

Abstract
Neonatal dried blood spots (NDBS) are a widely banked sample source that enables retrospective investigation into early life molecular events. Here, we performed low-pass whole genome bisulfite sequencing (WGBS) of 86 NDBS DNA to examine early life Down syndrome (DS) DNA methylation profiles. DS represents an example of genetics shaping epigenetics, as multiple array-based studies have demonstrated that trisomy 21 is characterized by genome-wide alterations to DNA methylation. By assaying over 24 million CpG sites, thousands of genome-wide significant (q < 0.05) differentially methylated regions (DMRs) that distinguished DS from typical development and idiopathic developmental delay were identified. Machine learning feature selection refined these DMRs to 22 loci. The DS DMRs mapped to genes involved in neurodevelopment, metabolism, and transcriptional regulation. Based on comparisons with previous DS methylation studies and reference epigenomes, the hypermethylated DS DMRs were significantly (q < 0.05) enriched across tissues while the hypomethylated DS DMRs were significantly (q < 0.05) enriched for blood-specific chromatin states. A ~28 kb block of hypermethylation was observed on chromosome 21 in the RUNX1 locus, which encodes a hematopoietic transcription factor whose binding motif was the most significantly enriched (q < 0.05) overall and specifically within the hypomethylated DMRs. Finally, we also identified DMRs that distinguished DS NDBS based on the presence or absence of congenital heart disease (CHD). Together, these results not only demonstrate the utility of low-pass WGBS on NDBS samples for epigenome-wide association studies, but also provide new insights into the early life mechanisms of epigenomic dysregulation resulting from trisomy 21.
Introduction
Down syndrome (DS) is caused by trisomy 21 and is the most common chromosomal aneuploidy present in live births, where it affects approximately 1 in 691 (1). DS is also the most common genetic cause of intellectual disability and is characterized by distinct facial features and immune system abnormalities. Congenital heart defects (CHD) occur in approximately half of DS patients. Furthermore, DS displays a distinct cancer risk profile, which includes an increased risk for childhood leukemia and a decreased risk for solid tumors (2,3). While the genetic basis of DS is well understood, molecular profiling of DS offers insight into the variability in secondary clinical phenotypes, particularly how epigenetic variation can result from a primary genetic change and reflect variable clinical features.
At the molecular level, DS is characterized by differences in gene expression and epigenetic modifications not only on chromosome 21 but also across the entire genome (4). Global or region-specific DNA CpG hypermethylation has been observed in a number of DS studies and is most pronounced in the brain and placenta (5–8). A meta-analysis of DS DNA methylation array studies uncovered a pan- and multi-tissue CpG methylation signature of DS, where 24 out of the 25 differential genes were hypermethylated in at least 3 of the following tissues: adult brain, fetal brain, placenta, buccal epithelial, and adult blood (4). While global hypermethylation (~1% difference) has been observed in DS adult and fetal brain tissue and sorted cells using arrays, the same study demonstrated that sorted T-lymphocytes (CD3+) from adult DS peripheral blood showed the opposite; a trend for global hypomethylation (~0.3% difference, P = 0.186) in DS compared to control (9). The sorted DS T-lymphocytes (CD3+) showed an approximately equal ratio of hypermethylated and hypomethylated CpGs when compared to controls. Furthermore, the top transcription factor binding site identified within the DS hypomethylated CpGs for the sorted T-lymphocytes (CD3+) was the RUNX1 motif (4,9). More recently, a separate DS study assayed whole-blood from neonates using array technologies (10). In this study, differentially methylated regions (DMRs) distinguishing DS from healthy controls showed a slight but significant bias for hypomethylation, although hypermethylation was observed at RUNX1 (<1 kb in size). RUNX1 is a developmental transcription factor located on chromosome 21 that is associated with acute myeloid leukemia and classically known to regulate the development of blood cells (hematopoiesis), however, it also regulates neurodevelopment (11,12).
While reduced representation methods, such as arrays, have provided invaluable insight into the DS methylome, they only assay less than 1 million out of the ~30 million CpG sites present in the human genome. Previously, in order to assay a larger proportion of the DS methylome, we performed the first whole genome bisulfite sequencing (WGBS) analysis of human DS samples, specifically postmortem brain (7). Our low-pass sequencing analysis of post-mortem brains identified 3152 DMRs, where 75% were hypermethylated and involved in neurodevelopment and metabolism. The hypermethylated DMRs showed a cross-tissue signal, where we replicated many DS pan- and multi-tissue genes and expanded them to larger DMRs, while the hypomethylated DMRs showed a more tissue-specific profile. In this study, we present, to our knowledge, the first WGBS of neonatal dried blood spots (NDBS). We assayed more than 24 million CpGs from the NDBS of 86 children of both sexes with either DS, idiopathic developmental delay (DD), or typical development (TD) enrolled in the Childhood Autism Risks from Genetics and Environment (CHARGE) study (13). Our analyses demonstrate that the DS newborn blood methylome is distinguishable from that of both TD and DD. We also characterized the profiles of hyper- and hypo-methylated DMRs, which diverge by tissue specificity and suggest a key role for RUNX1 in shaping early life epigenetic alterations in DS.
Results
Low-pass WGBS of NDBS DNA detects both trisomy 21 and a trend of global hypomethylation
DNA was isolated from 21 DS (13 Male, 8 Female), 33 DD (16 Male, 17 Female), and 32 (16 Male and 16 Female) TD archived NDBS. WGBS sequencing libraries prepared from 10 ng of DNA per sample were indexed and pooled for sequencing across a single NovaSeq 6000 S4 flow cell (4 lanes) to obtain ~5× coverage (Supplementary Material, Table S1). By comparing read depth across each chromosome, a novel copy number variation (CNV) calling algorithm was utilized to confirm trisomy 21 in the DS samples (Fig. 1A) and to rule out the possibility of other large CNVs on chromosome 21 in the DD and TD samples. Smoothed methylomes from the 24 456 995 CpGs assayed in all groups were first used to detect possible differences in global methylation levels. The DS group showed the lowest global CpG methylation levels (82.1%), followed by DD (82.5%) and TD (82.7%) (Fig. 1B). When compared to the DD and TD groups, there was a trend (PDiagnosis = 0.1, two-way ANOVA) for a slight (~0.5%) decrease in global CpG methylation levels in DS samples.
Figure 1.
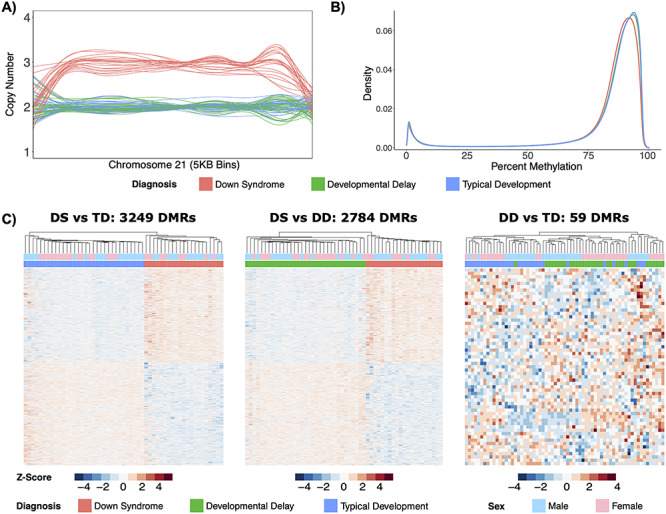
Distinct DS methylome profiles. (A) Line plot of normalized copy number based on read depth over 5 kb bins for chromosome 21 in all 86 samples. (B) Density plot of average percent smoothed methylation for CpGs covered in the 3 diagnostic groups. (C) Heatmaps of significant (q < 0.05) DMRs from the DS versus TD comparison, significant (q < 0.05) DMRs from the DS versus DD comparison, and significant (p < 0.05) DMRs from the DD versus TD comparison. All heatmaps display hierarchal clustering of Z-scores, which are the number of standard deviations from the mean of non-adjusted percent smoothed individual methylation values for each DMR.
Genes associated with DMRs specific to DS reflect differences in neurodevelopment, metabolism and transcriptional regulation
In order to identify DMRs that distinguished DS from TD and DD, three comparisons with adjustments for sex were performed. The DS versus TD comparison identified 3249 genome-wide significant (q < 0.05) DMRs (47% hypermethylated, 53% hypomethylated) from 22 608 background regions that were assembled from the 24 659 362 CpGs covered in these groups (Supplementary Material, Table S2A). On average, the DS versus TD DMRs were 740 bp long and contained 18 CpGs. The DS versus DD comparison identified 2784 genome-wide significant (q < 0.05) DMRs (48% hypermethylated, 52% hypomethylated) from 20 540 background regions that were assembled from the 24 770 743 CpGs covered in these groups (Supplementary Material, Table S2B). On average, the DS versus DD DMRs were 733 bp long and contained 18 CpGs. In contrast to the other comparisons, the DD versus TD comparison only identified 59 nominally significant (p < 0.05) DMRs (54% hypermethylated, 46% hypomethylated) from 1861 background regions that were assembled from the 25 122 420 CpGs covered in these groups (Supplementary Material, Table S2C). On average, the DD versus TD DMRs were 498 bp long and contained 24 CpGs. Hierarchal clustering analysis also showed that while DS DMRs clearly distinguished DS from either DD or TD groups, they did not distinguish DD from TD (Fig. 1C). DMRs from all comparisons were distributed throughout the genome with no evident preference for a particular chromosome (Supplementary Material, Fig. S1).
Given that the adjustment for sex removed sex-specific effects, we also performed sex-stratified analyses for each of the three main comparisons. The majority of DS DMRs from the sex-stratified analyses overlapped with the sex-adjusted analyses and there were fewer DMRs overall in the sex-stratified analyses (Supplementary Material, Fig. S2). This finding suggests that the adjustment for sex provided increased power to detect differences by enabling larger sample sizes and that there are relatively few sex-specific differences in DS associated NDBS methylation changes. In contrast, DD versus TD sex-stratified analyses detected a larger number of DMRs than the sex-adjusted analysis, which suggests a more sex-specific profile in idiopathic neurodevelopmental disorders when compared to DS.
Next, we examined the functional relevance of the DS DMRs. First, we tested for enrichments within known CpG and gene region annotations. For the CpG annotations, the hypermethylated DS DMRs were significantly (q < 0.05) enriched within CpG islands and shores and the hypomethylated DS DMRs were enriched within the open sea (Supplementary Material, Fig. S3A). Genic annotation testing revealed that the hypermethylated DS DMRs were significantly (q < 0.05) enriched within promoters, exons, and 3′ untranslated regions (UTRs) and the hypermethylated DS versus TD DMRs were also enriched within 5′ UTRs (Supplementary Material, Fig. S3B). The hypomethylated DS versus TD DMRs were significantly (q < 0.05) enriched within exons and introns, while the hypomethylated DS versus DD DMRs were enriched within 3′ UTRs.
DMRs from all comparisons were mapped to genes, where the top significantly (p < 0.05, dispensability ≤0.25) enriched Gene Ontology (GO) terms were identified (Fig. 2). Some neurodevelopmental GO terms were enriched in all comparisons (behavior, juxtaparanode region of axon, and GABA-ergic synapse); however, GO terms unique to DS DMRs included some neurodevelopmental terms (autonomic nervous system development and semaphorin receptor complex/activity) as well as terms related to metabolism (choline metabolic process, hemoglobin metabolic process, pyridoxal phosphate binding, and transaminase activity) and transcriptional regulation (nucleosome and DNA-binding transcription factor activity). The GO terms were relatively similar when split by hyper- and hypo-methylation (Supplementary Material, Fig. S4).
Figure 2.
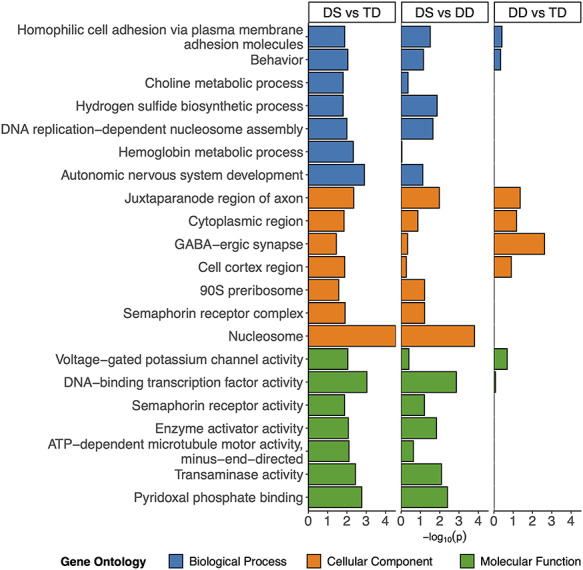
GO enrichments. Bar plot of the least dispensable slimmed significant (p < 0.05, dispensability ≤0.25) GO enrichments for DS versus TD comparison with corresponding values from the DS versus DD and DD versus TD comparison. NAs in the DD versus TD comparison were replaced with 0.
Consensus DMRs and machine learning predictors distinguish DS from DD and TD NDBS samples
In order to determine if DS DMRs that accurately distinguish DS from idiopathic DD as well as TD samples could be identified, we used two approaches. First, we examined the overlap of DMRs identified in each of the three comparisons (Fig. 3A and Supplementary Material, Fig. S5A). Merging the overlaps into single regions that spanned all combined DMRs produced a consensus DMR profile of 4205 DMRs whose smoothed methylation distinguished DS from DD and TD by the first principle component (Fig. 3B and Supplementary Material, Fig. S5B). Second, machine learning feature selection was performed on the consensus DMRs and identified a minimal set of 22 DMRs that distinguished DS from DD and TD (Fig. 3C). Notably, only one of the DMRs was intergenic and only one was located on chromosome 21 (RUNX1). Finally, a machine learning analysis to predict the binary diagnosis class of all 86 samples with the minimal 22 DMRs as predictors performed with an accuracy of 100% and kappa of 1.
Figure 3.
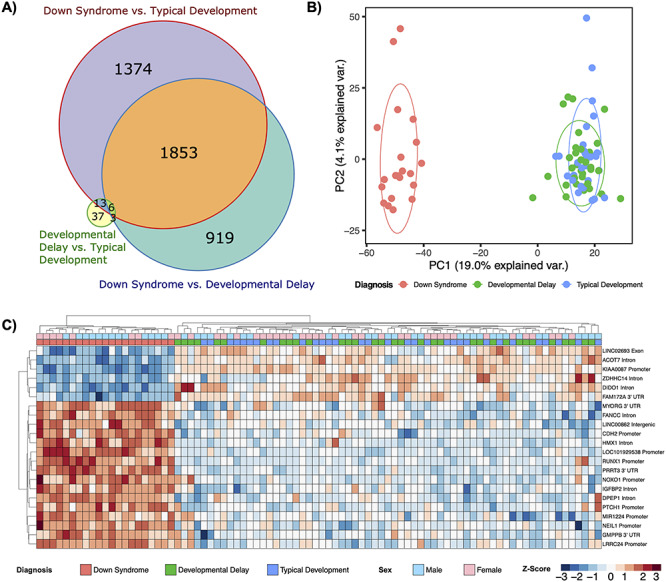
Consensus DMR profiles. (A) Area-proportional Venn diagram of sequence overlaps for DMRs from all comparisons used to assemble the consensus DMRs. (B) Principal component analysis of consensus DMRs. Ellipses represent the 68% confidence interval, which is 1 standard deviation from the mean for a normal distribution. (C) Hierarchal clustering heatmap of the machine learning feature selection analysis of the consensus DMRs.
Hypermethylated DS DMRs are enriched across multiple tissues while hypomethylated DS DMRs are enriched for blood-specific regions and chromatin states
In order to determine if the DS DMRs we identified in newborn blood are similar to those found in other DS tissues and studies, we performed enrichment analyses separately for hyper- and hypo-methylated DS DMRs for each comparison group (Fig. 4A and Supplementary Material, Table S3). DS hyper- or hypo-methylated CpGs or DMRs were identified from 14 datasets from 9 different studies: 1) Neonatal whole-blood CpGs assayed by Illumina’s Infinium Human Methylation 450 K BeadChip array (450 K) from Henneman et al. (10), 2) whole-blood CpGs within DMRs assayed by the 450 K from Bacalini et al. (14), 3) Myocardium (heart muscle) CpGs assayed on the 450 K from Cejas et al. (15), 4) sorted adult peripheral T-lymphocytes (CD3+), adult frontal cortex, sorted adult frontal cortex neurons (NeuN+), sorted adult frontal cortex glia (NeuN−), adult cerebellar folial cortex, and mid-gestation fetal cerebrum CpGs assayed by 450 K from Mendioroz et al. (9), 5) buccal epithelium CpGs assayed by 450 K from Jones et al. (16), 6) placenta DMRs assayed by reduced representation bisulfite sequencing from Jin et al. (5), 7) neural induced pluripotent stem cell (iPSC) derivative CpGs assayed by 450 K from Laan et al. (17), 8) fetal frontal cortex CpGs assayed by 450 K from El Hajj et al. (6), and 9) adult frontal cortex DMRs assayed by WGBS from Laufer et al. (7).
Figure 4.
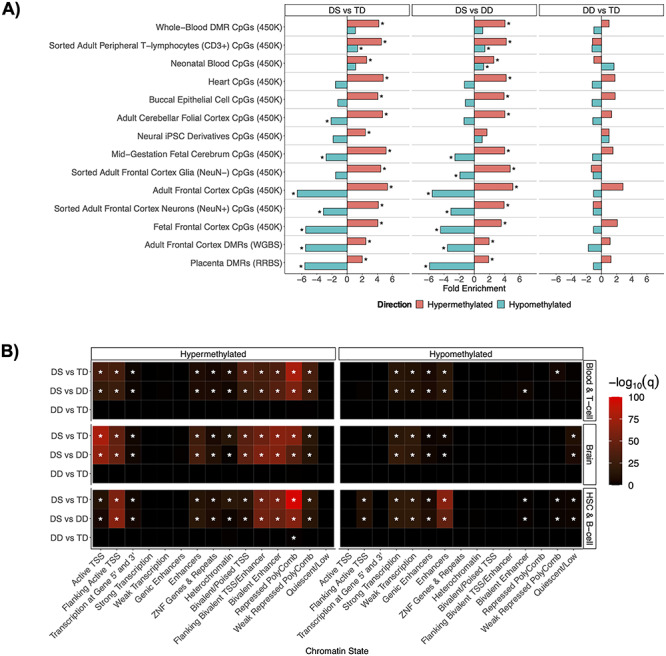
Divergent DNA hyper- and hypo-methylation profiles. (A) DS cross-tissue enrichments for differential sites from existing DS studies. (B) Summary heatmap of top q-values for Roadmap epigenomics 127 reference epigenomes chromHMM chromatin state enrichments for all comparisons within the blood and brain tissue reference datasets. *q < 0.05.
As expected, the strongest overall significant (q < 0.05) enrichments for the DS NDBS DMRs were within differentially methylated sites from previous DS blood studies, where whole blood, sorted adult peripheral T-lymphocytes (CD3+), and neonatal blood were the top ranked. However, in tissues other than blood, the hyper- and hypo-methylated DMRs displayed divergent enrichment profiles with sites from previous DS studies. The DS NDBS hypermethylated DMRs were significantly (q < 0.05) enriched for multiple tissues and studies. Additionally, 12 out of the 25 previously known DS pan- and multi-tissue genes were present in our hypermethylated NDBS DMRs, including RUNX1, the clustered protocadherins, the HOXA cluster, LRRC24, GLI4, TEX14, RYR1, CYTH2, ZNF837, MZF1, CPT1B, and CELSR3 (4). In contrast to our hypermethylated DS NDBS DMRs, our hypomethylated DS NDBS DMRs showed significant (q < 0.05) de-enrichments in DS differentially methylated sites from non-blood tissues. These results indicate an interesting divergence between hypermethylated regions in DS, which are observed across tissue-type, and hypomethylated regions in DS, which are blood-specific.
To gain further insight into the tissue patterns of DS methylation changes, we performed functional annotation of DMRs using chromatin state segmentations from the chromHMM core 15-state model (based on 5 histone post-translational modifications from 127 reference epigenomes) (Fig. 4B) (18,19). DS hypermethylated DMRs showed significant enrichments (q < 0.05) across numerous chromatin states, where the strongest enrichments were for repressed polycomb, active transcription start sites (TSS), and bivalent chromatin states. Unlike the hypermethylated DMRs, the hypomethylated DMRs were enriched in a more tissue-specific manner for chromatin states known to vary by tissue type, particularly enhancers in hematopoietic stem cells.
Further investigation of the 5-core histone post-translational modifications from the 127 reference epigenomes also revealed divergence of the hyper- and hypo-methylated DMRs (19). Although the top enrichment for the hypermethylated DMRs is from the heterochromatin associated mark H3K9me3 in the thymus (Supplementary Material, Fig. S6A), which is where T-lymphocytes mature, the hypomethylated DS DMRs in NDBS showed a prominent enrichment for the enhancer associated mark H3K4me1 across blood, and H3K36me3, a mark associated with actively transcribed gene bodies, across multiple cell types (Supplementary Material, Fig. S6B). Together, these results demonstrate a divergence between the hypermethylated DS DMRs, which contain a cross-tissue signature, and the hypomethylated DS DMRs, which are primarily tissue-specific.
Early life RUNX1 dysregulation and hypermethylation is reflected in hypomethylated DS DMRs
In order to identify larger genomic ‘blocks’ of differential methylation in DS, a separate analysis with smoothing parameters optimized to detect regions >5 kb was performed. The DS versus TD analysis identified 3 significant (q < 0.05) blocks from 4 background blocks (Supplementary Material, Table S4A) and the DS versus DD analysis identified 2 significant (q ≤ 0.05) blocks from 3 background blocks (Supplementary Material, Table S4B). Two of the previously known pan- and multi-tissue hypermethylated DS DMRs also overlapped within the only significant (q ≤ 0.05) blocks in common between the DS versus TD and DS versus DD comparisons, while no blocks were detected in DD versus TD. These two blocks were located in RUNX1 (Fig. 5A) and the clustered protocadherins. Overall, RUNX1 was the highest ranked block and among the top two highest ranked DMRs in the DS comparisons.
Figure 5.
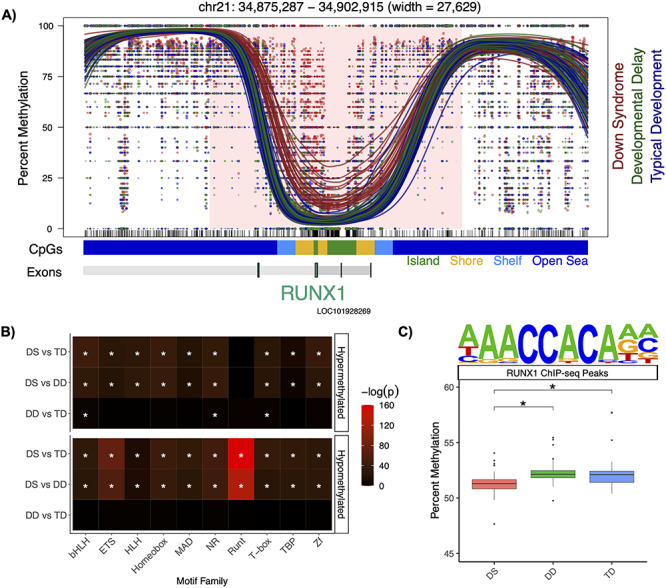
RUNX1 profile. (A) Significant (q < 0.05) hypermethylation within the RUNX1 block. The lines represent individual smoothed methylation level estimates for DS (red), DD (green) or TD (blue). The dots represent the methylation level estimates of an individual CpG, and the size of each dot is representative of coverage. CpG and genic annotation tracks are shown below each plot, and the RUNX1 gene is encoded on the negative strand. (B) Summary heatmap of top P-values for top 10 transcription factor motif family enrichments for all comparison groups (*q < 0.05). (C) Mean percent smoothed DNA methylation levels in RUNX1 binding sites alongside the motif (*Padjusted < 0.05).
An enrichment analysis of transcription factor binding motifs was performed for NDBS DMRs across comparison groups. This analysis identified the Runt motif family as the most significantly (q < 0.05) enriched overall, and enrichment for this motif was specific to hypomethylated DS DMRs (Fig. 5B). The top Runt motif identified belonged to RUNX1 and was from a chromatin immunoprecipitation sequencing (ChIP-seq) experiment that assayed Jurkat cells, which are an immortalized human T-lymphocyte cell line (Fig. 5C) (20). A genome-wide analysis, which was not restricted to the DMRs, revealed that diagnosis had a significant (P < 0.05, two-way ANOVA) effect on the mean of the smoothed DNA methylation levels within the RUNX1 ChIP-seq peaks. The DS samples had a mean of 51.4% methylation, which was significantly (Padjusted < 0.05, post-hoc Tukey HSD tests) lower than the 52.4% methylation of both DD and TD. Taken together, these results indicate that large-scale hypermethylation of RUNX1 (~28 kb) is the strongest signal within the DS DMRs, while its binding sites are specifically associated with the hypomethylated DMR profile as the top transcription factor motif.
DS patients with CHD display a distinct DMR profile
Among the DS cases in our study, there were 11 with CHD (5 Males, 6 Females) and 10 without CHD (8 males, 2 females). In order to identify DMRs that distinguished DS with CHD from DS without CHD, a comparison with an adjustment for sex was performed. There were 1588 nominally significant (p < 0.05) DMRs (35% hypermethylated, 65% hypomethylated) from 50 330 background regions that were assembled from the 22 372 366 CpGs covered in these groups (Supplementary Material, Table S5). The DMRs distinguished DS with CHD from DS without CHD (Supplementary Material, Fig. S7A). Notably, there was an 880 bp hypomethylated DMR that mapped to RUNX1 in CHD compared to non-CHD DS cases. The only other CHD DMRs that mapped to the known DS pan- and multi-tissue genes were VPS37B and the HOXA cluster locus, both of which were also hypomethylated, as well as the clustered protocadherin locus, where DMRs distinguishing CHD DS cases were observed in both directions. GO enrichment analyses of the DS CHD distinguishing DMRs revealed significant (P < 0.05, dispensability ≤ 0.03) enrichments for terms related to the heart (atrial cardiac muscle tissue development and actin-based cell projection) as well as similar terms to the previous DS versus TD and DS versus DD comparisons, specifically those related to neurodevelopment and metabolism (Supplementary Material, Fig. S7B). Machine learning feature selection was performed on the DS CHD DMRs and identified a minimal set of 7 DMRs that distinguished DS with CHD from DS without CHD (Supplementary Material, Fig. S7C). Additionally, a final machine learning analysis to predict the diagnosis class of all 21 samples with the 7 minimal DMRs as predictors performed with an accuracy of 100% and kappa of 1. The top overall significant (q < 0.05) transcription factor motif enrichment for the DS CHD DMRs was for the homeobox transcription factor BAPX1, which was specific to the hypomethylated DMRs (Supplementary Material, Fig. S7D) (21). Notably, an ETS:RUNX motif, which refers to regions co-occupied by the two factors, was enriched within the hypermethylated DS CHD DMRs and also within the hypomethylated DS DMRs from the previous comparisons (22).
Discussion
Overall, the results of the first WGBS analysis of NDBS DNA samples demonstrate that the DS NDBS methylome is characterized by a genome-wide profile consisting of thousands of DMRs that distinguish it from not only TD but also, for the first time, idiopathic DD. The DS DMRs mapped to genes that are enriched for processes related to neurodevelopment, metabolism, and transcriptional regulation. Furthermore, the hyper- and hypo-methylated DMRs distinguishing DS from DD or TD showed divergent profiles, where the hypermethylated DMRs contained a cross-tissue signature and the hypomethylated DMRs reflected a blood-specific profile related to RUNX1 downstream targets.
One limitation of our study was that neonatal whole-blood is a heterogenous mixture of different cell types, which includes nucleated red blood cells, and alterations in cell composition could influence some of the observed differences. Since our study was, to our knowledge, the first WGBS of NDBS, we utilized multiple existing cell composition estimation methods and reference datasets; however, none accurately estimated the expected cell composition or alterations. Therefore, it was not yet possible to correct for cell type composition due to a lack of a combination of appropriate neonatal whole-blood reference datasets and low-pass WGBS cell composition estimation methods. It is our hope that by demonstrating the feasibility of this assay of a low input sample source for epigenome-wide association studies (EWAS) that future research will generate specific neonatal whole-blood reference datasets and low-pass WGBS cell-type composition estimation methods. However, we note that our analyses replicate many known DS specific differences that have been observed in analyses of bulk tissues (whole-blood) (14) with cell-type composition correction and purified cell types (T-lymphocytes) (9), which both showed the strongest enrichments within our DS discriminating DMRs, a result that would not be expected if cell type shifts had a large effect on our analyses.
Notably, the GO analyses are consistent with the literature and replicate known differences in prior DS metabolome and methylome studies. Previous analyses of plasma metabolites at childhood from DS patients in the CHARGE study revealed distinct alterations to methylation metabolism, specifically choline, which are consistent with our enrichment for DS differentially methylated genes involved in choline metabolism and pyridoxal phosphate (the active form of vitamin B6) binding (23). Additionally, our findings replicate those of DS differentially methylated genes from whole-blood, which were involved in processes related to hematopoiesis, neurodevelopment, and chromatin (14). Finally, the GO terms identified from DMRs discriminating DS cases with CHD reflect not only heart development, but also neurodevelopmental functions, consistent with the observation that DS infants with CHD are known to show an increased severity of neurodevelopmental disabilities (24,25).
RUNX1 is a strong candidate for being a primary dysregulated driver of the epigenetic changes in DS blood. The hypermethylation of RUNX1 is reproducible across multiple blood studies (9,10,14). In our study, hypermethylation of RUNX1 was the strongest DS signal and it was one of the 22 machine learning predictors of DS in NDBS, where it distinguished DS from not only TD but also idiopathic DD. RUNX1 was also the top overall transcription factor motif, specifically enriched within the hypomethylated DS DMRs but not the hypermethylated DMRs. The enrichment for RUNX1 binding motifs in hypomethylated sites has been previously reported in DS T-lymphocytes (4,9). Mechanistically, RUNX1 binding results in demethylation of the bound region during hematopoietic development through the recruitment of DNA demethylation enzymes (TET2, TET3, TDG, and GADD45) (26). Since RUNX1 is located on chromosome 21, it is overexpressed in DS (4). Taken together, these observations suggest that the tissue-specific hypomethylated DMRs we observed in DS NDBS are downstream of a primary alteration to RUNX1, where they may have been demethylated by excess RUNX1 binding. This mechanism may also contribute to the observed trend of genome-wide hypomethylation, which has also been previously reported in DS T-lymphocytes (9). Notably, the block of hypermethylation overlaps with an enhancer in the first intron of RUNX1, which regulates RUNX1 expression in hematopoietic stem cells and is also part of a larger super-enhancer involved in hematopoiesis (27–29).
Our findings are consistent with a two-stage mechanism to explain the complex RUNX1 DNA methylation profile (Fig. 6). Initially, during early prenatal development, the increased dosage of RUNX1 may result in genome-wide hypomethylation of its binding sites through recruitment of TET2. Then, during later development and apparent at birth, the RUNX1 super-enhancer may become hypermethylated as a compensatory response to attenuate the overexpression of RUNX1. This hypermethylation may be driven by the catalytically inactive but stimulatory DNA methyltransferase DNMT3L, which is also located on chromosome 21 and known to methylate key DS genes (30). Ultimately, the observed alterations in RUNX1 could influence blood cell function and/or composition as well as neurodevelopment (11,12). Finally, RUNX1 methylation could also reflect the severity of secondary clinical features, as a smaller region within RUNX1 was hypomethylated in DS cases with CHD compared to those without.
Figure 6.
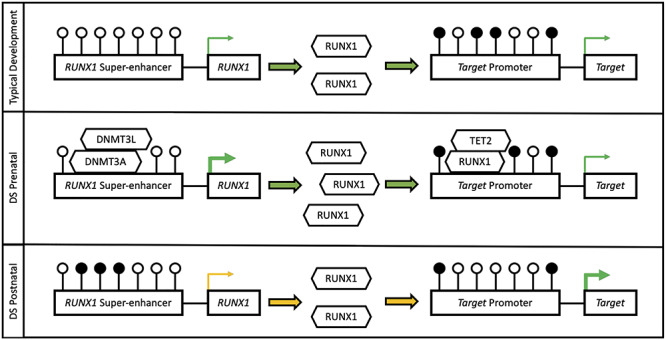
Putative RUNX1 mechanism. During early development the increased dosage of RUNX1 results in genome-wide hypomethylation of its binding sites through recruitment of the TET2 demethylase. Then, during later development and apparent at birth, the RUNX1 enhancer becomes hypermethylated by DNMT3A and DNMT3L to attenuate the increased dosage. White lollipops represent unmethylated CpG sites and black lollipops represent methylated CpG sites.
Taken together, our findings represent one of the largest sample sizes for a DS methylome study, define a DMR profile that distinguishes DS not only from TD but also from idiopathic DD, and provide a novel and general framework for the design and analysis of low-pass WGBS to detect genome-wide methylation changes in NDBS that can be applied to other genetic disorders and environmental exposures.
Materials and Methods
Cohort and DNA extraction from NDBS
Study protocols were approved by the Institutional Review Board at the University of California, Davis (IRB #226028 and #952089) and the Committee for the Protection of Human Subjects at the State of California’s Health and Human Services Agency (Federalwide Assurance #00000681, Project #2018-088). Formal written informed consent from parents or guardians of the participants was obtained prior to collection of any data or specimens. The uploading of the raw or processed sequencing data derived from NDBS of individuals to an external bank or repository was prohibited, as the NDBS were property of the Genetic Diseases Screening Program and subject to restrictions in accordance with California Health and Safety Code, Sections 124980 (j), 124991 (b), (g), (h) and 103850 (a) and (d).
The California Department of Developmental Services’ Regional Center database was used to identify research subjects from the three groups (DS, DD, and TD) within the CHARGE study. The CHARGE participants were matched to the California Newborn Dried Bloodspot Registry, which archives NDBS, also known as Guthrie cards, from heel pricks of newborns obtained 24–72 h after birth. DNA was extracted from 86 NDBS using protocol GQ, which was established for DNA methylation array profiling of NDBS (31). This protocol utilized the GenSolve DNA COMPLETE (GSC-100A, GenTegra) and QIAamp DNA Micro (Qiagen, 56 304) kits and was modified to follow the updated manufacturer’s instructions. DNA quality and quantity were assessed via spectrophotometry on a Nanodrop instrument and fluorometry on a Qubit instrument.
Low-pass WGBS
All DNA samples were sonicated to ~350 bp on a Covaris E220 with a peak power of 175, a duty factor of 10, a cycle/burst of 200 and a time of 47 s. A 1.8× SPRI size selection was performed after sonication. Sonication traces were assessed using a Caliper LabChip GX Analyzer. Approximately 10 ng of the sonicated and size selected DNA was bisulfite converted using the EZ DNA Methylation-Lightning Kit (Zymo Research, D5031) according to the manufacturer’s instructions. Bisulfite converted DNA was eluted into Low EDTA TE (Swift Biosciences, 90 296). Libraries were prepared by post-bisulfite adapter tagging with terminal deoxyribonucleotidyl transferase-assisted adenylate connector-mediated single-stranded-DNA ligation using the Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences, 30 096) with the Methyl-Seq Combinatorial Dual Indexing Kit (Swift Biosciences, 38 096) according to the manufacturer’s instructions with 12 cycles of indexing PCR (32,33). Library traces were assessed using a Caliper LabChip GX Analyzer. The libraries were quantified by fluorometry on a Qubit instrument and pooled in equimolar ratios. The library pool was sequenced across 1 Illumina NovaSeq 6000 S4 flow cell (4 lanes) for 150 bp paired end reads to generate ~100 million read-pairs (~5× coverage) of the genome per sample.
Sequencing alignment
Raw sequencing reads were demultiplexed and sample specific FASTQ files were merged across lanes. The FASTQ files were then aligned to the human genome (hg38) using CpG_Me (https://github.com/ben-laufer/CpG_Me) with the default parameters. The pipeline consisted of trimming adapters and methylation bias, screening for contaminating genomes, aligning to the reference genome, removing PCR duplicates, calculating coverage, calculating insert size, extracting CpG methylation, generating a genome-wide cytosine report (CpG count matrix), as well as examining quality control metrics (34–36).
CNV calling algorithm
A novel read depth based CNV calling algorithm (https://github.com/hyeyeon-hwang/CNV_Me) was utilized to detect large-scale structural variation among the DS, DD, and TD samples. The coverage of 5 kb bins in the chromosomes of each sample was calculated by dividing the sum of the number of reads times the read lengths in each bin by the bin size of 5 kb. Each bin coverage was then normalized by dividing the sum of the number of reads times read lengths in each bin. To calculate the copy number, the normalized bin coverage value was divided by a normalization factor. To anchor the normal copy number value of autosomal chromosomes to be 2, the normalization factor of autosomes was calculated by multiplying 0.5 by the sum of the normalized bin coverage of all the TD control samples and dividing by the total number of control samples. The normalization factors of the sex chromosomes were calculated similarly, except the normal copy number of the sex chromosomes was anchored to 1 for male samples. The sex of each sample was verified using k-means clustering on the ratios of the number of reads aligned to the sex chromosomes.
DMR and block analyses
DMR and block calling as well as some of the downstream enrichment analyses were performed using DMRichR (https://github.com/ben-laufer/DMRichR). To find DMRs that consisted of at least 5 CpGs with at least a 5% difference, the default parameters were used, aside from directly adjusting for sex to remove sex-specific effects, setting perGroup to 0.75 to ensure that a CpG is covered in 75% of samples in each group, and setting the block permutations to 50. DMRichR utilizes the dmrseq (37) and bsseq (38) algorithms to infer methylation levels from CpG count matrices and identify DMRs. These algorithms utilize smoothing and weighting based approaches to infer DMRs from low-pass WGBS, where CpGs with higher coverage are given higher weight. Therefore, the statistical approaches benefit from additional biological replicates more so than deeper sequencing. DMRichR utilizes the dmrseq algorithm to identify DMRs (a few hundred bp to several kb) as well as blocks (>5 kb), where the coverage filtering settings exceeded the established minimum requirements (37). The dmrseq algorithm first assembles candidate background regions, which show a difference between groups, and then performs a statistical analysis to estimate a region statistic. Finally, permutation testing of the pooled null distribution is utilized to identify significant DMRs by calculating empirical P-values that are then FDR corrected (q-values). Individual smoothed methylation levels for downstream analyses and data visualization were obtained using bsseq (38).
Machine learning
Random forest and support vector machine algorithms within the Boruta (39) and sigFeature (40) packages, respectively, were used to build binary classification models and generate two lists of the DMRs ranked by variable importance for the feature selection analyses. From the two lists, the common DMRs within the top 1% of each were selected as minimal DMRs. The follow-up machine learning analyses to predict the class of diagnosis from the minimal DMRs identified in the feature selection analyses utilized the random forest algorithm and 5-fold cross validation. The ntree (number of trees) and mtry (number of predictors sampled at each tree split) model parameters were 500 and 2, respectively.
Enrichment testing
GO enrichment testing was performed using a customized version of GOfuncR, which was based on genomic coordinates and relative to background regions with regions being annotated to a gene if they were between 5 kb upstream to 1 kb downstream of the gene body (41,42). The identified significant (Punadjusted < 0.05) GO terms were then slimmed using REVIGO and ranked by dispensability (43). The hypergeometric optimization of motif enrichment (HOMER) toolset was utilized to test for enriched transcription factor motifs in DMRs relative to background regions through the findMotifsGenome.pl script, where the region size was set to size given and the normalization was set to CpG content (44). The genomic association tester (GAT) was utilized to test for sequence specific overlap relative to background regions with GC content correction for the pan- and multi-tissue enrichment testing (45). A total of 10 000 random samplings were used for all GAT analyses. Previous DS datasets were obtained from their respective publications. The locus overlap analysis (LOLA) (46) program was also utilized for DMR enrichment testing, relative to background regions, for the reference epigenome histone post-translational modifications (5 marks, 127 epigenomes) and the related chromHMM chromatin states from the core 15-state model (18,19). The scripts to reproduce the main figures and analyses are available online (https://github.com/ben-laufer/Low-Pass-WGBS-of-DS-NDBS).
Supplementary Material
Acknowledgements
The authors would like to thank Yunin Ludena Rodriguez for assistance with selecting the samples, Emily Kumimoto for preparing the sequencing libraries, and Matthew Settles for advice about some of the bioinformatic approaches.
Conflict of Interest statement. The authors declare no competing interests.
Contributor Information
Benjamin I Laufer, Department of Medical Microbiology and Immunology, School of Medicine, University of California, Davis, CA 95616, USA; Genome Center, University of California, Davis, CA 95616, USA; MIND Institute, University of California, Davis, CA 95616, USA.
Hyeyeon Hwang, Department of Medical Microbiology and Immunology, School of Medicine, University of California, Davis, CA 95616, USA; Genome Center, University of California, Davis, CA 95616, USA; MIND Institute, University of California, Davis, CA 95616, USA.
Julia M Jianu, Department of Medical Microbiology and Immunology, School of Medicine, University of California, Davis, CA 95616, USA; Genome Center, University of California, Davis, CA 95616, USA; MIND Institute, University of California, Davis, CA 95616, USA.
Charles E Mordaunt, Department of Medical Microbiology and Immunology, School of Medicine, University of California, Davis, CA 95616, USA; Genome Center, University of California, Davis, CA 95616, USA; MIND Institute, University of California, Davis, CA 95616, USA.
Ian F Korf, Genome Center, University of California, Davis, CA 95616, USA; Department of Molecular and Cellular Biology, College of Biological Sciences, University of California, Davis, CA 95616, USA.
Irva Hertz-Picciotto, MIND Institute, University of California, Davis, CA 95616, USA; Department of Public Health Sciences, School of Medicine, University of California, Davis, CA 95616, USA.
Janine M LaSalle, Department of Medical Microbiology and Immunology, School of Medicine, University of California, Davis, CA 95616, USA; Genome Center, University of California, Davis, CA 95616, USA; MIND Institute, University of California, Davis, CA 95616, USA.
Funding
National Institutes of Health (NIH) (3R01ES015359-10S1 to I.H.P. and J.M.L); Canadian Institutes of Health Research (CIHR) postdoctoral fellowship (MFE-146824 to B.I.L.); CIHR Banting postdoctoral fellowship (BPF-162684 to B.I.L.), and the UC Davis Intellectual and Developmental Disabilities Research Center (IDDRC) (1U54HD079125-01). The sequencing was carried out by the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center and was supported by a NIH Shared Instrumentation (1S10OD010786-01).
Authorship Contributions
J.M.L., I.H.P., and B.I.L. designed the study. I.H.P. and J.M.L. acquired funding. J.M.L. and I.F.K. supervised the project. J.M.J. and B.I.L. performed the DNA extractions. B.I.L., H.H., and C.E.M. performed the bioinformatic analyses. B.I.L. and J.M.L. interpreted the results and wrote the manuscript with intellectual contributions and edits from all other authors. All authors reviewed and approved the final manuscript.
References
- 1. Parker S.E., Mai C.T., Canfield M.A., Rickard R., Wang Y., Meyer R.E., Anderson P., Mason C.A., Collins J.S., Kirby R.S. et al. (2010) Updated national birth prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. A Clin. Mol. Teratol., 88, 1008–1016. [DOI] [PubMed] [Google Scholar]
- 2. Hasle H., Haunstrup Clemmensen I. and Mikkelsen M. (2000) Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet, 355, 165–169. [DOI] [PubMed] [Google Scholar]
- 3. Yang Q., Rasmussen S.A. and Friedman J.M. (2002) Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet, 359, 1019–1025. [DOI] [PubMed] [Google Scholar]
- 4. Do C., Xing Z., Yu Y.E. and Tycko B. (2017) Trans-acting epigenetic effects of chromosomal aneuploidies: lessons from Down syndrome and mouse models. Epigenomics, 9, 189–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jin S., Lee Y.K., Lim Y.C., Zheng Z., Lin X.M., Ng D.P.Y., Holbrook J.D., Law H.Y., Kwek K.Y.C., Yeo G.S.H. et al. (2013) Global DNA hypermethylation in Down syndrome placenta. PLoS Genet., 9, e1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. El Hajj N., Dittrich M., Böck J., Kraus T.F.J., Nanda I., Müller T., Seidmann L., Tralau T., Galetzka D., Schneider E. et al. (2016) Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics, 11, 563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laufer B.I., Hwang H., Vogel Ciernia A., Mordaunt C.E. and LaSalle J.M. (2019) Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics, 14, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim J.H., Kang Y.J., Lee B.Y., Han Y.J., Chung J.H., Kim M.Y., Kim M.H., Kim J.W., Cho Y.H. and Ryu H.M. (2019) Epigenome-wide base-resolution profiling of DNA methylation in chorionic villi of fetuses with Down syndrome by methyl-capture sequencing. Clin. Epigenetics, 11, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mendioroz M., Do C., Jiang X., Liu C., Darbary H.K., Lang C.F., Lin J., Thomas A., Abu-Amero S., Stanier P. et al. (2015) Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol., 16, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henneman P., Bouman A., Mul A., Knegt L., Van Der Kevie-Kersemaekers A.M., Zwaveling-Soonawala N., Meijers-Heijboer H.E.J., van Trotsenburg A.S.P. and Mannens M.M. (2018) Widespread domain-like perturbations of DNA methylation in whole blood of Down syndrome neonates. PLoS One, 13, e0194938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Inoue K.I., Shiga T. and Ito Y. (2008) Runx transcription factors in neuronal development. Neural Dev., 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Halevy T., Biancotti J.C., Yanuka O., Golan-Lev T. and Benvenisty N. (2016) Molecular characterization of Down syndrome embryonic stem cells reveals a role for RUNX1 in neural differentiation. Stem Cell Reports, 7, 777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hertz-Picciotto I., Croen L.A., Hansen R., Jones C.R., Water J. and Pessah I.N. (2006) The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environ. Health Perspect., 114, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bacalini M.G., Gentilini D., Boattini A., Giampieri E., Pirazzini C., Giuliani C., Fontanesi E., Scurti M., Remondini D., Capri M. et al. (2015) Identification of a DNA methylation signature in blood cells from persons with Down syndrome. Aging, 7, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cejas R.B., Wang J., Hageman-Blair R., Liu S. and Blanco J.G. (2020) Comparative genome-wide DNA methylation analysis in myocardial tissue from donors with and without Down syndrome. Gene, 764, 145099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones M.J., Farré P., McEwen L.M., MacIsaac J.L., Watt K., Neumann S.M., Emberly E., Cynader M.S., Virji-Babul N. and Kobor M.S. (2013) Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in Down syndrome. BMC Med. Genet., 6, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laan L., Klar J., Sobol M., Hoeber J., Shahsavani M., Kele M., Fatima A., Zakaria M., Annerén G., Falk A. et al. (2020) DNA methylation changes in Down syndrome derived neural iPSCs uncover co-dysregulation of ZNF and HOX3 families of transcription factors. Clin. Epigenetics, 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ernst J. and Kellis M. (2012) ChromHMM: automating chromatin-state discovery and characterization. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods, 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanda T., Lawton L.N., Barrasa M.I., Fan Z.P., Kohlhammer H., Gutierrez A., Ma W., Tatarek J., Ahn Y., Kelliher M.A. et al. (2012) Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic Leukemia. Cancer Cell, 22, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatterjee S., Sivakamasundari S., Yap S.P., Kraus P., Kumar V., Xing X., Lim S.L., Sng J., Prabhakar S. and Lufkin T. (2014) In vivo genome-wide analysis of multiple tissues identifies gene regulatory networks, novel functions and downstream regulatory genes for Bapx1 and its co-regulation with Sox9 in the mammalian vertebral column. BMC Genomics, 15, 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hollenhorst P.C., Chandler K.J., Poulsen R.L., Johnson W.E., Speck N.A. and Graves B.J. (2009) DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet., 5, e1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Orozco J.S., Hertz-Picciotto I., Abbeduto L. and Slupsky C.M. (2019) Metabolomics analysis of children with autism, idiopathic-developmental delays, and Down syndrome. Transl. Psychiatry, 9, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marino B.S., Lipkin P.H., Newburger J.W., Peacock G., Gerdes M., Gaynor J.W., Mussatto K.A., Uzark K., Goldberg C.S., Johnson W.H. et al. (2012) Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management a scientific statement from the american heart association. Circulation, 126, 1143–1172. [DOI] [PubMed] [Google Scholar]
- 25. Homsy J., Zaidi S., Shen Y., Ware J.S., Samocha K.E., Karczewski K.J., DePalma S.R., McKean D., Wakimoto H., Gorham J. et al. (2015) De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science, 350, 1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki T., Shimizu Y., Furuhata E., Maeda S., Kishima M., Nishimura H., Enomoto S., Hayashizaki Y. and Suzuki H. (2017) RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv., 1, 1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kwiatkowski N., Zhang T., Rahl P.B., Abraham B.J., Reddy J., Ficarro S.B., Dastur A., Amzallag A., Ramaswamy S., Tesar B. et al. (2014) Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature, 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hnisz D., Weintrau A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A. et al. (2016) Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science, 351, 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liau W.S., Ngoc P.C.T. and Sanda T. (2017) Roles of the RUNX1 enhancer in normal hematopoiesis and leukemogenesis. Adv. Exp. Med. Biol., 962, 139–147. [DOI] [PubMed] [Google Scholar]
- 30. Lu J., Mccarter M., Lian G., Esposito G., Capoccia E., Delli-Bovi L.C., Hecht J. and Sheen V. (2016) Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet., 25, 1714–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghantous A., Saffery R., Cros M.P., Ponsonby A.L., Hirschfeld S., Kasten C., Dwyer T., Herceg Z. and Hernandez-Vargas H. (2014) Optimized DNA extraction from neonatal dried blood spots: application in methylome profiling. BMC Biotechnol., 14, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miura F., Enomoto Y., Dairiki R. and Ito T. (2012) Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res., 40, e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miura F., Shibata Y., Miura M., Sangatsuda Y., Hisano O., Araki H. and Ito T. (2019) Highly efficient single-stranded DNA ligation technique improves low-input whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res., 47, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wingett S.W. and Andrews S. (2018) FastQ screen: a tool for multi-genome mapping and quality control. F1000Research, 7, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krueger F. and Andrews S.R. (2011) Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ewels P., Magnusson M., Lundin S. and Käller M. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korthauer K., Chakraborty S., Benjamini Y. and Irizarry R.A. (2018) Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics, 20, 367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hansen K.D., Langmead B. and Irizarry R.A. (2012) BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol., 13, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kursa M.B. and Rudnicki W.R. (2010) Feature selection with the boruta package. J. Stat. Softw., 36, 1–13. [Google Scholar]
- 40. Das P., Roychowdhury A., Das S., Roychoudhury S. and Tripathy S. (2020) sigFeature: novel significant feature selection method for classification of gene expression data using support vector machine and t statistic. Front. Genet., 11, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al. (2000) Gene ontology: tool for the unification of biology. Gene ontology: tool for the unification of biology. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prüfer K., Muetzel B., Do H.H., Weiss G., Khaitovich P., Rahm E., Pääbo S., Lachmann M. and Enard W. (2007) FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics, 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Supek F., Bošnjak M., Škunca N. and Šmuc T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H. and Glass C.K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heger A., Webber C., Goodson M., Ponting C.P. and Lunter G. (2013) GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics, 29, 2046–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sheffield N.C. and Bock C. (2015) LOLA: enrichment analysis for genomic region sets and regulatory elements in R and bioconductor. Bioinformatics, 32, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.