

Abstract
The COMPASS protein family catalyzes histone H3 lysine 4 (H3K4) methylation and its members are essential for regulating gene expression. MLL2/COMPASS methylates H3K4 on many developmental genes and bivalent clusters. To understand MLL2-dependent transcriptional regulation, we performed a CRISPR-based screen with an MLL2-dependent gene as a reporter in mouse embryonic stem cells (mESCs) and found that MLL2 functions in gene expression by protecting developmental genes from repression via repelling PRC2 and DNA methylation machineries. Accordingly, repression in the absence of MLL2 is relieved by inhibition of PRC2 and DNA methyltransferases. Furthermore, recruitment of DNA demethylation machineries on such loci leads to reactivation of MLL2-dependent genes not only by removing DNA methylation but also by opening up previously CpG methylated regions for PRC2 recruitment, diluting PRC2 at Polycomb-repressed genes. These findings reveal how the context and function of these three epigenetic modifiers of chromatin can orchestrate transcriptional decisions and demonstrate that prevention of active repression by the context of the enzyme and not H3K4me3 underlies transcriptional regulation on MLL2/COMPASS targets.
Introduction
Posttranslational modifications of chromatin play diverse roles in eukaryotic cell function1. One of these posttranslational marks of histone on chromatin, H3K4me3, is a broadly evolutionary conserved modification that has been extensively studied2. In the budding yeast Saccharomyces cerevisiae, H3K4 methylation is deposited by Set1/COMPASS, a complex containing the lysine methyltransferase (KMT) Set1 and several accessory proteins3–7. In Drosophila melanogaster, three variants of COMPASS have been reported, each with a distinct KMT: dSet1, Trithorax (Trx) or Trithorax-related protein (Trr)8. Mammals have six forms of COMPASS with two close relatives each for the Drosophila enzymes: SET1A (SETD1A) and SET1B (SETD1B), related to dSet1, MLL1 (KMT2A) and MLL2 (KMT2B), related to Trx, and MLL3 (KMT2C) and MLL4 (KMT2D), related to Trr3. Each of these COMPASS family members appear to have unique properties and exhibit both redundant and nonredundant functions. In mammalian cells, SET1A/B reportedly implement most of the H3K4me3 in cells9,10, while MLL3/4 implement monomethylation of H3K4 at enhancers11–13. MLL1/2 implement H3K4me2 and H3K4me3 at developmental genes14,15. MLL1 is required for hematopoiesis and its gene is frequently translocated in leukemia16–18, while MLL2 is required for early development19 and heterozygous variants in the MLL2 gene are causative of childhood dystonia20,21.
The extent to which the overall biological function of COMPASS is related to its lysine methyltransferase activity has been re-evaluated during the past few years in mammals. Indeed, the SET domain of SET1A protein has been shown to be dispensable for mESC proliferation and self-renewal22. Similarly, H3K4 monomethylation catalyzed by Trr and mammalian MLL3/4 COMPASS-like proteins at enhancers was shown to be dispensable for enhancer activation23 and for development24; however, Trr and MLL3/4 proteins are required for viability and development24. Therefore, the role of H3K4 methylation and COMPASS complexes in regulating transcription and development remains a complex issue that requires further molecular investigations.
Our previous studies demonstrated that although MLL2 is required broadly for the implementation of H3K4me3 at bivalent genes in mESCs14,25, surprisingly only few genes’ expression depended on H3K4 methylation function of MLL214. In the present study, we employed a genome-wide genetic screening approach to identify factors required for the proper regulation of transcription of these MLL2-dependent target genes to understand the molecular basis for this epigenetic process. In so doing, we endogenously tagged Magohb (the most downregulated gene in the absence of MLL214,19) and performed a CRISPR/Cas9-mediated screen for regulators of Magohb expression in the presence or absence of MLL2. Based on this molecular screen, we found that loss of SET1A/B COMPASS leads to upregulation of ≈1,200 genes downregulated as the result of MLL2 loss. Our mechanistic studies demonstrated that in the absence of MLL2, these loci are repressed either by DNA methylation and/or H3K27me3. The effect of SET1A/B COMPASS loss in upregulation of these genes is due to its positive regulation of DNA methyltransferase gene transcription, which in turn can influence H3K27me3. Therefore, our study demonstrates the existence of a molecular interplay between the COMPASS family, H3K27me3 and DNA methylation for the proper developmental regulation of MLL2-dependent target genes.
Results
Genome-wide CRISPR screen to identify factors required for MLL2-dependent Magohb expression
Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) for MLL2 and H3K4me3 demonstrated that MLL2 binds to the Magohb promoter and that, H3K4me3 is strongly reduced at this locus upon knocking out MLL2 (MLL2 KO) (Fig. 1a), in accordance to previously reported data14,19. RNA-seq analysis confirmed that Magohb expression was lost in MLL2 knockout (KO) mESCs compared to wild type (WT) mESCs (Fig. 1b and Extended Data Fig. 1a). Since Magohb transcription was fully dependent on MLL2 binding to its promoter, we chose this gene to create a reporter to study MLL2-dependent transcriptional regulation.
Fig. 1: Generation of endogenously tagged mCherry-Magohb mESCs.

(a) ChIP-seq tracks of MLL2 and H3K4me3 occupancy at the Magohb locus in WT and MLL2 KO mESCs. The experiment was repeated two times independently with similar results. (b) RNA-seq tracks for Magohb in WT and MLL2 KO mESCs. Two biological replicates are displayed. n = 2 independent biological replicates. (c) Top: Strategy for integration of a 3×FLAG-mCherry tag at the N-terminus of Magohb. Primers (P1 to P4) used for genotyping are shown as arrows. Bottom: PCR genotyping showing WT and a homozygous mCherry-Magohb knockin (KI) clone. (d) Diagram of the CRISPR screen.
An endogenous reporter was generated in WT and MLL2 KO mESCs by inserting a 3×Flag-mCherry sequence at the N-terminus of Magohb via CRISPR/Cas9-mediated genome editing (Fig. 1c). To ensure our reporter was functional, we complemented MLL2 KO mESCs using a MLL2-expressing vector and could effectively rescue the mCherry-MAGOHB signal (Extended Data Fig. 2a). In contrast, knockdown of MLL2 in WT mESCs strongly decreased the mCherry-MAGOHB signal (Extended Data Fig. 2b), confirming that our fusion gene maintained its MLL2-dependent expression.
We subsequently performed a CRISPR screen using our generated reporter cell line by isolating populations of cells based on their mCherry fluorescence by FACS (Fig. 1d). Briefly, Cas9-expressing cells were selected (Extended Data Fig. 2c) and transduced with the Brie CRISPR-knockout library26 at a multiplicity of infection (MOI) rate of 0.3 and a library representation of at least 400 cells infected per single-guide RNA (sgRNA). After 7 days of puromycin selection, a fraction of the population was harvested to assess the presort sgRNA library coverage (“Total population”). The rest of the population was sorted (“Sorted population”) to isolate cells carrying a mCherry signal lower than the negative control for the WT cells or higher than the negative control for MLL2 KO cells (Extended Data Fig. 2d). Genomic DNA was extracted and sgRNA abundance was measured by deep sequencing. Each sgRNA with a log2FC > 1 in the “Sorted population” vs. the “Total population” was considered as an enriched sgRNA, and the total number of enriched sgRNA per gene was calculated. As expected, Magohb and Mll2 were identified as top hits in the cells sorted for lower MAGOHB expression in the WT mESCs (Extended Data Fig. 3a and Supplementary Table 1) demonstrating the specificity of our screen. Moreover, MAGOH and MAGOHB were previously identified as being paralog dependent, as only the simultaneous depletion of MAGOH and MAGOHB was shown to impair viability27,28. In accordance, sgRNAs targeting Magoh were depleted from our CRISPR screen performed in MLL2 KO mESCs that do not express MAGOHB, further validating our approach (Supplementary Table 2).
Knockdowns of SET1A/B COMPASS members rescue Magohb expression in MLL2 KO cells
To identify repressors of Magohb expression in the absence of MLL2, we examined the candidates from the screen performed in MLL2 KO mESCs and identified eight potential hits that met our stringent threshold (4 out of 4 sgRNA enriched) (Fig. 2a and Supplementary Table 2). To individually test each candidate, the strongest scoring sgRNA per gene was used to infect MLL2 KO Magohb-knockin cells and was then selected for 7 days, before assessing their effect on Magohb expression by Western blot and qRT-PCR (Fig. 2b–c and Extended Data Fig. 3b). Note that we only found one out of the eight hits as impacting on Magohb expression, suggesting that the seven remaining potential hits were either false positives or that the sgRNA used were ineffective to fully deplete its target.
Fig. 2: Knockdowns of SET1A/B COMPASS complex members rescue Magohb expression in MLL2 KO cells.
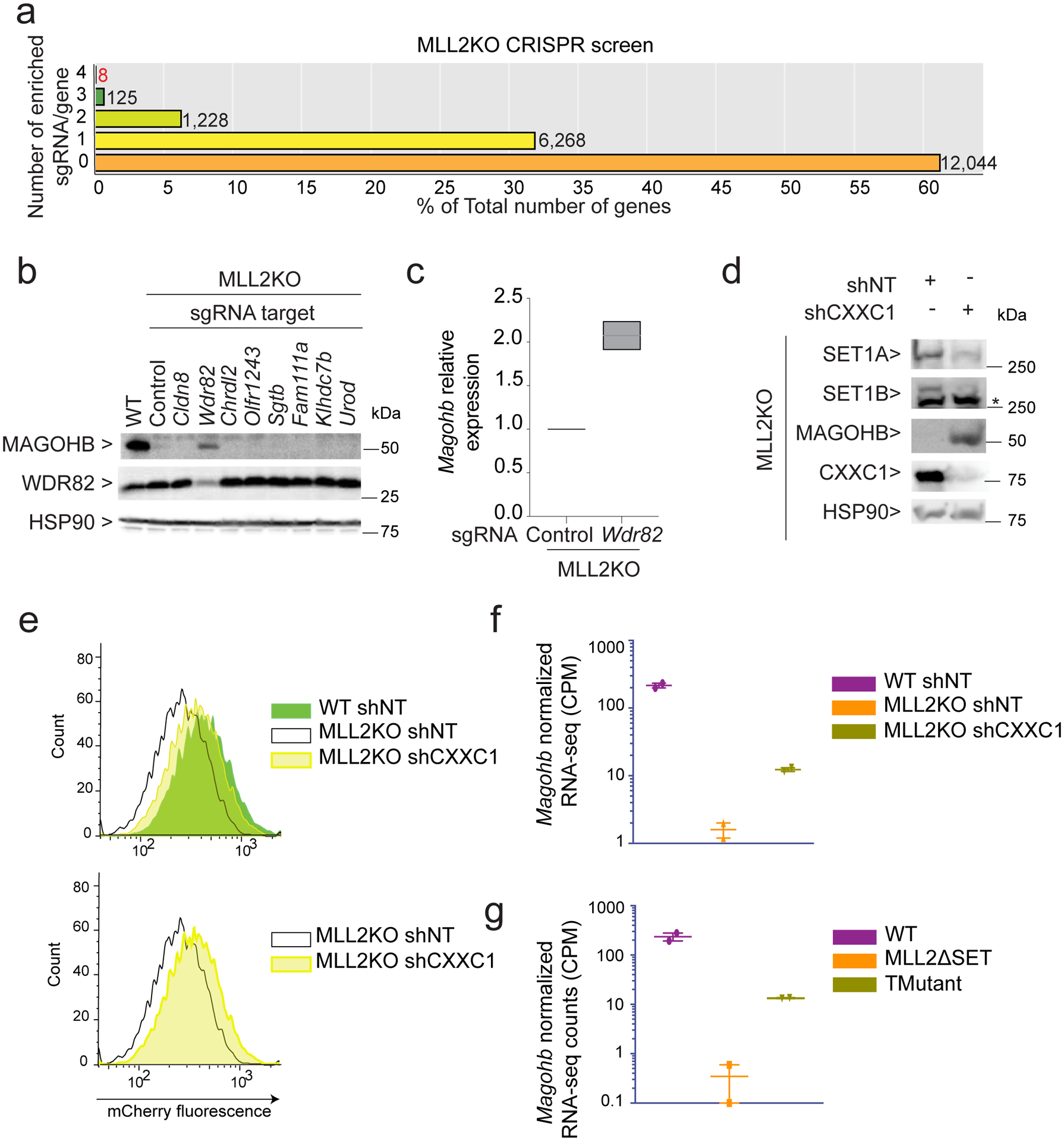
(a) CRISPR screen results showing the number of enriched sgRNAs in the ‘Sorted Population’ compared to the ‘Total population’ per gene. The majority of genes fall into the category ‘0 sgRNA enriched out of 4’. (b) Western blot of MAGOHB, WDR82 and HSP90 (loading control) protein levels upon sgRNA targeting each of the 8 candidates. The experiment was repeated two times independently with similar results. (c) MAGOHB expression level of as assessed by real-time qPCR in MLL2 KO mESCs expressing a control sgRNA (Control) or a sgRNA targeting Wdr82 (n = 2). (d) Western blot of SET1A, SET1B, FLAG-MAGOHB, CXXC1 and HSP90 (loading control) protein levels in MLL2 KO mESCs expressing a control shRNA (shNT) or a shRNA targeting CXXC1 (shCXXC1). The experiment was repeated two times independently with similar results. (e) Flow cytometry on mCherry-Magohb KI WT or MLL2 KO mESCs expressing a control shRNA (shNT) or a shRNA targeting CXXC1 (shCXXC1). The experiment was repeated two times independently with similar results. (f) Interleaved scatter plot of Magohb normalized RNA-seq counts in WT mESCs expressing a control shRNA (shNT), MLL2 KO mESCs expressing a control shRNA (shNT) or a shRNA targeting CXXC1 (shCXXC1) (n = 2). (g) Interleaved scatter plot of Magohb normalized RNA-seq counts in WT mESCs, MLL2ΔSET mESCs and TMutant mESCs (n = 2). Uncropped gels are available as source data.
Strikingly, Wdr82 knockdown led to a significant rescue of Magohb expression in MLL2 KO mESCs at the protein level (Fig. 2b) and the RNA level (Fig. 2c and Extended Data Fig. 3b). WDR82 is primarily found in two complexes: 1) SET1A/B COMPASS implicated in H3K4me3 deposition2,9,29 and 2) the PNUTS/PP1 complex implicated in transcription termination of active enhancers30. To pinpoint the specific function of WDR82 implicated in Magohb transcriptional regulation, we targeted individual members of the SET1A/B COMPASS or PNUTS/PP1 complex via CRISPR sgRNAs. We found that sgRNAs targeting Cxxc1 or Set1a and Set1b rescued Magohb expression in MLL2 KO cells (Extended Data Fig. 3c), suggesting that SET1A/B COMPASS members, rather than PNUTS/PP1 complex, are implicated in Magohb transcription inhibition in the absence of MLL2.
While WDR82 is reported to be a subunit of two major complexes, CXXC1 has mainly been described as a subunit of SET1A/B COMPASS31 and thus, to target SET1A/B COMPASS specifically in the following experiments, we decided to use a short hairpin (shRNA) strategy against Cxxc1 (Fig. 2d). Upon CXXC1 knockdown, both SET1A and SET1B protein levels were largely decreased, similar to previous findings after WDR82 knockdown30, while, in contrast, MAGOHB protein level was considerably increased. Consistently, by flow cytometry, the whole population of MLL2 KO cells exhibited a shift in mCherry-MAGOHB fluorescence signal upon CXXC1 knockdown (Fig. 2e) and Magohb expression rescue was recapitulated in our RNA-seq experiments (Fig. 2f). Finally, we validated the direct implication of SET1A/B in Magohb transcriptional inhibition by using mESCs knocked out for SET1B and deleted for the SET domain of SET1A and MLL2, referred as ‘TMutant’ hereafter, previously generated in our laboratory (Christie C. Sze, Patrick A. Ozark, Kaixiang Cao, Michal Ugarenko, Siddhartha Das et al.). TMutant mESCs exhibited a significant increase of Magohb transcription compared to MLL2ΔSET cells, akin to effects observed in CXXC1 knockdown (Fig. 2g). Taken together, our results identify an underlying balance within the COMPASS family, where SET1A/B are required for inhibition of Magohb transcription in the absence of MLL2 in mESCs.
MLL2 KO transcriptional defects are rescued by SET1A/B COMPASS knockdown
We next extended our analysis genome-wide to examine if Magohb’s transcriptional behavior was specific or shared by a whole set of genes. We focused on genes bound by MLL2 after partitioning MLL2 and H3K4me3 ChIP-seq data into clusters, with ‘cluster1’ containing loci bound by MLL2 and where H3K4me3 was strongly decreased in MLL2 KO compared to WT mESCs, representing 2,021 associated genes (Fig. 3a). Our analysis revealed that the majority of cluster1-associated genes exhibited a strongly reduced transcription in MLL2 KO compared to WT mESCs (Fig. 3b and Extended Data Fig. 4a). Strikingly, the reduction in expression of cluster1-associated genes was significantly rescued upon CXXC1 knockdown for more than 82% of cluster1-associated genes (Fig. 3b and Extended Data Fig. 4a–b). This indicates that the regulatory balance between MLL2 and SET1A/B complexes does not only apply to Magohb, but rather is shared among approximatively 1,200 MLL2-bound genes. Noticeably, while Magohb expression was only partially rescued, the expression of a majority of genes in cluster1 were fully rescued upon CXXC1 knockdown (Extended Data Fig. 4b–c and Extended Data Fig. 1b), indicating that Magohb does not fully exemplify the bulk of MLL2-dependent genes and potentially explains the low rate of confirmed hits from our CRISPR screen. Finally, our precision nuclear run-on sequencing (PRO-seq)32 analyses, which measures engaged RNA polymerase, indicated that the effect on gene expression could be imputed to active transcription since our results complemented our RNA-seq data (Fig. 3c). Note that the expression of cluster2-associated genes is globally stable in all tested conditions, indicating that effects of CXXC1 knockdown are specific to cluster1 and not a reflection of a global change in transcription.
Fig. 3: MLL2 KO transcriptional defects are rescued by SET1A/B knockdown.

(a) Left panel: MLL2 occupancy is shown in WT cells in read per million. Note that ChIP-seq signal found in MLL2 KO cells have been subtracted for analysis. Profile is centered on TSS (± 3 kb). Right panel: log2 fold-changes of H3K4me3 ChIP-seq signal in MLL2 KO mESCs compared to WT mESCs (n = 2). K-means clustering was used to partition all genes with Pol II signal in WT mESCs by their H3K4me3 log2 fold-changes upon MLL2 KO and by their MLL2 occupancy. (b) Heatmaps show the corresponding log2 fold-changes in gene expression in MLL2 KO shNT mESCs compared to WT shNT mESCs and MLL2 KO shCXXC1 mESCs compared to MLL2 KO shNT mESCs. (c) Boxplots quantifying PRO-seq signal around TSS (± 3 kb) are shown. The boxplots indicate the median (middle line), the third and first quartiles (box) and the first and fourth quartiles (error bars). P values were computed using Wilcoxon test (two-sided), cluster 1 n = 2,357, cluster 2 n = 10,605. The experiment was repeated two times independently with similar results. (d) Heatmaps show the corresponding log2 fold-changes in gene expression in TMutant shNT mESCs vs. MLL2ΔSET shNT mESCs, TMutant shNT mESCs vs. TMutant shCXXC1 mESCs, and MLL2 KO shCXXC1 mESCs vs. TMutant shNT mESCs. (e) GO terms shown for genes at least partially rescued upon CXXC1 knockdown in MLL2 KO mESCs compared to MLL2 KO mESCs. P values were computed using metascape that utilizes the hypergeometric test and Benjamini-Hochberg P value correction algorithm, n = 942.
We then analyzed the TMutant mESCs and observed that gene expression of cluster1 genes was significantly rescued compared to MLL2ΔSET mESCs (Fig. 3d and Extended Data Figs. 1c, 4c–d), in agreement with the CXXC1 knockdown effects discussed earlier, further indicating that SET1A/B COMPASS negatively impact the transcription of cluster1 genes in MLL2 KO mESCs. To further validate that the effect of CXXC1 knockdown was due to SET1A/B COMPASS activity loss, we performed epistasis experiments by knocking down CXXC1 in the TMutant cells. While TMutant cells with a shRNA control (shNT) rescued cluster1 expression, the knockdown of CXXC1 on top of the TMutant cells did not have a global additive effect (Fig. 3d and Extended Data Fig. 4d). This implicates that the rescue of MLL2-dependent transcription was specifically due to SET1A/B COMPASS function. Gene ontology analysis of cluster1 genes that were at least partially rescued by CXXC1 knockdown, showed enrichment for genes involved in ‘protein localization to cell periphery’ and ‘regulation of neuron projection development’ (Fig. 3e). To conclude, our studies demonstrate that in the absence of MLL2, SET1A/B COMPASS are necessary to maintain the transcriptional inhibition of ~1,200 MLL2-dependent genes.
Transcription in the absence of H3K4me3 in MLL2 KO CXXC1 knockdown cells
Previous studies demonstrated that CXXC1 is required for deposition of H3K4me3 at many promoters genome-wide10. Therefore, we decided to further analyze H3K4me3 changes in our conditions. Unlike cluster2, where H3K4me3 level was globally unchanged between WT and MLL2 KO cells, we observed a significant decrease of H3K4me3 levels in cluster1 gene promoters (Fig. 4a–b and Extended Data Fig. 1d–e). This pattern was expected since cluster1-associated genes are downregulated in MLL2 KO mESCs and reduction of transcription is generally correlated with a decrease of H3K4me3 levels around the transcription start site (TSS)33. However, H3K4me3 remained low in cluster1 upon CXXC1 knockdown, despite the strong rescue in gene expression (Fig. 4a–b–Fig. 3b–d). Thus, in MLL2 KO shCXXC1 cells, we observed an uncoupling of H3K4me3 and transcription. We conclude that increasing H3K4me3 is not required to increase transcription under these conditions, thereby challenging the existing notion of an instructional role of H3K4me3 for transcription, at least on these loci.
Fig. 4: H3K4me3 levels at MLL2-dependent genes are not rescued by CXXC1 knockdown.
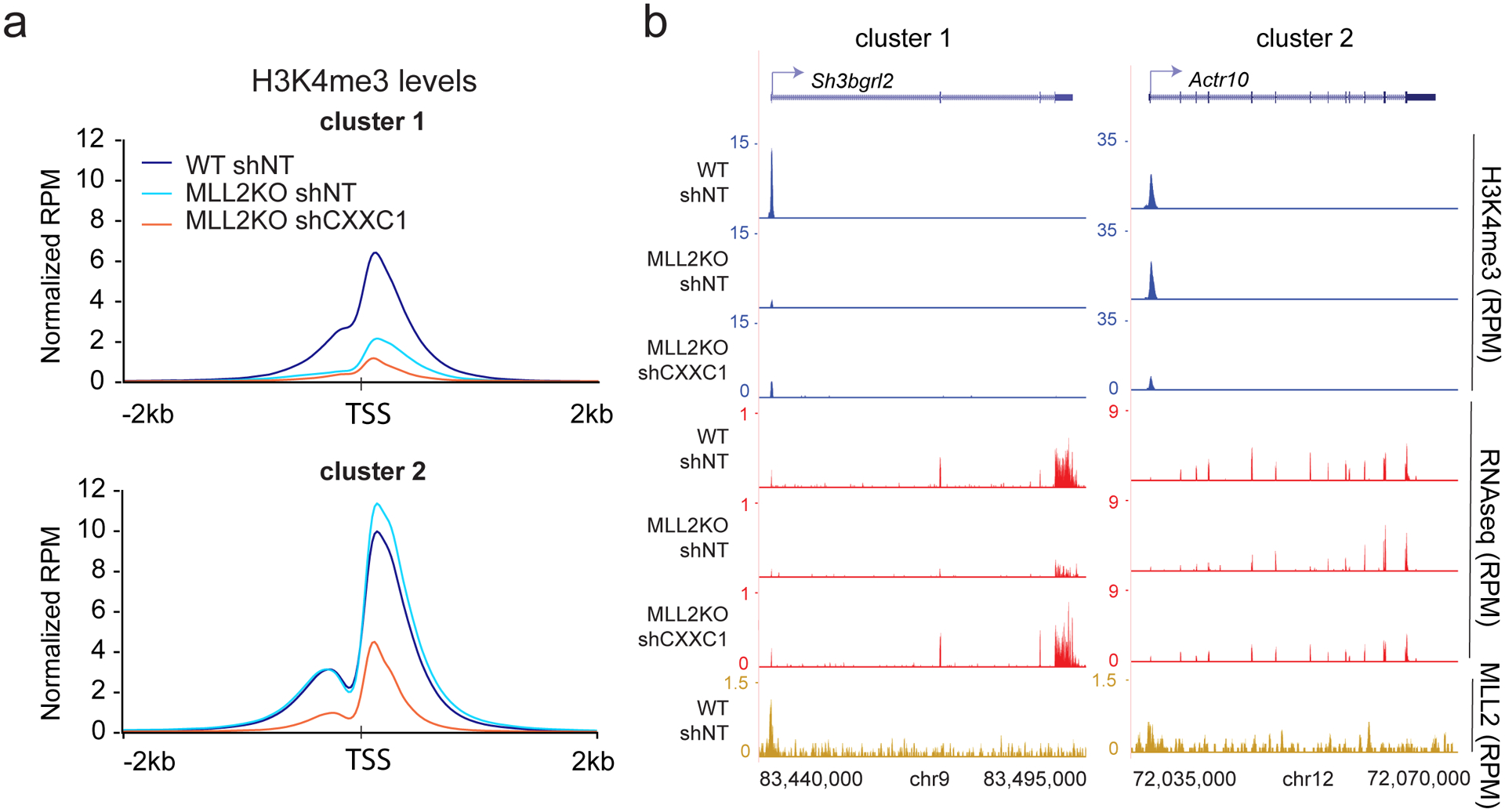
(a) Metaplots comparing H3K4me3 levels at clusters defined in Figure 3a. Plots are centered on TSS (± 2 kb). The experiment was repeated two times independently with similar results. (b) H3K4me3 ChIP-seq, RNA-seq and MLL2 ChIP-seq track examples are shown for each cluster in read per million.
MLL2/COMPASS functions to counteract DNA methylation inhibition of transcription
We next addressed the question of how CXXC1 knockdown rescues MLL2-dependent transcriptional loss in MLL2 KO mESCs. Since CXXC1 KO mESCs were previously shown to exhibit reduced DNA Methyltransferase 1 (DNMT1) expression34,35, we investigated the impact of CXXC1 knockdown on DNA methylation in the context of MLL2 KO mESCs. We found that Dnmt1 transcription was significantly reduced upon CXXC1 knockdown in MLL2 KO mESCs (Fig. 5a and Extended Data Fig. 1f). Interestingly, Dnmt1 expression was correlated with SET1A (subunit of COMPASS) binding at this locus (Fig. 5a): a strong SET1A peak was observed in WT and MLL2 KO mESCs, while this SET1A peak was highly reduced upon CXXC1 knockdown at the Dnmt1 promoter. Immunoblotting showed that reduced Dnmt1 transcription was reflected at the protein level (Extended Data Fig. 5a). Noticeably, Dnmt3a expression was decreased in MLL2 KO mESCs compared to WT mESCs, though its expression appears to be unrelated to SET1A expression (Extended Data Figs. 1g, 5a–b). Overall, these data indicate that SET1A is necessary for Dnmt1 transcription and suggest that this regulation could be through direct binding of SET1A/COMPASS to the Dnmt1 promoter.
Fig. 5: Inhibition of DNA methylation is sufficient to restore cluster1 gene expression in MLL2 KO mESCs.
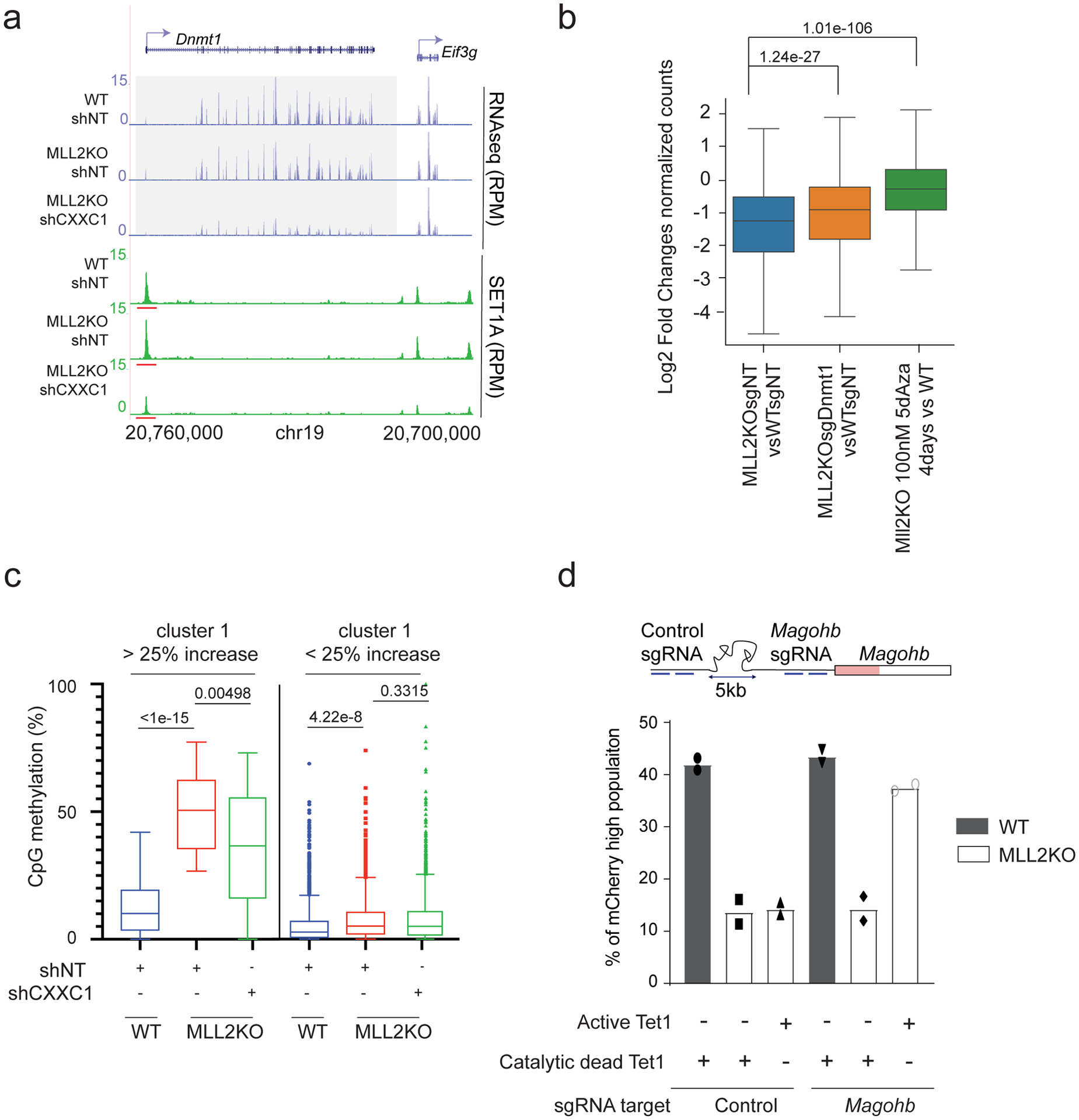
(a) RNA-seq and SET1A ChIP-seq tracks for Dnmt1. The experiment was repeated two times independently with similar results. (b) Boxplot analysis of cluster1 gene expression in 1) MLL2 KO vs. WT mESCs, 2) MLL2 KO expressing a sgRNA targeting Dnmt1 vs. WT mESCs expressing a control sgRNA and 3) MLL2 KO treated for 4 days with 100 nM 5dAza vs. WT mESCs. The boxplots indicate the median (middle line), the third and first quartiles (box) and the first and fourth quartiles (error bars). P values were computed using the Wilcoxon test (two-sided), n = 8,662. (c) Box-and-whisker plot quantifying changes in CpG methylation around TSS (± 1 kb) for cluster1 that was sub-clustered into genes with more or less than 25% increase in DNA methylation in MLL2 KO vs WT (n = 3 biological replicates). P values were calculated using a Brown-Forsythe and Welch ANOVA test assuming unequal variances with the Dunnett T3 correction for multiple comparisons (two-sided). (d) Percentage of mCherry-high cells were calculated by flow cytometric analyses 2 days post infection and shown as the mean percentages of mCherry-high cells of two biological replicates.
Next, to determine if the reduction in DNMT1 expression was sufficient to recapitulate CXXC1 knockdown, we directly targeted DNMT1 in MLL2 KO mESCs (Extended Data Fig. 5c). Our genome-wide analyses indicated that DNMT1 knockdown resulted in a partial rescue of cluster1 expression (Fig. 5b and Extended Data Fig. 5c). DNMT1 transfers methyl groups to cytosine nucleotides and its knockout has been shown to significantly reduce DNA methylation36. Thus, we then directly targeted DNA methylation using a widely used DNA-demethylating agent37, the cytosine nucleoside analog 5-Aza-2′-deoxycytidine (5dAza), and observed a significant rescue of cluster1 transcription in MLL2 KO mESCs (Fig. 5b and Extended Data Fig. 5c). Noticeably, 5dAza treatment led to a higher rescue than the knockdown of DNMT1 indicating either that the knockdown efficiency of DNMT1 was not sufficient to recapitulate CXXC1 knockdown or that other actors regulating DNA methylation were implicated.
To further investigate the relationship between MLL2-dependent transcription and DNA methylation, we performed modified Reduced Representation Bisulfite Sequencing (mRRBS) to map CpG methylation genome-wide38. Our analysis demonstrated that DNA methylation was slightly decreased for cluster2 genes in MLL2 KO compared to WT mESCs, presumably reflecting the reduced DNMT3A expression in these cells (Extended Data Fig. 5d). Interestingly, we found that cluster1 promoter regions exhibited two distinct patterns of DNA methylation changes, with one sub-cluster (41 genes including Magohb) exhibiting a strong increase of DNA methylation upon MLL2 KO (more than 25% increase of CpG methylation) that was rescued by CXXC1 knockdown (Fig. 5c). These results suggested that, for these genes, loss of DNA methylation was sufficient to restore MLL2-dependent transcription. To test this hypothesis, we next performed targeted DNA demethylation recruitment using a dCas9-Tet1 Catalytic Domain (CD) fusion39. In MLL2 KO mESCs, targeting Tet1CD to Magohb promoter led to a significant increase of MAGOHB expression, while targeting a dead Tet1CD to the promoter of Magohb or targeting a region 5 kb upstream of the Magohb promoter did not affect MAGOHB expression, as assessed by flow cytometry (Fig. 5d and Extended Data Fig. 5e). These results confirmed a direct repressive role of DNA methylation for approximatively 40 genes, including Magohb, in MLL2 KO cells that can be rescued by CXXC1 knockdown. Noticeably, the rest of cluster1 genes, representing the vast majority of the cluster, exhibited low levels of DNA methylation on their promoter regions and their DNA methylation levels remained globally constant among all three conditions, suggesting the existence of a third actor implicated in the repression of these MLL2-dependent genes in MLL2 KO cells.
DNA methylation and H3K27me3 deposition are counteracted by MLL2
We hypothesized that H3K27me3, deposited by Polycomb Repressive Complex 2 (PRC2) and classically linked to transcriptional silencing40, might be at the basis of this repression. Our H3K27me3 ChIP-seq analyses demonstrated that H3K27me3 signal was indeed increased around the TSS of MLL2-dependent genes upon MLL2 KO (Fig. 6a), while in contrast H3K27me3 signal was significantly rescued upon CXXC1 knockdown. In order to test if this effect was directly associated with gene expression changes, we next deleted the Suz12 gene (one of the four core proteins of PRC2) in MLL2 KO mESCs to generate double mutant MLL2 and SUZ12 KO cells that lose H3K27me3 (Fig. 6b). Our RNA-seq analyses revealed that SUZ12 KO was sufficient to significantly rescue the loss of gene expression in the MLL2 KO cells in all the tested MLL2 and SUZ12 KO clones (Fig. 6c–d and Extended Data Figs. 1h, 5a). When comparing the effect of CXXC1 knockdown, 5dAza treatment or SUZ12 KO on MLL2-dependent gene rescue in MLL2 KO cells, we observed a large overlap among the genes at least partially rescued (Fig. 6e).
Fig. 6: Interplay of H3K27me3, DNA methylation and MLL2 for gene transcription.
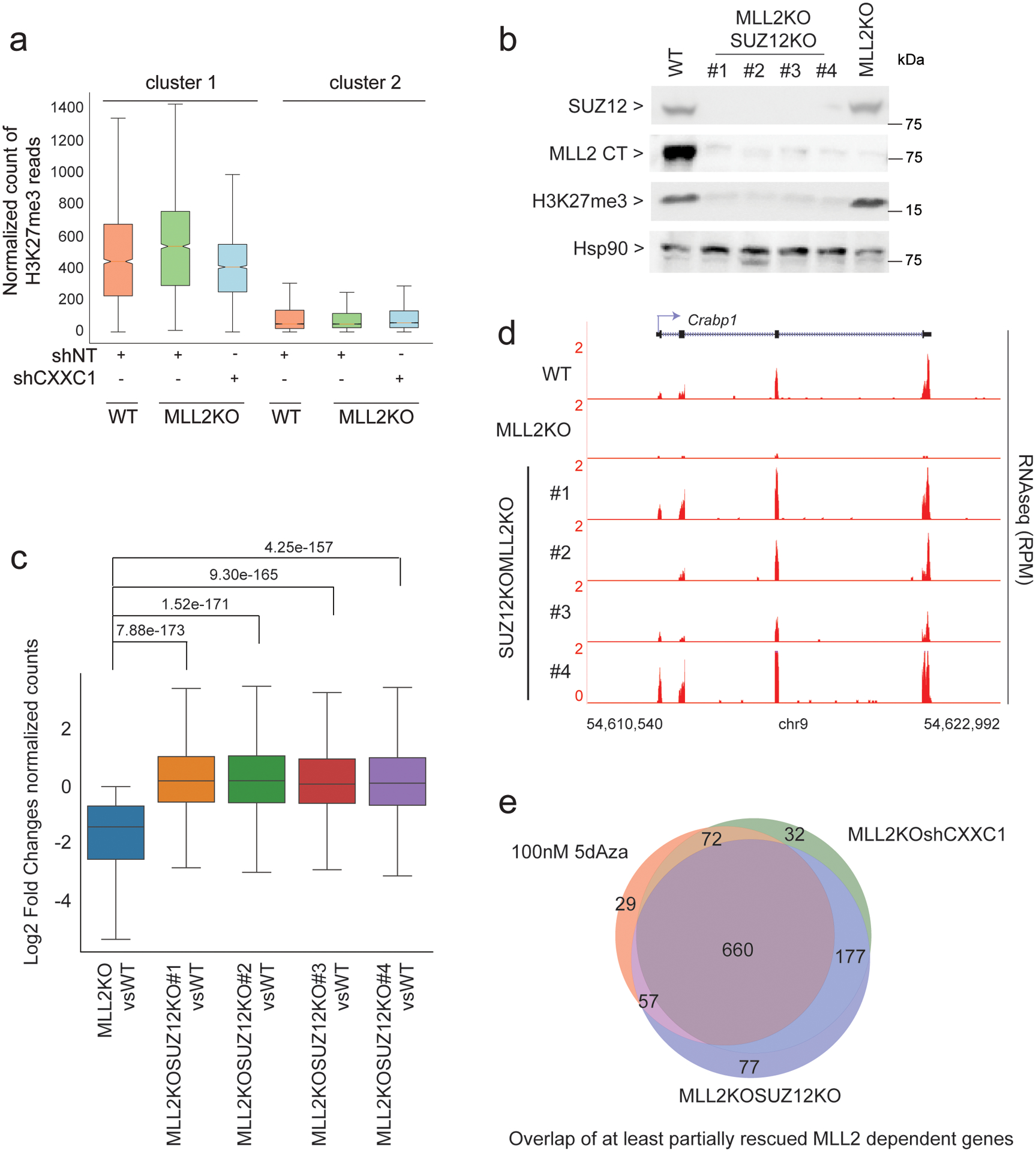
(a) Boxplots quantifying H3K27me3 ChIP-seq signal around TSS (± 3 kb) are shown (n = 2). (b) Western blot of SUZ12, MLL2, H3K27me3 and HSP90 (loading control) protein levels in WT, MLL2 and SUZ12 double KO (4 clones) and MLL2 KO. The experiment was repeated two times independently with similar results. (c) Boxplot analysis of cluster1 gene expression in 1) MLL2 KO vs. WT mESCs and 2–5) MLL2 and SUZ12 double KO vs. WT mESCs (clone #1 to #4). P values were computed using Wilcoxon test (two-sided), n = 1,311. The boxplots indicate the median (middle line), the third and first quartiles (box) and the first and fourth quartiles (error bars). (d) RNA-seq track examples are shown for each clone. The experiment was repeated two times independently with similar results. (e) Venn diagram of RNA-seq comparing MLL2/SUZ12 KO or MLL2 KO shCXXC1 or MLL2 KO 5dAza-treated vs MLL2 KO. Left panel shows the overlap of partially rescued genes (< wild type expression) and right panel shows the overlap of fully rescued genes (= wild type expression level). Uncropped gels are available as source data.
These data point toward two different mechanisms of regulation for MLL2-dependent genes: for 2% of MLL2-dependent genes, DNA methylation directly represses genes expression in the absence of MLL2. In contrast, for the vast majority of MLL2-dependent genes (98%), it is H3K27me3 that represses those genes in the absence of MLL2. Unexpectedly, this larger set of genes repressed by H3K27me3 in the absence of MLL2, can also be rescued by DNA methylation removal. We hypothesized that this overlap is due to the fact that DNA methylation impacts on H3K27me3 deposition, as previously reported41, and thus, explains why either removal of H3K27me3 or 5dAza treatment can rescue most of the MLL2-dependent gene expression in MLL2 KO mESCs.
An MLL2, H3K27me3 and DNA methylation balance regulates developmental gene expression
To test our model proposed above, we next assessed the effect of 5dAza treatment on H3K27me3 deposition (Fig. 7a–b and Extended Data Fig. 5a). First, we confirmed the efficiency of our 5dAza treatment on DNA methylation reduction by mRRBS experiments: 5dAza treatment led to a significant decrease of DNA methylation for both sub-clusters of MLL2-dependent genes (Fig. 7a). In fact, for the genes with more than 25% increase in DNA methylation (as defined in Fig. 5c) (2% of MLL2-dependent genes), we observed up to a 4.5-fold reduction of DNA methylation upon 5dAza treatment, confirming that DNA methylation directly regulates those genes’ expression. For the second sub cluster (98 % of MLL2-dependent genes), we observed no major changes in DNA methylation in all conditions tested. In fact, our H3K27me3 ChIP-seq analyses showed an increase of H3K27me3 levels in MLL2 KO mESCs compared to WT mESCs, while, in contrast, 5dAza treatment led to a significant decrease of H3K27me3 signal (Fig. 7b and Extended Data Fig. 5a). These results confirmed that altering DNA methylation affects H3K27me3 deposition. Taken together our results establish that for most MLL2-dependent genes, removal of H3K27me3 or 5dAza treatment can restore MLL2-dependent genes expression in the absence of MLL2 through direct loss of H3K27me3 or through indirect dilution of H3K27me3.
Fig. 7: Interplay of H3K27me3, DNA methylation and MLL2 and its impact on MLL2 KO differentiation defects.

(a) Box-and-whisker plot quantifying changes in CpG methylation around TSS (± 1 kb) for cluster1 that was sub-clustered into genes with more or less than 25% increase in DNA methylation in MLL2 KO vs WT (n = 3 biological replicates). P values were calculated using a Brown-Forsythe and Welch ANOVA test assuming unequal variances with the Dunnett T3 correction for multiple comparisons (two-sided). The boxplots indicate the median (middle line), the third and first quartiles (box) and the first and fourth quartiles (error bars). (b) Boxplots quantifying H3K27me3 ChIP-seq signal around TSS (± 3 kb) are shown (n = 2). (c) Heatmap of gene expression in WT, MLL2 KO, MLL2 KO 5dAza-treated and MLL2/SUZ12 double KO EBs at day 6. Differentially expressed genes were selected for genes down-regulation in MLL2 KO EBs compared to WT EBs and rescued upon 5dAza treatment as well as SUZ12 KO. (d) GSEA of genes deregulated between WT vs MLL2 KO EBs, MLL2 KO vs MLL2 KO 5dAza-treated and MLL2 KO vs MLL2/SUZ12 double KO at day 6 (normalized enrichment score (NES); false discovery rate-adjusted P value (FDR q)), n = 434. P values were computed using GSEA that utilizes an empirical phenotype-based permutation test procedure (two-sided).
Next, we decided to explore the conservation of our proposed molecular mechanism of MLL2-dependent gene regulation during differentiation. WT, MLL2 KO, MLL2 KO treated with 5dAza, and MLL2/SUZ12 double KO mESCs were differentiated into embryoid bodies (EBs) for 6 days and RNA was collected for sequencing. Our analysis demonstrated that up to 434 genes were 1) downregulated in MLL2 KO versus WT EBs and 2) rescued in both MLL2 KO treated with 5dAza and MLL2/SUZ12 double KO EB conditions (Fig. 7c). The fact that DNA methylation reduction or loss of H3K27me3 both led to this rescue, indicated that our presented molecular mechanism for MLL2-dependent transcriptional regulation, implicating COMPASS, H3K27me3 and DNA methylation, was at least partially conserved during differentiation. Moreover, 5dAza treatment led to a larger rescue than SUZ12 KO, in accordance to our model where 5dAza treatment affects DNA methylated sites as well as H3K27me3 deposition, while SUZ12 KO only affects H3K27me3 sites. Finally, our gene set enrichment analysis (GSEA) on this specific set of genes, comparing MLL2 KO to WT EBs, showed that genes associated with spermatogenesis were among the most downregulated genes (Fig. 7d), in accordance to previously reported data42. Interestingly, 5dAza treatment or SUZ12 KO were both sufficient to rescue that same gene set associated with spermatogenesis (Fig. 7d).
In summary, our results demonstrate the existence of a molecular interplay between DNA methylation, H3K27me3 and MLL2-dependent function in transcription. In the absence of MLL2, SET1A/B are required to inhibit the transcription of a cluster of ~1,200 genes normally bound by MLL2. This inhibition can be rescued by inhibiting DNA methylation or H3K27me3 deposition and unexpectedly, this transcriptional reactivation is not linked to the re-establishment of H3K4me3. These data suggest that the function of MLL2 at these loci is to overcome or repel transcriptional silencing exerted by H3K27me3 and/or DNA methylation and that in the absence of these inhibitory marks, transcription can proceed without H3K4me3. Finally, we demonstrate that prevention of active repression by MLL2 and not H3K4me3 underlies transcriptional regulation on MLL2 targets and that this molecular mechanism is at least partially conserved during differentiation.
Discussion
Our study identifies a regulatory circuit between the COMPASS family, DNA methylation machinery and the Polycomb-deposited H3K27me3, which becomes apparent in the absence of MLL2 (member of the COMPASS family). Our genome-scale screen indicated that depletion of SET1A/B in MLL2 KO mESCs was sufficient to rescue the loss of expression of ~1,200 MLL2-dependent genes. To our surprise, this rescue in gene expression as the result of SET1A/B loss in MLL2 KO cells did not require H3K4me3. These findings are in accordance with the growing body of evidence interrogating the existence of an instructional role for H3K4 methylation in active transcription10,25,33,43,44. Deletion of the only H3K4 methyltransferase in yeast, SET1, had no extensive transcriptional alteration43. Accordingly, deletion of CPS40 (SPP1) or CPS35 (SWD2), resulting in severe loss of H3K4me3, had no effects on coding gene transcription43. Similarly, in mammals, loss of H3K4me3 at active genes only had very limited effect on gene expression44 or transcriptional induction25,44.
Our study has uncovered that affecting global DNA methylation or altering DNA methylation of targeted regions were both sufficient to restore MLL2-dependent transcriptional loss in MLL2 KO mESCs. Similarly, several recent studies using CRISPR/Cas9-based systems to modulate DNA methylation have demonstrated that targeted DNA demethylation of methylated promoters is associated with gene re-expression45–47. The mechanism by which the removal of DNA methylation is sufficient to induce transcription remains an intriguing question and determining the factors that are recruited upon DNA demethylation for gene activation will be required to better understand this cis-regulatory mechanism.
To this end, we found that the loss of H3K27me3 somewhat phenocopies DNA methylation loss for MLL2-dependent gene expression in MLL2 KO mESCs, indicating that DNA methylation or H3K27me3 can inhibit MLL2-dependent gene transcription in the absence of MLL2. This is particularly interesting considering the fact that an intact DNA methylome was shown to be required for proper H3K27me3 deposition41: at the CpG islands that contain high level of H3K27me3, loss of DNA methylation (TKO (Dnmt[1kd,3a−/−,3b−/−]) mESCs) causes a concomitant decrease of H3K27me3 by dilution of this mark. According to this model, in the absence of DNA methylation, PRC2 activity can access newly unmethylated CpG sites and therefore H3K27me3 is redistributed in the genome, which results in de-repression of PRC2 targets. Our data show that the vast majority of genes that are rescued by inhibition of DNA methylation can also be rescued by PRC2 inhibition (Figs. 6e, 8). These results are in agreement with DNA methyltransferase inhibition inducing a reduction of H3K27me3 by dilution, as confirmed by our H3K27me3 ChIP-seq and bisulfite sequencing experiments (Fig. 7a–b). Therefore, inhibition of DNA methyltransferases can activate the transcription of MLL2-dependent genes by indirect reduction of H3K27me3 acquired upon MLL2 loss, thus explaining how perturbations of mutually exclusive epigenetics marks can end up affecting the same set of genes. Interestingly, loss of H3K27me3 or reduction of DNA methylation were both able to rescue the most downregulated gene set (“hallmark_spermatogenesis”) of MLL2 KO EBs at day 6 (Fig. 7d). Thus, indicating that the molecular mechanism uncovered in this study for MLL2-dependent transcriptional regulation in normal growth condition is at least partially conserved during differentiation. This is particularly interestingly in regard to the fact that MLL2 was recently shown to be mutated in early onset-dystonia20,21. Our finding that the activity of MLL2-dependent genes can be modulated by DNA methylation and H3K27me3 raises the possibility that MLL2 haploinsufficiency could be potentially rescued by inhibiting DNA methylation and/or PRC2 activity.
Fig. 8: Model for an epigenetic equilibrium between Polycomb, COMPASS and DNA methylation machineries at most MLL2-dependent genes.

In WT mESCs (top panel), MLL2 COMPASS binds to the promoters of MLL2-dependent genes and deposits H3K4me3 on these promoter regions. In the absence of MLL2 (2nd panel, MLL2 KO), H3K27me3 is deposited on the regions that are now free of MLL2, while DNA methylation remains for most of the genes at the shore of H3K27me3 marks. When DNA methylation is lost (3rd panel), either by targeting CXXC1 that affects Dnmt1 expression or by 5dAza treatment (MLL2 KO shCXXC1 or MLL2 KO 5dAza), H3K27me3 marks are spread to these newly CpG hypomethylated DNA regions, and consequently, the H3K27me3 signal previously observed on the promoter regions of MLL2-dependent genes is diluted and becomes insufficient to repress these genes’ expression. Similarly, after knockout of SUZ12 in MLL2 KO mESCs (bottom panel, MLL2 and SUZ12 double KO), MLL2-dependent genes are transcribed in the absence of MLL2 due to the absence of the repressive H3K27me3 mark.
Finally, Magohb encodes a core member of the exon-junction complex that is redundant with Magoh gene function28. Interestingly, a recent paper determined that hemizygous loss of MAGOH is observed in a large number of tumors, establishing MAGOHB as a promising therapeutic target in this context27. This unexpected dependency of cancer cells to paralog genes during tumor growth emphasize the importance of performing genome-wide CRISPR screens to gain molecular insights into context-dependent transcription regulation.
Methods
mESCs culture, knockin, shRNA knockdown, complementation, knockout and EB formation.
V6.5 mESCs were grown in N2B27 medium supplemented with two inhibitors and LIF, as described previously48. MLL2 KO cells were generated in the laboratory as previously described14. TMutant cells will be described in another manuscript (Christie C. Sze, Patrick A. Ozark, Kaixiang Cao, Michal Ugarenko, Siddhartha Das et al.). For 3×FLAG-mCherry knockin, oligonucleotides encoding the desired sgRNA sequence (5’-CGGTATAGCGGGCTTTACCG-3’) were annealed and cloned into the pX459 plasmid according to a published protocol49. Donor plasmid containing homology arms flanking Magohb promoter and a 3×FLAG-mCherry sequence where cloned in the pUC57 vector. mESCs were transfected with pX459 containing the desired sgRNA-coding sequences and donor plasmid using Nucleofector 2b (Lonza) according to the manufacturer’s instructions. Transfected mESCs were selected with 2 μg/ml puromycin (Life Technologies) for 48 h and grown for 10 days until cell clones were pickable. Cell clones were screened by PCR and the genotypes were confirmed by sequencing. For MLL2/SUZ12 double KO, mESCs were transfected with pX459 containing the desired sgRNA-coding sequences using Nucleofector 2b (Lonza) according to the manufacturer’s instructions. Transfected mESCs were selected with 2 μg/ml puromycin (Life Technologies) for 48 h and grown for 10 days until cell clones were pickable. Cell clones were screened by PCR and the genotypes were confirmed by sequencing. Guide RNA sequences used are listed in Supplementary Table 3.
shRNA knockdown was performed by transducing mESCs with lentiviruses expressing shRNA for 24 h before selection with 2 μg/ml puromycin for 3 days. The lentiviral construct containing shRNA against CXXC1 (TRCN0000241395) was purchased from Sigma.
MLL2 re-expression in MLL2 KO cells was performed by nucleo-transfecting 2 × 107 mESCs with 40 μg MLL2 plasmid (Promega, FHC12727). Flow cytometry experiments were performed 5 days after transfection.
Fuw-dCas9-dead Tet1CD-P2A-BFP and Fuw-dCas9-Tet1CD-P2A-BFP were a gift from Rudolf Jaenisch (Addgene plasmid #108246 and Addgene plasmid #108245). mESCs were transduced with sgRNA expressing vector (2 sgRNAs targeting a control region or 2 sgRNAs targeting Magohb promoter, see Supplementary Table1 for sequences) and selected for 2 days with puromycin. mESCs were then transfected with Fuw-dCas9-dead Tet1CD-P2A-BFP or FuwdCas9-Tet1CD-P2A-BFP and flow cytometric measurements were performed 2 days later.
EBs were generated as described previously22 using the hanging drop method. Briefly, 1,500 ESCs were cultured in 25 μl of EB differentiation medium on the lid of 150-mm culture plates for 6 days. EB differentiation medium was composed of DMEM supplemented with 15% FBS (Gemini Bio-Products), 1× GlutaMAX (Gibco), 1× MEM nonessential amino acids (Gibco), 1× β-mercaptoethanol (Gibco), and 1× penicillin–streptomycin (Gibco).
RT-qPCR.
RNA was extracted from approximatively 1 × 106 cells using RNeasy Kit (Qiagen), followed by DNase digestion using RNase-Free DNase (Qiagen). 1 μg of total RNA was reverse transcribed using Superscript II RT according to the manufacturer protocol (Invitrogen) and quantitative PCR was performed using SYBR Green dye (Roche Scientific) and a CFX96 instrument (BioRad). For data analysis, normalized gene expression values were calculated using the comparative CT Method (ΔΔCT Method). Ct values of target gene (Magohb) were normalized to Ct values of 16S rRNA and to a control sample (Control) to obtain ΔΔCt values. Fold-differences in gene expression compared to controls (calculated as 2−ΔΔCt) are represented in Figure 2c.
Chromatin immunoprecipitation and RNA extraction.
ESCs were fixed with 1% formaldehyde for 10 min. After quenching and cell lysis, chromatin was fragmented to 200–600 bp using a Covaris E220 bath sonicator (Covaris). Chromatin was incubated with antibodies overnight at 4 °C, immunoprecipitated the following day by incubating with Protein A/G agarose beads (Santa Cruz Biotechnology), washed six times in RIPA buffer (25 mM Tris pH 7.5, 140 mM NaCl, 1% Triton X-100, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 0.5 mM DTT), and eluted (0.1 M NaHCO3, 1% SDS). After cross-link reversal and proteinase K digestion overnight at 65 °C, DNA was purified using Qiagen PCR purification spin columns. ChIP-seq libraries were generated with KAPA HTP library preparation kit (KAPA Biosystems) following the manufacturer’s instruction and loaded onto NextSeq 500 sequencer or NovaSeq (Illumina) for sequencing. At least two biological replicates were performed for ChIP-seq of H3K4me3, H3K27me3, MLL2 and SET1A under each experimental condition. RNA was isolated using RNeasy mini kit (Qiagen) following the manufacturer’s instructions, and RNA-seq libraries were made using the TruSeq Stranded Total RNA with Ribo-Zero kit (Illumina).
Precision nuclear run-on sequencing.
We performed PRO-seq following a previously described protocol50 with some modifications. Nuclei were isolated by Dounce homogenizer with loose pestle. Approximately 107 nuclei were subjected to nuclear run-on (30 °C, 3 min) in the presence of 25 μM biotin-11-ATP/GTP/CTP/UTP (PerkinElmer) and 6 × 105 Drosophila S2 spike-in nuclei. Total RNAs were extracted and hydrolyzed in 0.2 M NaOH (on ice, 10 min). Biotinylated nascent RNA was purified by Dynabeads M-280 streptavidin (Invitrogen). After 3′ VRA3 adapter ligation and the second purification by Dynabeads, 5′ cap and 5′ hydroxyl RNA were converted to 5′ monophosphorylated RNA by RppH (NEB) and PNK (NEB), respectively. After 5′ VRA5 adapter ligation and the third purification by Dynabeads, complementary DNA was generated by reverse transcription with SuperScript III (Invitrogen) and RP1 primer. PCR by Phusion Hot Start (Thermo) and RP1/RPIx primer sets in 10 cycles amplified indexed DNA libraries. DNA libraries of 140–350 bp were size-selected by Pippin HT with 2% gel cassette 20B (Sage Science). Two independent cell cultures per condition were then sequenced by NextSeq 500 system (Illumina) with single-read runs.
Modified reduced representation bisulfite sequencing.
mRRBS was performed as previously reported38,51–53. Briefly, genomic DNA was digested with MspI (New England BioLabs) before size selection of 100- to 250-bp fragments with solid-phase reversible immobilization beads (MagBio Genomics). DNA was bisulfite converted with the EZ DNA Methylation-Lightning Kit (Zymo Research), resulting in a conversion efficiency of 99.6% ± 0.05% SD based on the observed CpG methylation frequency of an unmethylated λ-bacteriophage spike-in control genome (New England BioLabs, N3013S). Libraries were prepared with the Pico Methyl-Seq Library Prep Kit (Zymo Research) using Illumina TruSeq indices and sequenced using single-end reads (NextSeq 500, Illumina) with a 500/550 V2 High Output reagent kit (1 × 75 cycles). Bioinformatic processing and alignment of the sequenced libraries to the mm9 reference genome were performed as previously reported38,51–53. The code used for mRRBS data processing is reported elsewhere54. CpG methylation was quantified using the SeqMonk platform (v1.40.1) with the bisulfite feature methylation pipeline. Between-group comparisons of individual CpG values were performed using the Brown-Forsythe and Welch ANOVA method and the Games-Howell multiple comparisons test with individual variances computed for each comparison.
Antibodies.
The following antibodies were generated in-house: H3K4me3 rabbit #519 (1:2,000), H3K27me3 rabbit #67 (1:2,000), MLL2Nterm rabbit #9170 (1:1,000), MLL2Cterm rabbit #9173 (1:1,000), WDR82 rabbit affinity purified) (1:1,000), SET1A rabbit #919 (1:1,000) and SET1B rabbit #386 (1:2,000). Antibody to CXXC1 was purchased from Abcam (ab56035), antibodies to DNMT1 and CAS9 were purchased from CST (5032S, 14697), antibody to DNMT3A was purchased from Abcam (ab2850), antibody to FLAG was purchased from Sigma (F3165) and antibodies to SUZ12, HSP90 and β-TUBULIN were purchased from Santa Cruz (#sc-9104, #sc-13119 and #sc-46264).
Genome-wide CRISPR–Cas9 screen.
Brie library amplification, lentiviral production, MOI determination and transduction were performed as described previously55. Briefly, ~300 million MLL2 KO cells stably expressing Cas9 were infected with the genome wide sgRNA Brie library26 at a MOI < 0.3. Infected cells underwent puromycin selection (2 μg/ml) for 7 days. 4–6 million cells were pelleted and frozen (‘Total population’) and ~10 million cells were sorted for cells with an increased mCherry fluorescence compared to control MLL2 KO cells (‘Sorted cells’). To achieve a better selection and reduce the number of false positive cells, cells were sorted twice.
Genomic DNA was extracted from ‘Total population’ and ‘Sorted cells’ and the sgRNA library was amplified by PCR as previously described25. Deep sequencing on an Illumina Nextseq platform was used to monitor library composition. Guide composition between ‘Total population’ or ‘Sorted cells’ was compared: briefly, the enrichment of individual guides was calculated as the log2 ratios ‘Sorted cells’ and ‘Total population’, with each sgRNA with a log2FC > 1 considered as enriched. Each gene was then classified in one of the 5 categories: 0 sgRNA enriched out of 4 sgRNAs, 1 sgRNA enriched out of 4 sgRNAs, 2 sgRNAs enriched out of 4 sgRNAs, 2 sgRNAs enriched out of 4 sgRNAs or 4 sgRNAs enriched out of 4 sgRNAs.
Knockout of Candidate Genes in Pooled ESCs.
For the 8 top candidate genes (4 out of 4 sgRNA enriched), individual targeted sgRNA were cloned into lentiCRISPRv2 plasmid (Addgene #52961). These constructs were used to produce lentivirus and transduce MLL2 KO cells, followed by puromycin selection for 7 days and western blot analyses. Guide RNA sequences used are listed in Supplementary Table 3.
Sequencing data processing.
RNA-seq and ChIP–seq samples were sequenced with Illumina technology, and output data were processed with the bcl2fastq software tool. Sequence quality was assessed using FastQC v0.11.2 (Andrew 2010), and quality trimming was performed using the FASTX toolkit. RNA-seq and ChIP–seq reads were aligned to the mm9 genome using TopHat v2.0.956 and Bowtie v0.12.957, respectively, and only uniquely mapped reads meeting a two-mismatch threshold were considered for downstream analysis. Gene annotations from Ensembl 67 were used. Output BAM files were converted into BigWig track files to display coverage throughout the genome (in RPM) using the GenomicRanges package. For Fig. 7D, the total number of ChIP-seq reads was normalized to the number of Drosophila spike-in reads.
ChIP–seq analysis and RNA-seq analysis.
Peaks were called with MACS v1.4.258 software using default parameters. Meta-plots and heat maps were generated using deeptools59. k-means clustering was also performed using deeptools, and nearest-gene log fold changes in gene expression corresponding to the clustered peaks in the heat maps were determined using in-house scripts (Gene count tables were used as input for edgeR 3.0.860 to analyze RNA-seq data, the in-house script can be found here: https://github.com/ebartom/NGSbartom) and visualized with Java TreeView61.
PRO-seq analysis.
PRO-seq raw data were trimmed by cutadapt 1.1462 and Trimmomatic 0.3363. Each sample was next mapped to the mouse and fruit fly genome assemblies (mm9 and dm3, respectively) using Bowtie. The number of fly reads on each experiment was used to normalize the corresponding genome-wide strand-specific profiles. Boxplots quantifying PRO-seq level were generated by quantifying the PRO-seq level around TSS, considering the direction of transcription (± 3 kb) and normalized using the total number of reads per sample.
Statistical analysis.
For statistical analyses, R, Python and GraphPad were used. Appropriate statistical tests were used for all data where a statistical analysis was reported. All experiments were conducted in unblinded conditions and statistical tests used were reported in the figure legends.
GO term.
P values for Gene Ontology were determined with the hypergeometric test using the software Metascape64. GSEA of the pre-ranked lists of genes by EdgeR stat value was performed with the GSEA software65. Mouse genes were ranked by the ratio between WT and MLL2 KO RNA-seq expression on EBs at day 6, MLL2 KO and MLL2 KO 5dAza RNA-seq expression on EBs at day 6 and MLL2 KO and MLL2/SUZ12 double KO RNA-seq expression on EBs at day 6.
Reporting Summary.
Further information on research design is available in the Life Science Reporting Summary linked to this paper.
Data availability.
RNA-seq, ChIP–seq, bisulfite sequencing raw data are available in the Gene Expression Omnibus (GEO) database under accession GSE129037. Additional data supporting the findings of this study are available from the corresponding author upon request. Source data (full scans of the immunoblots) for Figures 2 and 6 and Extended Data Figures 2, 3 and 5 are provided with the paper.
Code availability
We have made use of publicly available software and tools (as referenced in the Methods section, the in-house script used for ChIP-seq and RNA-seq analyses can be found here: https://github.com/ebartom/NGSbartom.
Extended Data
Extended Data Fig. 1. Quantification of RNAseq changes.

(a-h) Interleaved scatter plot of indicated genes’ normalized RNA-seq counts in the indicated conditions for the genes Magohb (a), Zdhhc2 (b), Zdhhc2(c), Sh3bgrl2 (d), Actr10 (e), Dnmt1 (f), Dnmt3a (g) and Crabp1 (h) (n=2).
Extended Data Fig. 2. CRISPR-Cas9 screen using MAGOHB knock in in mESCs.
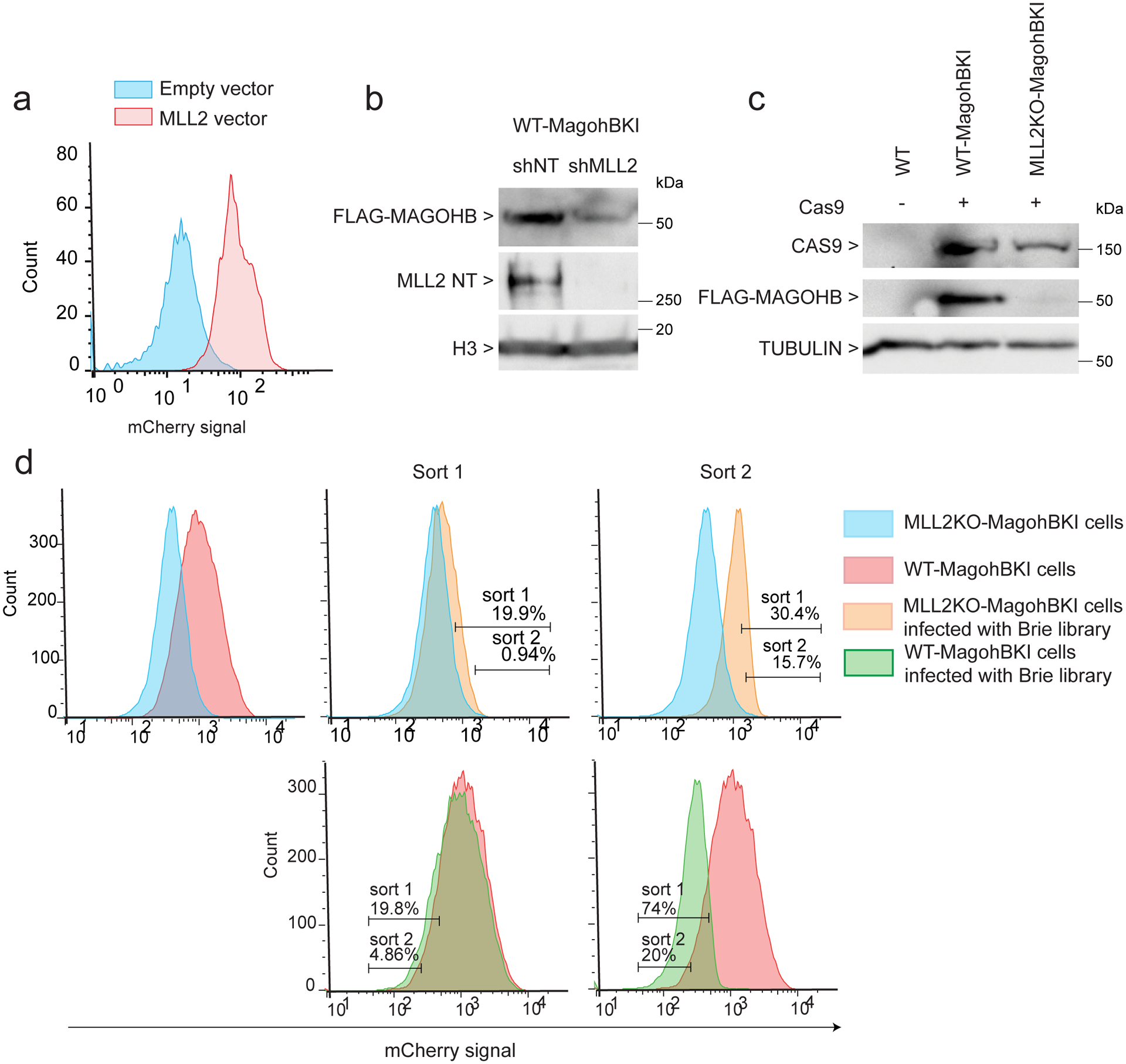
(a) FACS analysis of mCherry-Magohb MLL2KO mESCs transformed with an empty vector or a MLL2 expressing vector. The experiment was repeated three times independently with similar results. (b) Western blot for FLAG-MAGOHB expression in WT cells expressing a shRNA control (shNT) or a shRNA targeting MLL2 (shMLL2). The experiment was repeated two times independently with similar results. (c) Western blot for CAS9, FLAG-MAGOHB and TUBULIN (loading control) protein levels in WT, Magohb KI WT cells (WT-MagohbKI) and Magohb KI MLL2KO cells (MLL2KO-MagohbKI). The experiment was repeated two times independently with similar results. (d) Top panel: mCherry high cells were isolated via two successive rounds of FACS in MLL2KO mESCs. Bottom panel: mCherry low cells were isolated via two successive rounds of FACS in WT mESCs. The experiment was repeated three times independently with similar results. Uncropped gels are available as source data.
Extended Data Fig. 3. CRISPR screen validation.
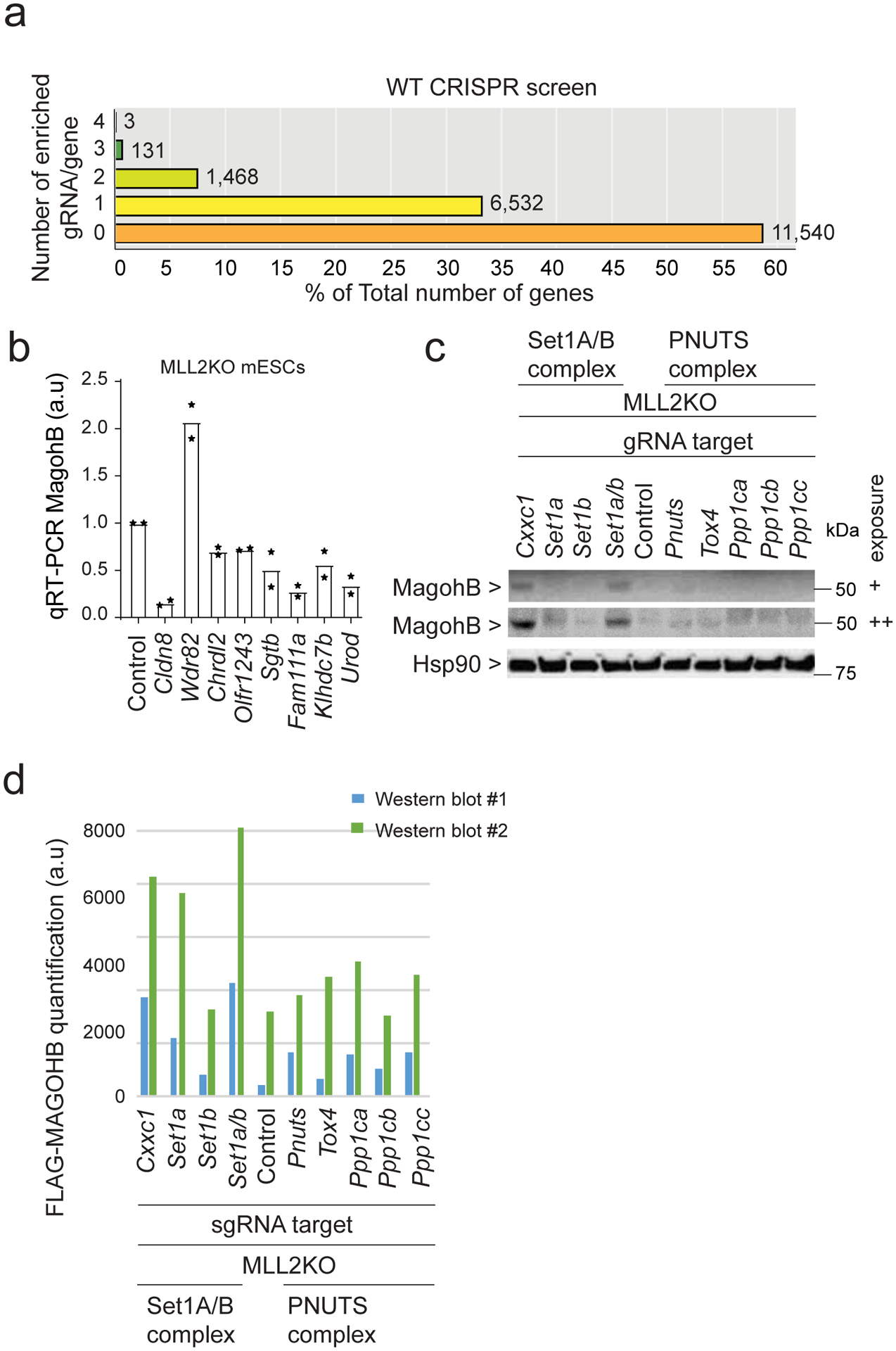
(a) CRISPR screen results representing the number of enriched sgRNA in the ‘Sorted Population’ compared to the ‘Total population’ per gene for WT screen. The majority of the genes fall into the category ‘0 sgRNA enriched out of 4’. (b) Expression levels of Magohb as assessed by real-time PCR in MLL2KO mESCs expressing a control sgRNA (Control) or sgRNAs targeting various genes (n = 2). (c) Western blot of MAGOHB and HSP90 (loading control) protein levels. Two different exposures are shown. The experiment was repeated two times independently with similar results. (d) Quantification of two independent replicates of Extended Data Figure 2c western blots. Uncropped gels are available as source data.
Extended Data Fig. 4. Cluster-1 gene expression rescue by CXXC1 knockdown.
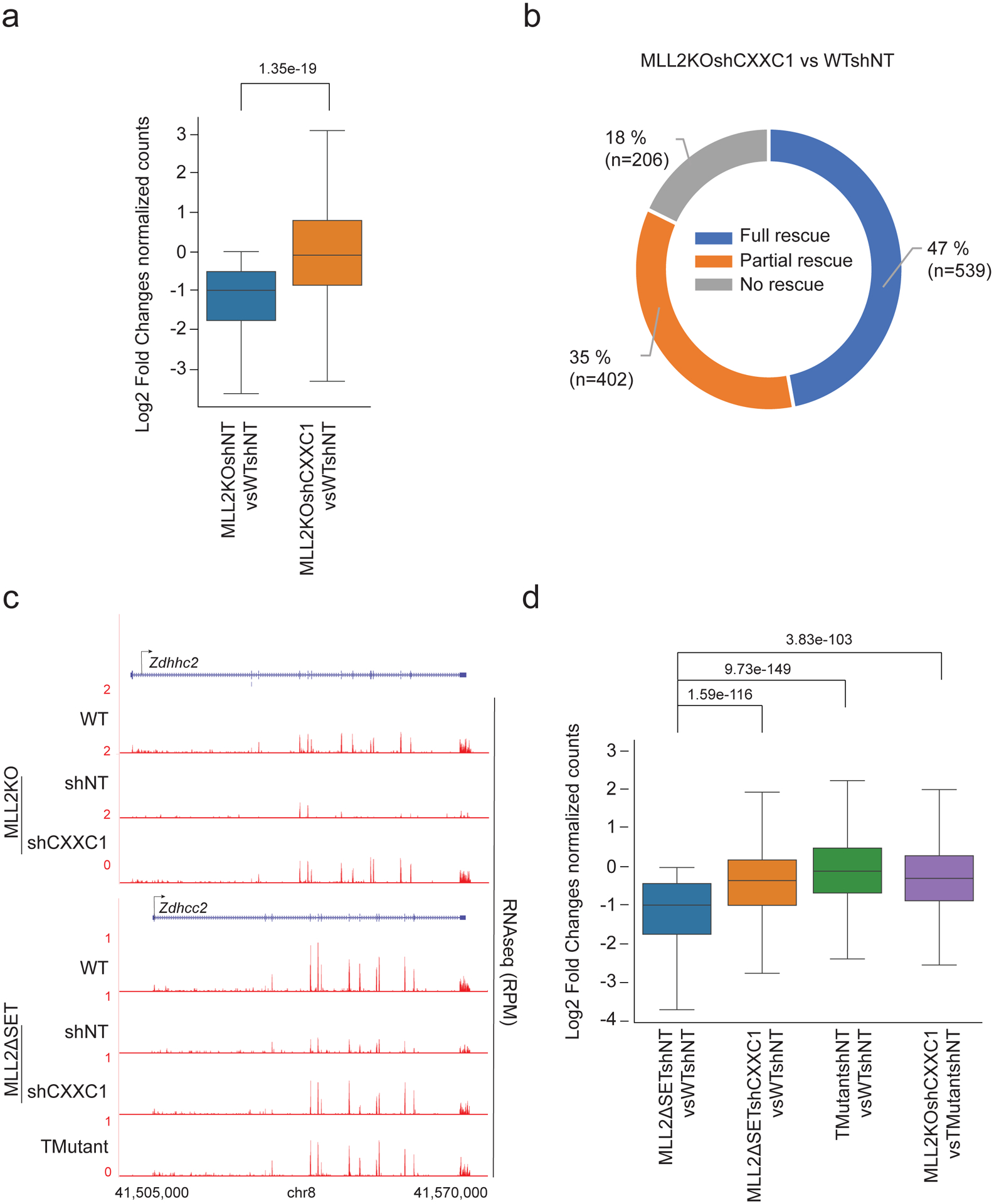
(a) Boxplot analysis of cluster1 gene expression in 1) MLL2KOshNT vs. WTshNT mESCs and 2) MLL2KOshCXXC1 vs. WTshNT mESCs. P values were computed using Wilcoxon test (two-sided), n=941. The boxplots indicate the median (middle line), the third and first quartiles (box) and the first and fourth quartiles (error bars). (b) Donut chart of cluster 1 distribution of genes based on their level of rescue in MLL2KO shCXXC1 cells compared to WT cells. ‘Full rescue’ genes were characterized by a log2FC ≥ 0 comparing MLL2KOshCXXC1 to WT mESCs, ‘Partial rescue’ genes were characterized by a log2FC > 0 comparing MLL2KOshCXXC1 and MLL2KO mESCs and ‘No rescue’ were characterized by a log2FC ≤ 0 comparing MLL2KOshCXXC1 and MLL2KO mESCs. (c) RNA-seq tracks for Zdhhc2 in WT, MLL2KOshNT, MLL2KOshCXXC1, MLL2ΔSETshNT, MLL2ΔSETshCXXC1 and TMutant. The experiment was repeated two times independently with similar results. (d) Boxplot analysis of cluster1 gene expression in 1) MLL2ΔSETshNT vs. WTshNT mESCs and 2) MLL2ΔSETshCXXC1 vs. WTshNT mESCs, 3) TMutantshNT vs. WTshNT mESCs, 4) TMutantshCXXC1 vs. WTshNT mESCs and 5) MLL2KOshCXXC1 vsTMutantshNT mESCs (n=2).
Extended Data Fig. 5. DNA methylation and MLL2 dependent transcription.
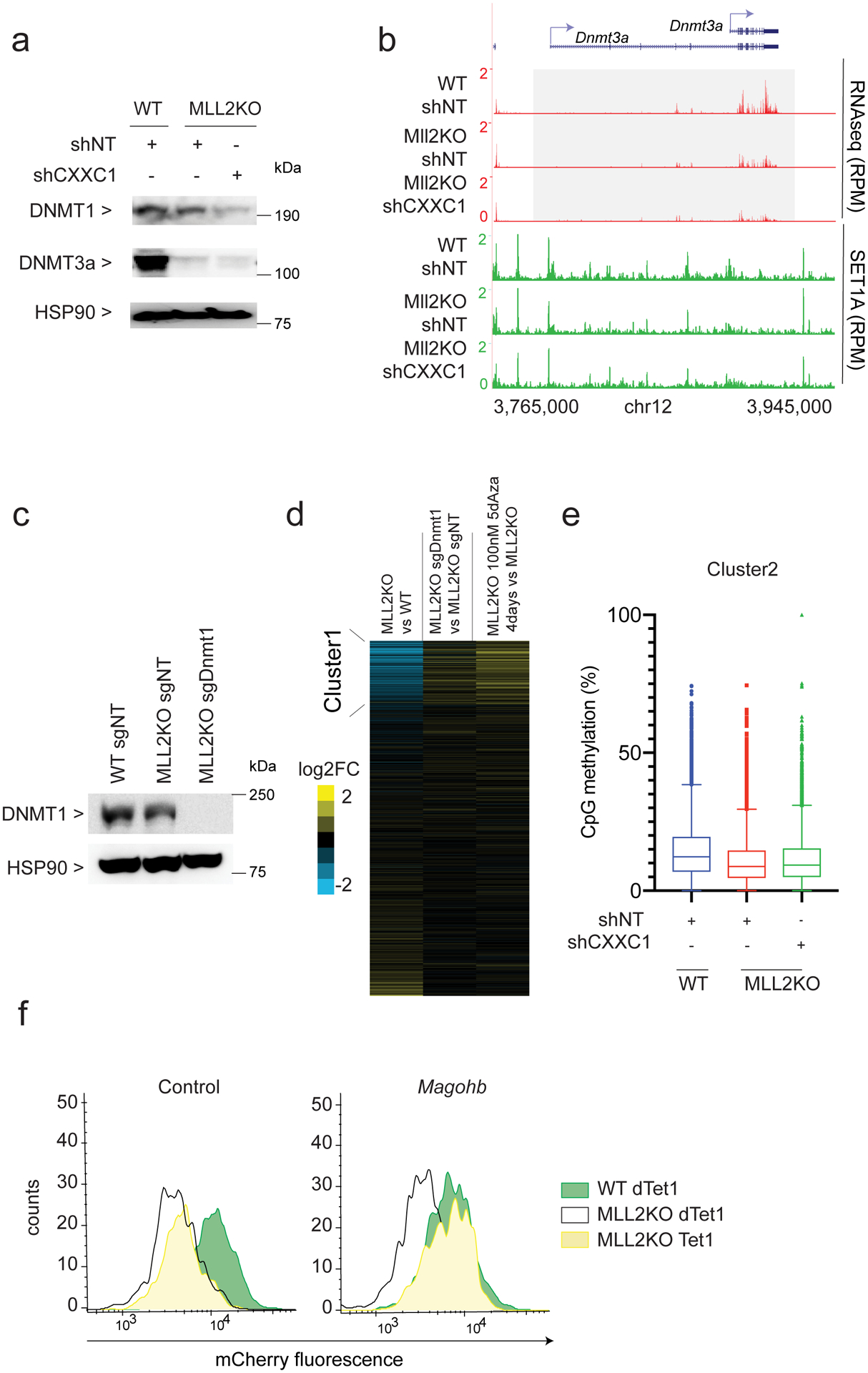
(a) Western blot of DNMT1, DNMT3A and HSP90 (loading control) protein levels in WTshNT, MLL2KOshNT and MLL2KOshCXXC1 mESCs. The experiment was repeated two times independently with similar results. (b) RNA-seq and SET1A ChIP-seq tracks for Dnmt3a. The experiment was repeated two times independently with similar results. (c) Western blot of DNMT1 and HSP90 (loading control) protein levels in WTsgNT, MLL2KOsgNT and MLL2KOsgDnmt1. The experiment was repeated two times independently with similar results. (d) Heatmaps show the corresponding log2 fold changes in gene expression in 1) MLL2KO vs. WT mESCs, 2) MLL2KO sgDnmt1 vs. MLL2KOsgNT mESCs and 3) MLL2KO treated for 4 days with 100 nM 5dAza vs. MLL2KO mESCs. (e) Box-and-Whisker plot quantifying changes in CpG methylation around TSS (±3kb) of cluster2 clusters identified in Figure 3A (N=3 biological replicates, n=13420). The boxplots indicate the median (middle line), the third and first quartiles (box) and the first and fourth quartiles (error bars). (f) Flow cytometry on mCherry-Magohb KI WT or MLL2KO mESCs expressing a dead Tet1 catalytic domain (dTet1) or an active Tet1 catalytic domain (Tet1) targeted to a control region 5kb upstream Magohb or Magohb promoter. The experiment was repeated two times independently with similar results. Uncropped gels are available as source data.
Extended Data Fig. 6. H3K27me3 and MLL2 dependent transcription.
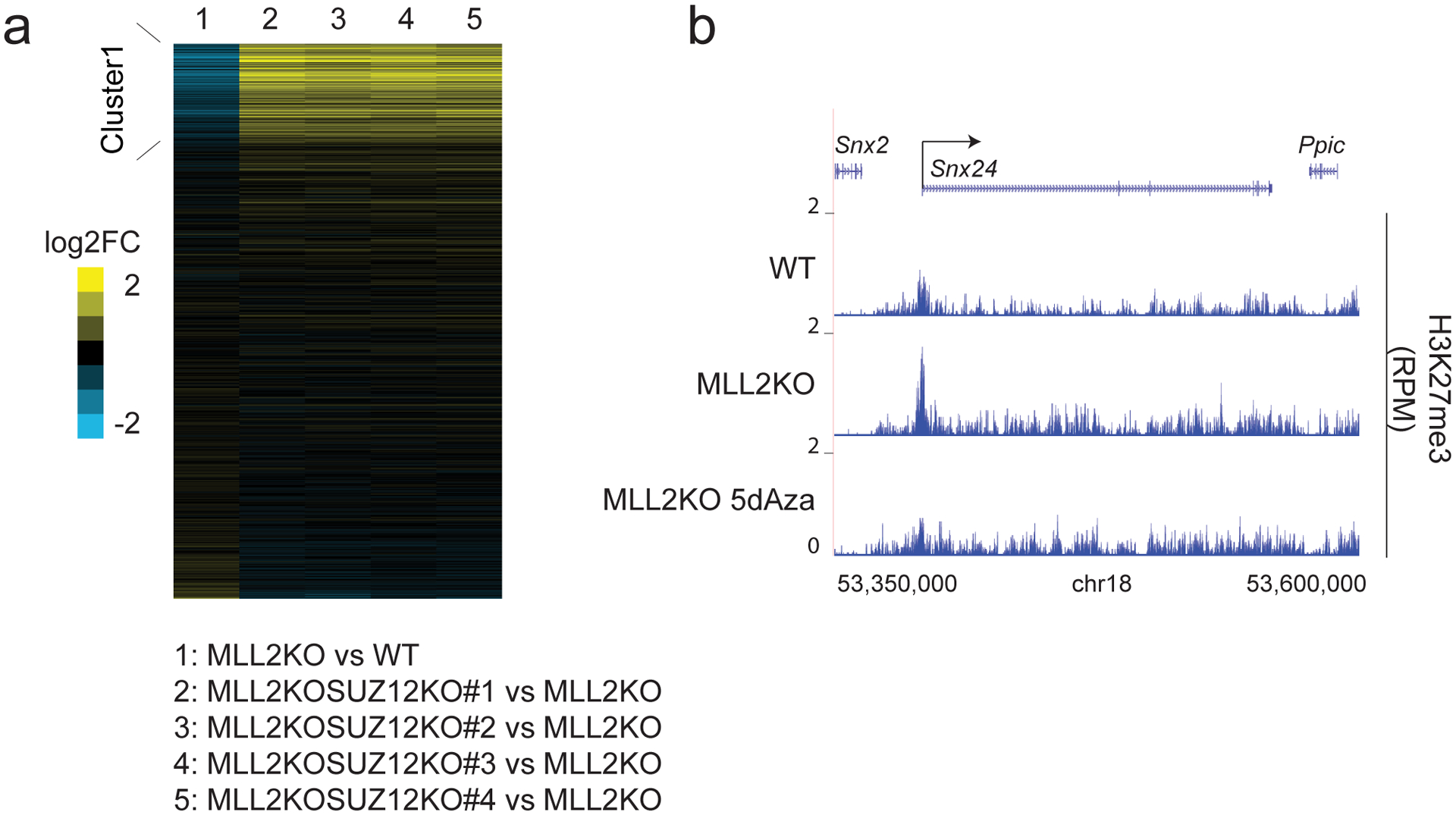
(a) Heatmaps show the corresponding log2 fold changes in gene expression in 1) MLL2KO vs. WT mESCs, 2) MLL2KOSUZ12KO#1 vs. MLL2KO mESCs, 3) MLL2KOSUZ12KO#2 vs. MLL2KO mESCs, 4) MLL2KOSUZ12KO#3 vs. MLL2KO mESCs and 5) MLL2KOSUZ12KO#4 vs. MLL2KO mESCs. The experiment was repeated two times independently with similar results. (b) ChIP-seq tracks of H3K27me3 occupancy at the Snx24 locus in WT, MLL2KO and MLL2KO 100 nM 5dAza for 4 days treated mESCs. The experiment was repeated two times independently with similar results.
Supplementary Material
Acknowledgements
We thank the Shilatifard laboratory members for helpful suggestions and discussions. K.A.H. and B.S. are supported by NIH K08HL128867. C.C.S. is supported, in part, by NIH Predoctoral to Postdoctoral Transition Award F99CA234945. K.C. is supported, in part, by NIH Pathway to Independence Award K99HD094906. A.P. is supported by NIH Pathway to Independence Award K99CA234434. E.R.S. is supported by NIH R50CA211428. We thank Nicole J. Ethen for the graphical representation of the model (Fig. 8). Studies in the Shilatifard laboratory related to COMPASS are supported by NCI’s Outstanding Investigator Award R35CA197569.
Footnotes
Competing interests
The authors declare no competing interests.
References.
- 1.Kouzarides T Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Meeks JJ & Shilatifard A Multiple Roles for the MLL/COMPASS Family in the Epigenetic Regulation of Gene Expression and in Cancer. Annual Review of Cancer Biology 1, 425–446 (2017). [Google Scholar]
- 3.Shilatifard A The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81, 65–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller T et al. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proceedings of the National Academy of Sciences 98, 12902–12907 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider J et al. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 19, 849–56 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Roguev A et al. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. The EMBO Journal 20, 7137–7148 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krogan NJ et al. COMPASS, a Histone H3 (Lysine 4) Methyltransferase Required for Telomeric Silencing of Gene Expression. Journal of Biological Chemistry 277, 10753–10755 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Mohan M et al. The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol 31, 4310–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu M et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol 28, 7337–44 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clouaire T et al. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev 26, 1714–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan MA & Shilatifard A Chromatin signatures of cancer. Genes Dev 29, 238–49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu D et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 33, 4745–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herz HM et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26, 2604–20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu D et al. Not All H3K4 Methylations Are Created Equal: Mll2/COMPASS Dependency in Primordial Germ Cell Specification. Mol Cell 65, 460–475.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rickels R et al. An Evolutionary Conserved Epigenetic Mark of Polycomb Response Elements Implemented by Trx/MLL/COMPASS. Mol Cell 63, 318–328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tkachuk DC, Kohler S & Cleary ML Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71, 691–700 (1992). [DOI] [PubMed] [Google Scholar]
- 17.Gu Y et al. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 71, 701–8 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Djabali M et al. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet 4, 431 (1993). [DOI] [PubMed] [Google Scholar]
- 19.Glaser S et al. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin 2, 5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer E et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet 49, 223–237 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Zech M et al. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. Am J Hum Genet 99, 1377–1387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sze CC et al. Histone H3K4 methylation-dependent and -independent functions of Set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev 31, 1732–1737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorighi KM et al. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol Cell 66, 568–576.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rickels R et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat Genet 49, 1647–1653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu D et al. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol 20, 1093–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viswanathan SR et al. Genome-scale analysis identifies paralog lethality as a vulnerability of chromosome 1p loss in cancer. Nat Genet 50, 937–943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh KK, Wachsmuth L, Kulozik AE & Gehring NH Two mammalian MAGOH genes contribute to exon junction complex composition and nonsense-mediated decay. RNA Biol 10, 1291–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JH, Tate CM, You JS & Skalnik DG Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J Biol Chem 282, 13419–28 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Austenaa LM et al. Transcription of Mammalian cis-Regulatory Elements Is Restrained by Actively Enforced Early Termination. Mol Cell 60, 460–74 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Lee JH & Skalnik DG CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem 280, 41725–31 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Kwak H, Fuda NJ, Core LJ & Lis JT Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howe FS, Fischl H, Murray SC & Mellor J Is H3K4me3 instructive for transcription activation? Bioessays 39, 1–12 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Tate CM, Lee JH & Skalnik DG CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. Febs j 277, 210–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlone DL et al. Reduced genomic cytosine methylation and defective cellular differentiation in embryonic stem cells lacking CpG binding protein. Mol Cell Biol 25, 4881–91 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Z et al. Distinct roles of DNMT1-dependent and DNMT1-independent methylation patterns in the genome of mouse embryonic stem cells. Genome Biol 16, 115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christman JK 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–95 (2002). [DOI] [PubMed] [Google Scholar]
- 38.McGrath-Morrow SA et al. DNA methylation regulates the neonatal CD4(+) T-cell response to pneumonia in mice. J Biol Chem 293, 11772–11783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu XS et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172, 979–992.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piunti A & Shilatifard A Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352, aad9780 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Brinkman AB et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22, 1128–38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mas G et al. Promoter bivalency favors an open chromatin architecture in embryonic stem cells. Nat Genet 50, 1452–1462 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Margaritis T et al. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3’-end antisense transcription. PLoS Genet 8, e1002952 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clouaire T, Webb S & Bird A Cfp1 is required for gene expression-dependent H3K4 trimethylation and H3K9 acetylation in embryonic stem cells. Genome Biol 15, 451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu X et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov 2, 16009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morita S et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol 34, 1060–1065 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Liu XS et al. Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao K et al. SET1A/COMPASS and shadow enhancers in the regulation of homeotic gene expression. Genes Dev 31, 787–801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahat DB et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weinberg SE et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L et al. TET2 coactivates gene expression through demethylation of enhancers. Sci Adv 4, eaau6986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walter JM, Helmin KA, Abdala-Valencia H, Wunderink RG & Singer BD Multidimensional assessment of alveolar T cells in critically ill patients. JCI Insight 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singer BD A Practical Guide to the Measurement and Analysis of DNA Methylation. Am J Respir Cell Mol Biol 61, 417–428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pusapati GV et al. CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev Cell 44, 271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–40 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saldanha AJ Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–8 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Journal M-M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal (2011). [Google Scholar]
- 63.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ChIP–seq, bisulfite sequencing raw data are available in the Gene Expression Omnibus (GEO) database under accession GSE129037. Additional data supporting the findings of this study are available from the corresponding author upon request. Source data (full scans of the immunoblots) for Figures 2 and 6 and Extended Data Figures 2, 3 and 5 are provided with the paper.