

Abstract
Several antibody-drug conjugates (ADCs) showing strong clinical responses in solid tumors target high expression antigens (HER2, TROP2, Nectin-4, and folate receptor alpha/FRα). Highly expressed tumor antigens often have significant low-level expression in normal tissues, resulting in the potential for target-mediated drug disposition (TMDD) and increased clearance. However, ADCs often do not cross-react with normal tissue in animal models used to test efficacy (typically mice), and the impact of ADC binding to normal tissue antigens on tumor response remains unclear. An antibody that cross-reacts with human and murine FRα was generated and tested in an animal model where the antibody/ADC bind both human tumor FRα and mouse FRα in normal tissue. Previous work has demonstrated that a ‘carrier’ dose of unconjugated antibody can improve the tumor penetration of ADCs with high expression target-antigens. A carrier dose was employed to study the impact on cross-reactive ADC clearance, distribution, and efficacy. Co-administration of unconjugated anti-FRα antibody with the ADC improved efficacy, even in low expression models where co-administration normally lowers efficacy. By reducing target-antigen mediated clearance in normal tissue, the co-administered antibody increased systemic exposure, improved tumor tissue penetration, reduced target-antigen mediated uptake in normal tissue, and increased ADC efficacy. However, payload potency and tumor antigen saturation are also critical to efficacy, as shown with reduced efficacy using too high of a carrier dose. The judicious use of higher antibody doses, either through lower DAR or carrier doses, can improve the therapeutic window by increasing efficacy while lowering target-mediated toxicity in normal tissue.
Keywords: target mediated drug-disposition, carrier dosing, cross-reactive antibody, ADC efficacy
Introduction
Antibody drug conjugates have the potential to combine the high tumor selectivity of antibodies with the extraordinary potency of small molecule drugs(1). With the recent approval of enfortumab vedotin (anti-nectin-4), trastuzumab deruxtecan (anti-HER2), and sacituzumab govitecan (anti-Trop-2) for solid tumors, there are currently nine FDA-approved ADCs, several more in late stage clinical trials, and a large number in early clinical testing and preclinical development. Despite their promise, this class of anti-cancer agents has been hampered by a relatively narrow therapeutic index(2). Many approaches have been taken to increase this window, but several of these have resulted in shifts in the therapeutic window (i.e. increased efficacy with increased toxicity) rather than a true improvement/expansion of the window. For example, higher potency payloads can lower the efficacious dose, but they often cause a greater degree of toxicity resulting in a lower maximum tolerated dose (MTD). Likewise, the selection of ‘cleaner’ targets with less normal-tissue expression can shift the dose-limiting toxicity from a target-mediated effect to non-target mediated toxicity(3) without a major improvement in the MTD. In fact, nearly all ADCs have a toxicity profile determined by the payload at the MTD in first-in-human studies(4,5). A key challenge is delivering this maximum dose of payload in a manner that reaches and kills as many cancer cells as possible while avoiding concentrations that are toxic in healthy tissues. More stable linkers and/or site-specific conjugation can often reduce non-target-antigen mediated payload release and associated hematological toxicity (e.g. from deconjugation). However, the longer circulation of intact ADC can potentially increase toxicity from non-target-antigen mediated internalization (e.g. macropinocytosis(6,7)), resulting in side effects, such as ocular toxicity(8,9)) and/or other toxicities from a target-antigen mediated mechanism(10). Higher potency payloads can increase in vitro cell killing, but this can be offset by increased toxicity necessitating lower doses. Newer approaches to expand the therapeutic window are greatly needed, particularly those that can increase cancer cell delivery/killing and reduce normal tissue uptake in the dose-limiting organ at a fixed payload dose.
Recent ADCs with clinical responsiveness have targeted antigens with significant normal tissue expression. Unfortunately, most antibodies have been developed in murine systems, and thus do not cross-react with mouse antigen. While the role of cross reactivity and expression can be tested in cynomolgus monkeys for toxicity, the effect of normal tissue expression on efficacy cannot be gauged in these systems. To expand the therapeutic window and achieve clinical success, development of ADCs must account for the impact of normal tissue expression on both toxicity and efficacy, particularly for the type of high expression targets that are showing significant clinical relevance.
To examine the role of normal tissue expression, a cross-reactive antibody was generated in rabbit that binds both mouse folate receptor alpha (FRα) in normal tissues and the human FRα in tumor xenografts. The lead rabbit monoclonal antibody was chimerized into an IgG2a murine backbone and designated rmFR1–12. In this work, we look at the impact of a ‘carrier dose’, the co-administration of unconjugated rmFR1–12 with the rmFR1–12-ADC, on systemic clearance, local antibody distribution in the tumor, uptake of the ADC by normal tissues, and efficacy using this cross-reactive antibody. The effects of a carrier dose also depend on factors such as target expression and payload potency, so both high (4 million FRα/cell) and low (40,000 FRα/cell) antigen expression models were used along with a high potency DM4 payload (microtubule inhibitor) and very high potency DGN549 payload (DNA alkylator). Overall, cross-reactive ADC shows decreased efficacy due to increased plasma clearance as compared to a non-cross-reactive ADC, which can be improved by a carrier dose of unconjugated antibody by blocking target-mediated uptake in normal tissue.
Materials and Methods
Krogh Cylinder Model
Computational simulations to mechanistically predict intratumoral distribution of the ADC with and without a carrier dose was performed using a previously published Krogh Cylinder Model(11). Differential equations and ADC kinetic parameters have been detailed in Supplementary Information.
Antibody Generation
Rabbits were immunized with recombinant 300–19/Folr1 cells expressing murine FRα. Hybridomas were screened by flow cytometry with mouse and human FRα-positive and -negative cell lines for production of anti-FRα antibodies. The resulting lead antibody was chimerized to a mouse IgG2a (muIgG2a) backbone and designated rmFR1–12. Anti-human FRα ADCs, M-SPDB-DM4 and M-s-SPDB-DM4 have been characterized previously(12). DGN549 ADC of anti-human FRα antibody M9346A was prepared by the method previously described(13).
Cell Lines
M109 (murine lung cancer) cells were cultured from ImmunoGen’s cell bank for binding affinity studies without further characterization. KB (ATCC CCL-17 – containing HeLa chromosomal markers, human carcinoma/papilloma cell line)(14–16) and OV90(17) human ovarian cancer cells were obtained from ATCC. All lines were characterized by the vendor using routine DNA profiling; no further authentication was performed by the authors. M109 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), KB cells in Eagle’s Minimum Essential medium with 10%, and OV-90 cells in a 1 :1 mixture of MCDB-105 and Medium-199 with 15%. Cells were expanded and maintained as recommended in a humidified 37 °C, 5% CO2 incubator.
Binding Assay
Binding of rmFR1–12 and its DM4 and DGN549 conjugates to M109 and KB cells was evaluated flow-cytometrically by indirect immunofluorescence. Briefly, 2 × 104 cells per well in a 96-well plate were incubated for 2 hours at 4°C with an anti-FRα antibody diluted to various concentrations in assay medium [RPMI-1640 supplemented with 2% (w/v) bovine serum albumin (Sigma)]. The cells were then washed with cold assay medium, stained with fluorescein isothiocyanate–labeled goat anti-murine or anti-human immunoglobulin G (IgG) antibody for 40 minutes at 4°C in the dark, washed with cold assay medium, fixed in a solution of phosphate-buffered saline, pH 7.4 (PBS) containing 1% formaldehyde, and analyzed using a FACSCalibur flow cytometer (BD Bioscience). Data was analyzed via GraphPad Prism sigmoidal dose-response (variable slope) function. The concentration of the antibody achieving half-maximal binding was taken as its apparent affinity (Kd).
Cytotoxicity Assay
Dilutions of conjugates in the appropriate culture medium were added to wells of 96-well flat-bottomed plates containing 1 × 103 cells per well. The plates were incubated at 37°C, 5% CO2 for 5 days. Cell viability was determined by the WST-8 assay (Donjindo Molecular Technologies, Inc.) in accordance with the manufacturer’s protocol, and IC50 values were generated using a sigmoidal dose–response (variable slope) nonlinear regression curve fit (GraphPad Software Inc.).
In vivo Studies
All procedures were carried out in accordance with the “Guide for the Care and Use of Laboratory Animals” (National Research Council). Eight to twelve-week-old female CB-17 severe combined immunodeficient (SCID) mice were obtained from Charles River Laboratories and quarantined for 7 days prior to study initiation. Mice were inoculated subcutaneously with cells (1 × 107 cells per mouse) resuspended in serum-free culture media (KB cells) or 50% Matrigel in serum-free culture media (OV90 cells). When tumor volume reached approximately 100 mm3, mice were randomized (N = 6 per group) and received a single intravenous bolus injection of PBS (control), DM4 conjugate or DGN549 conjugate at the indicated doses (based on antibody concentration). For biodistribution studies, radiolabeled DM4 conjugate was administered at the indicated doses. Unconjugated mFR1–12 was used in co-administration studies at the indicated doses. Mice received a single intravenous bolus injection. Tumor dimensions were measured two or three times per week and volume was calculated as ½ (L × W × H), where L is the length, W is the width, and H is the height of the tumor. A partial tumor regression was defined as a reduction in tumor volume by 50% or greater. A complete tumor regression was scored when no palpable tumor could be detected. Termination criteria was set at tumor volumes exceeding 1500mm3 in volume, > 20% body weight loss (BWL), or clinical observations of the mice being in distress. Statistical differences in tumors sizes was assessed using Prism following the nadir during regrowth. Antigen density in vitro was assessed via flow cytometry with indirect fluorescence using a FITC-labeled secondary antibody with phycoerythrin-labeled anti-FRα antibody using QuantiBRITE PE kit (Becton Dickinson), and in KB and OV-90 tumor models was assessed by ex-vivo immunohistochemical staining of formalin-fixed paraffin-embedded tumor tissues.
DM4 ADCs Cellular Processing
DM4 catabolites within normal tissue and FRα-positive cancer cells were quantified using tritiated DM4 ADC (18), wherein [3H] was incorporated in the methoxy group at the C20 position of the payload.. Briefly, protein-free radioactivity (processed ADC) and protein-associated radioactivity (unprocessed ADC) were separated by acetone extraction, and the associated radioactivity measured via liquid scintillation counting, and the data were used to calculate %ID/g, % processed ADC, and pmol processed ADC.
Immunofluorescence Studies
Mice were inoculated subcutaneously with 1 × 107 KB cells per mouse resuspended in serum-free culture media. Tumors grew to ~200mm3 before the mice were injected intravenously with saline or 2.5 mg/kg Alexa488-labeled rmFR1–12 antibody alone or concurrently with either 10 or 25 mg/kg unlabeled rmFR1–12 antibody. Twenty-four and 48 hours after treatment, mice were injected with Tomato Lectin DyLight594 (Vector Laboratories, CA) for visualization of active blood vessels. After 5 minutes, mice were euthanized, tumors were collected and fixed in 10% neutral buffered formalin for paraffin-embedding.
For staining, all chemicals were procured from Sigma Aldrich (St. Louis, MO), unless noted otherwise. Formalin-fixed paraffin embedded (FFPE) tumors were cut for histology into 5μm sections using a microstat. Tumor sections were deparaffinized and antigen-retrieved prior to imaging (Supplementary Methods). Immunofluorescence studies were performed by blocking tumor sections with 0.5–2% BSA for 15 minutes, following by staining with 6μg/mL AlexaFluor647-labeled chicken anti-mouse antibody (Thermo Fisher) and 15μg/mL AlexaFluor555-labeled rat anti-mouse CD31 antibody in 0.5% BSA for 40 min at room temperature in the dark. Slides were washed for 5 minutes in PBS, followed by staining with Hoechst 33342 for 1min, then washed again for 3 min in PBS. Stained slides were imaged using a 20x objective on an upright Olympus FV1200 confocal microscope in the 405 (Hoechst 33342), 488 (ADC), 542 (CD31), and 635 (chicken anti-mouse antibody) lasers. Images were analyzed using ImageJ.
Results
Matching Cellular Delivery with Payload Potency Results in Highest Efficacy
To first demonstrate the impact of receptor expression and payload potency on cell killing in vitro, a non-cross-reactive antibody to human FRα, the M antibody (M9346A), and cross-reactive antibody binding to both human and mouse antigen were conjugated to DM4 (“low” potency) or DGN549 (“high” potency) payloads and tested in a ‘high’ expression and ‘low’ FRα expression model (14,19)(Fig. S1). Quotation marks emphasize that the potency is context dependent, denoting a relative comparison in this case. Likewise, expression levels require absolute quantification since target receptors per cell vary over many orders of magnitude (e.g. 105 receptors/cell is considered low for HER2 but high for HER3). KB cells express ~4 million folate receptors per cell in vitro and form xenografts with “high” homogeneous target expression, while OV90 cells express ~40,000 folate receptors per cell with “low” heterogeneous membrane expression. Low and high levels in this case are relative to the range of FRα expression observed in various xenograft models and ovarian carcinoma patients.
The antibody M9436A (M antibody) does not bind mouse FRα and only targets the xenograft human cancer cells. In the high expression KB model, the more potent DGN549 payload, which must be administered at low antibody doses (0.29 mg/kg M-DGN549) due to the high potency/low tolerability, is likely not able to penetrate the tumor tissue far, reaching few cells (Fig 1A), and resulting in modest efficacy (Fig. 1B). In contrast, cures were achieved in all animals treated with ~2.5 mg/kg of M-s-SPDB-DM4, since the higher dose can penetrate deeper in the tissue while still delivering lethal amount of payload to cells. In low expression models, ADC tumor penetration (regardless of payload used) is often fairly efficient. However, low receptor expression causes fewer total ADC molecules to internalize into tumor cells, limiting cellular payload accumulation. Treatment of OV90 with M-s-SPDB-DM4 likely results in subtherapeutic concentrations of the “low potency” DM4 that are insufficient to kill tumor cells. However, treatment with the high potency M-DGN549 ADC could result in complete regression of tumors from delivery of lethal concentration of DGN549. (Fig. 1C). In the low expression OV90 model, the ADC with the lower potency DM4 payload only achieved modest efficacy given at the same dose that achieved complete regression in the high expression KB tumors. The ADC with the more potent DGN549 showed limited activity at the 0.29 mg/kg dose against the high expressing xenograft, while a two-fold lower dose led to complete responses in the low expressing OV90 model (Fig. 1B, 1D).
Fig. 1. The tissue and cellular potency of an ADC payload must be matched to delivery.
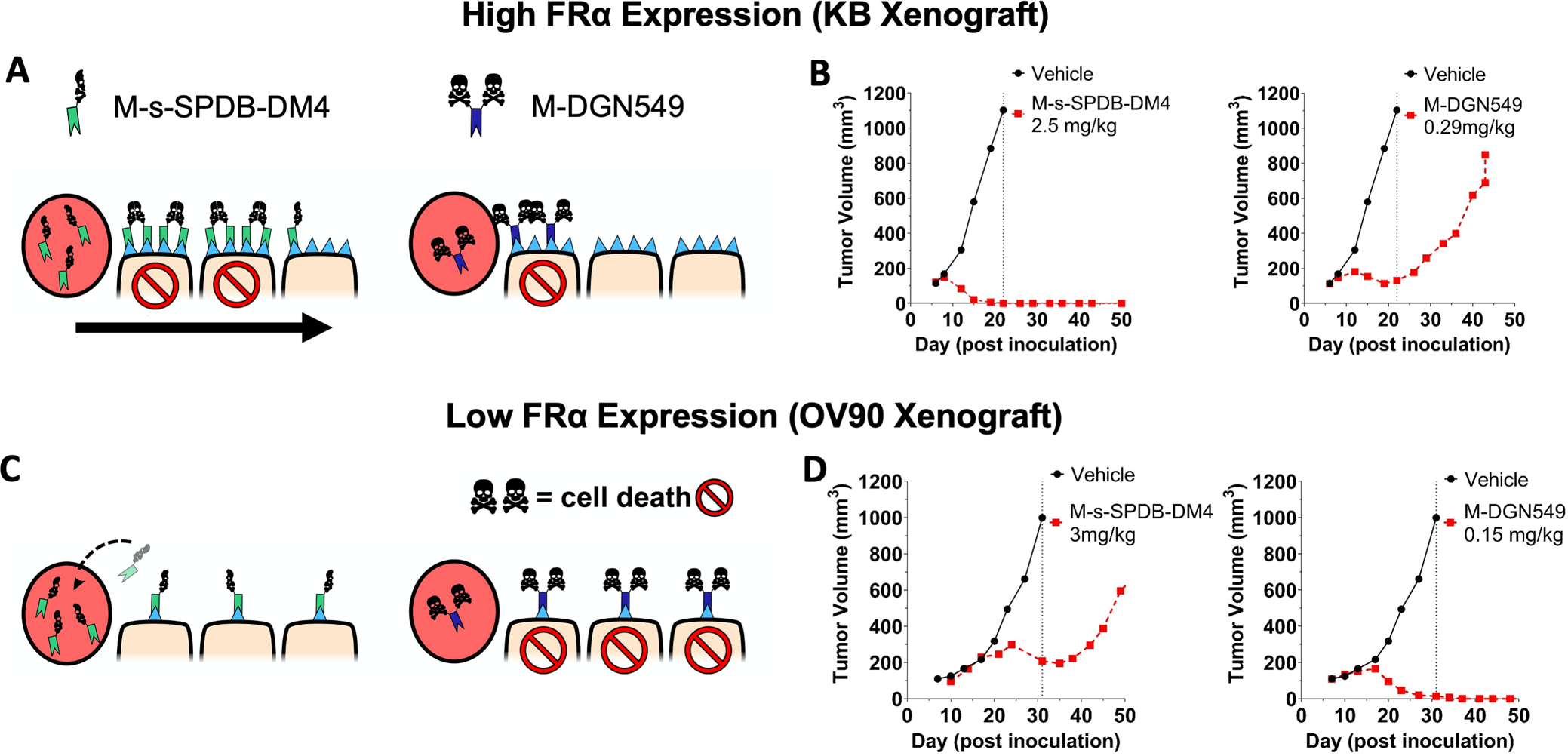
The schematic of half versus two skull and crossbones represents payload potency, not DAR. The high dose of potent DM4 payload in a high FRα expression (KB) tumor model can target more cells with lethal payload concentration (A, left) compared to a lower dose of highly potent DGN549 ADC (A, right). This causes complete tumor regression at clinically tolerable doses of DM4 (B, left) but only a partial response with DGN549, which is tolerated at lower total antibody doses (B, right). This is due to the lower tissue penetration from a reduced antibody dose In a low expression xenograft model (OV90), the FRα-targeting DM4 ADC saturates the tumor (C, left) but can only deliver enough DM4 payload per cell to invoke a partial response (D, left). At super-saturating antibody doses, a significant amount of the ADC washes out of the tumor without being taken up by cells. In contrast, the highly potent DGN549-ADC can penetrate deeper into the tumor tissue while maintaining a lethal dose to the targeted cells (C, right), there by efficiently kill cells with low expression at clinically tolerable doses (D, right). Animals dosed 7 days post inoculation; ADC doses were based on the payload dose (μg payload per kg body weight) but are reported as mg ADC per kg body weight.
A Carrier Dose Can Increase or Decrease Efficacy Depending on Target Expression
Unconjugated antibody that is co-administered with the ADC will block some of the perivascular binding sites, allowing deeper penetration into the tissue (at the cost of fewer ADC molecules delivered per cell). As the antibody dose is increased in the high expression KB model (Fig. 1B), the ADC is “diluted” and spreads out deeper into the tumor tissue. Based on the poor tissue penetration of M-DGN549 in the high expression model, a carrier dose is expected to improve tissue penetration and increase efficacy. According to predictive simulations(11), the addition of 2.5 mg/kg of unconjugated antibody to 0.29 mg/kg of M-DGN549 results in similar overall payload uptake in tumor as 0.29 mg/kg of M-DGN549-ADC but distributed more homogeneously throughout the tumor (Fig. 2B). This translates to improved efficacy in vivo as expected (Fig. 2C). A super-saturating dose of 10 mg/kg unconjugated antibody, however, reduces total payload uptake in the tumor and therefore efficacy. Once the tumor is saturated with antibody, competition of any additional unconjugated antibody with the ADC is antagonizing and can reduce efficacy. This phenomenon is apparent in the low expression model OV90 (Fig. 2D), where 0.29mg/kg M-DGN549 is predicted to show efficient tissue penetration, consistent with previous observations(17), and high payload uptake (Fig. 2E), thereby demonstrating maximum efficacy (Fig. 2F). Both the 2.5 and 10 mg/kg carrier doses super-saturate the tumor and antagonize payload uptake and efficacy. Conceptually, efficacy is driven by the cellular payload delivery versus payload potency; the carrier dose changes the cellular payload delivery, which can be beneficial or detrimental depending on how well-matched the original ADC dose is to receptor expression and payload potency.
Fig. 2. A carrier dose of unconjugated antibody can improve tissue penetration and impact efficacy.
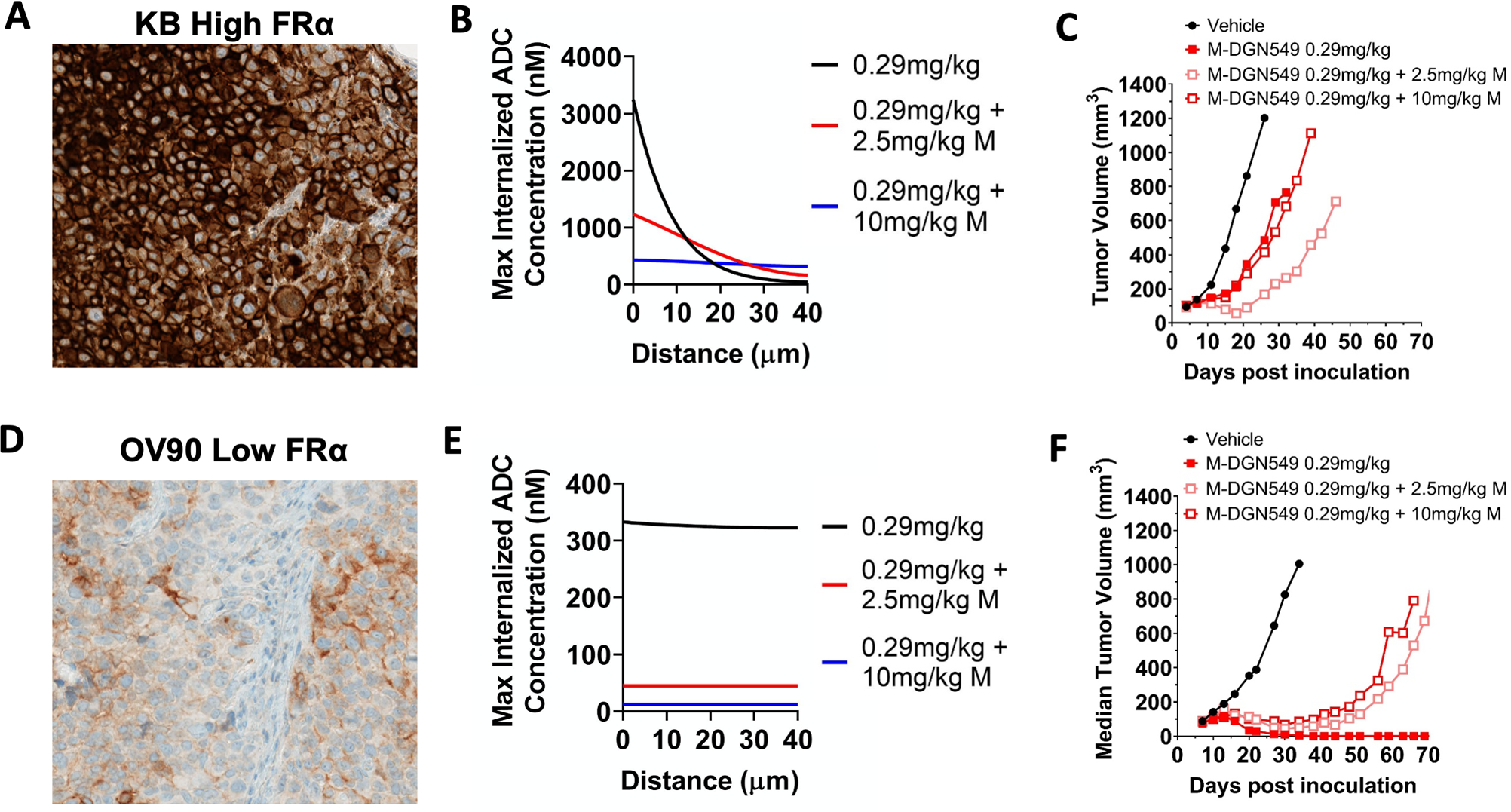
In a high expression model (A), adding 10 mg/kg of unconjugated antibody to a 0.29 mg/kg dose of ADC saturates the tumor and makes tumor penetration more homogeneous (B) but at the cost of decreased total ADC uptake (half the payload AUC as 0.29mg/kg). However, a 2.5 mg/kg dose of unconjugated antibody improves tissue penetration of the ADC without affecting total uptake of the DGN549 ADC (payload AUC similar to 0.29mg/kg). This trend correlates to in vivo efficacy, with 2.5mg/kg carrier dose improving tumor growth inhibition, but at a higher 10 mg/kg carrier dose, the ADC is ‘diluted’ too much, resulting in lower efficacy (C). This can occur by reduced ADC uptake from tumor saturation and/or reducing payload delivery per cell below a toxic threshold. In a low expression OV90 model (D), the payload potency is already matched to the delivery with no carrier dose (E), and co-administration of 2.5 or 10 mg/kg of unconjugated antibody uniformly lowers efficacy (F).
Cross-Reactivity of Antibody/ADC Increases Plasma Clearance and Reduces Efficacy
To test the impact of normal tissue expression and binding, a rabbit anti-mouse FRα antibody was generated. The variable domain of the rabbit antibody was then grafted onto a murine framework to generate a chimera, denoted rmFR1–12 (Fig. S2). Fortuitously, the chimera binds human and mouse folate receptor with similar affinity and does not compete with folate at even supraphysiological concentrations (Fig. S3). When injected into CD-1 mice, the plasma clearance of rmFR1–12-s-SPDB-DM4 significantly increased resulting in a 3-fold reduction in AUC (Fig. 3A) presumably due to significant binding in normal tissue leading to target mediated drug disposition(20). To determine the impact of faster clearance on tumor uptake, the DM4 payload component was radiolabeled, and SCID mice with KB xenografts were administered 5 mg/kg of rmFR1–12-s-SPDB-DM4 or M-s-SPDB-DM4. Tumor uptake was significantly lower for the cross-reactive rmFR1–12-s-SPDB-DM4 compared to the non-cross-reactive M-s-SPDB-DM4, indicating the faster plasma clearance reduced tumor uptake (Fig. 3B). Next, tumor growth was measured to see if the increased plasma clearance and lower tumor uptake had an impact on efficacy. At a 2.5 mg/kg dose, which resulted in complete cures for the non-cross-reactive M-s-SPDB-DM4 (Fig. 3C), efficacy of rmFR1–12-s-SPDB-DM4 was significantly reduced, presumably due to binding to normal tissues that increased clearance and lowered tumor uptake (Fig. 3D).
Fig. 3. Impact of target expression in normal tissue – target mediated drug disposition (TMDD).
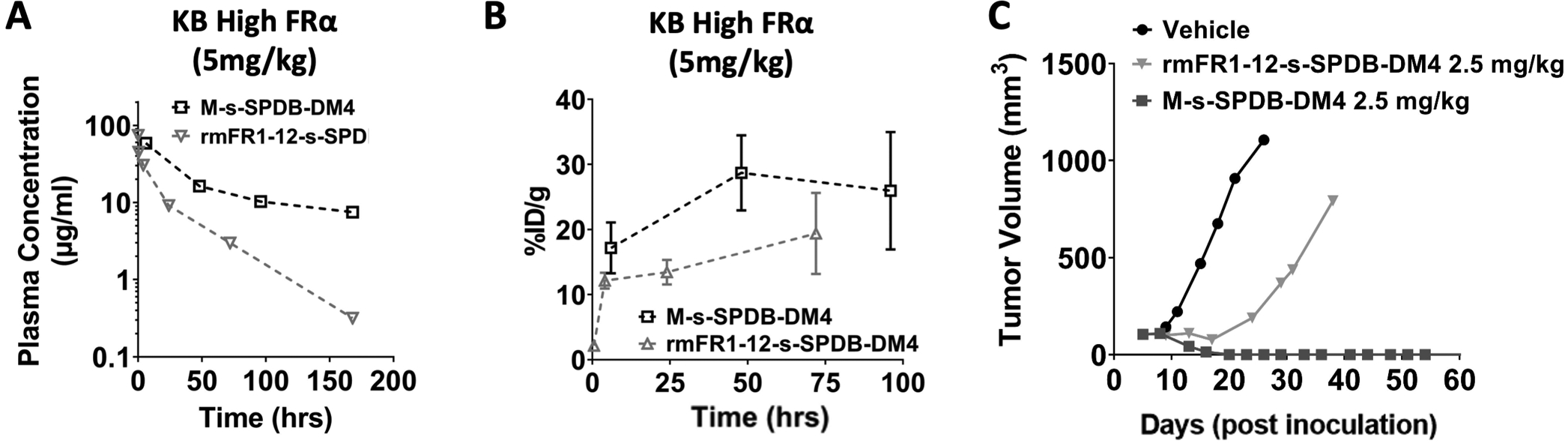
Using a cross-reactive antibody that binds both human (in the tumor xenograft) and mouse (endogenous FR in multiple tissues) receptor, the ADC at the same dose showed significantly increased plasma clearance (A) because of binding FR in normal tissues. The faster clearance reduced tumor exposure, resulting in lower payload levels within the tumor (B). While a 2.5 mg/kg dose of the non-cross-reactive DM4-ADC (M-s-SPDB-DM4) results in complete responses in a high expression KB xenograft mouse system(14), the faster clearance and lower tumor uptake of the cross-reactive DM4-ADC (rmFR1–12-s-SPDB-DM4) contributes to the decreased efficacy (C). Note M-s-SPDB-DM4 data is replotted from Fig. 1 for comparison purposes. Between 6 and 10 mg/kg doses are needed for complete responses with the cross-reactive ADC (Fig. S4 and Fig. S5).
A Carrier Dose Can Improve Tumor Exposure and Efficacy in Low and High Expression Models
Because FRα is expressed in multiple tissues, even low-level expression can result in significant target mediated uptake and clearance due to the large normal tissue ‘sink.’ These effects are typically dose-dependent, and an increase in antibody dose can saturate the target-mediated clearance, resulting in linear kinetics. Although ADCs are usually administered at doses that are limited by payload toxicity, unconjugated antibodies are often well-tolerated even at very high doses. Increasing doses of the cross-reactive rmFR1–12 antibody resulted in a higher Cmax and slower clearance as expected (Fig. 4A), and increasing doses of the rmFR1–12 ADC showed both dose-dependent pharmacokinetics and efficacy response (Fig. S4). By combining carrier doses with a constant 2.5 mg/kg dose of ADC, the Cmax of the ADC does not change. However, the unconjugated antibody blocks normal tissue receptors, slowing clearance of the ADC and increasing tumor exposure (Fig. 4B).
Fig. 4. Higher antibody doses can overcome target-mediated drug disposition (TMDD).
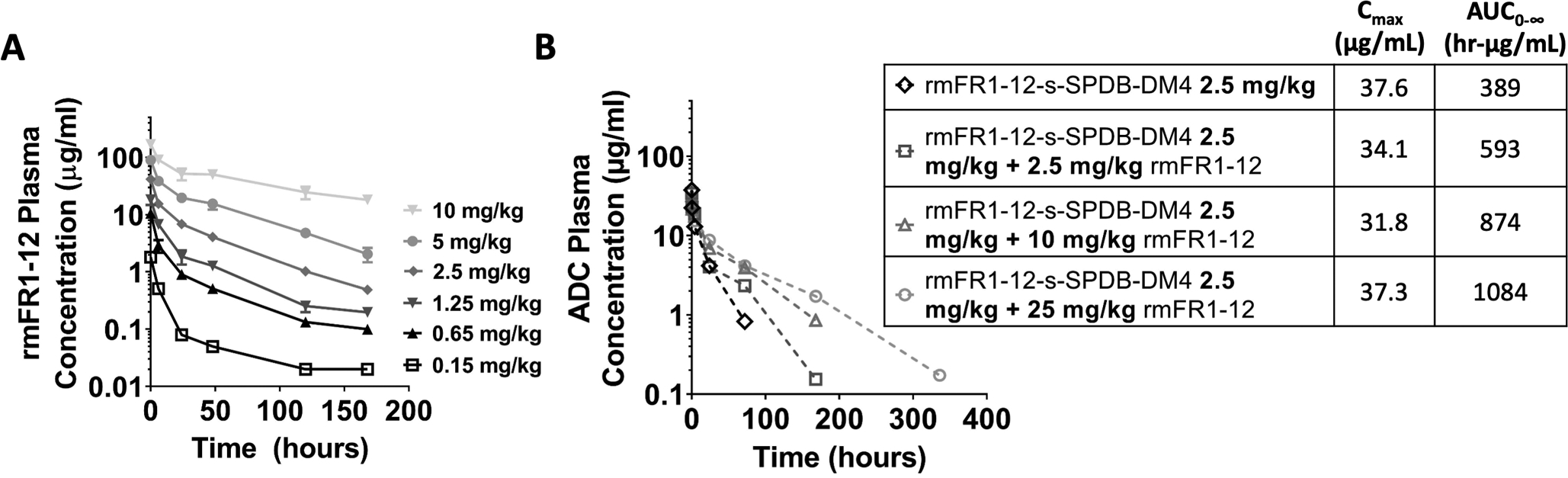
TMDD gives rise to nonlinear plasma clearance but can be overcome by higher antibody doses that saturate the normal tissue (mice bearing no tumors) (A). The higher antibody doses reduce TMDD of the ADC. Co-administration of increasing doses of unconjugated antibody (2.5 to 25 mg/kg) with a fixed 2.5 mg/kg dose of ADC results in the same Cmax of the ADC but reduced plasma clearance (mice bearing KB tumors) (B). Therefore, the tumor exposure of the ADC is increased by the carrier dose.
To determine if this increased exposure had an impact on efficacy, increasing doses of the unconjugated rmFR1–12 antibody were co-administered with rmFR1–12-DGN549 and tumor growth was monitored. Notably, the unconjugated antibody has no detectable efficacy in these tumor models by itself (Fig. S4). The addition of 5 and 10 mg/kg of the cross-reactive rmFR1–12 antibody to 0.59 mg/kg of the DGN549-ADC resulted in a significant increase in efficacy in the high expressing KB model (Fig. 5A). The increased efficacy at 10 mg/kg is notable, because the same 10 mg/kg carrier dose decreased efficacy in the non-cross-reactive system (Fig. 2B). To image the impact of dose on tissue distribution, 2.5 mg/kg of fluorescently labeled rmFR1–12 was injected intravenously into mice bearing high expression KB tumors with or without 10 mg/kg of non-fluorescent rmFR1–12. At 48 hrs post injection, the 2.5 mg/kg dose was heterogeneously distributed while the addition of 10 mg/kg carrier dose resulted in efficient tissue penetration and homogeneous targeting (Fig. 5B). Interestingly, the carrier doses improved efficacy in the low expression OV90 model at all doses tested (up to 10 mg/kg, Fig. 5C). This is in direct contrast to the non-cross-reactive ADC where any carrier dose decreased efficacy (Fig. 2C). Therefore, the impact of a carrier dose on TMDD and increasing tumor uptake can be equally important as tissue penetration and cellular delivery effects.
Fig. 5. Carrier dose increases the efficacy in both high and low expression models when using a cross-reactive antibody that exhibits TMDD.
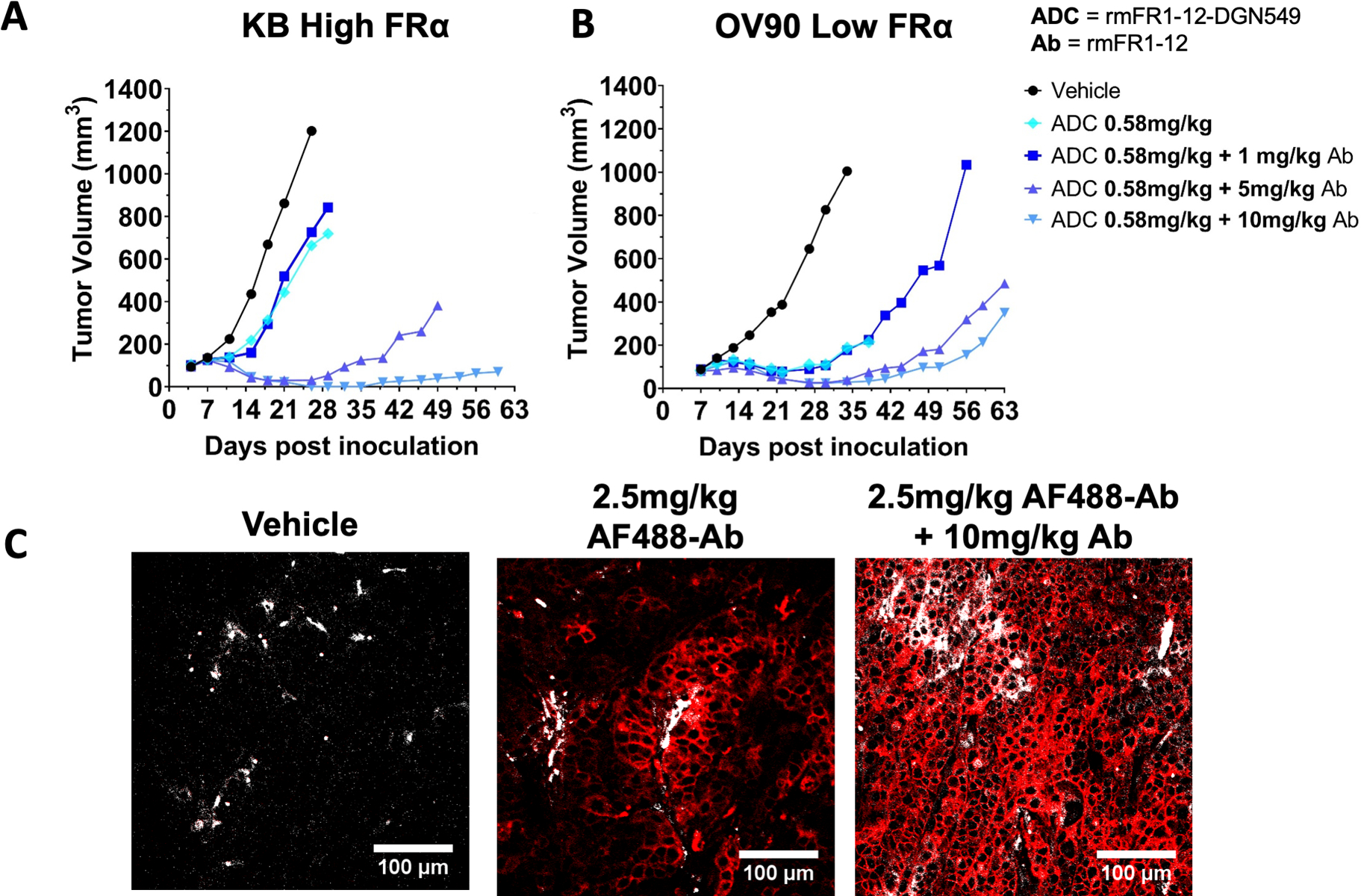
The increased clearance due to binding FR in normal tissue lowered the efficacy of the DGN549-ADC (Fig. 3D) but adding a carrier dose to this animal model significantly improves efficacy (A). The 10 mg/kg carrier dose, which lowered efficacy in the non-cross-reactive antibody (Fig. 2B), has the highest response. Immunofluorescence histology confirms that low doses of AF488-rmFR1–12 (~2.5mg/kg) are sub-saturating in the high expression system (KB), as seen by the heterogeneous distribution of the fluorescent ADC, while adding a 10 mg/kg unlabeled rmFR1–12 carrier dose (~12.5mg/kg) is close to saturating, as seen by the homogeneous fluorescence signal (B). red – AF488 antibody, gray – blood vessels. Even in the low expression tumor model (OV90), where a carrier dose uniformly decreased efficacy in the non-crossreactive ADC (Fig. 2C), a carrier dose up to 10 mg/kg improves efficacy (C).
Optimal Carrier Dose Depends on Both Systemic (Plasma) and Local (Tumor) Effects
Administration of carrier antibody improved the efficacy of the high potency rmFR1–12-DGN549 in high and low expressing xenograft models. Next, we tested whether a carrier dose also had an impact on the activity of the lower potency rmFR1–12-s-SPDB-DM4 that can be administered at higher doses (e.g. 2.5 mg/kg of ADC). The cross-reactive rmFR1–12-s-SPDB-DM4 at 2.5 mg/kg had reduced efficacy in the high expression KB model (Fig. 3D) and resulted in heterogeneous distribution (Fig. 5B). The addition of 2.5 mg/kg of rmFR1–12 antibody to 2.5 mg/kg of ADC improved efficacy, but a higher carrier antibody dose (10 mg/kg) resulted in the greatest efficacy with 4/6 complete responses (Fig. 6A).
Fig. 6. The optimal carrier dose (or antibody loading) depends on both the local and systemic ADC distribution.
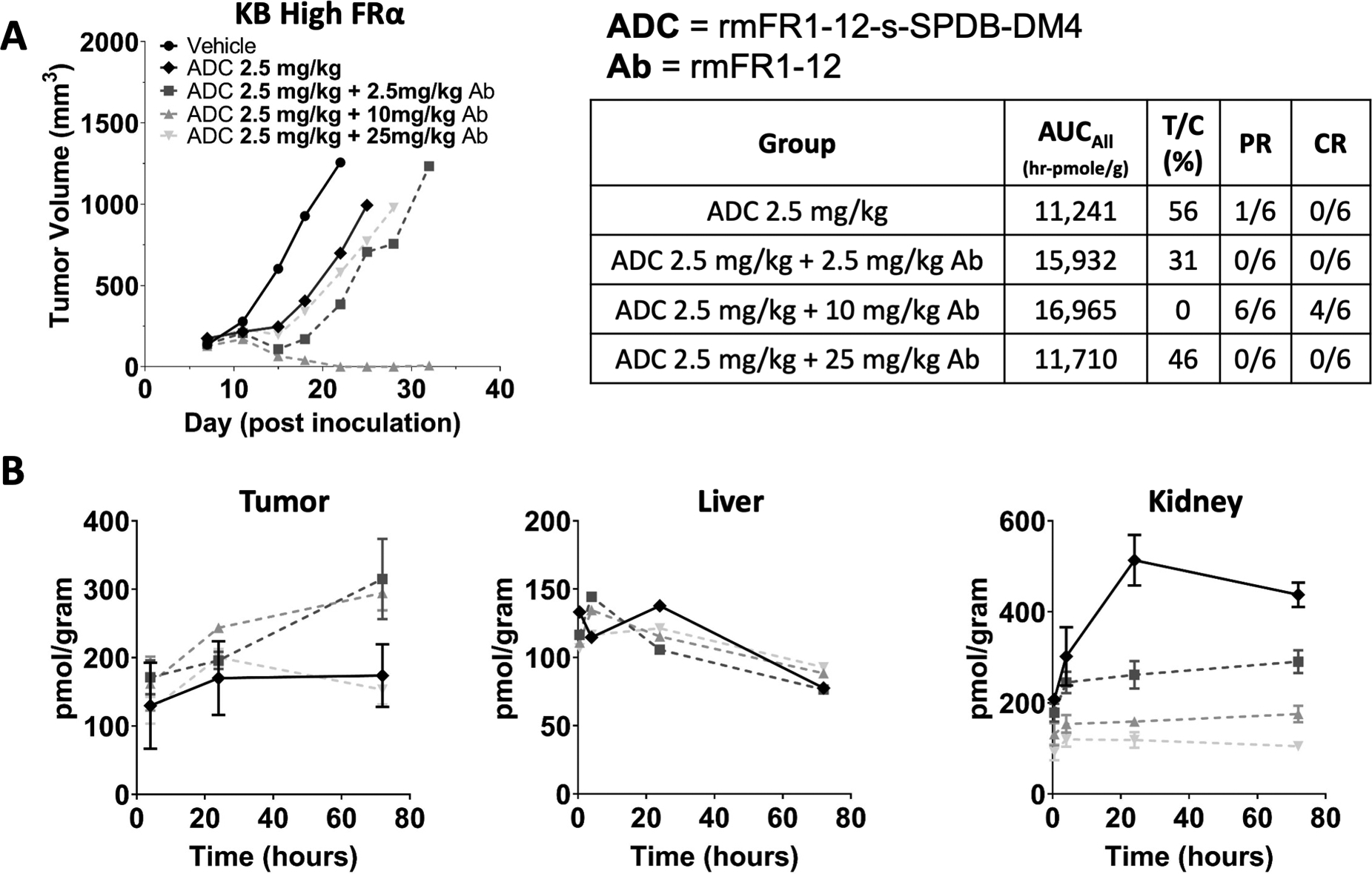
Increasing doses of unconjugated antibody co-administered with the DM4-ADC also improved efficacy (A) as they did with the DGN549 ADC (Fig. 5). However, with a carrier dose of 25 mg/kg of antibody, the efficacy dropped back down to a level seen with the ADC alone. While carrier doses of 2.5 and 10 mg/kg reduced plasma clearance and increased tumor uptake, at 25 mg/kg, the tumor is super-saturated, and tumor uptake is reduced (B). The 2.5 and 10 mg/kg carrier doses resulted in the same total tumor uptake (B), but the higher carrier dose was more effective, likely due to improved tissue penetration. Therefore, both the local (tumor distribution) and systemic (tumor exposure/uptake) effects determine the maximum ADC efficacy. For toxicity, the carrier dose had minimal impact on non-target mediated uptake in the liver (C). However, the carrier dose significantly reduced target-mediated uptake in the kidney (D). PR = partial response, CR = complete response
Despite the improvement noted from the carrier dose for both payloads and in the high and low expression models, it is anticipated that too large of a carrier dose will decrease efficacy by blocking too much cellular uptake (similar to Fig. 2C and F). To test this hypothesis, a 25 mg/kg dose of rmFR1–12 antibody was co-administered with 2.5 mg/kg of rmFR1–12-s-SPDB-DM4. This indeed lowered efficacy close to levels seen without any carrier dose. Tumor uptake of ADC payload measured using radiolabeled DM4 (Fig. 6B) was found to increase with the addition of a 2.5 or 10 mg/kg carrier dose of antibody from slower plasma clearance. However, a 25 mg/kg carrier dose reduced the tumor uptake to less than that of the ADC-only dose (although not significantly different). It is interesting to note that total payload uptake was similar for both the 2.5 and 10 mg/kg carrier dose (Fig. 6B), but the 10 mg/kg carrier dose was much more effective (Fig. 6A). This is consistent with the 10 mg/kg carrier dose improving efficacy by two mechanisms: increasing the total tumor uptake of ADC (relative to no carrier dose) by blocking normal tissue uptake and improving tissue penetration (relative to the 2.5 mg/kg carrier dose, Fig. 5B, Fig. S6).
Potential Impact of Carrier Doses on Toxicity
ADCs often suffer from a narrow therapeutic window due to dose-limiting toxicity, which can be either target-mediated or non-target-mediated depending on the particular target, antibody, and payload among other factors. Even with cross-reactive antibodies, mice are not ideal models for tissue specific uptake given differences in expression levels relative to humans, but the impact of a carrier dose on biodistribution can highlight the expected effect in other species (e.g. cynomolgus monkeys, humans). The biodistribution of rmFR1–12-s-SPDB-DM4 (radiolabeled DM4) was measured as a function of the carrier dose in both liver and kidney. The liver uptake was largely independent of the carrier dose (Fig. 6C), indicating primarily non-target mediated uptake. However, folate receptor is expressed at a high level in the kidney. The carrier dose significantly decreased radiolabeled DM4 uptake in the kidney proportional to the carrier dose, with 25 mg/kg reducing uptake almost 5-fold (Fig. 6D).
Discussion
Many ADCs currently in late stage clinical trials or recently FDA approved, such as enfortumab vedotin (anti-nectin-4), sacituzumab govitecan (anti-Trop-2), trastuzumab deruxtecan (anti-HER2), and mirvetuximab soravtansine (anti-FRα), target tumors expressing high receptor levels(21–24). Target antigens overexpressed on tumor cells are often accompanied by lower but still significant expression in normal tissue. Because antibodies are frequently generated using mouse systems, cross-reactivity between mouse and human targets is rare. This can create a disconnect between estimates of therapeutic doses, typically measured in mouse models, and tolerable doses based on toxicity, typically measured in non-human primates. The impact of normal tissue uptake and increased clearance seen in primate models is not recapitulated in the mouse models used for efficacy, where the ADC only binds to cells in the tumor. However, target-mediated drug disposition (TMDD) and clearance is a widespread and important phenomenon that must be taken into account during drug development(20,25,26). In this work, we generated a cross-reactive murine antibody and ADC to test the impact of antibody dosing, target expression, and payload potency on efficacy in a FRα-targeted system where normal tissue expression is relevant.
One method to overcoming the sink for ADCs caused by expression of receptors in normal tissues is to increase the dose. Once the normal tissue receptors are saturated, the pharmacokinetics become linear and clearance is reduced. Indeed, several ADCs recently approved or in late stage clinical trials are administered at high doses, exceeding that of the first ADC for solid tumors (ado-trastuzumab emtansine). Co-administration of unconjugated antibody can also improve tissue penetration and efficacy as demonstrated by adding trastuzumab to the clinical dose of ado-trastuzumab emtansine(27). However, adding too much unconjugated antibody (at a fixed ADC dose) could block cellular uptake to an extent that results in a decrease in efficacy. The appropriate amount of antibody to maximize efficacy depends on several factors, most prominently the receptor expression (the major driver of cell delivery) and payload potency (the major driver of cell death). To first examine how increased antibody doses impact efficacy without the confounding effects of normal tissue binding, a non-cross-reactive anti-folate antibody was tested with two different payloads in a high and low expression model.
In a ‘clean’ system, where the ADC only binds to human FRα in the xenograft, the highest efficacy was seen when the cellular delivery matches the potency as we and others have reported(2,28,29). DM4-conjugates are typically tolerated at several mg/kg doses in the clinic, while DNA-alkylating conjugates are often tolerated at <0.5 mg/kg. Treatment with the DM4-ADC resulted in complete responses in the high expressing tumor model at the 2.5 mg/kg antibody dose due to high cellular uptake rate (Fig. 1A). In contrast, the lower antibody dose of the DGN549-ADC gave poor responses despite the high potency due to the lower antibody tissue penetration (Fig. 1B). However, in a low expressing model, the opposite was true. The DM4-conjugate was ‘potency-limited’ due to cellular accumulation of DM4 being below the concentration required for cell death, but the high potency DGN549 gave complete responses (Fig. 1C and D).
Comparing the DGN549-ADC treated tumors in the high and low expression system, the efficacy of this ADC actually improves with lower expression despite the ADC being more potent on the high expressing cells in vitro (Fig. S1). This is similar to the result seen in a prostate cancer model with an ADC targeting PSMA(30). Together with the different sensitivities between clinical patient tumors, this is likely a major confounding factor for why a clear correlation between target expression and efficacy has not been found. Since lower expression can result in increased ADC tissue penetration even at low doses, using high potency payloads can lead to higher efficacy by allowing better penetration of lethal concentrations of the payload into the tissue.
To investigate the effects of a carrier antibody dose in a more representative scenario where the antibody and ADC bind receptors expressed in normal tissue, a cross-reactive antibody was developed. A rabbit antibody that binds with comparable affinity to mouse and human folate receptor was grafted onto a murine framework for these studies. When administered at the same dose as the non-cross reactive version, plasma clearance increased resulting in a 3-fold drop in AUC and significantly lower tumor uptake. The efficacy of the ADC was appreciably compromised compared to the ‘clean’ system with no normal tissue binding. By co-administering increasing doses of the antibody with the ADC (both cross-reacting), the plasma concentration of antibody/ADC increases, presumably due to saturation of the normal tissue, resulting in linear kinetics. Although Cmax of the ADC (given at a fixed dose) is not affected, the AUC increased due to lowered target-mediated clearance. Note that these are SCID mice, so the reduced clearance is attributed to blocking folate receptor rather than Fc-receptors(31). However, the conclusions are the same in either case. The improvement in systemic exposure with the addition of unconjugated antibody resulted in increased efficacy for all carrier doses tested with the high potency DGN549-ADC and high expression model. Interestingly, even in the low expression model, where the expectation is that a carrier dose would reduce cellular delivery too much (and lower efficacy)(32), the improvement in tumor uptake was enough to overcome this limitation, resulting in higher efficacy with the carrier dose (Fig. 5). Therefore, even targets with expression as low as 40,000 receptors per cell can benefit from carrier doses depending on the potency of the payload and normal tissue receptor levels.
The impact of a carrier dose depends on a variety of factors, including the dose administered, so adding a carrier dose does not always improve distribution and/or efficacy. This is apparent when increasing the dose after the tumor is already saturated; any increase in a carrier dose will not improve tissue penetration since all accessible antigen within the tumor is already targeted. A more subtle but important effect occurs at lower doses, particularly in high expression models. In order to allow deeper tissue penetration, the carrier dose must block a sufficient amount of antigen on perivascular cells. In essence, the antibody must ‘fill up’ the binding sites on the first cell layer in order to reach the next. Therefore, adding a small carrier dose at the low end of the dose range may not improve penetration, and this may contribute to the lack of improvement with only 1 mg/kg carrier dose in Fig. 5A.
Carrier doses improved the efficacy of the lower potency DM4-ADC in the high expression model when using the cross-reactive antibody. Despite the improvement from the carrier doses, there still exists a limit beyond which increased antibody doses start to lower efficacy. In this system, 2.5 mg/kg of ADC administered with a 10-fold higher antibody dose (25 mg/kg) super-saturated the tumor, lowered uptake, and reduced efficacy to levels seen with no carrier dose. Taken together, minimal efficacy is seen with the ADC alone; addition of the 2.5 mg/kg carrier dose increases the tumor uptake (systemic effect, Fig. 6B) but only marginally increases efficacy; addition of a 10 mg/kg carrier dose provides the same improvement in tumor uptake (systemic effect) but also improves tissue distribution (local effect) to result in a strong response. Note that prior to saturation, no decrease in tumor uptake of the ADC was seen, similar to Kadcyla(28) and consistent with theoretical predictions(33). Beyond this dose (25 mg/kg carrier dose in the cross-reactive system), there is no improvement in tissue penetration, and increased carrier doses simply reduce the tumor and cellular uptake of the payload, lowering efficacy. This is one of several(34,35) considerations necessary when optimizing DAR and conjugation chemistry. The optimal results can differ depending on whether a saturating dose or subsaturating dose is used(36,37).
Mouse models are not ideal for measuring toxicity, and the doses used here did not significantly impact mouse body weight (Fig. S8, S9). However, the biodistribution of the payload in this system provides some potential insight on the impact of carrier doses. The non-target-mediated uptake in the liver is the same at all carrier doses while the target-mediated kidney uptake is dramatically reduced. This observation matches computational models(28) and experimental data (e.g. antibody pre-dosing with anti-TENB2 ADCs reduces intestinal uptake while maintaining tumor efficacy(38,39)), and could help lower target-mediated toxicity.
Antibody distribution in clinical tumors is highly heterogeneous(40). Clinical tumors often show different characteristics than cell line xenografts, including greater stromal tissue and lower vascularity. The higher vascularity and permeability of mouse models could mask the importance of drug tissue penetration in human tumors which may be even lower due to reduced vascularity. Computational simulations of clinical tumors, supported by clinical observations, can help predict the impact of dosing in patients to guide drug design. Increasing the antibody dose of ADCs (e.g. by reducing the DAR) has the potential to improve human tumor tissue penetration and enhance tumor uptake, and these results could explain some clinical data. The anti-MUC16 ADC sofituzumab vedotin (DAR 3.5) showed a 17% response rate when administered at 2.4 mg/kg every 3 weeks(41). When the antibody was modified to a site-specific DAR 2 ADC, it could be dosed at 5.2 mg/kg every 3 weeks, generating a 45% response rate(42).
There are also several regulatory considerations regarding co-administration of unconjugated antibodies with ADCs. Separate approval of an antibody and ADC is unlikely for several reasons, particularly for an antibody that isn’t expected to have efficacy on its own. However, current ADC formulations already have a distribution of different DARs with some unconjugated antibody present. In fact, gemtuzumab ozogamicin (Mylotarg) consists of a 1:1 mixture of unconjugated antibody and ADC(43,44). While this formulation was selected to avoid ADC aggregation(45), the evident role of tissue penetration may require a re-evaluation of the prevailing view that unconjugated antibody is always undesirable(1). Rather, the optimal amount of unconjugated antibody (if any) needs to be selected based on target expression/internalization, payload potency, etc. Alternatively, the DAR could be reduced to the point of maximum efficacy. Typically, the DAR has been reduced only to the point where DAR-dependent clearance is minimized(34). However, depending on the target and payload, a further reduction could result in an improved therapeutic window, and this approach is under investigation(46,47). Likewise, higher DARs and associated technologies may be needed for lower expression and/or lower potency payloads for appropriate ‘matching’. From an intratumoral payload delivery and antibody dosing perspective, adding unconjugated antibody to an ADC is similar to lowering the DAR. However, from the perspective of systemic distribution, effects such as DAR-dependent deconjugation and DAR-dependent clearance(34,48,49) can result in disparities from the variable DAR distribution despite the same average DAR; these must be taken into account when investigating a carrier dose versus lowering the DAR.
Finally, it is important to point out that both DM4 and DGN549 exhibit bystander killing, where the released payload can diffuse out of the targeted cell and kill adjacent cells(50–52). Tissue penetration is still an important consideration even with bystander-capable payloads however, as evidenced experimentally in the current study (e.g. Fig. 1) and previous examples(28,32). Improved tissue penetration of a single-domain anti-PSMA antibody also showed improved efficacy over larger constructs(30) when using the DGN549 payload. A computational analysis by Khera et al. indicates these results are due to the higher efficiency of direct cell killing relative to bystander killing(11).
In summary, increased antibody dosing with ADCs can improve both local delivery (better tissue penetration) and systemic delivery (reducing target-mediated clearance) resulting in increased efficacy. Importantly, blocking target-mediated uptake in normal tissues can also reduce toxicity in these tissues. Combined with the potential for improvements in other mechanisms of action, such as antibody signaling receptor blockade and/or Fc-effector functions, this can genuinely expand the therapeutic window rather than simply shift the bar. The utility of this technique is a function of many factors, including receptor expression in the target tissue, normal tissue expression, target internalization rate, and payload potency. Therefore, a judicious, quantitative pharmacology approach is necessary to clearly define the potential benefits of clinical application. These complex molecules require equally sophisticated simulations to identify optimal parameters for clinical efficacy. As demonstrated by current clinical ADCs and promising agents in the pipeline, these factors can be taken into account to gain both the targeting specificity of antibodies and the potency of small molecules for effective therapies.
Supplementary Material
Acknowledgements
Financial support was provided by Immunogen, NIH R35 GM128819 (GMT), and the National Cancer Institute of the National Institutes of Health under Award Number P30CA046592 by the use of the following Cancer Center Shared Resource(s): histology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest Statement: JFP, LL, RL, OA, CE, NK, BM, YS, MLM, TK, RC, JP, and RG were employees of ImmunoGen at the time of this work. GMT has served as a consultant for AstraZeneca/MedImmune, Advanced Proteome Therapeutics, Abbvie, Bristol Myers Squibb, Crescendo Biologics, CytomX Therapeutics, Eli Lilly, ImmunoGen, Immunomedics, InVicro, Lumicell, Nodus Therapeutics, Novartis, Mersana Therapeutics, Roche/Genentech, Seattle Genetics, and Takeda Pharmaceuticals. EK has no conflicts of interest to disclose.
REFERENCES
- 1.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017;16(5):315–37 doi 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 2.Coats S, Williams M, Kebble B, Dixit R, Tseng L, Yao NS, et al. Antibody-Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clinical cancer research : an official journal of the American Association for Cancer Research 2019;25(18):5441–8 doi 10.1158/1078-0432.CCR-19-0272. [DOI] [PubMed] [Google Scholar]
- 3.Mahalingaiah PK, Ciurlionis R, Durbin KR, Yeager RL, Philip BK, Bawa B, et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol Ther 2019;200:110–25 doi 10.1016/j.pharmthera.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Saber H, Leighton JK. An FDA oncology analysis of antibody-drug conjugates. Regul Toxicol Pharmacol 2015;71(3):444–52 doi 10.1016/j.yrtph.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 5.Saber H, Simpson N, Ricks TK, Leighton JK. An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul Toxicol Pharmacol 2019;107:104429 doi 10.1016/j.yrtph.2019.104429. [DOI] [PubMed] [Google Scholar]
- 6.Donaghy H Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016;8(4):659–71 doi 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lyon R Drawing lessons from the clinical development of antibody-drug conjugates. Drug discovery today Technologies 2018;30:105–9 doi 10.1016/j.ddtec.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Eaton JS, Miller PE, Mannis MJ, Murphy CJ. Ocular Adverse Events Associated with Antibody-Drug Conjugates in Human Clinical Trials. J Ocul Pharmacol Ther 2015;31(10):589–604 doi 10.1089/jop.2015.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao H, Atkinson J, Gulesserian S, Zeng Z, Nater J, Ou J, et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody-Drug Conjugates. Cancer Res 2018;78(8):2115–26 doi 10.1158/0008-5472.CAN-17-3202. [DOI] [PubMed] [Google Scholar]
- 10.Strop P, Tran TT, Dorywalska M, Delaria K, Dushin R, Wong OK, et al. RN927C, a Site-Specific Trop-2 Antibody-Drug Conjugate (ADC) with Enhanced Stability, Is Highly Efficacious in Preclinical Solid Tumor Models. Mol Cancer Ther 2016;15(11):2698–708 doi 10.1158/1535-7163.MCT-16-0431. [DOI] [PubMed] [Google Scholar]
- 11.Khera E, Cilliers C, Bhatnagar S, Thurber GM. Computational transport analysis of antibody-drug conjugate bystander effects and payload tumoral distribution: implications for therapy. Molecular Systems Design & Engineering 2018;3(1):73–88 doi 10.1039/C7ME00093F. [DOI] [Google Scholar]
- 12.Ab O, Whiteman KR, Bartle LM, Sun X, Singh R, Tavares D, et al. IMGN853, a Folate Receptor-alpha (FRalpha)-Targeting Antibody-Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRalpha-Expressing Tumors. Mol Cancer Ther 2015;14(7):1605–13 doi 10.1158/1535-7163.MCT-14-1095. [DOI] [PubMed] [Google Scholar]
- 13.Miller ML, Fishkin NE, Li W, Whiteman KR, Kovtun Y, Reid EE, et al. A New Class of Antibody-Drug Conjugates with Potent DNA Alkylating Activity. Mol Cancer Ther 2016;15(8):1870–8 doi 10.1158/1535-7163.MCT-16-0184. [DOI] [PubMed] [Google Scholar]
- 14.Ab O, Whiteman KR, Bartle LM, Sun X, Singh R, Tavares D, et al. IMGN853, a Folate Receptor-α (FRα)–Targeting Antibody–Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRα-Expressing Tumors. Molecular Cancer Therapeutics 2015;14(7):1605 doi 10.1158/1535-7163.MCT-14-1095. [DOI] [PubMed] [Google Scholar]
- 15.Reddy JA, Dorton R, Bloomfield A, Nelson M, Dircksen C, Vetzel M, et al. Pre-clinical evaluation of EC1456, a folate-tubulysin anti-cancer therapeutic. Sci Rep 2018;8(1):8943 doi 10.1038/s41598-018-27320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy JA, Dorton R, Bloomfield A, Nelson M, Vetzel M, Guan J, et al. Rational combination therapy of vintafolide (EC145) with commonly used chemotherapeutic drugs. Clin Cancer Res 2014;20(8):2104–14 doi 10.1158/1078-0432.CCR-13-2423. [DOI] [PubMed] [Google Scholar]
- 17.Ponte JF, Ab O, Lanieri L, Lee J, Coccia J, Bartle LM, et al. Mirvetuximab Soravtansine (IMGN853), a Folate Receptor Alpha-Targeting Antibody-Drug Conjugate, Potentiates the Activity of Standard of Care Therapeutics in Ovarian Cancer Models. Neoplasia 2016;18(12):775–84 doi 10.1016/j.neo.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, Wilhelm SD, et al. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem 2010;21(1):84–92 doi 10.1021/bc900315y. [DOI] [PubMed] [Google Scholar]
- 19.Bai C, Reid EE, Wilhelm A, Shizuka M, Maloney EK, Laleau R, et al. Site-Specific Conjugation of the Indolinobenzodiazepine DGN549 to Antibodies Affords Antibody-Drug Conjugates with an Improved Therapeutic Index as Compared with Lysine Conjugation. Bioconjug Chem 2020;31(1):93–103 doi 10.1021/acs.bioconjchem.9b00777. [DOI] [PubMed] [Google Scholar]
- 20.Mager DE. Target-mediated drug disposition and dynamics. Biochemical Pharmacology 2006;72(1):1–10. [DOI] [PubMed] [Google Scholar]
- 21.Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N Engl J Med 2019;380(8):741–51 doi 10.1056/NEJMoa1814213. [DOI] [PubMed] [Google Scholar]
- 22.Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res 2016;76(10):3003–13 doi 10.1158/0008-5472.CAN-15-1313. [DOI] [PubMed] [Google Scholar]
- 23.Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med 2019. doi 10.1056/NEJMoa1914510. [DOI] [Google Scholar]
- 24.Moore KN, O’Malley DM, Vergote I, Martin LP, Gonzalez-Martin A, Malek K, et al. Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRalpha)-targeting antibody-drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecol Oncol 2018;151(1):46–52 doi 10.1016/j.ygyno.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 25.Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med 1990;31(7):1191–8. [PubMed] [Google Scholar]
- 26.Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res 1992;52(19):5144–53. [PubMed] [Google Scholar]
- 27.Cilliers C, Menezes B, Nessler I, Linderman J, Thurber GM. Improved Tumor Penetration and Single-Cell Targeting of Antibody-Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res 2018;78(3):758–68 doi 10.1158/0008-5472.CAN-17-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cilliers C, Guo H, Liao J, Christodolu N, Thurber GM. Multiscale Modeling of Antibody-Drug Conjugates: Connecting Tissue and Cellular Distribution to Whole Animal Pharmacokinetics and Potential Implications for Efficacy. The AAPS journal 2016. doi 10.1208/s12248-016-9940-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakada T, Sugihara K, Jikoh T, Abe Y, Agatsuma T. The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem Pharm Bull (Tokyo) 2019;67(3):173–85 doi 10.1248/cpb.c18-00744. [DOI] [PubMed] [Google Scholar]
- 30.Nessler I, Khera E, Vance S, Kopp A, Qiu Q, Keating TA, et al. Increased Tumor Penetration of Single-Domain Antibody Drug Conjugates Improves In Vivo Efficacy in Prostate Cancer Models. Cancer Res 2020. doi 10.1158/0008-5472.CAN-19-2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma SK, Chow A, Monette S, Vivier D, Pourat J, Edwards KJ, et al. Fc-Mediated Anomalous Biodistribution of Therapeutic Antibodies in Immunodeficient Mouse Models. Cancer Res 2018;78(7):1820–32 doi 10.1158/0008-5472.CAN-17-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh AP, Guo L, Verma A, Wong GG, Thurber GM, Shah DK. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: a Quantitative Investigation. The AAPS journal 2020;22(2):28 doi 10.1208/s12248-019-0387-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhatnagar S, Deschenes E, Liao JS, Cilliers C, Thurber GM. Multichannel Imaging to Quantify Four Classes of Pharmacokinetic Distribution in Tumors. Journal of Pharmaceutical Sciences 2014;103(10):3276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clinical cancer research : an official journal of the American Association for Cancer Research 2004;10(20):7063–70 doi 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 35.Sun X, Ponte JF, Yoder NC, Laleau R, Coccia J, Lanieri L, et al. Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug Chem 2017;28(5):1371–81 doi 10.1021/acs.bioconjchem.7b00062. [DOI] [PubMed] [Google Scholar]
- 36.Yoder NC, Bai C, Tavares D, Widdison WC, Whiteman KR, Wilhelm A, et al. A Case Study Comparing Heterogeneous Lysine- and Site-Specific Cysteine-Conjugated Maytansinoid Antibody-Drug Conjugates (ADCs) Illustrates the Benefits of Lysine Conjugation. Mol Pharm 2019;16(9):3926–37 doi 10.1021/acs.molpharmaceut.9b00529. [DOI] [PubMed] [Google Scholar]
- 37.Bai C, Reid EE, Wilhelm A, Shizuka M, Maloney EK, Laleau R, et al. Site-Specific Conjugation of the Indolinobenzodiazepine DGN549 to Antibodies Affords Antibody-Drug Conjugates with an Improved Therapeutic Index as Compared with Lysine Conjugation. Bioconjug Chem 2019. doi 10.1021/acs.bioconjchem.9b00777. [DOI] [PubMed] [Google Scholar]
- 38.Boswell CA, Yadav DB, Mundo EE, Yu SF, Lacap JA, Fourie-O’Donohue A, et al. Biodistribution and efficacy of an anti-TENB2 antibody-drug conjugate in a patient-derived model of prostate cancer. Oncotarget 2019;10(58):6234–44 doi 10.18632/oncotarget.27263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boswell CA, Mundo EE, Zhang C, Stainton SL, Yu SF, Lacap JA, et al. Differential effects of predosing on tumor and tissue uptake of an 111In-labeled anti-TENB2 antibody-drug conjugate. J Nucl Med 2012;53(9):1454–61 doi 10.2967/jnumed.112.103168. [DOI] [PubMed] [Google Scholar]
- 40.Lu G, Fakurnejad S, Martin BA, van den Berg NS, van Keulen S, Nishio N, et al. Predicting Therapeutic Antibody Delivery into Human Head and Neck Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 2020. doi 10.1158/1078-0432.CCR-19-3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JF, Moore KN, Birrer MJ, Berlin S, Matulonis UA, Infante JR, et al. Phase I study of safety and pharmacokinetics of the anti-MUC16 antibody-drug conjugate DMUC5754A in patients with platinum-resistant ovarian cancer or unresectable pancreatic cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2016;27(11):2124–30 doi 10.1093/annonc/mdw401. [DOI] [PubMed] [Google Scholar]
- 42.Moore K, Hamilton EP, Burris HA, Barroilhet LM, Gutierrez M, Wang JS, et al. Abstract CT036: Targeting MUC16 with the THIOMABTM-drug conjugate DMUC4064A in patients with platinum-resistant ovarian cancer: A phase I expansion study. Cancer Research 2018;78(13 Supplement):CT036–CT doi 10.1158/1538-7445.Am2018-ct036. [DOI] [Google Scholar]
- 43.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011;3(2):161–72 doi 10.4161/mabs.3.2.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2001;7(6):1490–6. [PubMed] [Google Scholar]
- 45.Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem 2002;13(1):47–58 doi 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 46.Demetri GD, Luke JJ, Hollebecque A, Powderly JD, Spira AI, Subbiah V, et al. First-in-human phase 1 study of ABBV-085, an antibody-drug conjugate (ADC) targeting LRRC15, in sarcomas and other advanced solid tumors. Journal of Clinical Oncology 2019;37(15_suppl):3004- doi 10.1200/JCO.2019.37.15_suppl.3004. [DOI] [PubMed] [Google Scholar]
- 47.Sands JM, Shimizu T, Garon EB, Greenberg J, Guevara FM, Heist RS, et al. First-in-human phase 1 study of DS-1062a in patients with advanced solid tumors. Journal of Clinical Oncology 2019;37(15_suppl):9051- doi 10.1200/JCO.2019.37.15_suppl.9051. [DOI] [Google Scholar]
- 48.Sukumaran S, Gadkar K, Zhang C, Bhakta S, Liu L, Xu K, et al. Mechanism-Based Pharmacokinetic/Pharmacodynamic Model for THIOMAB Drug Conjugates. Pharm Res 2015;32(6):1884–93 doi 10.1007/s11095-014-1582-1. [DOI] [PubMed] [Google Scholar]
- 49.Bender B, Leipold DD, Xu K, Shen BQ, Tibbitts J, Friberg LE. A mechanistic pharmacokinetic model elucidating the disposition of trastuzumab emtansine (T-DM1), an antibody-drug conjugate (ADC) for treatment of metastatic breast cancer. The AAPS journal 2014;16(5):994–1008 doi 10.1208/s12248-014-9618-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res 2016;76(9):2710–9 doi 10.1158/0008-5472.CAN-15-1795. [DOI] [PubMed] [Google Scholar]
- 51.Miller ML, Shizuka M, Wilhelm A, Salomon P, Reid EE, Lanieri L, et al. A DNA-Interacting Payload Designed to Eliminate Cross-Linking Improves the Therapeutic Index of Antibody-Drug Conjugates (ADCs). Mol Cancer Ther 2018;17(3):650–60 doi 10.1158/1535-7163.MCT-17-0940. [DOI] [PubMed] [Google Scholar]
- 52.Chari RV, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angewandte Chemie 2014;53(15):3796–827 doi 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.