

Abstract
Endothelial cells (ECs) maintain vascular integrity and mediate vascular repair and angiogenesis, by which new blood vessels are formed from pre-existing blood vessels. Hyperglycemia has been shown to increase EC angiogenic potential. However, few studies have investigated effects of fatty acids (FAs) on EC angiogenesis. Cluster of differentiation 36 (CD36) is a FA transporter expressed by ECs, but its role in EC proliferation, migration, and angiogenesis is unknown. We sought to determine if circulating FAs regulate angiogenic function in a CD36-dependent manner. CD36-dependent effects of FAs on EC proliferation and migration of mouse heart ECs (MHECs) and lung ECs (MLECs) were studied. We used both silencing RNA and antisense oligonucleotides to reduce CD36 expression. Oleic acid (OA) did not affect EC proliferation, but significantly increased migration of ECs in wound healing experiments. CD36 knockdown prevented OA-induced increases in wound healing potential. In EC transwell migration experiments, OA increased recruitment and migration of ECs, an effect abolished by CD36 knockdown. Phospho-AMP-activated protein kinase (AMPK) increased in MHECs exposed to OA in a CD36-dependent manner. To test whether in vivo CD36 affects angiogenesis, we studied 21-day recovery in post-hindlimb ischemia. EC-specific CD36 knockout mice had reduced blood flow recovery as assessed by laser Doppler imaging. EC content in post-ischemic muscle, assessed from CD31 expression, increased in ischemic muscle of control mice. However, mice with EC-specific CD36 deletion lacked the increase in CD31 and matrix metalloprotease 9 expression observed in controls. EC expression of CD36 and its function in FA uptake modulate angiogenic function and response to ischemia, likely due to reduced activation of the AMPK pathway.
Keywords: Angiogenesis, endothelium, ischemia, lipids, peripheral vascular disease
Introduction
Endothelial cells (ECs) line vessel walls, maintain vascular integrity, and form a permeability barrier that allows selective molecule transport. ECs also regulate vascular tone and release factors that mediate blood clotting, thrombolysis, and cell infiltration through the endothelium [1]. ECs promote vascular repair and angiogenesis. Unlike vasculogenesis, which occurs at the developmental stages, angiogenesis allows new vessels to form by sprouting from pre-existing vessels, promoting blood supply to expanding tissues [2]. Under pathological ischemic injury, angiogenesis is required for injury repair. Vascular endothelial growth factors (VEGFs) regulate angiogenesis [3] by controlling proliferation and migration of ECs and production of matrix metalloproteases (MMPs) [4]. MMPs drive matrix degradation required for EC invasion and vessel sprouting. VEGFα and its receptor VEGFR2 are stimulators of angiogenesis, whereas VEGFβ and its receptor VEGFR1 are negative regulators that limit angiogenic effects of VEGFα/VEGFR2 [5,6]. MMP9 and MMP2 are regulated by a family of tissue inhibitors of MMPs (TIMPs) [7,8], composed of TIMP1-TIMP4 [9,10]. TIMP1 and TIMP3 regulate MMP9, whereas TIMP2-TIMP4 regulate MMP2 [11].
Hyperglycemic effects on EC function have been widely studied [12,13], showing increased EC angiogenesis, particularly in pathological disorders such as diabetic retinopathy [14,15]. However, few studies have investigated the effects of lipid metabolism on angiogenesis. Cluster of differentiation 36 (CD36) is a multi-ligand receptor that functions as a fatty acid (FA) transporter [16,17] and influences cytokine release and signaling [18,19], calcium flux, insulin signaling [20,21], and platelet function [22,23]. The role of CD36 as a FA transporter was identified in adipocytes (named fatty acid translocase, FAT) [24-26]. However, CD36 is also expressed in ECs, monocytes, macrophages, platelets, epithelial cells, cardiomyocytes, and skeletal muscle [19]. CD36 expression is regulated by lipidactivated peroxisome proliferator-activated receptors (PPARs), particularly PPARγ [27]. Increased FA uptake results in binding of these molecules to PPARs at the surface of the nucleus. In turn, PPARs translocate and dimerize with the nuclear receptor RXR resulting in the modulation of CD36 gene transcription [27]. Cellular CD36 is localized in specialized cholesterol-rich membrane microdomains. Phosphatidylcholine and phosphatidylserine on lipoproteins and apoptotic cells are also ligands for CD36. Moreover, the Src family of non-receptor tyrosine kinases interacts with CD36 to alter several downstream processes [28].
CD36 is more abundant in microvascular than macrovascular ECs [29], but how CD36 levels impact the functions of the two cell types remains unknown. A substantial number of studies have investigated the role of CD36 in microvascular ECs based on its function as a thrombospondin-1 (TSP-1) receptor, showing that CD36 is anti-angiogenic [30]. Inhibition of CD36 in microvascular ECs prevents the formation of a SHP-1/VEGFR2 complex, thereby blocking EC migration and tube formation. SHP-1 mediates an anti-angiogenic pathway that is induced by the interaction of CD36 with TSP-1, which blocks VEGFR2 signaling. However, there is a lack of knowledge on the role of CD36 in FA metabolism in vascular ECs. We tested whether CD36 interactions with long-chain FAs (LCFAs) affect microvascular and macrovascular EC functions by studying the role of CD36 in vascular repair in peripheral vascular disease. Using cultured ECs and mice with EC-specific CD36 deletion, we show that CD36 affects EC-mediated angiogenesis and that these actions are likely due to interactions with LCFAs rather than effects on cellular lipid oxidation.
Materials and methods
Mouse heart and lung endothelial cell culture
Mouse heart ECs (MHECs, mVera-hrt-01) and mouse lung ECs (MLECs, mVera-lng-01) (Angiocrine Bioscience) were cultured in 12-well tissue culture plates (Sigma Aldrich) in complete endothelial cell growth media containing 10% FBS (Gibco). In functional assays, MHECs and MLECs were treated with bovine serum albumin (BSA) (Sigma Aldrich) (control), BSA-bound oleic acid (OA, 300 µmol/L), BSA-bound linoleic acid (LA, 300 µmol/L), or BSA-bound palmitic acid (PA, 300 µmol/L, Agilent) for 16 hours. In CD36 knockdown experiments, CD36 silencing RNA (siRNA) (50 nmol/L, Dharmacon) and CD36 antisense oligonucleotide (ASO) (50 nmol/L, Ionis Pharmaceuticals) were used with Lipofectamine RNAiMax (Invitrogen) transfection reagent.
mRNA expression by qRT-PCR
MHEC and MLEC mRNA expression was assessed by quantitative real-time PCR (qRT-PCR) for mouse endothelial, angiogenic, and metabolic markers (Supplementary Table 1).
Proliferation assay
Proliferation of MHECs and MLECs treated with CD36 siRNA or CD36 ASO and OA was assessed by Ki67 mRNA expression using qRT-PCR and measured using a Fluorometric Proliferation Assay kit (BioVision). Relative fluorescent units (RFUs) are directly proportional to the number of cells. The fluorometric assay used in this study is based on a nuclear dye which binds with high specificity to the nucleic acids in the cell and generates a green fluorescent signal which was detected using a fluorescence microplate reader (Ex/Em: 480/538 nm). This assay provides sensitivity to detect a wide range of cell number, 25 to 60,000 cells, which allows for more precise assessment of the effects on both cell survival and cell proliferation.
Wound scratch assay
Migration potential of MHECs and MLECs was assessed using a wound healing assay system, as reported previously [31]. A scratch was manually made on seeded ECs using a 100 μL pipet tip. Average wound scratch area was 20% of the surface of ECs seeded. After 16 hours of incubation in the presence of BSA, OA, LA, or PA, the wound area was imaged and measured. In a second set of experiments, MHECs and MLECs were treated with OA in the presence of CD36 siRNA or CD36 ASO in order to compare and confirm the effects of CD36 knockdown using two methods. For imaging, MHECs and MLECs were stained with Calcein-AM (Molecular Probes, 30 mins, 37°C), and percentage wound closure was calculated between the 0 hour and 16 hour time point. Three replicates per condition were analyzed in every experiment and three field images were taken for each condition.
Transwell migration assay
MHEC and MLEC migration was evaluated by transwell migration assay. ECs transfected with CD36 siRNA or CD36 ASO were seeded in the top chamber of a 24-well plate insert, and migration towards OA (300 µmol/L) or control BSA was assessed after 16 hours. In a second set of experiments, ECs transfected with CD36 ASO were pre-treated with OA for 16 hours prior to the transwell migration assay and seeded on the top chamber in order to determine whether chronic presence of FAs affects EC angiogenic potential. Migration towards media containing 2% fetal bovine serum (FBS) (bottom chamber) was monitored. The inserts were stained with Calcein-AM dye (Molecular Probes 30 mins, 37°C), and data was collected using a fluorescence plate reader. RFUs were compared between the different conditions.
Matrigel tube formation assay
MHECs and MLECs were incubated for 16 hours in the presence of OA or control BSA, and tube formation was evaluated using a Matrigel matrix invasion assay. In a second set of experiments, ECs transfected with CD36 ASO were pre-treated with OA for 16 hours prior to tube formation assay in order to determine whether chronic presence of FAs affects EC angiogenic potential. Tubes were then stained with Calcein-AM dye, and the well-defined closed tube structures formed were counted to generate a ratio of fold change in number of tubes compared to control conditions (BSA).
MMP release assay
Supernatants of MHECs and MLECs treated with BSA or OA for 16 hours were assessed in mouse MMP9 (MyBiosource) and MMP2 (Abcam) ELISA colorimetric assays according to manufacturer’s instructions.
Western blot
MHECs and MLECs treated with BSA or OA with or without insulin for 16 hours were subjected to SDS-PAGE protein analysis for phospho-AMPK (AMP-activated protein kinase) α, AMPKα, and α-tubulin.
Hindlimb ischemia mouse model
C57BL/6J control LoxP (EC-Cd36fl/fl named LoxP) and C57BL/6J Tie2 promoter-driven EC-specific CD36 knockout mice (EC-Cd36-/-) were used in hindlimb ischemia (HLI) studies performed by femoral artery ligation. All protocols were approved by the New York University Animal Research Committee (IACUC) and based on NIH guidelines. Animals were anesthetized using ketamine (100 mg/kg) and xylazine (10 mg/kg) by intraperitoneal injection during the time of surgery and using 2% isoflurane inhalation at the time of Laser Doppler imaging. Buprenorphine (0.05 mg/kg) was administered subcutaneously following the surgery every 12 hours for a total of 48 hours (total of 4 doses) per animal. Vascular recovery was monitored over 7 and 21 days post-ischemia. Muscle tissues were collected for mRNA expression analysis and immunohistological staining. At the end of the study, mice were euthanized by inhaled CO2 and cervical dislocation.
Laser Doppler imaging
Blood flow imaging of EC-Cd36fl/fl and EC-Cd36-/- mice was performed at day 0 (immediately following ischemic injury), day 7, and day 21 to assess collateral vessel formation and the restoration of flow in the hindlimb muscles.
Protein expression by immunohistochemistry
Immunohistochemistry was performed on OCT embedded muscle tissues to assess CD36 and CD31 protein expression at 21-day post-ischemia.
Statistical analysis
All results are presented as the mean ± SEM of at least 3 independent experiments. Statistical comparisons were performed using either paired student’s t-test followed by a Mann-Whitney post-test, one-way or two-way ANOVA as well as a Dunnett’s post-test for comparison against the control group or the Bonferroni post-test for comparison between different groups. Statistical comparisons with P < 0.05 and P < 0.01 were considered statistically significant. Comparisons between all treatment groups in gene expression analyses were used to show the effect of OA on EC gene expression both with and without the effect of CD36 inhibition. This approach allows for more specific measurement and deciphering of the effects of OA and CD36 inhibition separately. Moreover, we performed statistical analyses on the combination of CD36 inhibition and OA treatment. Statistical comparisons for each study have been specified in the Figure legends.
Results
CD36 knockdown does not affect cell survival and proliferation
ECs derived from different tissues show different angiogenic potential and responses to VEGFs and inflammatory molecules, including interleukin-1β (IL-1β) [32]. We used MLECs and MHECs; the majority of MLECs are from capillaries, which are non-angiogenic vessels, while MHECs contain a mixture of ECs predominantly derived from larger blood vessels [29]. In vivo differences in gene and protein expression of these ECs have been described, including endothelial markers VE-Cadherin, CD34, CD36, VEGFR2, and VCAM [29]. Microvascular ECs are organ specific and regulate metabolism. Microvascular ECs express higher levels of VCAM-1 than macrovascular heart ECs [29].
We first asked whether CD36 knockdown affects survival and proliferation of MHECs and MLECs. Microvascular MLECs had slightly higher CD36 mRNA expression compared to macrovascular MHECs, data not shown, as previously reported [29]. CD36 siRNA and ASO reduced CD36 expression by 80% in MHECs and 60% in MLECs compared to control ECs and non-targeting (NT) siRNA or NT ASO (Figure 1A). It is important to note that EC transfection utilizing siRNA does not achieve 100% knockdown of genes; 60-80% knockdown is widely reported in many studies for this cell type [33]. As assessed by measuring Ki67 mRNA and a proliferation assay, CD36 knockdown did not affect EC proliferation (Figure 1B). We treated MHECs and MLECs with BSA-bound OA and compared with BSA alone. No changes were noted in CD36-inhibited and OA-treated cells, suggesting that FAs do not induce cell death or increase EC proliferation (Figure 1B).
Figure 1.

Effect of oleic acid on cell proliferation in CD36-deficient MHECs and MLECs (A) siRNA- and ASO-mediated CD36 knockdown in MHECs and MLECs. mRNA expression of CD36 assessed by qRT-PCR. Histograms show fold change in CD36 mRNA expression normalized to 18S mRNA expression of non-targeting (NT) and CD36 siRNA or ASO-treated ECs compared to control (CTRL) ECs. Data represent mean ± SEM, n = 8, *P < 0.05, **P < 0.01 vs. CTRL. (B) Effect of oleic acid (OA, 300 µmol/L) on siRNA-mediated CD36 knockdown on MHEC and MLEC proliferation assessed by Ki67 mRNA expression and fluorescence-based cell proliferation assay. Histograms show fold change in Ki67 mRNA expression normalized to 18S mRNA expression compared to ECs treated with non-targeting siRNA (NT siRNA). Data represent mean fold change ± SEM, n = 6; Histograms show fold change in RFU compared to NT siRNA ECs. Data represent ± SEM, n = 6. One-way ANOVA statistical tests were used to determine statistical significance.
CD36 knockdown hinders EC migration during exposure to FAs
We then examined EC angiogenic functions by assessing wound healing potential of MHECs and MLECs transfected with CD36 siRNA, CD36 ASO, or control NT siRNA using a wound scratch assay. Wound closure in the presence of different BSA-bound FAs (OA, LA, or PA) was measured at 0 and 16 hours. Time-course experiments allowed us to determine that 16 hours is the optimal migration time (data not shown). OA and LA stimulated both MHEC and MLEC migration with OA being more efficient (Figure 2A). PA significantly reduced EC wound healing potential and was toxic, resulting in observable EC death. We elected to treat ECs with OA for the remaining experiments in this study. We tested whether CD36 expression is required for improved migration with FAs. We found that CD36 knockdown impaired OA-mediated increases in wound healing potential in MHECs and MLECs (Figure 2B, 2C).
Figure 2.

Effect of oleic acid on cell migration in CD36-deficient MHECs and MLECs. A. Effect of oleic acid (OA), linoleic acid (LA), combination of OA+ LA or palmitic acid (PA) (300 µmol/L) on MHEC and MLEC migration compared to control (CTRL) ECs. Representative images of wound healing assays imaged at 0 h and 16 h. White lines were added to show the width of the wound. Histograms show percent wound closure over 16 hours. Data represent mean ± SEM, n = 6, *P < 0.05 vs. CTRL; B. Effect of oleic acid (OA, 300 µmol/L) on siRNA-mediated CD36 knockdown on MHEC and MLEC migration compared to NT siRNA ECs. Representative images of wound healing assays imaged at 0 h and 16 h. White lines were added to show the width of the wound. Histograms show percent wound closure over 16 hours. Data represent mean ± SEM, n = 6, *P < 0.05 vs. NT siRNA; C. Effect of oleic acid (OA, 300 µmol/L) on ASO-mediated CD36 knockdown on MHEC and MLEC migration compared to NT ASO ECs. Representative images of wound healing assays imaged at 0 h and 16 h. White lines were added to show the width of the wound. Scale bars represent 100 μm. Histograms show percent wound closure over 16 hours. Data represent mean ± SEM, n = 6, *P < 0.05 vs. NT ASO. Two-way ANOVA statistical tests were used to determine statistical significance.
To assess EC migratory potential towards high FA environments, we performed a migration assay using transwell inserts where MHECs or MLECs, transfected with CD36 siRNA or ASO, were included in top chambers, while BSA-bound OA or control BSA were added to bottom chambers. We did not utilize a migration-inducing factor such as VEGF or serum as we did not want to drive MHEC migration by activating VEGFRs and consequently mask the potential impact of CD36 on EC migration. Control ECs, labelledNT siRNA- or ASO-treated, significantly migrated towards OA whereas CD36-deficient ECs did not show increased migratory potential when stimulated with OA (Figure 3A, 3B). In parallel, we pretreated ASO-transfected ECs with OA (16 hours), removed OA before the migration assay, and assessed EC migration towards media with 2% FBS. In these pretreatment studies, both ECs with CD36 knockdown and control ECs showed no differences in EC migration. These results confirm that OA-stimulated ECs lose their migratory potential in the absence of continued OA uptake (Figure 3C). We associated OA-induced migratory effect to CD36 expression rather than a proliferative effect as no changes in Ki67 expression and cell proliferation were measured (Figure 1B).
Figure 3.

Effect of oleic acid on transwell migration and tube formation in CD36-deficient MHECs and MLECs. A. Effect of siRNA-mediated CD36 knockdown on MHEC and MLEC migration compared to NT siRNA ECs in transwell migration assays stimulated by oleic acid (OA, 300 µmol/L) or control BSA (bottom chamber). Histograms show fold change in RFUs of fluorescent-labelled EC migration. Data represent mean ± SEM, n = 6, *P < 0.05 vs. NT siRNA; B. Effect of ASO-mediated CD36 knockdown on MHEC and MLEC migration compared to NT ASO ECs in transwell migration assays stimulated by oleic acid (OA, 300 µmol/L) or control BSA (bottom chamber). Histograms show fold change in RFUs of fluorescent-labelled EC migration. Data represent mean ± SEM, n = 6, *P < 0.05 vs. NT ASO; C. Effect of ASO-mediated CD36 knockdown and pre-treatment with oleic (OA, 300 µmol/L) on MHEC and MLEC migration compared to NT ASO ECs in transwell migration assays stimulated by 2% fetal bovine serum (FBS) (bottom chamber). Histograms show fold change in RFUs of fluorescent-labelled EC migration. Data represent mean ± SEM, n = 6. D. Effect of oleic acid (OA, 300 µmol/L) on ASO-mediated CD36 knockdown on MHEC and MLEC new vessel formation in Matrigel tube formation assays. Scale bars represent 100 μm. Histograms show fold change in a number of well-defined closed tube structures. Data represent mean ± SEM, n = 5, *P < 0.05 vs. NT ASO; E. Effect of ASO-mediated CD36 knockdown and pre-treatment with oleic acid (OA, 300 µmol/L) on MHEC and MLEC new vessel formation compared to control (CTRL) ECs in Matrigel tube formation assays. Scale bars represent 100 μm. Histograms show fold change in number of well-defined closed tube structures. Data represent mean ± SEM, n = 5, *P < 0.05 vs. NT ASO. Two-way ANOVA statistical tests were used to determine statistical significance.
FAs and CD36 modulate EC tube formation
To evaluate how CD36 influences MHEC and MLEC tube formation, ECs transfected with CD36 siRNA or ASO were plated on Matrigel matrix in the presence of BSA-bound OA or control BSA. OA promoted EC Matrigel invasion and tube formation, whereas CD36 knockdown impaired tube formation (Figure 3D). In ASO-transfected ECs pretreated with OA and then subjected to the tube formation assay, we noted loss of OA-induced closed-loop tubes (Figure 3E). These data together with those of transwell migration assays suggest that EC angiogenic potential is stimulated by chronic presence of high FA levels via CD36.
CD36 knockdown downregulates FA-induced increases in EC angiogenic factors
The effects of CD36 knockdown on EC migration could be due to altered EC metabolism or activation of a signaling process consequent to CD36 ligation. To understand the mechanism by which high FAs drive EC angiogenic function, we measured mRNA levels of CD36, CD31, and endothelial nitric oxide synthase (eNOS). As our goal is to investigate CD36 functions in vascular repair in peripheral vascular disease, we focused on MHEC gene expression because these macrovascular ECs function more similarly to the ECs found in large vessels of the peripheral muscle tissues in a hindlimb ischemia mouse model. It has been established that increased FAs can contribute to atherosclerotic changes and result in macrovascular complications including insufficiency of blood flow to the extremities, which is a common manifestation of peripheral vascular disease [34]. MLEC gene expression data is included in the supplementary material (Supplementary Figures 1, 2). OA increased CD36 expression. CD36 knockdown in MHECs reduced OA-mediated increases in CD31 expression (Figure 4A). OA upregulated expression of VEGFα, VEGFβ, and VEGFR1, while VEGFR2 trended higher (Figure 4B). CD36 knockdown increased mRNA for VEGFα, VEGFR2, and VEGFR1 without affecting VEGFβ and reversed all regulatory effects of OA treatment, as outlined in statistical comparisons between OA-treated MHECs in which CD36 has been inhibited and MHECs which are CD36-deficient and were not treated with OA (Figure 4B). These comparisons provide relevant insight as it provides a way to decipher the role of CD36 in FA metabolism in ECs and the associated effects on angiogenic function. Previous studies suggest VEGFβ regulates EC uptake of FAs by transcriptional regulation of vascular FA transport proteins whereby VEGFβ-deficient mice display less lipid accumulation in heart and muscle [35]. We suggest that the inhibition of CD36 promotes an upregulation of VEGFα as a compensatory mechanism in order to maintain EC angiogenic function. Moreover, although CD31 is commonly used as an endothelial marker, it is specifically an adhesion molecule associated with platelet-endothelial adhesion. It may be used as a marker to detect both mature and immature/new ECs; thus, CD31 may be used as an adequate marker in in vivo studies. However, CD31 is also expressed in hematopoietic cells [36]. We chose to investigate effects on VEGFα and VEGFR as they are angiogenic drivers that stimulate cell proliferation and impact downstream players such as MMPs in the process of angiogenesis whereas CD31 does not directly affect the angiogenic pathway. We have also previously shown that CD36 and CD31 co-localize in heart tissue capillaries [37]. Hence, in our in vivo studies we chose to assess CD31, VEGF and MMP expression in muscle tissues of a hindlimb ischemia mouse model.
Figure 4.
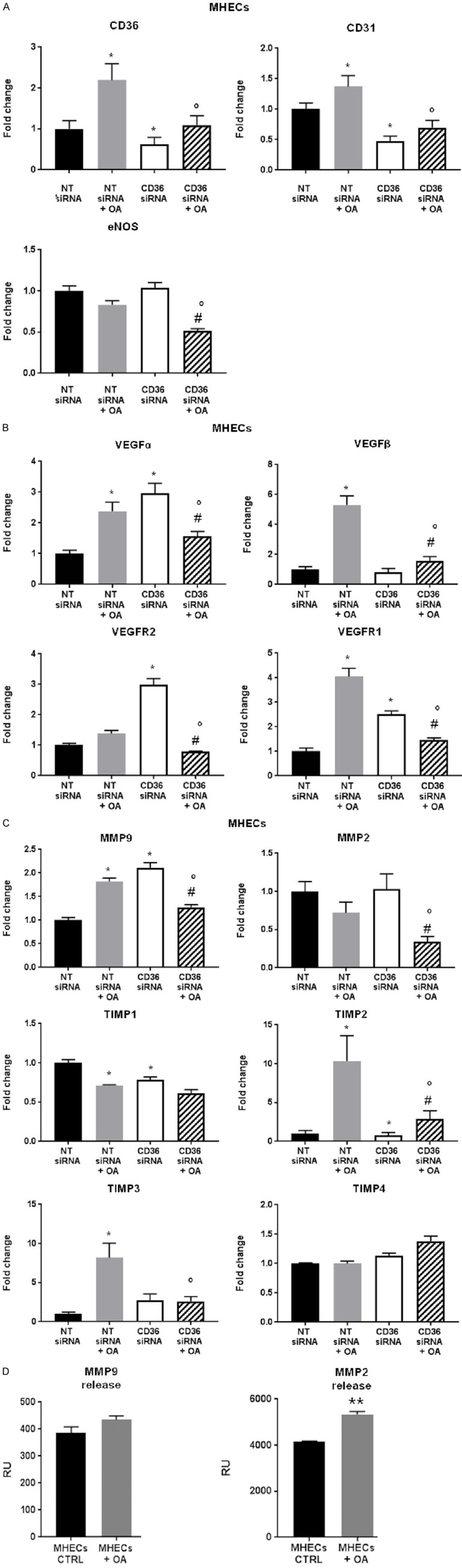
Effect of oleic acid on gene expression of endothelial and angiogenic markers in CD36-deficient MHECs. Effect of oleic acid (OA, 300 µmol/L) and siRNA-mediated CD36 knockdown on MHEC endothelial and angiogenic marker mRNA expression: (A) CD36, CD31, eNOS (B) VEGFα, VEGFR2, VEGFβ, VEGFR1 and (C) MMP9, MMP2, TIMP1, TIMP2, TIMP3, TIMP4 assessed by quantitative realtime PCR (qRT-PCR) normalized to 18S mRNA expression. Histograms show fold change in mRNA expression compared to NT siRNA ECs. Data represent mean ± SEM, n = 6, *P < 0.05 vs. NT siRNA, °P < 0.05 vs. NT siRNA + OA, #P < 0.05 vs. CD36 siRNA; (D) MMP9/MMP2 release assessed by colorimetric ELISA assays determined at 450 nm. Histograms show relative units (RU) of MMP release in supernatant samples of MHECs and MLECs treated with BSA (CTRL) or BSA-bound OA (OA) for 16 hours. Data represent mean ± SEM, n = 7, **P < 0.01 vs. MHECs CTRL. Two-way ANOVA statistical tests were used to determine statistical significance.
We measured gene expression levels of MMPs and their inhibitors, TIMPs. MMP9 expression increased in MHECs treated with OA and CD36 siRNA compared to NT siRNA (Figure 4C). OA, in combination with CD36 siRNA, could not further increase MMP expression (Figure 4C). Although OA had no effect on MMP2 in MHECs, knockdown of CD36 in MHECs treated with OA significantly reduced MMP2 expression (Figure 4C). Hence, CD36 inhibition increases MMP9 expression. In treatment with OA, CD36 knockdown reduced expression of MMP9 and MMP2. Moreover, OA-treated MHECs increased TIMP2 and TIMP3 and reduced TIMP1 mRNA levels, whereas CD36 knockdown reduced TIMP1 and TIMP2. Although no differences were noted in MMP2 gene expression, we show OA increased MMP2 release by MHECs (Figure 4D).
The data suggest that CD36 influences expression of some VEGFs and MMPs in MHECs, which mediate EC migration and invasion potential and drive the ability to form new vasculature. We examined whether these functional alterations are associated with metabolic changes in ECs by assessing metabolic gene expression in MHECs treated with OA and in CD36 knockdown. Metabolic genes carnitine palmitoyltransferase 1 (Cpt1) a, Cpt1b, glucose transporter 1 (GLUT1), and perilipin 2 (PLIN2) were upregulated in OA-treated MHECs. CD36 knockdown reduced Cpt1a, but further increased Cpt1b, GLUT1, PLIN2, and AMPKα (Figure 5B). These genes were all further upregulated in the presence of OA, compared to MHECs treated with NT siRNA and OA alone (Figure 5).
Figure 5.
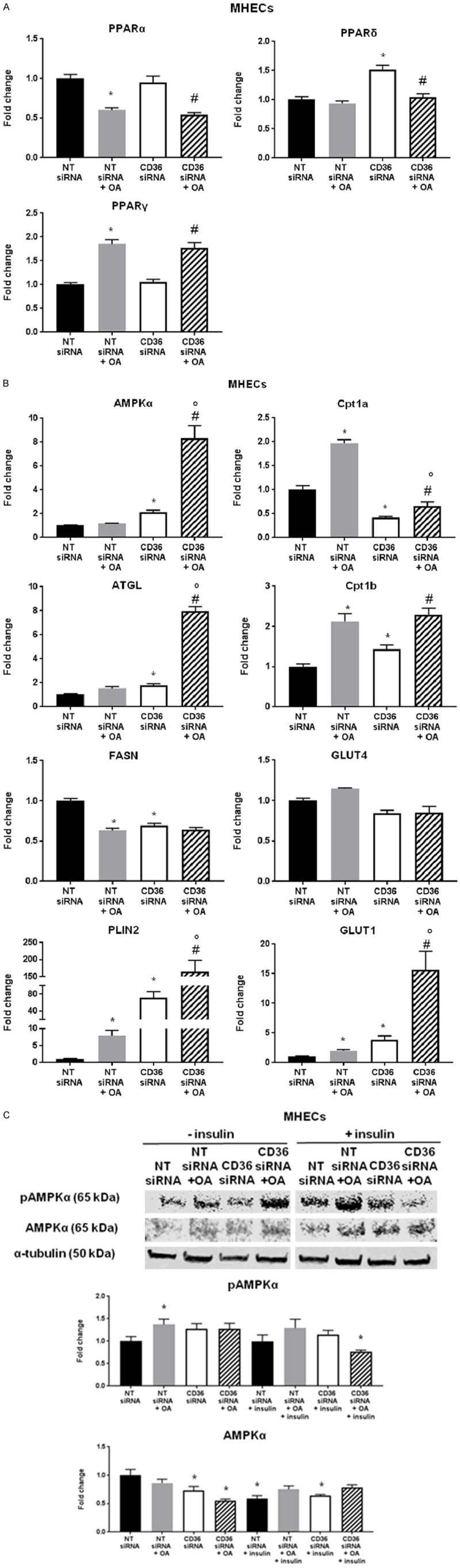
Effect of oleic acid on gene expression of metabolic markers and protein expression of AMPK in CD36-deficient MHECs. Effect of oleic acid (OA, 300 µmol/L) and siRNA-mediated CD36 knockdown on MHEC metabolic gene mRNA expression: (A) PPARα, PPARδ, PPARγ (B) AMPKα, ATGL, FASN, PLIN2, Cpt1a. Cpt1b, GLUT4, and GLUT1. Histograms show fold change in mRNA expression compared to NT siRNA ECs. Data represent mean ± SEM, n = 5, *P < 0.05 vs. NT siRNA, °P < 0.05 vs. NT siRNA + OA, #P < 0.05 vs. CD36 siRNA; (C) Effect of oleic acid (OA, 300 µmol/L) and insulin (50 units/mL) and siRNA-mediated CD36 knockdown on MHEC intracellular signaling proteins as assessed by western blot. Images of SDS-PAGE gels represent protein expression of phospho-AMPK, AMPK and housekeeping gene α-tubulin. Histograms show fold change in protein expression compared to NT siRNA ECs. Data represent mean ± SEM, n = 3, *P < 0.05 vs. NT siRNA. Two-way ANOVA statistical tests were used to determine statistical significance.
Alterations in metabolic gene expression in FA-treated MHECs suggest that CD36-dependent effects on EC angiogenic function correlate with changes in EC metabolism. Protein expression data showed phospho-AMPK was significantly increased in macrovascular MHECs exposed to high levels of OA, whereas total AMPK expression tended to decrease with CD36 knockdown in conditions with and without insulin. Upon interaction of FAs with the CD36 receptor, Fyn is released from the complex, allowing cytosolic liver kinase B1 (LKB1) to activate AMPK [38]. Our data support these findings, as treating MHECs with OA caused increases in AMPK phosphorylation (Figure 5C).
We looked at the same endothelial angiogenic and metabolic factors in both our in vitro and in vivo systems. The mRNA expression data in our in vivo model provide evidence that in EC-Cd36-/- mice muscle tissues, there is upregulation of angiogenic factors by day 21 (not seen by day 7 - Supplementary Figure 4). CD36 inhibition hinders the reparatory mechanism in muscle in vivo as it hinders similarly the migratory function of MHECs in vitro.
Vascular recovery of EC-Cd36-/- mice under ischemic conditions
We have recently shown that in heart tissues of EC-Cd36-/- mice, mRNA levels of PPARα are increased [37]. This study concluded that EC CD36 plays an important role in the selection of tissue fuel as it regulates heart and skeletal muscle FA delivery as well as glucose utilization. We were interested in studying the effects of endothelial-specific CD36 knockdown on muscle tissue expression of metabolic genes. We assessed whether CD36 loss in ECs affects post-ischemic recovery. Following HLI surgery in control EC-Cd36fl/fl and EC-Cd36-/- mice, muscle tissues were collected at days 7 and 21 (Supplementary Figure 3). Using laser Doppler imaging, we compared relative blood flow recovery in the post-ischemic and sham limb (labeled normal muscle). Control EC-Cd36fl/fl mice showed some recovery by day 7 and much more collateral vessel formation by day 21 (Figure 6A), whereas EC-Cd36-/- mice had impaired vascular recovery at days 7 and 21 post-ischemic injury (Figure 6A). Control and EC-Cd36-/- mice were compared to determine differences in blood flow recovery and collateral vessel formation. EC-Cd36fl/fl and EC-Cd36-/- mice showed increased blood flow recovery and new vessel formation over 7 and 21 days post-ischemia compared to day 0. However, EC-Cd36-/- mice had only 50% recovery by day 21 compared to control EC-Cd36fl/fl mice (Figure 6A). At day 21, control mice had significantly increased CD31 protein in HLI muscle compared to normal muscle (Figure 6B).
Figure 6.
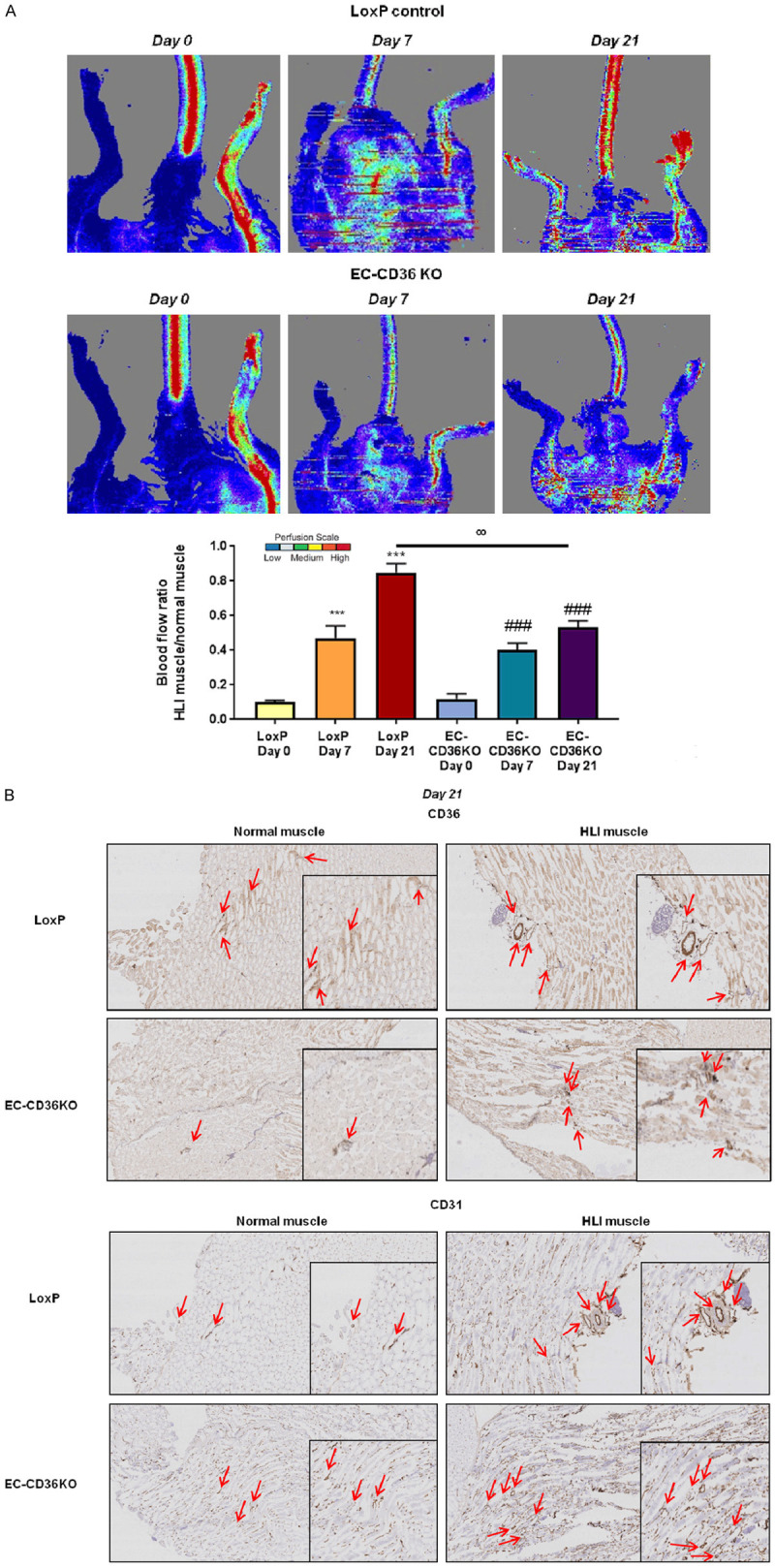
In vivo hindlimb ischemia in EC-specific CD36 knockout mouse model. A. Representative images of blood flow by Laser Doppler imaging of normal and HLI muscle tissues of LoxP and EC-CD36 KO mice at day 0 (immediately after HLI) and days 7 and 21 post-ischemia, n = 7. Quantification of laser Doppler images of blood flow recovery in normal and HLI muscle tissues of LoxP and EC-CD36 KO mice at day 0 (immediately after HLI) and days 7 and 21 post-ischemia. Histograms show blood flow ratios of HLI muscle compared to normal muscle in control LoxP and EC-CD36 KO mice. Data represent mean ± SEM, n = 7, ***P < 0.001 vs. LoxP Day 0, ###P < 0.001 vs. EC-CD36 KO Day 0, °°P < 0.01 vs. LoxP Day 21; B. Representative images of immunohistochemical staining for CD36 and CD31 expression in normal muscle and HLI muscle from control LoxP and EC-CD36 KO mice at day 21 post-ischemia, n = 4. Scale bars represent 100 μm and 200 μm. Two-way ANOVA statistical tests were used to determine statistical significance.
Factors regulating angiogenesis in control EC-Cd36fl/fl and EC-Cd36-/- mice
We compared baseline differences in endothelial and angiogenic marker gene expression in muscle tissues of EC-Cd36-/- and EC-Cd36fl/fl mice. Considering most significant differences in blood flow recovery were at day 21 post-ischemia (Figure 6B), we focused on gene and protein regulation at day 21.
This yielded similar results to the gene expression studies, which suggested reduced CD36 expression limits post-ischemic CD31 increases in HLI muscle of EC-Cd36-/- mice compared to control mice, reflecting less EC content and collateral vessel formation (Figure 7B-D). Gene regulation comparisons between normal and HLI muscle tissues from EC-Cd36-/- and EC-Cd36fl/fl mice at day 7 post-ischemia have been compiled in the supplementary data (Supplementary Figure 4A-C).
Figure 7.

Effect of EC-CD36 knockout in HLI mouse muscle tissues on endothelial, vascular and metabolic gene expression at 21 days post-HLI. In vivo hindlimb ischemia (HLI) mouse tissue mRNA expression in normal and HLI muscle tissues of LoxP and EC-CD36 KO mice of (A) baseline (day 0) vascular endothelial and angiogenic marker mRNA expression of CD36, CD31, Ki67, eNOS, MMP9, MMP2, VEGFα, VEGFR2, VEGFβ, VEGFR1, Cpt1a, GLUT1, PLIN2, and AMPKα assessed by quantitative realtime PCR (qRT-PCR). Histograms represent fold changes in mRNA expression normalized to RPS3 mRNA expression. Data represent mean ± SEM, n = 8, *P < 0.05 vs. normal muscle LoxP; (B) Day 21 post-ischemia mRNA expression of CD36, CD31, eNOS, Ki67, VEGFα, VEGFR2, VEGFβ, and VEGFR1. Histograms represent fold changes in mRNA expression normalized to RPS3 mRNA expression. Data represent mean ± SEM, n = 8, *P < 0.05, **P < 0.01, ***P < 0.001 vs. normal muscle LoxP; (C) Day 21 post-ischemia mRNA expression of MMP9, MMP2, TIMP1, TIMP2, TIMP3, and TIMP4. Histograms represent fold changes in mRNA expression normalized to RPS3 mRNA expression. Data represent mean ± SEM, n = 8, *P < 0.05, **P < 0.01 vs. normal muscle LoxP; (D) Day 21 post-ischemia mRNA expression of Cpt1a, GLUT1, and PLIN2. Histograms represent fold changes in mRNA expression normalized to RPS3 mRNA expression. Data represent mean ± SEM, n = 8, *P < 0.05, **P < 0.01 vs. normal muscle LoxP, °P < 0.01 vs. normal muscle EC-CD36 KO. One-way and two-way ANOVA statistical tests were used to determine statistical significance.
At day 21, control mice had significantly increased CD31 expression in HLI muscle compared to normal muscle. EC-Cd36-/- mice showed increased CD31 in normal muscle compared to control mice (Figure 7B); however, there were no further increases in CD31 expression in HLI muscle 21 days post-ischemia. Assessment of CD31 protein levels by immunohistochemistry (Figure 6B) paralleled gene expression results, suggesting reduced CD36 expression limits post-ischemic CD31 increases in HLI muscle of EC-Cd36-/- mice compared to control mice, reflecting less EC content and collateral vessel formation. CD31, MMP2, VEGFα, VEGFβ, GLUT1, and AMPKα were significantly upregulated in muscle tissues of EC-Cd36-/- mice compared to control mice, whereas VEGFR1 and PLIN2 were downregulated at baseline (day 0) (Figure 7A).
We next asked whether post-ischemic increases in EC content were associated with changes in cell proliferation or reflected increased EC recruitment to sites of vascular injury. In control mice, HLI muscle at day 21 showed reduced Ki67 expression, suggesting increases in CD31 and eNOS expression were not associated with higher EC proliferative potential (Figure 7B). This downregulation in Ki67 is expected, due to EC vascular-associated injury. EC-Cd36-/- mice did not show increased Ki67 expression at day 21 post-ischemia (Figure 7B). CD36 deficiency is not associated with post-ischemic cell proliferation, which is consistent with our in vitro data showing that CD36 knockdown does not alter cell proliferation (Figure 1B). Rather, collateral vessel formation upon post-ischemic recovery may be due to recruitment of ECs from pre-existing vessels.
Having shown no changes in cell proliferation post-ischemia, we examined regulation of growth factors that drive EC recruitment to sites of vascular injury. At day 21 post-ischemia, VEGFβ and VEGFR1 expression increased in EC-Cd36-/- mice compared to control mice. Although knockout mice showed upregulation of pro-angiogenic VEGFα in normal muscle, no further increase occurred in HLI muscle (Figure 7B). Moreover, upregulation of anti-angiogenic VEGFβ and VEGFR1 may contribute to the hindered recovery of EC-Cd36-/- mice, supporting that VEGFβ upregulation impairs angiogenesis (Figure 7A).
MMP and TIMP regulation in EC-Cd36-/- mice
At day 21 post-ischemia, HLI muscle of EC-Cd36-/- mice only showed significant increases in MMP2 activity, but not MMP9 (Figure 7C). This supports our in vitro results showing that muscle-derived ECs (MHECs) release higher levels of MMP2 compared to MMP9 and may contribute more to repair processes (Figure 4D). Hence, CD36 deficiency hinders angiogenic potential of ECs.
In contrast, HLI muscle of control mice at day 21 post-ischemia showed increased TIMP2, whereas HLI muscle in EC-Cd36-/- mice show increased TIMP1 and TIMP3, which explains the lack of MMP9 increase (Figure 7C). Altogether, these data suggest that TIMP upregulation and the consequent MMP inhibition contribute to limiting EC angiogenic potential during vascular recovery.
AMPK and metabolic gene regulation in EC-Cd36-/- mice
Another pathway known to modulate angiogenesis is via AMPK [39-42]. Under hypoxic conditions, angiogenesis switches from VEGF- to AMPK-mediated angiogenesis [39,40]. Moreover, CD36 dysfunction is associated with reduced lipid-sensing activity of AMPK [38], a metabolic regulator during nutrient deprivation, hypoxia, ischemia, and exercise [41]. EC-Cd36-/- mice show increased muscle AMPKα mRNA expression at baseline compared to control mice (Figure 7A), correlating with our in vitro data that shows MHECs display increased AMPKα mRNA expression upon CD36 knockdown (Figure 5B). In vitro, we show that MHECs exposed to high levels of OA increase protein expression of phospho-AMPK (Figure 5C). These data suggest that CD36 is tightly involved in positively regulating EC angiogenic migratory function since regardless of the injury-induced increase in AMPK activity through phosphorylation, ECs did not overcome the anti-angiogenic effects of CD36 inhibition.
Moreover, we investigated metabolic genes and noted no changes in Cpt1a at day 21 post-ischemia. However, there was a significant increase in PLIN2 in HLI muscle of control mice and an even larger increase in PLIN2 in HLI muscle of EC-Cd36-/- mice, compared to their normal muscle (Figure 7D). GLUT1 was increased in normal muscle of EC-Cd36-/- mice and downregulated in HLI muscle of both EC-Cd36-/- and control mice (Figure 7D).
Discussion
ECs regulate vascular repair by releasing autocrine and paracrine factors to maintain vascular integrity. Their interaction with other vascular cells is central to vascular function. ECs drive neovascularization through angiogenesis, a process allowing for vascular repair and extension of the vascular system. However, dysfunctional ECs contribute to disease pathogenesis by excessively upregulating or downregulating angiogenesis [7,43].
We have uncovered a novel pro-angiogenic role for CD36, particularly as it relates to the function of CD36 as a FA transporter in ECs. Many studies investigated EC functional dysregulation in metabolic syndromes, diabetes, and obesity to better understand how this disruption promotes cardiovascular diseases [44]. Most studies focused on effects of high circulating glucose on EC function, leaving gaps in knowledge pertaining to the impact of high circulating FAs. Thus, we investigated the role of FA metabolism in EC repair. Our in vitro studies showed that LCFAs promote EC migration and invasion without modulating proliferation. These effects were mediated via CD36, as CD36 knockdown negated them. In addition, EC-Cd36-/- mice showed defective angiogenesis after HLI, as assessed by blood flow imaging and endothelial angiogenic gene expression profiles.
FAs induced EC migration and invasion; however, FA-pretreated ECs lost their FA-induced increase in angiogenic properties and effects with CD36 inhibition. Moreover, MHEC and MLEC angiogenic functions were differentially regulated by VEGFs and MMPs. Angiogenic functions of muscle-derived macrovascular ECs (MHECs) are mainly driven by VEGFα and MMP2 release.
Vascular ECs from different tissues show differential angiogenic functions. We show that not all ECs react similarly to FA uptake through CD36. Microvascular ECs release more MMP9 and TIMP1, whereas macrovascular ECs release more MMP2 [45,46]. Our data support these claims since MHECs showed significantly higher MMP2 release compared to MLECs, and MMP2 was increased in the presence of OA in MHECs. Hence, we associated increased uptake of FAs through CD36 to the upregulation of VEGFα whereby VEGFα, in turn, upregulates MMPs, providing a positive feedback communication between VEGFs and MMPs, as previously reported [4,47]. Blocking MMPs has also been previously reported to cause inhibition of EC migration [48,49].
To translate our in vitro findings, we studied whether EC-Cd36-/- mice had a repair defect after HLI. CD36 deficiency hinders post-ischemic recovery compared to normal mice. By day 21 post-ischemia, HLI muscle of control mice showed upregulation of endothelial and angiogenic markers such as CD31, MMP9, and MMP2, whereas EC-Cd36-/- mice showed similar increases in normal muscle, but no further increase in the HLI muscle (Figure 7B, 7C). Our data suggest that although CD36-deficient mice have angiogenic factors required to induce vascular repair, they do so to a lesser extent compared to control mice. We associated the inhibition of further recovery to an upregulation of TIMP1 and TIMP3, which regulate MMP9 and MMP2 function.
Several metabolic pathways modulate angiogenesis. Insulin regulates vascular cell functions and drives EC angiogenesis through stimulation of glucose transport, release of VEGF, induction of eNOS, and proliferation of cells via a mechanism that involves PI3K-Akt pathway activation [50].
Metabolic genes affect angiogenesis in ECs. Although ECs derive their energy mainly from glycolysis, during injury or in the absence of adequate glucose supply, ECs switch their metabolic flux to uptake and utilize FAs in an AMPK-dependent manner for energy [40]. An important factor shown to drive angiogenesis in ECs is insulin. Insulin potentiates the release of vascular growth factors and promotes EC proliferation, migration, and new vessel formation [50]. Insulin activates PI3K-Akt through a phosphorylation mechanism, resulting in eNOS activation and NO production; this, in turn, regulates VEGF [50]. Insulin receptor knockout mice display a deficiency in neovascularization [51,52]. A similar effect is observed in FOXO1-deficient mice [53,54], which show significant defects in embryonic vascular formation [55]. FOXO1 is a checkpoint regulator of vascular homeostasis, as it regulates genes involved in cell growth and survival [55]. Regarding metabolic gene regulation and its relation to angiogenesis, studies correlated FA oxidation and Cpt1a expression to increased lymphatic EC embryonic angiogenesis. In this study, we associate a novel role to metabolic genes in regulating angiogenesis during vascular repair [55]. Recently, it has been shown that skeletal muscle CD36 deficiency is associated with reduced cellular insulin signaling and increased phosphorylation of the insulin receptor [21].
AMPK is a cellular energy sensor that regulates ATP homeostasis and balances ATP production and consumption. AMPK regulates EC angiogenesis by protecting cells from stressors like hypoxia, oxidative stress, and increased levels of glucose and FAs. AMPK mediates VEGF levels and VEGF-mediated angiogenesis [49,56]. Under vascular injury, the AMPK pathway is activated in ECs as a protective survival mechanism and mediates vascular homeostasis [40,57]. Moreover, the activation of endothelial AMPK promotes mitochondrial biogenesis and adaptation to stress via eNOS signaling [58]. We found that CD36 deficiency upregulated AMPKα gene expression and activity in macrovascular ECs, possibly as a compensatory mechanism to maintain EC angiogenic function. However, changes in baseline AMPK expression did not overcome the defective response to ischemia observed with EC CD36 deficiency. Rather, CD36 inhibition hindered pro-migratory changes reflected in normal cells exposed to FAs and pro-angiogenic reparative effects induced in normal tissues post-ischemia, regardless of AMPK increase. These data suggest that although CD36 deficiency may promote AMPK activity in vascular repair, this defect in CD36-mediated FA uptake simultaneously upregulates anti-angiogenic regulators such as VEGFβ/VEGFR1 and TIMPs. We suggest this is due to increased insulin sensitivity.
Our data demonstrate a novel role for CD36 in EC angiogenic function and vascular repair. We propose a mechanism for the effects of FA uptake on ischemic injury via CD36. Although ECs primarily utilize glucose for energy under quiescent conditions [59], we suggest that ECs utilize FAs to promote their migration and invasion, allowing for new vessel formation following vascular injury. Our study provides insights into the pro-angiogenic role of CD36 in ECs as a therapeutic target in peripheral vascular disease, supporting that ECs utilize FA oxidation likely in an AMPK-dependent manner during glucose deprivation (Figure 8) [59,60]. Further studies would be needed to decipher the alterations observed in the AMPK mechanistic pathway as a result of ECs exposed to chronically high levels of FAs.
Figure 8.
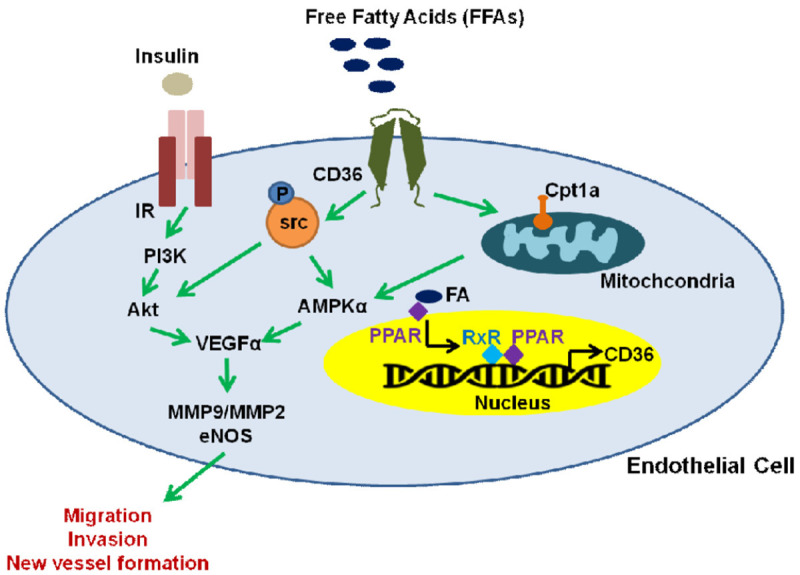
Mechanism of FA-mediated stimulation of EC angiogenic function via CD36. Increased FA uptake results in binding of these molecules to PPARs at the surface of the nucleus. In turn, PPARs translocate and dimerize with the nuclear receptor RXR resulting in the modulation of CD36 gene transcription. Functionally, FA uptake through the CD36 receptor results in the upregulation of mitochondrial Cpt1a and activation of AMPKα, which in turn increases VEGF and eNOS expression in ECs under ischemic conditions. This increase in angiogenic factors promotes EC recruitment, migration, invasion and the formation of new vessels in muscle tissues during post-ischemic recovery. On the other hand, insulin receptor (IR) actions are modulated in a CD36-dependent manner preventing src-mediated phosphorylation leading to reduced PI3K/Akt signaling, which results in decreased VEGF and angiogenesis in ECs [38,39].
Acknowledgements
We would like to acknowledge the NYU Experimental Pathology Immunohistochemistry Core Laboratory. Sources of funding include R01-HL135987 and HL07039 (IJG), R01 DK111175 (NAA) and support of the Core Laboratory from the Laura and Isaac Perlmutter Cancer Center Support Grant; NIH/NCI P30CA01608 and the National Institutes of Health S10 Grants; NIH/ORIP S10OD01058 and S10OD018338.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Boulanger CM. Endothelium. Arterioscler Thromb Vasc Biol. 2016;36:e26–31. doi: 10.1161/ATVBAHA.116.306940. [DOI] [PubMed] [Google Scholar]
- 2.Bulgin D. Therapeutic angiogenesis in ischemic tissues by growth factors and bone marrow mononuclear cells administration: biological foundation and clinical prospects. Curr Stem Cell Res Ther. 2015;10:509–522. doi: 10.2174/1574888x10666150519094132. [DOI] [PubMed] [Google Scholar]
- 3.Henning RJ. Therapeutic angiogenesis: angiogenic growth factors for ischemic heart disease. Future Cardiol. 2016;12:585–599. doi: 10.2217/fca-2016-0006. [DOI] [PubMed] [Google Scholar]
- 4.Mahecha AM, Wang H. The influence of vascular endothelial growth factor-A and matrix metalloproteinase-2 and -9 in angiogenesis, metastasis, and prognosis of endometrial cancer. Onco Targets Ther. 2017;10:4617–4624. doi: 10.2147/OTT.S132558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malik AK, Baldwin ME, Peale F, Fuh G, Liang WC, Lowman H, Meng G, Ferrara N, Gerber HP. Redundant roles of VEGF-B and PlGF during selective VEGF-A blockade in mice. Blood. 2006;107:550–557. doi: 10.1182/blood-2005-05-2047. [DOI] [PubMed] [Google Scholar]
- 6.Cao Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci Signal. 2009;2:re1. doi: 10.1126/scisignal.259re1. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Wang S, He Y, Yao B, Zhang Y. Regulation of matrix metalloproteinases 2 and 9 in corneal neovascularization. Chem Biol Drug Des. 2020;95:485–492. doi: 10.1111/cbdd.13529. [DOI] [PubMed] [Google Scholar]
- 8.Berg G, Schreier L, Miksztowicz V. Circulating and adipose tissue matrix metalloproteinases in cardiometabolic risk environments: pathophysiological aspects. Horm Mol Biol Clin Investig. 2014;17:79–87. doi: 10.1515/hmbci-2013-0069. [DOI] [PubMed] [Google Scholar]
- 9.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44-46:247–254. doi: 10.1016/j.matbio.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Sena CM, Pereira AM, Seica R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013;1832:2216–2231. doi: 10.1016/j.bbadis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman RP. Hyperglycemic endothelial dysfunction: does it happen and does it matter? J Thorac Dis. 2015;7:1693–1695. doi: 10.3978/j.issn.2072-1439.2015.10.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawada N, Arany Z. Metabolic regulation of angiogenesis in diabetes and aging. Physiology (Bethesda) 2017;32:290–307. doi: 10.1152/physiol.00039.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubsam A, Parikh S, Fort PE. Role of inflammation in diabetic retinopathy. Int J Mol Sci. 2018;19:942. doi: 10.3390/ijms19040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenwalt DE, Scheck SH, Rhinehart-Jones T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J Clin Invest. 1995;96:1382–1388. doi: 10.1172/JCI118173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 18.Silverstein RL. Inflammation, atherosclerosis, and arterial thrombosis: role of the scavenger receptor CD36. Cleve Clin J Med. 2009;76(Suppl 2):S27–30. doi: 10.3949/ccjm.76.s2.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. 2014;46:e99. doi: 10.1038/emm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abumrad NA, Goldberg IJ. CD36 actions in the heart: lipids, calcium, inflammation, repair and more? Biochim Biophys Acta. 2016;1861:1442–1449. doi: 10.1016/j.bbalip.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samovski D, Dhule P, Pietka T, Jacome-Sosa M, Penrose E, Son NH, Flynn RC, Shoghi KI, Hyrc KL, Goldberg IJ, Gamazon ER, Abumrad NA. Regulation of insulin receptor pathway and glucose metabolism by CD36 signaling. Diabetes. 2018;67:1272–1284. doi: 10.2337/db17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silverstein RL. Type 2 scavenger receptor CD36 in platelet activation: the role of hyperlipemia and oxidative stress. Clin Lipidol. 2009;4:767. doi: 10.2217/clp.09.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nergiz-Unal R, Lamers MM, Van Kruchten R, Luiken JJ, Cosemans JM, Glatz JF, Kuijpers MJ, Heemskerk JW. Signaling role of CD36 in platelet activation and thrombus formation on immobilized thrombospondin or oxidized low-density lipoprotein. J Thromb Haemost. 2011;9:1835–1846. doi: 10.1111/j.1538-7836.2011.04416.x. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2009;50(Suppl):S86–90. doi: 10.1194/jlr.R800085-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abumrad N, Coburn C, Ibrahimi A. Membrane proteins implicated in long-chain fatty acid uptake by mammalian cells: CD36, FATP and FABPm. Biochim Biophys Acta. 1999;1441:4–13. doi: 10.1016/s1388-1981(99)00137-7. [DOI] [PubMed] [Google Scholar]
- 26.Glatz JF, Luiken JJ. From fat to FAT (CD36/SR-B2): understanding the regulation of cellular fatty acid uptake. Biochimie. 2017;136:21–26. doi: 10.1016/j.biochi.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Marechal L, Laviolette M, Rodrigue-Way A, Sow B, Brochu M, Caron V, Tremblay A. The CD36-PPARγ pathway in metabolic disorders. Int J Mol Sci. 2018;19:1529. doi: 10.3390/ijms19051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silverstein RL, Li W, Park YM, Rahaman SO. Mechanisms of cell signaling by the scavenger receptor CD36: implications in atherosclerosis and thrombosis. Trans Am Clin Climatol Assoc. 2010;121:206–220. [PMC free article] [PubMed] [Google Scholar]
- 29.Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding BS, Schachterle W, Liu Y, Rosenwaks Z, Butler JM, Xiang J, Rafii A, Shido K, Rabbany SY, Elemento O, Rafii S. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. 2013;26:204–219. doi: 10.1016/j.devcel.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramakrishnan DP, Hajj-Ali RA, Chen Y, Silverstein RL. Extracellular vesicles activate a CD36-dependent signaling pathway to inhibit microvascular endothelial cell migration and tube formation. Arterioscler Thromb Vasc Biol. 2016;36:534–544. doi: 10.1161/ATVBAHA.115.307085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 32.Mohr T, Haudek-Prinz V, Slany A, Grillari J, Micksche M, Gerner C. Proteome profiling in IL-1beta and VEGF-activated human umbilical vein endothelial cells delineates the interlink between inflammation and angiogenesis. PLoS One. 2017;12:e0179065. doi: 10.1371/journal.pone.0179065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 34.Huang D, Refaat M, Mohammedi K, Jayyousi A, Al Suwaidi J, Abi Khalil C. Macrovascular complications in patients with diabetes and prediabetes. BioMed Res Int. 2017;2017:1–9. doi: 10.1155/2017/7839101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, Klar J, Genove G, Pietras K, Stone-Elander S, Claesson-Welsh L, Yla-Herttuala S, Lindahl P, Eriksson U. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- 36.Gogiraju R, Bochenek ML, Schafer K. Angiogenic endothelial cell signaling in cardiac hypertrophy and heart failure. Front Cardiovasc Med. 2019;6:20. doi: 10.3389/fcvm.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Son NH, Basu D, Samovski D, Pietka TA, Peche VS, Willecke F, Fang X, Yu SQ, Scerbo D, Chang HR, Sun F, Bagdasarov S, Drosatos K, Yeh ST, Mullick AE, Shoghi KI, Gumaste N, Kim K, Huggins LA, Lhakhang T, Abumrad NA, Goldberg IJ. Endothelial cell CD36 optimizes tissue fatty acid uptake. J Clin Invest. 2018;128:4329–4342. doi: 10.1172/JCI99315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, Stahl PD, Abumrad NA. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes. 2015;64:353–359. doi: 10.2337/db14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, Kassab GS, Garland T Jr, Shyy JY. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. 2006;26:1281–1287. doi: 10.1161/01.ATV.0000221230.08596.98. [DOI] [PubMed] [Google Scholar]
- 40.Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–31006. doi: 10.1074/jbc.M300643200. [DOI] [PubMed] [Google Scholar]
- 41.Xu Q, Si LY. Protective effects of AMP-activated protein kinase in the cardiovascular system. J Cell Mol Med. 2010;14:2604–2613. doi: 10.1111/j.1582-4934.2010.01179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spengler K, Grosse S, Kryeziu N, Knierim A, Heller R. Studying the role of AMPK in angiogenesis. Methods Mol Biol. 2018;1732:519–537. doi: 10.1007/978-1-4939-7598-3_33. [DOI] [PubMed] [Google Scholar]
- 43.Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, Murfee WL, Pacher P, Csiszar A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. 2018;15:555–565. doi: 10.1038/s41569-018-0030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bierhansl L, Conradi LC, Treps L, Dewerchin M, Carmeliet P. Central role of metabolism in endothelial cell function and vascular disease. Physiology (Bethesda) 2017;32:126–140. doi: 10.1152/physiol.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackson CJ, Nguyen M. Human microvascular endothelial cells differ from macrovascular endothelial cells in their expression of matrix metalloproteinases. Int J Biochem Cell Biol. 1997;29:1167–1177. doi: 10.1016/s1357-2725(97)00061-7. [DOI] [PubMed] [Google Scholar]
- 46.Jayawardena DP, Kulkarni NP, Gill SE. The role of tissue inhibitors of metalloproteinases in microvascular endothelial cell barrier dysfunction during sepsis. Metalloproteinases in Medicine. 2019;6:1–12. [Google Scholar]
- 47.Hollborn M, Stathopoulos C, Steffen A, Wiedemann P, Kohen L, Bringmann A. Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest Ophthalmol Vis Sci. 2007;48:4360–4367. doi: 10.1167/iovs.06-1234. [DOI] [PubMed] [Google Scholar]
- 48.Oh J, Seo DW, Diaz T, Wei B, Ward Y, Ray JM, Morioka Y, Shi S, Kitayama H, Takahashi C, Noda M, Stetler-Stevenson WG. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res. 2004;64:9062–9069. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- 49.Li W, Li N, Song D, Rong J, Qian A, Li X. Metformin inhibits endothelial progenitor cell migration by decreasing matrix metalloproteinases, MMP-2 and MMP-9, via the AMPK/mTOR/autophagy pathway. Int J Mol Med. 2017;39:1262–1268. doi: 10.3892/ijmm.2017.2929. [DOI] [PubMed] [Google Scholar]
- 50.Escudero CA, Herlitz K, Troncoso F, Guevara K, Acurio J, Aguayo C, Godoy AS, Gonzalez M. Pro-angiogenic role of insulin: from physiology to pathology. Front Physiol. 2017;8:204. doi: 10.3389/fphys.2017.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kondo T, Vicent D, Suzuma K, Yanagisawa M, King GL, Holzenberger M, Kahn CR. Knockout of insulin and IGF-1 receptors on vascular endothelial cells protects against retinal neovascularization. J Clin Invest. 2003;111:1835–1842. doi: 10.1172/JCI17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rask-Madsen C, Kahn CR. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2012;32:2052–2059. doi: 10.1161/ATVBAHA.111.241919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Nakayama K, Ikeda K, Motoyama N, Mori N. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- 54.Rudnicki M, Abdifarkosh G, Nwadozi E, Ramos SV, Makki A, Sepa-Kishi DM, Ceddia RB, Perry CGR, Roudier E, Haas TL. Endothelial-specific FoxO1 depletion prevents obesity-related disorders by increasing vascular metabolism and growth. ELife. 2018;7:e39780. doi: 10.7554/eLife.39780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Draoui N, de Zeeuw P, Carmeliet P. Angiogenesis revisited from a metabolic perspective: role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017;7:170219. doi: 10.1098/rsob.170219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stahmann N, Woods A, Spengler K, Heslegrave A, Bauer R, Krause S, Viollet B, Carling D, Heller R. Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase. J Biol Chem. 2010;285:10638–10652. doi: 10.1074/jbc.M110.108688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Omura J, Satoh K, Kikuchi N, Satoh T, Kurosawa R, Nogi M, Otsuki T, Kozu K, Numano K, Suzuki K, Sunamura S, Tatebe S, Aoki T, Sugimura K, Miyata S, Hoshikawa Y, Okada Y, Shimokawa H. Protective roles of endothelial AMP-activated protein kinase against hypoxia-induced pulmonary hypertension in mice. Circ Res. 2016;119:197–209. doi: 10.1161/CIRCRESAHA.115.308178. [DOI] [PubMed] [Google Scholar]
- 58.Li C, Reif MM, Craige SM, Kant S, Keaney JF Jr. Endothelial AMPK activation induces mitochondrial biogenesis and stress adaptation via eNOS-dependent mTORC1 signaling. Nitric Oxide. 2016;55-56:45–53. doi: 10.1016/j.niox.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–1244. doi: 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dagher Z, Ruderman N, Tornheim K, Ido Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ Res. 2001;88:1276–1282. doi: 10.1161/hh1201.092998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.