

Abstract
Chimeric antigen receptor (CAR) T-cell therapy remains limited to select centers that can carefully monitor adverse events. To broaden use of CAR T-cells in community clinics and in a frontline setting, we developed a novel CD8+ CAR T-cell product, Descartes-08, with predictable pharmacokinetics for treatment of multiple myeloma. Descartes-08 is engineered by mRNA transfection to express anti-BCMA CAR for a defined length of time. Descartes-08 express anti-BCMA CAR for 1 week, limiting risk of uncontrolled proliferation; produce inflammatory cytokines in response to myeloma target cells; and are highly cytolytic against myeloma cells regardless of presence of myeloma-protecting bone marrow stromal cells, exogenous a proliferation-inducing ligand, or drug resistance including IMiDs. The magnitude of cytolysis correlates with anti-BCMA CAR expression duration, indicating a temporal limit in activity. In the mouse model of aggressive disseminated human myeloma, Descartes-08 induces BCMA CAR-specific myeloma growth inhibition and significantly prolongs host survival (P<.0001). These preclinical data, coupled with an ongoing clinical trial of Descartes-08 in relapsed/refractory myeloma (NCT03448978) showing preliminary durable responses and a favorable therapeutic index, have provided the framework for a recently initiated trial of an optimized/humanized version of Descartes-08 (i.e., Descartes-11) in newly diagnosed myeloma patients with residual disease after induction therapy.
Keywords: multiple myeloma, MM; plasma cell, PC; bone marrow, BM; microenvironment; B cell maturation antigen, BCMA; chimeric antigen receptor (CAR) T cell; cellular immunotherapy; mRNA transient transfection; non-viral mRNA CAR T
Introduction
Multiple myeloma (MM) is a clonal expansion of malignant bone marrow plasma cells (PCs) characterized by dynamic genetic and epigenetic abnormalities that occur as consequences of complex interactions between PCs and a variety of accessory cells in the bone marrow (BM) microenvironment [1–9]. For the past decade, therapeutic advancements including proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), and monoclonal antibodies have significantly improved prognosis and survival in patients [2, 10]. However, MM remains incurable, with most patients eventually succumbing to progressive disease. It is thus crucial to develop novel effective immunotherapies with improved efficacy and safety for MM patients throughout all disease stages.
Chimeric antigen receptor (CAR) T cells that redirect patient T cells to recognize and kill cancer cells have recently been a major breakthrough for relapsed/refractory (RR) B-leukemia and lymphoma [11–16]. The CAR is a synthetic fusion protein with an antigen recognition domain derived from a monoclonal antibody and an intracellular T cell signaling domain (CD3ζ) linked with costimulatory domains such as 4–1BB or CD28 [16, 17]. By permanently engineering T cells via viral transduction to express a CAR, CAR T-cells become activated upon binding to specific antigens on tumor cells.
The first CAR T-cells for MM were developed to target B cell maturation antigen (BCMA), which is only present on the surface of myeloma and plasma cells, but no other normal tissues and essential organs [18–24]. BCMA has the most restricted tissue expression pattern among several validated MM targets such as SLAMF7 and CD38, and BCMA expression is upregulated during MM progression irrespective of cytogenetic abnormalities [19, 20, 23, 24]. Impressively, autologous T-cells permanently modified by gene transfer to express a CAR against BCMA have shown high complete response rates (CRs) in relapsed/refractory MM (RRMM) [25–28]. Their efficacy, however, is dependent on in vivo proliferation. This requires lymphodepletive chemotherapy, and uncontrolled proliferation often leads to cytokine release syndrome (CRS). Use of these breakthrough therapies therefore remains limited to select tertiary centers that can carefully monitor side effects from lymphodepletion, CRS, and neurotoxicity. Despite these adverse effects, patients relapse, albeit after up to 12–18 months of progression-free survival. Further extended survival may require earlier treatment, likely in the context of induction therapy in a frontline setting, where it is possible to eliminate tumor burden prior to acquiring a highly treatment-resistant phenotype.
We have developed an engineered T-cell product, Descartes-08, that transiently modifies a purified population of autologous CD8+ T-cells with anti-BCMA CAR mRNA. CD8+ T cells are the main effector cells to execute target cell lysis in vitro and in vivo. Descartes-08 has potent activity against primary myeloma cells and MM cell lines, including those resistant to immunomodulatory agents, i.e., lenalidomide, pomalidomide, and inhibits tumor growth in an aggressive in vivo model of myeloma. Preliminary clinical enabling studies from an ongoing Phase I/II clinical trial in patients with RRMM (NCT03448978) suggest that it may be possible to achieve deep, durable response (e.g., stringent complete response, sCR) without significant CAR T-cell related toxicity such as CRS or neurotoxicity. These data support a recently initiated Phase II study of Descartes-11, a humanized anti-BCMA mRNA CAR T-cell, in frontline myeloma.
Materials and methods
Manufacturing of Descartes-08 cells for preclinical experiments
CD8+ T cells were selected from peripheral blood of healthy donors or patients. T cells were activated and expanded over time to achieve a high yield of activated CD8+ cells followed by transfection with anti-BCMA mRNA. The mRNA was a codon-optimized sequence containing the following sections (5’ to 3’); 3’UTR, human CD8 signal peptide, mouse kappa variable region, scFv linker, mouse IgH variable region, human CD8 hinge/transmembrane region, human CD28 signal domain, human CD3 signal domain, mouse alpha globin 5’UTR, and poly(A) tail (Figure 1A). The mRNA is synthesized by in vitro transcription from a linearized DNA plasmid. Mock RNA electroporated T cells are used as controls. Upon manufacturing, cells were frozen in cryomedia containing 10% DMSO, and were stored at −80°C until the day of use. Upon thawing, cells were incubated with LGM3 (Lonza), 5% Serum, and 2 ng/ml of human IL-2 (I2644, Sigma).
Fig. 1. Descartes-08 cells are highly viable and significantly express BCMA CAR following mRNA transfection and freezing-thawing procedure.
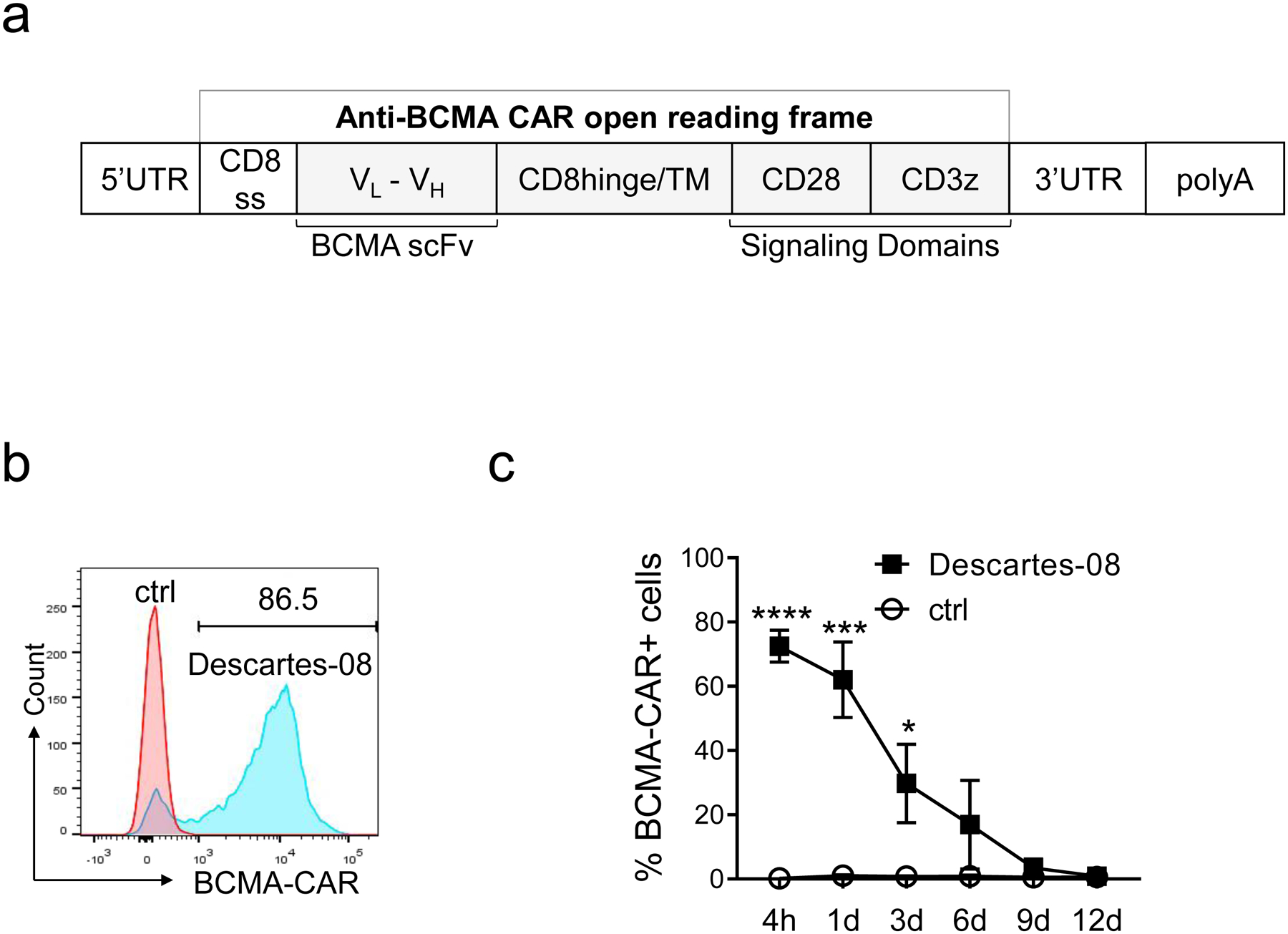
a Schematic representation of in vitro transcribed mRNA encoding anti-BCMA-CAR (Descartes-08). b The CD8+ T cells were activated and then transfected with mRNA encoding BCMA CAR for 4h, followed by flow cytometry (FCM) analysis to evaluate the percentages of BCMA-CAR on Descartes-08 and the paired control (ctrl) CD8+ T cells. Shown are histograms from a representative transfection among three experiments using three healthy donor T cells. c Frozen Descartes-08 cells generated via mRNA transfection (n=3) were thawed and the expression of BCMA CAR was then evaluated by FCM analysis from 4 hours (4h) to 12 day (12d). Data are shown as means ± SDs (error bars) from three independent experiments using three healthy donors, with each condition in triplicate. *P<.05, ***P<.001, ****P<.0001
Analysis of Descartes-08 cells
Viability of Descartes-08 cells was assessed using Acridine Orange and Propidium Iodide staining on the K2 Cellometer (Nexcelom Biosciences). CAR expression was determined by incubating CAR T-cells with 0.4 μg/ml of PE-conjugated recombinant BCMA (Recombinant human BCMA/TNFRSF17 protein, Fc/His-tagged, R-PE labeled; Creative Biomart, Shirley, NY) for 20 minutes at room temperature in FACS Buffer (DPBS, 0.5% BSA, 0.01% Sodium Azide). Propidium Iodide was added at 1 μg/ml to stain non-viable cells. Cells were washed with FACS Buffer and resuspended before analysis on a Guava® EasyCyte® 12HT Flow cytometer (EMD Millipore). More than 3000 events were acquired using InCyte flow cytometry software. Electronic gating was used to exclude dead cells (PI fluorescence in the Near Infrared channel), and CAR expression was evaluated by BCMA-PE staining (PE fluorescence in the Yellow channel).
Cytotoxicity assay
Cells (BM mononuclear cells (BMMCs) or CD138+ cells from MM patients, MM cell lines were co-incubated with Descartes-08 or control CD8+ cells. After indicated incubation periods, cells were stained with antibodies (CD138 and CD8) and analyzed by flow cytometry analysis. The LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Invitrogen) was routinely used to determine the viability of cells and in conjunction with PE-Annexin V (BioLegend) to determine if cells were apoptotic. Cytotoxicity was determined by the depletion of the percentage of viable CD138+ cells. The lysis of MM cells was determined by the summary (means ± SDs) of the following formula: Percentage of lysis (%) = 100 x (1 - (viable CD138+ cell fraction of the indicated T cell group/viable CD138+ cell fraction in control T cell group)).
Animal model study
MM1S-luc cells were harvested and injected on day 0 at 2 × 106 cells per mouse intravenous (i.v.) into 6–9-week-old male NSG mice (NOD.Cg-PrkdcSCIDIl2rgTM1wJ1/SzJ, Jackson labs). Tumor growth was monitored during the study using bioluminescence imaging (BLI). BLI measurement and randomization into treatment groups (n=4 per group) was performed on day 6. On days 7, 14, 21 and 28, CAR T-cells were thawed from cryovials, washed, and resuspended for administration via i.v. injection with vehicle, mock-transfected CD8+ or Descartes-08 cells (20 × 106 cells per mouse). Pre-conditioning of mice was performed on days 13, 20, and 27 by administration of cyclophosphamide at 1.2 mg/mouse via intraperitoneal (i.p.) injection [29, 30]. Descartes-08 cells were further engineered to reduce expression of T cell receptor (TCR) by CRISPR/Cas9 deletion of the Tcra gene (with ~95% knockout efficiency) to inhibit non-specific xenogeneic and allogeneic responses with repeat-dose CAR T-cell infusions [31]. Animals were monitored twice daily. Animals showing obvious signs of distress or pain including inability to drink or feed or a >20% reduction in body weight were humanely euthanized prior to study end according to Institutional Animal Care and Use Committee-approved protocol.
Soluble BCMA (sBCMA) analysis
During the course of an IRB approved ongoing clinical study NCT03448978 in RRMM, clinical specimens were collected for free immunoglobulin light chain and soluble BCMA (sBCMA) analysis. Specimens were analyzed for subject 103–016 who achieved an ongoing (i.e. >9 month) sCR by International Myeloma Working Group (IMWG) criteria. Circulating sBCMA was determined by ELISA (Human BCMA/TNFRSF17 kit, R&D Systems).
Statistical analysis
The results obtained from in vitro experiments were expressed as the means ± standard deviations (SDs). All experiments were performed in triplicate at each condition with T cells from multiple donors or patients (n ≥ 3), as well as multiple MM cell lines and patient MM samples. Multiple groups (≥3) were analyzed by one-way ANOVA, and paired groups were analyzed by two-way ANOVA or Student t test. All data were graphed and analyzed using GraphPad Prism 8.1.2 (San Diego, CA). P<.05 was considered statistically significant.
Tumor growth curves in vivo were analyzed by 2-way ANOVA after log-transformation of data. The survival curve in our animal model was plotted by using the Kaplan-Meier method, and compared by the log-rank test.
Results
Decartes-08 T cells express high levels of BCMA CAR following mRNA transfection
Using mRNA electroporation, highly activated CD8 T cells from healthy donors (n=3) were generated and transduced with BCMA-CAR (Descartes-08) or control mRNA (Fig. 1a). Transfection efficiency remained > 72.48 ± 4.9%, ranging from 57.1% to 86.5% for three donors, as determined by the percentage of BCMA CAR expression on Descartes-08 cells after being thawed (Fig. 1b, Fig. S1A–B). The expression of BCMA CAR on Descartes-08 cells was sustained for approximately 1 week, and gradually declined until 9 days (9d) after thawing following their generation (Fig. 1c). At the end of 9 days, the viability of Descartes-08 and the paired control CD8+ T cells were reduced but comparable (Fig. S1C). These data defined the time interval of functional Descartes-08 cells in vitro.
Descartes-08 demonstrates selective cytolytic function to deplete MM cells
We next evaluated degranulation of lytic cytosolic vesicles by staining for upregulation of the CD107a degranulation marker [32–34] on the surface of Descartes-08 cells vs paired control CD8+ T cells co-cultured with MM cells expressing various (up to 6-fold) levels of surface BCMA [20, 35, 36] (Fig. S2A). All BCMA-expressing MM target cells (n=4) triggered degranulation of Descartes-08, as evidenced by significant induction of surface CD107a expression in Descartes-08 compared with the paired control CD8+ T cells (n=3) (Fig. 2a, Fig. S2B–D). At the low Effector: Target (E:T) ratio of 1:1 for 6h co-incubation, degranulation (CD107a) was increased to >5–12 fold in Descartes-08 vs the paired control T cells, regardless of BCMA levels on MM target cells.
Fig. 2. Descartes-08 cells induced potent cytolytic function to deplete MM cells in the ex vivo co-cultures.
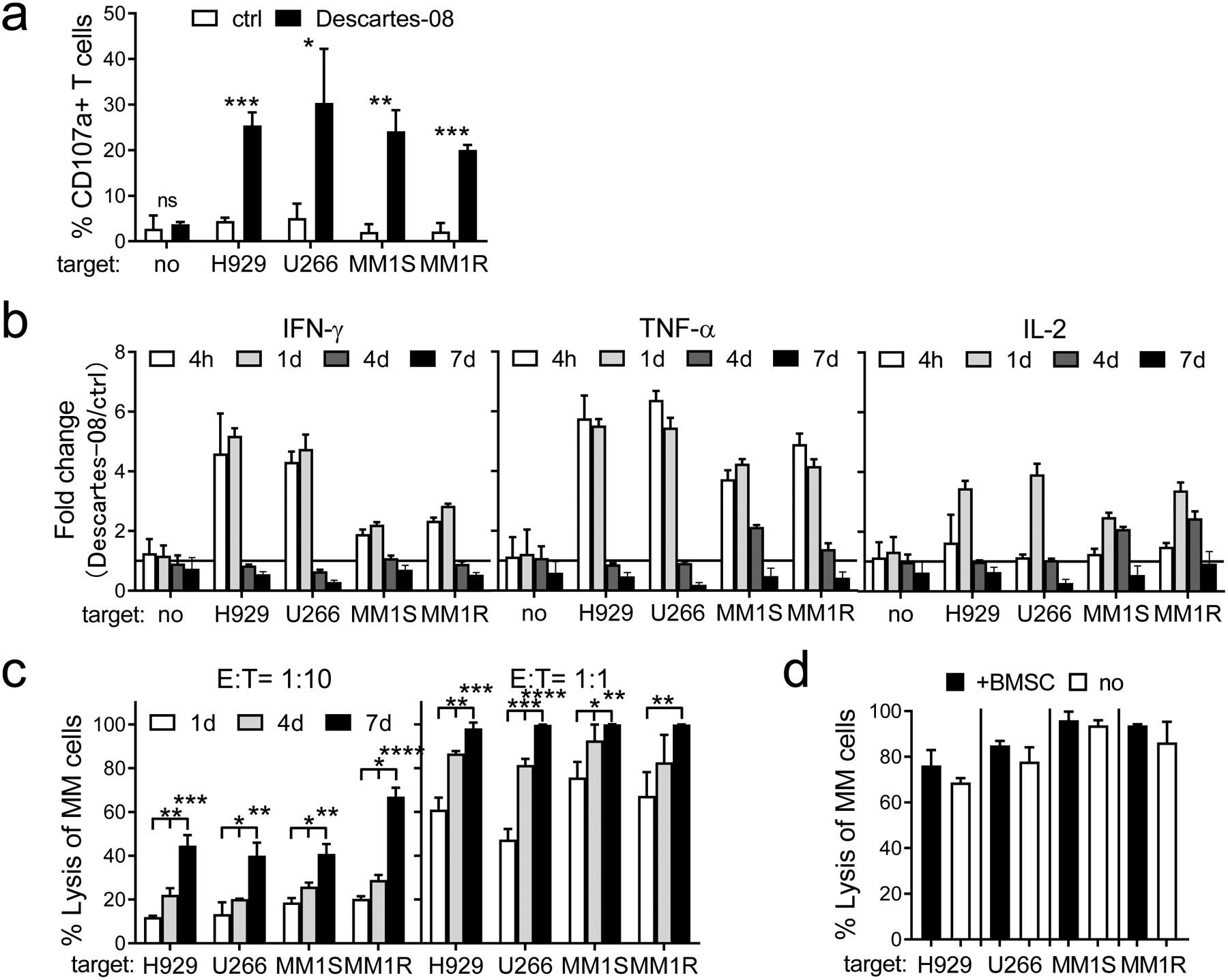
a Descartes-08 and the paired control (ctrl) CD8+ T cells (n=3) were co-cultured for 6h with 4 indicated target MM cell lines at the low E:T ratio of 1:1, followed by FCM analysis to determine the percentages (means ± SDs) of % CD107a+ cells in Descartes-08 vs ctrl T cells. b Indicated cytokines produced in Descartes-08 vs the paired ctrl CD8+ T cells (n=4) were evaluated by FCM analysis at indicated time periods (4h, 1d, 4d, 7d) following the co-culture with indicated target MM cell lines (n=4) (E:T=1:10). Fold changes of % indicated cytokines in Descartes-08 vs the paired ctrl T cells were determined by % in Descartes-08 with indicated MM cell lines vs % in the paired ctrl T cells co-incubated with the same target cell lines at the same time periods. c Descartes-08 or the matched ctrl CD8+ T cells (n=3) were co-cultured with MM cell lines at the E:T ratio of 1:10 (left) or 1:1 (right). MM cell lysis in the co-cultures was determined at indicated time points (1d, 4d, 7d). d Descartes-08-induced MM cell lysis, in the presence (+) or absence (no) of BMSCs, was determined at the E:T ratio of 1:5 following 7d-co-culture. All data are presented as means ± SDs of three independent experiments, with triplicates for each condition using 3 healthy donors. ns, not significant; *P<.05, **P<.01, ***P<.001, ****P<.0001
We also examined Th1 cytokine induction in Descartes-08 cells (n=4) co-cultured with indicated target MM cells (n=4). Interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), key cytokines for effector cell function, were significantly increased (P<.05) after 6h and continued at d1 (Fig. 2b, Fig. S2E). Descartes-08 induced IFN-γ and TNF-α by 1.7–6.1-fold and 3.4–6.6-fold, respectively. Both cytokines gradually declined to baseline between d4 and d7. Interleukin-2 (IL-2), a marker of CAR T-cell potency and persistence [37–39], gradually increased at d1 by 2.3–4.2 fold and then diminished by d7. The normalization of these key inflammatory cytokines by day 7 to the baseline coincided with the time-limited expression of the BCMA CAR in Descartes-08 cells (Fig. 1c).
Next, specific MM cell killing by Descartes-08 was determined by staining with the CD138 MM cell marker and quantification of depleted live CD138+ cells by flow cytometry analysis. One day after co-culture at low E:T ratios of 1:1 and 1:10, Descartes-08 cells (n=4 healthy donors) triggered lysis of 45–75% and 10–20% of MM cell lines (n=4), respectively (Fig. 2c). Descartes-08 cells induced MM cell lysis in the E:T ratio- and time-dependent manner. MM cell lysis was cumulative and dose-dependent over 7 days in the co-cultures (98–99.9% and 40–67% at the E:T ratios of 1:1 and 1:10, respectively, at the end of 7 days). In contrast, Descartes-08 T cells induce no significant degranulation and cytolytic function when co-cultured with BCMA-negative target cells with non-plasma cell lineages (293T, HL-60) (Fig. S3A–C).
We further co-incubated Descartes-08 cells with indicated target MM cell lines (n=4) at an E:T ratio of 1:5, in the presence or absence of patient-derived BM stromal cells (BMSCs) which promote growth, survival and drug resistance of MM cells in the BM microenvironment. At day 7, Descartes-08 cells induced comparable MM cell lysis in the co-cultures with or without BMSCs, suggesting that Descartes-08 cells could induce MM cell lysis in the BM milieu (Fig. 2d). In addition, 100 ng/ml APRIL, a major PC survival factor binding to BCMA in the MM BM microenvironment [33, 35, 40, 41], did not alter Descartes-08 cell-induced cytolysis of MM target cells in 7 day-co-cultures (Fig. S3D).
Key T cell activation and checkpoint proteins are transiently induced on Descartes-08 cells via a BCMA-specific manner
Expression of key T cell activation and checkpoint molecules was followed in the 7d-co-cultures with various MM target cells (n=4). Compared with the paired control CD8+ T cells (n=3), Descartes-08 cells showed significantly (P<.05) enhanced expression of CD25 (24.7–48.7-fold), PD1 (3.4–5.5-fold), TIM3 (1.3–2.2-fold), and LAG3 (5.9–12.9-fold) after 1d co-incubation with all MM target cells (n=4, Fig. 3, Fig. S4). Levels of these T cell proteins remained low and unchanged in paired control CD8+ T cells in the presence or absence of target MM cells during co-cultures. The expression of CD25, PD1, and LAG3 decreased on subsequent days to baselines between d4 and d7. TIM3 and CD38 expression continued to increase until d4 (1.3–4.5-fold in TIM3, 1.7–2.7-fold in CD38), and then decreased until d7 (Fig. S4). Thus, a transient induction of T cell activation and checkpoint proteins also confirmed the time course of functional Descartes-08 cells for approximately a week after their generation.
Fig. 3. Descartes-08 T cells showed transient upregulation in T cell activation and checkpoint proteins in a BCMA-selective manner.
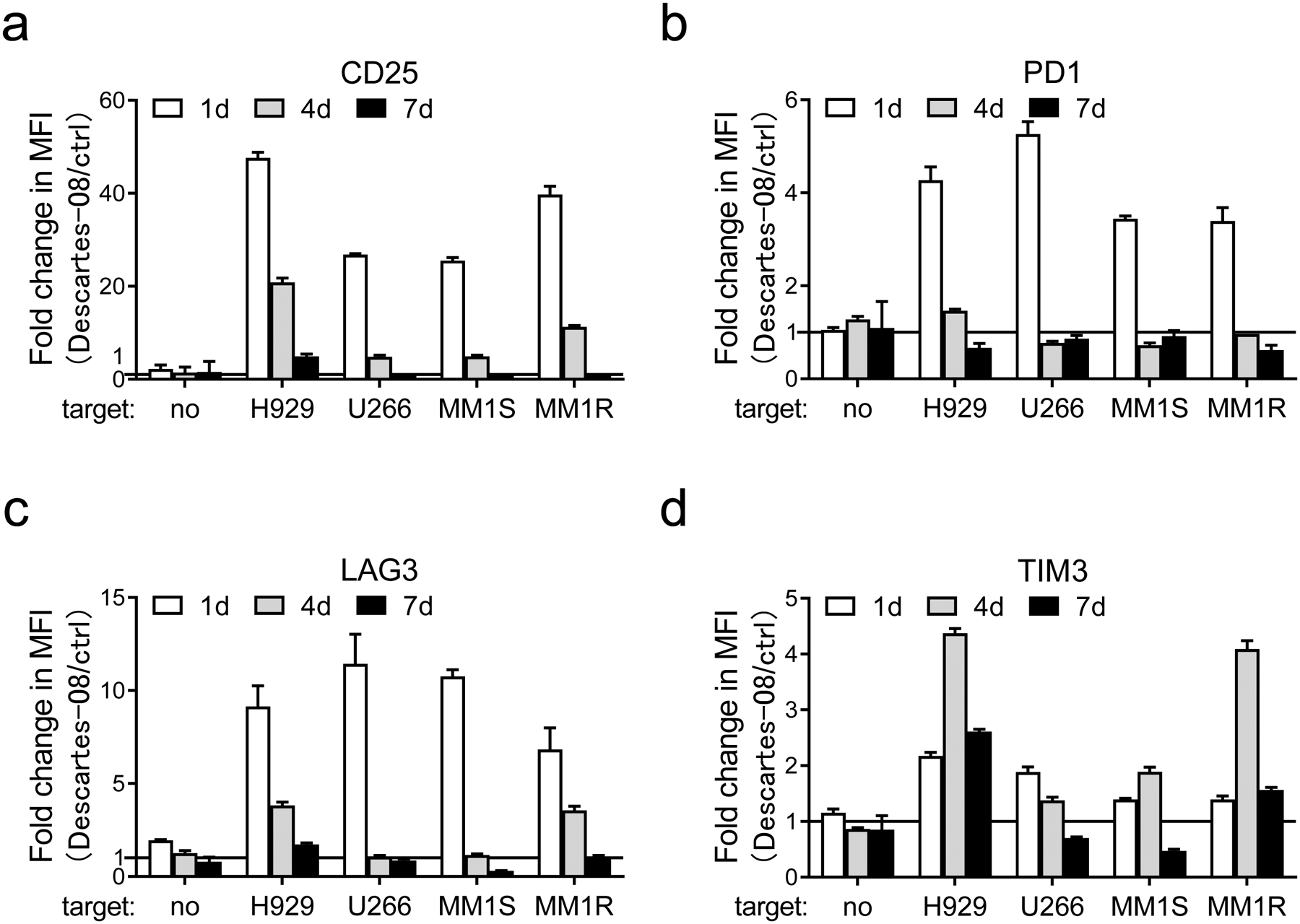
a-d Descartes-08 and the paired control (ctrl) CD8+ T cells (n=3) were co-cultured with various target MM cell lines (n=4) (E:T=1:10) or no target MM cells (no). Using FCM analysis, changes in CD25 (a), PD1 (b), LAG3 (c), and TIM3 (d) on T cells were determined by values of median fluorescence intensity (MFI) for the individual protein marker at indicated time periods (1d, 4d, 7d). Fold changes of MFI values of indicated proteins in Descartes-08 vs the paired ctrl T cells were determined by MFI values in Descartes-08 co-incubated with indicated MM cell lines vs those in the paired ctrl T cells co-incubated with the same target cell lines at the same time periods. Three independent experiments were done in triplicates at each condition using T cells from 3 individuals. All data were expressed as means (fold changes) ± SDs.
Decartes-08 significantly induces cytotoxic effect on IMiDs-resistant MM cells, alone or in the presence of IMiDs
The activity of Descartes-08 cells against IMiDs-resistant MM cells was assayed in co-cultures of lenalidomide/pomalidomide (Len/Pom)-resistant MM cells with Descartes-08. Len/Pom-resistant H929(R) and MM1S(R) were derived from H929 and MM1S cell lines, respectively [36]. Compared to the paired control CD8+ T cells, Descartes-08 cells showed elevated CD107a membrane expression following 6h co-culture at the E:T ratio of 1:1, and depleted Len/Pom-resistant cells to a similar extent as they did to eliminate sensitive parental MM cells after 1d co-incubation (Fig. 4a–b, Fig. S5). We further incubated Descartes-08 cells with these paired MM cell lines at an even lower E:T ratio of 1:10, in the presence of Len or Pom, and observed a cumulative cytotoxic effect of Len or Pom with Descartes-08 to deplete MM cells (P<.05, Fig. 4c). These results indicated that Descartes-08 cells effectively lysed IMiDs-resistant MM cells and were highly active in the presence of IMiDs.
Fig. 4. IMiDs-resistant MM cell lines were highly susceptible to Descartes-08 cells.
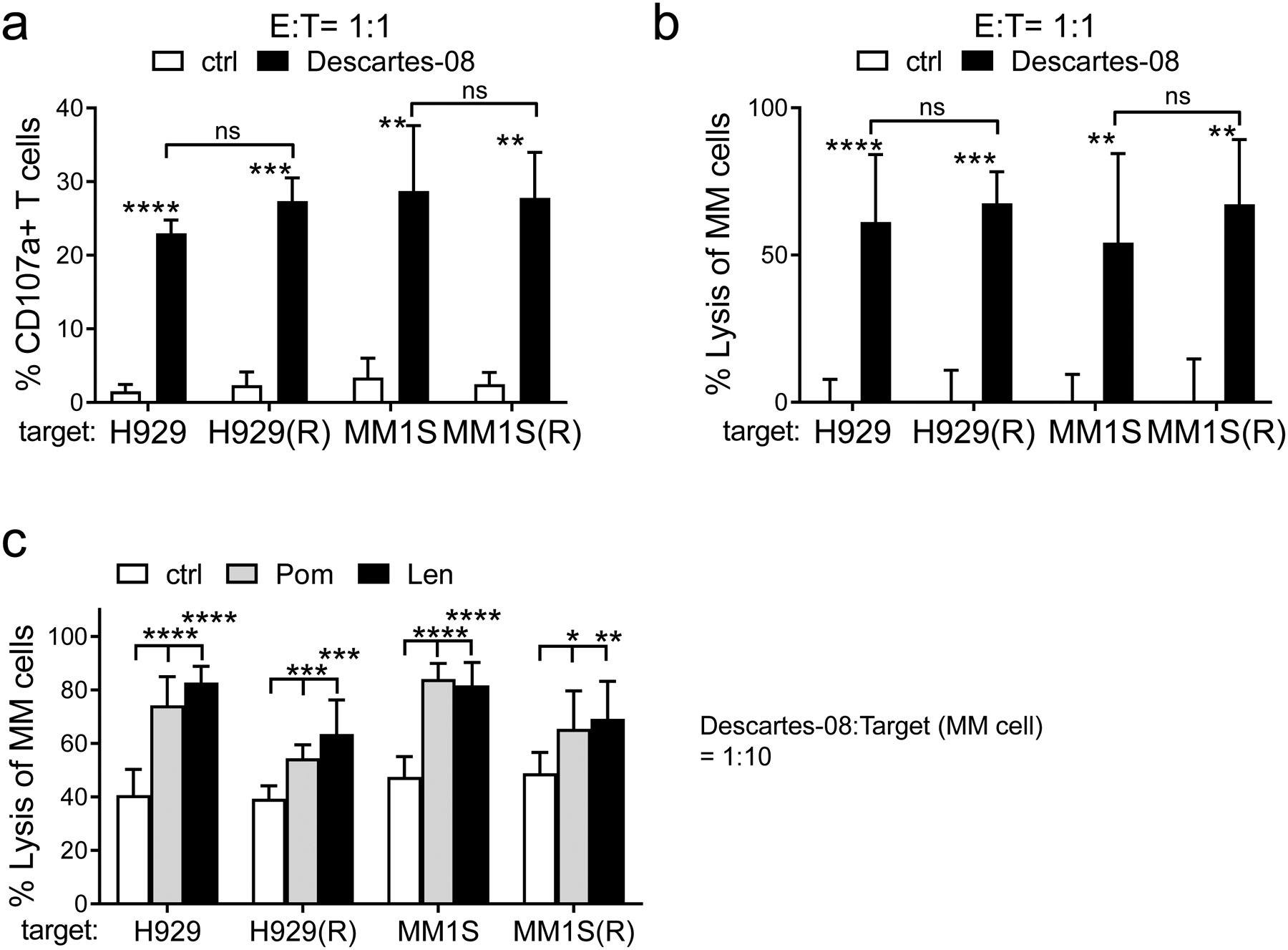
a-b Descartes-08 or paired control (ctrl) CD8+ T cells (n=3) were co-cultured with IMiDs-resistant (H929(R) or MM1S(R)) or their parental sensitive (H929 or MM1S) target MM cells [36] (E:T=1:1). The percentages of CD107a+ (a) and the cytolysis activity (b) of indicated T cells were determined at 6h (a) and 1d (b), respectively. c Descartes-08 T cells were co-cultured with indicated MM cells (E:T=1:10), in the presence of 5μM of Len or Pom or control medium (ctrl). All data are presented as means ± SDs from three independent experiments, using T cells from 3 individuals. ns, not significant; *P<.05, **P<.01, ***P<.001, ****P<.0001
Decartes-08 cells effectively lyse primary MM patient cells derived from bone marrow samples
To study the anti-MM effect of Descartes-08 cells on patient MM samples, we performed T cell-mediated cytolytic functional assays using target cells as total BMMCs as well as purified CD138+ cells from patients with newly diagnosed (n=4) and RRMM (n=4). Upon co-culture with patient cells in each group, Descartes-08 cells significantly upregulated cell membrane CD107a degranulation (>5-fold) compared with paired control CD8+ T cells (P<.01, Fig. 5a, Fig. S6A). CD138+ patient MM cells were selectively and significantly eliminated, with depletion of the CD138+ fraction by 82 ± 14% and 66 ± 17% from BMMC and CD138+ cell groups, respectively (P<.01, Fig. 5b). Since accessory cells supporting MM cell growth, survival, and drug resistance were present within BMMCs from patient BM aspirate samples, these results further suggest that Descartes-08 could kill patient MM cells in the MM BM microenvironment.
Fig. 5. Descartes-08 significantly induced cytolysis of primary MM patient samples.
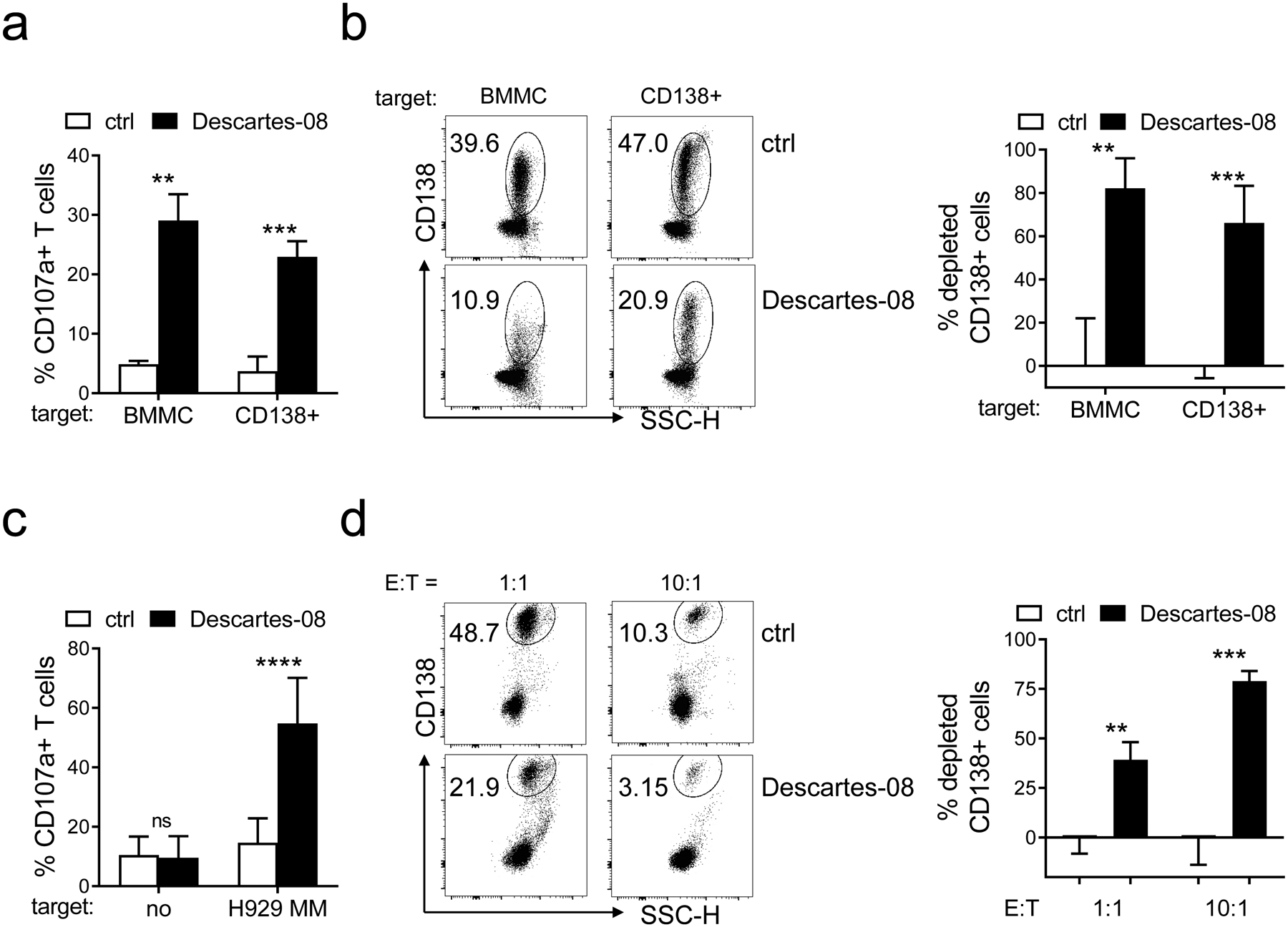
a-b Descartes-08 or the paired control (ctrl) CD8+ T cells were co-cultured with BMMCs (n=4) or purified CD138+ cells collected from MM patient samples (n=4) (E:T=1:1), respectively. Patient samples include newly diagnosed (n=2) and RRMM (n=2) for each BMMC and CD138+ cell test groups. The percentages of CD107a+ (means ± SDs) in indicated T cells were evaluated at 6h after co-culture (a). b The cytolytic ability of Descartes-08 to deplete CD138+ cell was evaluated overnight after co-culture with target patient cells from BMMC or purified CD138+ cell groups. Shown are the FCM dot plots of one representative sample from each group (left) and summarized results of % depleted CD138+ cells (means ± SDs) (right). c-d CD8+ T cells were purified from peripheral blood of MM patients (n=6) followed by transfection with Descartes-08 mRNA. Cytolytic activity of patient-derived Descartes-08 was evaluated by percentages of CD107a upregulation (c) and depletion of H929 target MM cells (CD138+) (d), respectively. CD138 was used to identified viable patient MM and H929 MM cells. All data are presented as means ± SDs. ns, not significant; **P<.01, ***P<.001, ****P<.0001
Functional anti-MM activity of Descartes-08 generated from patient-derived CD8+ cells
We next confirmed Descartes-08 derived from patients induced anti-MM lytic activity with comparable potency. Patient PBMCs were used to generate Decartes-08 cells following mRNA transfection, and their cytolytic activity against MM cells was evaluated in the co-culture. Following co-incubation of H929 MM target cells, cell surface CD107a expression was upregulated > 6-fold on patient-derived Descartes-08 cells compared to the paired control patient T cells (n=6) (P<.0001, Fig. 5c, Fig. S6B). Patient-derived Descartes-08 cells effectively eliminated H929 target cells in overnight co-cultures in an E:T ratio-dependent manner (P<.01, Fig. 5d). Percentages of depleted MM cells were 39±21% and 79±12% at E:T ratios of 1:1 and 10:1, respectively.
Descartes-08 cells potently inhibit in vivo tumor growth and prolong host survival in a therapeutic mouse model of human MM
We finally tested cytolytic activity of Descartes-08 cells in vivo in NSG mice bearing the aggressive disseminated human MM model of MM1S-luc cells [20, 42]. We and others had previously observed that unmodified control T cells or other human leukocytes contribute to the control of MM1S-luc in SCID mice models [31] (Fig. S7A). The xenogeneic environment of the mouse enables activation of transferred human T cells and may contribute to this control. In order to remove both xenogeneic graft versus host disease (GVHD) reactivity and any potential T cell receptor (TCR)-mediated anti-MM response, Descartes-08 and the paired control CD8+ T cells were generated which had been modified to lack the TCR by CRISPR/Cas9 genetic engineering [43]. NSG mice were injected with 2 × 106 MM1S-luc cells through i.v. administration and growth of MM1S-luc was monitored by BLI imaging (Fig. 6a). Descartes-08 or control CD8+ T cells were administered once per week from day 7 (Fig. 6b–c). BLI signal was highly consistent between mice within each group, with the signal by supine imaging concentrated in the lower leg and confirming MM growth in the bones. Mice in all groups remained healthy throughout the study to day 27, with no differences in body weight seen between groups (Fig. S7B). Animals showed clinical signs of MM progression, as expected, after day 28, with reduced body weight evident in control vehicle and control CD8+T cell groups. Median survival for mice in vehicle-treated and control CD8+ T cell-treated groups was 43 and 44 days, respectively (Fig. 6c). The cohort treated with Descartes-08 with deleted TCR showed a statistically significant increase in median survival to 69 days (Kaplan-Meier; P<.0001). Thus, Descartes-08 CAR T-cells demonstrated in vivo anti-MM activity in the MM1S-luc NSG mouse model, in which confounding effects caused by the TCR on these cells have been eliminated. Importantly, Descartes-08 CAR T-cells when given intermittently could control the growth of MM1S-luc MM in NSG mice and significantly prolong host survival.
Fig. 6. Descartes-08 cells with TCR-knockout significantly blocked disseminated MM1S tumor growth in NSG mice, leading to prolonged host survival.
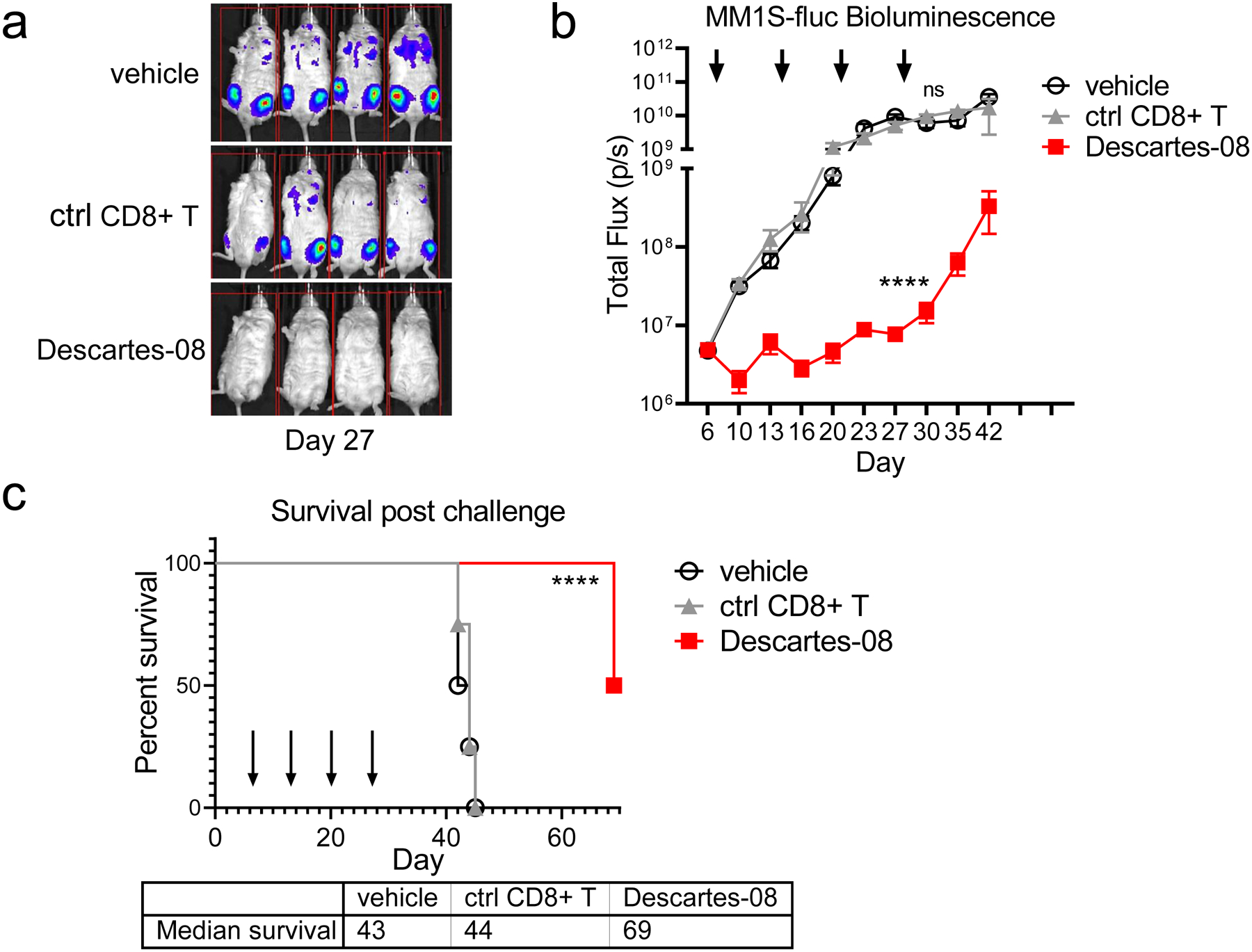
NSG mice receiving MM1S-luc cells (2 × 106 cells per mouse i.v.) at day 0 were randomized into treatment groups (n=4 per group) on day 6. On days 7, 14, 21 and 28, T-cells were thawed from cryovials, washed, and resuspended for administration via i.v. injection with vehicle, 20 × 106 mock-transfected control TCR knockout (KO) CD8+ (ctrl CD8+) or Descartes-08 cells (showing 67% BCMA CAR expression by FCM analysis). On day 13, 20 and 27, all mice were treated with 60 mg/kg cyclophosphamide. a Bioluminescence (BLI) images (MM1S-fluc growth) of mice in each group on day 27 (after 3 doses of Descartes-08 cells or controls). Tumor growth (b, means ± SDs) and survival (c) of animals are shown through timeframe of study. b-c Each of the 4 dosing days was marked with an arrow. Median survival days are 43 (vehicle), 44 (ctrl CD8+ T cells) and 69 (Descartes-08 CAR T) days. Survival curves of TCR KO Descartes-08 (Descartes-08) was significantly different (P<.0001) compared with paired control TCR KO CD8+ T cells (ctrl CD8+ T) by log-rank (Mantel-Cox) test. Tumor growth curves were analyzed by 2-way ANOVA after log-transformation of data. Survival curve in animal model was plotted using the Kaplan-Meier method and compared by the log-rank test. ****P<.0001
Descartes-08 cells can induce deep and durable clinical response with favorable side effect profile
A 76-year-old female patient with plasma cell leukemia and relapsed/refractory disease was enrolled into an IRB approved clinical trial of an mRNA anti-BCMA CAR T-cell therapy in RRMM (NCT03448978). At the time of enrollment, the patient had a free kappa was 1445.4 mg/L (K/L ratio=438), BM biopsy showed 60–90% cellularity, and BM aspirate demonstrated 60% myeloma cells. The patient received lymphodepletive therapy followed by 3 infusions of 2.4×109 viable CAR+ cells (Descartes-08) over 2 weeks. There were no associated adverse events related to the Descartes-08 CAR T-cell infusion. At 4 weeks, the patient was noted to have a very good partial response (VGPR) by IMWG criteria. At 12 weeks, the patient achieved a sCR with undetectable serum M-spike, free kappa 1.8 mg/L (kappa/lambda ratio 1.2), and negative urine immunofixation. BM biopsy and FISH analysis did not detect MM cells. Serum sBCMA levels, known to be a highly correlative marker of disease response [23, 27], correlated with clinical response (Figure 7).
Fig. 7. Biomarkers of disease burden in a patient treated with Descartes-08.
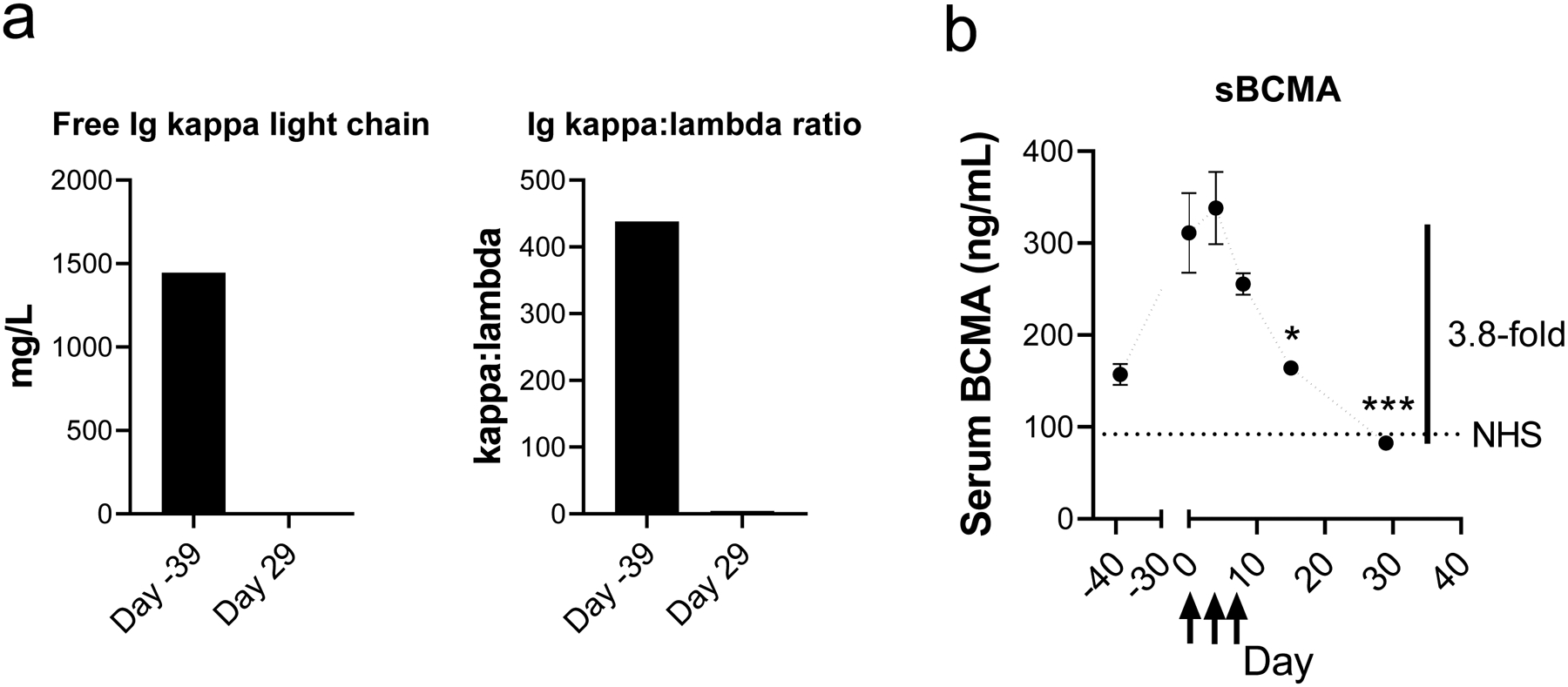
Serum samples were collected from patient 103–016 in NCT03448978 and analyzed for free immunoglobulin light chains (a) or by soluble BCMA (sBCMA) ELISA (b). Arrows indicate days of infusion of Descartes-08 on days 0, 3, and 7. NHS indicates measured level in a sample of normal human serum from pooled normal donors. A 3.8-fold drop in sBCMA level was observed from peak level till Day 29 post infusion. *P<.05, ***P<.001
Discussion
CAR T-cells targeting BCMA are positioned to transform treatment of MM [24, 27, 44, 45]. Specifically, virally-generated anti-BCMA CAR T-cells have shown impressive clinical results with high frequency and deep extent of response in heavily-pretreated RRMM patients [25–27, 46]. Thus, adoptive BCMA CAR T-cells may induce specific anti-MM immunity to extend remission. However, the utility of CAR T-cells for all but the most advanced cases of MM has been limited by serious toxicities such as CRS and neurotoxicity, which are common and can be life-threatening, as well as durability of response. In fact, the benefits of conventional virally generated CAR T-cells have appeared to be inseparably correlated with their toxicities. To address these variables affecting clinical potency and safety of CAR T therapies, several prediction models and newer techniques are under development to improve CAR constructs exerting high anti-tumor capacity, while reducing the risk of CRS and improving the treatment protocol [39, 47–49]. Novel forms of structure-modified CARs are being explored, including those incorporating humanized binding domains to reduce immunogenicity, alternative CAR spacer designs and costimulatory domains, and multiple epitopes against target antigens to enhance reactivity [24, 44]. Also explored are alternative gene transfer vectors, as well as devising other gene transduction methods to rationally control quality and quantity of CAR T-cells, while reducing off-target acute toxicity.
Descartes-08 is the first BCMA CAR T-cell therapy to use mRNA delivery to engineer CAR protein with a defined half-life. Using in vitro transcribed mRNA [49–51] to transfect CD8 T cells, Descartes-08 is rapidly generated with high transfection efficiency. As mRNA does not integrate into the host genome, there is no risk of transgene-mediated mutagenesis, and there is no unidentified long-term risk, as inherited by DNA and viral vector transduced CAR T-cells. Importantly, Descartes-08 cells express BCMA CAR at high levels in >80% expanded CD8 T cells and exhibit a limited duration of active phase for a maximum of 5–7 days in vitro. After stimulation with target MM cells, CRS-related cytokines including IFN-γ and TNF-α were induced and then decreased to baseline levels at 7 days. Such temporal changes are tightly associated with levels of BCMA CAR expression on Descartes-08. These results indicate that Descartes-08 will have restricted proliferation in vivo, and that its expansion kinetics will differ from other products, where early and height of expansion post infusion correlates with acute toxicity. The limited one-week active phase of Descartes-08 may reduce risk of acute complications by CRS, a distinct feature of Descartes-08 versus viral vector-modified CAR T-cells. Moreover, since Descartes-08 is efficient and transient, it can be dosed repetitively.
CD8 T cells were used to generate Descartes-08 with rapid and robust MM cell killing in vitro and in vivo. In order to provide meaningful preclinical data in the immunocompromised NSG mouse, it proved beneficial to remove confounding effects from the TCR of the transferred cells. Indeed, a well-established graft-versus-myeloma effect occurs in immunocompromised mouse models where major histocompatibility complex (MHC) reactivity of graft T cells against the allogeneic myeloma cell and/or the xenogeneic tissues of the mouse provides a large anti-tumor effect [31]. As a consequence, these myeloma models have major limitations when studying autologous cell gene therapy and attempting to assign efficacy to direct interaction between a CAR and target cells. Our approach of genetically deleting the TCR of transferred CD8+ T cells through CRISPR/Cas9 editing completely eliminated the baseline graft-versus-myeloma effect, thus providing a novel and highly specific method to evaluate effects of cell and gene therapy that are likely to provide more translational relevance for autologous cell products.
The manufacturing process is clinically scalable, with high purity and viability of Descartes-08 cells following cryopreservation. Descartes-08 cells, after being manufactured, frozen, and thawed, significantly lyses MM cells. Descartes-08 cells exhibits rapid CD107a degranulation and IFN-γ production followed by effective lysis of MM cell lines and primary MM cells from patients, regardless of BCMA expression levels and resistance to current anti-MM therapies including dexamethasone, PIs, lenalidomide, and pomalidomide. Co-treatment of Descartes-08 with IMiDs induce higher cytotoxic effects, suggesting the potential for combination treatment strategies to enhance its efficacy and durability. Moreover, in animal model studies, Descartes-08 cells significantly and potently inhibited progression of MM cell growth and survival, prolonged host overall survival, and illustrated a potential advantage of continual serial treatment. The short half-life of Descartes-08 cells may make it amenable to repeat dosing. While this question is best answered in the ongoing clinical trials, it is anticipated that the product, when integrated into a frontline setting, will benefit from a short (e.g., 2–3 week) repeat dosing regimen between the end of induction therapy, but prior to transplantation. We believe that if lymphodepetion is required during this time frame, it would not need to be re-administered to achieve minimal residual disease (MRD)-negative responses. Furthermore, Descartes-08 safety profile enables its use in a frontline setting. Its potential use following induction therapy and before auto-transplantation may lead to higher rates of durable MRD-negative responses in high-risk patients.
In conclusion, Descartes-08 cells demonstrate high levels of degranulation, cytokine secretion, proliferation, and cytotoxicity against MM cell lines and patient MM cells, regardless of drug resistance and in a specific, dose-, and time- dependent manner. Descartes-08 can be administered at high numbers (e.g., several billion cells) and at fixed levels selected to provide clinical efficacy while avoiding side effects attendant to conventional viral CAR T-cells, thereby improving the therapeutic index. Likewise, the duration of exposure to Descartes-08 can be controlled by repetitive dosing since Descartes-08 has defined pharmacokinetics, including a defined half-life. The pre-clinical efficacy and tolerability observed in our study provides the framework for therapeutic evaluation of Descartes-08 in both relapsed refractory MM and as initial therapy of newly diagnosed MM.
Supplementary Material
Key points:
Descartes-08 is the first anti-BCMA CAR T for use in MM patients that is manufactured using non-viral mRNA.
Descartes-08 significantly and potently inhibited progression of myeloma in vivo and prolonged overall host survival.
Acknowledgements
The authors acknowledge the technical assistance from the flow cytometry facility at Dana-Farber Cancer Institute (DFCI). We thank Dr. Jiye Liu, Dr. Wenjuan Yang, and all other lab members, as well as the clinical research coordinators at the Jerome Lipper Multiple Myeloma Center and the LeBow Institute for Myeloma Therapeutics of the DFCI, for support and help in providing primary tumor specimens for this study.
Funding
This work was supported in part by grants from the National Institutes of Health Specialized Programs of Research Excellence (SPORE) P50 CA100707, P01CA155258, RO1 CA 207237, and RO1 CA 050947. This work was supported in part by Dr Miriam and Sheldon G Adelson Medical Research Foundation and the Riney Family Multiple Myeloma Initiative. Dr. Kenneth C. Anderson is an American Cancer Society Clinical Research Professor.
Footnotes
Conflict of interest disclosure:
M.K., Y.Z. and C.A.S. are employees of and hold stock interest in Cartesian Therapeutics. N.C.M. serves on advisory boards to Millennium-Takeda, Celgene, and Novartis. K.C.A. serves on advisory boards Celgene, Millennium-Takeda, Bristol-Myers Squibb, Gilead Sciences, Janssen, and Sanofi-Aventis and is a Scientific founder of OncoPep and C4 Therapeutics. All other authors declare no competing financial interests.
References
- 1.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nature communications. 2014;5:2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar SK, Anderson KC. Immune therapies in multiple myeloma. Clin Cancer Res. 2016;22:5453–60. [DOI] [PubMed] [Google Scholar]
- 3.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–13. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Tai YT, Ho M, Xing L, Chauhan D, Gang A, et al. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017;7:e547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tai YT, Cho SF, Anderson KC. Osteoclast immunosuppressive effects in multiple myeloma: role of programmed cell death ligand 1. Front Immunol. 2018;9:1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghobrial IM, Detappe A, Anderson KC, Steensma DP. The bone-marrow niche in MDS and MGUS: implications for AML and MM. Nat Rev Clin Oncol. 2018;15:219–33. [DOI] [PubMed] [Google Scholar]
- 7.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nature communications. 2019;10:3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan Y, Mao X, Liu J, Fan H, Du C, Li Z, et al. The impact of response kinetics for multiple myeloma in the era of novel agents. Blood Adv. 2019;3:2895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An G, Yan Y, Xu Y, Mao X, Liu J, Fan H, et al. Monitoring the cytogenetic architecture of minimal residual plasma cells indicates therapy-induced clonal selection in multiple myeloma. Leukemia. 2020;34:578–88. [DOI] [PubMed] [Google Scholar]
- 10.Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018;32:252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. 2018;15:31–46. [DOI] [PubMed] [Google Scholar]
- 14.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45–56. [DOI] [PubMed] [Google Scholar]
- 16.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–65. [DOI] [PubMed] [Google Scholar]
- 17.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tai YT, Li XF, Breitkreutz I, Song W, Neri P, Catley L, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006;66:6675–82. [DOI] [PubMed] [Google Scholar]
- 19.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19:2048–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy. 2015;7:1187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee L, Bounds D, Paterson J, Herledan G, Sully K, Seestaller-Wehr LM, et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br J Haematol. 2016;174:911–22. [DOI] [PubMed] [Google Scholar]
- 23.Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (BCMA) in multiple myeloma: potential uses of BCMA-based immunotherapy. Front Immunol. 2018;9:1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tai YT, Anderson KC. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin Biol Ther. 2019;19:1143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36:2267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129:2210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380:1726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen AD, Garfall AL, Dogan A, Lacey SF, Martin C, Lendvai N, et al. Serial treatment of relapsed/refractory multiple myeloma with different BCMA-targeting therapies. Blood Adv. 2019;3:2487–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24:717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasian SK, Kenderian SS, Shen F, Ruella M, Shestova O, Kozlowski M, et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood. 2017;129:2395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozemuller H, van der Spek E, Bogers-Boer LH, Zwart MC, Verweij V, Emmelot M, et al. A bioluminescence imaging based in vivo model for preclinical testing of novel cellular immunotherapy strategies to improve the graft-versus-myeloma effect. Haematologica. 2008;93:1049–57. [DOI] [PubMed] [Google Scholar]
- 32.Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23:4290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tai YT, Lin L, Xing L, Cho SF, Yu T, Acharya C, et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: therapeutic implications. Leukemia. 2019;33:426–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tai YT, Horton HM, Kong SY, Pong E, Chen H, Cemerski S, et al. Potent in vitro and in vivo activity of an Fc-engineered humanized anti-HM1.24 antibody against multiple myeloma via augmented effector function. Blood. 2012;119:2074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tai YT, Acharya C, An G, Moschetta M, Zhong MY, Feng X, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127:3225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xing L, Lin L, Yu T, Li Y, Cho SF, Liu J, et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23:2130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res. 2018;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130:2295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood. 2016;128:1590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidts A, Ormhoj M, Choi BD, Taylor AO, Bouffard AA, Scarfo I, et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019;3:3248–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28:155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Agostino M, Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia. 2020;34:21–34. [DOI] [PubMed] [Google Scholar]
- 45.Cho SF, Lin L, Xing L, Li Y, Yu T, Anderson KC, et al. BCMA-targeting therapy: driving a new era of immunotherapy in multiple myeloma. Cancers (Basel). 2020;12:1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116:9543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer discovery. 2016;6:664–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sadelain M CD19 CAR T Cells. Cell. 2017;171:1471. [DOI] [PubMed] [Google Scholar]
- 49.Foster JB, Barrett DM, Kariko K. The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol Ther. 2019;27:747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiesinger M, Marz J, Kummer M, Schuler G, Dorrie J, Schuler-Thurner B, et al. Clinical-scale production of CAR-T cells for the treatment of melanoma patients by mRNA transfection of a CSPG4-specific CAR under full GMP compliance. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster JB, Choudhari N, Perazzelli J, Storm J, Hofmann TJ, Jain P, et al. Purification of mRNA encoding chimeric antigen receptor is critical for generation of a robust T-cell response. Hum Gene Ther. 2019;30:168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.