

Abstract
In the past few decades, the studies of extrachromosomal DNA (ecDNA), which existed independently of chromosomes, were tepid. However, recent studies on ecDNA rekindled the enthusiasm of oncologists for further studying ecDNA. In this review, we summarized the recent advances of ecDNA in oncogenesis and oncotherapy. ecDNA consists of highly open chromatin, and its circular structure enables ultra-long-range chromatin contacts. ecDNA is not inherited in accordance with Mendel’s laws. Furthermore, ecDNA is widely existed in cancer cells, but almost never found in normal cells. It has been found that ecDNA played important roles in tumorigenesis and tumor progression, including oncogene amplification, tumor heterogeneity, enhancer hijacking and genomic rearrangement. More importantly, ecDNA is closely related to cancer treatment resistance. In hence, further understanding of ecDNA would contribute to developing innovative targeting ecDNA therapies.
Keywords: Extrachromosomal DNA, double minutes, tumorigenesis, therapeutic resistance
Introduction
Extrachromosomal DNA (ecDNA), which was first described over 50 years ago, has gradually attracted scientists’ extensive attention recently [1-3]. In 1965, during the examination of chromosomes, which were directly prepared from human tumors, COX D et al. encountered a curious phenomenon: in addition to the apparently structurally intact chromosomes, there were some, sometimes in large numbers, very small double chromatin bodies [3]. Afterwards, researchers named them ecDNAs, this refers to DNAs those exist independently of chromosomes according to the definition. Although ecDNA had been readily observable, technical limitations seriously hampered the detailed and further studies of ecDNA, for instance, because ecDNA is so small that it was hard to detect it under a conventional microscope, the real face of ecDNA and where it came from were still a mystery. With the rapid development and wide application of various advanced experimental technologies and equipment such as whole genome sequencing (WGS), structural modelling, cytogenetic analyses and ECdetect (a semi-automated image analysis software package for cytogenetic analyses), numerous studies had been conducted to investigate the structure, features and biological functions of ecDNA [4-7], Studies on the relationship between ecDNA and cancer biology presented some breathtaking findings, which were highly valued in the field of oncology. For example, ecDNA played crucial roles in driving tumor evolution [4,8], accelerating tumor progression [9] and cancer treatment resistance [10,11].
In this review, we focused exclusively on ecDNA and its special and important roles in tumorigenesis and therapeutic resistance of cancers, and expected to provide novel insights for developing new approaches to improve anticancer outcomes.
Biogenesis and characteristics of ecDNA
Since ecDNA was been exposed, a few research groups have attempted to investigate its biogenesis and characteristics, thus facilitated the emergence of some advanced technologies and easy-to-use tools.
Initially, CsCl gradient purification and electron microscopy imaging were used to detect the existence of ecDNAs [3,12-14]. Jeon Y et al. demonstrated that fluorescence in situ hybridization (FISH) was a sensitive and useful method in discovering and monitoring double minutes (DMs, a small fragment of ecDNA) [15]. In 2017, Turner KM et al. developed a software package called ECdetect which could provide insights into the biology of ecDNA in human cancers [4,7]. Soon afterwards, Pu L et al. constructed the SDquest algorithm for segmental duplication finding, and they found that some segmental duplications might originate from ecDNA, not dissimilar to ecDNA that contributed to accelerating cancer evolution [16]. Møller HD et al. introduced an innovative method entitled Circle-Seq for purifying ecDNA with high sensitivity [17], Khatami F et al. deemed that isolation and characterization of ecDNA would be possible by Circle-Seq [18]. With the use of computational analysis of WGS data from cancer patients, Kim H et al. found that oncogenes were highly enriched on amplified ecDNA, and the most common recurrent oncogene amplifications arose on ecDNA [6].
By means of the above experimental methods and detection techniques, the biogenesis of ecDNA was described but not fully elucidated. Previous studies indicated that ecDNA might derive from some form of micro homology directed repair, because a large percentage of ecDNAs, whose levels had been known to increase with the addition of carcinogens [19], contained or were proximal to short direct repeats [20,21]. However, van Loon N et al. observed that a significant portion of ecDNA fragments cloned from HeLa S3 cells were composed entirely of nonrepetitive or low-copy DNA sequences [22]. Hull RM et al. demonstrated that yeast aged under environmental copper accumulated high levels of ecDNA containing the copper-resistance gene CUP1 [23]. In 2015, Meng X et al. revealed that depletion or inhibition of DNA-PKcs, a key protein participated in non-homologous end joining (NHEJ), caused the reduction of dihydrofolate reductase (DHFR) amplification, the disappearance of DMs, and the increased formation of micronuclei or nuclear buds, which increased the sensitivity of colon cancer to methotrexate (MTX), their results indicated for the first time that NHEJ played a specific role in ecDNA formation [24]. In 2019, Cai M et al. found that, compared with MTX-sensitive colon cancer cells, DM-containing MTX-resistant colon cancer cells had significantly increased homologous recombination (HR) activity, and the inhibition of HR through BRCA1 silencing led to the decreased numbers of ecDNA, but had no effect on intrachromosomal amplification in MTX-resistant colon cancer cells [25].
Furthermore, under the painstaking research of scientists, characteristics of ecDNA had been gradually thoroughly studied and summarized as follows [5,10,26-29]: First, unlike DNA, which is compressed like a cookie in chromosomes, ecDNA is circular with sizes ranging from several hundred kilobases to five megabases, it’s kind of like a plasmid in a bacterium. Second, ecDNA enables ultra-long-range chromatin contacts. The circular structure of ecDNA causes two genes that might be far apart in linear DNA suddenly meet, and this situation will undoubtedly disrupt the original regulatory mechanisms of DNA expression and also lead to abnormal expression of some genes. Third, ecDNA consists of highly open chromatin. Although ecDNA has histone partners and chromatin structures, it is relatively open and particularly easy to express, which leads to the expression of oncogenes on ecDNA in large quantities. Furthermore, relative to chromosomal amplicon, ecDNA is less stable. Moreover, because of the lack of centromeres, ecDNA is not inherited in accordance with Mendel’s laws, and its genetic materials segregate unequally to daughter cells, this could lead to a daughter cell acquiring a large amount of ecDNAs, which may contain all oncogenic genes, and therefore the daughter cell is even more harmful (Figure 1) [30].
Figure 1.
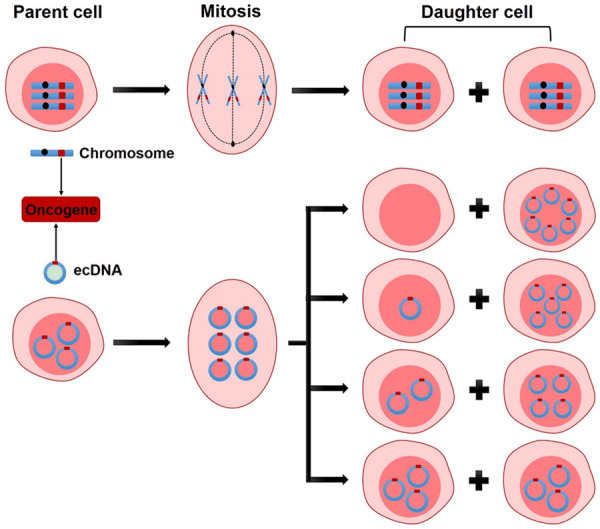
Schematic diagram of hereditary differences between chromosome and ecDNA. When the oncogene is on a chromosome, the sister chromatids attached to the centromeres are equally divided into two daughter cells because of the pull of spindle fibers during mitosis, this is a classic Mendel’s law. But when the oncogene is located on ecDNA, the ecDNA replicates with the chromosome, the spindle fibers can’t hold on to it because there is no centromere, which leads to the ecDNAs after replication being randomly divided into two daughter cells during cell cleavage.
The role of ecDNA in tumorigenesis and tumor progression
In the past few decades, the studies of ecDNA were tepid. Until 2014, the team of Paul S. Mischel, a professor of the Ludwig cancer institute at the University of California, restudied ecDNA, they found that ecDNA was related with drug resistance in cancer treatment, and could affect the effectiveness of targeted therapies related to the epidermal growth factor receptor (EGFR) gene [10]. The exciting results put ecDNA back to the sights of oncologists.
ecDNA was frequently founded across multiple cancers
In 2017, professor Paul S. Mischel’s team integrated WGS of multitudinous cancer cell lines, patient-derived tumor cell cultures and tumor tissues from a range of cancer types with bioinformatic and cytogenetic analysis of numerous cancer cells and normal cells in metaphase, they found that nearly half of human cancers owned ecDNAs (Figure 2), approximately 30% of the ecDNAs were paired DMs, and their frequency varied by cancer types, with substantially higher levels in patient-derived cultures, glioblastoma had a high proportion of ecDNAs, while colon cancer had a low proportion of ecDNAs, but ecDNA was almost never found in normal cells. Moreover, the research team discovered that, there were no significant associations between ecDNA level and primary tumor or metastatic status; untreated or treated samples; un-irradiated or post-irradiated tumors [4]. Analogously, Kim H et al. found that ecDNA amplification frequently occurred in most cancer types but not in blood or normal tissue [6]. It was a wonder that, the ecDNA-positive proportion of established cell lines passed through multiple generations was only 40%, but cultures derived from cancer patients contained higher levels of ecDNA, and the ecDNA-positive proportion could be up to 90% [4], suggesting that the in vivo tumor environment may somehow contribute to ecDNA maintenance [31].
Figure 2.
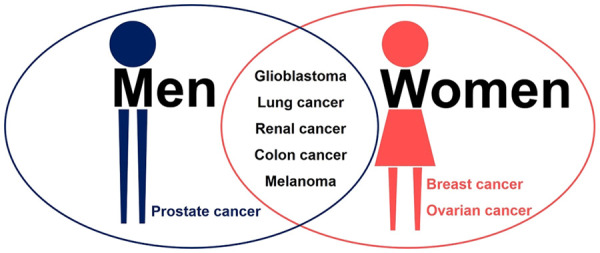
Prensence of ecDNA in human cancers. ecDNA is existed extensively in multiple cancers. In addition to glioblastoma, lung cancer, renal cancer, colon cancer and melanoma own ecDNA, men with prostate cancer and women with breast cancer or ovarian cancer also possess ecDNA.
ecDNA relieved heredity constraints and contributed to dynamic cancer evolution
Studies have shown that the complexity of cancer was induced by the tumor heterogeneity emanating at different levels including at the molecular, genomic and epigenomic level [31-34]. Tandon I et al. holden the opinion that, besides genomic instability within the chromosomal linear DNAs, the extra heterogeneity within cancer cells in the form of a great deal of ecDNAs added another dimension to the expression of precancerous players acting as a driver for cancer cell survival and proliferation [31]. deCarvalho AC and his colleagues performed a comprehensive genomic and transcriptomic analysis of tumor samples from patients diagnosed with glioblastoma and orthotopic xenograft models established from early-passage neurospheres, their results showed that oncogenic ecDNA was frequently retained throughout the course of glioblastoma, extrachromosomal elements allowed rapid increase of genomic heterogeneity during the evolution of glioblastoma, independently of chromosomal DNA alterations [35]. Xu K et al. performed in-depth analyses of the populations of different DMs in the paired tumors in glioblastoma patients, their results suggested that DMs readily evolved and increased tumor heterogeneity rapidly [36]. Wu S et al. [27] integrated RNA sequencing with WGS from cancer cell lines and from The Cancer Genome Atlas (TCGA) clinical tumor samples of diverse histological types, they revealed that genes encoded on ecDNA, especially authentic oncogenes such as EGFR, MYC, CDK4 and MDM2, were among the top 1% of genes expressed in cancer genomes, besides, compared with the same genes when they were not amplified by circularization, oncogenes amplified on ecDNA had markedly increased numbers of transcripts, owing to its increased DNA copy numbers, and thus increased the intratumoral heterogeneity and accelerated cancer evolution.
To sum up, the heterogeneity provided tumors with a pool of genomic alterations that might help them to response to microenvironment-induced and therapy-induced stress factors and perhaps provided an evolutionary advantage [37-39].
Enhancer hijacking of ecDNA promoted cancer development
Enhancer hijacking is an efficient mechanism driving oncogene activation in cancer [40,41], whereas the studies on its role in the biological function of ecDNA is relatively few.
In 2019, professor Peter C. Scacheri of Case Western Reserve University and professor Jeremy N. Rich of the University of California discovered that the ecDNA presented in glioblastoma contained not only the EGFR gene, which promoted cancer development [42,43], but also a number of regulatory elements such as enhancer sequences. Even more surprising, some of the sequences were not originally around the EGFR gene, they were more likely to be hijacked from various parts of the genome to specifically enhanced oncogenes. In order to understand the function of these regulatory elements, the researchers silenced them one by one using clustered regularly interspaced short palindromic repeats (CRISPR) gene-editing technology, and found that almost every regulatory element promoted tumor growth. Similar phenomena were found in a variety of cancer types, most commonly the MYC gene in medulloblastoma and the MYCN gene in neuroblastoma [44]. This study is the first to reveal the important role of enhancer hijacking in the carcinogenic effects mediated by cycled amplification of oncogenes, and this mechanism greatly expand the dynamic plasticity of regulation of oncogene expression in space, showing the key role of the global regulatory network as a functional unit in the occurrence and development of cancer that transcends commonly defined genetic boundaries.
Hence, the view of cancer treatment should be broadened in the future, besides targeting oncogenes, more attention need to be paid to how to turn off the enhancer switches that turn on oncogenes.
Extrachromosomal oncogene amplification accelerated cancer progression
Amplification, a mutation by which a cell acquires multiple copies of part of its genome, is one of the mechanisms by which proto-oncogenes may be activated in cancer cells [45,46]. Oncogene amplification, one of the most common drivers of tumorigenesis by facilitating cancer cells with specific growth advantages through overexpression of oncogenes and functional elements [47,48], is often mediated through focal amplification of genomic segments [49,50].
Accumulation of ecDNA is often responsible for gene amplification in cancers, and the potential mechanisms were partially elucidated. Zou HY et al. identified that platelet derived growth factor receptor α gene was frequently amplified and maintained on ecDNAs as DMs in brain tumors and cell lines derived from brain tumor tissues, suggesting its occurrence as an early mutational event contributing to the malignant transformation of oligodendrocyte precursor cells [51]. In the study of Turner KM et al., they detected that driver oncogenes were amplified most commonly in ecDNA, thereby increasing transcript levels, and the mathematical modelling predicted that ecDNA amplification would not only enhance the intratumoral heterogeneity, but also increase the copy number of oncogene more effectively than chromosomal amplification, which was validated by quantitative analyses of cancer samples [4].
Previous studies of ecDNA focused almost exclusively on its positive effect on the abundance of the oncogenes themselves, while less attention has been paid to the potential value of the non-coding sequences amplified with oncogenes, especially the enhancers characterized by flexible mode and wide range of action [52,53]. In 2019, using a combination of ChIP-seq, 4C-seq and CRISPR interference screening, Morton AR et al. performed a comprehensive survey of the patterns of coamplification around oncogenes and investigated the role of co-amplifications in gene regulation, regulatory element acquisition, chromatin topology, and their impact on cell fitness, they found that oncogene amplifications were shaped by regulatory dependencies in the non-coding genome, the oncogenes amplified on ecDNA selected for existing and new regulatory interactions that promoted cancer growth [44].
In summary, oncogene amplification on ecDNA is a frequent occurrence in many cancer types, and the presence of amplified oncogenes on ecDNA has clinical significance.
ecDNA was an unanticipated major source of genomic rearrangements in cancer
Genomic rearrangements are alterations of large genomic segments, sometimes spanning megabases [54]. Somatic rearrangements of the cancer genome are important drivers of oncogenesis. For example, some translocations lead to oncogenic gain-of-function that can act as critical cancer drivers and potential therapeutic targets [55,56].
In order to investigate the relationship between ecDNA and genomic rearrangement, some studies were conducted recently. In 2014, Vogt N et al. studied a xenografted human oligodendroglioma where the co-amplification of the EGFR and MYC loci was present in the form of DMs at early passages and of homogeneously staining regions (HRS) at later passages, they uncovered that, during the formation of DMs and their transformation into HRS, the amplified regions underwent multiple rearrangements and deletions [57]. In the process of describing the landscape of ecDNA in neuroblastoma, Koche RP et al. accidentally detected that ecDNA was an unanticipated major source of somatic rearrangements, contributing to oncogenic remodeling through chimeric circularization and reintegration of circular DNA into the linear genome, cancer-causing lesions could emerge out of circle-derived rearrangements and were associated with adverse clinical outcomes [58]. It was highly probable that circle-derived rearrangements represent an ongoing mutagenic process. Thus, ecDNA represented a multihit mutagenic process, with important functional and clinical implications for the origins of genomic remodeling in cancer.
However, the research on this topic should be further extended to more cancer types in the future, so as to provide new strategies for further elucidating other functions of ecDNA.
ecDNA may be closely related to cancer treatment resistance
When it comes to tumor therapeutic resistance, the most common mechanisms, excluding pharmacokinetic factors, are that tumors develop new genetic mutations or activate a compensatory survival pathway [59,60]. Surprisingly, several high-quality articles published recently identified a key code, the presence of ecDNA promoted the invasiveness of cancer cells and played an important role in resisting external threats, such as chemotherapy, radiotherapy and other treatments.
In 2014, Nathanson DA et al. demonstrated that glioblastoma cells resistance to EGFR tyrosine kinase inhibitors was due to cancer cells reversibly eliminated mutant EGFR from ecDNA, after drug withdrawal, reemergence of clonal EGFR mutations on ecDNA followed, thus conferred distinct cellular phenotypes to reach an optimal equilibrium for growth. These results indicated that cancer cells could evade therapies that targeted oncogenes maintained on ecDNA by a highly specific, dynamic and adaptive pathway [10]. Study has shown that significant increasement in the number of DHFR copies, up to several dozen copies, can be found in MTX-resistant tumor samples [61], however, these increased copy numbers could not only appear on chromosomes, but also exist as ecDNA, as shown in the research results of Miguel A Peinado’s team. They evaluated the association between different genetic features and the capacity to develop MTX resistance in three aneuploid cell lines (HT-29, SW480 and SK-CO-1) representative of alternative genetic pathways, and found that only HT29 cell developed MTX resistance, showing amplification of the DHFR gene at 5q12-14 (>20-fold amplification and presence of ecDNA) [62]. Meng X et al. demonstrated that the depletion or inhibition of DNA-PKcs of DM-containing cells caused disappearance of DMs and increased cells’ sensitivity to MTX (Figure 3) [24].
Figure 3.
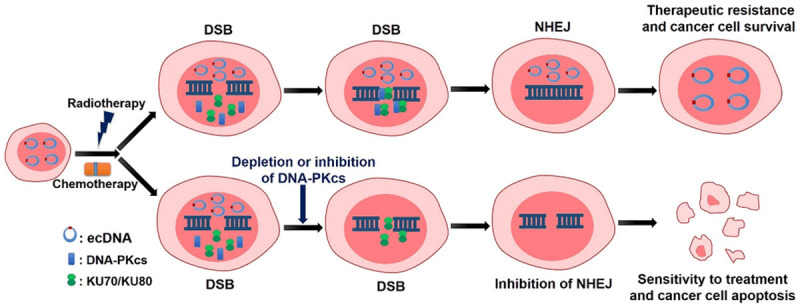
Novel role of NHEJ in formation of ecDNA in therapeutic resistance of cancers. ecDNA-containing cancer cells express higher level of proteins associated with NHEJ, which can promote the repair of DNA double-strand break (DSB) induced by radiotherapy and chemotherapy, and then lead to the occurrence of therapeutic resistance. However, depletion or inhibition of DNA-PKcs, a key NHEJ protein, can cause the disappearance of ecDNA and the inhibition of DSB repair by NHEJ, thus increase cancer’s sensitivity to treatment and induce cancer cell apoptosis.
Therefore, innovative targeting ecDNA therapy may be the fourth revolution in cancer treatments after radio-chemotherapy, targeted therapy and immunotherapy.
Conclusions
In recent years, tremendous and major breakthroughs have been made in the field of ecDNA. As more and more study results were reported, ecDNA is revealing itself in fascinating ways. There are growing evidences support that ecDNA is existed extensively in multiple cancers and play distinctive and important roles in tumorigenesis and cancer’s therapeutic resistance (Figure 4). However, knowledge of the characteristics of ecDNA and its functions in development and treatment of malignant tumors merely represent the tip of the iceberg, and the above unprecedented glimpses into ecDNA open up new questions about their roles in malignancy. For example, by which specific mechanism tumors maintain ecDNA homeostasis? In which way do cancer cells dynamically regulate the amount of ecDNA? Unfortunately, there is no research provides a definitive answer to the questions so far. As an ideal target for tumor therapy, it is strongly necessary to unveil the mysteries of ecDNA. Therefore, numerous endeavors and unremitting explorations are required to explore the underlying molecular mechanisms of ecDNA, and this might help to develop excellent anti-tumor strategies that either prevent carcinoma progression or overcome therapy resistance through directly targeting ecDNA.
Figure 4.
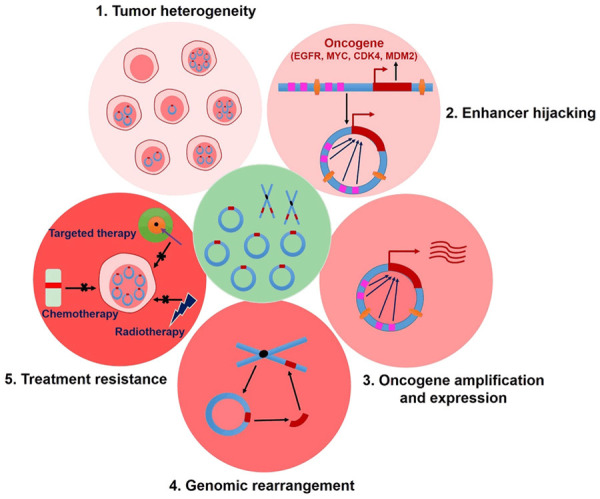
Functions of ecDNA in tumors. ecDNA plays important roles in tumorigenesis and tumor progression by virtue of several approaches, for example, ecDNA relieves heredity constraints and contributes to dynamic cancer evolution; enhancer hijacking and oncogene amplification of ecDNA can accelerate cancer development. Besides, ecDNA is an unanticipated major source of genomic rearrangements in cancer. More importantly, cancer cells potentially evade therapies that targeted oncogenes maintained on ecDNA by a highly specific, dynamic and adaptive pathway and thus induce treatment resistance.
Acknowledgements
This work was supported by the Jiangsu Provincial Medical Innovation Team under Grant CXTDA2017034; National Natural Science Foundation of China under Grant 81972845.
Disclosure of conflict of interest
None.
References
- 1.Hotta Y, Bassel A. Molecular size and circularity of DNA in cells of mammals and higher plants. Proc Natl Acad Sci U S A. 1965;53:356–362. doi: 10.1073/pnas.53.2.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lubs HA Jr, Salmon JH. The chromosomal complement of human solid tumors. Ii. karyotypes of glial tumors. J Neurosurg. 1965;22:160–168. doi: 10.3171/jns.1965.22.2.0160. [DOI] [PubMed] [Google Scholar]
- 3.Cox D, Yuncken C, Spriggs AI. Minute chromatin bodies in malignant tumours of childhood. Lancet. 1965;1:55–58. doi: 10.1016/s0140-6736(65)90131-5. [DOI] [PubMed] [Google Scholar]
- 4.Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, Kornblum HI, Taylor MD, Kaushal S, Cavenee WK, Wechsler-Reya R, Furnari FB, Vandenberg SR, Rao PN, Wahl GM, Bafna V, Mischel PS. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017;543:122–125. doi: 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deshpande V, Luebeck J, Nguyen ND, Bakhtiari M, Turner KM, Schwab R, Carter H, Mischel PS, Bafna V. Exploring the landscape of focal amplifications in cancer using ampliconarchitect. Nat Commun. 2019;10:392. doi: 10.1038/s41467-018-08200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, Liu J, Deshpande V, Rajkumar U, Namburi S, Amin SB, Yi E, Menghi F, Schulte JH, Henssen AG, Chang HY, Beck CR, Mischel PS, Bafna V, Verhaak RGW. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet. 2020;52:891–897. doi: 10.1038/s41588-020-0678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starling S. Cancer genomics: ECdetect hunts extrachromosomal DNA. Nat Rev Genet. 2017;18:212. doi: 10.1038/nrg.2017.13. [DOI] [PubMed] [Google Scholar]
- 8.Bailey C, Shoura MJ, Mischel PS, Swanton C. Extrachromosomal DNA-relieving heredity constraints, accelerating tumour evolution. Ann Oncol. 2020;31:884–893. doi: 10.1016/j.annonc.2020.03.303. [DOI] [PubMed] [Google Scholar]
- 9.Liao Z, Jiang W, Ye L, Li T, Yu X, Liu L. Classification of extrachromosomal circular DNA with a focus on the role of extrachromosomal DNA (ecDNA) in tumor heterogeneity and progression. Biochim Biophys Acta Rev Cancer. 2020;1874:188392. doi: 10.1016/j.bbcan.2020.188392. [DOI] [PubMed] [Google Scholar]
- 10.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, Paucar A, Yang H, Ohashi M, Zhu S, Wykosky J, Reed R, Nelson SF, Cloughesy TF, James CD, Rao PN, Kornblum HI, Heath JR, Cavenee WK, Furnari FB, Mischel PS. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343:72–76. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan Y, Guo G, Huang J, Gao M, Zhu Q, Zeng S, Gong Z, Xu Z. Current understanding of extrachromosomal circular DNA in cancer pathogenesis and therapeutic resistance. J Hematol Oncol. 2020;13:124. doi: 10.1186/s13045-020-00960-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radloff R, Bauer W, Vinograd J. A dye-buoyant-density method for the detection and isolation of closed circular duplex DNA: the closed circular DNA in HeLa cells. Proc Natl Acad Sci U S A. 1967;57:1514–1521. doi: 10.1073/pnas.57.5.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buongiorno-Nardelli M, Amaldi F, Lava-Sanchez PA. Electron microscope analysis of amplifying ribosomal DNA from Xenopus laevis. Exp Cell Res. 1976;98:95–103. doi: 10.1016/0014-4827(76)90467-5. [DOI] [PubMed] [Google Scholar]
- 14.VanDevanter DR, Tseng JC, Yirdaw G. Electrophoretic isolation of extrachromosomal DNA from tumor cells. Genes Chromosomes Cancer. 1995;12:262–271. doi: 10.1002/gcc.2870120405. [DOI] [PubMed] [Google Scholar]
- 15.Jeon Y, Kim SY, Kim M, Park HK, Lee SH, See CJ, Kwon J, Lee DS. Fluorescence in situ hybridization panel for monitoring of minimal residual disease in patients with double minute chromosomes. Blood Cells Mol Dis. 2014;52:208–213. doi: 10.1016/j.bcmd.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Pu L, Lin Y, Pevzner PA. Detection and analysis of ancient segmental duplications in mammalian genomes. Genome Res. 2018;28:901–909. doi: 10.1101/gr.228718.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moller HD, Bojsen RK, Tachibana C, Parsons L, Botstein D, Regenberg B. Genome-wide purification of extrachromosomal circular DNA from eukaryotic cells. J Vis Exp. 2016:e54239. doi: 10.3791/54239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khatami F, Larijani B, Tavangar SM. The presence of tumor extrachomosomal circular DNA (ecDNA) as a component of liquid biopsy in blood. Med Hypotheses. 2018;114:5–7. doi: 10.1016/j.mehy.2018.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Cohen S, Regev A, Lavi S. Small polydispersed circular DNA (spcDNA) in human cells: association with genomic instability. Oncogene. 1997;14:977–985. doi: 10.1038/sj.onc.1200917. [DOI] [PubMed] [Google Scholar]
- 20.Jones RS, Potter SS. L1 sequences in HeLa extrachromosomal circular DNA: evidence for circularization by homologous recombination. Proc Natl Acad Sci U S A. 1985;82:1989–1993. doi: 10.1073/pnas.82.7.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misra R, Matera AG, Schmid CW, Rush MG. Recombination mediates production of an extrachromosomal circular DNA containing a transposon-like human element, THE-1. Nucleic Acids Res. 1989;17:8327–8341. doi: 10.1093/nar/17.20.8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Loon N, Miller D, Murnane JP. Formation of extrachromosomal circular DNA in HeLa cells by nonhomologous recombination. Nucleic Acids Res. 1994;22:2447–2452. doi: 10.1093/nar/22.13.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hull RM, King M, Pizza G, Krueger F, Vergara X, Houseley J. Transcription-induced formation of extrachromosomal DNA during yeast ageing. PLoS Biol. 2019;17:e3000471. doi: 10.1371/journal.pbio.3000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng X, Qi X, Guo H, Cai M, Li C, Zhu J, Chen F, Guo H, Li J, Zhao Y, Liu P, Jia X, Yu J, Zhang C, Sun W, Yu Y, Jin Y, Bai J, Wang M, Rosales J, Lee KY, Fu S. Novel role for non-homologous end joining in the formation of double minutes in methotrexate-resistant colon cancer cells. J Med Genet. 2015;52:135–144. doi: 10.1136/jmedgenet-2014-102703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai M, Zhang H, Hou L, Gao W, Song Y, Cui X, Li C, Guan R, Ma J, Wang X, Han Y, Lv Y, Chen F, Wang P, Meng X, Fu S. Inhibiting homologous recombination decreases extrachromosomal amplification but has no effect on intrachromosomal amplification in methotrexate-resistant colon cancer cells. Int J Cancer. 2019;144:1037–1048. doi: 10.1002/ijc.31781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wahl GM. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989;49:1333–1340. [PubMed] [Google Scholar]
- 27.Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, Zhang W, Jameson N, Corces MR, Granja JM, Chen X, Coruh C, Abnousi A, Houston J, Ye Z, Hu R, Yu M, Kim H, Law JA, Verhaak RGW, Hu M, Furnari FB, Chang HY, Ren B, Bafna V, Mischel PS. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019;575:699–703. doi: 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanda T, Otter M, Wahl GM. Mitotic segregation of viral and cellular acentric extrachromosomal molecules by chromosome tethering. J Cell Sci. 2001;114:49–58. doi: 10.1242/jcs.114.1.49. [DOI] [PubMed] [Google Scholar]
- 29.Von Hoff DD, Needham-VanDevanter DR, Yucel J, Windle BE, Wahl GM. Amplified human MYC oncogenes localized to replicating submicroscopic circular DNA molecules. Proc Natl Acad Sci U S A. 1988;85:4804–4808. doi: 10.1073/pnas.85.13.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ott CJ. Circles with a point: new insights into oncogenic extrachromosomal DNA. Cancer Cell. 2020;37:145–146. doi: 10.1016/j.ccell.2020.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Tandon I, Pal R, Pal JK, Sharma NK. Extrachromosomal circular DNAs: an extra piece of evidence to depict tumor heterogeneity. Future Sci OA. 2019;5:FSO390. doi: 10.2144/fsoa-2019-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 33.Khoo BL, Chaudhuri PK, Ramalingam N, Tan DS, Lim CT, Warkiani ME. Single-cell profiling approaches to probing tumor heterogeneity. Int J Cancer. 2016;139:243–255. doi: 10.1002/ijc.30006. [DOI] [PubMed] [Google Scholar]
- 34.Di J, Yang H, Jiang B, Wang Z, Ji J, Su X. Whole exome sequencing reveals intertumor heterogeneity and distinct genetic origins of sporadic synchronous colorectal cancer. Int J Cancer. 2018;142:927–939. doi: 10.1002/ijc.31140. [DOI] [PubMed] [Google Scholar]
- 35.deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, Koeman J, Seth S, Protopopov A, Felicella M, Zheng S, Multani A, Jiang Y, Zhang J, Nam DH, Petricoin EF, Chin L, Mikkelsen T, Verhaak RGW. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet. 2018;50:708–717. doi: 10.1038/s41588-018-0105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu K, Ding L, Chang TC, Shao Y, Chiang J, Mulder H, Wang S, Shaw TI, Wen J, Hover L, McLeod C, Wang YD, Easton J, Rusch M, Dalton J, Downing JR, Ellison DW, Zhang J, Baker SJ, Wu G. Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol. 2019;137:123–137. doi: 10.1007/s00401-018-1912-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verhaak RGW, Bafna V, Mischel PS. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer. 2019;19:283–288. doi: 10.1038/s41568-019-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, Shih DJ, Hovestadt V, Zapatka M, Sturm D, Jones DT, Kool M, Remke M, Cavalli FM, Zuyderduyn S, Bader GD, VandenBerg S, Esparza LA, Ryzhova M, Wang W, Wittmann A, Stark S, Sieber L, Seker-Cin H, Linke L, Kratochwil F, Jager N, Buchhalter I, Imbusch CD, Zipprich G, Raeder B, Schmidt S, Diessl N, Wolf S, Wiemann S, Brors B, Lawerenz C, Eils J, Warnatz HJ, Risch T, Yaspo ML, Weber UD, Bartholomae CC, von Kalle C, Turanyi E, Hauser P, Sanden E, Darabi A, Siesjo P, Sterba J, Zitterbart K, Sumerauer D, van Sluis P, Versteeg R, Volckmann R, Koster J, Schuhmann MU, Ebinger M, Grimes HL, Robinson GW, Gajjar A, Mynarek M, von Hoff K, Rutkowski S, Pietsch T, Scheurlen W, Felsberg J, Reifenberger G, Kulozik AE, von Deimling A, Witt O, Eils R, Gilbertson RJ, Korshunov A, Taylor MD, Lichter P, Korbel JO, Wechsler-Reya RJ, Pfister SM. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haller F, Bieg M, Will R, Korner C, Weichenhan D, Bott A, Ishaque N, Lutsik P, Moskalev EA, Mueller SK, Bahr M, Woerner A, Kaiser B, Scherl C, Haderlein M, Kleinheinz K, Fietkau R, Iro H, Eils R, Hartmann A, Plass C, Wiemann S, Agaimy A. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat Commun. 2019;10:368. doi: 10.1038/s41467-018-08069-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055–4060. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O’Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L, Network TR. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, Piazza MS, Allan KC, Mack SC, Wang X, Gimple RC, Wu Q, Rubin BP, Shetty S, Angers S, Dirks PB, Sallari RC, Lupien M, Rich JN, Scacheri PC. Functional enhancers shape extrachromosomal oncogene amplifications. Cell. 2019;179:1330–1341. e1313. doi: 10.1016/j.cell.2019.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alt FW, Kellems RE, Bertino JR, Schimke RT. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem. 1978;253:1357–1370. [PubMed] [Google Scholar]
- 46.Knudson AG. Chasing the cancer demon. Annu Rev Genet. 2000;34:1–19. doi: 10.1146/annurev.genet.34.1.1. [DOI] [PubMed] [Google Scholar]
- 47.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stutz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, Efthymiopoulos T, Erkek S, Siegl C, Brenner H, Brustugun OT, Dieter SM, Northcott PA, Petersen I, Pfister SM, Schneider M, Solberg SK, Thunissen E, Weichert W, Zichner T, Thomas R, Peifer M, Helland A, Ball CR, Jechlinger M, Sotillo R, Glimm H, Korbel JO. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49:65–74. doi: 10.1038/ng.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luebeck J, Coruh C, Dehkordi SR, Lange JT, Turner KM, Deshpande V, Pai DA, Zhang C, Rajkumar U, Law JA, Mischel PS, Bafna V. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nat Commun. 2020;11:4374. doi: 10.1038/s41467-020-18099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajkumar U, Turner K, Luebeck J, Deshpande V, Chandraker M, Mischel P, Bafna V. EcSeg: semantic segmentation of metaphase images containing extrachromosomal DNA. iScience. 2019;21:428–435. doi: 10.1016/j.isci.2019.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou H, Feng R, Huang Y, Tripodi J, Najfeld V, Tsankova NM, Jahanshahi M, Olson LE, Soriano P, Friedel RH. Double minute amplification of mutant PDGF receptor alpha in a mouse glioma model. Sci Rep. 2015;5:8468. doi: 10.1038/srep08468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16:483–493. doi: 10.1038/nrc.2016.62. [DOI] [PubMed] [Google Scholar]
- 53.Chen H, Li C, Peng X, Zhou Z, Weinstein JN Cancer Genome Atlas Research Network. Liang H. A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell. 2018;173:386–399. e312. doi: 10.1016/j.cell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greer SU, Nadauld LD, Lau BT, Chen J, Wood-Bouwens C, Ford JM, Kuo CJ, Ji HP. Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome Med. 2017;9:57. doi: 10.1186/s13073-017-0447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 56.Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, Li X, Gumin J, Zheng H, Hu L, Yli-Harja O, Haapasalo H, Visakorpi T, Liu X, Liu CG, Sawaya R, Fuller GN, Chen K, Lang FF, Nykter M, Zhang W. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Invest. 2013;123:855–865. doi: 10.1172/JCI67144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vogt N, Gibaud A, Lemoine F, de la Grange P, Debatisse M, Malfoy B. Amplicon rearrangements during the extrachromosomal and intrachromosomal amplification process in a glioma. Nucleic Acids Res. 2014;42:13194–13205. doi: 10.1093/nar/gku1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, Chamorro R, Munoz-Perez N, Puiggros M, Dorado Garcia H, Bei Y, Roefzaad C, Bardinet V, Szymansky A, Winkler A, Thole T, Timme N, Kasack K, Fuchs S, Klironomos F, Thiessen N, Blanc E, Schmelz K, Kunkele A, Hundsdorfer P, Rosswog C, Theissen J, Beule D, Deubzer H, Sauer S, Toedling J, Fischer M, Hertwig F, Schwarz RF, Eggert A, Torrents D, Schulte JH, Henssen AG. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet. 2020;52:29–34. doi: 10.1038/s41588-019-0547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov. 2020;19:39–56. doi: 10.1038/s41573-019-0044-1. [DOI] [PubMed] [Google Scholar]
- 60.Alvarez-Calderon F, Gregory MA, DeGregori J. Using functional genomics to overcome therapeutic resistance in hematological malignancies. Immunol Res. 2013;55:100–115. doi: 10.1007/s12026-012-8353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Banerjee D, Mayer-Kuckuk P, Capiaux G, Budak-Alpdogan T, Gorlick R, Bertino JR. Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim Biophys Acta. 2002;1587:164–173. doi: 10.1016/s0925-4439(02)00079-0. [DOI] [PubMed] [Google Scholar]
- 62.Morales C, Ribas M, Aiza G, Peinado MA. Genetic determinants of methotrexate responsiveness and resistance in colon cancer cells. Oncogene. 2005;24:6842–6847. doi: 10.1038/sj.onc.1208834. [DOI] [PubMed] [Google Scholar]