

ABSTRACT
Although less prevalent than its relative Candida albicans, the yeast Candida glabrata is a successful pathogen of humans, which causes life-threatening candidiasis. It is thus vital to understand the pathogenicity mechanisms and contributing genes in C. glabrata. However, gene complementation as a tool for restoring the function of a previously deleted gene is not standardized in C. glabrata, and it is less frequently used than in C. albicans.
In this study, we established a gene complementation strategy using genomic integration at the TRP1 locus. We prove that our approach can not only be used for integration of complementation cassettes, but also for overexpression of markers like fluorescent proteins and the antigen ovalbumin, or of potential pathogenicity-related factors like the biotin transporter gene VHT1. With urea amidolyase Dur1,2 as an example, we demonstrate the application of the gene complementation approach for the expression of sequence-modified genes. With this approach, we found that a lysine-to-arginine mutation in the biotinylation motif of Dur1,2 impairs urea-dependent growth of C. glabrata and C. albicans. Taken together, the TRP1-based gene complementation approach is a valuable tool for investigating novel gene functions and for elucidating their role in the pathobiology of C. glabrata.
Keywords: Candida glabrata, TRP1, gene complementation, overexpression, green fluorescent protein, ovalbumin, urea amidolyase
Introduction of a gene complementation and reporter gene expression strategy for the human-pathogenic yeast Candida glabrata, based on genomic integration at the TRP1 locus.
INTRODUCTION
The opportunistic pathogen Candida glabrata is a haploid fungus, which belongs to the Nakaseomyces clade (Kurtzman and Robnett 2003) and is closely related to the baker's yeast Saccharomyces cerevisiae (Bolotin-Fukuhara and Fairhead 2014; Gabaldón and Carrete 2016). Candida glabrata is normally associated with a mammalian host and is the second most prevalent Candida species to cause life-threatening candidiasis, after Candida albicans (Diekema et al. 2012; Galocha et al. 2019). It is therefore necessary to understand and characterize mechanisms and genes contributing to the pathogenicity of this fungus.
The molecular Koch's postulates define criteria that must be satisfied to prove the contribution of a certain gene to pathogenesis (Falkow 1988). One of these criteria states that reintegration of the original gene into a mutated organism (complementation) should restore its pathogenic potential. Thus, complementation is an important step in elucidating gene functions and to exclude potential pleiotropic effects that originate from genetic manipulation. However, to date, gene complementation strategies in C. glabrata are less well developed than e.g. in C. albicans, less frequently used, and each have certain disadvantages. The first strategy used for C. glabrata is based on episomal replicative vectors (Kitada, Yamaguchi and Arisawa 1996; Frieman, McCaffery and Cormack 2002; Zordan et al. 2013), which contain autonomously replicating sequences (ARS) and a centromere (CEN) together with the gene of interest (GOI). An example is the centromere-based plasmid pCgACT which contains the nutritional marker TRP1 to restore prototrophy of a trp1Δ deletion mutant (Miyazaki et al. 2011; Hosogaya et al. 2013; Noble et al. 2013). A strong disadvantage of episomal vectors is, however, unpredictable multimerizations or recombinations with the genome, causing unwanted genetic alterations (Pla et al. 1996). Additionally, plasmid loss (for example, in non-selective environments where tryptophan is readily available) or copy number variations could increase population heterogeneity (Zordan et al. 2013).
A second strategy for the generation of C. glabrata complementation strains relies on genomic integration, either by re-introducing the original gene of interest (GOI) together with specific selection markers at its native locus (Yánez-Carrillo et al. 2015), or by replacing the URA3 gene by the GOI and the dominant NAT1 marker (Nevitt and Thiele 2011). Both strategies have the potential to restore the gene of interest to a wild type-like state. The NAT1 selection marker is, however, widely used for gene deletions in C. glabrata (Schwarzmüller et al. 2014) and therefore often not available anymore for later gene complementation. In addition, changes in URA3 expression levels, depending on the gene locus, might influence virulence phenotypes (Brand et al. 2004).
Gene complementation in the diploid yeast C. albicans often uses the CIp10 strategy (Murad et al. 2000), whereby the selection marker URA3 is ectopically expressed at one of the alleles of the neutral RPS1 locus. In contrast to C. albicans, the haploid genome of C. glabrata makes it more difficult to find suitable loci, since single-allele knock-outs and dosage effects of ectopic expression of nutritional selection markers cannot be compensated by a second allele, with unknown influences on the phenotype.
In this study, we describe a novel gene complementation strategy for C. glabrata based on transformation with and stable genomic integration of a complementation cassette into the TRP1 (CAGL0C04092g) locus. We show that re-integration of TRP1 at this native chromosomal location can restore tryptophan prototrophy to wild type-like levels. This strategy can not only be used for gene complementation but also for reporter gene expression, gene overexpression, heterologous expression and expression of genetically modified gene variants in the haploid yeast C. glabrata.
We validate the functionality of our complementation system by heterologous expression of fluorescence reporter genes and of ovalbumin (OVA). Artificial OVA expression by pathogens is frequently used as a tool to investigate the cross-reactivity between effector and memory T cells in response to the microbe (Ishizuka et al. 2009; Krummey et al. 2014; Harms et al. 2018). Further, we use the complementation system to overexpress the C. glabrata biotin importer gene VHT1 (Sprenger et al. 2020) and to introduce point mutations into the biotinylation motif of the urea amidolyase gene DUR1,2 (Roon and Levenberg 1972; Navarathna et al. 2012).
RESULTS AND DISCUSSION
Complementation of tryptophan auxotrophy in C. glabrata by genomic re-integration
Gene complementation in the best-studied Candida species, C. albicans, is often achieved by genomic integration into coding genes like ADH1 (Hünniger et al. 2014), RPS1 (Murad et al. 2000), or ACT1 (Morschhäuser, Michel and Hacker 1998; Vandeputte et al. 2011; Xu et al. 2014). Integration in these loci disrupts one allele of the target gene in the diploid C. albicans genome which, in most cases, does not significantly affect growth or fitness of transformants.
In contrast, similar approaches in the haploid genome of C. glabrata lead to a complete knock-out of the original gene at the target locus. For selection of a target gene for complementation in C. glabrata, we therefore relied on previously published data. We chose the tryptophan biosynthesis gene TRP1 (CAGL0C04092g), as genetic manipulation in this locus or even complete lack of TRP1 did not impair C. glabrata survival in a systemic C. glabrata infection model (Jacobsen et al. 2010) or in macrophages (Schwarzmüller et al. 2014; Seider et al. 2014). In addition, the TRP1 gene is, in addition to HIS3, LEU2 and URA3, widely used as selection marker for different plasmid vectors in S. cerevisiae and C. glabrata.
As a basis for complementation constructs, we created a pTRP1 plasmid vector based on the plasmid pUC19 (Norrander, Kempe and Messing 1983). This new plasmid harbors the TRP1 gene as the selection marker together with its up- and downstream regions for homologous recombination at the native TRP1 locus (Sprenger et al. 2020). A multiple cloning site remains downstream of the TRP1 terminator to allow easy insertion of any GOI (Fig. 1A). A trp1Δ mutant, which can be used as the parental strain for integration of pTRP1, has been previously created using the SAT1 flipper protocol (Jacobsen et al. 2010). In this strain only a single recombination site (FRT) and no selection marker remain at the TRP1 locus (Fig. 1B).
Figure 1.
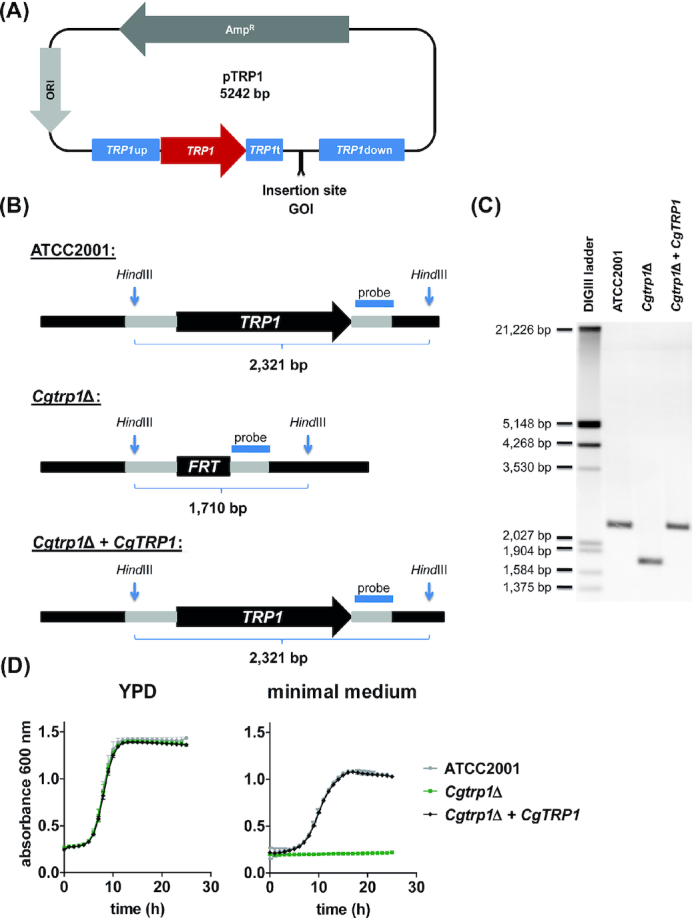
Construction and verification of TRP1 complemented strains. (A), Overview of the pTRP1 plasmid used for gene complementation in C. glabrata. The insertion site allows cloning of gene of interest (GOI) sequences with promotor and terminator regions. (B), Depiction of the TRP1 gene locus in the C. glabrata ATCC2001 reference genome, the trp1 deletion mutant (Cgtrp1Δ) and our complementation strain (Cgtrp1Δ + CgTRP1). A DNA probe was designed for the terminator region to detect gene-specific fragments by Southern blotting. (C), Southern blot of HindIII-digested genomic DNA of ATCC2001, Cgtrp1Δ, and Cgtrp1Δ + CgTRP1 (Cg wt). The expected sizes from (b) correspond to the visible bands. (D), Growth analysis of C. glabrata ATCC2001, Cgtrp1Δ and Cgtrp1Δ + CgTRP1 in nutrient-rich medium (YPD) and minimal medium (SD) at 37°C for 24 h. Values are absorbance at 600 nm and are shown as mean ± SD of at least three replicates.
We started by generating a TRP1 complementation strain from this strain. The TRP1 construct was PCR-amplified from pTRP1 and used to transform trp1Δ, replacing the FRT sequence in the original TRP1 locus. We used a PCR-amplified complementation cassette instead of the whole plasmid to rule out any unspecific genomic integration of additional vector DNA. Correct integration into the TRP1 locus was then verified by Southern blotting (Fig. 1C). Phenotypically, the growth of the complemented strain (trp1Δ + TRP1) was fully restored in minimal medium lacking tryptophan (Fig. 1D). Based on these data, we conclude that the TRP1 locus is suitable for expression of target genes. Our approach is therefore similar to the CIp10 strategy in C. albicans (Murad et al. 2000), but uses the native chromosomal location of the TRP1 locus.
To apply our approach for the analysis of gene functions, a mutant strain lacking the GOI has to be generated in a trp1Δ strain background. This mutant will then be transformed with the TRP1 cassette with or without the GOI, to restore TRP1 prototrophy or create a TRP1-isogenic complemented strain. This method then allows the direct comparison of three prototrophic strains—the trp1Δ + TRP1 wild type-like strain (Fig. 1), the trp1ΔGOIΔ + TRP1 mutant strain and the trp1ΔGOIΔ + TRP1 + GOI complemented strain. We have successfully applied this strategy for complementation of C. glabrata mutants with the biotin metabolism genes VHR1 and VHT1, for which recovery of wild type phenotypes after complementation was proven (Sprenger et al. 2020).
Gene complementation approaches help to verify the results of functional genomics approaches like large-scale screening of deletion mutant libraries (Janbon et al. 2019). Of note, our strategy of restoring the tryptophan prototrophy of a TRP1-deleted parental strain can be used to complement genes in a published triple auxotrophic his3Δ leu2Δ trp1Δ C. glabrata mutant library (Schwarzmüller et al. 2014). In this case, the leucine and histidine auxotrophies remain for further genetic manipulations. An alternative strategy to our TRP1-based method can be the integration of complementation cassettes into intergenic or promotor regions, which is already established in C. albicans (Davis et al. 2002; Staab, Bahn and Sundstrom 2003; Gerami-Nejad et al. 2013) and in C. glabrata (Ueno et al. 2011). However, genomic integration in these regions can raise the problem of inefficient transcriptional activity due to a silenced chromatin structure (Rando and Winston 2012; Gartenberg and Smith 2016).
Heterologous expression of yEGFP, mCherry, and ovalbumin in C. glabrata
We next determined whether the TRP1 locus is suitable for heterologous expression of markers like fluorescent proteins or ovalbumin, an antigen often used in immunological studies. We used the In-Fusion HD cloning system to fuse different promotors with these GOIs and into the XbaI-digested pTRP1 vector (Fig. 2A). This technique allows the combination of promotor, protein-encoding, and terminator sequences in a single cloning step.
Figure 2.
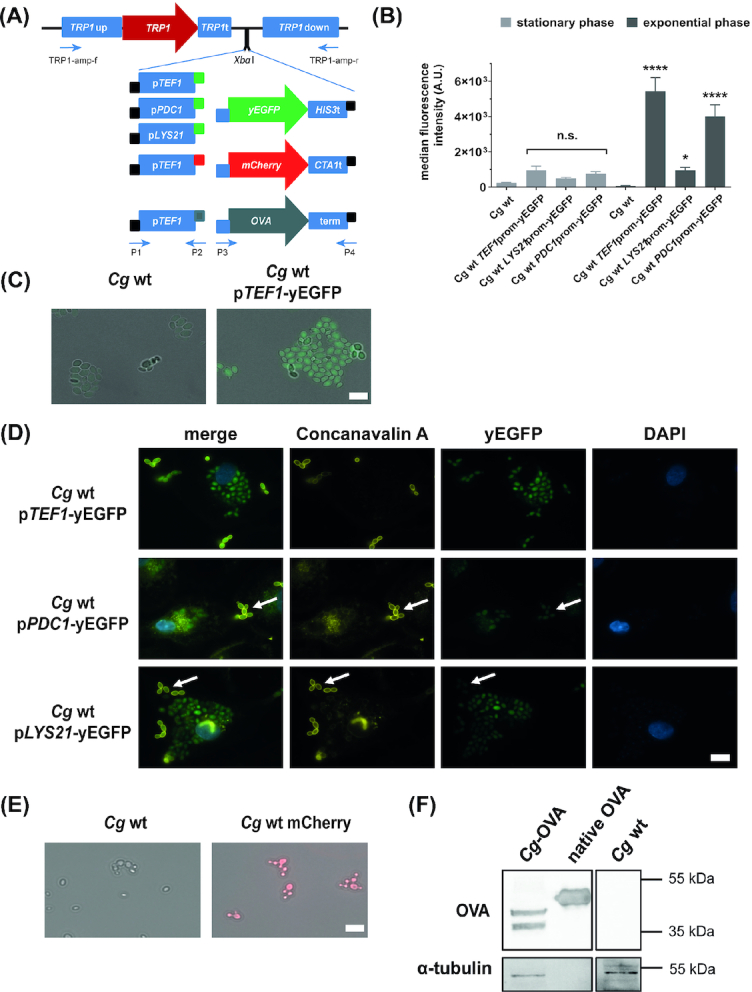
Heterologous expression of yEGFP, mCherry, and ovalbumin in C. glabrata. (A), Overview of constructed plasmids for yEGFP, mCherry, and ovalbumin (OVA) expression in C. glabrata. Three different promotors were PCR-amplified and combined with PCR-amplified yEGFP. Similarly, the constitutive CgTEF1 promotor was combined with PCR-amplified mCherry or OVA. Narrow bars: Amplification primers introduced ≥15 bp overlaps to the pTRP1 plasmid (black), promotor (blue) or yEGFP/mCherry/OVA (green/red/grey). Promotor and coding sequences were fused and cloned into an XbaI-linearized pTRP1 plasmid via the InFusion Cloning system. Primers used for PCR amplification are indicated. (B), Flow cytometric analysis of median fluorescence intensity in arbitrary units (A.U.). Cells grown in YPD at 37°C and 180 rpm (stationary phase) were diluted 1:20 in fresh YPD and incubated 2 h under the same growth conditions (exponential phase). Yeast cells of both growth phases were analyzed for green fluorescence (FITC channel) using the BD FACS Verse. Values are shown as mean ± SD of four replicates. For statistical analysis, a repeated measures ANOVA with Bonferroni's multiple comparison test was performed, comparing fluorescent strains to wt (*P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001). (C), Representative fluorescence microscopy image of stationary phase yeast cells grown in minimal medium. (D), Representative fluorescence microscopy image of primary human macrophages (MDMs) after infection with yeast cells (MOI 5) for 3 h at 37°C and 5% CO2. Non-phagocytosed yeast cells were counterstained with Alexa Fluor 647-coupled concanavalin A (representative examples shown by arrows). (E), Representative fluorescence microscopy image of stationary phase yeast cells containing mCherry grown in minimal medium. Scale bar (C-E): 10 µm. (F), Representative Western Blot image of OVA expression. Equal amounts of protein extracts of the OVA-expressing C. glabrata (Cg-OVA), the isogenic strain (Cg wt) and isolated egg white ovalbumin (OVA) were blotted and probed for OVA. A separate blot with the same amounts was probed for α-tubulin (loading control). Uncropped images are shown in Fig. S1.
Three well-investigated promotors (Zordan et al. 2013) were each fused to the gene encoding yEGFP (yeast-enhanced green fluorescent protein (Cormack et al. 1997)). The promotors were selected to achieve constitutive expression under in vitro conditions (TEF1 and PDC1 promotors), but also induced expression upon phagocytosis by macrophages (LYS21 promotor). The correct integration into the TRP1 locus of the trp1Δ strain was verified by PCR, and positive clones were analyzed for yEGFP fluorescence by microscopy and FACS.
As expected from a previous study (Zordan et al. 2013), we observed differences in fluorescence levels depending on the strength of the promotor and promotor-specific induction conditions: Expression of yEGFP under control of the TEF1 promotor led to the highest median fluorescence intensity among all tested promotors in culture medium (Fig. 2B and C). Fluorescence was strongly induced in exponentially growing cells compared to stationary cells (Fig. 2B). The TEF1 promotor-controlled construct was furthermore highly expressed by C. glabrata cells internalized by macrophages, but also by non-internalized cells (Fig. 2D). Candida glabrata cells containing the PDC1 promotor-controlled yEGFP expression construct showed fluorescence levels similar to cells containing the TEF1 promotor construct in culture medium (Fig. 2B). Fluorescence was detected in macrophage-internalized and in non-internalized C. glabrata cells (Fig. 2D). When yEGFP expression was controlled by the LYS21 promotor, the fluorescence signal was higher in macrophage-internalized than in extracellular C. glabrata cells (Fig. 2D, as expected from a previous study (Zordan et al. 2013)). In contrast to the other promotors, the expression of yEGFP remained low in the exponential growth phase when controlled by the LYS21 promotor (Fig. 2B).
To further validate the usefulness of the strategy, the proven strong constitutive TEF1 promotor was fused with mCherry (red fluorescent protein), cloned into the XbaI-digested pTRP1, and used to transform the trp1Δ strain. The expression of mCherry controlled by the TEF1 promotor led to a strong fluorescence signal in the stationary phase (Fig. 2E).
With this, we provide a set of plasmids for fluorescence reporter-based analysis that integrate into the TRP1 locus of C. glabrata. The expression of fluorescent markers under control of constitutive or inducible promotors is not only useful for generating fluorescently labeled fungal wild type or mutant strains that can be traced during infection. It also generates reporter strains that can be used to identify conditions that induce the respective promotors during infection (Miramón et al. 2012). In addition, the efficiency of transcription from promotors of interest can be quantified by measuring the fluorescence intensity even on a single cell level. This technique also opens up the possibility to distinguish C. glabrata strains with different fluorescent markers in combined infection or competition experiments.
The heterologous expression of ovalbumin (OVA) in pathogens is used as a tool to induce OVA-specific immune responses for investigation of the immune recognition of these pathogens and T cell activation (Ishizuka et al. 2009; Krummey et al. 2014; Harms et al. 2018). To generate an OVA-expressing C. glabrata strain, we fused an OVA gene version, which had been codon-optimized for C. glabrata, with the TEF1 promotor and cloned it into the XbaI-digested pTRP1. Codon optimization was done to increase OVA expression in C. glabrata, as yeast and Gallus gallus have different codon usage frequencies (Branlant and Branlant 1985). The expression of OVA in C. glabrata was verified by Western blot (Fig. 2F). The heterologously expressed OVA in C. glabrata was smaller in size than native ovalbumin from egg white and was detected as two bands. A previous study in the yeast Pichia pastoris demonstrated that these two bands correspond to mono- and diglycosylated OVA. The smaller size of C. glabrata-expressed OVA is likely due to missing N-acetylation and phosphorylation in yeasts as opposed to chicken (Ito and Matsudomi 2005).
Overexpression of the biotin transporter gene VHT1 modulates intracellular survival within macrophages
To evaluate the suitability of our TEF1, LYS21 and PDC1 promotor constructs for overexpression of C. glabrata’s own genes, we used the system to overexpress the C. glabrata biotin transporter gene VHT1. Candida glabrata is auxotrophic for biotin and strictly relies on Vht1, the ortholog of an S. cerevisiae biotin importer, for biotin uptake from the environment (Sprenger et al. 2020). The native promotor of VHT1 is activated by the transcriptional regulator Vhr1 when biotin in the environment is limiting (Sprenger et al. 2020).
Similar constructs as described for yEGFP were created and used to transform a trp1Δ strain (wild type) and a trp1Δ/vhr1Δ strain (mutant). The resulting strains were analyzed for their VHT1 transcript abundance in nutrient-rich medium—a condition in which VHT1 expression from its native promotor is low (Sprenger et al. 2020). The transcript abundance of VHT1 was strongly increased compared to the wild type when the gene was controlled by constitutive TEF1 or PDC1 promotors, whereas LYS21-controlled VHT1 expression was only marginally increased (Fig. 3A). Overexpression of VHT1 at the TRP1 locus was independent of its native regulator, Vhr1, as the deletion of VHR1 had no impact on the transcript abundance of VHT1.
Figure 3.
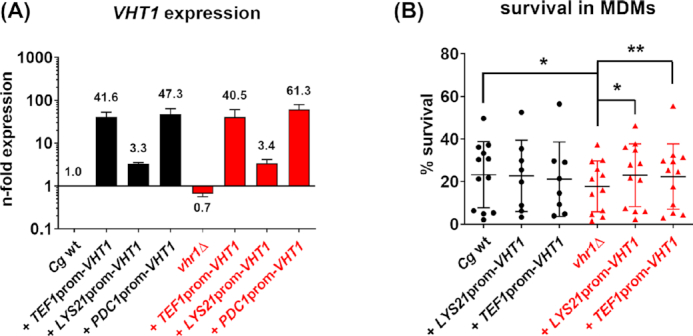
The overexpression of CgVHT1 increases intracellular fitness of Cgvhr1Δ. (A), Candida glabrata wt (black letters), mutant (Cgvhr1∆, red letters) and corresponding overexpression strains (+LYS21prom-VHT1, +TEF1prom-VHT1, +PDC1prom-VHT1) were cultivated in YPD for 90 min at 37°C and 180 rpm. Target gene expression was analyzed by qRT-PCR and normalized to ACT1 and EFB1. Expression is shown relative to the Cg wt strain. Data are shown as mean ± SD of two independent experiments. (B), Candida glabrata wt (black letters), mutant (vhr1∆, red letters), and corresponding overexpression strains (+pLYS21-VHT1, +pTEF1-VHT1) were pre-cultured in YPD + 2 mg/L biotin before confrontation with MDMs. Survival of C. glabrata strains after 3 h co-incubation with MDMs at an MOI of 1 is shown as survival in % of inoculum (mean ± SD). Each single dot represents one individual blood donor (in total at least two independent experiments with four donors each). For statistical analysis, a repeated measures ANOVA with Bonferroni's multiple comparison test was performed comparing all strains (*P ≤ 0.05, **P ≤ 0.01).
A vhr1Δ mutant has reduced survival in macrophages (Sprenger et al. 2020). To test whether artificial up-regulation of its regulatory target VHT1 can rescue this survival defect, we confronted monocyte-derived macrophages (MDMs) with vhr1Δ strains overexpressing VHT1 and evaluated survival of phagocytosed fungal cells. As previously described, the vhr1Δ strain showed a lower intracellular survival than the wild type. In contrast, the vhr1Δ strains overexpressing VHT1 reached higher survival rates, which were comparable to the wild type (Fig. 3B). These data imply that the overexpression of VHT1 alone can indeed compensate the deletion of VHR1, whereas the wild type with its functional Vhr1 does not benefit from higher VHT1 expression (Fig. 3B).
Mutations in the biotinylation motif of Dur1,2 prevent the utilization of urea
Biotin is needed for central metabolic pathways, where it acts as a cofactor of biotin-dependent enzymes (Knowles 1989; Zempleni, Wijeratne and Hassan 2009; Tong 2013). Biotin is covalently linked to lysine residues of target proteins (Lane et al. 1964; Sternicki et al. 2017). Most of these lysine residues can be found in highly conserved AMKM sequence motifs of biotin-dependent enzymes (Samols et al. 1988; Chapman-Smith and Cronan 1999). The enzyme, urea amidolyase Dur1,2 possesses such a conserved biotinylation motif in the C-terminal carboxylase domain in both, C. glabrata and C. albicans (Fig. 4A, (Samols et al. 1988; Chapman-Smith and Cronan 1999)). Dur1,2 is important for using urea as a nitrogen source (Navarathna et al. 2011) and is necessary for the pathogenicity of C. albicans (Ghosh et al. 2009; Navarathna et al. 2012) and C. glabrata (Brunke et al. 2015). To study whether biotinylation is important for Dur1,2 function, we mutated the lysine codon AAA into the arginine-coding AGA (amino acid position 1798 in C. glabrata and 1779 in C. albicans (Fig. 4B)), using our newly developed complementation strategy in C. glabrata and the established CIp10-based strategy in C. albicans. As expected, C. glabrata and C. albicans deletion mutants lacking DUR1,2 showed growth defects in liquid and solid media containing urea as the sole nitrogen source, while the wild type or a mutant strain with re-integration of one wild type copy of DUR1,2 grew well (Fig. 4C–F). The mutants failed to grow in urea-containing liquid media and formed smaller colonies than the wild type on solid media (although some growth rescue seems to take place in close proximity to urea-metabolizing colonies, Fig 4F). The C. albicans dur1,2Δ/Δ mutant also failed to induce colony wrinkling at 37°C (Fig. 4F). Mutant strains with re-integration of DUR1,2 carrying a K→R point mutation in the biotinylation motif phenocopied the dur1,2 deletion mutants in both species (Fig. 4C–F). These data suggest that biotinylation of Dur1,2 is indeed essential for urea utilization in C. glabrata and C. albicans.
Figure 4.

Biotinylation of Dur1,2 is essential for urea utilization. (A), Dur1,2 contains a highly conserved biotinylation motif (AMKT) in C. glabrata and C. albicans. (B), Upper part: DNA sequence, which is transcribed into the lysine (K) residue essential for biotinylation and biotin structure coupled to lysine. Lower part: The adenine of the triplet codon was mutated to guanine to induce an amino acid exchange to arginine. (C and D), Growth analysis of C. glabrata wt, Cgdur1,2Δ, +CgDUR1,2WT, and + CgDUR1,2K1798R in liquid (c) or on solid (d) minimal medium at 37°C. (E, F), Growth analysis of C. albicans wt, Cadur1,2Δ/Δ, +CaDUR1,2WT and + CaDUR1,2K1779R in liquid (e, 30°C) or on solid (f, 37°C) minimal medium. Media contained either 0.5% ammonium sulfate or 0.5% urea as sole nitrogen source. Values are represented as mean ± SD of at least three replicates and representative pictures are shown.
MATERIALS AND METHODS
Ethics statement
Human blood was taken from healthy volunteers with written, informed consent. The blood donation protocol and use of blood for this study were approved by the institutional ethics committee of the university hospital Jena (Ethik-Kommission des Universitätsklinikums Jena, Permission No. 2207–01/08).
Strains and growth conditions
All strains used in this study are listed in Table 1. Escherichia coli was cultured in liquid lysogeny broth (LB) at 37°C with shaking at 180 rpm and with 50 µg mL−1 ampicillin for plasmid propagation. Candida strains were cultured in liquid Yeast Peptone Dextrose (YPD) broth (2% glucose, 2% peptone, 1% yeast extract) at 30°C (C. albicans) or 37°C (C. glabrata), with shaking at 180 rpm for 14–16 h. For growth on solid medium, 2% agar was added. Routinely, stationary phase yeast cells from pre-cultures were washed three times in PBS, and the cell number was adjusted in PBS or culture medium.
Table 1.
Strains used in this study.
| Strains | Internal ID | Designation/genotype | Reference |
|---|---|---|---|
| C. glabrata | |||
| ATCC2001 | C94 | Clinical isolate | (Schwarzmüller et al. 2014) |
| Cgtrp1∆ | G17 | Parental strain (derivative of ATCC2001) trp1::FRT | (Jacobsen et al. 2010) |
| Cgtrp1∆ + CgTRP1 (Cg wt) | G203 | Isogenic wild type trp1::TRP1 | (Sprenger et al. 2020) |
| Cg wt + OVA | G205 | trp1::(TRP1-CgTEF1prom(-1 to -1000)-OVA) | This study |
| Cg wt + CgLYS21p-CgVHT1 | G247 | trp1::(TRP1-CgLYS21prom(-1 to -1157)- CAGL0K04609g-CAGL0K04609gterm) | This study |
| Cg wt + CgTEF1p-CgVHT1 | G249 | trp1::(TRP1-CgTEF1prom(-1 to -1000)-CAGL0K04609g-CAGL0K04609gterm) | This study |
| Cg wt + CgPDC1p-yEGFP | G257 | trp1::(TRP1-CgPDC1prom(-1 to -973)-yEGFP-CgHIS3term) | This study |
| Cg wt + CgLYS21p- yEGFP | G259 | trp1::(TRP1-CgLYS21prom(-1 to -1157)-yEGFP-CgHIS3term) | This study |
| Cg wt + CgTEF1p- yEGFP | G261 | trp1::(TRP1-CgTEF1prom(-1 to -1000)-yEGFP-CgHIS3term) | This study |
| Cg wt + CgTEF1p-mCherry | G325 | trp1::(TRP1-CgTEF1prom(-1 to -1000)-mCherry- CgCTA1term) | This study |
| Cgtrp1∆vhr1∆ | G237 | trp1::FRT CAGL0M12496g::NAT1 | This study |
| Cgvhr1∆ | G241 | trp1::TRP1 CAGL0M12496g::NAT1 | (Sprenger et al. 2020) |
| Cgvhr1∆ + CgLYS21p-CgVHT1 | G253 | CAGL0M12496g::NAT1 trp1::(TRP1-CgLYS21prom(-1 to -1157)- CAGL0K04609g-CAGL0K04609gterm) | This study |
| Cgvhr1∆ + CgTEF1p-CgVHT1 | G255 | CAGL0M12496g::NAT1 trp1::(TRP1-CgTEF1prom(-1 to -973)- CAGL0K04609g-CAGL0K04609gterm) | This study |
| Cgtrp1∆dur1,2∆ | G330 | trp1::FRT CAGL0M05533g::NAT1 | This study |
| Cgdur1,2∆ | G366 | trp1::TRP1 CAGL0M05533g::NAT1 | This study |
| Cgdur1,2∆ + CgDUR1,2 | G367 | CAGL0M05533g::NAT1; trp1::(TRP1-CAGL0M05533g) | This study |
| Cgdur1,2∆ + CgDUR1,2K1798R | G368 | CAGL0M05533g::NAT1; trp1::(TRP1-CAGL0M05533g with codon 1798: AAA → AGA) | This study |
| C. albicans | |||
| BWP17 | M130 | Parental strain (derivative of SC5314) ura3::λimm434/ura3::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | (Wilson, Davis and Mitchell 1999) |
| BWP17 CIp30 (Ca wt) | M1477 | Isogenic wildtype ura3::λimm434/ura3::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG RPS1/rps1::CIp30-URA3-HIS1-ARG4 | (Citiulo et al. 2012) |
| Caura3∆/∆dur1,2∆/∆ | M2671 | orf19.7468::ARG4/orf19.7468::HIS1 | This study |
| Cadur1,2∆/∆ | M2672 | orf19.7468::ARG4/orf19.7468::HIS1 RPS1/rps1::CIp10-URA3 | This study |
| Cadur1,2∆/∆ + CgDUR1,2 | M2674 | orf19.7468::ARG4/orf19.7468::HIS1 RPS1/rps1::CIp10-URA3-orf19.7468 | This study |
| Cadur1,2∆/∆ + CgDUR1,2K1779R | M2721 | orf19.7468::ARG4/orf19.7468::HIS1 RPS1/rps1::CIp10-URA3-orf19.7468 with (codon 1779: AAA → AGA) | This study |
| E. coli | |||
| Stellar™ Competent Cells | F–, endA1, supE44, thi-1, recA1, relA1, gyrA96, phoA, Φ80d lacZΔ M15, Δ (lacZYA-argF) U169, Δ (mrr—hsdRMS—mcrBC), mcrA, λ– | In-Fusion® HD Cloning Plus Kit |
For growth analyses in liquid media, 20 µL of a yeast cell suspension (5 × 106 cells mL−1) were added to 180 µL media in a 96-well plate (Tissue Culture Test Plate, TPP Techno Plastic Products AG). In some experiments, minimal medium (0.19% yeast nitrogen base (YNB) without biotin, amino acids and ammonium sulfate [Formedium]; 2% glucose) with varying biotin concentrations and different nitrogen sources (0.5% ammonium sulfate, 0.5% urea) was used. Growth was monitored by measuring the absorbance at 600 nm every 30 min for 100 cycles at 30°C or 37°C using a microplate reader (Plate Reader infinite M200 PRO, Tecan Group GmbH) with orbital shaking (30 s, amplitude: 6 mm, wait: 10 s) before each measurement and multiple reads per well. All experiments were performed in technical duplicates and, at least, biological triplicates. Growth of Candida on solid agar was tested by spotting serial dilutions (1 × 106 to 1 × 101) of yeast cells on YNB plates (0.19% YNB without amino acids and biotin; 2% glucose; either 0.5% ammonium sulfate or 0.5% urea; 2% oxoid agar).
Construction of VHT1 overexpression, OVA overexpression and fluorescently labelled strains
Plasmids and primers used for strain construction are listed in Table 2 and Table S1 (Supporting Information), respectively. For generation of overexpression strains, promotor and coding sequence fragments were introduced into the pTRP1 plasmid (Sprenger et al. 2020) by In-Fusion HD cloning (Takara), generating overexpression plasmids pTRP1-TEF1prom-yEGFP, pTRP1-PDC1prom-yEGFP, pTRP1-LYS21 prom-yEGFP, pTRP1-TEF1prom-mCherry, pTRP1-TEF1prom-OVA, pTRP1-TEF1prom-VHT1, pTRP1-PDC1prom-VHT1 and pTRP1-LYS21prom-VHT1 (Table 2). First, promotor sequences of TEF1, LYS21 or PDC1 were PCR-amplified from C. glabrata genomic DNA with P1 and P2 primers (Fig. 2A; Table S1, Supporting Information). P1 primers introduced ≥15 bp overlaps to the pTRP1 plasmid sequence, while P2 primers introduced overlaps to the respective coding sequences (see below). In a second PCR-reaction, coding sequences of yEGFP, mCherry, ovalbumin (OVA) or VHT1 were amplified using P3 and P4 primers (Fig. 2A; Table S1, Supporting Information). pCN-PDC1-GFP (Zordan et al. 2013) served as a PCR template for yEGFP, pYC56 (Yánez-Carrillo et al. 2015) for mCherry, and C. glabrata genomic DNA for VHT1. For OVA, a codon-optimized version of chicken ovalbumin synthesized by GeneArt Gene Synthesis (Thermo Fisher Scientific) was used as a PCR template. The constructs contained native (VHT1) or foreign terminator sequences (HIS3 terminator for yEGFP, CTA1 terminator for mCherry, and Ashbya gossypii TEF1 terminator for OVA). P3 primers introduced ≥15 bp overlaps to the respective promotor sequences (see above), and P4 primers generated overlaps to the pTRP1 plasmid sequence. Promotor and coding sequence fragments were combined and cloned with the help of the In-Fusion HD cloning system (Takara Bio) into XbaI-digested pTRP1. An exception was pTRP1-PDC1prom-yEGFP, for which promotor and coding sequence was directly amplified form plasmid pCN-PDC1-GFP, using P1 and P4 primers, and cloned into pTRP1. The resulting plasmids were confirmed by PCR and sequencing, using primers listed in Table S1 (Supporting Information). The complete cassettes containing the TRP1 selection marker together with the overexpression constructs were then PCR-amplified from pTRP1 overexpression plasmids using primers TRP1-amp-f and TRP1-amp-r (Table S1, Supporting Information; Fig. 2A). PCR products were purified and used to transform a tryptophan-auxotrophic trp1∆ strain (Jacobsen et al. 2010). The VHT1 constructs were additionally transformed into a tryptophan-auxotrophic vhr1∆ strain (Sprenger et al. 2020). Transformation of C. glabrata strains was done using the modified heat-shock method ((Sanglard et al. 1996) with heat shock for 15 min at 45°C). Tryptophan-prototrophic clones with integration of the cassette into the native TRP1 locus were selected after plating on solid minimal medium that lacks tryptophan. The correct integration into the TRP1 locus was verified by PCR, and positive clones were analyzed for yEGFP and mCherry fluorescence by microscopy and FACS (only yEGFP).
Table 2.
Plasmids used in this study.
| Plasmid | Used for | Reference |
|---|---|---|
| pFA-HIS1 | Amplification of CaHIS1 cassette | (Gola et al. 2003) |
| pFA-ARG4 | Amplification of CaARG4 cassette | (Gola et al. 2003) |
| CIp10 | Integration of CaURA3 into RPS1 locus | (Murad et al. 2000) |
| pTRP1 | Amplification of CgTRP1 cassette, Basis for TRP1 complementation constructs | (Sprenger et al. 2020) |
| pCN-PDC1-GFP | CEN/ARS episomal plasmid for yEGFP expression in C. glabrata under control of the PDC1 promotor | (Zordan et al. 2013) |
| pYC56 | Recipient integrative vector harboring mCherry | (Yánez-Carrillo et al. 2015) |
| pTRP1-LYS21prom-yEGFP | Amplification of CgTRP1 and yEGFP under control of the LYS21 promotor | This study |
| pTRP1-LYS21prom-VHT1 | Amplification of CgTRP1 and CAGL0K04609g under control of the LYS21 promotor | This study |
| pTRP1-PDC1prom-yEGFP | Amplification of CgTRP1 and yEGFP under control of the PDC1 promotor | This study |
| pTRP1-PDC1prom-VHT1 | Amplification of CgTRP1 and CAGL0K04609g under control of the PDC1 promotor | This study |
| pTRP1-TEF1prom-yEGFP | Amplification of CgTRP1 and yEGFP under control of the TEF1 promotor | This study |
| pTRP1-TEF1prom-VHT1 | Amplification of CgTRP1 and CAGL0K04609g under control of the TEF1 promotor | This study |
| pTRP1-TEF1prom-OVA | Amplification of CgTRP1 and OVA under control of the TEF1 promotor | This study |
| pTRP1-TEF1prom-mCherry | Amplification of CgTRP1 and mCherry under control of the TEF1 promotor | This study |
| CIp10-CaDUR1,2 | Integration of CaURA3 and CaDUR1,2 (orf19.780) into RPS1 locus | This study |
| CIp10-CaDUR1,2 (K1779R) | Integration of CaURA3 and orf19.780 (lysine 1779 mutated to arginine) into RPS1 locus | This study |
| pTRP1-CgDUR1,2 | Amplification of CgTRP1 and CgDUR1,2 (CAGL0M05533g) | This study |
| pTRP1-CgDUR1,2 (K1798R) | Amplification of CgTRP1 and CAGL0M05533g (lysine 1798 mutated to arginine) | This study |
In experiments that required comparisons to a wild type, the strain Cgtrp1Δ + TRP1 was used as an isogenic wild type strain (Cg wt). This strain was generated by re-introducing TRP1, amplified from pTRP1 into the native TRP1 locus into Cgtrp1Δ (Sprenger et al. 2020). The Cgtrp1Δvhr1∆ mutant strain carrying the TRP1 complementation insert (Cgvhr1∆; (Sprenger et al. 2020)) was constructed in a similar way.
Construction of DUR1,2 deletion mutants
The C. glabrata dur1,2∆ mutant (CAGL0M05533gΔ) was generated in the trp1Δ parental strain. A deletion cassette was PCR-amplified from genomic DNA of a previously generated triple-auxotrophic dur1,2∆ mutant (Schwarzmüller et al. 2014) using primers CgDUR1,2amp fwd and rev (Table S1, Supporting Information)), followed by purification with the QIAquick® PCR purification kit (Qiagen). The transformation was performed as described above.
For interspecies comparison, a C. albicans dur1,2∆/∆ (C1_04660W∆/∆) mutant was generated by a standard gene disruption method (Wilson, Davis and Mitchell 1999) using lithium acetate transformation (Walther and Wendland 2003). Briefly, the Arg-, His-, and Ura-auxotrophic parental strain BWP17 was sequentially transformed with 10 µg of purified HIS1 and ARG4 deletion cassettes (amplified with primers CaDUR1,2 del fwd and rev from pFA-HIS1 and -ARG4 plasmids, respectively; Table 2 and Table S1, Supporting Information), which were flanked by 100 bp of the target homology region. Transformation resulted in disruption of the open reading frames of both alleles of C1_04660W, yielding strain Caura3Δ/Δdur1,2Δ/Δ. The correct deletion of DUR1,2 in C. glabrata and C. albicans was confirmed by PCR and Southern blot.
Construction of DUR1,2 complementation strains
Candida glabrata DUR1,2 complementation strains were constructed as follows: The DUR1,2 wild type allele together with its up- and downstream intergenic regions was PCR-amplified from C. glabrata genomic DNA with primers pTRP1-CgDUR1,2 fwd (P1) and pTRP1-CgDUR1,2 rev new (P4) that introduced ≥15 bp overlaps to the pTRP1 sequence (Table S1). The mutated version DUR1,2K1798R was created by combining P1 and P4 (see above) with internal primers P2 and P3, which introduce a single nucleotide change resulting in a codon exchange from lysine to arginine. The primer combinations P1 + P2 and P3 + P4 generated two PCR fragments with ≥15 bp overlaps to each other and to pTRP1. The wild-type allele (one fragment) or the mutated allele (two fragments) were cloned into XbaI-digested pTRP1 by In-Fusion HD cloning, generating plasmids pTRP1-DUR1,2 and pTRP1-DUR1,2 (K1798R), which were verified by sequencing. Cassettes containing only TRP1 or TRP1-DUR1,2WT and TRP1-DUR1,2K1798R were then amplified from the complementation plasmids by PCR, purified, and used to transform a Cgtrp1∆/dur1,2∆ mutant background strain to generate tryptophan prototrophic mutants and complemented strains with the native or mutated biotinylation motif, respectively. Resulting clones were selected for on solid minimal medium lacking tryptophan.
To generate C. albicans DUR1,2 complementation strains, the coding sequence of DUR1,2 was PCR-amplified together with the native up- and downstream intergenic regions from C. albicans genomic DNA with primers pCIp10-CaDUR1,2 fwd (P1) and pCIp10-CaDUR1,2 rev (P4) that introduced ≥15 bp overlaps to the pCIp10 sequence (Table S1). The mutated version DUR1,2K1779R was created using additional internal primers P2 and P3, as described above for C. glabrata. The wild type allele (one fragment) or the mutated allele (two fragments) were cloned by In-Fusion HD cloning into the CIp10 plasmid using XhoI, generating plasmids CIp10-DUR1,2 or CIp10-DUR1,2 (K1779R) (Table 2). Constructed plasmids were verified by sequencing. Homozygous uridine-auxotrophic deletion mutants (Caura3Δ/Δdur1,2Δ/Δ) were transformed with the StuI-linearized empty CIp10 plasmid (Murad et al. 2000) to generate prototrophic mutants, or with StuI-linearized CIp10-DUR1,2 or CIp10-DUR1,2 (K1779R) plasmids to generate complemented strains with the native or mutated biotinylation motif. Resulting clones were selected for on solid minimal medium lacking uridine.
The correct integration of C. glabrata and C. albicans complementation cassettes was confirmed by PCR and Southern blot.
Genomic DNA isolation and Southern blot
Genomic DNA was isolated as described in (Millon et al. 1994). Briefly, cells form overnight cultures were protoplasted in lysis buffer (1 M sorbitol; 100 mM sodium citrate, pH 5.8; 50 mM EDTA, pH 8.0; 0.6 mg mL−1 lyticase and 2.5% β-mercaptoethanol) at 37°C for 45 min. Protoplasts were then pelleted, resuspended in proteinase buffer (10 mM Tris-HCl, pH 7.5; 50 mM EDTA, pH 7.5; 0.5% SDS; 1 mg mL−1 proteinase K) and incubated at 60°C for 30 min. After addition of phenol/chloroform/isoamylalcohol (25:24:1), vortexing for 4 min and centrifugation (20 000g, 5 min, RT), the aqueous phase was collected, and DNA was precipitated using isopropanol, washed with 70% ethanol and reconstituted in ddH2O containing 10 mg mL−1 RNase A. The DNA quantity was evaluated using a NanoDrop Spectrophotometer (ND-100, Peqlab). HindIII (NEB)-digested genomic DNA (30 µg) and the DNA Molecular Weight Marker III, DIG-labelled (50 pg; Roche) was separated by agarose gel electrophoresis. The DNA was depurinated with 0.25 M HCl and denatured with 0.5 M NaOH, 1.5 M NaCl. The gel was neutralized with 1 M Tris-HCl pH 7.5; 1.5 M NaCl and equilibrated for at least 10 min in 20 × SSC (3 M NaCl, 0.3 M sodium citrate; pH 7.0).
The DNA was vacuum blotted 1.5 h at 50–100 mbar (Biometra) onto activated nylon membrane (positively charged, Roche). Following, the membrane was rinsed 30 sec in 0.4 M NaOH, 30 sec in 0.2 M Tris-HCl pH 7.5 and dried 10–15 min on a piece of Whatman™ filter paper. The DNA was crosslinked to the membrane by UV light (120 mJ/cm2) for at least 60 sec and then rinsed with 2 × SSC. Further steps were performed according the DIG nonradioactive nucleic acid labeling and detection system (Roche), using the PCR DIG Probe Synthesis Kit, DIG Easy Hyb™, Anti-Digoxigenin-AP, Fab fragments, and CDP-Star.
Fungal RNA isolation and quantitative reverse transcription PCR (qRT-PCR)
For preparation of RNA from in vitro cultures of Candida, stationary phase yeast cells were washed three times in PBS, and 2 × 107 Candida cells per mL were inoculated into minimal medium with different biotin concentrations. At indicated time points, cells were harvested and centrifuged (4000 ×g, 10 min and 4°C). The cell pellet was washed with ice cold water, centrifuged again and immediately frozen in liquid nitrogen. The isolation of the fungal RNA was performed as previously described (Lüttich, Brunke and Hube 2012). The quantity of the RNA was determined using the NanoDrop Spectrophotometer ND-1000 (NRW International GmbH). DNase-treated RNA (600 ng) was transcribed into cDNA using 0.5 µg oligo-dT12–18, 100 U Superscript™ III Reverse Transcriptase, and 20 U RNaseOUT™ Recombinant RNase Inhibitor (all: Thermo Fischer Scientific) in a total volume of 35 µL for 2 h at 42°C, followed by heat-inactivation for 15 min at 70°C. The cDNA was diluted 1:20 in DEPC-treated water and used for quantitative PCR with EvaGreen® QPCR Mix II (Bio&SELL) performed in a CFX96 thermocycler (Bio-Rad). Primers (Table S1, Supporting Information) were used at a final concentration of 500 nM. Target gene expression was calculated using the ∆∆Ct method (Pfaffl 2001), with normalization to the housekeeping genes ACT1 and EFB1.
Protein isolation and Western blot
For preparation of whole protein extracts, stationary phase cells were washed three times in PBS, diluted to OD600 0.2 into minimal medium and incubated at 37°C at 180 rpm overnight. The cells were harvested and mechanically lysed in PBS-KMT (PBS + 3 mM KCl, 2.5 mM MgCl2, 0.1% Triton-X-100) with a protease inhibitor cocktail (Roche) using bead beating with acid-washed glass beads in a Precellys 24 homogenizer (Peqlab; 6500 rpm, 2 cycles, each 30 s, 15 s pause). The lysate was centrifuged (14 000 rpm, 4°C for 5 min) and the protein concentration of the supernatant was determined by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Two aliquots of 20 µg protein of each sample were denatured in one-fourth volume of 4 × Lämmli buffer (125 mM Tris-HCl pH 6.8, 50% glycerol, 4% SDS, 0.02% bromophenol blue, 10% β-mercaptoethanol) at 95°C for 5 min and separated by denaturizing SDS-PAGE with Rotiphorese® Gel 30 (final 12% acrylamide mix (Roth)). Proteins were electro-transferred to nitrocellulose membranes (Whatman) in blotting buffer (25 mM Tris, 192 mM glycine, 10% methanol and 0.1% SDS) and free binding sites were blocked with 5% milk powder in TBS (0.5 M Tris, 1.5 M NaCl, pH 7.6) + 0.05% Tween®-20 (TBS-T) for 3 h at room temperature. One membrane was incubated with monoclonal mouse anti-chicken egg albumin antibody (Sigma), diluted 1:2000 in TBS-T containing 2% milk powder. The other membrane was incubated with rat anti-α-tubulin antibody (AbD Serotec), diluted 1:1000 in TBS-T containing 2% BSA. Both were incubated at 4°C overnight with gentle shaking. The membranes were rinsed three times in TBS-T, incubated with goat anti-mouse IgG-HRP (1:1000 in 2% milk powder) or goat anti-rat IgG-HRP (1:2000 in 2% BSA) (both Santa Cruz Biotech) to detect ovalbumin and α-tubulin, respectively. Finally, the membranes were rinsed three times in TBS-T and two times in TBS, followed by chemiluminescence detection using Pierce™ ECL Plus Western Blotting Substrate (Thermo Fisher Scientific) according to the manufactures’ instructions. Protein sizes were estimated using PageRuler Plus prestained (Fisher Scientific), which was visible on the blot membrane. Marker band positions were determined by overlaying marker and chemiluminescence images. Full size scans of Western blots are provided in Fig. S1 (Supporting Information).
Experiments with human monocyte-derived macrophages (MDMs)
Primary macrophages were differentiated from CD14-positive monocytes originating from independent blood donors as previously described (Sprenger et al. 2020), seeded in 24-well plates (2 × 105 MDMs/well) in RPMI with 50 ng mL−1 M-CSF, and incubated overnight. Prior to macrophage infection, the medium was exchanged to serum- and biotin-free RPMI with l-glutamine (Thermo Fisher Scientific) to exclude biotin uptake by Candida prior to phagocytosis.
Fungal survival was determined as described previously (Sprenger et al. 2020). Briefly, macrophages were infected with C. glabrata at a multiplicity of infection (MOI) of 1 (one well per Candida strain and donor). After 3 h, non-internalized Candida cells were removed by washing with PBS. Intracellular yeast cells were then released by lysing the macrophages with 0.1% Triton‐X‐100, and lysates were plated on YPD agar plates in duplicate. In parallel, culture inocula were plated on YPD. Survival rates were then calculated based on CFU numbers in macrophages lysates relative to CFU numbers of initial inocula.
For fluorescence microscopy after macrophage phagocytosis, 1.5 × 105 MDMs/well were allowed to adhere onto coverslips in a 24-well plate overnight in RPMI containing 50 ng mL−1 M-CSF and 10% FBS, and then infected with C. glabrata at an MOI of 2 for the indicated time points. Phagocytosis was synchronized on ice for 30 min followed by two washing steps with pre-warmed medium to remove unbound Candida cells.
Fluorescence analysis by flow cytometry and microscopy
Stationary phase cells were diluted 1:20 into fresh YPD medium and incubated at 37°C for 2 h and 180 rpm. Then, cells were harvested and washed once with PBS, fixed with 500 µL Roti®-Histofix 4% for 20 min and washed again with PBS. The median fluorescence intensity (MFI) of C. glabrata yeast cells was evaluated with BD FACS Verse® (BD Biosciences, Franklin Lakes (USA)) counting 50 000 events with the same instrument settings. Data analysis was performed using the FlowJO™ 10.2 software (FlowJO LLC, Ashland (USA)). The gating strategy was based on blotting forward (FSC) and side scatter (SSC) to exclude cellular debris, followed by gating for single cells by using FSC-W and FSC-H.
The staining of phagocytosed yeast cells was adapted from (Kasper et al. 2018). Infected macrophages were fixed with Roti®-Histofix 4% and stained with 50 µg mL−1 Concanavalin A conjugated with Alexa Fluor™ 647 (ConA-AF647; Thermo Fisher Scientific) at 37°C for 30 min to visualize external Candida cells. Coverslips were mounted with ProLong™ Gold Antifade Mountant with DAPI and fluorescence images were recorded using a Zeiss AXIO Observer.Z1 (Carl Zeiss Microscopy). Stationary phase cells expressing yEGFP and mCherry under the control of the TEF1 promotor were diluted 1:20 into fresh YPD medium and incubated at 37°C for 2 h and 180 rpm. Washed and fixed cells were imaged on glass slides using a Zeiss AXIO Observer.Z1 (Carl Zeiss Microscopy). Yeast cells of different strain backgrounds were imaged with the same exposure time and fluorescence channel settings, and image processing was conducted with the same parameters.
Statistics
Data are reported as scatterplot, line charts, or bar charts, each showing mean + standard deviation (SD). Presented results are from at least three independent biological replicates (growth assays), four independent biological replicates (flow cytometry analysis), and two independent biological replicates (qRT-PCR analysis). Macrophage survival experiments were done with immune cells originating from four independent blood donors per day, on at least two independent days. Data were analyzed using GraphPad Prism 5 (GraphPad Software, San Diego, USA). For statistical analysis of matched observations (macrophage experiments), a repeated measures ANOVA, and for non-matched observations (GFP fluorescence intensity experiments), a one-way ANOVA was performed. In both cases a Bonferroni's multiple comparison test was included. Statistically significant results are marked with asterisks. (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
SUMMARY
Taken together, we have constructed plasmids for fluorescence reporter-based analysis that integrate into the TRP1 locus of C. glabrata. We show that fluorescence levels depend on the strength of the promotor and promotor-specific induction conditions. Moreover, we used the overexpression system for heterologous expression of the well-characterized model antigen OVA. This provides a basis for future measurements of specific immune responses of T cells against C. glabrata. Finally, we showed that the TRP1 locus is a suitable locus to (re-)integrate genes of interest, either with their native DNA sequence, with specific sequence variations or with alternative promotors, to investigate the role and function of these genes for fungal (patho)biology. The set of plasmids presented here expands the genetic toolbox available for use with C. glabrata.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge Isabell Nold, Patrick Schmerer, Yi Enn Cheong, Aylina Kulle and Simone Schiele for their technical help with genotypic mutant characterization and CFU counting, Matthias Brock for helpful discussions in the development of the complementation strategy and Annemarie Landmann for the ovalbumin construct and her advice on the ovalbumin project. We thank Nadja Jablonowski, Stephanie Wisgott, Daniela Schulz and Dorothee Eckardt for isolation of PBMCs and excellent technical assistance; Annika König, Daniel Fischer, Fabrice Hille and Sophie Austermeier for their support during cultivation of MDMs. Use of the auto-MACS system was kindly provided by the research group Fungal Septomics, ZIK Septomics Jena.
Contributor Information
Marcel Sprenger, Department Microbial Pathogenicity Mechanisms, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Adolf-Reichwein-Straße 23, 07745 Jena, Germany.
Sascha Brunke, Department Microbial Pathogenicity Mechanisms, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Adolf-Reichwein-Straße 23, 07745 Jena, Germany.
Bernhard Hube, Department Microbial Pathogenicity Mechanisms, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Adolf-Reichwein-Straße 23, 07745 Jena, Germany; Institute of Microbiology, Friedrich Schiller University, Neugasse 25, 07743 Jena, Germany.
Lydia Kasper, Department Microbial Pathogenicity Mechanisms, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Adolf-Reichwein-Straße 23, 07745 Jena, Germany.
FUNDING
This work was supported by Deutsche Forschungsgemeinschaft SPP 1580 [Grant number Hu 528/17–1 to B.H.].
Conflicts of Interest
None declared.
REFERENCES
- Bolotin-Fukuhara M, Fairhead C. Candida glabrata: a deadly companion? Yeast. 2014;31:279–88. [DOI] [PubMed] [Google Scholar]
- Brand A, MacCallum DM, Brown AJ et al. Ectopic expression of URA3 can influence the virulence phenotypes and proteome of Candida albicans but can be overcome by targeted reintegration of URA3 at the RPS10 locus. Eukaryot Cell. 2004;3:900–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branlant G, Branlant C. Nucleotide sequence of the Escherichia coli gap gene. Different evolutionary behavior of the NAD+-binding domain and of the catalytic domain of D-glyceraldehyde-3-phosphate dehydrogenase. Eur J Biochem. 1985;150:61–66. [DOI] [PubMed] [Google Scholar]
- Brunke S, Quintin J, Kasper L et al. Of mice, flies–and men? Comparing fungal infection models for large-scale screening efforts. Dis Model Mech. 2015;8:473–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman-Smith A, Cronan JE Jr. Molecular biology of biotin attachment to proteins. J Nutr. 1999;129:477S–84S. [DOI] [PubMed] [Google Scholar]
- Citiulo F, Jacobsen ID, Miramón P et al. Candida albicans scavenges host zinc via Pra1 during endothelial invasion. PLoS Pathog. 2012;8:e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Bertram G, Egerton M et al. Yeast-enhanced green fluorescent protein (yEGFP): a reporter of gene expression in Candida albicans. Microbiology. 1997;143(Pt 2):303–11. [DOI] [PubMed] [Google Scholar]
- Davis DA, Bruno VM, Loza L et al. Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics. 2002;162:1573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekema D, Arbefeville S, Boyken L et al. The changing epidemiology of healthcare-associated candidemia over three decades. Diagn Microbiol Infect Dis. 2012;73:45–48. [DOI] [PubMed] [Google Scholar]
- Falkow S. Molecular Koch's postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10 Suppl 2:S274–276. [DOI] [PubMed] [Google Scholar]
- Frieman MB, McCaffery JM, Cormack BP. Modular domain structure in the Candida glabrata adhesin Epa1p, a beta1,6 glucan-cross-linked cell wall protein. Mol Microbiol. 2002;46:479–92. [DOI] [PubMed] [Google Scholar]
- Gabaldón T, Carrete L. The birth of a deadly yeast: tracing the evolutionary emergence of virulence traits in Candida glabrata. FEMS Yeast Res. 2016;16:fov110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galocha M, Pais P, Cavalheiro M et al. Divergent Approaches to Virulence in C. albicans and C. glabrata: Two Sides of the Same Coin. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartenberg MR, Smith JS. The Nuts and Bolts of Transcriptionally Silent Chromatin in Saccharomyces cerevisiae. Genetics. 2016;203:1563–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerami-Nejad M, Zacchi LF, McClellan M et al. Shuttle vectors for facile gap repair cloning and integration into a neutral locus in Candida albicans. Microbiology. 2013;159:565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Navarathna DH, Roberts DD et al. Arginine-induced germ tube formation in Candida albicans is essential for escape from murine macrophage line RAW 264.7. Infect Immun. 2009;77:1596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gola S, Martin R, Walther A et al. New modules for PCR-based gene targeting in Candida albicans: rapid and efficient gene targeting using 100 bp of flanking homology region. Yeast. 2003;20:1339–47. [DOI] [PubMed] [Google Scholar]
- Harms JS, Khan M, Hall C et al. Brucella Peptide Cross-Reactive Major Histocompatibility Complex Class I Presentation Activates SIINFEKL-Specific T Cell Receptor-Expressing T Cells. Infect Immun. 2018;86:e00281–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogaya N, Miyazaki T, Nagi M et al. The heme-binding protein Dap1 links iron homeostasis to azole resistance via the P450 protein Erg11 in Candida glabrata. FEMS Yeast Res. 2013;13:411–21. [DOI] [PubMed] [Google Scholar]
- Hünniger K, Lehnert T, Bieber K et al. A virtual infection model quantifies innate effector mechanisms and Candida albicans immune escape in human blood. PLoS Comput Biol. 2014;10:e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka J, Grebe K, Shenderov E et al. Quantitating T cell cross-reactivity for unrelated peptide antigens. J Immunol. 2009;183:4337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Matsudomi N. Structural characteristics of hen egg ovalbumin expressed in yeast Pichia pastoris. Biosci Biotechnol Biochem. 2005;69:755–61. [DOI] [PubMed] [Google Scholar]
- Jacobsen ID, Brunke S, Seider K et al. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect Immun. 2010;78:1066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janbon G, Quintin J, Lanternier F et al. Studying fungal pathogens of humans and fungal infections: fungal diversity and diversity of approaches. Genes Immun. 2019;20:403–14. [DOI] [PubMed] [Google Scholar]
- Kasper L, Konig A, Koenig PA et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat Commun. 2018;9:4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada K, Yamaguchi E, Arisawa M. Isolation of a Candida glabrata centromere and its use in construction of plasmid vectors. Gene. 1996;175:105–8. [DOI] [PubMed] [Google Scholar]
- Knowles JR. The mechanism of biotin-dependent enzymes. Annu Rev Biochem. 1989;58:195–221. [DOI] [PubMed] [Google Scholar]
- Krummey SM, Floyd TL, Liu D et al. Candida-elicited murine Th17 cells express high Ctla-4 compared with Th1 cells and are resistant to costimulation blockade. J Immunol. 2014;192:2495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman CP, Robnett CJ. Phylogenetic relationships among yeasts of the ‘Saccharomyces complex’ determined from multigene sequence analyses. FEMS Yeast Res. 2003;3:417–32. [DOI] [PubMed] [Google Scholar]
- Lane MD, Rominger KL, Young DL et al. The Enzymatic Synthesis of Holotranscarboxylase from Apotranscarboxylase and (+)-Biotin. Ii. Investigation of the Reaction Mechanism. J Biol Chem. 1964;239:2865–71. [PubMed] [Google Scholar]
- Lüttich A, Brunke S, Hube B. Isolation and amplification of fungal RNA for microarray analysis from host samples. Methods Mol Biol. 2012;845:411–21. [DOI] [PubMed] [Google Scholar]
- Millon L, Manteaux A, Reboux G et al. Fluconazole-resistant recurrent oral candidiasis in human immunodeficiency virus-positive patients: persistence of Candida albicans strains with the same genotype. J Clin Microbiol. 1994;32:1115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miramón P, Dunker C, Windecker H et al. Cellular responses of Candida albicans to phagocytosis and the extracellular activities of neutrophils are critical to counteract carbohydrate starvation, oxidative and nitrosative stress. PLoS One. 2012;7:e52850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Izumikawa K, Yamauchi S et al. The glycosylphosphatidylinositol-linked aspartyl protease Yps1 is transcriptionally regulated by the calcineurin-Crz1 and Slt2 MAPK pathways in Candida glabrata. FEMS Yeast Res. 2011;11:449–56. [DOI] [PubMed] [Google Scholar]
- Morschhäuser J, Michel S, Hacker J. Expression of a chromosomally integrated, single-copy GFP gene in Candida albicans, and its use as a reporter of gene regulation. Mol Gen Genet. 1998;257:412–20. [DOI] [PubMed] [Google Scholar]
- Murad AM, Lee PR, Broadbent ID et al. CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast. 2000;16:325–7. [DOI] [PubMed] [Google Scholar]
- Navarathna DH, Das A, Morschhauser J et al. Dur3 is the major urea transporter in Candida albicans and is co-regulated with the urea amidolyase Dur1,2. Microbiology. 2011;157:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarathna DH, Lionakis MS, Lizak MJ et al. Urea amidolyase (DUR1,2) contributes to virulence and kidney pathogenesis of Candida albicans. PLoS One. 2012;7:e48475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevitt T, Thiele DJ. Host iron withholding demands siderophore utilization for Candida glabrata to survive macrophage killing. PLoS Pathog. 2011;7:e1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble JA, Tsai HF, Suffis SD et al. STB5 is a negative regulator of azole resistance in Candida glabrata. Antimicrob Agents Chemother. 2013;57:959–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrander J, Kempe T, Messing J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene. 1983;26:101–6. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pla J, Gil C, Monteoliva L et al. Understanding Candida albicans at the molecular level. Yeast. 1996;12:1677–702. [DOI] [PubMed] [Google Scholar]
- Rando OJ, Winston F. Chromatin and transcription in yeast. Genetics. 2012;190:351–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roon RJ, Levenberg B. Urea amidolyase. I. Properties of the enzyme from Candida utilis. J Biol Chem. 1972;247:4107–13. [PubMed] [Google Scholar]
- Samols D, Thornton CG, Murtif VL et al. Evolutionary conservation among biotin enzymes. J Biol Chem. 1988;263:6461–4. [PubMed] [Google Scholar]
- Sanglard D, Ischer F, Monod M et al. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob Agents Chemother. 1996;40:2300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzmüller T, Ma B, Hiller E et al. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 2014;10:e1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seider K, Gerwien F, Kasper L et al. Immune evasion, stress resistance, and efficient nutrient acquisition are crucial for intracellular survival of Candida glabrata within macrophages. Eukaryot Cell. 2014;13:170–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger M, Hartung TS, Allert S et al. Fungal biotin homeostasis is essential for immune evasion after macrophage phagocytosis and virulence. Cell Microbiol. 2020. [DOI] [PubMed] [Google Scholar]
- Staab JF, Bahn YS, Sundstrom P. Integrative, multifunctional plasmids for hypha-specific or constitutive expression of green fluorescent protein in Candida albicans. Microbiology. 2003;149:2977–86. [DOI] [PubMed] [Google Scholar]
- Sternicki LM, Wegener KL, Bruning JB et al. Mechanisms Governing Precise Protein Biotinylation. Trends Biochem Sci. 2017;42:383–94. [DOI] [PubMed] [Google Scholar]
- Tong L. Structure and function of biotin-dependent carboxylases. Cell Mol Life Sci. 2013;70:863–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno K, Matsumoto Y, Uno J et al. Intestinal resident yeast Candida glabrata requires Cyb2p-mediated lactate assimilation to adapt in mouse intestine. PLoS One. 2011;6:e24759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte P, Ischer F, Sanglard D et al. In vivo systematic analysis of Candida albicans Zn2-Cys6 transcription factors mutants for mice organ colonization. PLoS One. 2011;6:e26962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A, Wendland J. An improved transformation protocol for the human fungal pathogen Candida albicans. Curr Genet. 2003;42:339–43. [DOI] [PubMed] [Google Scholar]
- Wilson RB, Davis D, Mitchell AP. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol. 1999;181:1868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu QR, Yan L, Lv QZ et al. Molecular genetic techniques for gene manipulation in Candida albicans. Virulence. 2014;5:507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yánez-Carrillo P, Orta-Zavalza E, Gutierrez-Escobedo G et al. Expression vectors for C-terminal fusions with fluorescent proteins and epitope tags in Candida glabrata. Fungal Genet Biol. 2015;80:43–52. [DOI] [PubMed] [Google Scholar]
- Zempleni J, Wijeratne SS, Hassan YI. Biotin. Biofactors. 2009;35:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Ren Y, Pan SJ et al. Expression plasmids for use in Candida glabrata. G3 (Bethesda). 2013;3:1675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.