

Abstract
A large variety of fusion tags have been developed to improve protein expression, solubilization, and purification. Nevertheless, these tags have been combined in a rather limited number of composite tags and usually these composite tags have been dictated by traditional commercially‐available expression vectors. Moreover, most commercially‐available expression vectors include either N‐ or C‐terminal fusion tags but not both. Here, we introduce TSGIT, a fusion‐tag system composed of both N‐ and a C‐terminal composite fusion tags. The system includes two affinity tags, two solubilization tags and two cleavable tags distributed at both termini of the protein of interest. Therefore, the N‐ and the C‐terminal composite fusion tags in TSGIT are fully orthogonal in terms of both affinity selection and cleavage. For using TSGIT, we streamlined the cloning, expression, and purification procedures. Each component tag is selected to maximize its benefits toward the final construct. By expressing and partially purifying the protein of interest between the components of the TSGIT fusion, the full‐length protein is selected over truncated forms, which has been a long‐standing problem in protein purification. Moreover, due to the nature of the cleavable tags in TSGIT, the protein of interest is obtained in its native form without any additional undesired N‐ or C‐terminal amino acids. Finally, the resulting purified protein is ready for efficient ligation with other proteins or peptides for downstream applications. We demonstrate the use of this system by purifying a large amount of native fluorescent mRuby3 protein and bacteriophage T7 gp2.5 ssDNA‐binding protein.
Keywords: biotin, fusion tag, Intein, IPL, protein cleavage, protein degradation, protein expression, protein ligation, purification tag, SUMO, truncated protein
1. INTRODUCTION
The use of composite tags fused to a protein of interest to improve protein expression, solubility, and purification has gained increasing popularity in protein science. 1 , 2 , 3 , 4 Despite their advantages, fusion tags can introduce a various number of undesired amino acids to the N‐terminus and/or to the C‐terminus in the protein of interest. To solve this problem, cleavable fusion tags have been developed. 5 , 6 The nature of these cleavable tags however limits their usability to either the N‐ or the C‐terminus. Moreover, cleavage of the vast majority of these tags, for example the ones derived from the sequence‐specific TEV technology, 7 leaves undesired amino acids that may interfere with protein function.
Composite fusion tags typically include a mixture of one or more small affinity tags, an optional solubilization tag and a cleavable tag. Ideally, the cleavable tag is located immediately after the protein of interest to reduce the number of amino acids leftover after cleavage. In general, N‐terminal fusion tags leave a smaller number of amino acids than their C‐terminal counterparts. 5 , 6 , 7 Nevertheless, in practice, it can often be beneficial to include both N‐ and C‐terminal tags. The main motivation for this approach is to select for full‐length proteins over truncated and degraded protein forms, 8 which have caused a long‐standing problem in protein purification. Three main mechanisms can generate these unwanted truncated protein forms. First, they can be generated directly during protein expression and in some cases the truncated form remains soluble and consequently can be carried on to the purification step. 9 , 10 , 11 This scenario increases dramatically when the protein of interest is also fused to a solubilization tag. Second, proteolytic cleavage can occur at various positions inside the protein's backbone during expression or even during purification. 12 , 13 , 14 Third, based on the protein's structure and its amino acid sequence, sequential degradation of the N‐terminus 14 , 15 , 16 or C‐terminus 14 , 17 , 18 can occur. Depending on the amount of similarity between the full‐length protein and its truncated forms, the truncated forms may or may not be removed during purification. 8 , 9 , 10 , 11
To overcome the aforementioned limitations, we developed a system that employs two different composite tags at the N‐ and C‐termini, each containing a different affinity tag. Sequential selection for the N‐ and then the C‐terminal affinity tags would therefore select for the full‐length protein over its truncated versions. Moreover, we combined these affinity tags with cleavable tags to allow for their removal. To achieve these versatile features, we focused on selecting optimal composite tags that would maximize the benefits of each individual tag and also of the construct as a whole.
For the N‐terminal part of our fusion system, we selected a double His6‐tag (two consecutive hexa‐histidine tags connected by a glycine2‐serine2 flexible linker) 19 , 20 as the purification tag, Trx (Thioredoxin) 21 , 22 as the solubilization tag and SUMO (small ubiquitin‐like modifier, Smt3p) 23 , 24 , 25 as the cleavable tag. His‐tag‐capturing media exhibits high dynamic binding capacity 26 and therefore can capture large amounts of the protein of interest, whereas the SUMO‐tag is efficiently cleaved via Ulp1 SUMO protease without leaving any additional N‐terminal amino acids. The redundancy in adding both Trx and SUMO at the N‐terminus may have an additive effect on enhancing protein expression and solubility. 24 , 27 For the C‐terminal part of the fusion system, we selected a Twin Strep‐tag (two consecutive Strep‐tag IIs connected by an optimized flexible linker) 28 , 29 as the purification tag, Trx as the solubilization tag and a contiguous mini‐intein 30 , 31 , 32 , 33 , 34 (optimized Mxe GyrA intein containing N198A mutation; this mutant version will be referred to as Intein for simplicity) as the cleavable tag. The Twin Strep‐tag exhibits high specificity and results in sharp chromatographic peaks, which makes its elution ideal for subsequent purification steps. The Intein‐tag is efficiently cleaved by reducing agents without leaving any additional C‐terminal amino acids. Moreover, SUMO and Intein cleavage can proceed simultaneously for optimizing the duration of the protocol. In addition, the use of the Intein‐tag allows the target protein to be ready for intein‐mediated protein ligation (IPL). 30 , 31 , 32 We demonstrate the use of this system, which we called TSGIT for Trx‐SUMO‐Gene of interest‐Intein‐Trx, by expressing and purifying native mRuby3 fluorescent protein 35 and bacteriophage T7 ssDNA‐binding protein gp2.5. 36 We also streamlined TSGIT's cloning, expression and purification protocols for maximum ease of use and time efficiency.
2. RESULTS AND DISCUSSION
We tested our composite TSGIT system in five steps. Initially, we verified if the N‐ and C‐terminal parts of the fusion expressed correctly (Figure 1). Second, we purified mRuby3 fluorescent protein using this optimized system as an example (Figure 3). Next, we verified that the IPL reaction proceeded efficiently for the purified mRuby3 (Figure 3). Further, we monitored mRuby3 fluorescence using an energy‐transfer assay that involved confirming both the functionality of mRuby3 through its fluorescence and the presence of the biotin moiety attached through IPL (Figure 4). Finally, we purified gp2.5 using a tag‐free protocol and using the TSGIT system, and showed that TSGIT had no effect on its oligomerization and activity (Figures 5 and 6).
FIGURE 1.
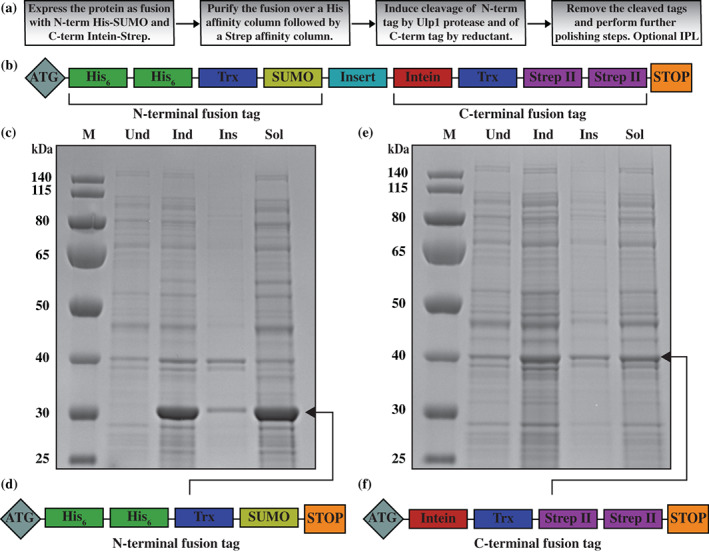
Design of the TSGIT system. (a) Workflow diagram for protein expression and purification using TSGIT. (b) Schematic representation of component tags of the TSGIT fusion system. Induction and solubility controls for the (c) N‐terminal and (e) C‐terminal parts of TSGIT. 10% SDS‐PAGE gels showing the uninduced (Und), induced (Ind), insoluble (Ins), and soluble (Sol) samples for TSGIT‐N and TSGIT‐C. The marker (M) is PageRuler Prestained Protein Ladder. Schematic representation of the components of the independently tested (d) N‐terminal and (f) C‐terminal fusion tags
2.1. Design of the TSGIT system
The general scheme for using TSGIT is shown in Figure 1a; a schematic representation and an amino acid sequence of TSGIT are provided in Figure 1b and Figure 2a, respectively. Several points were taken into consideration during the design of the fusion system. First, both the His and Strep affinity tags were included in their double form to improve the purification step (Figure 1b). Second, a variety of flexible linkers were introduced between the individual elements of the fusion to increase the exposure and accessibility of each tag (Figure 2a). Third, Trx‐tags were introduced to both the N‐ and C‐terminal parts of the fusion to ensure maximum solubility of these parts and of the fusion as a whole (Figures 1b and 2a). Finally, SUMO and Intein are used as cleavable tags since they are two well‐characterized structure‐specific cleavable tags that do not leave additional undesired amino acids on the target protein after cleavage (traceless cleavage). The sequence encoding the gene of interest can be assembled together with the N‐ and C‐terminal fusion tags of TSGIT using Gibson assembly cloning between the SUMO and Intein regions into any desired destination plasmid (Figure 2a).
FIGURE 2.
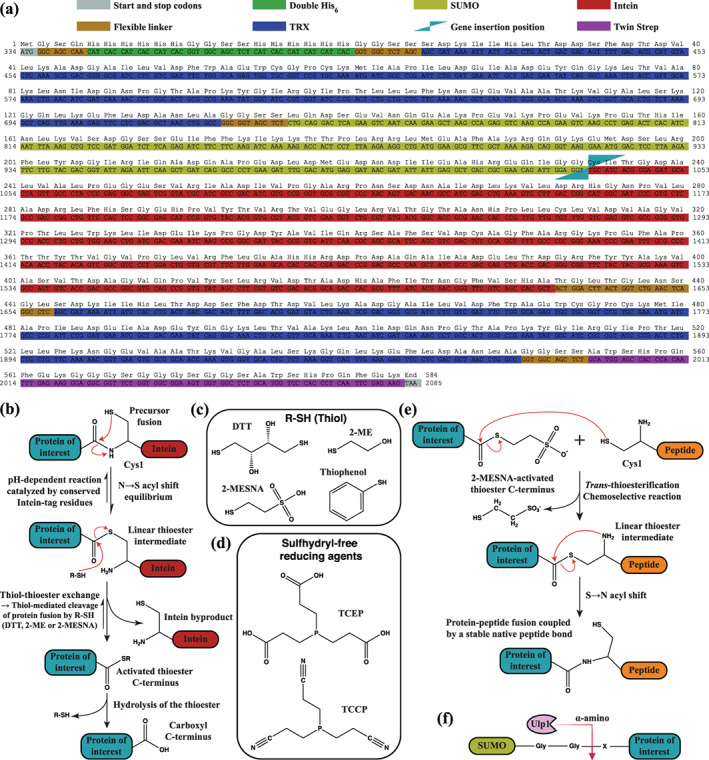
Amino acid sequence of empty TSGIT and cleavage mechanisms of its component cleavable tags. (a) The amino acid sequence and the corresponding nucleotide sequence are presented in FASTA format. The different tags that compose TSGIT are identified by their corresponding color code. The sequence encoding the protein of interest is inserted between the SUMO and Intein regions. (b) Schematic representation of the cascade of chemical reactions that leads to the cleavage of the C‐terminal fusion tag via traceless Intein cleavage at its N‐terminal junction; based on the reaction schemes presented in 30 , 61 . (c) Chemical structures of commonly employed thiol reagents for Intein‐tag cleavage at its N‐terminal junction. (d) Chemical structures of sulfhydryl‐free reducing agents. (e) Schematic representation of the IPL reaction between a C‐terminal 2‐MESNA‐activated protein of interest and a peptide that contains an N‐terminal cysteine residue. Additional details about thiol‐mediated Intein cleavage and IPL can be found in Supporting Information Material. (f) Schematic representation of the traceless cleavage of the SUMO‐tag and therefore of the N‐terminal fusion tag by the specific SUMO protease Ulp1 between the first amino acid of the protein of interest (X) and the second glycine residue of the C‐terminal diglycine motif of SUMO
The N‐terminal SUMO cleavable tag was selected due to its small size and its efficient traceless cleavage reaction at its C‐terminal junction (Figure 2f), which occurs over a wide range of buffer conditions and relatively independent of the N‐terminal amino acid sequence of the target protein of interest. 23 , 24 , 25 N‐terminal intein‐fusions could also be considered since their C‐terminal cleavage does not leave any additional undesired amino acids on the target protein. Nevertheless, as opposed to SUMO, they present several drawbacks. Thiol‐cleavable inteins at their C‐terminal junction such as the Sce VMA intein are much larger in size. The smaller Ssp DnaB intein, on the other hand, is cleaved by pH and temperature shift rather than reductants, 33 which might be unsuitable for certain proteins. As for most inteins, their cleavage efficiency is influenced by the terminal amino acids of the target protein. 32 , 33 Moreover, another limitation is imposed by their premature cleavage in an undesired fashion in vivo during protein expression. 32 , 33 Premature cleavage of inteins can be avoided by using split inteins (reviewed in 37 , 38 ) rather than contiguous inteins. However, split inteins introduce other specific limitations such as low cleavage rates, low solubility and inefficient assembly of the two split intein parts. 37 Finally, the N‐ and C‐terminal combinations of the Ssp DnaB and Mxe GyrA or Mth RIR1 inteins (implemented in the pTWIN1 and pTWIN2 expression vectors, New England BioLabs) have slightly different optimal cleavage pH preferences, which renders their efficient cleavage to be performed sequentially rather than simultaneously. 33
For the C‐terminal part of the fusion, two complementary small‐size contiguous inteins can be used depending on the last amino acid of the target protein: Mxe GyrA and Mth RIR1. 33 Some C‐terminal amino acids of the target result in better intein cleavage than others. 33 These two inteins are cleaved at their N‐terminal junction (Figure 2b) by sulfhydryl‐containing reagents (thiols, Figure 2c), among which 2‐Mercaptoethanesulfonic acid (2‐MESNA) and Thiophenol are preferred if a subsequent IPL reaction is required (Figure 2e). 30 , 32 While some of the aforementioned general drawbacks of inteins are also true for Mxe GyrA and Mth RIR1, these represent the most competitive contiguous tag options available for traceless C‐terminal cleavable tags. We incorporated Mxe GyrA rather than Mth RIR1 in our TSGIT design, since its cleavage reaction occurs with a relatively high efficiency for C‐terminal lysine or phenylalanine residues, 32 such as the ones in our mRuby3 (Figure S2) or gp2.5 (Figure S3) target proteins. As most inteins, naturally occurring Mxe GyrA 39 can undergo cleavage at both of its termini. To block cleavage at its C‐terminal junction, Mxe GyrA was engineered to carry the previously described 30 N198A mutation (Figure 2a). Removal of the C‐terminal cyclization‐capable asparagine residue blocks intermediate succinimide formation 30 and consequently the cleavage at the Intein‐tag C‐terminal junction.
We next proceeded to verify the expression and solubility levels of the individual N‐ and C‐terminal parts of TSGIT. Cultures of Escherichia coli transformed with pTSGIT‐N and pTSGIT‐C were prepared as described in Section 4. Upon induction of expression of pTSGIT‐N, the N‐terminal part of TSGIT (Figure 1d), consisting of a double His‐tag, Trx, and SUMO, was immediately visible as a strong band in SDS‐PAGE (Figure 1c). After cell lysis and separation of the soluble and insoluble fractions, this part of the fusion was found to be mostly in the soluble form as desired. The same observations are made about the C‐terminal part of the fusion (Figure 1f), consisting of Intein, Trx, and Twin Strep (Figure 1e). Nevertheless, the expression level of the TSGIT C‐terminal part was relatively lower than that of the N‐terminal part, suggesting that the N‐terminal Trx and SUMO tags were efficient in driving upward the expression level.
Together, these experiments showed that both the N‐ and C‐terminal parts of TSGIT were not limiting for protein expression and solubility. The high expression level of the N‐terminal part of the fusion was particularly desirable, as it could potentially drive forward the expression of the whole fusion. Moreover, the solubility of the individual parts of the fusion was also critical, both during the protein extraction phase and during purification, especially after cleavage. If the tags were insoluble on their own, cleavage might result in protein precipitation, which would negatively interfere with the downstream protein purification steps. It is worth noting that in cases where the protein of interest is large and reduction of the size of the construct is needed, the size of the fusion can be reduced by sequential removal of the Trx‐tags, starting with the N‐terminal one since SUMO can already cover on its own some of the benefits offered by Trx. 24 , 25 If further reduction is needed, the His and Strep affinity tags can be reduced from their double to single versions.
2.2. Purification of IPL‐ready mRuby3 using the TSGIT system
While SUMO has been used in a variety of applications in combination with His‐tag, the C‐terminal Intein, especially in commercially available plasmids, is often fused to a Chitin‐binding domain tag (CBD‐tag). 40 , 41 Although the CBD‐tag is suitable for on‐resin bulk cleavage or denaturing elution conditions, its relatively strong binding makes it unsuitable for FPLC‐based protein purification under native conditions when on‐column cleavage is not desirable. To build a purification scheme that was fully FPLC‐compatible, we replaced this tag with Twin Strep. The resulting purification scheme based on chromatography with His‐ and Strep‐tags is shown in Figure 3a. To demonstrate the success of this scheme, we first purified mRuby3 fluorescent protein using TSGIT.
FIGURE 3.
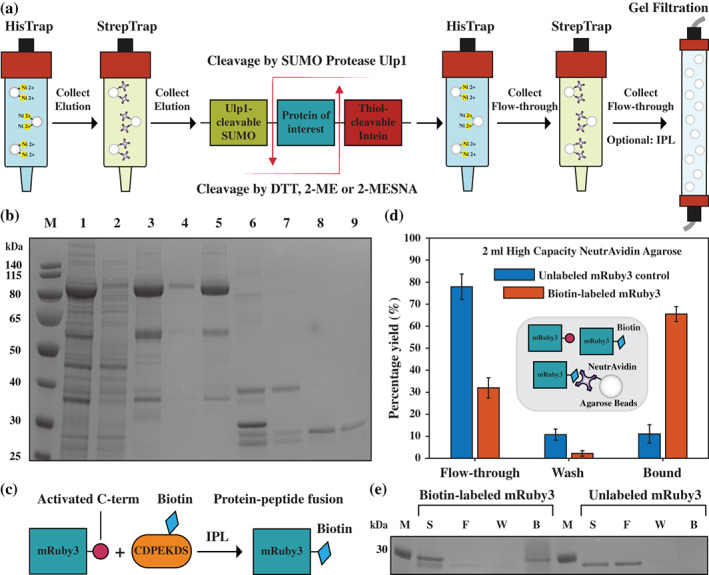
Purification of mRuby3 by employing TSGIT. (a) A schematic of the procedure employed for purification of mRuby3 using TSGIT. (b) Image of a 10% SDS‐PAGE gel showing the different steps of purification of mRuby3 starting from E. colily sate containing the expression product of the pTSGIT‐mRuby3 expression vector: Lane 1, soluble fraction of the lysate; Lane 2, flow‐through of the first HisTrap affinity column; Lane 3, TSGIT‐mRuby3 elution from the HisTrap affinity column; Lane 4, flow‐through of the first StrepTrap affinity column; Lane 5, elution of TSGIT‐mRuby3 from StrepTrap affinity column; Lane 6, release of mRuby3 from the TSGIT‐mRuby3 fusion via SUMO protease and 2‐MESNA cleavage; Lane 7, flow‐through of cleaved mRuby3 through the second HisTrap affinity column; Lane 8, flow‐through of cleaved mRuby3 through the second StrepTrap affinity column; Lane 9, mRuby3 after Superdex 75 pg size‐exclusion elution. The marker (M) is PageRuler Prestained Protein Ladder. (c) Schematic representation of the IPL reaction of activated mRuby3 produced by TSGIT and the biotin‐containing BioP peptide. (d) Bar chart illustrating the percentage of flow‐through, wash and bound fractions of unlabeled and IPL biotin‐labeled mRuby3 from NeutrAvidin resin. The inset schematic shows the three coexisting populations: unlabeled mRuby3, free and NeutrAvidin‐bound biotin‐labeled mRuby3. (e) Image of an SDS‐PAGE gel showing the sample (S), flow‐through (F), wash (W), and bound (B) fractions of unlabeled and IPL biotin‐labeled mRuby3 from NeutrAvidin resin. The marker (M) is the 30 kDa band of PageRuler Prestained Protein Ladder
We included 0.1 mM TCEP in the lysis and purification buffers in order to maintain a reducing environment prior to initiating Intein‐tag cleavage by the addition of thiol reagents (Figure 2b,c). Sulfhydryl‐free reducing agents such as TCEP and TCCP (Figure 2d) can provide efficient reducing conditions to prevent undesired disulfide bond formation, while their lack of sulfhydryl groups ensures that Intein cleavage reaction is not initiated prematurely. On the other hand, most of the thiols used to initiate the Intein cleavage reaction are also reducing agents (Figure 2c) that will provide reducing conditions during the cleavage reaction.
The soluble fraction of the cell lysates for TSGIT‐mRuby3 (Lane 1 in Figure 3b) was applied at a flow rate of 3 ml/min onto a 5‐ml HisTrap affinity column pre‐equilibrated with buffer A. This column was then washed with 15 column volumes (CV) of buffer A at a flow rate of 5 ml/min. The protein fusions were eluted with a 15 CV linear gradient of buffer B from 30 mM to 350 mM Imidazole (gradient of buffer B at a flow rate of 5 ml/min). The protein fusion eluted in a peak centered at ~210 mM Imidazole concentration (Lane 3 in Figure 3b). The eluted protein was then applied at a flow rate of 2 ml/min onto a 5‐ml StrepTrap affinity column pre‐equilibrated with buffer C. This column was then washed with 15 CV of buffer C at a flow rate of 5 ml/min. The protein fusions were eluted with a 15 CV linear gradient of buffer C from 0 to 2.5 mM d‐Desthiobiotin (gradient of buffer D at a flow rate of 5 ml/min). The protein fusion eluted in a peak centered at ~0.85 mM d‐Desthiobiotin concentration (Lane 5 in Figure 3b).
The protein was then concentrated to 2.5 ml using 10 kDa cut‐off spin concentrators. Two PD‐10 desalting columns were equilibrated three times with 2‐MESNA‐cotnaining cleavage buffer. The concentrated protein was then passed through the PD‐10 columns for rapid exchange to the cleavage buffer to initiate cleavage of the C‐terminal part of the fusion via Intein. Cleavage of the N‐terminal SUMO‐containing part was initiated by the addition of His‐tagged SUMO protease Ulp1; the purification of the His‐tagged SUMO protease Ulp1 (Figure S1) is described in Supporting Information Materials and Methods section. Cleavage was allowed to continue overnight at 4°C with gentle rotation.
Following incubation, cleavage was confirmed by SDS‐PAGE (Lane 6 in Figure 3b) and 2‐MESNA was removed by rapid buffer exchange to buffer A via two PD‐10 columns. The cleaved His‐tagged N‐terminal part of the fusion, the His‐tagged SUMO protease and any N‐terminal uncleaved products were removed by passing the cleaved protein products through a second HisTrap affinity column and collecting the flow‐through (Lane 7 in Figure 3b). The cleaved Strep‐tagged C‐terminal part of the fusion and any C‐terminal uncleaved products were removed by passing the cleaved protein products through a second StrepTrap affinity column and collecting the flow‐through (Lane 8 in Figure 3b).
The flow‐through of this second StrepTrap affinity column contained more than 90% pure mRuby3 with both N‐ and C‐terminal parts of the fusion cleaved and removed from the protein. The yield of the purified mRuby3 was ~2.7 ± 0.2 mg from 50 ml of resuspended expression cells. The yields and purities of the protein obtained from each purification step are summarized in Table S1. Following quantification, the protein prep was split into two equal fractions. One fraction was concentrated to 1 ml using 10 kDa cut‐off spin concentrators and applied at a flow rate of 1 ml/min to a 120 ml Superdex 75 pg size‐exclusion column that was pre‐equilibrated with 1.5 CV of storage buffer. The pure fractions were selected (Lane 9 in Figure 3b), concentrated to 1 ml using 10 kDa cut‐off spin concentrators and flash‐frozen for subsequent experiments. This size‐exclusion step increased the purity of the protein to greater than 95%. The functionality of the protein was tested by verifying its emission spectra (Figure S4a). This protein served as the negative control for the subsequent experiments involving the IPL‐treated protein. We denoted it as unlabeled mRuby3.
The second fraction was used to verify whether the purified mRuby3 was IPL‐ready, as offered post‐cleavage by the C‐terminal Intein‐tag cleavage mechanism. 32 , 33 Cleavage of this tag, especially in the presence of 2‐MESNA as reducing agent, generates a reactive C‐terminus for the protein of interest that can efficiently react with a peptide or protein containing an N‐terminal cysteine residue forming a continuous backbone (Figures 2e and 3c). To illustrate this IPL reaction for the proteins generated by TSGIT, we chose to fuse mRuby3 to a small BioP peptide, containing a modified lysine residue fused to a biotin moiety and an N‐terminal cysteine residue for attachment.
The second half of the fraction obtained from the flow‐through of the second StrepTrap affinity column was concentrated to 2.5 ml using 10 kDa cut‐off spin concentrators and buffer was rapidly exchanged to IPL buffer by using two PD‐10 columns. The concentration of the protein was adjusted to 10 μM with IPL buffer and 1 mM BioP peptide in IPL buffer was then added. The IPL reaction was allowed to continue overnight at 4°C with gentle rotation. Following incubation, the excess unreacted peptide was removed by applying the reaction mixture at a flow rate of 1 ml/min onto a 120 ml Superdex 75 pg size‐exclusion column that was pre‐equilibrated with 1.5 CV of storage buffer. The pure fractions were selected, concentrated to 1 ml using 10 kDa cut‐off spin concentrators and flash‐frozen for subsequent experiments. The functionality of the protein was tested by verifying its emission spectra (Figure S4b). We denoted this protein as biotin‐labeled mRuby3.
To test the efficiency of the IPL reaction and therefore the addition of the biotin moiety to mRuby3, two preparations of 2 ml of NeutrAvidin Agarose resin were equilibrated three times with storage buffer; each time, the resin was settled by centrifugation and the supernatant was discarded. Solutions of 1 ml of 1 μM unlabeled mRuby3 and biotin‐labeled mRuby3 were freshly prepared from the protein stocks in storage buffer. These protein solutions were then incubated with the NeutrAvidin Agarose resin for 2 hr at 4°C. The flow‐through was separated by centrifugation. The resin containing the bound protein fusions was washed with storage buffer and then separated by centrifugation.
With the exception of the bound fraction, the yield of protein in each fraction was quantified by NanoDrop reading of A280 and converted to a percentage of the starting protein yield. The percentage yield for the bound fraction was estimated by subtracting the yield of the flow‐through and wash fractions from the starting fraction because the presence of the large Agarose beads can interfere with its direct absorbance reading (Figure 3d). Biotin‐labeled mRuby3 bound with ~65% efficiency to the NeutrAvidin Agarose resin, while the unlabeled mRuby3 control bound with only ~10% efficiency probably through nonspecific binding. This binding enhancement clearly demonstrated the presence of the biotin moiety in the biotin‐labeled mRuby3 due to the IPL reaction. Moreover, the same results could be directly visualized using SDS‐PAGE (Figure 3e). Additionally, SDS‐PAGE showed that the ~35% of unbound biotin‐labeled mRuby3 was generated by an incomplete IPL reaction. The IPL reaction and Intein cleavage have been previously shown to be strongly dependent on the C‐terminal amino acid of the protein of interest. 32 , 42 Moreover, the efficiency of cleavage and IPL were shown to be able to increase with longer (up to 40 hr) incubation times. Nevertheless, we intentionally restricted the IPL reaction time to only 12 h to capture this limitation.
To directly test the fluorescence functionality of mRuby3 purified using TSGIT and the efficiency of the IPL reaction, we next designed an assay based on sensitized emission Förster resonance energy transfer (FRET) 43 between mRuby3 and NeutrAvidinDyLight650. DyLight650 was selected as acceptor since its excitation spectrum exhibits significant overlap with the emission spectrum of mRuby3. Solutions of 150 μl of 100 nM unlabeled mRuby3 and biotin‐labeled mRuby3 were freshly prepared from the protein stocks in storage buffer. Solutions of 150 μl of various concentrations (10–1,000 nM monomer) of NeutrAvidinDyLight650 were freshly prepared from protein stock in storage buffer. To account for the direct excitation at 520 nm of NeutrAvidinDyLight650, the emission spectra of the solutions of various concentrations, mixed in equal volume with storage buffer, were recorded (Figure 4a). Next, the solutions of unlabeled mRuby3 and biotin‐labeled mRuby3 were mixed with solutions of various concentrations of NeutrAvidinDyLight650. The emission spectra were collected for each sample, corrected for the direct excitation of NeutrAvidinDyLight650 at the given concentration and normalized to a total area of 1 A.U., as described in Supporting Information Materials and Methods section.
FIGURE 4.
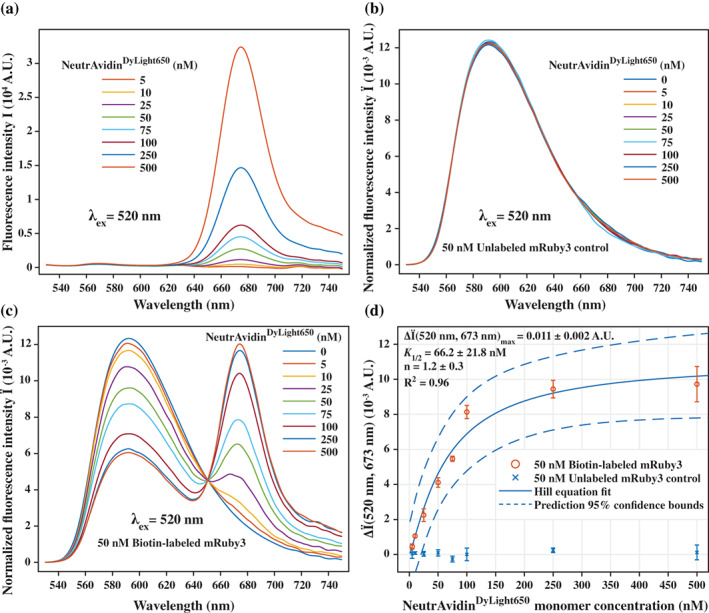
Fluorescence functionality of the TSGIT‐purified mRuby3. (a) Emission spectra of various concentrations of NeutrAvidinDyLight650. Corrected and normalized emission spectra of various concentrations of NeutrAvidinDyLight650 in the presence of (b) 50 nM unlabeled mRuby3 or (c) 50 nM IPL biotin‐labeled mRuby3. All data points represent the average of three independent acquisitions. All spectra were collected between 530 and 750 nm upon excitation at 520 nm. All spectra follow the color codes presented in the inset tables. Correction and normalization of the emission spectra were performed as described inSupporting Information Materials and Methods section. (d) Plot of the enhancement in corrected and normalized emission intensity at 673 nm upon addition of various concentrations of NeutrAvidinDyLight650 to 50 nM unlabeled mRuby3 (blue crosses) or to 50 nM IPL biotin‐labeled mRuby3 (red circles). All error bars represent the standard deviation of three independent acquisitions. For IPL biotin‐labeled mRuby3, the experimental datapoints were fit to a Hill equation, as described inSupporting Information Materials and Methods section. The parameters of the fit are described in the inset table together with their standard deviations. The 95% confidence bounds of the fitted model are depicted by the dashed lines. In all panels, the concentration of NeutrAvidinDyLight650 is indicated as the concentration of NeutrAvidin monomers, that is, biotin binding sites
The signals from the samples containing unlabeled mRuby3 remained largely unchanged, beyond variations expected from experimental noise, upon addition of NeutrAvidinDyLight650 (Figure 4b). This suggests the absence of factors that would spatially constrain mRuby3 and NeutrAvidinDyLight650 to be within FRET distance. On the contrary, for biotin‐labeled mRuby3 produced by the IPL reaction, addition of increasing concentrations of NeutrAvidinDyLight650 resulted in the emergence of a second emission peak centered at ~673 nm (Figure 4c). Simultaneously, the amplitude of the direct emission of mRuby3, centered at ~592 nm, decreased in an anti‐corelated manner with the increase in the emission of NeutrAvidinDyLight650 (Figure 4c), suggesting the efficient occurrence of FRET. When we plotted the increase in emission at 673 nm as a function of NeutrAvidinDyLight650 concentration, the totally different behaviors of unlabeled mRuby3 and IPL biotin‐labeled mRuby3 became immediately obvious (Figure 4d). Taken together, our FRET experiments clearly show that mRuby3 purified through TSGIT maintained its fluorescence functionality and that the IPL reaction proceeded properly allowing for biotin‐labeling of the protein.
2.3. TSGIT‐purification of gp2.5 ssDNA‐binding protein
We next tested some of the potential limitation of TSGIT with respect to the effect of the tags on oligomerization and protein folding. We opted to use gp2.5 since it forms a stable dimer in solution 36 and its ssDNA binding activity has been extensively characterized. 36 , 44 , 45 TSGIT‐gp2.5 was expressed and purified (Figure 5a) as described for mRuby3 with two differences; cleavage of the C‐terminal fusion via Intein‐tag was initiated by DTT rather than 2‐MESNA since no subsequent IPL was intended and the cleavage time was increased to 36 hr.
FIGURE 5.
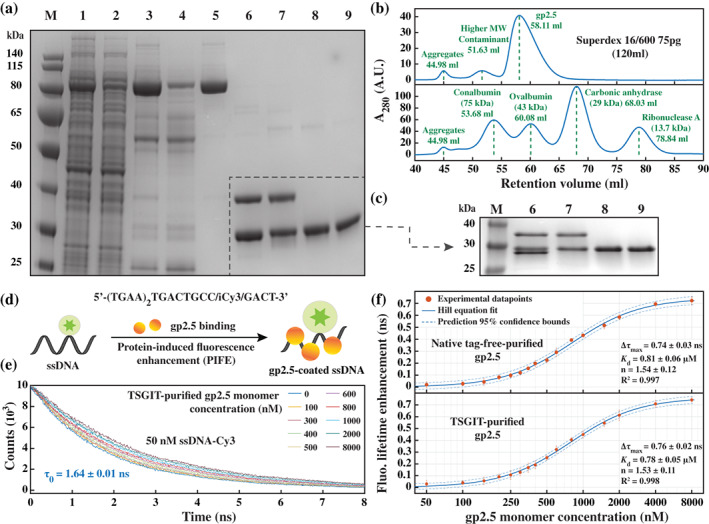
Purification of gp2.5 by employing TSGIT and its microscopic characterization. (a) Image of a 10% SDS‐PAGE gel showing the different steps of purification of gp2.5 starting from E. colily sate containing the expression product of the pTSGIT‐gp2.5 expression vector: Lane 1, soluble fraction of the lysate; Lane 2, flow‐through of the first HisTrap affinity column; Lane 3, TSGIT‐gp2.5 elution from the HisTrap affinity column; Lane 4, flow‐through of the first StrepTrap affinity column; Lane 5, elution of TSGIT‐gp2.5 from StrepTrap affinity column; Lane 6, release of gp2.5 from the TSGIT‐gp2.5 fusion via SUMO protease and DTT cleavage; Lane 7, flow‐through of cleaved gp2.5 through the second HisTrap affinity column; Lane 8, flow‐through of cleaved gp2.5 through the second StrepTrap affinity column; Lane 9, gp2.5 after Superdex 75 pg size‐exclusion elution. The marker (M) is PageRuler Prestained Protein Ladder. (b) Chromatograms showing the elution of TSGIT‐purified gp2.5 (top) and four different molecular weight markers (bottom) from the Superdex 75 pg size‐exclusion column. (c) Image of a 12% SDS‐PAGE gel showing a better separation of the proteins in the gray dashed area from the 10% SDS‐PAGE shown in panel (a) with the same numbering of the lanes. (d) Schematic representation of the PIFE assay used to monitor the binding affinity of gp2.5 to short ssDNA. (e) Examples of time‐resolved fluorescence decays of the Cy3‐labelled ssDNA obtained in the presence of various concentrations of TSGIT‐purified gp2.5. (f) Binding isotherms (log‐linear) of native tag‐free‐purified gp2.5 (top) and TSGIT‐purified gp2.5 (bottom) to the Cy3‐labelled ssDNA as determined from time‐resolved fluorescence measurements. The y‐axis represents the increase in Cy3 lifetime upon gp2.5 binding as compared to the lifetime of the oligo alone. All the elements of the plots have the same meaning as in Figure 4d
TSGIT‐gp2.5 was eluted from the first HisTrap affinity column together with several contaminants (Lane 3 in Figure 5a), which were removed efficiently by the subsequent StrepTrap affinity step (Lane 5 in Figure 5a). The fusion was cleaved with high efficiency (Lane 6 in Figure 5a,c) and the released N‐ and C‐terminal fusion tags were removed by second HisTrap (Lane 7 in Figure 5a,c) and StrepTrap (Lane 8 in Figure 5a,c) affinity steps. Finally, gp2.5 purity was polished using a Superdex 75 pg column (Lane 9 in Figure 5a,c).
It is worth noting that due to their similar size and electrophoretic properties, the N‐terminal fusion tag and gp2.5 could not be separated using the standard SDS‐PAGE conditions (Lane 6 in Figure 5a). To separate them, the post‐cleavage samples were analyzed using an increased percentage SDS‐PAGE that was run at lower voltage for an increased amount of time as described in Supporting Information Materials and Methods section. The N‐terminal fusion tag appeared as a slightly lower band than gp2.5 in Lane 6 in Figure 5c, which was efficiently removed by the second HisTrap affinity step (Lane 7 in Figure 5c.). TSGIT‐based purification of gp2.5 yielded ~4.3 ± 0.1 mg from 35 mL of resuspended expression cells (7 g of dry cells) with a purity higher than 98% (Table S2). Although this yield was ~3‐fold lower than the one described in Reference 36, it was obtained without using very rich media or fermenter expression conditions.
The solution dimerization state of TSGIT‐purified gp2.5 was assessed by size‐exclusion analysis using a Superdex 75 pg column pre‐equilibrated and run with analysis buffer (Supporting Information Materials and Methods section). The gp2.5 monomer has a molecular weight of ~25.56 kDa as estimated from its amino acid sequence, yet is known to run higher in SDS‐PAGE. 36 The retention volume of TSGIT‐purified gp2.5 (Figure 5b) was situated between the elution peaks of Conalbumin (~75 kDa) and Ovalbumin (~43 kDa), demonstrating that it formed a dimer in solution. Furthermore, using a calibration curve generated by the four known molecular weight calibration markers predicts a relative molecular weight of TSGIT‐purified gp2.5 of ~53.62 kDa (Figure S5a), which is consistent with the previously reported value of 53.7 kDa. 36 These experiments demonstrate that TSGIT methodology did not interfere with gp2.5 dimerization.
The complete size of the TSGIT‐gp2.5 uncleaved fusion is ~88.42 kDa as estimated from its amino acid sequence. Performing a similar analysis as described above for the cleaved gp2.5 placed the uncleaved TSGIT‐gp2.5 fusion molecular weight (Figure S5b) in the vicinity of the Aldolase molecular weight calibration marker (~158 kDa) on Superdex 200 pg column. A calibration curve analysis estimated the relative molecular weight of the uncleaved TSGIT‐gp2.5 to be ~184.6 kDa (Figure S5c), which is consistent with a dimer fusion protein rather than monomer. Remarkably, these results show that TSGIT fusion tags do not impair gp2.5 dimer formation neither before nor after fusion tags cleavage.
Next, we sought to investigate if the TSGIT purification method had any impact on gp2.5 ssDNA binding activity. For comparison, we purified native tag‐free gp2.5 as described previously. 36 First, we investigated gp2.5 microscopic dissociation constant (K d) as an indication of its affinity for ssDNA through a previously‐established protein‐induced fluorescence enhancement (PIFE) assay. 46 We selected a 21 nucleotide (nt) ssDNA sequence labeled internally with Cy3, which undergoes an increase in Cy3 fluorescence lifetime upon gp2.5 biding (Figure 5d). Previous reports show that gp2.5 binds with a stoichiometry of one monomer per ~7 nt 36 and requires ~23 nt for stable binding. 45 Therefore, our Cy3‐ssDNA substrate can support the binding of up to three gp2.5 molecules.
The lifetime of the oligo in the absence of protein was found to be ~1.64 ns (Figures 5e and S5d). Increased PIFE was observed upon protein titration for both TSGIT‐purified gp2.5 and native tag‐free‐purified gp2.5 (Figures 5e and S5d). A plot of fluorescence lifetime versus ligand concentration servers as a binding isotherm, where the isotherm saturation value is proportional to the maximum achievable PIFE. 47 Such plots were generated for both TSGIT‐purified gp2.5 and native tag‐free‐purified gp2.5 and the experimental datapoints were fit to a PIFE‐adjusted Hill‐type dependence (Figure 5f) described by Equation (5) in Supporting Information Materials and Methods section. The K d was calculated as ~0.78 μM for TSGIT‐purified gp2.5 and ~0.81 μM for native tag‐free‐purified gp2.5 (Figure 5f). These values are consistent with each other and with the previously reported value of ~0.8 μM. 36 , 48
Unlike other ssDNA‐binding proteins, gp2.5 was previously reported to exhibit little binding cooperativity 36 , 48 to ssDNA at any salt concentration. In accordance with this finding, the binding isotherms of both TSGIT‐purified gp2.5 and native tag‐free‐purified gp2.5 were characterized by a Hill coefficient of only ~1.5 (Figure 5f) despite the length of the employed ssDNA which can support the binding of up to three gp2.5 molecules. It is worth noting that the sigmoidal shape of the binding isotherms is exaggerated since a log‐linear plot is used to better illustrate the whole titration concentration range; even a perfect Langmuir hyperbolic isotherm would appear sigmoidal in log‐linear plots. 49 The last parameter of interest of the binding isotherms is the maximum increase in Cy3 fluorescence lifetime at saturating gp2.5 concentrations of ~0.76 ns for TSGIT‐purified gp2.5 and ~0.74 ns for native tag‐free‐purified gp2.5 (Figure 5f). Since the fluorescence lifetime is highly specific and sensitive to the nearby protein residues, their conformation as well as the overall structure of the DNA‐dye‐protein complex, 46 this finding further strengthens the similarity between TSGIT‐purified gp2.5 and native tag‐free‐purified gp2.5 at microscopic scale.
In the last set of experiments, we investigated TSGIT‐purified gp2.5 binding to ssDNA at a larger scale and its power to stretch collapsed ssDNA upon binding. The experiments are based on a well‐established single‐molecule flow‐stretching bead assay, 44 , 50 , 51 which can monitor the conversion of free ssDNA to gp2.5‐coated ssDNA via their length difference through the observation of the time‐position dependence of a large bead attached to the free DNA end (Figure 6a). The assay was performed as described in Supporting Information Materials and Methods section using our previously described protocols 52 , 53 . Native tag‐free‐purified gp2.5 (Figures 6b and S6a) and TSGIT‐purified gp2.5 (Figure 6c and S6b) produced similar ssDNA stretching events; the stretching length is represented as equivalent length of dsDNA in base pairs (bp). Statistically, the distribution of the stretching lengths produced by native tag‐free‐purified gp2.5 and TSGIT‐purified gp2.5 are highly similar (Figure 6d) with their empirical cumulative distribution functions exhibiting less than 10% difference at any length. Both gp2.5 proteins stretched the ssDNA to an equivalent dsDNA length of ~6.5 kbp. The length of the ssDNA region of the employed substrate is ~7.2 knt. Therefore, we conclude that both TSGIT‐purified gp2.5 and native tag‐free‐purified gp2.5 perform highly similar in this assay and stretch the ssDNA region of the substrate to ~90% of the contour length of its equivalent dsDNA form, which is consistent with the previously reported value. 44
FIGURE 6.
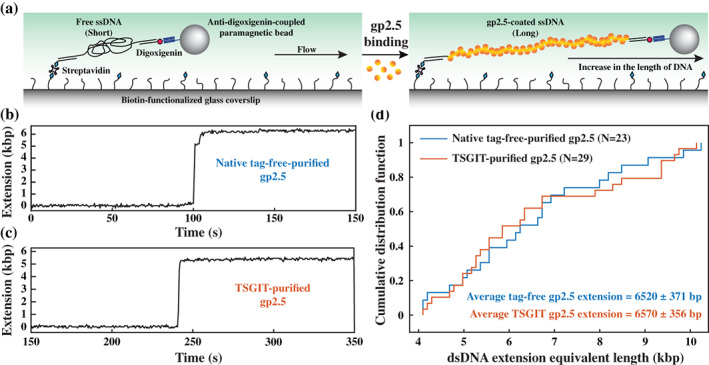
ssDNA stretching power of TSGIT‐purified gp2.5. (a) Schematic representation of the single‐molecule flow‐stretching bead assay used to monitor the elongation of collapsed long ssDNA upon binding of gp2.5. The position of the DNA‐attached bead is monitored over time and converted to dsDNA extension equivalent length as described in Supporting Information Materials and Methods section. (b) Example of a single‐molecule time‐trace showing the stretching of the ssDNA‐containing substrate upon injection of native tag‐free‐purified gp2.5. (c) Example of a single‐molecule time‐trace showing the stretching of the ssDNA‐containing substrate upon injection of TSGIT‐purified gp2.5. For both panels, the maximum extension was calculated between the initial and final basslines along the y‐axis. (d) A plot of the empirical cumulative distribution function, obtained from the indicated number (N) of individual time‐traces, of the maximum dsDNA equivalent stretching length by native tag‐free‐purified gp2.5 (blue) and TSGIT‐purified gp2.5 (red). The average stretching length together with its standard deviation is indicated for both proteins
Taken together, the gp2.5 experiments show that the protein obtained through TSGIT purification is virtually indistinguishable from the one obtained through native tag‐free purification. Therefore, TSGIT purification, despite the presence of its large fusion tags did not disturb gp2.5 proper folding, its dimeric solution form or its ssDNA‐binding activity. Nevertheless, as in the case of any fusion system, these results should be extrapolated with care to other proteins, for which the presence of the N‐ and C‐terminal fusion tags may have a negative effect depending on the overall three‐dimensional structure and folding of the fusion.
3. CONCLUSION
During protein expression and purification, a variety of mechanisms can generate soluble truncated forms of the protein of interest that coexist with the full‐length form. These truncated forms can vary by different degrees from the protein of interest, and small N‐ or C‐terminal degradations are typically inseparable from the full‐length target protein under such conditions. A possible solution to this problem is the addition of at least one N‐terminal and one different C‐terminal affinity tag to form a fusion protein. Sequential selection for the two different affinity tags results in the exclusive retention of the full‐length protein. Nevertheless, the presence of these tags can considerably interfere with the function of the protein and their removal is often desirable. By surveying existing affinity and cleavable tags, we designed TSGIT, a unified system for protein expression and purification that simultaneously resolves these problems. TSGIT also includes an N‐terminal and a C‐terminal Trx for increased solubility. TSGIT isolates the target protein between its N‐terminal and C‐terminal composite tags that are designed to maximize the benefits of each individual component. Upon simultaneous cleavage of these tags, the protein of interest is released without any undesired additional N‐ or C‐terminal amino acids. The main limitation of the TSGIT system is the relatively high concentration of thiol‐containing reductant that is required for Intein‐mediated cleavage of the C‐terminal fusion tag. Such strong reducing conditions may be harmful for certain proteins; for example in cases where natural disulfide bonds must occur within the protein structure 54 or in cases where reductants participate, even indirectly, in Peptide Backbone Fragmentation. 55 , 56 , 57 The purification strategy proposed for TSGIT resulted in a high yield of more than 95% pure native and active proteins of interest. Additionally, the proteins produced by TSGIT are ready for IPL with other peptides or proteins for downstream applications. Through custom‐synthesis, the peptides used for IPL can offer a large variety of modifications such as fluorophores, attachment groups (e.g., biotin and digoxigenin), glycans, localization sequences, and orthogonal reactive chemical groups for general coupling to different moieties (e.g., click chemistry), while having only the simple requirement to contain an N‐terminal cysteine residue.
4. MATERIALS AND METHODS
4.1. Plasmid design and construction
For the component tags of the TSGIT fusion, their amino acid sequences were selected from well‐established available sources. For the N‐terminal part of the fusion, the two His‐tags consist of six consecutive histidine residues 58 , 59 and the SUMO‐tag sequence was chosen from the pE‐SUMO expression vector (LifeSensors; plasmid 1001 K). For the C‐terminal part of the fusion, the Intein‐tag sequence and its subsequent linker were chosen from the pTXB1 expression vector (IMPACT system, New England BioLabs; plasmid N6707S) and the sequence of the Twin Strep‐tag was selected as the one described previously. 28 , 29 The Trx‐tag sequence used for both parts of the fusion was selected from the 2T‐T expression vector (MacroBac system; 60 Addgene plasmid 29712). All these amino acid sequences were converted to coding DNA sequences by the codon optimization tool available on‐line from Integrated DNA Technologies (IDT).
The genes encoding the empty full‐length TSGIT construct (Figure 2a), its N‐terminal part only or its C‐terminal part only were custom synthesized by IDT as gBlocks. The cloning primers were also custom synthesized by IDT. The pRSF‐1b plasmid was purchased from Novagen. The gene inserts encoding mRuby3 and gp2.5 were codon optimized and custom synthesized by IDT as gBlocks. The three TSGIT genes were independently cloned by Gibson assembly into empty pRSF‐1b plasmids, yielding plasmids that we denote as pTSGIT, pTSGIT‐N and pTSGIT‐C, respectively. The gene inserts encoding mRuby3 (Figure S2) and gp2.5 (Figure S3) were assembled together with the TSGIT N‐ and C‐terminal fusion tags between the SUMO and Intein regions by Gibson assembly into pRSF‐1b plasmids. We denote these plasmids as pTSGIT‐mRuby3 and pTSGIT‐gp2.5. All final constructs were verified by sequencing.
4.2. TSGIT fusion proteins expression and purification
pTSGIT‐N, pTSGIT‐C, pTSGIT‐mRuby3, and pTSGIT‐gp2.5 were independently expressed in BL21(DE3) E. coli expression strain (Novagen) in LB media containing Kanamycin (50 μg/ml). Cell growth and lysis were performed as described previously. 24 Briefly, cells were grown at 37°C to an OD600 of 0.8 and then protein expression was induced by the addition of 0.5 mM isopropyl β‐d‐thiogalactopyranoside (IPTG) and incubated further for 4 hr at 37°C. The cells were collected by centrifugation and pellets were re‐suspended in 5 ml of lysis buffer [50 mM HEPES pH (8), 500 mM NaCl, 30 mM Imidazole, 0.1 mM Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP), 1 mM PMSF, 5% Glycerol and one EDTA‐free protease inhibitor cocktail tablet per 50 ml (Roche, UK)] per 1 g of cells. The cells were then lysed by sonicating them twice on ice using a Qsonica 500 sonicator operating at 20 kHz, 37% amplitude with a cycle on‐time of 10 s, a cycle off‐time of 15 s and a total on‐time of 2 min. Next, the lysates were passed twice through a French press operating at 20,000 psi and cleared by centrifugation at 95,834g for 45 min at 4°C. All the subsequent protein purification steps were performed at 4°C as described in detail in Section 2.
All the affinity columns used for protein purification were HisTrap HP 5 ml and StrepTrap HP 5 ml affinity columns (GE Healthcare). The buffers used for protein binding, washing and elution for the HisTrap steps were buffer A [50 mM HEPES pH (8), 500 mM NaCl, 30 mM Imidazole, 0.1 mM TCEP, and 5% Glycerol] and buffer B [50 mM HEPES pH (8), 500 mM NaCl, 350 mM Imidazole, 0.1 mM TCEP, and 5% Glycerol]. The buffers used for protein binding, washing and elution for the StrepTrap steps were buffer C [50 mM HEPES pH (8.3), 500 mM NaCl, 0.1 mM TCEP, and 5% Glycerol] and buffer D [50 mM HEPES pH (8.3), 500 mM NaCl, 2.5 mM d‐Desthiobiotin, 0.1 mM TCEP, and 5% Glycerol]. Cleavage reaction of the fusion protein was performed in 2‐MESNA cleavage buffer [50 mM HEPES pH (8.5), 500 mM NaCl, 50 mM 2‐Mercaptoethanesulfonic acid (2‐MESNA) as Sodium 2‐Mercaptoethanesulfonate, and 5% Glycerol] or DTT cleavage buffer [50 mM HEPES pH (8.5), 500 mM NaCl, 40 mM Dithiothreitol (DTT), and 5% Glycerol]. All the final protein fractions were stored in storage buffer [50 mM HEPES pH (8), 250 mM NaCl, 1 mM Dithiothreitol (DTT), and 5% Glycerol]. The same storage buffer was used to equilibrate and elute the proteins from a 120 ml Superdex 16/600 75 pg (GE Healthcare) column. Buffer exchange steps were performed using PD‐10 desalting columns (8.3 ml of Sephadex G‐25 medium; Amersham Biosciences). Capturing of the biotin‐tagged proteins was performed using High Capacity NeutrAvidin Agarose Resin (Thermo Scientific). The buffer used to equilibrate, wash and elute the proteins from the NeutrAvidin Agarose Resin was storage buffer. An ÄKTA purifier fast protein liquid chromatography (FPLC) system (GE Healthcare) was used to perform all the column‐based chromatographic steps.
4.3. Intein‐mediated protein ligation reaction
Intein‐mediated protein ligation between activated mRuby3 protein and BioP (CDPEKBiotinDS) peptide 32 was performed in IPL buffer [50 mM HEPES pH (8.5), 500 mM NaCl, 10 mM 2‐Mercaptoethanesulfonic acid (2‐MESNA) as Sodium 2‐Mercaptoethanesulfonate and 5% Glycerol] at 4°C for 12 hr with gentle rotation. The BioP peptide‐containing biotin attached to an internal lysine residue was custom synthesized by GenScript (85.2% purity). The ligation reaction contained 10 μM 2‐MESNA‐activated mRuby3 and 1 mM BioP.
AUTHOR CONTRIBUTIONS
Samir M. Hamdan: Conceptualization; funding acquisition; investigation; methodology; project administration; supervision; writing‐original draft. Vlad‐Stefan Raducanu: Conceptualization; data curation; formal analysis; investigation; methodology; validation; writing‐original draft; writing‐review and editing. Daniela‐Violeta Raducanu: Data curation; investigation; methodology. Yujing Ouyang: Data curation; formal analysis. Muhammad Tehseen: Data curation. Masateru Takahashi: Data curation.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Appendix S1: Supporting Information consists of a single file named “Supplementary Material.pdf” that contains the Supporting Information Materials and Methods section, two Supporting Information explanation sections, six Supporting Information Figures, and two Supporting Information Table.
ACKNOWLEDGMENTS
The authors are grateful to Prof. Stefan T. Arold (KAUST) for providing access to time‐resolved fluorescence spectrofluorometer. The authors thank Dr. Fahad Rashid for the expression clone used to purify Ulp1 SUMO protease. pET His6 Thioredoxin TEV LIC cloning vector (2T‐T) was a gift from Scott Gradia (Addgene plasmid 29712). This work was supported by King Abdullah University of Science and Technology through baseline funding and a Competitive Research Award [Grant CRG6 (URF/1/3432‐01‐01) to Samir M. Hamdan].
Raducanu V‐S, Raducanu D‐V, Ouyang Y, Tehseen M, Takahashi M, Hamdan SM. TSGIT: An N‐ and C‐terminal tandem tag system for purification of native and intein‐mediated ligation‐ready proteins. Protein Science. 2021;30:497–512. 10.1002/pro.3989
Funding information King Abdullah University of Science and Technology, Grant/Award Number: CRG6 (URF/1/3432‐01‐01)
REFERENCES
- 1. Kimple ME, Brill AL, Pasker RL. Overview of affinity tags for protein purification. Curr Protoc Protein Sci. 2013;73:9.9.1–9.9.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Terpe K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. [DOI] [PubMed] [Google Scholar]
- 3. Costa S, Almeida A, Castro A, Domingues L. Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: The novel Fh8 system. Front Microbiol. 2014;5:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Young CL, Britton ZT, Robinson AS. Recombinant protein expression and purification: A comprehensive review of affinity tags and microbial applications. Biotechnol J. 2012;7:620–634. [DOI] [PubMed] [Google Scholar]
- 5. Waugh DS. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif. 2011;80:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frey S, Gorlich D. A new set of highly efficient, tag‐cleaving proteases for purifying recombinant proteins. J Chromatogr A. 2014;1337:95–105. [DOI] [PubMed] [Google Scholar]
- 7. Raran‐Kurussi S, Cherry S, Zhang D, Waugh DS. Removal of affinity tags with TEV protease. Methods Mol Biol. 2017;1586:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ryan BJ, Henehan GT. Overview of approaches to preventing and avoiding proteolysis during expression and purification of proteins. Curr Protoc Protein Sci. 2013;71:525.1–5.25.7. [DOI] [PubMed] [Google Scholar]
- 9. Jennings MJ, Barrios AF, Tan S. Elimination of truncated recombinant protein expressed in Escherichia coli by removing cryptic translation initiation site. Protein Expr Purif. 2016;121:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whitaker WR, Lee H, Arkin AP, Dueber JE. Avoidance of truncated proteins from unintended ribosome binding sites within heterologous protein coding sequences. ACS Synth Biol. 2015;4:249–257. [DOI] [PubMed] [Google Scholar]
- 11. Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: Advances and challenges. Front Microbiol. 2014;5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Narayanan N, Chou CP. Alleviation of proteolytic sensitivity to enhance recombinant lipase production in Escherichia coli . Appl Environ Microbiol. 2009;75:5424–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wingfield PT. Overview of the purification of recombinant proteins. Curr Protoc Protein Sci. 2015;80:6.1.1–6.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller CG. Peptidases and proteases of Escherichia coli and salmonella typhimurium. Annu Rev Microbiol. 1975;29:485–504. [DOI] [PubMed] [Google Scholar]
- 15. Jankiewicz U, Bielawski W. The properties and functions of bacterial aminopeptidases. Acta Microbiol Pol. 2003;52:217–231. [PubMed] [Google Scholar]
- 16. Gonzales T, Robert‐Baudouy J. Bacterial aminopeptidases: Properties and functions. FEMS Microbiol Rev. 1996;18:319–344. [DOI] [PubMed] [Google Scholar]
- 17. Lykkemark S, Mandrup OA, Friis NA, Kristensen P. Degradation of C‐terminal tag sequences on domain antibodies purified from E. coli supernatant. MAbs. 2014;6:1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parsell DA, Silber KR, Sauer RT. Carboxy‐terminal determinants of intracellular protein degradation. Genes Dev. 1990;4:277–286. [DOI] [PubMed] [Google Scholar]
- 19. Khan F, He M, Taussig MJ. Double‐hexahistidine tag with high‐affinity binding for protein immobilization, purification, and detection on ni‐nitrilotriacetic acid surfaces. Anal Chem. 2006;78:3072–3079. [DOI] [PubMed] [Google Scholar]
- 20. Fischer M, Leech AP, Hubbard RE. Comparative assessment of different histidine‐tags for immobilization of protein onto surface plasmon resonance sensorchips. Anal Chem. 2011;83:1800–1807. [DOI] [PubMed] [Google Scholar]
- 21. LaVallie ER, Lu Z, Diblasio‐Smith EA, Collins‐Racie LA, McCoy JM. Thioredoxin as a fusion partner for production of soluble recombinant proteins in Escherichia coli . Methods Enzymol. 2000;326:322–340. [DOI] [PubMed] [Google Scholar]
- 22. Xie YG, Luan C, Zhang HW, et al. Effects of thioredoxin: SUMO and intein on soluble fusion expression of an antimicrobial peptide OG2 in Escherichia coli . Protein Pept Lett. 2013;20:54–60. [PubMed] [Google Scholar]
- 23. Panavas T, Sanders C, Butt TR. SUMO fusion technology for enhanced protein production in prokaryotic and eukaryotic expression systems. Methods Mol Biol. 2009;497:303–317. [DOI] [PubMed] [Google Scholar]
- 24. Raducanu VS, Tehseen M, Shirbini A, Raducanu DV, Hamdan SM. Two chromatographic schemes for protein purification involving the biotin/avidin interaction under native conditions. J Chromatogr A. 2020;1621:461051. [DOI] [PubMed] [Google Scholar]
- 25. Tehseen M, Raducanu VS, Rashid F, Shirbini A, Takahashi M, Hamdan SM. Proliferating cell nuclear antigen‐agarose column: A tag‐free and tag‐dependent tool for protein purification affinity chromatography. J Chromatogr A. 2019;1602:341–349. [DOI] [PubMed] [Google Scholar]
- 26. Bornhorst JA, Falke JJ. Purification of proteins using polyhistidine affinity tags. Methods Enzymol. 2000;326:245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guerrero F, Ciragan A, Iwai H. Tandem SUMO fusion vectors for improving soluble protein expression and purification. Protein Expr Purif. 2015;116:42–49. [DOI] [PubMed] [Google Scholar]
- 28. Schmidt TG, Batz L, Bonet L, et al. Development of the Twin‐Strep‐tag(R) and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr Purif. 2013;92:54–61. [DOI] [PubMed] [Google Scholar]
- 29. Ivanov KI, Basic M, Varjosalo M, Makinen K. One‐step purification of twin‐strep‐tagged proteins and their complexes on strep‐tactin resin cross‐linked with bis(sulfosuccinimidyl) suberate (BS3). J Vis Exp. 2014;20:51536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans TC Jr, Benner J, Xu MQ. Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 1998;7:2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Southworth MW, Amaya K, Evans TC, Xu MQ, Perler FB. Purification of proteins fused to either the amino or carboxy terminus of the mycobacterium xenopi gyrase A intein. Biotechniques. 1999;27:110–114, 116, 118–120. [DOI] [PubMed] [Google Scholar]
- 32. Ghosh, I , Considine, N , Maunus, E , et al. Site‐Specific Protein Labeling by Intein‐Mediated Protein Ligation Thomas CE Jr., Ming‐Qun X, (eds.), Heterologous Gene Expression in E.coli, Methods in Molecular Biology 705, (pp. 87–107). New York, NY: Humana Press; 2011. [DOI] [PubMed] [Google Scholar]
- 33. Xu MQ, Evans TC Jr. Purification of recombinant proteins from E. coli by engineered inteins. Methods Mol Biol. 2003;205:43–68. [DOI] [PubMed] [Google Scholar]
- 34. Xu MQ, Paulus H, Chong S. Fusions to self‐splicing inteins for protein purification. Methods Enzymol. 2000;326:376–418. [DOI] [PubMed] [Google Scholar]
- 35. Bajar BT, Wang ES, Lam AJ, et al. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci Rep. 2016;6:20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim YT, Tabor S, Bortner C, Griffith JD, Richardson CC. Purification and characterization of the bacteriophage T7 gene 2.5 protein. A single‐stranded DNA‐binding protein. J Biol Chem. 1992;267:15022–15031. [PubMed] [Google Scholar]
- 37. Lahiry A, Fan YM, Stimple SD, Raith M, Wood DW. Inteins as tools for tagless and traceless protein purification. J Chem Technol Biotechnol. 2018;93:1827–1835. [Google Scholar]
- 38. Shah NH, Muir TW. Inteins: Nature's gift to protein chemists. Chem Sci. 2014;5:446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Telenti A, Southworth M, Alcaide F, Daugelat S, Jacobs WR Jr, Perler FB. The mycobacterium xenopi GyrA protein splicing element: Characterization of a minimal intein. J Bacteriol. 1997;179:6378–6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mitchell SF, Lorsch JR. Protein affinity purification using intein/chitin binding protein tags. Methods Enzymol. 2015;559:111–125. [DOI] [PubMed] [Google Scholar]
- 41. Bernard MP, Cao D, Myers RV, Moyle WR. Tight attachment of chitin‐binding‐domain‐tagged proteins to surfaces coated with acetylated chitosan. Anal Biochem. 2004;327:278–283. [DOI] [PubMed] [Google Scholar]
- 42. Goulatis LI, Ramanathan R, Shusta EV. Impacts of the −1 amino acid on yeast production of protein‐intein fusions. Biotechnol Prog. 2019;35:e2736. [DOI] [PubMed] [Google Scholar]
- 43. Qian J, Yao B, Wu C. Fluorescence resonance energy transfer detection methods: Sensitized emission and acceptor bleaching. Exp Ther Med. 2014;8:1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamdan SM, Loparo JJ, Takahashi M, Richardson CC, van Oijen AM. Dynamics of DNA replication loops reveal temporal control of lagging‐strand synthesis. Nature. 2009;457:336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marintcheva B, Hamdan SM, Lee SJ, Richardson CC. Essential residues in the C terminus of the bacteriophage T7 gene 2.5 single‐stranded DNA‐binding protein. J Biol Chem. 2006;281:25831–25840. [DOI] [PubMed] [Google Scholar]
- 46. Rashid F, Raducanu VS, Zaher MS, Tehseen M, Habuchi S, Hamdan SM. Initial state of DNA‐Dye complex sets the stage for protein induced fluorescence modulation. Nat Commun. 2019;10:2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Raducanu VS, Rashid F, Zaher MS, Li YY, Merzaban JS, Hamdan SM. A direct fluorescent signal transducer embedded in a DNA aptamer paves the way for versatile metal‐ion detection. Sens Actuators B. 2020;304:127376. [Google Scholar]
- 48. Hernandez AJ, Richardson CC. Gp2.5, the multifunctional bacteriophage T7 single‐stranded DNA binding protein. Semin Cell Dev Biol. 2019;86:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Salahudeen MS, Nishtala PS. An overview of pharmacodynamic modelling, ligand‐binding approach and its application in clinical practice. Saudi Pharm J. 2017;25:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439:621–624. [DOI] [PubMed] [Google Scholar]
- 51. van Oijen AM, Blainey PC, Crampton DJ, Richardson CC, Ellenberger T, Xie XS. Single‐molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–1238. [DOI] [PubMed] [Google Scholar]
- 52. Elshenawy MM, Jergic S, Xu ZQ, et al. Replisome speed determines the efficiency of the Tus‐Ter replication termination barrier. Nature. 2015;525:394–398. [DOI] [PubMed] [Google Scholar]
- 53. Takahashi M, Takahashi E, Joudeh LI, Marini M, Das G, Elshenawy MM, Akal A, Sakashita K, Alam I, Tehseen M and others. Dynamic structure mediates halophilic adaptation of a DNA polymerase from the deep‐sea brines of the Red Sea. FASEB J 2018;32:3346–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Braakman I, Helenius J, Helenius A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J. 1992;11:1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Understanding GFP posttranslational chemistry: Structures of designed variants that achieve backbone fragmentation, hydrolysis, and decarboxylation. J Am Chem Soc. 2006;128:4685–4693. [DOI] [PubMed] [Google Scholar]
- 56. Mormann M, Eble J, Schwoppe C, et al. Fragmentation of intra‐peptide and inter‐peptide disulfide bonds of proteolytic peptides by nanoESI collision‐induced dissociation. Anal Bioanal Chem. 2008;392:831–838. [DOI] [PubMed] [Google Scholar]
- 57. Gorman JJ, Wallis TP, Pitt JJ. Protein disulfide bond determination by mass spectrometry. Mass Spectrom Rev. 2002;21:183–216. [DOI] [PubMed] [Google Scholar]
- 58. Block H, Maertens B, Spriestersbach A, et al. Immobilized‐metal affinity chromatography (IMAC): A review. Methods Enzymol. 2009;463:439–473. [DOI] [PubMed] [Google Scholar]
- 59. Spriestersbach A, Kubicek J, Schafer F, Block H, Maertens B. Purification of His‐tagged proteins. Methods Enzymol. 2015;559:1–15. [DOI] [PubMed] [Google Scholar]
- 60. Gradia SD, Ishida JP, Tsai MS, Jeans C, Tainer JA, MacroBac FJO. New technologies for robust and efficient large‐scale production of recombinant multiprotein complexes. Methods Enzymol. 2017;592:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Worrell BT, Mavila S, Wang C, et al. A user's guide to the thiol‐thioester exchange in organic media: Scope, limitations, and applications in material science. Polymer Chem. 2018;9:4523–4534. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information consists of a single file named “Supplementary Material.pdf” that contains the Supporting Information Materials and Methods section, two Supporting Information explanation sections, six Supporting Information Figures, and two Supporting Information Table.