

Abstract
Melanoma is one of the most serious forms of skin cancer, and its increasing incidence coupled with non-lasting therapeutic options for metastatic disease highlights the need for additional novel approaches for its management. In this study, we determined the potential interactions between polo-like kinase 1 (PLK1, a serine/threonine kinase involved in mitotic regulation) and NOTCH1 (a type I transmembrane protein deciding cell fate during development) in melanoma. Employing an in-house human melanoma tissue microarray (TMA) containing multiple cases of melanomas and benign nevi, coupled with high-throughput, multispectral quantitative fluorescence imaging analysis, we found a positive correlation between PLK1 and NOTCH1 in melanoma. Further, TCGA database analysis of melanoma patients showed an association of higher mRNA levels of PLK1 and NOTCH1 with poor overall as well as disease-free survival. Next, utilizing small-molecule inhibitors of PLK1 and NOTCH (BI 6727 and MK-0752, respectively), we found a synergistic anti-proliferative response of combined treatment in multiple human melanoma cells. To determine the molecular targets of the overall and synergistic responses of combined PLK1 and NOTCH inhibition, we conducted RNA-sequencing analysis employing a unique regression model with interaction terms. We identified the modulations of several key genes relevant to melanoma progression/metastasis, including MAPK, PI3K, and RAS, as well as some new genes such as Apobec3G, BTK and FCER1G which have not been well-studied in melanoma. In conclusion, our study demonstrated a synergistic anti-proliferative response of a concomitant targeting of PLK1 and NOTCH in melanoma, unraveling a potential novel therapeutic approach for detailed preclinical/clinical evaluation.
Keywords: PLK1, NOTCH, Melanoma, Tissue Microarray, RNA-Sequencing
INTRODUCTION:
Melanoma is one of the most aggressive types of cancer and extremely difficult to treat when metastatic, with a median overall survival of less than one year. In the USA, 100,350 new melanoma cases and 6,850 melanoma-related deaths are predicted in 2020 (1). Current treatment options against metastatic melanoma have either failed to achieve significant response or the responses are short-lived due to the emergence of resistance to therapy in most patients (2). In melanoma, tumor progression and metastasis are complex processes involving multiple cellular events, including cell proliferation, enhanced survival, migration and invasion (3), suggesting a need for simultaneous targeting of multiple signaling pathways to effectively manage this deadly neoplasm. Therefore, instead of monotherapeutic approaches with modest therapy responses, many researchers are now shifting their attention towards combinatorial approaches to overcome resistance, and there is growing interest in the development of combinations of molecularly targeted therapies to obtain superior anti-tumor response (4).
Mammalian polo-like kinase 1 (PLK1) is a serine/threonine kinase that plays key roles in cell proliferation with critical regulatory functions in mitosis and cytokinesis (5). We have previously shown that PLK1 is overexpressed in melanoma cells as well as clinical melanoma tissues, and lentiviral shRNA or small molecule-targeted inhibition of PLK1 resulted in significant i) decrease in growth and clonogenic survival, ii) induction of G2/M phase cell-cycle arrest, iii) increase in apoptosis, and iv) altered metabolic regulation in melanoma cells (6–8). Another study from our laboratory demonstrated that Numb, an antagonist of the NOTCH pathway, regulates stability and localization of PLK1 and is required for transit through mitosis (9). Thus, based on the oncogenic functions of PLK1 in melanoma and other cancer types, inhibition of PLK1 has been suggested as a potential strategy for cancer therapy. Several studies have evaluated PLK1 inhibitors in both preclinical and clinical studies (10). Volasertib (BI 6727) is one of the most specific commercially available ATP-competitive PLK1 inhibitors, with an IC50 of 0.87 nM, and has been shown to induce mitotic arrest and apoptosis (10). In addition, a recent phase I clinical trial of BI 6727 in solid tumors resulted in a partial response in melanoma patients, and found that BI 6727 was generally well tolerated with a favorable pharmacokinetic profile (11). These studies suggest that further studies evaluating BI 6727 (or other PLK1 inhibitors) in conjunction with other agents may provide effective anti-melanoma therapies.
The NOTCH pathway is a highly evolutionally conserved molecular pathway that plays important roles in cell fate determination, proliferation, differentiation and survival (12). NOTCH signaling is deregulated in human hematological malignancies and solid tumors (12). In humans, there are four NOTCH receptors (NOTCH1-4), and upon each receptors’ activation, the NOTCH intracellular domain (NICD) is cleaved and translocated into the nucleus where it functions as a transcriptional regulator. Interestingly, the NOTCH1 pathway is activated in human melanoma and has been shown to enable primary melanoma cells to gain malignant phenotype (13). Recently, NOTCH1 signaling in melanoma has been shown to facilitate tumor immune escape and promote melanoma progression via TGF-β1 secretion (14). These above discussed studies provided us the scientific premise of this current investigation where we hypothesized that PLK1 and NOTCH pathways may correlate with each other and the simultaneous inhibition of PLK1 and NOTCH may provide a superior anti-proliferative response against melanoma. Overall, our data provides a basis for potential use of concomitant inhibition of PLK1 and NOTCH for melanoma management.
MATERIALS AND METHODS:
Tissue microarray immunostaining and analysis.
We used a human melanoma tissue microarray (TMA) constructed in-house under the University of Wisconsin IRB-approved protocol (described previously (15)) and processed and analyzed as depicted in Fig. 1A. Immunostaining was performed at the UW Translational Research in Pathology (TRIP) core facility using a Lab Vision Autostainer 360 (ThermoFisher). For immunostaining, the TMA slide was heated at 60°C for 30 minutes, then deparaffinized and rehydrated. Slides were then pretreated for 20 minutes with citrate buffer (pH 6.0) in Labvision PT module, rinsed with TBS-T, and loaded into Autostainer 360. This was followed by rinsing the slides with TBS-T again and blocking with Sniper (BioCare Medical) for 10 minutes. The slides were then sequentially incubated with antibodies for NOTCH1, AlexaFluor 555 secondary, PLK1, AlexaFluor 647 secondary, and Zenon 488-labeled S100 with TBS-T rinses between incubations. Details of antibodies used in the study are provided in Table S1. Stained slides were removed from Autostainer and rinsed in deionized water before coverslipping with ProLong Gold with DAPI and allowing to cure for at least 24 hours before imaging.
Figure 1. Study design and model visualization for differential gene expression analysis.
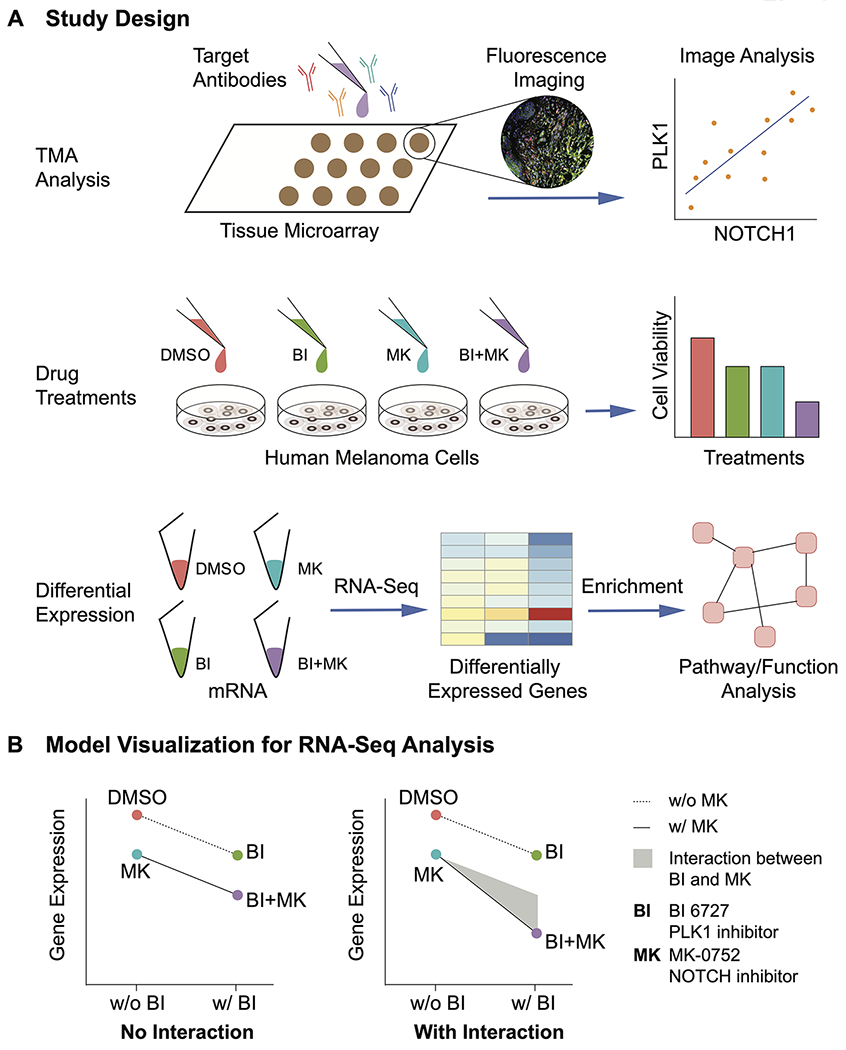
A, Experimental design for our studies. Our in-house tissue microarray (TMA) was stained with target antibodies including PLK1 and NOTCH1 followed by imaging, quantification and analysis. In vitro, human melanoma cells were treated with PLK1 inhibitor BI 6727 (BI) and/or NOTCH inhibitor MK-0752 (MK). The cell viability was assessed to evaluate the synergism between the two inhibitors. Next, we used RNA-seq to examine differential expression between the different treatment groups, followed by pathway and function analysis of these genes. B, Model visualization for drug interactions. A negative binomial model with an interaction term was implemented to compare the gene expression across all treatments. In the scenario without the interaction between BI and MK, the drug effects for the BI+MK combined treatment result solely from the addition of single drug effects. In the second scenario with drug interaction, the drug effects for the BI+MK combination came from i) the addition of single drug effects, and ii) the interaction or synergism between BI and MK shown as grey area, aka BI:MK.
Next, Vectra automated quantitative tissue imaging system coupled with an automated slide scanner and state-of-the-art software (Nuance and InForm; PerkinElmer) was used to image the TMA. This system takes advantage of the unique spectral characteristics (curve) of each chromogen via creation of a spectral library from single-chromogen slides to determine the relative contribution of each chromogen to the stained area on a slide. The stained TMA was imaged with the Vectra slide scanner, using a scanning protocol that was created based on the core size and layout. Nuance software was used to develop the spectral library to be imported into the InForm software. For each slide, an 8-bit image cube from each of the TMA tissue cores was acquired and the InForm advanced image analysis software was used to segment tissues (melanoma versus others) and cells (nucleus versus cytoplasm) to analyze protein levels. Then the target signals were quantified within the tissue and subcellular compartment(s) after unmixing the spectral curves with InForm software and removing signal noise and cross-talk. Continuous signal intensity data (mean optical density per pixel) was generated for each TMA core or cellular compartment and was used to assess IHC staining levels on a cell-by-cell and sub-cellular basis.
The statistical analysis of TMA data was performed in R, a publicly available programming language and environment using freely available analysis “packages” of commands. Protein expression in the nucleus and cytoplasm were considered paired data, and thus were compared using a nonparametric Wilcoxon signed-rank test using ggpubr package (v0.4.0) with p<0.05 indicating a significant difference of such protein expression between nucleus and cytoplasm. Pairwise simple linear regression of protein expression in nucleus and cytoplasm was analyzed by PerformanceAnalytics package (v1.5.3). A correlation coefficient matrix was generated from the previous step, and plotted by pheatmap package (v1.0.12).
The Cancer Genome Atlas (TGCA) survival analysis.
Relevant data from the TCGA database containing the mRNA expression and clinical information of 479 melanoma patients was downloaded from cBioPortal (TCGA, Firehose Legacy, accessed on April 6, 2020) (16, 17). The following columns of TCGA melanoma database were extracted and loaded to survival analysis: PLK1 expression, NOTCH1 expression, overall survival (OS) and disease-free survival (DFS) status, as well as OS and DFS months. Melanoma patients were divided into two groups according to the expression levels of PLK1 and NOTCH1: i) High PLK1+High NOTCH1 (mRNA levels ≥median) and ii) Others (mRNA levels <median) to study the cumulative effects of these two genes on survival outcome as described by Amelio et al (18). Using R, the OS and DFS between these two groups were analyzed by survival package (v2.43-3). The Kaplan–Meier curves between these two groups were plotted by survminer (v0.4.3) within R.
Cell culture and small-molecule inhibitors.
Human melanoma cell lines A375, SK-MEL-2 and SK-MEL-28 were purchased from ATCC. A375 cells were cultured in DMEM (Corning) with 10% FBS (Sigma-Aldrich). SK-MEL-2 and SK-MEL-28 cells were cultured in EMEM (Corning) supplemented with 10% FBS at standard cell culture conditions (37°C, 5% CO2 in a humidified incubator). All cell lines were routinely authenticated (upon thawing or cultured for >3 months) using STR testing. BI 6727, the small-molecule inhibitor used against PLK1, and MK-0572, the γ-secretase inhibitor used to inhibit NOTCH (19), were purchased from Selleck Chemicals and dissolved in DMSO (ThermoFisher) before use.
Trypan blue exclusion assay.
For trypan blue exclusion assay, 48 hours following drug treatments, melanoma cells were trypsinized and an aliquot was stained with trypan blue dye (BioRad). Viable, dead, and total cells were determined using the Bio-Rad TC10 Automated Cell Counter. Significance was analyzed by one-way ANOVA with Tukey’s multiple comparison test using the Prism software package (GraphPad). The data are expressed as the mean ± standard error of the mean with statistical significance (*P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001). The combination index (CI) assessing drug synergism was calculated via CalcuSyn software (Biosoft) employing the Chou-Talalay method (20).
Clonogenic survival assay.
Forty-eight hours following treatments, 1000 viable cells from each treatment group were plated in separate wells of a 6-well plate in duplicates. Cells were maintained under standard tissue culture conditions for ~10 days. Cells were then stained at room temperature for 30 minutes with 0.5% crystal violet solution in 1:1 methanol:water, rinsed 2 times with PBS (pH 7.4), and air-dried followed by digital photography.
Immunoblot analysis.
For immunoblot analyses, the protein lysates were prepared following treatment and harvesting of cells using standard protocol (9). The protein was quantified, and equal protein was subjected to SDS-PAGE followed by transfer to nitrocellulose membranes. The proteins were probed using primary antibodies (details in Table S1) and appropriate secondary antibodies followed by chemiluminescence detection with Pierce ECL Western Blotting Substrate (Thermo Scientific). Blots were imaged using a Li-COR Odyssey Fc.
RNA sequencing and differential gene expression analyses.
For RNA-seq analysis, total RNA was isolated from cells in 4 replicates in each treatment group. A total of 16 samples were submitted to the University of Wisconsin Biotechnology Center Gene Expression Center for quality control, library construction and further analysis. The purity and integrity of total RNA were assessed on the NanoDrop One Spectrophotometer and Agilent 2100 BioAnalyzer, respectively. Samples that met the TruSeq Stranded mRNA Sample Preparation Guide (Rev. E) input guidelines were prepared using the Illumina TruSeq Stranded mRNA Sample Preparation kit. The quality and quantity of prepared libraries were assessed by analysis on the Agilent 2100 Bioanalyzer and Qubit fluorimeter. An equimolar pool of all libraries with 30 million targeted reads per sample was sequenced on the Illumina NovaSeq S4 flowcell with 2x150bp sequencing. Images were analyzed using the standard Illumina Pipeline (v1.8.2).
From the RNA-seq data, gene abundances were estimated using RSEM (v1.3.0) (21), with read mapping performed using the STAR aligner (v2.5.3a) (22). Reads were mapped to the GRCh38 assembly of the human genome with the GENCODE v31 comprehensive transcript annotation. The “--forward-prob 0” option was used for RSEM to reflect the fact that the RNA-seq libraries were strand-specific. RSEM-estimated gene counts and effective lengths were compiled via tximport (v1.10.1) (23) and used for differential expression analyses with DESeq2 (v1.22.2) (24) within R. Read counts were modeled using a negative binomial generalized linear model (GLM) in DESeq2 with terms for each drug and an interaction term for effects resulting from the combination of the two drugs (Fig. 1B). Differentially expressed genes (DEGs) for each contrast of interest were identified as those with an adjusted p-value <0.05 after applying a multiple test correction using a false discovery rate of 0.05. RNA-Seq data were deposited into GEO data repository with accession number GSE159095.
Gene Ontology and Pathway Enrichment.
To analyze biological function and pathways affected by drug treatments, DEGs with log2-fold change ≥1 and log2-fold change ≤−1 were separately loaded to GOstats (v2.48) (25) and KEGGprofile (v1.24) (26) packages within R, respectively. The significantly up- and down-regulated GO terms and pathways with their p-values were separately uploaded to the ClueGO application (v2.5.6) in Cytoscape (27). To reduce redundancy, the GO Term Fusion box was checked to fuse GO terms with shared genes.
RESULTS:
PLK1 and NOTCH1 are positively correlated in melanoma and have a significant impact on patient survival.
Employing an in-house human melanoma TMA coupled with high-throughput, multispectral Vectra scanning and InForm analysis, we analyzed 110 melanoma tissues and 16 benign nevi (Fig. S1A). The TMA was immunostained for PLK1, NOTCH1 (antibody used had a high affinity for the “activated” intracellular form of NOTCH1 over the full-length form), the melanoma biomarker S100, and DAPI, followed by Vectra scanning and analysis with InForm software. A representative image with merged staining is shown in Fig. 2A. Using this system, we determined the levels of PLK1 and NOTCH1 in both the nucleus and cytoplasm of melanoma cells (as determined by positive S100 staining). We found that while PLK1 and NOTCH1 are detected in both the nucleus and cytoplasm, NOTCH1 levels are higher in the nucleus, and PLK1 levels are higher in the cytoplasm (Fig. 2B). To compare the correlative expression between PLK1 and NOTCH1, we generated a correlation matrix that demonstrated positive correlations between PLK1 and NOTCH1 in both the nucleus and cytoplasm (Fig. 2C–D).
Figure 2. PLK1 and NOTCH1 are positively correlated in melanoma and associated with survival of melanoma patients.
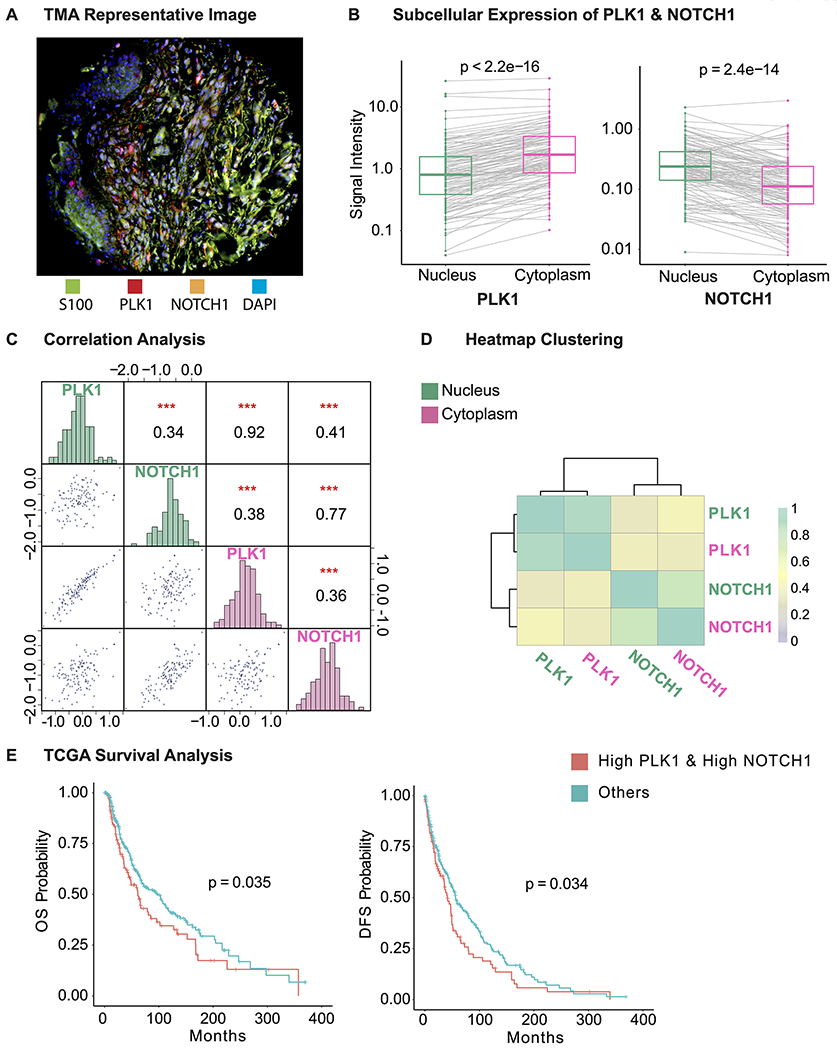
A, Representative image of a tissue core from our TMA. The TMA was stained with S100, PLK1, NOTCH1, and DAPI (green, red, orange, and blue fluorophores, respectively). B, Comparison of PLK1 and NOTCH1 expression in nucleus and cytoplasm. Gray line indicates expression from same core. C, Pairwise correlation analysis between PLK1 and NOTCH1 in nucleus (green) and cytoplasm (pink). PLK1 and NOTCH1 expression are shown in the histograms along the diagonal, while scatterplots of pairwise correlations and correlation coefficient and significance levels are represented on opposite sides of the diagonal. Axis is in natural log scale. D, Heatmap clustering of the correlation coefficient displayed in Fig. 2C. PLK1 and NOTCH1 are first clustered with themselves and then clustered together. E, Overall (OS) and disease-free survival (DFS) analysis in 479 melanoma patients using TCGA database. The overall and disease-free survival are compared between patients with “High PLK1 and High NOTCH1” (red, 125 patients, levels>median) and “Others” (green, 334 patients, levels<median). ***P < 0.001. S100, melanoma marker; DAPI, 4′,6-diamidino-2-phenylindole, a fluorescent DNA stain.
This association between PLK1 and NOTCH1 in our TMA analysis led us to further explore the correlation of these proteins with melanoma clinical parameters. Therefore, we determined if Breslow thickness, a measure of melanoma aggressiveness (28), is correlated with PLK1 and NOTCH1. Interestingly, we found a positive correlation between Breslow thickness and expression of both PLK1 and NOTCH1 (Fig. S1B).
Further, we determined the relationship between the mRNA expression of PLK1 and NOTCH1 and melanoma patient survival using the TCGA database, where a total of 459 patients and 402 patients with available clinical information were selected for overall survival and disease-free survival analysis, respectively. By comparing the survival probability between patients with highly expressed (≥median) PLK1 and NOTCH1 versus the rest of patients, we found that higher levels of PLK1 and NOTCH1 were associated with poor overall and disease-free survival (Fig. 2E). In addition, we performed survival analysis to correlate with PLK1 and NOTCH1 expression individually using TCGA data available from cBioportal and Protein Atlas platforms. Our results using Protein Atlas showed that high expression of both PLK1 and NOTCH1 correlate significantly with poor overall survival, however, with cBioPortal, only PLK1 expression was found to be significantly associated with poor overall survival but not NOTCH1 expression, which may be due to the difference in sample size and populations used between the platforms (Fig. S2). Taken together, these findings combined with previously published studies suggest that targeting PLK1 and NOTCH1 together may provide a novel means for melanoma management.
Combined inhibition of PLK1 and NOTCH results in synergistic anti-proliferative effects on melanoma cells.
To determine the effects of combined inhibition of PLK1 and NOTCH, we used PLK1 inhibitor BI 6727 and NOTCH pathway inhibitor MK-0752 (a potent γ-secretase inhibitor) in A375 (BRAF mutant (V600E) with wild type TP53), SK-MEL-2 (wild type BRAF and mutant TP53), and SK-MEL-28 (both BRAF and TP53 mutant) human melanoma cell lines. These lines were used as they represent the genetic backgrounds and mutations in the most common genetically altered genes in melanoma (Table S2). Employing trypan blue exclusion assays, we found that BI 6727 (10 and 20 nM) and MK-0752 (50 and 100 μM) treatment resulted in significant decreases (60-80%) in viability and growth of human melanoma cells (Fig. 3A). The Combination Index (CI), a metric to determine if drugs behave synergistically, was calculated using the Chou-Talalay theorem for the combination treatments. Our data demonstrated that the CI was less than 1 in all the tested combination doses of BI 6727 and MK-0752 in A375 and SK-MEL-2 cells, and at 10 nM BI 6727 and 50 μM MK-0752 in the SK-MEL-28 cells (Fig. 3A), indicating potential synergistic responses between these two drugs in melanoma. This was further confirmed using RealTime-Glo and MTT assays (Fig. S3). Immunoblotting of protein lysates from the treated cells for HES1, a key downstream NOTCH pathway protein (29), showed that the MK-0752 treatment alone as well as in combination with BI 6727 was associated with reduced HES1, confirming the efficient targeting of NOTCH pathway (Fig. S4). To determine the effects of PLK1 and/or NOTCH inhibition on clonogenic survival of melanoma cells, we used a colony formation assay. Our data demonstrated that compared to single drug treatments, the combination treatment resulted in a marked decrease in colony formation ability of the melanoma cells (Fig. 3B). Taken together, our data demonstrated that combined targeting of PLK1 and NOTCH signaling pathways impart synergistic anti-proliferative responses against melanoma.
Figure 3. PLK1 and NOTCH inhibition imparts synergistic anti-proliferative response against melanoma cells.
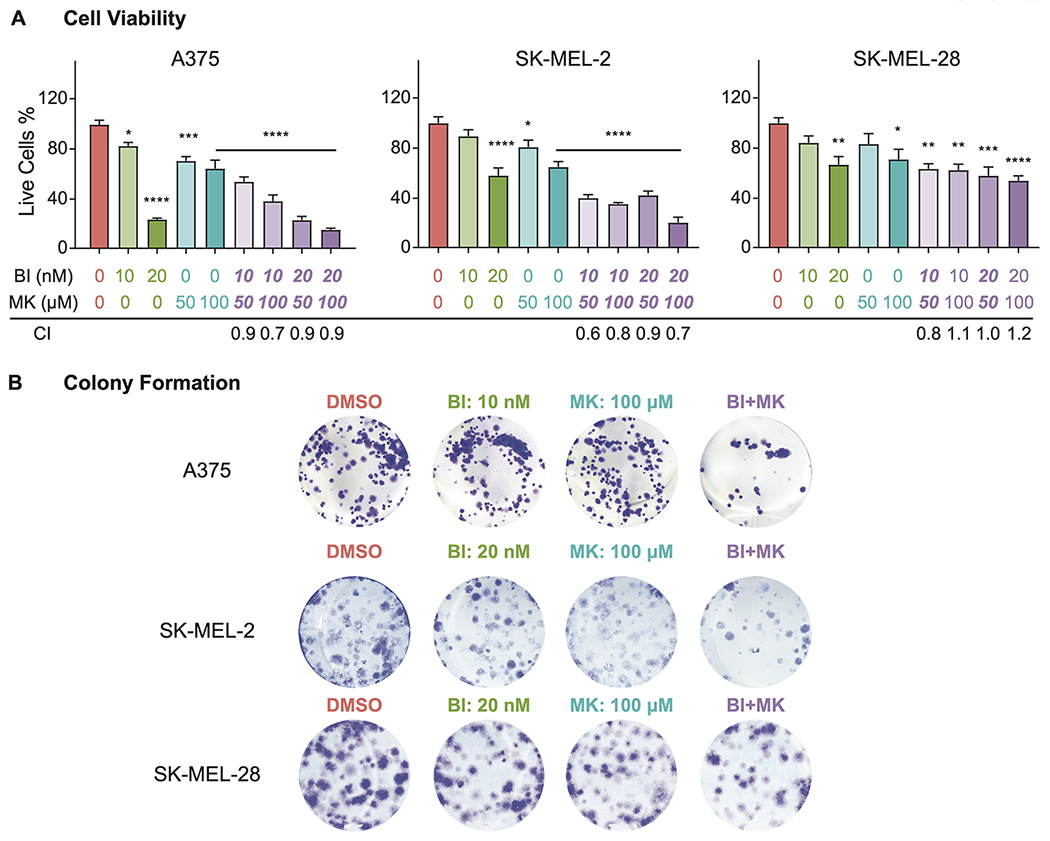
A, Cell viability assays evaluating the response of PLK1 and NOTCH inhibitors. Percent of live A375, SK-MEL-2, and SK-MEL-28 cells treated by different doses of PLK1 inhibitor BI 6727 (BI) and/or NOTCH inhibitor MK-0752 (MK) are shown as mean ±SEM of at least three biological replicates in duplicates. The combination index (CI) for to each treatment is listed below the dose. B, Representative images of colony formation assays. A375, SK-MEL-2 and SK-MEL-28 melanoma cells treated with PLK1 inhibitor BI 6727 and/or NOTCH inhibitor MK-0752 followed by colony staining using crystal violet of at least three biological replicates in duplicate. *P <0.05, **P <0.01, ***P <0.001, and ****P<0.0001. DMSO: dimethyl sulfoxide; BI: BI 6727; MK: MK-0752; CI: Combination Index.
RNA-seq analysis following combined inhibition of PLK1 and NOTCH in SK-MEL-2 melanoma cells shows differential regulation of multiple genes.
To determine the possible mechanisms behind the observed synergistic anti-proliferative response of combined PLK1-NOTCH inhibition, we used RNA-seq to measure the differential gene expression following treatment of SK-MEL-2 human metastatic melanoma cells with PLK1 inhibitor BI 6727 (20 nM) and/or NOTCH inhibitor MK-0752 (100 μM), with 4 replicates in each treatment (Fig. S5A). While most samples generated around 30 million sequencing reads and ~79% of them were aligned to human genome, it appears that the MK1 sample was an outlier (Fig. S5B), which was further confirmed by unsupervised clustering and the sample was removed from further analysis. Correlation analysis and principal component analysis (PCA) on the rest of the samples showed a favorable consistency between replicates within each treatment (Fig. S5C–D).
After evaluating the consistency of the samples, we identified differentially expressed genes (DEGs, |log2-fold change| ≥1, false-positive rate ≤0.05) when comparing the individual and combined treatments to vehicle (DMSO), as well as the interaction between BI 6727 and MK-0752. Specifically, we implemented a negative binomial regression model where the mean for each gene, after scaled by a sample-specific normalization factor, is modeled as:
where XBI and XMK are indicator variables that are 1 if the samples were treated with BI 6727 and MK-0752 and 0 otherwise; and XBI:MK is the interaction effect for BI 6727 and MK-0752. Herein, we define that the term BI:MK represents the interaction effect between BI 6727 and MK-0752 (i.e. transcriptomic responses different than the sum of the responses to the monotherapies), while BI+MK represents the overall effects resulting from the comparison between the combined treatment and vehicle control (i.e. overall effects of the dual treatments) (Fig. 1B).
Our data identified 909 DEGs from BI 6727 treatment, 675 DEGs from MK-0752 treatment, 2142 DEGs from the combined treatment of BI 6727 and MK-0752, as well as 304 DEGs from the synergistic interaction of BI 6727 and MK-0752 (Fig. 4A). Interestingly, as seen in the Venn Diagram (Fig. 4B), 1245 unique genes were found solely in the combined treatment, suggesting that combined PLK1 and NOTCH inhibition may have unique and broad treatment response mechanisms compared to the individual agents. Fig. 4C and Fig. S6 illustrate the top 100 most significant DEGs with |log2-fold change| ≥1 in the BI+MK combination and BI:MK interaction effects, along with their log2 fold change in the individual treatments. The list also includes significant decrease in NOTCH target gene HES1, further confirming the immunoblotting analysis for HES1 protein presented in Fig. S4. These exciting results led us to further explore the functionality of these DEGs in melanoma.
Figure 4. PLK1 and/or NOTCH inhibition in SK-MEL-2 melanoma cells via small molecule inhibitors leads to differential expression of genes as measured using RNA-seq analysis.
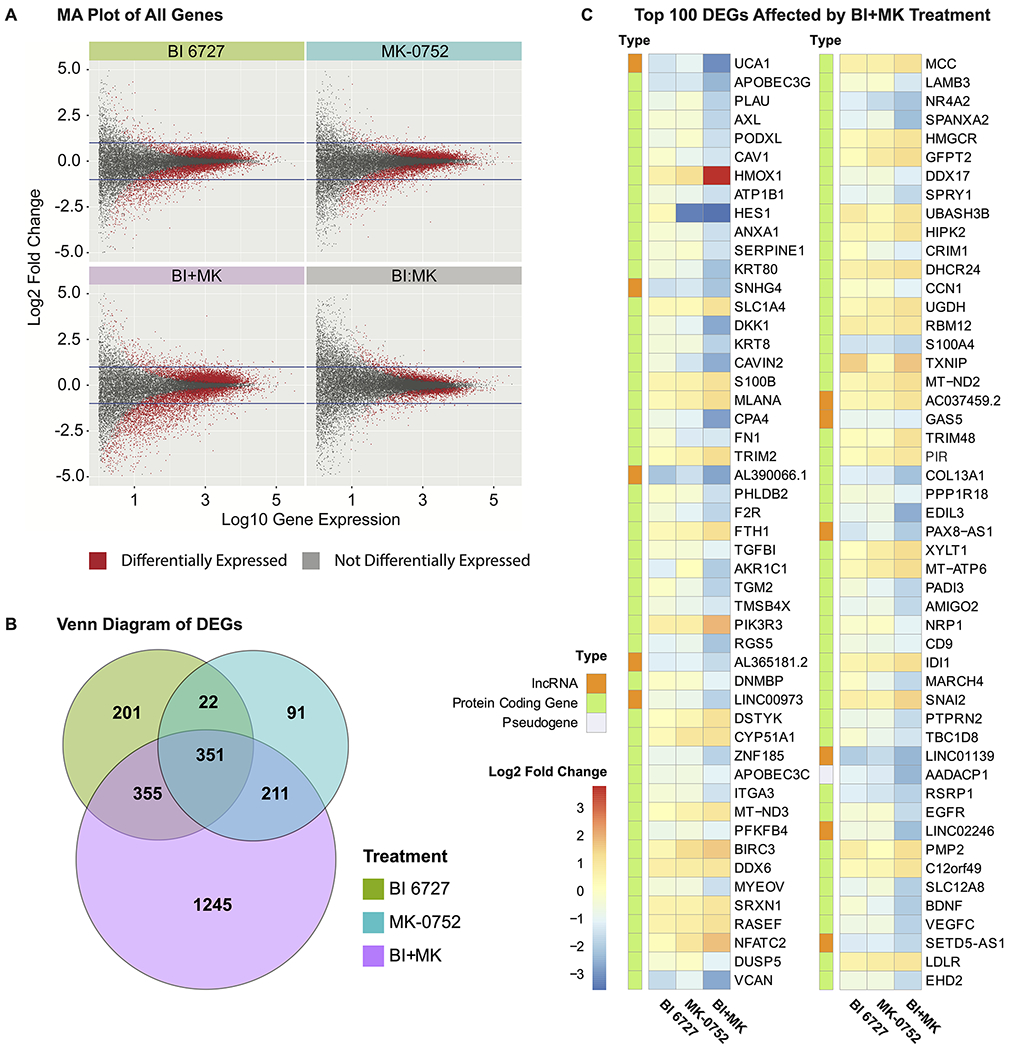
A, MA (log ratio - mean average) plot of all genes. Plots of gene expression against fold change for all genes affected by BI 6727, MK-0752, the combination of the two drugs (BI+MK), as well as the interaction of the two drugs (BI:MK). The differentially expressed genes (DEGs) are depicted as red dots, while the non-DEGs are depicted as gray dots. In each plot, two horizontal lines are drawn to represent |log2 fold change| =1. B, Venn diagram summarizing the relationships between the DEGs for each treatment. The diagram depicts the DEGs with |log2 fold change| ≥1 in each treatment group and their overlap. C, Heatmap of top 100 most significant DEGs with |log2 fold change| ≥1 in the treatment of BI+MK. The fold change for BI 6727 and MK-0752 single drug treatment, as well as the type of each gene, are also displayed.
Functional and pathway analysis of RNA-seq data suggests pleiotropic mechanisms of combined inhibition of PLK1 and NOTCH.
We conducted Gene Ontology (GO) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of the various DEGs. Specifically, we were interested in the overall transcriptomic responses (BI+MK), as well as the synergistic responses (BI:MK) of combined drug treatments. In GO analysis (counts ≥2, p ≤10−5), we identified 38 downregulated and 4 upregulated GO terms affected by BI+MK, including metabolism, cell proliferation, and migration (Fig. 5A). Further, we identified 10 downregulated and 6 upregulated GO terms affected by the interaction effects (BI:MK) of combined drug treatment, including increased hypersensitivity and apoptosis as well as decreased cell proliferation (Fig. 5B). In KEGG pathway analysis, the response of BI+MK was found to be associated with modulation of multiple pathways shared with single drug treatments, such as PI3K-AKT, ECM (extracellular matrix)-receptor interaction, and protein digestion and absorption, as well as pathways uniquely affected by combined treatment, such as MAPK, RAS, and RAP1 pathways (Fig. 5C). Further, we found that BI:MK was associated with multiple affected pathways, including MAPK, RAS, proteoglycans and phospholipase D (Fig. 5D).
Figure 5. Small molecule inhibition of PLK1 and NOTCH in melanoma cells affects key Gene Ontology (GO) and KEGG pathways.
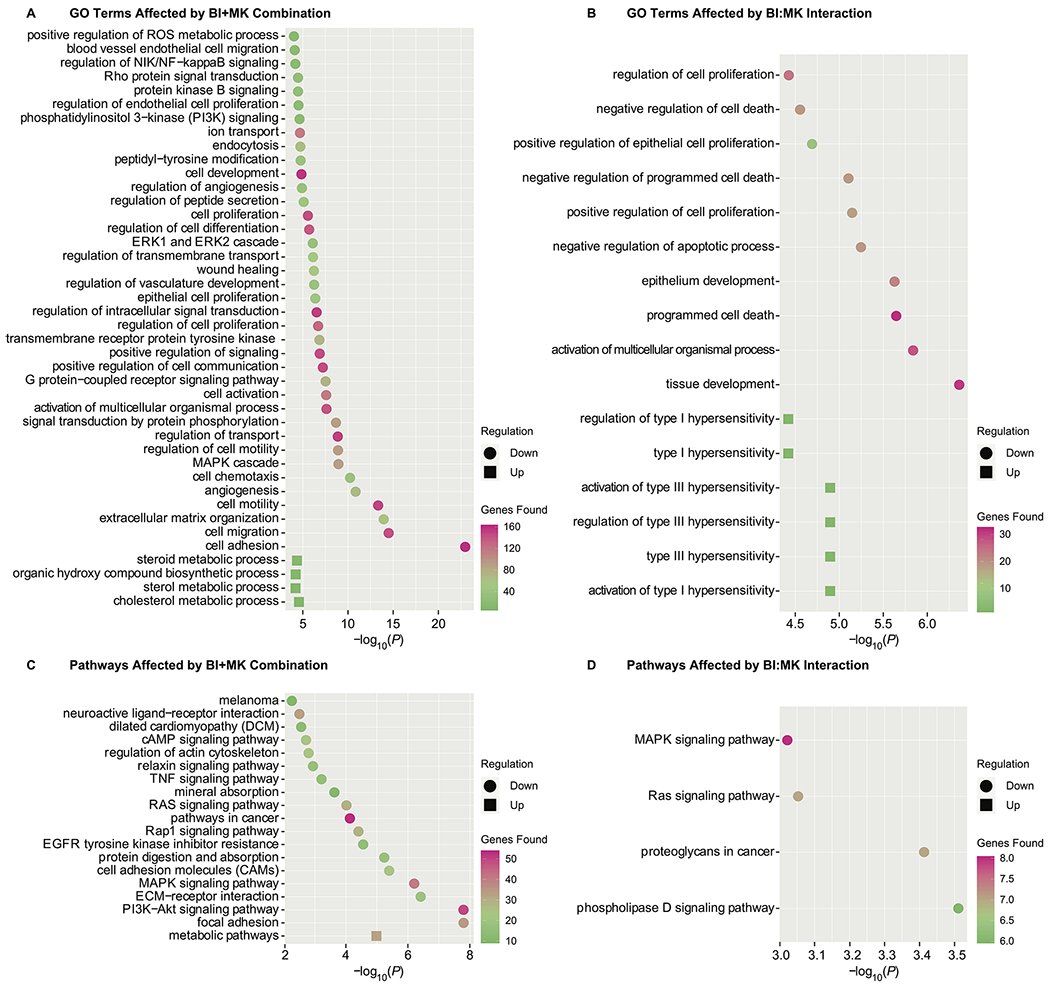
Each plot is ranked by p-values, and the upregulated and downregulated Gene Ontology GO terms or pathways are shown in circle and square, respectively. The color scale represents the number of DEGs found in the corresponding GO terms or pathways. A, GO terms affected by the combination of BI 6727 and MK-0752. B, GO terms affected by the interaction of BI 6727 and MK-0752. C, KEGG pathways affected by the combination of BI 6727 and MK-0752. D, KEGG pathways affected by the interaction of BI 6727 and MK-0752.
In order to get a broader look at how the networks integrate with each other, we utilized the ClueGO application in Cytoscape to plot the dysregulated biological functions and pathways due to the combined inhibition of PLK1 and NOTCH. The hierarchical structures and the connections between nodes reflect the interplay among the altered biological function and pathways. The combined inhibition of PLK1 and NOTCH (BI+MK) resulted in a comprehensive modulation of melanoma-related networks, including PI3K-AKT, NIK/NF-κB, and MAPK signaling (Fig. 6A and Fig. 6C). These networks are related to cell proliferation and development, which were found to be synergistically modulated as well (Fig. 6B and Fig. 6D). Interestingly, our data demonstrated that the combined inhibition of PLK1 and NOTCH upregulated pathways associated with the hypersensitivity of melanoma cells (Fig. 6B).
Figure 6. Inhibition of PLK1 and NOTCH in SK-MEL-2 melanoma cells results in modulation of several GO and KEGG pathway networks.
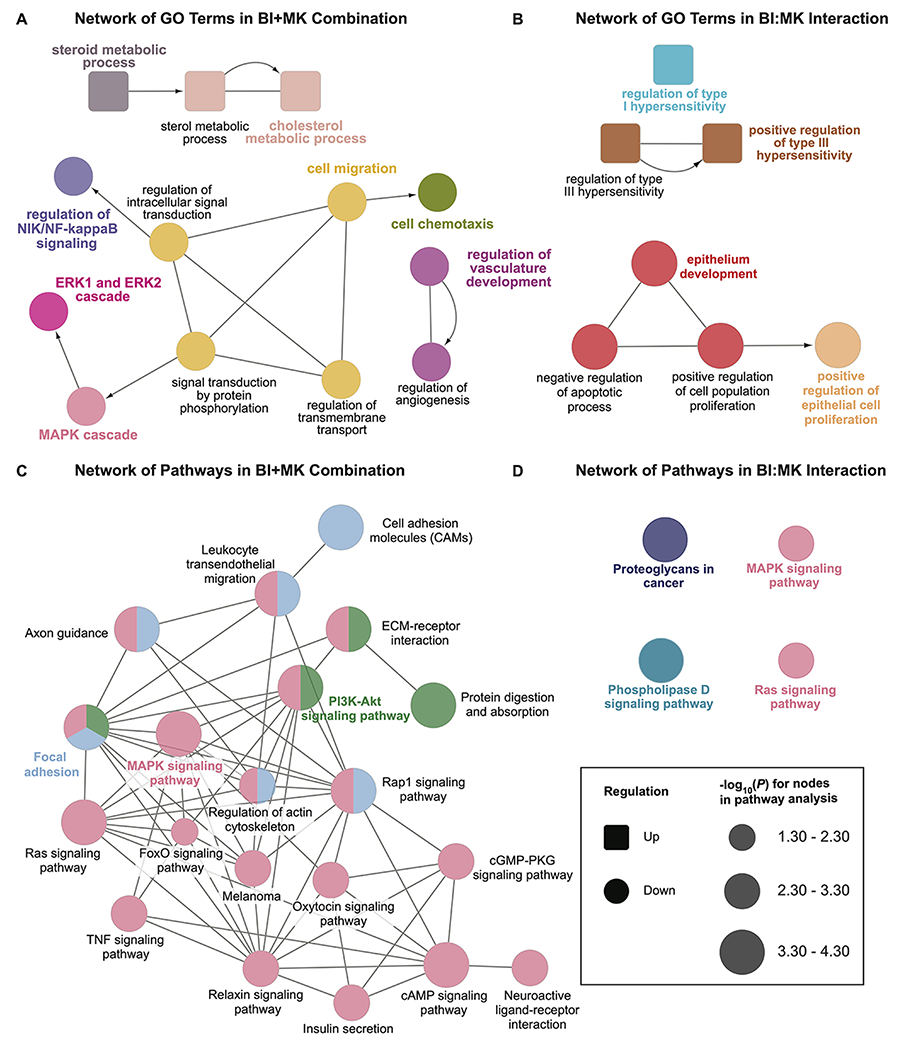
Nodes with same color belong to the same cluster of GO terms or pathways. Nodes connected by undirected edge indicate the two nodes are relevant to each other. Nodes connected by arrows indicates a hierarchical relationship in Gene Ontology. A, Network of GO terms affected by the combination of BI 6727 and MK-0752. B, Network of GO terms affected by the interaction of BI 6727 and MK-0752. C, Network of KEGG pathways affected by combination of BI 6727 and MK-0752. D, Network of KEGG pathways affected by interaction of BI 6727 and MK-0752.
Combined inhibition of PLK1 and NOTCH results in enhanced apoptosis of human melanoma cells.
Our cell culture data and RNA-seq results showed that the combined targeting of PLK1 and NOTCH led to enhanced apoptosis and decreased cell proliferation. To verify our findings, we analyzed several apoptosis-related proteins in treated A375 and SK-MEL-2 melanoma cells using immunoblotting. Our data demonstrated that BI 6727 and MK-0752 combination caused a marked increase in cleaved PARP (poly ADP ribose polymerase) and cleaved caspase-3 in both melanoma cell lines, as well as increased expression of tumor suppressor protein TP53 in A375 melanoma cells, which contains wild-type TP53 (Fig. 7A). Interestingly, we also observed increased apoptosis in SK-MEL-2 melanoma cells, which carry mutant TP53 (Fig. 7B).
Figure 7. Combined inhibition of PLK1 and NOTCH using small molecule inhibitors results in enhanced apoptosis in A375 and SK-MEL-2 human melanoma cells.
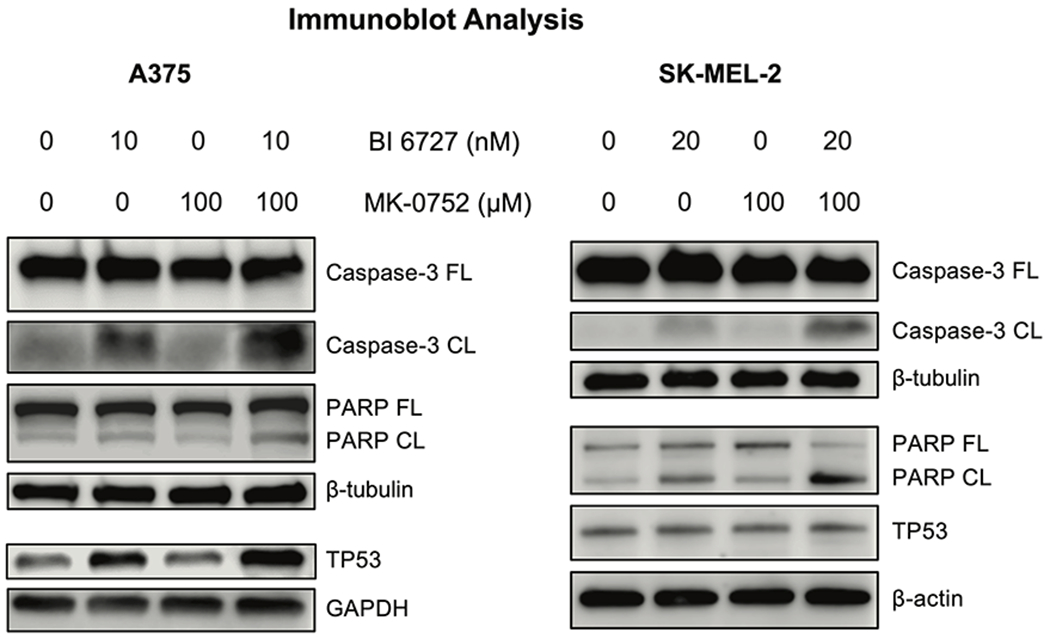
The cells were treated with vehicle (DMSO), BI 6727, MK-0752, and the combination of BI 6727 and MK-0752 before protein isolation and immunoblotting. Images shown are representative of three biological replicates. β-tubulin, β-actin, and GAPDH were used as loading controls. DMSO: dimethyl sulfoxide; PARP: Poly (ADP-ribose) polymerase; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase. FL, full-length; CL, cleaved.
DISCUSSION:
Since metastatic melanoma is notoriously resistant to existing therapeutic approaches, combinatorial therapeutic regimens may be a better strategy towards effective relapse-free melanoma management. Our study focuses on targeting two complementary and connected pathways (PLK1 and NOTCH pathways) in melanoma cells to obtain a superior anti-proliferative response. These pathways have been shown either by our lab or other groups to be important in melanoma development and progression (6, 7, 13, 30). However, to our knowledge, combined inhibition of these key pathways has not been investigated in melanoma before. In this study, we used two small molecule inhibitors that have been individually exploited for their anticancer effects in clinical trials, BI 6727 (11) and MK-0752 (31), and combined them and found a synergistic antiproliferative response in human melanoma cells.
The rationale for dual targeting was based on previous research discussed above and was further strengthened by our TMA and TCGA analysis (Fig. 2). In our TMA data, we observed a significant positive correlation between the expression of PLK1 and NOTCH1 in melanoma tissues. Our TMA results were supported by the TCGA analysis of large melanoma cohorts where we found that high mRNA expression of both PLK1 and NOTCH1 is associated with poorer overall and disease-free survival. In our cell culture experiments with multiple melanoma cell lines with varying genetic makeup, we observed synergistic anti-proliferative effects of small-molecule inhibition of PLK1 (BI 6727) in combination with NOTCH inhibition (MK-0752) on cell viability and clonogenic survival analysis (Fig. 3).
We also studied the potential mechanisms of combined inhibition utilizing RNA sequencing. To fully address the mechanisms of additive and/or synergistic effects between two inhibitors, we implemented a negative binomial regression with interaction terms for differential gene expression analysis. In terms of comparing differential gene expression in combination treatments, many existing analytical approaches only focus on comparing the individual or combined drugs versus vehicle control, but have ignored the potential synergistic interactions between the drugs (32, 33). Thus, our statistical model provides us with two advantages: i) it enables the evaluation of genes affected by the interaction responses of the two drugs (the BI:MK term), and ii) it allows us to study the overall landscape of differentially expressed genes (DEGs) due to the combination of the two drugs (the BI+MK term). Although the interaction term has been used to evaluate drug combinations at the cell level (34), to our knowledge, thus far it has not been applied to differential gene expression analysis in RNA-seq.
Using this RNA-seq analysis, we have identified the top 100 most significant DEGs due to the combined treatment with BI 6727 and MK-0752 (Fig. 4C). The top 5 genes in this list are associated with cancer metastasis, including Uca1 (Urothelial carcinoma-associated 1) (35), Apobec3G (Apolipoprotein B mRNA Editing Enzyme Catalytic Subunit 3G) (36), Plau (plasminogen activator urokinase) (37), Axl (AXL receptor tyrosine kinase) (38), and Podxl (podocalyxin like) (39). Additionally, Axl has been implicated in the regulation of acquired resistance against BRAF inhibitors by activating the PI3K-AKT pathway (40). Together, the fact that the top 5 most-downregulated genes indicate reduced metastasis supports the findings of our study suggesting a superior response of combined inhibition of PLK1 and NOTCH against melanoma metastasis. Moreover, these data also open new avenues for investigations on Apobec3G, which has not been associated with metastatic melanoma previously.
Our RNA-Seq analysis has also shown that the inhibition of PLK1 and NOTCH imparted significant downregulation of multiple melanoma-related pathways, especially the MAPK pathway (Fig. 5 and 6). Abnormities of MAPK pathway such as BRAF and RAS mutations account for more than 80% of melanoma cases (41), making it an important target for melanoma chemo- and immunotherapy (42). Our findings were supported by previous studies regarding the relationship between PLK1/NOTCH1 and MAPK. PLK1 has recently been shown to activate ERK1/2 signaling through phosphorylation of CRAF in prostate cancer, indicating a non-canonical function of PLK1 as a MAPK pathway stimulator (43). Additionally, it has been shown that the activation of MAPK/PI3K-AKT pathways by NOTCH1 results in the progression of primary melanoma (44).
To understand the mechanism(s) of action of drug-combination, we also determined top 100 most significant DEGs due to the BI:MK interaction effects (Fig. S5). The result suggests that the interaction of these two drugs produce significant additional gene modulations in melanoma cells when compared to individual drugs (Fig. 5 and 6). Interestingly, the inhibition of PLK1 and NOTCH increased type I and type III hypersensitivity GO terms, defined as inflammatory responses by Gene Ontology (45). The major contributors to these GO terms in our data are BTK (Bruton tyrosine kinase) and FCER1G (Fc Fragment of IgE Receptor Ig). Although these two genes have not been studied in melanoma, they have been shown to follow a context-dependent pattern in cancer progression. BTK, a Tec family kinase, has been shown to increase cell proliferation and survival signals or induce senescence and apoptosis depending on the tissue type (reviewed in (46)). The two available reports on FCER1G in cancer suggest its tumor promotor role in renal cell carcinoma (47) and tumor suppressor in multiple myeloma (48). Despite the lack of research on type I and type III hypersensitivity in melanoma, there are a few studies regarding PLK1 or NOTCH1 and melanoma immune activity. For example, Li et al. have found a negative correlation between PLK1 and immune activities in many cancer types including melanoma, and the inhibition of PLK1 led to the upregulation of MHC Class I in melanoma cell lines (49). Moreover, NOTCH1 in melanoma was shown to create an immune-suppressive tumor microenvironment (50), while NOTCH1 inhibition improved the efficacy of immune checkpoint inhibitors against melanoma. This may provide us with a novel strategy of coupling this combined therapy with immune therapy.
Furthermore, we validated one of the anti-proliferative pathways obtained from RNA-seq analysis in two melanoma cell lines with different mutation landscapes. We observed modulation of several apoptosis-related proteins after combined drug treatment (Fig. 7). In melanoma cells with mutant TP53, it is likely that the observed apoptosis following drug treatments is due to a TP53-independent process. However, in melanoma cells with wild-type TP53, a TP53-dependent phenomenon cannot be ruled out. Further experiments are needed to explore the role of TP53 in this process in greater detail. Overall, our molecular assays and RNA-seq data indicate that combined targeting of PLK1 and NOTCH pathways imparts synergistic anti-proliferative responses against melanoma cells and should be explored further for melanoma treatment.
Although our in vitro study utilized multiple melanoma cell lines and RNA-seq after single or combined treatments to study the inhibition of PLK1 and/or NOTCH, the results need to be further validated in in vivo models of melanoma relevant to human disease. Additionally, as the RNA-seq data was obtained in one mutant TP53 melanoma cell line, the results should be further verified in cell and mouse lines of varying genetic backgrounds. We have uncovered several genes and pathways affected by combined inhibition of PLK1 and NOTCH in TP53 mutated metastatic melanoma cells, which may or may not be linked to BRAF or NRAS mutated melanoma. Even with more work to be done, combined inhibition of the PLK1 and NOTCH pathways provides a potential novel therapeutic regimen to help in the fight against melanoma.
Taken together, our data suggest synergistic anti-proliferative responses of combined inhibition of PLK1 and NOTCH in melanoma cells. We have found that combined targeting of PLK1 and NOTCH signaling imparts downregulation of multiple pathways related to melanoma progression and metastasis, as well as potentially increases sensitivity to other therapeutic targets. Our study also identified potential new targets which have not been explored previously in detail for melanoma management. As the tested inhibitors, BI 6727 and MK-0752, have been used in clinical trials against certain cancer types, the combination studies with these two inhibitors have a high translational value that could be quickly developed into a successful strategy for melanoma management. Overall, our results suggest that combinatorial targeting of PLK1 and NOTCH shows promise and warrants additional studies in in vivo melanoma models as well as clinical investigations.
Supplementary Material
ACKNOWLEDGEMENTS:
The authors would like to thank the UW Department of Pathology & Laboratory Medicine’s Translational Research Initiatives in Pathology (TRIP) lab for their TMA staining services, as well as the UW Biotechnology Center’s Gene Expression Center for their RNA-sequencing services. We would also like to thank Dr. Cecile Ane for advice on data analysis and Lauren Benedict for experimental assistance. The results shown here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
This work was partially supported by funding from the NIH (R01AR059130 and R01CA176748 to NA; R21CA125091 to VS), and the Department of Veterans Affairs (VA Merit Review Awards I01BX001008 and I01CX001441 to NA and 1I01BX004921 to VS; and a Research Career Scientist Award IK6BX003780 to NA). This work was also supported by the core facilities in the Skin Diseases Research Center (SDRC) Core Grant P30AR066524 from NIH/NIAMS, the University of Wisconsin Translational Research Initiatives in Pathology laboratory (TRIP), supported by the UW Department of Pathology and Laboratory Medicine, the Office of The Director- NIH (S10OD023526), as well as the University of Wisconsin Carbone Cancer Center Support Grant P30CA014520.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
REFERENCES:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Domingues B, Lopes JM, Soares P, Populo H. Melanoma treatment in review. Immunotargets Ther 2018;7:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damsky WE, Theodosakis N, Bosenberg M. Melanoma metastasis: new concepts and evolving paradigms. Oncogene 2014;33:2413–22. [DOI] [PubMed] [Google Scholar]
- 4.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol 2012;30:679–92. [DOI] [PubMed] [Google Scholar]
- 5.Cholewa BD, Liu X, Ahmad N. The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res 2013;73:6848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cholewa BD, Ndiaye MA, Huang W, Liu X, Ahmad N. Small molecule inhibition of polo-like kinase 1 by volasertib (BI 6727) causes significant melanoma growth delay and regression in vivo. Cancer Lett 2017;385:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmit TL, Zhong W, Setaluri V, Spiegelman VS, Ahmad N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small-molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J Invest Dermatol 2009;129:2843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutteridge RE, Singh CK, Ndiaye MA, Ahmad N. Targeted knockdown of polo-like kinase 1 alters metabolic regulation in melanoma. Cancer Lett 2017;394:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmit TL, Nihal M, Ndiaye M, Setaluri V, Spiegelman VS, Ahmad N. Numb regulates stability and localization of the mitotic kinase PLK1 and is required for transit through mitosis. Cancer Res 2012;72:3864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther 2016;15:1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin CC, Su WC, Yen CJ, Hsu CH, Su WP, Yeh KH, et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br J Cancer 2014;110:2434–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013;34:1420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinnix CC, Lee JT, Liu ZJ, McDaid R, Balint K, Beverly LJ, et al. Active Notch1 confers a transformed phenotype to primary human melanocytes. Cancer Res 2009;69:5312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z, Qi Y, Lai N, Zhang J, Chen Z, Liu M, et al. Notch1 signaling in melanoma cells promoted tumor-induced immunosuppression via upregulation of TGF-beta1. J Exp Clin Cancer Res 2018;37:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez CI, Castro-Perez E, Longley BJ, Setaluri V. Elevated cyclic AMP levels promote BRAF(CA)/Pten(−/−) mouse melanoma growth but pCREB is negatively correlated with human melanoma progression. Cancer Lett 2018;414:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amelio I, Tsvetkov PO, Knight RA, Lisitsa A, Melino G, Antonov AV. SynTarget: an online tool to test the synergetic effect of genes on survival outcome in cancer. Cell Death Differ 2016;23:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Gong L, Ou R, Zheng Z, Chen J, Xie F, et al. Sequential combination therapy of ovarian cancer with cisplatin and gamma-secretase inhibitor MK-0752. Gynecol Oncol 2016;140:537–44. [DOI] [PubMed] [Google Scholar]
- 20.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 21.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007;23:257–8. [DOI] [PubMed] [Google Scholar]
- 26.Zhao S, Guo Y, Shyr Y. KEGGprofile: An annotation and visualization package for multi-types and multi-groups expression data in KEGG pathway. R package version 1.30.0. . 2020. [Google Scholar]
- 27.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25:1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marghoob AA, Koenig K, Bittencourt FV, Kopf AW, Bart RS. Breslow thickness and clark level in melanoma: support for including level in pathology reports and in American Joint Committee on Cancer Staging. Cancer 2000;88:589–95. [PubMed] [Google Scholar]
- 29.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 2003;194:237–55. [DOI] [PubMed] [Google Scholar]
- 30.Bedogni B Notch signaling in melanoma: interacting pathways and stromal influences that enhance Notch targeting. Pigment Cell Melanoma Res 2014;27:162–8. [DOI] [PubMed] [Google Scholar]
- 31.Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 2012;30:2307–13. [DOI] [PubMed] [Google Scholar]
- 32.Regan-Fendt KE, Xu J, DiVincenzo M, Duggan MC, Shakya R, Na R, et al. Synergy from gene expression and network mining (SynGeNet) method predicts synergistic drug combinations for diverse melanoma genomic subtypes. NPJ Syst Biol Appl 2019;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foidart P, Yip C, Radermacher J, Blacher S, Lienard M, Montero-Ruiz L, et al. Expression of MT4-MMP, EGFR, and RB in Triple-Negative Breast Cancer Strongly Sensitizes Tumors to Erlotinib and Palbociclib Combination Therapy. Clin Cancer Res 2019;25:1838–50. [DOI] [PubMed] [Google Scholar]
- 34.Silva A, Lee BY, Clemens DL, Kee T, Ding X, Ho CM, et al. Output-driven feedback system control platform optimizes combinatorial therapy of tuberculosis using a macrophage cell culture model. Proc Natl Acad Sci U S A 2016;113:E2172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tian Y, Zhang X, Hao Y, Fang Z, He Y. Potential roles of abnormally expressed long noncoding RNA UCA1 and Malat-1 in metastasis of melanoma. Melanoma Res 2014;24:335–41. [DOI] [PubMed] [Google Scholar]
- 36.Lan H, Jin K, Gan M, Wen S, Bi T, Zhou S, et al. APOBEC3G expression is correlated with poor prognosis in colon carcinoma patients with hepatic metastasis. Int J Clin Exp Med 2014;7:665–72. [PMC free article] [PubMed] [Google Scholar]
- 37.Mahmood N, Mihalcioiu C, Rabbani SA. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front Oncol 2018;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Revach OY, Sandler O, Samuels Y, Geiger B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res 2019;79:2634–48. [DOI] [PubMed] [Google Scholar]
- 39.Wenzina J, Holzner S, Puujalka E, Cheng PF, Forsthuber A, Neumuller K, et al. Inhibition of p38/MK2 Signaling Prevents Vascular Invasion of Melanoma. J Invest Dermatol 2020;140:878–90 e5. [DOI] [PubMed] [Google Scholar]
- 40.Zuo Q, Liu J, Huang L, Qin Y, Hawley T, Seo C, et al. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018;37:3275–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell 2015;161:1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amaral T, Sinnberg T, Meier F, Krepler C, Levesque M, Niessner H, et al. MAPK pathway in melanoma part II-secondary and adaptive resistance mechanisms to BRAF inhibition. Eur J Cancer 2017;73:93–101. [DOI] [PubMed] [Google Scholar]
- 43.Wu J, Ivanov AI, Fisher PB, Fu Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, et al. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res 2006;66:4182–90. [DOI] [PubMed] [Google Scholar]
- 45.The Gene Ontology C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 2019;47:D330–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rada M, Barlev N, Macip S. BTK: a two-faced effector in cancer and tumour suppression. Cell Death Dis 2018;9:1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Yuan L, Wang Y, Wang G, Zhu Y, Cao R, et al. Co-expression network analysis identified FCER1G in association with progression and prognosis in human clear cell renal cell carcinoma. Int J Biol Sci 2017;13:1361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu L, Cheng Z, Dong F, Quan L, Cui L, Liu Y, et al. Enhanced expression of FCER1G predicts positive prognosis in multiple myeloma. J Cancer 2020;11:1182–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li M, Liu Z, Wang X. Exploration of the Combination of PLK1 Inhibition with Immunotherapy in Cancer Treatment. J Oncol 2018;2018:3979527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu H, Zmina PM, Huang AY, Askew D, Bedogni B. Inhibiting Notch1 enhances immunotherapy efficacy in melanoma by preventing Notch1 dependent immune suppressive properties. Cancer Lett 2018;434:144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.