

Abstract
Population genetic theory and empirical evidence indicate that deleterious alleles can be purged in small populations. However, this viewpoint remains controversial. It is unclear whether natural selection is powerful enough to purge deleterious mutations when wild populations continue to decline. Pheasants are terrestrial birds facing a long-term risk of extinction as a result of anthropogenic perturbations and exploitation. Nevertheless, there are scant genomics resources available for conservation management and planning. Here, we analyzed comparative population genomic data for the three extant isolated populations of Brown eared pheasant (Crossoptilon mantchuricum) in China. We showed that C. mantchuricum has low genome-wide diversity and a contracting effective population size because of persistent declines over the past 100,000 years. We compared genome-wide variation in C. mantchuricum with that of its closely related sister species, the Blue eared pheasant (C. auritum) for which the conservation concern is low. There were detrimental genetic consequences across all C. mantchuricum genomes including extended runs of homozygous sequences, slow rates of linkage disequilibrium decay, excessive loss-of-function mutations, and loss of adaptive genetic diversity at the major histocompatibility complex region. To the best of our knowledge, this study is the first to perform a comprehensive conservation genomic analysis on this threatened pheasant species. Moreover, we demonstrated that natural selection may not suffice to purge deleterious mutations in wild populations undergoing long-term decline. The findings of this study could facilitate conservation planning for threatened species and help recover their population size.
Keywords: conservation genetics, deleterious alleles, genetic diversity, inbreeding depression, pheasant, purifying selection
Introduction
Genetic diversity is a key factor in planning conservation measures for threatened species as it provides information that is vital for conservation efforts (Frankham et al. 2014; Xue et al. 2015; Feng et al. 2019). Because demographic bottlenecks lead to increased levels of inbreeding and genetic drift, it is expected that a population contraction may result in the accumulation or even fixation of deleterious mutations (Xue et al. 2015; Feng et al. 2019). Recent evidence has shown that threatened species with small effective population sizes and low genetic diversity may nonetheless persist for thousands of years after a demographic bottleneck (Robinson et al. 2016). Purifying selection may exert a relatively greater influence on removing deleterious alleles in small, stable populations (Robinson et al. 2016; Grossen et al. 2020). Nevertheless, it is uncertain whether genetic drift reduces the efficacy of purified selection in mitigating the detrimental genetic consequences when wild populations continuously decline.
Pheasants (Phasianidae; Galliformes) are widely distributed terrestrial birds (Johnsgard 1986). They have played important roles in the economy, culture, and ecology of various human societies (Peng et al. 2016; Peters et al. 2016; Barton et al. 2020). As their plumage is bright, their meat is palatable, and their mobility is limited, pheasants have been extensively hunted and exploited by humans (Fuller and Garson 2000; Barton et al. 2020). Consequently, certain pheasant species have declined or become extinct altogether (Keane et al. 2005; McGowan et al. 2012). For example, the New Zealand quail (Coturnix novaezelandiae) rapidly declined to extinction by 1875 (BirdLife International 2016a). However, there are few published studies documenting the population genomic consequences of historical population fluctuations and recent anthropogenic activity for threatened pheasant populations.
The Brown eared pheasant (Crossoptilon mantchuricum) is a vulnerable and declining species (BirdLife International 2016c). It appears on the 2020 IUCN Red List of threatened species and is confined to three fragmented areas in Shaanxi, Shanxi, and Hebei Provinces and Beijing Municipality in north central China (fig. 1a) (Zheng 2015). These areas have undergone continuous anthropogenic perturbations for the last 40,000 years (40 ka) (Shang et al. 2007; Barton et al. 2020). The capitals of the Qin (221–207 BC), Ming, and Qing (AD 1368–1912) dynasties were situated in Shaanxi and Beijing. Massive deforestation occurred during these periods to accommodate the construction of palaces and houses. The Travels of Marco Polo and other literature sources recorded the use of C. mantchuricum tail feathers to decorate the official hats of generals. This extended period of exploitation and habitat destruction may have restricted the distribution of C. mantchuricum and caused its low nuclear and mitochondrial genetic diversity (Wang et al. 2017). Thus, the extensive threatened history of C. mantchuricum makes this species suitable for investigating the genomic consequences incurred by populations under long-term threat.
Fig. 1.

Distribution and genetic population structure of Crossoptilon mantchuricum and C. auritum. (a) Distributions of C. mantchuricum and partial C. auritum populations and sampling locations in the current study. N is number of samples. Pictures at upper right and upper left show C. mantchuricum and C. auritum, respectively. The two portraits were from Wang et al. (2017). (b) Genetic structure estimated using clustering algorithm ADMIXTURE. Cross-validation supports existence of four genetic clusters (K = 4) corresponding to C. auritum and three geographical C. mantchuricum populations. (c) Top three principle component axes of genetic variation. Percentage variation explained by each principle component indicated in brackets. Blue is C. auritum. Brown-W, Brown-C, and Brown-E indicate Western, Central, and Eastern C. mantchuricum populations, respectively.
The Blue eared pheasant (C. auritum) is a sister species of C. mantchuricum and native to northwestern China (Zheng 2015; Wang et al. 2017). It is distributed in Qinghai, Ningxia, Gansu, and Sichuan Provinces. The C. auritum is allopatrically distributed with C. mantchuricum within the geographic desert barriers of the Loess Plateau (Zheng 2015). Hence, a recent gene flow between these species is unlikely. Historical gene flow between these species was documented and might have been associated with their relatively late divergence (≥0.11 Ma) (Wang et al. 2017). The C. auritum populations are stable and this species is of no conservation concern according to the IUCN Red List (BirdLife International 2016b). Multilocus nuclear data revealed high genetic diversity in C. auritum, unlike C. mantchuricum (Wang et al. 2017). Therefore, C. auritum is suitable for comparison with C. mantchuricum in terms of conservation genomics and we can apply a comparative population genomic approach to investigate the consequences of population decline in C. mantchuricum.
Here, we assembled and annotated a de novo reference genome for C. mantchuricum. We resequenced the genomes of 40 individuals from three isolated wild C. mantchuricum populations and 11 C. auritum individuals. We then investigated the genomic diversity, relative role of purifying selection and genetic drift, historical demography, and genetic consequences of population declination, that is, level of inbreeding and genetic load in C. mantchuricum. The analyses herein clarified the genomic legacy of a threatened bird species and could facilitate its conservation management.
Results
Genome Assembly and Annotation
The draft genome assembly comprised 2,416 scaffolds with a total length of 1.01 Gb and 1.3% gaps and constituting 96.06% of the genome length (1.05 Gb) estimated by Kmer spectrum analysis. The chicken (Gallus gallus) genome was 1.06 Gb long (GRCg6a; GenBank: GCA_000002315.5) and was, therefore, similar in size to the C. mantchuricum genome analyzed here. The genome sequence had 40.5% GC content. The scaffold and contig N50 were 3,632.75 and 112.76 kb, respectively. This draft genome covered 95.1% of all avian single-copy orthologs (supplementary fig. S1 and data set S1, Supplementary Material online) and was, therefore, highly contiguous. We annotated 19,304 coding genes using the combined evidence of homologous and RNA sequences and ab initio prediction. This gene set was comparable to most published vertebrate genomes (Sackton et al. 2019) and covered 89.6% of all avian single-copy orthologs. Moreover, 94.2% of its genes had AED <0.5. More than 76.6% of the proteomes produced by this coding gene set had recognizable domains (supplementary fig. S2, Supplementary Material online). The foregoing statistics suggest that this genome annotated to a high standard (Holt and Yandell 2011).
Strong Structuring among Fragmented Populations
We identified the three allopatric C. mantchuricum populations as Brown-W, Brown-C, and Brown-E. These represented the Shaanxi (Western), Shanxi (Central), and Hebei & Beijing (Eastern) populations, respectively (fig. 1a). Blue was used as an abbreviation for C. auritum. The admixture algorithm supported four clusters when it was run with cross-validation (fig. 1b and supplementary fig. S3, Supplementary Material online). The principal component analysis (PCA) also supported four clusters (fig. 1c and supplementary fig. S4, Supplementary Material online). Its first three eigenvectors separated Blue, Brown-W, Brown-C, and Brown-E with 66.81% variance in the explained single-nucleotide polymorphism (SNP) data. Evidence from the haplotype network and phylogeny relationships was consistent with the results of the admixture algorithm and the PCA (supplementary fig. S5, Supplementary Material online).
Low Magnitude of Genetic Diversity and Elevated Level of Genetic Drift in C. mantchuricum
We calculated the genetic diversity of C. mantchuricum and C. auritum using the proportions of heterozygous SNPs per callable base per species. Among bird species for which genome-wide estimates are available, C. mantchuricum had lower genetic diversity than the White-tailed eagle (Haliaeetus albicilla), the Crested ibis (Nipponia nippon), and the Dalmatian pelican (Pelecanus crispus) (fig. 2a and supplementary data set S2, Supplementary Material online). The genetic diversity values of C. mantchuricum and C. auritum were 9.60 × 10−5 (SD=0.03 × 10−5) and 8.25 × 10−4 (SD=0.6 × 10−4), respectively. What was worse, there was minimal variation in the genetic diversity values across the genomes of C. mantchuricum (fig. 2b).
Fig. 2.

Comparison of genetic diversity among species differing in conservation status. (a) Crossoptilon mantchuricum exhibits lowest level of genetic diversity among bird species based on available estimates from genome-wide sequencing data. Genetic diversity was measured as proportion of heterozygous SNPs per base pair and plotted in rank order for 23 bird species including C. auritum and C. mantchuricum (bold text and asterisk). Colored bars indicate 2020 IUCN Red List threatened species status. References for each species provided in supplementary data set S2, Supplementary Material online. (b) Nucleotide diversity in 100-kb windows with 25-kb step size across chromosomes. Number under abbreviated name indicates average nucleotide diversity marked by dotted line.
To map C. mantchuricum heterozygosity, we calculated its population genetics summary statistics (pairwise nucleotide diversity, Θπ, and Watterson’s theta, Θw) for synonymous 4-fold degeneration variants. Brown-W had the lowest Θπ (0.09 × 10−3, 95% CI=8.64 × 10−5–8.68 × 10−5) and Θw (0.07 × 10−3, 95% CI=6.89 × 10−5–6.92 × 10−5). Brown-C had higher Θπ (0.17 × 10−3, 95% CI=17.19 × 10−5–17.24 × 10−5) and Θw (0.14 × 10−3, 95% CI=14.15 × 10−5–14.18 × 10−5) than Brown-W and Brown-E. All three isolated C. mantchuricum populations had lower Θπ and Θw than Blue (fig. 3a and b; supplementary data set S3, Supplementary Material online). The low diversity pattern in C. mantchuricum was reflected in its nonsynonymous mutations (fig. 3c and d; supplementary data set S3, Supplementary Material online). This finding aligned with the relative differences among species in terms of genomic variation (fig. 2). To identify whether C. mantchuricum accumulated missense mutations, we computed the Θπ (0-fold degeneration variants)/Θπ (4-fold degeneration variants) ratio. It was higher for all three C. mantchuricum populations than it was for C. auritum (fig. 3e and supplementary data set S3, Supplementary Material online). For all comparisons among C. mantchuricum and C. auritum populations, the nonadjusted P < 10−3.
Fig. 3.

Summary of population genetics statistics. (a) Nucleotide diversity (Θπ) on 4-fold degeneration sites. (b) Watterson’s theta (Θw) on 4-fold degeneration sites. (c) Nucleotide diversity (Θπ) on 0-fold degeneration sites. (d) Watterson’s theta (Θw) on 0-fold degeneration sites. (e) Nucleotide diversity (Θπ) of 0-fold degeneration sites over nucleotide diversity (Θπ) of 4-fold degeneration sites. (f) Genome-wide Tajima’s D. For each statistic, all three Crossoptilon mantchuricum populations were compared with the C. auritum population. Three asterisks mean nonadjusted P < 10−3.
To determine whether genetic drift relaxed purifying selection strength in C. mantchuricum, we calculated the genome-wide Tajima’s D and estimated the folded site frequency spectrum (SFS) of unlinked autosomal SNPs. The Tajima’s D values were positive and significantly higher in three populations of C. mantchuricum than C. auritum (fig. 3f), indicating balancing selection and population contraction. Crossoptilon mantchuricum also exhibited deficiency in low-frequency alleles but had excess of medium-frequency alleles. In contrast, folded SFS displayed a “L-shaped” in C. auritum, illustrating an excess of singletons (supplementary fig. S6, Supplementary Material online). Hence, both Tajima’s D and folded SFS suggested an elevated level of genetic drift and no sign of purifying selection in threatened C. mantchuricum as compared with its sister species with least conservation concern.
Genetic Signatures of Inbreeding
There were substantial differences between C. mantchuricum and C. auritum in terms of genome-wide linkage disequilibrium (LD) (fig. 4a). The average distance for the LD to decay until R2=0.2 was >500 kb for C. mantchuricum. In contrast, it was only ∼36.5 kb for C. auritum. The fraction of runs of homozygosity (ROH) was far greater for the autosomes of C. mantchuricum than C. auritum (fig. 4b and supplementary fig. S7a, Supplementary Material online). The ROH fractions in the three C. mantchuricum populations were 90.39% (Brown-W, SD=1.53%), 83.37% (Brown-C, SD=1.19%), and 88.93% (Brown-E, SD=1.62%), respectively. For Blue, however, it was only 20.78% (SD=5.16%). The ROH length indicates the relative timing of inbreeding as haplotype blocks become fragmented after successive recombination events. ROHs >0.5 Mb reflected recent inbreeding and comprised approximately ≥50% of the C. mantchuricum genome (Brown-W: 68.94±3.95%; Brown-C: 49.38±2.23%; Brown-E: 67.93±4.10%) (fig. 4b). However, the same fraction in C. auritum was only 2.96±1.34% (fig. 4b). Short ROHs reflect ancient inbreeding and had relatively larger fractions in C. mantchuricum (Brown-W: 21.45±2.67%; Brown-C: 34.00±1.45%; Brown-E: 20.99±2.76%) than C. auritum (17.82±4.54%) (fig. 4b). The extensive genome-wide LD and high ROH fraction in C. mantchuricum may be the results of population bottlenecks and recent and ancient inbreeding. The inbreeding coefficient, Fis also indicated that C. mantchuricum underwent a higher level of inbreeding than C. auritum (supplementary fig. S7b, Supplementary Material online).
Fig. 4.

Genetic linkage disequilibrium (LD) and runs of homozygosity (ROHs). (a) LD decay is represented by change in average correlation (R2) between SNPs among all individuals per population as physical distance increases between SNPs. (b) Percentages of ROH on autosomes. Integral bars indicate fraction of all ROHs. Solid bars show fractions of long ROH ≥0.5 Mb. Blank bars show fraction of short ROH <0.5 Mb. Error bars are ±1 SD. Blue indicates Crossoptilon auritum. Brown-W, Brown-C, and Brown-E are three isolated C. mantchuricum populations.
Evidence of Genetic Load in C. mantchuricum
There were relatively more missense mutations in all three C. mantchuricum populations than in C. auritum (fig. 5a). This finding was consistent with the low genetic diversity (fig. 2), strong genetic drift (fig. 3f), and extensive inbreeding (fig. 4) observed in C. mantchuricum. Brown-E had relatively more loss-of-function (LOF) variants than Blue (fig. 5a). However, the number of LOF variants in Brown-C and Brown-W did not significantly differ from those in Blue (fig. 5a). Though C. mantchuricum accumulated more missense mutations than C. auritum (fig. 5a), the test did not segregate the deleterious and neutral variants. Therefore, we calculated the ratio of missense to synonymous mutations at the homozygous and heterozygous sites. There were significantly more missense mutation rates at the heterozygous than the homozygous sites for all three C. mantchuricum populations (fig. 5b). However, missense mutations occurred at equal rates at the heterozygous and homozygous sites for C. auritum. As lethal recessive genes are more likely to be heterozygous in living organisms, this result suggests that all three C. mantchuricum populations accumulated deleterious recessive mutations whereas C. auritum did not.
Fig. 5.

Characterization of genetic load and extent of inbreeding depression in Crossoptilon mantchuricum. (a) Relative number of derived alleles at missense (red) and LOF (orange) variations that are frequent in one population of C. mantchuricum but not in C. auritum (RA/B). Error bars represent ±2 SD. (b) Ratio of derived missense variants to synonymous variations in homozygous (circle) and heterozygous (square) tracts per sample. Horizontal bars represent average values. P values for each population obtained from Kolmogorov–Smirnov tests comparing homozygous and heterozygous tracts. (c) Effects of inbreeding on accumulation of deleterious mutations. Rnhom was calculated by dividing ratio of missense to synonymous counts of homozygous genotypes inside ROH by corresponding ratio outside ROH. Upward-pointing triangles represent long ROH. Downward-pointing triangles represent short ROH. (d) Genetic diversity (π) estimated for each population in different genomic regions. MHC, major histocompatibility complex region; CDS, coding region; CNEE, conserved nonexonic elements; conserved introns; conserved intergenics; putative nonfunction regions; nonconserved intergenics; nonconserved introns. Error bars represent 95% CI estimated from 1,000-bootstrap resampling.
We tested whether the inbreeding common to all three C. mantchuricum populations caused the accumulation of deleterious recessive mutations. For all three C. mantchuricum populations, the ROHs had fewer homozygous missense alleles than the other regions (fig. 5c). Hence, inbreeding did in fact contribute to the accumulation of deleterious recessive mutations. We then compared the genetic diversity values for various genomic regions of C. mantchuricum and C. auritum. The functional genomic regions (major histocompatibility complex, conserved nonexonic elements, conserved introns, conserved intergenic regions, and coding regions) and the nonfunctional regions (putative nonfunctional regions, nonconserved introns, and nonconserved intergenic regions) of C. mantchuricum had less genetic diversity than the corresponding regions of C. auritum (fig. 5d and supplementary fig. S8, Supplementary Material online). This finding was consistent with the observation that C. mantchuricum had the lower genetic diversity (fig. 2). Taken together, these results indicate that C. mantchuricum has a higher genetic load and lower adaptive genetic variation.
We examined putatively functional genes in order to assess the extent of the genetic load. We selected 308 genes according to genome-wide dXY and genetic variation (π) (supplementary fig. S9 and data sets S4 and S5, Supplementary Material online). Two had derived LOF mutations and 121 had derived missense mutations. One of the derived LOF variants was linked with DNA repair function and fixed in all three C. mantchuricum populations. However, no derived LOF variants were fixed in C. auritum (supplementary data set S4, Supplementary Material online). Of the derived missense mutations on the genes putatively under positive selection, 48 were fixed in C. mantchuricum. Nine were deleterious according to their Provean scores. In contrast, only 27 derived missense mutations were fixed on the putatively adaptive genes in C. auritum and of these, six were deleterious (supplementary data set S5, Supplementary Material online). The genes putatively under positive selection were linked mainly with digestion according to the KEGG pathway and GO enrichment analyses. The average genetic diversity of these genes in C. mantchuricum was lower than that for the corresponding genes in C. auritum (supplementary data set S6, Supplementary Material online). For C. mantchuricum, the relatively high fixation rate for deleterious mutations on potentially functional genes indicated low purifying selection power.
Long-Term Population Contraction in C. mantchuricum
To explore the population size dynamics of C. mantchuricum, we inferred the long-term demographic history of the three populations using multiple sequential Markovian coalescence (MSMC). The effective population sizes (Ne) started to decline ∼37.5 ka (fig. 6). The most recent (∼0.5 ka) Ne of the three C. mantchuricum populations were 1,420 (Brown-W), 24,041 (Brown-C), and 730 (Brown-E). Over the past ∼1.64 × 104 years, effective population sizes for C. mantchuricum were always less than those for C. auritum (fig. 6). This discovery is consistent with the genetic diversity pattern (fig. 2). The bootstrap results of the Ne fluctuations are shown in supplementary figure S10, Supplementary Material online.
Fig. 6.
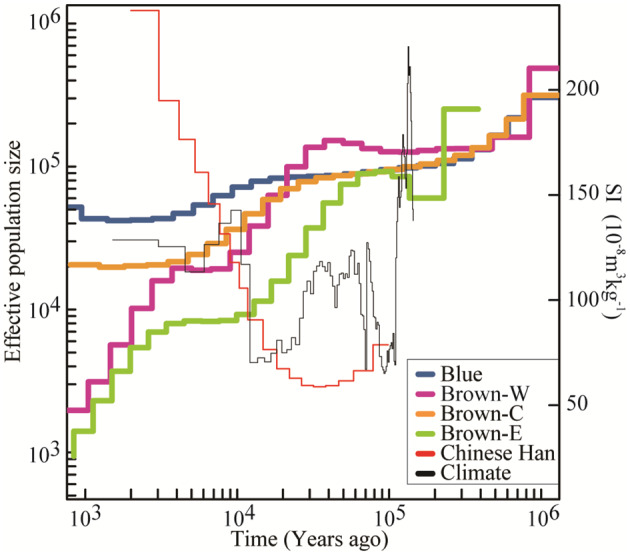
Demographic history of Crossoptilon mantchuricum and C. auritum reconstructed from autosomes using the multiple sequential Markovian coalescence (MSMC) model. Inferred fluctuations in effective population size (Ne) from 1 ka to 0.8 Ma based on the 2-year generation time and 4.02 × 10−9 per site per generation mutation rate assumptions. Blue line represents estimated Ne of C. auritum (blue). Purple, brown, and green lines represent Western (Brown-W), Central (Brown-C), and Eastern (Brown-E) C. mantchuricum populations, respectively. Red line represents Chinese Han population (CH). Black line shows magnetic susceptibility (SI) of central Chinese Loess Plateau. SI was a proxy for temperature.
In addition, coalescent simulations run in fastsimcoal26 corroborated the population history inferred by MSMC that long-term contraction inclusively occurred in the three C. mantchuricum populations based on the empirical site frequency spectra (supplementary data set S7 and fig. S11, Supplementary Material online). In addition, there was no gene flow between adjacent C. mantchuricum populations suggested by the best model in fastsimcoal26, which is consistent with gene flow estimation using Migrate-N (supplementary data set S8, Supplementary Material online).
Discussion
To the best of our knowledge, this study is the first to perform a population genomic survey of the C. mantchuricum. This threatened species is now restricted to the Montane forests of northern China. Here, we used a de novo genome and population genomic data from three fragmented C. mantchuricum populations and one population of its sister species, C. auritum. The present study elucidated the genomic consequences of long-term population decline including extremely low level of genetic diversity and evidence of inbreeding depression. The comparative analysis between a highly threatened species and its demographically stable sister species demonstrated that, genetic drift lower efficacy of natural selection, and can lead the accumulation of deleterious mutations in the endangered species. Together, this study furnishes genomic resources that facilitate the conservation management of C. mantchuricum.
The C. mantchuricum population started to contract during the most recent glacial period ∼12.1–34.8 ka (fig. 6). The population size trajectory of C. mantchuricum and the Chinese Han population (CH) exhibited the opposite trends (fig. 6). The latter began to expand ∼30 ka (Schiffels and Durbin 2014) whereas the former began to contract simultaneously. Such a population trend of C. mantchuricum also roughly coincided with long-term cooling of the central Chinese Loess Plateau 34.8–12.1 ka (Song et al. 2007). However, climate change was probably not the sole reason for the long-term decline of C. mantchuricum. Unlike other pheasant species such as the black grouse (Tetrao tetrix) (Kozma et al. 2016), the population of C. mantchuricum did not recover even after the temperature trajectory reversed (fig. 6). The earliest record of human activity in the geographic region of the present study is 39 ka (Shang et al. 2007; Barton et al. 2020) and roughly corresponds to acceleration of the decline in the C. mantchuricum population (fig. 6). Over the past millennium, the human impact on C. mantchuricum distribution has increased in the form of events such as the construction of the Great Wall in Shanxi, Shaanxi, Hebei, and Beijing during the Ming and Qing dynasties. Hence, recently expanding anthropogenic activities such as deforestation, land-use changes, and hunting may have contributed to the steady population decline of C. mantchuricum. The strong population genetic structure in isolated C. mantchuricum populations (fig. 1) may have been the consequence of loss and fragmentation of suitable habitats between them and consequently blocking gene flow. Thus, the three isolated populations with different Ne can be treated as independent management units, separately.
We found that C. mantchuricum underwent inbreeding during its population decline as there were LD patterns and homozygous tracts in its genome (fig. 4). LD rapidly decays to very low levels in wild bird populations (Poelstra et al. 2013; Kawakami et al. 2014) as it did for C. auritum (fig. 4a). In contrast, C. mantchuricum exhibited extremely high LD (fig. 4a). Population decline and fragmentation in C. mantchuricum might have increased inbreeding frequency which, in turn, would reduce the effective population size further still. ROH widely occurred in C. mantchuricum chromosomes (fig. 4b). ROHs are the result of high rates of inheritance of chromosomal segments that are identical by descent (Ceballos et al. 2018). All C. mantchuricum populations had several times more fractions of autosomal sequences within ROH than C. auritum (fig. 4b). At least half the C. mantchuricum genome was contained in long ROHs and its LD rate was extremely high (fig. 4). Both attributes are genomic consequences of population decline and inbreeding, which will reduce the genetic diversity and increase the genetic load in wild populations.
The genomic signatures of reduction in genetic diversity and accumulation of deleterious alleles were detected in C. mantchuricum. Compared with numerous other bird species studied using comparable data sets, C. mantchuricum has extremely low genetic diversity (fig. 2a). This discovery was consistent with an earlier study using multiexon sequences (Wang et al. 2017). In general, genetic diversity is negatively correlated with population fitness (Frankham 2004). Hence, together with low genetic diversity in the functional genomic region of C. mantchuricum (figs. 3c and d and 5d), these findings indicate that C. mantchuricum is at an increased risk of extinction. Here, we found that C. mantchuricum has accumulated more missense variants than C. auritum, and Brown-E has more LOF than C. auritum (fig. 5a). The missense variants are deleterious as the ratio of missense mutations in the homozygous sites was significantly lower than that in the heterozygous sites (fig. 5b). Therefore, these genomic signatures suggest C. mantchuricum is likely suffering inbreeding depression. The uncovered patterns are documented in some endangered species with extremely low level of genetic diversity, for example, Channel Island fox (Urocyon littoralis) (Robinson et al. 2016), and snub-nosed monkeys (Rhinopithecus spp.) (Zhou et al. 2016). An obvious question arises, in small and highly inbred populations, whether purifying selection is sufficient to purge deleterious mutations?
Inbreeding exposes lethal recessive and detrimental partially recessive mutations to purifying selection. In this way, it reduces the frequency of detrimental mutation and the level of inbreeding depression in populations (genetic purging) (Lande and Schemske 1985; García-Dorado 2015). However, this trend might not apply to extremely small populations. According to the nearly neutral theory, alleles are invisible to purifying selection when the selective effect is less than 1/2 Ne (Ohta 1972; Akashi et al. 2012). Populations may decline until purifying selection no longer purges deleterious mutations. Previous studies showed that purifying selection efficiency varies among wild populations. The Alpine ibex (Capra ibex) has experienced numerous bottlenecks but few highly deleterious mutations, which suggested selection has purged deleterious mutations (Grossen et al. 2020). In contrast, there was severe inbreeding depression and relaxation of purifying selection by genetic drift in some cases. For instance, the burden of deleterious alleles in two subspecies of mountain gorilla (Gorilla spp.) increased after a prolonged population decline (Xue et al. 2015). The results of the present study are consistent with the gorilla case, which are indicative of strong genetic drift in C. mantchuricum. These detrimental genetic consequences can be strengthened if populations of C. mantchuricum keep declining. Even if populations would re-expand, signature of genetic erosion would still be persistent (Kirkpatrick and Jarne 2000). In this regard, viable conservation strategies need to target at restoring effective population size of C. mantchuricum (Peart et al. 2020).
Crossoptilon mantchuricum is currently listed as “vulnerable” by IUCN Red List (BirdLife International 2016c) and a first-class national protected animal in China (Zheng 2015). Thus, hunting is strictly prohibited for wild populations. To allow its long-term persistence, some recommendations are proposed based on our findings. As we show that purifying selection is less efficient in all three isolated populations of C. mantchuricum, any measures aiding wild population augmentation and restoration suitable habitats, are most important as selection would be more effectively working on larger populations. Second, because we found evidence of the relatively high fixation rate for deleterious mutations in C. mantchuricum, monitoring population genetic health may be a necessary means to reduce negative effects caused by genetic erosion, and natural catastrophic events, such as diseases and forest fire. This is probably more critical for the western and eastern populations as they have a smaller geographical range and Ne, comparing the central population in Shanxi. Hence, in case genetic rescue program, such as population translocation and reintroduction programs are planned, the central population in Shanxi may be a source population. The differences in genetic profiles among these three populations also indicate refined conservation management programs should be considered.
Materials and Methods
DNA Sample Collection
Tissue for de novo whole-genome sequencing was obtained from a female C. mantchuricum. For genome resequencing, tissue samples were obtained from 40 C. mantchuricum and 11 C. auritum individuals. For genome annotation, the transcriptome of the C. mantchuricum individual used for de novo genome assembly was also sequenced.
Genome Assembly and Annotation
Fragment and jumping libraries were constructed for de novo genome sequencing. Low-quality data were removed and 1.37 billion reads (195.6× sequence coverage) were used for de novo C. mantchuricum genome assembly in ALLPATHS-LG v. 52488 (Butler et al. 2008). Repetitive elements were identified and an integrative approach was taken to identify a protein-coding gene set of the repeated-masked genome in MAKER v. 2.31.8 (http://www.yandell-lab.org/software/maker.html).
Read Mapping and SNP Calling
Fragment libraries were constructed for genome resequencing and targeted >15× coverage per individual. Clean reads in all fragment libraries per individual were mapped to the C. mantchuricum reference genome with BWA-MEM v. 0.7.9a (Li and Durbin 2009). Local realignment around indels, recalibration of base quality scores, and SNP filtering were performed in the GATK v. 3.7 pipeline according to best practice recommendations (McKenna et al. 2010).
Population Structure and PCA
Principal component analysis of the genotypes was performed in GCTA v. 1.91.3 using a filtered set of 49,701 autosomal SNPs (Yang et al. 2011). The same SNP set was used to infer the population structure in ADMIXTURE v. 1.3.0 with cross-validation and 100 bootstraps (Alexander and Lange 2011).
Genetic Diversity Estimation
To assess genetic diversity within and among C. mantchuricum populations, the heterozygosity of each sample was calculated and defined as the proportion of heterozygous sites in all callable sites across all autosomes. For each population or species, genetic diversity was defined as the mean heterozygosity per individual per population or species. Population genetics statistics (Θπ and ΘW) were computed on synonymous and nonsynonymous sites. To test whether C. mantchuricum accumulated more missense mutations than C. auritum, the Θπ (0-fold)/Θπ (4-fold) ratios were determined for each population.
Genetic Drift Power Evaluation
To assess the roles of purifying selection and genetic drift in C. mantchuricum, genome-wide Tajima’s D values were calculated in ANGSD v. 0.930 (Korneliussen et al. 2014) for each population. The window and step sizes were 100 and 25 kb, respectively. To test whether the C. mantchuricum and C. auritum populations significantly differed in terms of Tajima’s D, nonpaired Welch two-sample t-tests were performed. Genetic drift strength was also estimated from the folded SFS in ANGSD v. 0.930 (Korneliussen et al. 2014) using unlinked autosomal chromosome SNPs in the C. mantchuricum and C. auritum.
Inbreeding Pattern and Genetic Load Estimation
To determine the genomic extent of inbreeding in C. mantchuricum, genome-wide ROH, estimated LD, and inbreeding coefficient (Fis) were identified for the three C. mantchuricum populations and C. auritum.
Methods previously applied toward gorillas (Xue et al. 2015) were used to measure the genetic load. SnpEff was used to identify LOF, missense, and synonymous variations in coding regions (Cingolani et al. 2012). The relative numbers of derived LOF and missense variants in each C. mantchuricum population and C. auritum were estimated and compared according to the formula of Xue et al. (2015).
Inbreeding depression was assessed by determining whether ROH regions accumulate more homozygous missense mutations than the regions outside the ROH. The relative numbers of heterozygous and homozygous missense mutations were compared. The genetic diversity of the functional and nonfunctional regions between each C. mantchuricum population and C. auritum were also compared.
To establish whether purifying selection has power to remove deleterious mutations in C. mantchuricum, the genes putatively under positive selection were inferred and the Provean scores (Choi et al. 2012) of the derived missense mutations were calculated. These values indicated whether the derived missense mutations were deleterious to the genes putatively under positive selection. The genetic diversity of each candidate gene and the frequency of the derived missense mutations were also calculated. KOBAS v. 3.0 was used to perform KEGG and GO enrichment analyses on those genes (Wu et al. 2006).
Demographic History Reconstruction
The potential historical factors associated with the current genetic status of C. mantchuricum were examined by reconstructing demographic histories for each study population with the MSMC2 model (Schiffels and Durbin 2014). The mutation rate of C. mantchuricum and C. auritum was 4.02 × 10−9 per site per generation based on the comparison of the 4-fold degenerate sites among the Galliformes lineages (Zhang et al. 2014) and the generation time was 2 years based on field observation (Zheng 2015). The population history of C. mantchuricum was compared with that of the Chinese Han (Schiffels and Durbin 2014) and the Loess Plateau climatic history (Song et al. 2007).
To investigate whether there was gene flow between adjacent C. mantchuricum populations, Migrate-n v. 3.6.11 was used in this study (Beerli and Felsenstein 2001). It performed a Bayesian inference of the gene flow pattern. The simulation-based framework fastsimcoal26 (Excoffier et al. 2013) was run to infer the demographic history of all three C. mantchuricum populations and distinguish whether the C. mantchuricum populations continuously declined or simply remained small. Full details of the materials and methods are presented in Supplementary Material online.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors thank Tim Sackton, Sangeet Lamichhaney, Kadeem J. Gilbert, Allison Shultz, Alison Cloutier, Xukang Shen, Simon Yung Wa Sin, and Phil Grayson of Harvard University for their advice on bioinformatic analyses, Funing Xin, Lixia Zhang, and Jianguo Qiang for contributing samples, and the Pangquangou National Natural Reserve of Shanxi and the Beijing and Taiyuan Zoos for fieldwork assistance and sample provision. Computations were run on the Odyssey cluster supported by the FAS Division of Science Research Computing Group at Harvard University. This work was supported by the National Key Program of Research and Development from Ministry of Science and Technology of China (No. 2016YFC0503200 to Z.Z.), the National Natural Science Foundation of China (No. 31872244 to Z.Z. and No. 31601839 to D.C.), the China Postdoctoral Science Foundation (No. 2019M660044 to P.W.), and the Biodiversity Survey, Monitoring and Assessment Project (2019–2023) of the Ministry of Ecology and Environment, China (No. 2019HB2096001006 to Z.Z.). We are grateful to the two anonymous reviewers and associate editor, Fuwen Wei, for constructive comments on a previous draft of this article.
Data Availability
All the data have been deposited at the National Genomics Data Center (https://bigd.big.ac.cn/? lang=en), the BioProject number is PRJCA003284, the accession number of the whole genome sequence is GWHAOPW00000000, the accession number of each sample can be seen in supplementary data set S9, Supplementary Material online.
References
- Akashi H, Osada N, Ohta T.. 2012. Weak selection and protein evolution. Genetics 192(1):15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Lange K.. 2011. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics 12(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton L, Bingham B, Sankaranarayanan K, Monroe C, Thomas A, Kemp BM.. 2020. The earliest farmers of northwest China exploited grain-fed pheasants not chickens. Sci Rep. 10(1):2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli P, Felsenstein J.. 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci U S A. 98(8):4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BirdLife International. 2016. a. Coturnix novaezelandiae. The IUCN Red List of Threatened Species 2016: e.T22678955A92795779.
- BirdLife International. 2016. b. Crossoptilon auritum. The IUCN Red List of Threatened Species 2016: e.T22679307A92810024.
- BirdLife International. 2016. c. Crossoptilon mantchuricum. The IUCN Red List of Threatened Species 2016: e.T22679299A92809690.
- Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB.. 2008. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18(5):810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF.. 2018. Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet. 19(4):220–234. [DOI] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP.. 2012. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7(10):e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM.. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M.. 2013. Robust demographic inference from genomic and SNP data. PLoS Genet. 9(10):e1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Fang Q, Barnett R, Li C, Han S, Kuhlwilm M, Zhou L, Pan H, Deng Y, Chen G, et al. 2019. The genomic footprints of the fall and recovery of the Crested Ibis. Curr Biol. 29(2):340–349.e347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham R. 2004. A primer of conservation genetics. Cambridge: Cambridge University Press. [Google Scholar]
- Frankham R, Bradshaw CJA, Brook BW.. 2014. Genetics in conservation management: revised recommendations for the 50/500 rules, red List criteria and population viability analyses. Biol Conserv. 170:56–63. [Google Scholar]
- Fuller RA, Garson PJ.. 2000. Pheasants: status survey and conservation action plan 2000-2004. Oxford: Information Press. [Google Scholar]
- García-Dorado A. 2015. On the consequences of ignoring purging on genetic recommendations for minimum viable population rules. Heredity 115(3):185–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossen C, Guillaume F, Keller LF, Croll D.. 2020. Purging of highly deleterious mutations through severe bottlenecks in Alpine ibex. Nat Commun. 11(1):1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M.. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12(1):491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsgard. 1986. The pheasant of the world. Oxford: Oxford University Press. [Google Scholar]
- Kawakami T, Backström N, Burri R, Husby A, Olason P, Rice AM, Ålund M, Qvarnström A, Ellegren H.. 2014. Estimation of linkage disequilibrium and interspecific gene flow in Ficedula flycatchers by a newly developed 50k single-nucleotide polymorphism array. Mol Ecol Resour. 14(6):1248–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane A, Brooke ML, McGowan PJK.. 2005. Correlates of extinction risk and hunting pressure in gamebirds (Galliformes). Biol Conserv. 126(2):216–233. [Google Scholar]
- Kirkpatrick M, Jarne P.. 2000. The effects of a bottleneck on inbreeding depression and the genetic load. Am Nat. 155(2):154–167. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R.. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15(1):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R, Melsted P, Magnússon KP, Höglund J.. 2016. Looking into the past – the reaction of three grouse species to climate change over the last million years using whole genome sequences. Mol Ecol. 25(2):570–580. [DOI] [PubMed] [Google Scholar]
- Lande R, Schemske DW.. 1985. The evolution of self-fertilization and inbreeding depression in plants. I. Genetic models. Evolution 39(1):24–40. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan P, Owens L, Grainger M.. 2012. Galliformes science and species extinctions: what we know and what we need to know. Anim Biodivers Conserv Biol. 35:321–331. [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. 1972. Population size and rate of evolution. J Mol Evol. 1(4):305–314. [DOI] [PubMed] [Google Scholar]
- Peart CR, Tusso S, Pophaly SD, Botero-Castro F, Wu C-C, Aurioles-Gamboa D, Baird AB, Bickham JW, Forcada J, Galimberti F, et al. 2020. Determinants of genetic variation across eco-evolutionary scales in pinnipeds. Nat Ecol Evol. 4(8):1095–1104. [DOI] [PubMed] [Google Scholar]
- Peng M-S, Wu F, Murphy RW, Yang X-J, Zhang Y-P.. 2016. An ancient record of an avian hybrid and the potential uses of art in ecology and conservation. Ibis 158(2):444–445. [Google Scholar]
- Peters J, Lebrasseur O, Deng H, Larson G.. 2016. Holocene cultural history of Red jungle fowl (Gallus gallus) and its domestic descendant in East Asia. Quat Sci Rev. 142:102–119. [Google Scholar]
- Poelstra JW, Ellegren H, Wolf JBW.. 2013. An extensive candidate gene approach to speciation: diversity, divergence and linkage disequilibrium in candidate pigmentation genes across the European crow hybrid zone. Heredity 111(6):467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J A, Ortega-Del Vecchyo D, Fan Z, Kim Bernard Y, vonHoldt Bridgett M, Marsden Clare D, Lohmueller Kirk E, Wayne Robert K.. 2016. Genomic flatlining in the endangered island fox. Curr Biol. 26(9):1183–1189. [DOI] [PubMed] [Google Scholar]
- Sackton TB, Grayson P, Cloutier A, Hu Z, Liu JS, Wheeler NE, Gardner PP, Clarke JA, Baker AJ, Clamp M, et al. 2019. Convergent regulatory evolution and loss of flight in paleognathous birds. Science 364(6435):74–78. [DOI] [PubMed] [Google Scholar]
- Schiffels S, Durbin R.. 2014. Inferring human population size and separation history from multiple genome sequences. Nat Genet. 46(8):919–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang H, Tong H, Zhang S, Chen F, Trinkaus E.. 2007. An early modern human from Tianyuan Cave, Zhoukoudian, China. Proc Natl Acad Sci U S A. 104(16):6573–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Fang X, Torii M, Ishikawa N, Li J, An Z.. 2007. Late Neogene rock magnetic record of climatic variation from Chinese eolian sediments related to uplift of the Tibetan Plateau. J Asian Earth Sci. 30(2):324–332. [Google Scholar]
- Wang P, Liu Y, Liu Y, Chang Y, Wang N, Zhang Z.. 2017. The role of niche divergence and geographic arrangement in the speciation of Eared Pheasants (Crossoptilon, Hodgson 1938.). Mol Phylogenet Evol. 113:1–8. [DOI] [PubMed] [Google Scholar]
- Wu J, Mao X, Cai T, Luo J, Wei L.. 2006. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 34(Web Server):W720–W724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Prado-Martinez J, Sudmant PH, Narasimhan V, Ayub Q, Szpak M, Frandsen P, Chen Y, Yngvadottir B, Cooper DN, et al. 2015. Mountain gorilla genomes reveal the impact of long-term population decline and inbreeding. Science 348(6231):242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM.. 2011. GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet. 88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, et al. 2014. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346(6215):1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G-M. 2015. Pheasants in China. Beijing (China: ): Higher Education Press. [Google Scholar]
- Zhou X, Meng X, Liu Z, Chang J, Wang B, Li M, Wengel PO-T, Tian S, Wen C, Wang Z, et al. 2016. Population genomics reveals low genetic diversity and adaptation to hpoxia in Snub-nosed monkeys. Mol Biol Evol. 33(10):2670–2681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data have been deposited at the National Genomics Data Center (https://bigd.big.ac.cn/? lang=en), the BioProject number is PRJCA003284, the accession number of the whole genome sequence is GWHAOPW00000000, the accession number of each sample can be seen in supplementary data set S9, Supplementary Material online.