial response rates, however, also high relapse rates with reemerging CD19− tumor cell subpopulations.34 In order to tackle these obstacles, the idea of modular CARs targeting multiple tumor-specific antigens was proposed.32
Our recently developed adapter CAR (AdCAR) technology utilizes biotin-labeled antibodies (bAb) as adapter molecules (AM) to translate tumor-specific antigen recognition into immune effector cell activation. The AdCAR is based on the unique properties of a novel scFv targeting a “neo”-epitope-like structure consisting of the endogenous vitamin biotin in the context of monoclonal antibodies as linkers, referred to as linker-label-epitope (LLE), whose context is introduced in the patent application EP3315511A1.35–37
NK-92 cell-mediated target cell lysis is the result of a two-step process: antibody-specific binding to the target cell surface and binding of the AdCAR-modified NK-92 cells to the bAb (Figure 1a). AdCAR specificity relies solely on the bAb used, leading to almost unlimited possibilities in tumor antigen targeting.
Figure 1.
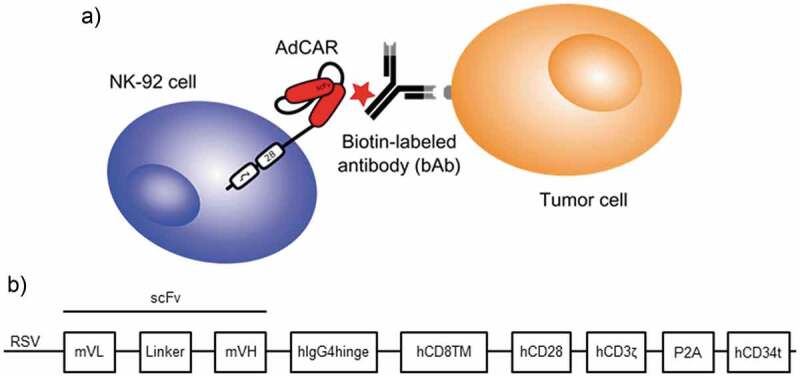
Schematic representation of AdCAR NK-92 function and structure. The NK-92 cells were generated with an adapter CAR recognizing a neoepitope on biotinylated antibodies which are directed against surface antigens on tumor cells (a). Schematic representation of the lentiviral transfer plasmid encoding the AdCAR construct comprising a CD28 co-stimulatory domain and a CD34 tag sequence (b), which was provided by Miltenyi Biotec.
Combining the flexible targeting and controllability of the AdCAR with the “off-the-shelf” properties of the NK-92 cells led to the creation of a universal, on-demand cellular CAR NK product which was tested with primary lymphoma cells, different lymphoma cell lines and a lymphoma antigen-loss model.
Methods
Design of the AdCAR system
The second-generation adapter CAR is based on the mAb “mBio3”-derived single-chain variable fragment (scFv). Further, it comprises an IgG4 hinge domain, a CD8 transmembrane domain, CD28 co-stimulatory, as well as CD3ζ signaling domains. Truncated CD34 (tCD34) is co-expressed after a furin P2A site for detection and enrichment.
Generation of AdCAR-construct and lentiviral vectors
Lentiviral particles (LVP) were produced in Lenti-X 293 T cells (Clontech) after lipofection (Lipofectamine 3000, Thermo Fisher) with a second-generation packaging plasmid, a VSV-G envelope plasmid, and the respective AdCAR transfer plasmid. LVP containing supernatants were concentrated using Lenti-X concentrator (TaKaRa) and stored at −80°C until further use.
Generation of AdCAR-engineered NK-92 cells
NK-92 cells were seeded at a concentration of 1.25 106 cells/ml of MEM Alpha Medium (Thermo Fisher Scientific), supplemented with 8 ng/µl of protamine sulfate (Sigma-Aldrich) and 2.5 µM of BX-795 (Cayman Chemical Company). Subsequently, cells were transduced with AdCAR lentiviral particles for 16 h. Transduced cells were cultivated in NK-92 complete medium. Transduction efficiency was determined by flow cytometric analysis of CD34 marker gene surface expression using a BD FACSCanto II flow cytometer. Cells were subsequently single-cell sorted and screened for highest CAR expression. Results were analyzed using FlowJo software V10.0.8 (BD Biosciences).
Cell lines and culturing conditions
NHL cell lines Raji and Daudi and mantle-cell lymphoma (MCL) cell line JeKo-1 were purchased from the European Collection of Authenticated Cell Cultures (ECACC) or the American Type Culture Collection (ATCC) and maintained in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific) containing stable L-glutamine (Glutamax, Thermo Fisher Scientific), referred to as RPMI complete medium. Lenti-X 293 T cells (Clontech) were cultivated in DMEM high glucose (4.5 g/L) medium (Thermo Fisher Scientific) containing GlutaMAX supplemented with 10% FBS and 1 mM sodium pyruvate (Thermo Fisher Scientific). NK-92 cells were purchased from ATCC and maintained at a concentration of 105 cells/ml in MEM Alpha Medium containing GlutaMAX supplemented with 20% FBS and 100 U/ml IL-2 (PROLEUKIN, Aldesleukin Chiron) referred to as NK-92 complete medium. Primary MCL cells were isolated from a blood sample from a patient diagnosed with a hematogenously disseminated mantle-cell lymphoma. Primary CLL cells were isolated from blood samples from three patients diagnosed with chronic lymphocytic leukemia. All primary cells were maintained in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% pooled AB-serum from healthy donors (Transfusion Medicine, University Hospital Tuebingen). The study was approved by the ethics committee at the Medical Faculty of the Eberhard Karls University and the University Hospital Tuebingen (reference number 13/2007 V). Human material was collected after obtaining informed consent in accordance with the Helsinki protocol.
All media contained 1x antibiotic-antimycotic solution (Thermo Fisher Scientific) consisting of 100 U/mL of penicillin, 100 µg/mL of streptomycin and 0.25 µg/ml amphotericin B. All cell lines were maintained at 37°C in a humidified 5% CO2 atmosphere and regularly tested for mycoplasma contamination.
Generation of CD19/CD20 JeKo-1 knockout variants using CRISPR/Cas9 technology
The JeKo-1 wildtype (CD19+/CD20+) cell line was used to generate three distinct knockout variants (CD19−/CD20+, CD19+/CD20− and CD19−/CD20−) using CRIPR/Cas9 technology and kindly provided by Miltenyi Biotec.
Antibody biotinylation
The FDA-approved CD20 antibody Rituximab was acquired from Hoffmann-La Roche, Basel, Switzerland. The chimeric CD19 antibody 4G7SDIE was acquired from SYNIMMUNE GmbH, Tuebingen, Germany. It was approved by the local ethics committee for experimental settings. Both, Rituximab and 4G7SDIE, were biotinylated by Davids Biotechnologie GmbH, Regensburg, Germany. All other bAb were acquired from Miltenyi Biotec, Bergisch Gladbach, Germany.
Flow cytometry
Staining of lymphoma and CLL cells was conducted using primary biotinylated or fluorophore-labeled mAb with antigen specificity of interest. Cells were co-incubated with antibodies at 4°C in flow cytometry buffer containing PBS (Sigma-Aldrich) supplemented with 2% FBS and 0.5 M EDTA (Sigma-Aldrich) for 15 min. Unbound antibody was washed off by centrifugation (4°C, 350 g, 5 min) and, for cells stained with bAb, a secondary, fluorophore-labeled anti-biotin antibody was added for 15 min followed by another washing step. Surface antigen expression was analyzed using a BD FACSCanto II flow cytometer.
Calcein release-based cytotoxicity assay (CRA)
Target cells were labeled with 10 µM of calcein acetoxymethyl (Calcein AM) (Thermo Fisher Scientific) at a concentration of 106 cells/ml in RPMI medium supplemented with 2% FBS. AdCAR NK-92 cells were thoroughly washed, resuspended in RPMI medium supplemented with 2% FBS and co-incubated with target cells with and without biotinylated antibodies for 2 h at various effector-to-target (E:T) ratios. AdCAR-specific cytotoxicity was determined by fluorescence measurement using the Spark microplate reader (Tecan).
Real-time label-free live cell analysis
Lymphoma cell lines were adjusted to a concentration of 105 cells/ml in RPMI complete medium and seeded in E-Plate 96 VIEW (OLS) micro-well plates that had been coated with 170 µg/ml of fibronectin (Corning) for 1 h. Effector AdCAR NK-92 cells were adjusted to an E:T ratio of 5:1 in NK-92 complete medium without IL-2 and co-incubated with the target cells. Utilizing the xCELLigence real-time cell analysis (RTCA) system, cells were monitored for over 24 h. Lymphoma cell viability was calculated using the RTCA 2.0 software and AdCAR-mediated cytotoxicity was subsequently determined.
Flow cytometry-based cytotoxicity assays using an antigen loss model
To visualize antigen-specific killing, JeKo-1 wildtype, JeKo-1 CD19 knockout (KO), JeKo-1 CD20 KO and JeKo-1 CD19/CD20 double KO variants were incubated at a 1:1:1:1 ratio with 25.000 cells per JeKo-1 variant (in total 100.000 cells) plus 500.000 AdCAR NK-92 cells (E:T 5:1) in equal parts of RPMI complete medium and NK-92 complete medium without IL-2 at 37°C. Furthermore, bCD19 or bCD20 mAb and combinations thereof were added. Cells were incubated for indicated time intervals and then analyzed using a BD CantoFACS II flow cytometer. AdCAR NK-92 cells were identified by CD34 CAR marker gene expression, JeKo-1 in total by GFP expression and JeKo-1 subpopulations by CD19 and CD20 expression. Trucount Tubes (BD Biosciences) were used for quantification and acquisition, with a stopping gate at 20.000 beads. Normalized viability of target cells was calculated using FlowJo software.
Quantification of cytokine release
Target and effector cells were co-incubated in equal parts of target cell complete medium and NK-92 complete medium without IL-2 at an E:T ratio of 5:1 at 37°C for 6 h, supernatants were collected and stored at −80°C until further use. AdCAR NK-92 maximum degranulation was achieved using the cell activation cocktail (BioLegend) containing PMA/Ionomycin.
Cytokine release was determined using the Bio-Plex Pro human cytokine 17-plex assay (Bio-Rad), the human perforin ELISA kit (Thermo Fisher Scientific) and the LEGEND MAX human granulysin and granzyme B ELISA kits (BioLegend).
Data analysis
All statistical analyses were performed with GraphPad Prism 8 software (GraphPad Software Inc.). Flow cytometry data were analyzed using FlowJo software V10.0.8 (FlowJo LLC).
Results
NK-92 cells were transduced with lentiviral vectors encoding the adapter CAR with a CD28 co-signaling domain (Figure 1b). Transduction efficiency of NK-92 was determined via flow cytometric analysis of CD34 marker gene surface expression which correlates with CAR expression. We found that the AdCAR was efficiently expressed on the surface of transduced NK-92 cells which were subsequently single-cell-sorted by FACS for high CAR expression (Figure 2a). Ten individual NK-92 clones were tested for CAR expression using flow cytometry. The NK-92 clone with the highest CAR expression and highest viability was chosen for further experiments. AdCAR NK-92 cells showed stable AdCAR surface expression for at least 150 d after cell sorting (Figure 2b) with an average viability of >90%. Notably, the proliferation rate was not impaired by the transduction process compared to untransduced, parental NK-92 cells (Figure 2c).
Figure 2.
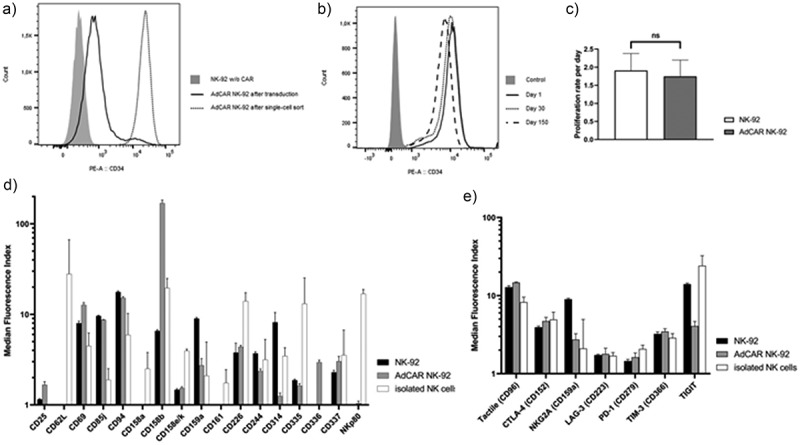
Generation and characterization of AdCAR NK-92 cells. CD34 marker gene expression was measured via flow cytometry on untransduced (gray, filled) and transduced NK-92 cells before (black line) and after (gray line) single-cell sort (a) and monitored for 150 d (b). Average proliferation rate of parental NK-92 and sorted AdCAR NK-92 was measured over the same time frame and calculated as mean ± SD (c). AdCAR-transduced and parental NK-92 cells were characterized for the expression of activating and inhibitory NK receptors as well as immune checkpoint receptors via flow cytometry. Results were compared to flow cytometric analysis of isolated NK cells from three healthy donors. Median fluorescence intensity ± SD was calculated using FlowJo software, n = 3 (d, e).
To compare the NK cell receptor profile of NK-92 cells and primary NK cells, as well as analyzing the effects of CAR transduction on the NK receptor expression of NK-92 cells, flow cytometry was used. Compared to primary NK cells, NK-92 and AdCAR NK-92 cells showed lower expression of DNAM-1 (CD226), one of the main activating NK receptors. Another important activating receptor, NKG2D (CD314) was almost completely absent on AdCAR NK-92 cells. Furthermore, AdCAR-transduced NK-92 cells displayed very high expression of KIR2DL2/DL3 (CD158b), a member of the killer-cell immunoglobulin-like receptor (KIR) family recognizing the human HLA-C1/2 peptide, and thus, inhibiting NK effector function. Interestingly, NKp80, an activation receptor on all activated NK cells, which induces NK cell-mediated cytotoxicity and cytokine production, was only present on the isolated NK cells and not on NK-92 cells (Figure 2d). Next, AdCAR NK-92 cells were characterized for the expression of important immune checkpoint receptors via flow cytometry and compared to parental NK-92 as well as isolated NK cells from three healthy donors (Figure 2e). AdCAR NK-92 cells showed a similar immune checkpoint receptor expression profile, compared to primary NK cells and parental NK-92 cells apart from a slight decrease in expression of TIGIT, a protein that directly inhibits NK cell cytotoxicity.
We then tested whether AdCAR NK-92 cells combined with different biotinylated antibodies targeting lymphoma cells could induce AdCAR-mediated antigen-specific cytotoxicity. As proof of principle for the functionality and specificity of AdCAR NK-92 cells in vitro, the lymphoma cell lines Raji, Daudi (both NHL) and JeKo-1 (MCL) were used. The cell lines were immunophenotyped via flow cytometry for lymphoma antigen expression (Table 1) and used as target cells for functional analyses.
Table 1.
Surface antigen expression of lymphoma cell lines. Raji, Daudi and JeKo-1 cell lines were screened for target antigen expression using flow cytometry. Lymphoma cells were co-incubated with primary biotinylated antibodies for 15 min at 4°C. Antigen expression was detected using a secondary PE-coupled anti-biotin antibody. Mean positivity and mean fluorescence index (MFI) values ± SD were calculated using staining with the secondary antibody alone as a negative control, n = 3.
| |
Raji |
Daudi |
JeKo-1 |
|||
|---|---|---|---|---|---|---|
| Positive cells | MFI | Positive cells | MFI | Positive cells | MFI | |
| CD19 | 99.97 ± 0.06% | 107.72 ± 22.13 | 99.90 ± 0.00% | 74.08 ± 4.50 | 97.93 ± 1.90% | 21.93 ± 3.51 |
| CD20 | 95.8 ± 3.99% | 44.57 ± 15.78 | 98.5 ± 0.69% | 85.85 ± 40.91 | 97.07 ± 3.35% | 40.42 ± 19.28 |
| CD22 | 99.1 ± 0.35% | 47.90 ± 6.80 | 98.60 ± 0.69% | 77.32 ± 26.56 | 24.07 ± 3.48% | 1.80 ± 0.09 |
| CD30 | 2.77 ± 0.92% | 1.10 ± 0.10 | 0.57 ± 0.21% | 1.04 ± 0.01 | 1.49 ± 0.49% | 1.07 ± 0.02 |
| CD38 | 98.43 ± 1.59% | 57.50 ± 20.20 | 95.07 ± 1.16% | 191.70 ± 11.45 | 81.07 ± 2.35% | 17.69 ± 4.47 |
| CD70 | 98.83 ± 1.68% | 92.02 ± 23.64 | 41.97 ± 8.38% | 2.93 ± 0.52 | 98.47 ± 0.29% | 35.11 ± 8.53 |
| CD79b | 66.23 ± 9.82% | 5.24 ± 2.08 | 98.30 ± 1.01% | 76.67 ± 24.13 | 96.30 ± 0.56% | 27.66 ± 0.59 |
Using a standard calcein release cytotoxicity assay (CRA), AdCAR NK-92 cells were co-incubated with calcein-labeled lymphoma cells with and without bAb for 2 h. For determination of the optimal antibody concentration lymphoma cells were co-incubated with varying amounts of bCD19 and AdCAR NK-92 cells. The AdCAR NK-92 cells induced cellular cytotoxicity of Raji cells in a bCD19-dose-dependent manner (Figure 3a). A concentration of 100 ng/ml was chosen for all biotinylated antibodies for further functional assays. Altering antibody concentration decreased the AdCAR NK-92-mediated cytotoxicity. Thus, the AdCAR NK-92 effect is tunable, directly depending on the concentration of bAb used. Since biotin, which is conjugated to the mAb, is an endogenous vitamin, we tested whether free biotin decreases AdCAR-mediated cytotoxicity. Notably, AdCAR NK-92 cells retained their specific cytotoxic ability even in the presence of unbound biotin that exceeded physiological concentrations which proves that the AdCAR specifically binds to the neoepitope on biotinylated antibodies and not biotin itself (Figure 3b). Raji cells were incubated with bCD19 and varying concentrations of NK-92 effector cells (Figure 3c). Interestingly, titration of effector cell concentration showed that at low E:T ratios the AdCAR still reliably enhanced NK-92 effector function in the presence of a specific bAb.
Figure 3.
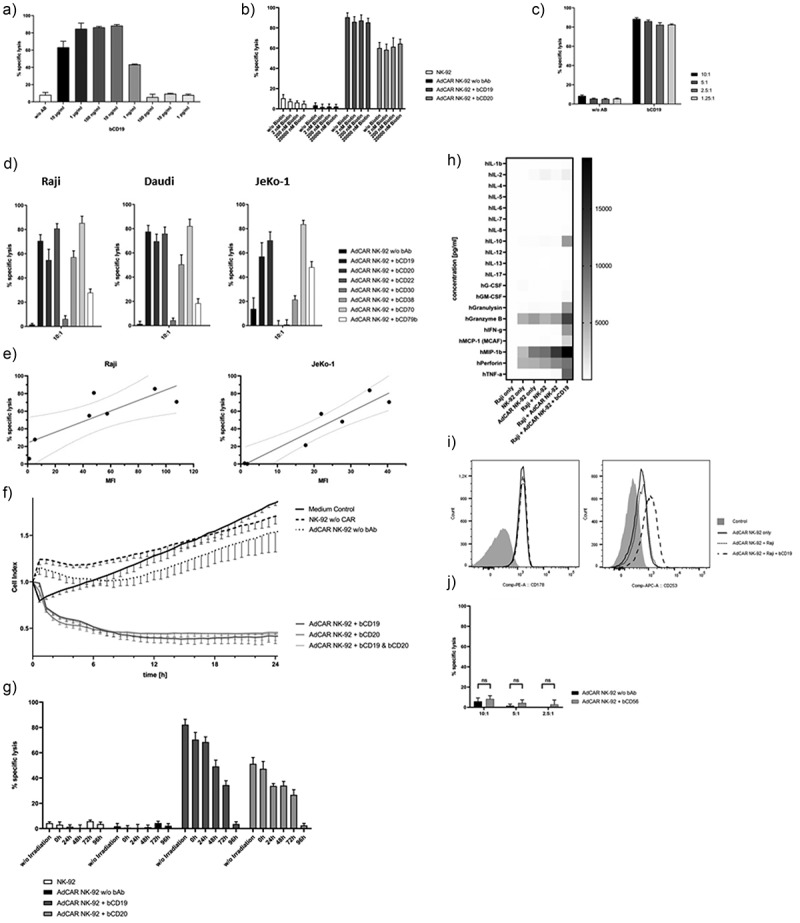
AdCAR NK-92-mediated tumor cell lysis. AdCAR NK-92 cells were co-incubated with calcein-labeled lymphoma cell lines in the presence or absence of indicated biotinylated antibodies for 2 h. Specific lysis is shown as mean ± SD, n = 3. Using the Raji cell line as target, an optimal bAb concentration of 100 ng/ml was chosen after titration of bCD19 (a). Free biotin added in excess of physiological concentrations showed no impairment of AdCAR-mediated lysis of lymphoma cells (b). Various E:T ratios utilizing AdCAR NK-92 cells as effectors and Raji cells as target were analyzed using a calcein release assay (c). Biotinylated antibodies, already utilized for the flow cytometry screening panel (Table 1), were tested with Raji, Daudi and JeKo-1 cells as target in a CRA (d). Target antigen expression levels were correlated with their respective AdCAR NK-92-mediated tumor cell lysis (e). Kinetics of AdCAR-mediated lysis of Raji cells was assessed using the xCELLigence real-time cell-analysis system at an E:T ratio of 5:1 (f). Cytotoxic effector function of AdCAR NK-92 cells irradiated with 10 Gy at indicated time points was assessed using Raji cells as target (g). The release of cytokines by NK-92 cells was measured using the Bio-Plex Pro human cytokine 17-plex assay and is shown as a heatmap (h). AdCAR NK-92 cells were co-incubated with Raji cells in the presence or absence of bCD19 for 6 h and screened for expression of FasL (CD178) and TRAIL (CD253) via flow cytometry (i). Finally, AdCAR NK-92-mediated lysis of calcein-labeled parental NK-92 cells using bCD56 as adapter molecule (j) was tested for the assessment of possible fratricide.
Most importantly, AdCAR NK-92 but not untransduced NK-92 cells significantly induced cellular cytotoxicity against all lymphoma cell lines but only in the presence of bAb targeting antigens expressed on lymphoma cell lines (Figure 3d). Biotinylated antibodies targeted against antigens that were not expressed on the cell surface were not capable of inducing AdCAR-mediated responses, thus proving AdCAR specificity. Moreover, NK-92 cytotoxicity and antigen expression levels (MFI) correlated significantly for the NHL cell line Raji (R2 = 0.695; 95%-CI: 0.2166–0.9748; P = .0198) and the MCL cell line JeKo-1 (R2 = 0.8942; 95%-CI: 0.6689–0.9922; P = .0013) (Figure 3e). Utilizing the xCELLigence real-time cell analysis (RTCA) assay, kinetics of NK-mediated cytotoxicity following co-incubation of AdCAR NK-92 cells and Raji cells were assessed (Figure 3f). The dimensionless cell index is indicative of target cell viability and therefore anti-proportional to tumor lysis. The assay demonstrated a rapid decrease in cell index of Raji cells following the addition of AdCAR NK-92 cells but only in the presence of the respective biotinylated antibodies bCD19 and/or bCD20 while no considerable AdCAR-effect was detectable in the absence of bAb.
Essentially, irradiation of NK-92 cells prior to testing, as required in all active clinical trials using NK-92, had no significant effect on target cell lysis when AdCAR NK-92 cells were used immediately after irradiation; however, AdCAR NK-92 cells gradually lost effector function and underwent apoptosis over time (Figure 3g).
Furthermore, an NK-92 cytokine secretion profile was established to screen for the secretion of a variety of cytokines, including NK cell effector molecules. Various cytokines were significantly increased after co-incubation of AdCAR-transduced NK-92 cells with Raji cells. IL-10 (36-fold; p < .0004), granulysin (27-fold; p < .002), IFN-γ (65-fold; p < .01), MCP-1 (40-fold; p < .04) and TNF-α (28-fold; p < .0002) showed significantly elevated levels but only upon AdCAR induction via specific biotinylated antibodies. Notably, NK-92 cells alone showed medium basal secretion of granzyme B, MIP-1b and perforin which was further augmented after AdCAR activation (2.2-fold, 1.5-fold and 1.3-fold, respectively) but did not reach statistical significance (Figure 3h). TNF-related apoptosis-inducing ligand (TRAIL, CD253) upregulation on AdCAR NK-92 cells could be observed after co-incubation with Raji cells and bCD19 for 6 h (MFI: AdCAR NK-92 only: 2.08 ± 0.18; AdCAR NK-92 + Raji: 2.36 ± 0.06; AdCAR NK-92 + Raji + bCD19: 4.48 ± 0.29) while expression of Fas ligand (FasL, CD178), another important apoptosis-inducing transmembrane protein, was not elevated (Figure 3i).
In order to determine whether AdCAR activation also induced NK-92 fratricide, AdCAR-engineered NK-92 cells were co-incubated with calcein-labeled, parental NK-92 cells. Favorably, co-incubation of bCD56 with NK-92 target cells and AdCAR-transduced NK-92 effector cells did not show a significant increase in lysis of parental NK-92 cells (Figure 3j).
To prove the specificity of our AdCAR system, a JeKo-1 CD19 and/or CD20-knockout (KO) model was used. JeKo-1 wildtype, JeKo-1 CD19 KO, JeKo-1 CD20 KO and JeKo-1 CD19/CD20 double KO variants were mixed at an equal ratio and subsequently co-incubated with AdCAR NK-92 cells at an E:T ratio of 5:1 in the presence or absence of bCD19 and/or bCD20 antibodies for 4 h and 24 h. Subsequently, cells were analyzed via flow cytometry.
Addition of AdCAR NK-92 cells alone to the JeKo-1 target cell mix did not decrease target cell count or alter the ratio of the respective subpopulations. However, co-incubation with specific biotinylated antibodies induced AdCAR-mediated lysis of respective antigen positive subpopulations while the antigen-negative subpopulations were spared (Figure 4a). With the combination of bCD19 and bCD20, AdCAR NK-92 cells eradicated CD19+ and CD20+ JeKo-1 cells almost completely (Figure 4b). Thus, the AdCAR NK-92 cells specifically lyse tumor cells only in the presence of a bAb targeting an antigen that is expressed on the surface of the tumor cell. If tumor cells down-regulate antigen expression levels in response to immune therapy, AdCAR NK-92-mediated activity is easily retained by switching target structures through the addition of a different bAb.
Figure 4.
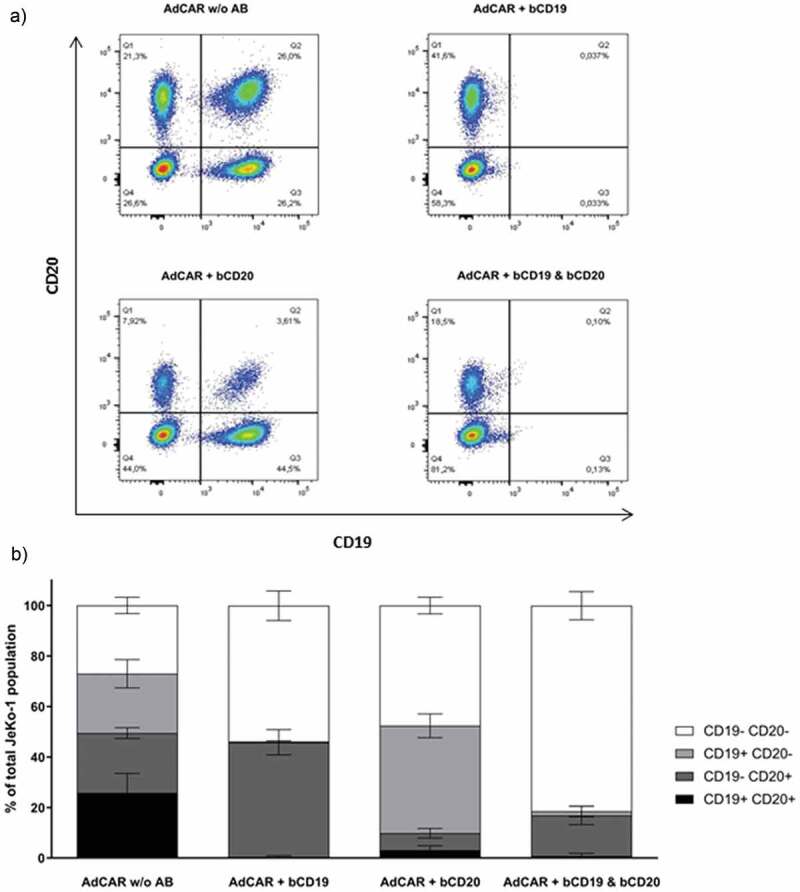
JeKo-1 cell line target antigen-loss model. JeKo-1 CD19 and/or CD20 knock-out (KO) cell lines were mixed at an equal 1:1:1:1 ratio and co-incubated with AdCAR NK-92 cells at an E:T ration of 5:1 in the presence and/or absence of indicated bAb for 24 h and analyzed using flow cytometry (a). Ratio of KO variants after cytotoxicity assay is shown as a mean of three independent experiments (b).
Next, we tested whether AdCAR activation by co-incubation with lymphoma cells would permanently decrease the cytotoxic effector function of AdCAR NK-92 cells due to immune exhaustion. Therefore, AdCAR NK-92 cells were co-incubated with Raji cells and bCD19 for 4, 24 or 48 h at an E:T ratio of 5:1 and screened for expression of the immune checkpoint receptors at indicated time points (Figure 5b). Notably, only CD96 (TACTILE), CD223 (LAG-3) and CD366 (TIM-3) were significantly elevated with peak expression levels after 24 h of stimulation. We then subjected AdCAR NK-92 cells to continuous re-stimulation by the addition of fresh Raji cells every 24 h for a total of 72 h and again screened for immune checkpoint receptor expression. Only slightly elevated CD96 expression levels could be observed, while CD223 and CD366 were moderately increased. For functional testing, we compared the continuously re-stimulated AdCAR NK-92 cells with unstimulated control AdCAR NK-92 cells using Raji cells as target and bCD19 as well as bCD20 as adapter molecules (Figure 5c). We found no significant decrease in CAR-mediated cytotoxic effector function after re-stimulation. Thus, AdCAR NK-92 cells did not show relevant signs of exhaustion after multiple killing events.
Figure 5.
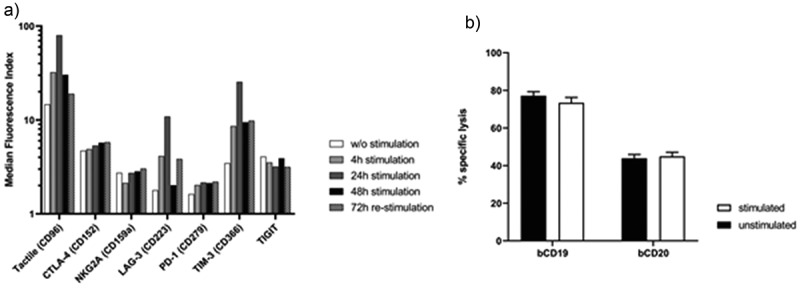
Immune checkpoint receptor expression profile and cytotoxic effector function of AdCAR NK-92 cells upon re-stimulation with Raji cells. AdCAR NK-92 cells were co-incubated with Raji cells at an E:T ratio of 5:1 either for indicated time points (filled bars) or re-stimulated every 24 h with fresh Raji cells for 72 h (checkered pattern) and screened for immune checkpoint receptor expression (a). Re-stimulated AdCAR NK-92 cells were co-incubated with calcein-labeled Raji cells at an E:T ratio of 5:1 for 2 h. Specific cytotoxicity was calculated and compared to un-stimulated control AdCAR NK-92 cells (b). Specific lysis is shown as mean ± SD, n = 3.
Further, we wanted to examine whether AdCAR NK-92 cells were also capable of inducing CAR-mediated cytotoxicity against patient-derived, primary tumor cells. Therefore, we used tumor cells that had been previously isolated from a blood sample from one patient diagnosed with a hematogenously disseminated mantle-cell lymphoma and three patients with chronic lymphocytic leukemia. These cells were immunophenotyped (Table 2) and used as target cells in a standard CRA. AdCAR NK-92 cells were able to induce significant cellular cytotoxicity in the presence of specific bAb (Figure 6). Likewise, the addition of bAb targeting non- or very low expressed antigens did not induce any AdCAR-related lysis. Evidently, AdCAR NK-92 cells are effective in specifically targeting various patient-derived, primary tumor cells.
Table 2.
Surface antigen expression of primary cells derived from different B cell malignancies. Patient-derived, primary MCL and CLL cells were co-incubated with primary biotinylated antibodies for 15 min at 4°C. Antigen expression was detected using a secondary, PE-coupled anti-biotin antibody. Mean positivity and mean fluorescence index (MFI) values ± SD were calculated using staining with the secondary antibody alone as a negative control.
| |
Primary MCL (W) |
Primary CLL (AWK) |
Primary CLL (FA) |
Primary CLL (M) |
||||
|---|---|---|---|---|---|---|---|---|
| Positive cells | MFI | Positive cells | MFI | Positive cells | MFI | Positive cells | MFI | |
| CD19 | 91.67% | 46.81 | 90.05% | 184.92 | 95.30% | 254.32 | 90.70% | 153.47 |
| CD20 | 94.27% | 300.12 | 90.90% | 41.04 | 66.07% | 12.1 | 81.11% | 12.42 |
Figure 6.
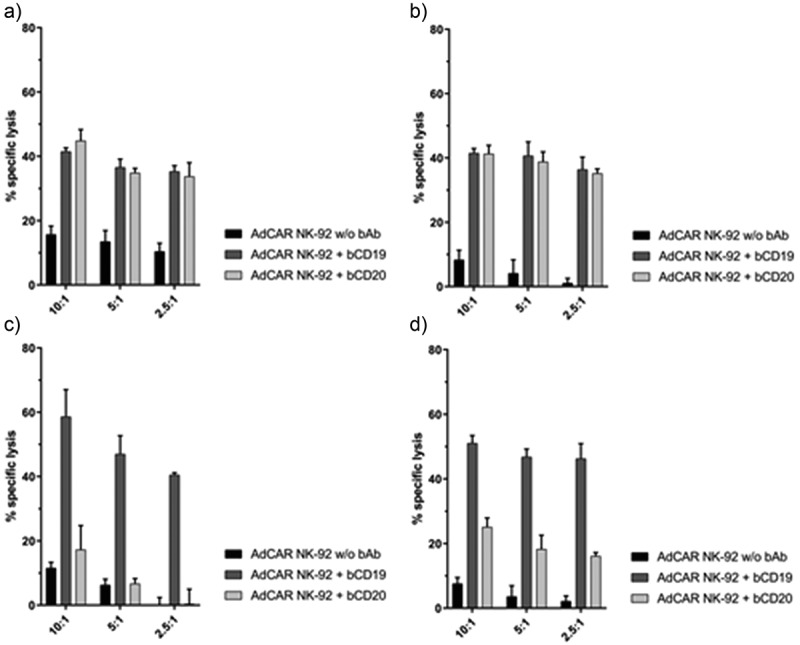
AdCAR NK-92-mediated lysis of primary cells of different B cell malignancies. AdCAR NK-92 cells were co-incubated with calcein-labeled patient-derived, primary MCL cells (one patient, (a) and CLL cells (three different patients, (b-d) in the presence or absence of indicated biotinylated antibodies for 2 h. Specific lysis is shown as mean ± SD, n = 3.
Discussion
Tumor heterogeneity remains a major obstacle even for advanced immunotherapeutic options such as CAR T cells. NK cells provide an interesting approach as an alternative cell source for CAR therapy.38,39 Most current pre-clinical and clinical trials work with either allogeneic donor NK cells or NK-derived cell lines since primary, autologous NK cells can easily be silenced by tumor cells through self-antigens.21,40,41 NK-92 is a continuously expandable cell line that lacks the expression of most inhibitory receptors.31 It constitutively expresses a variety of activating receptors.42 The lack of KIR expression and constitutive expression of activating receptors allows for the high cytotoxic nature of NK-92 cells which have been shown to be effective and safe in various pre-clinical trials.28,40,43–46 To avoid malignant expansion through in vivo proliferation of the NHL-derived NK-92, cells need to be γ-irradiated prior to administration.28,43 Irradiation with 10 Gy blocks NK-92 proliferation limiting its life span to only a few days while retaining most of their cytolytic activity. This increases safety since without permanent engraftment NK-92 cells there is no need for active elimination after application using a CAR T-like safety switch.47 Conversely, CAR-transduced NK-92 cells could safely be administered multiple times to further increase efficiency.48
In the present study, we demonstrated that the combination of the well-established NK-92 cell line with the controllability of our newly developed Adapter CAR system enables distinct therapeutic options for a multitude of malignancies while counteracting drawbacks of conventional CAR T cell therapy such as tumor evasion strategies like antigen loss. By targeting extracellular antigen structures using specific biotinylated antibodies (bAb) NK-mediated cell lysis could be successfully induced in multiple lymphoma cell lines as well as various primary tumor cells. The absence of either the bAb or its corresponding target antigen prevents AdCAR activity. Additional controllability of AdCAR NK-92 cells is demonstrated by the fact that cytolytic activity occurred in a concentration-dependent fashion. Contrary to primary NK cells, AdCAR NK-92 cells lack the CD16 (FcγRIII) expression and cannot be retargeted by IgG1 antibodies.40 Therefore, the formation of the immunological synapse is simply controlled by bAb titration.
Transduction of NK-92 cells with the AdCAR was shown to be stable for at least 150 d without limiting the NK-92 cells proliferative or cytotoxic activity. Thus, “off-the-shelf” AdCAR NK-92 cells provide a cost-effective alternative to standard CAR T cells which have to be produced individually for each patient.49 These direct CAR-mediated immune cell approaches also lack the flexibility of a universal CAR. In previous CAR T cell studies targeting CD19 tumor cells reacted to therapy by down-regulation of CD19.49–52 Using a JeKo-1 antigen-loss model AdCAR NK-92 cells demonstrated their ability to target multiple antigens simultaneously. They were able to react to tumor antigen loss by switching target structures simply utilizing a different bAb while retaining therapeutic efficacy. Other universal CAR approaches with T cells, including the presented adapter CAR, as well as NK cells have already shown promising results.32,35,36,53,54 However, CAR systems using adapter molecules that are foreign to the human body can cause immunogenic reactions and make clinical translation difficult. Even CAR systems utilizing endogenous molecules as tag such as biotin may face challenges due to the potential immunogenicity of avidin and streptavidin as well as their high affinity for physiological biotin in the clinical setting.54,55 Here, the AdCAR is based on an scFv targeting a “neo”-epitope-like structure, the linker-label-epitope, consisting of biotin in the context of a mAb, instead of biotin itself.37 Specificity was proven since we saw no interference of unbound biotin even at supraphysiological levels. Thus, using AdCAR NK-92 cells is a promising approach to circumvent adverse immunogenic reactions of other CAR systems and therefore decrease the risk of severe side effects during therapy. Moreover, this study proved that biotinylation of already FDA-approved antibodies such as Rituximab is capable of creating functional adapter molecules for AdCAR NK-92 therapy thus facilitating translation into clinical settings. Our data represent preliminary results which have to be further investigated in vivo using different mouse models of hematological malignancies as well as solid tumors to evaluate AdCAR efficacy and predict the response to AdCAR NK-92 immunotherapy in patients.
To conclude, AdCAR NK-92 cells can be manufactured as an “off-the-shelf, on demand” standardized product whose effector function can be tightly regulated. They may provide a universal and cost-effective therapeutic option for safe and tunable targeting of malignant cells.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377:2545–11. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadelain M, Brentjens R, Rivière I.. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, Kloss CC, Gunset G, Sadelain M.. CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 2013;8:1–14. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 6.Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. 2019;8. doi: 10.1002/cti2.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohty M, Gautier J, Malard F, Aljurf M, Bazarbachi A, Chabannon C, Kharfan-Dabaja MA, Savani BN, Huang H, Kenderian S, et al. CD19 chimeric antigen receptor-T cells in B-cell leukemia and lymphoma: current status and perspectives. Leukemia. 2019;2767–2778. doi:doi: 10.1038/s41375-019-0615-5. [DOI] [PubMed] [Google Scholar]
- 8.Subklewe M, Von Bergwelt-Baildon M, Humpe A. Chimeric antigen receptor T cells: A race to revolutionize cancer therapy. Transfus Med Hemotherapy. 2019;46:15–24. doi: 10.1159/000496870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma. N Engl J Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen ES, Stroncek DF, Ren J, Eder AF, West KA, Fry TJ, Lee DW, Mackall CL, Conry-Cantilena C. Autologous lymphapheresis for the production of chimeric antigen receptor T cells. Transfusion. 2017;57(5):1133–1141. doi: 10.1111/trf.14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, An G, Meng H, Yang L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9:64–78. [PMC free article] [PubMed] [Google Scholar]
- 12.Ceppi F, Rivers J, Annesley C, Pinto N, Park JR, Lindgren C, Mgebroff S, Linn N, Delaney M, Gardner RA, et al. Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion. 2018;58(6):1414–1420. doi: 10.1111/trf.14569. [DOI] [PubMed] [Google Scholar]
- 13.Mackall CL, Fleisher TA, Brown MR, Magrath IT, Shad AT, Horowitz ME, Wexler LH, Adde MA, McClure LL, Gress RE, et al. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood. 1994;84(7):2221–2228. doi: 10.1182/blood.V84.7.2221.2221. [DOI] [PubMed] [Google Scholar]
- 14.Mackall CL. T-cell immunodeficiency following cytotoxic antineoplastic therapy: A review. Stem Cells. 2000;18:10–18. doi: 10.1634/stemcells.18-1-10. [DOI] [PubMed] [Google Scholar]
- 15.Verma R, Foster RE, Horgan K, Mounsey K, Nixon H, Smalle N, Hughes TA, Carter CR. Lymphocyte depletion and repopulation after chemotherapy for primary breast cancer. Breast Cancer Res. 2016;18(1):1–12. doi: 10.1186/s13058-015-0669-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldmann A, Arndt C, Koristka S, Berndt N, Bergmann R, Bachmann MP. Conventional CARs versus modular CARs. Cancer Immunol Immunother. 2019;68:1713–1719. doi: 10.1007/s00262-019-02399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bachmann M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol Lett. 2019;211:13–22. [DOI] [PubMed] [Google Scholar]
- 18.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. [DOI] [PubMed] [Google Scholar]
- 19.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang W, Jiang J, Wu C. CAR-NK for tumor immunotherapy: clinical transformation and future prospects. Cancer Lett. 2020;472:175–180. doi: 10.1016/j.canlet.2019.11.033. [DOI] [PubMed] [Google Scholar]
- 21.Rezvani K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant. 2019;54:785–788. doi: 10.1038/s41409-019-0601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, Leung W, Campana D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14:830–840. doi: 10.3109/14653249.2012.671519. [DOI] [PubMed] [Google Scholar]
- 23.Oelsner S, Wagner J, Friede ME, Pfirrmann V, Genßler S, Rettinger E, Buchholz CJ, Pfeifer H, Schubert R, Ottmann OG, et al. Chimeric antigen receptor-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-cell acute lymphoblastic leukemia and enhance survival. Int J Cancer. 2016;139:1799–1809. doi: 10.1002/ijc.30217. [DOI] [PubMed] [Google Scholar]
- 24.Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33:1255–1259. doi: 10.1016/j.leukres.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutlu T, Nyström S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23:1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:1–17. doi: 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, Robertson P, Grzywacz B, Conlon KC, Waldmann TA, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019;3:1970–1980. doi: 10.1182/bloodadvances.2018028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, Seifried E, Wels WS, Tonn T. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. 2018;67:25–38. doi: 10.1007/s00262-017-2055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merker M, Pfirrmann V, Oelsner S, Fulda S, Klingebiel T, Wels WS, Bader P, Rettinger E. Generation and characterization of ErbB2-CAR-engineered cytokine-induced killer cells for the treatment of high-risk soft tissue sarcoma in children. Oncotarget. 2017;8(39):66137–66153. doi: 10.18632/oncotarget.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, Zeng T, Huang H, Zhang X, Sun W, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297–310. doi: 10.1016/j.molonc.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klingemann HG, Wong E, Maki G. A cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant. 1996;2:68–75. [PubMed] [Google Scholar]
- 32.Darowski D, Kobold S, Jost C, Klein C. Combining the best of two worlds: highly flexible chimeric antigen receptor adaptor molecules (CAR-adaptors) for the recruitment of chimeric antigen receptor T cells. MAbs. 2019;11(4):621–631. doi: 10.1080/19420862.2019.1596511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minutolo NG, Hollander EE, Powell DJ. The emergence of universal immune receptor t cell therapy for cancer. Front Oncol. 2019;9. doi: 10.3389/fonc.2019.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Chen W. Mechanisms of failure of chimeric antigen receptor T-cell therapy. Curr Opin Hematol. 2019;26:427–433. doi: 10.1097/MOH.0000000000000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seitz CM, Schlegel P, Hau J, Krahl AC, Schroeder S, Bender G, Reiter S, Schleicher S, Schilbach K, Ebinger M, et al. Novel adapter chimeric antigen receptor (aCAR) T cells for temporally controllable targeting of single and multiple tumor antigens. Blood. 2017;130:1912–1912. [Google Scholar]
- 36.Seitz CM, Kieble V, Illi C, Reiter S, Grote S, Mittelstaet J, Lock D, Kaiser A, Schleicher S, Handgretinger R, et al. Combinatorial targeting of multiple shared antigens by adapter-CAR-T cells (aCAR-Ts) allows target cell discrimination and specific lysis based on differential expression profiles. Blood. 2018;132:4543–4543. doi: 10.1182/blood-2018-99-115630. [DOI] [Google Scholar]
- 37.Kaiser A, Mittelstaet J, Huppert V, Miltenyi S, Schlegel P, Seitz C, Lang P, Handgretinger R, Inventors; Miltenyi Biotec GmbH, assignee . Adapter chimeric antigen receptor expressing cells for targeting of multiple antigens - EP3315511A1. Eur Pat Off. 2018. [Google Scholar]
- 38.Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. 2015;6:578. doi: 10.3389/fimmu.2015.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25:1769–1781. doi: 10.1016/j.ymthe.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91. doi: 10.3389/fimmu.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Zheng H, Diao Y.. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int J Mol Sci. 2019;20:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, Suttorp M, Seifried E, Ottmann OG, Bug G, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi: 10.1016/j.jcyt.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 43.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematotherapy Stem Cell Res. 2001;10:535–544. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 44.Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann H-G, Wels W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100:1265–1273. doi: 10.1182/blood.V100.4.1265.h81602001265_1265_1273. [DOI] [PubMed] [Google Scholar]
- 45.Maki G, Klingemann HG, Martinson JA, Tam YK. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J Hematotherapy Stem Cell Res. 2001;10:369–383. doi: 10.1089/152581601750288975. [DOI] [PubMed] [Google Scholar]
- 46.Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147. doi: 10.4161/onci.28147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a ‘safety switch’ to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. doi: 10.3389/fphar.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, Marschalek R, Dotti G, Jung G, Große‐Hovest L, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145:1935–1945. [DOI] [PubMed] [Google Scholar]
- 49.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topp MS, Gökbuget N, Zugmaier G, Degenhard E, Goebeler M-E, Klinger M, Neumann SA, Horst HA, Raff T, Viardot A, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120:5185–5187. doi: 10.1182/blood-2012-07-441030. [DOI] [PubMed] [Google Scholar]
- 51.Topp MS, Gökbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, Viardot A, Marks R, Diedrich H, Faul C, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32:4134–4140. doi: 10.1200/JCO.2014.56.3247. [DOI] [PubMed] [Google Scholar]
- 52.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, Loureiro LR, Bergmann R, Máthé D, Hegedüs N, et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci Rep. 2020;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lohmueller JJ, Ham JD, Kvorjak M, Finn OJ. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology. 2017. doi: 10.1080/2162402X.2017.1368604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72(7):1844–1852. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]