

Abstract
Background
Tandem repeats are highly mutable and contribute to the development of human disease by a variety of mechanisms. It is difficult to predict which tandem repeats may cause a disease. One hypothesis is that changeable tandem repeats are the source of genetic diseases, because disease-causing repeats are polymorphic in healthy individuals. However, it is not clear whether disease-causing repeats are more polymorphic than other repeats.
Methods
We performed a genome-wide survey of the millions of human tandem repeats using publicly available long read genome sequencing data from 21 humans. We measured tandem repeat copy number changes using tandem-genotypes. Length variation of known disease-associated repeats was compared to other repeat loci.
Results
We found that known Mendelian disease-causing or disease-associated repeats, especially CAG and 5′UTR GGC repeats, are relatively long and polymorphic in the general population. We also show that repeat lengths of two disease-causing tandem repeats, in ATXN3 and GLS, are correlated with near-by GWAS SNP genotypes.
Conclusions
We provide a catalog of polymorphic tandem repeats across a variety of repeat unit lengths and sequences, from long read sequencing data. This method especially if used in genome wide association study, may indicate possible new candidates of pathogenic or biologically important tandem repeats in human genomes.
Keywords: Nanopore long read sequencing, Tandem repeats, Triplet repeat disease, Genome-wide analysis
Background
There are more than 30 rare Mendelian diseases caused by tandem repeat expansions in human genomes [1]. Genome-wide surveys of tandem repeats in individual genomes are now feasible due to the development of high-throughput sequencing technologies, which enable direct identification of large pathogenic expansions [2–4]. However, it is still difficult to predict which tandem repeats cause disease, because there are thousands of tandem repeats in each individual that are different from the reference genome. Usually pathogenic expansions are + 100 to ~ 10,000 base-pairs, and the risk cutoff is beyond ~ 100 base-pairs [1, 2]. Some disease-causing repeats are polymorphic even in healthy individuals [5]. If disease-causing tandem repeats have distinct variation in the general population, compared to other repeats, that would help identify novel disease-causing repeat candidates.
Although tandem repeats are highly mutable and can affect phenotype, they are rarely considered in genome-wide association studies (GWAS). GWAS has found many polymorphisms that have significant but weak association with phenotypes, so far failing usually to give satisfying genetic explanations of the phenotypes. As tandem repeats’ rapid evolution causes them to have weak association with nearby polymorphisms, we may hypothesize that repeats explain these phenotypes, as represented in previous studies [6, 7].
Current genome-wide studies of tandem repeats using short read sequencers are mainly focusing on short repeats (repeat unit range: 1–6 bp) [8] due to the limitation of detecting long repeats. Current long read sequencing technologies (PacBio and Nanopore) have achieved reads longer than 10 kb on average, which have a high chance to cover whole tandem repeats including flanking unique sequences [9, 10]. However, to the best of our knowledge, there has been no study that characterizes the genotypic variation of disease-causing and other tandem repeats using only long reads.
Until recently, most of the known disease-causing tandem repeats are CAG or GGC triplet repeats [1], although there are a few exceptions; quadruplet repeat (CCTG) in Myotonic Dystrophy type 2 (MIM#602668), and sextuplet repeat (GGGGCC) in Frontotemporal dementia and/or amyotrophic lateral sclerosis (ALS) (MIM#614260). CAG and GGC triplet diseases have three major disease mechanisms: poly-glutamine diseases (CAG), poly-alanine diseases (GGC), or 5′UTR GGC expansion diseases [11–13]. In addition to triplet repeats, pathogenic expansions of quintuplet repeat loci (represented as AAAAT in hg38) are associated with myoclonic epilepsies. In 2018 and 2019, six AAAAT repeat loci were reported [4, 14–16] in addition to BEAN1 which causes spinocerebellar ataxia 31 (MIM#117210) [17]. We focus on these triplet and quintuplet repeats so that we can test several disease loci.
Our recently developed tool, tandem-genotypes, can robustly detect tandem repeat changes from whole genome long read sequencing data [18]. Here, we used this tool to measure tandem repeats in publicly available nanopore long read whole genome sequencing data. We show that certain types of disease-causing tandem repeats have greater length variation than other repeats.
Methods
Long read sequencing and mapping to the reference genome
We used 21 long read whole genome sequencing datasets, from 21 humans (Additional file 1: Table S1). Fifteen of these are from previous studies [10, 19, 20]. The other six were sequenced by our group, using Nanopore PromethION as previously described [3], with DNA obtained from lymphoblastoid cell lines from the Coriell Institute Cell Repository (coriell.org). Reads were mapped to the human reference genome GRCh38 using LAST according to the instructions (https://github.com/mcfrith/last-rna/blob/master/last-long-reads.md), with repeat-masked reference genome.
last-train GRCh38 data.fa > train-out
lastal -p train-out GRCh38 data.fa | last-split > alignment.maf
Tandem repeat detection
Tandem repeats in the human reference genome GRCh38 were detected using tantan (http://cbrc3.cbrc.jp/~martin/tantan/) [21], with this command:
tantan -f4 -w2000 GRCh38.fa > tantan-out
Prediction of tandem repeat copy number changes relative to the reference
Tandem-repeat copy number changes relative to the reference were predicted using tandem-genotypes. We used one non-default parameter, n = 10 instead of n = 60, to make it more specific but less sensitive. This is because the precise boundaries of (inexact) repeats are ambiguous: n = 10 makes it less likely to regard an insertion near a repeat as an expansion of the repeat, but more likely to miss expansions of repeats with fuzzy boundaries [18]. Disease-associated tandem repeats were analyzed separately, using the repeat annotations in Table 1.
Table 1.
Variability of triplet- and quintuplet-repeat disease locus in 21 individuals detected by long read nanopore sequencing. OMIM: Online Mendelian Inheritance in Men
| Chromosome | Start | End | Repeat | Gene | Function | Repeat copy number change from the reference | Absolute repeat length | Disease | OMIM | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Median | IQR | Q1 | Q3 | Mean length (bp) | Q1 length (bp) | Q3 length (bp) | ||||||||
| chr2 | 176,093,058 | 176,093,103 | GGC | HOXD13 | Coding | − 0.1 | 0 | 1.00 | − 1.00 | 0.00 | 45 | 42 | 45 | Syndactyly, type V | 186,300 |
| chr4 | 41,745,971 | 41,746,031 | GGC | PHOX2B | Coding | 0.0 | 0 | 2.00 | − 1.00 | 1.00 | 60 | 57 | 63 | Congenital central hypoventilation syndrome | 209,880 |
| chr6 | 45,422,750 | 45,422,801 | GGC | RUNX2 | Coding | − 0.7 | 0 | 1.00 | − 1.00 | 0.00 | 49 | 48 | 51 | Cleidocranial dysplasia, forme fruste, with brachydactyly | 119,600 |
| chr7 | 27,199,924 | 27,199,966 | GGC | HOXA13 | Coding | 0.0 | 0 | 2.00 | − 1.00 | 1.00 | 42 | 39 | 45 | Hand-foot-genital syndrome | 140,000 |
| chr13 | 99,985,448 | 99,985,493 | GGC | ZIC2 | Coding | 0.1 | 0 | 2.00 | − 1.00 | 1.00 | 45 | 42 | 48 | Holoprosencephaly | 609,637 |
| chr14 | 23,321,472 | 23,321,502 | GGC | PABPN1 | Coding | − 0.1 | 0 | 1.00 | − 1.00 | 0.00 | 30 | 27 | 30 | Oculopharyngeal muscular dystrophy | 164,300 |
| chrX | 25,013,649 | 25,013,697 | GGC | ARX | Coding | 0.2 | 0 | 1.00 | 0.00 | 1.00 | 49 | 48 | 51 | Early infantile epileptic encephalopathy | 308,350 |
| chrX | 140,504,316 | 140,504,361 | GGC | SOX3 | Coding | 0.0 | 0 | 2.00 | − 1.00 | 1.00 | 45 | 42 | 48 | Mental retardation with isolated growth hormone deficiency | 300,123 |
| chr1 | 149,390,802 | 149,390,842 | GGC | NOTCH2NLC | 5′UTR | 6.7 | 6 | 5.00 | 3.00 | 8.00 | 60 | 49 | 64 | Neuronal intranuclear inclusion disease | 603,472 |
| chr8 | 104,588,965 | 104,588,999 | CCG | LRP12 | 5′UTR | 0.3 | 0 | 2.00 | − 1.00 | 1.00 | 35 | 31 | 37 | Ocuropharyngodistal myopathy | 164,310 |
| chr10 | 79,826,315 | 79,826,404 | GGC | LOC642361,NUTM2B-AS1 | Non coding exon | 2.6 | 3 | 3.00 | 1.00 | 4.00 | 97 | 92 | 101 | Ocuropharyngodistal myopathy | 618,637 |
| chr19 | 14,496,042 | 14,496,085 | CCG | GIPC1 | 5′UTR | 1.6 | 0 | 3.00 | − 1.00 | 2.00 | 48 | 40 | 49 | Ocuropharyngodistal myopathy | 618,940 |
| chrX | 147,912,050 | 147,912,110 | GGC | FMR1 | 5′UTR | 8.7 | 9 | 5.00 | 6.00 | 11.00 | 86 | 78 | 93 | Fragile X syndrome/tremor-ataxia syndrome | 300,624/300,623 |
| chrX | 148,500,637 | 148,500,682 | GGC | AFF2 | 5′UTR | 2.2 | 2 | 5.00 | 0.00 | 5.00 | 52 | 45 | 60 | Fragile X syndrome | 309,548 |
| chr3 | 63,912,685 | 63,912,715 | CAG | ATXN7 | Coding | 0.2 | 0 | 2.00 | − 1.00 | 1.00 | 30 | 27 | 33 | Spinocerebellar ataxia 7 | 164,500 |
| chr4 | 3,074,876 | 3,074,939 | CAG | HTT | Coding | − 1.2 | − 2 | 3.00 | − 3.00 | 0.00 | 59 | 54 | 63 | Huntington disease | 143,100 |
| chr6 | 16,327,635 | 16,327,722 | CAG | ATXN1 | Coding | − 1.1 | − 1 | 4.00 | − 3.00 | 1.00 | 84 | 78 | 90 | Spinocerebellar ataxia 1 | 164,400 |
| chr6 | 170,561,907 | 170,562,021 | CAG | TBP | Coding | − 2.7 | − 3 | 3.00 | − 4.00 | − 1.00 | 106 | 102 | 111 | Spinocerebellar ataxia 17 | 607,136 |
| chr12 | 6,936,716 | 6,936,773 | CAG | ATN1 | Coding | − 2.5 | − 2 | 5.00 | − 5.00 | 0.00 | 49 | 42 | 57 | Dentatorubral-pallidoluysian atrophy | 125,370 |
| chr12 | 111,598,950 | 111,599,019 | CAG | ATXN2 | Coding | − 1.4 | − 1 | 1.00 | − 2.00 | − 1.00 | 65 | 63 | 66 | Spinocerebellar ataxia 2 | 183,090 |
| chr14 | 92,071,010 | 92,071,040 | CAG | ATXN3 | Coding | 7.2 | 8 | 10.00 | 1.00 | 11.00 | 51 | 33 | 63 | Spinocerebellar ataxia 3 | 109,150 |
| chr19 | 13,207,858 | 13,207,897 | CAG | CACNA1A | Coding | − 2.0 | − 2 | 2.00 | − 3.00 | -1.00 | 33 | 30 | 36 | Spinocerebellar ataxia 6 | 183,086 |
| chrX | 67,545,317 | 67,545,386 | CAG | AR | Coding | − 1.7 | − 2 | 5.00 | − 4.00 | 1.00 | 64 | 57 | 72 | Spinal and Bulbar Muscular Atrophy | 313,200 |
| chr2 | 190,880,868 | 190,880,920 | GCA | GLS | 5′UTR | − 4.3 | − 3 | 6.75 | − 8.00 | − 1.25 | 39 | 28 | 48 | Global developmental delay, progressive ataxia, and elevated glutamine | 618,412 |
| chr18 | 55,586,153 | 55,586,229 | AGC | TCF4 | 5′UTR | − 5.5 | − 7 | 9.00 | − 10.00 | − 1.00 | 59 | 46 | 73 | Fuchs corneal dystrophy | 602,272 |
| chr19 | 45,770,204 | 45,770,264 | CAG | DMPK | 3′UTR | − 9.4 | − 9 | 5.00 | − 12.00 | − 7.00 | 32 | 24 | 39 | Myotonic dystrophy 1 | 160,900 |
| chr2 | 96,197,066 | 96,197,124 | AAAAT | STARD7 | intron | 2.9 | 1 | 3.00 | 0.00 | 3.00 | 72 | 58 | 73 | Myoclonic epilepsy | 607,876 |
| chr3 | 183,712,187 | 183,712,226 | TTTTA | YEATS2 | intron | 11.0 | 2 | 30.00 | 0.00 | 30.00 | 94 | 39 | 189 | Myoclonic epilepsy | 615,127 |
| chr4 | 159,342,526 | 159,342,618 | AAAAT | RAPGEF2 | Intron | 0.5 | 1 | 1.00 | 0.00 | 1.00 | 94 | 92 | 97 | Epilepsy, familial adult myoclonic, 7 | 618,075 |
| chr5 | 10,356,339 | 10,356,411 | AAAAT | MARCHF6 | Intron | 0.4 | 0 | 1.00 | 0.00 | 1.00 | 74 | 72 | 77 | Myoclonic epilepsy | 613,608 |
| chr8 | 118,366,815 | 118,366,918 | AAAAT | SAMD12 | Intron | − 0.8 | − 1 | 3.00 | − 2.00 | 1.00 | 99 | 93 | 108 | Epilepsy, familial adult myoclonic, 1 | 601,068 |
| chr16 | 24,613,438 | 24,613,532 | AAAAT | TNRC6A | Intron | − 5.0 | − 5 | 2.00 | − 6.00 | − 4.00 | 69 | 64 | 74 | Epilepsy, familial adult myoclonic, 6 | 618,074 |
| chr16 | 66,490,396 | 66,490,466 | AAAAT | BEAN1 | Intron | 24.1 | 3 | 7.00 | − 1.00 | 6.00 | 190 | 65 | 100 | Spinocerebellar ataxia 31 | 117,210 |
tandem-genotypes -n10 -g refFlat.txt tantan-out alignnment.maf > out
All tandem-genotypes output files from 21 datasets were merged like this:
tandem-genotypes-join file1 file2 file3… > merged-file
IQR and mean length were calculated from tandem-genotypes output using GNU datamash (https://www.gnu.org/software/datamash/).
Repeat disease selection
We selected triplet-repeat and quintuplet-repeat diseases, because several diseases are known in this category. We took these repeats from a previously published article [1], and recently discovered repeat diseases were added by manual literature search.
Phasing the repeat and near-by GWAS SNP
Phasing of a disease-associated (ATXN3 or GLS) tandem-repeat and nearby GWAS SNP (< 10 kb) [22] was done from consensus sequences of the DNA reads. Briefly, a repeat’s copy number in each of the two alleles was estimated by tandem-genotypes, then the reads from the two alleles were merged into two consensus sequences, and re-aligned to the reference genome. tandem-genotypes-merge merges these reads using lamassemble [23, 24]:
tandem-genotypes -o2 -v repeat-locus alignnment.maf > out
tandem-genotypes-merge reads.fa train-out out > merged.fa
Results
We identified tandem repeats in a human reference genome (GRCh38) using tantan [21] (http://cbrc3.cbrc.jp/~martin/tantan/). In total, 3,347,418 loci were identified, with the repeat units ranging from 1 to 2000 bp. We used 21 publicly available long read whole genome sequencing datasets (we suppose they do not have pathogenic tandem repeat expansions), with average coverage of 27x (ranging 8x–48x, Additional file 1: Table S1). tandem-genotypes predicted lengths for more than 98% of the 3 million tandem repeats (Additional file 1: Table S1), including 215,561 triplet repeats.
We investigated 12 CAG and 14 GGC triplet repeat and 7 AAATA quintuplet repeat disease loci (Table 1), and plotted the distribution of copy number changes from the reference in all the reads. We found that disease-causing repeats show different distribution from other non-disease repeats (Additional file 2: Fig S1A–C). We randomly extracted the same number of non-disease repeat loci for comparison to the disease repeat loci (CAG: n = 12, GGC: n = 14, AAAAT: n = 7) (Additional file 2: Figure S1). This supports our hypothesis that disease-causing tandem repeats are more polymorphic among the normal population than other loci.
Given that different repeat sequences may have different mutation rates [25], we compared the ten kinds of non-disease triplet repeats (All triplet repeats can be categorized into 10 kinds. Note that AAC repeats includes AAC, ACA, CAA, GTT, TGT, TTG repeats) (Additional file 2: Figure S2). We plotted the variation of repeat length (interquartile range (IQR) of repeat-unit count from each read), and mean repeat length, at each exonic locus (including UTR). Most of the non-disease triplet repeats have little or no length polymorphism. A large fraction (> 94% of all repeats) have IQR 2 or less, while disease causing tandem repeats usually show more variation (always more than 2) (Table 1). It is of interest that GGC and CAG repeats have more polymorphic loci than other repeat structures (Additional file 2: Figure S2). In addition, shorter-unit repeats are more numerous and more variable (Additional file 2: Figure S3 A, B). Therefore, we analyzed the variation (IQR) and repeat length for disease causing repeats in comparison to other repeats considering the repeat unit and repeat location.
Disease-associating CAG repeats are longer and more variable than most other CAG repeats (Fig. 1a, b, Table 1). We showed coding and non-coding repeats separately (A: coding, B: non-coding). All disease-causing CAG repeats are located in protein-coding regions except for DMPK, GLS, and TCF4 which are in 5′UTR (Table 1). Next we tested GGC repeats. Disease-causing 5′-UTR GGC loci are long and variable (Fig. 2b) but protein-coding regions are long but show less variability (Fig. 2a). Gene names were used to indicate the disease-causing repeats because the pathogenic repeats are present only once in each gene. All known protein-coding GGC repeat diseases are located at poly-alanine tracts. This may reflect the difference in disease mechanisms of protein-coding versus 5′-UTR GGC repeats or protein-coding GGC versus CAG repeats. Next, we examined the variation and length of all intronic AAAAT repeat loci in 21 individuals, and found several highly polymorphic AAAAT repeats including disease loci (Fig. 3, Table 1).
Fig. 1.

Variation (IQR, interquartile range) and length of repeats with disease-associated sequences. coding CAG repeats (a), and non-coding exonic CAG repeats (b). x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median
Fig. 2.

Variation (IQR) and length of repeats with disease-associated sequences. coding GGC repeats (a), and non-coding exonic GGC repeats (b). x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median
Fig. 3.
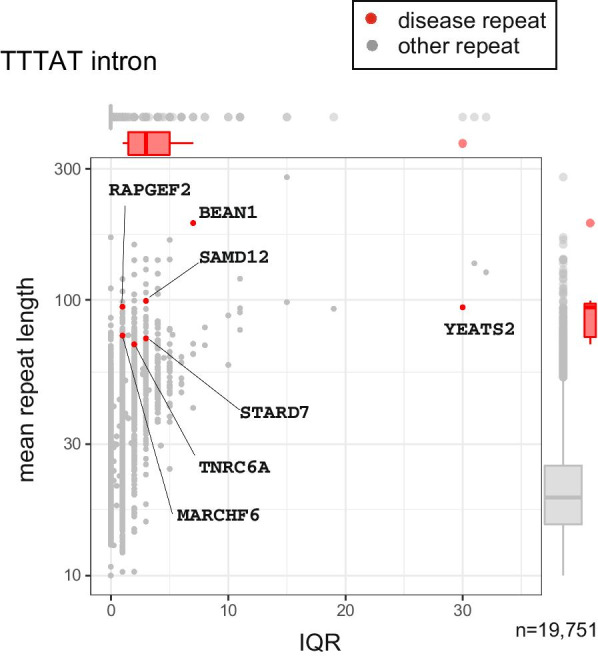
Variation (IQR) and length of repeats with disease-associated intronic AAAAT sequences. x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median
We repeated our analysis using repeat annotations from Tandem Repeats Finder (TRF, a.k.a. simpleRepeat.txt) [26]. TRF annotates fewer repeats than tantan (Additional file 2: Figure S4A), however, the proportion of triplet repeat sequences is similar (Additional file 2: Figure S4B). Numbers of intersections between these annotations were calculated using bedtools v2.27.1 (Additional file 2: Table S2). We analyzed disease-associated CAG and GGC repeats, and observed similar results to tantan-annotated repeats (Additional file 2: Figure S5: CAG, S6: GGC, S7: AAAAT).
Next, we tested if polymorphic disease-associated tandem repeats are correlated with reported GWAS SNPs. We tested ATXN3 and GLS disease-associated repeats because they are highly polymorphic among disease-associated CAG repeats. These repeats have two (rs12588287: coronary artery calcification [27], rs10143310: ALS [28]) and one (rs4853525: reticulocyte count [29]) near-by GWAS SNPs (< 10 kb) [22], respectively. Due to the limited coverage and read length, we could obtain genotypes in most but not all of the 21 cases (Additional file 1: Table S3 A, B, C). In each case, one of the two SNP alleles is significantly (p < 0.05, unpaired t test) associated with longer repeats (Additional file 2: Figure S8). Risk alleles tend to occur with shorter repeats for two SNPs: rs4853525-C and rs12588287-T. Risk allele for rs10143310 is not available [28]. This merits further investigation by genotyping a larger number of individuals.
Finally, we listed highly polymorphic repeats (IQR ≥ 5) which have very near GWAS signals (< 100 bp) from a GWAS catalog [22] (Additional file 1: Table S4). We found an interesting candidate, an intronic repeat in the CLN8 gene: a SNP within this repeat (rs11986414) and a near-by SNP (rs4875960) are reported to be associated with severity of Gaucher syndrome [30]. It is an intriguing possibility that this repeat genuinely acts as a driver of the GWAS signals and affects the disease severity. We found that the A genotypes of these two SNPs are correlated with shorter repeat (Additional file 2: Figure S9). It would be interesting to investigate functional consequences of changing these repeats. These speculative examples need further association studies targeting near-by tandem repeats together with functional studies to elucidate the mechanistic relation to the phenotype.
Discussion
We showed that CAG, non-coding GGC and intronic AAAAT disease-associated tandem-repeats are polymorphic and long compared to other repeats using whole genome long read sequencing data. However, coding GGC repeats did not show such variability, although the repeat lengths were longer than other repeats. It is known that poly-alanine is toxic to cells [31] and usually fewer than 10 additional alanine residues are enough to cause disease [2]. This may explain our observation that alanine-coding GGCs are less variable in the general population. In contrast, disease-associated 5′UTR GGCs are more polymorphic. One possible pathomechanism of 5′UTR GGC repeats is gene suppression as seen in fragile X syndrome [11]. Another envisioned mechanism is repeat associated non-AUG translation, which is suspected in the neurological symptoms in patients with FMR1 premutation (more than 55 GGC repeats) [32]. The different mechanisms may reflect different variation patterns of disease-causing GGC repeats. Quintuplet AAAAT repeat loci are associated with newly-discovered types of disease, and pathomechanisms of AAAAT repeat expansions are yet unclear [15]. We also showed that there are several highly polymorphic AAAAT repeats which may be undiscovered pathogenic repeats for epilepsy.
GWAS have identified numerous genomic markers over the past fifteen years, however their functional relation to the diseases or traits is usually unclear. It is plausible that tandem repeats near those GWAS markers actually have functional relation to the traits. Interestingly, some repeat expansion disease loci may be associated with multiple diseases or traits, even when the repeat length is within the normal range [33, 34]. It is reported that polymorphic tandem repeats contribute to gene expression variation [35]. A recent study showed that tandem repeats which can alter expression of near-by genes are potential drivers of published GWAS signals [36]. Fotsing et al. listed 1380 such tandem repeats as eSTR (repeats associated with the expression of nearby genes) [36], although no Mendelian disease-causing repeats are included in eSTR, possibly because most of the known repeat diseases may not be caused by altering gene expression levels but by changing protein products. However, there may be other diseases or traits caused by altering gene expression, like Fragile X syndrome.
Importantly, among disease associated CAG repeats, the noncoding repeat in TCF4 has high IQR. This triplet repeat was known to be highly polymorphic [37], in agreement with our result. This repeat has an association with Fuchs endothelial corneal dystrophy (FECD) (MIM#613267) [38]. Initially, GWAS showed an association of a SNP (rs613872), but later studies showed this disease has much higher association to a 43 kb-downstream CAG repeat which is in linkage disequilibrium with the GWAS SNP [6, 7]. It is intriguing to consider that further studies on polymorphic repeats may lead to the discovery of true pathogenic variants from GWAS SNPs. However, it is reported that tandem repeats with multiple genotypes are poorly tagged with SNPs [39]. Nevertheless, some repeat expansion diseases are known to be linked to certain haplotypes [40, 41], although there are repeat expansions that do not share haplotype or occur de novo [42]. We showed some examples in this study. The first example is a 5′UTR GCA repeat in the GLS gene, which is highly polymorphic and also listed as an eSTR [36]. Expansions (> ~ 680 repeats) are known to cause deficiency of GLS and linked to neurological disease [43]. Several lines of evidence show that an 8 kb-downstream SNP is associated with reticulocyte count (Additional file 1: Table S3 C). We showed that this SNP is correlated with repeat length. GLS encodes glutaminase, which catalyzes glutamine conversion to glutamate, has high activity in red blood cells (erythrocytes), and plays a role in glutathione metabolism [44, 45]. There is an intriguing possibility that this 5′UTR repeat actually acts as a driver of the GWAS signal and affects reticulocyte-erythrocyte maturation by altering the expression of GLS thus affecting glutathione metabolism. The next example is ATXN3. We found two near-by GWAS SNPs, including one associated with ALS, are significantly correlated with repeat length. Since another spinocerebellar ataxia repeat in ATXN2 is associated with ALS, this locus is of interest. A final example is the Gaucher disease severity associated SNPs in and near the polymorphic repeat in an intron of CLN8. These speculative examples need further association studies targeting near-by tandem repeats together with functional studies to elucidate the mechanistic relation to the phenotype.
Conclusion
In conclusion, our results indicate that known disease-associated coding CAG repeats, 5′UTR GGC repeats, and intronic AAAAT repeats are long and variable, but alanine-coding GGC repeats are stable (but long) among the 21 individuals. Our study is limited due to lack of a large number of healthy individuals from multiple ethnicities. Nevertheless, we provide a first example of applying long read sequencing to identify polymorphic tandem repeats. We believe further tandem-repeat surveys using a large number of individuals may provide more insights into human genomes and diseases.
Supplementary Information
Additional file 1. Table S1. Data sets used in this study.Detection rates are the number of tandem repeats whose length is predicted from at least one DNA read. *3,312,291 loci. Table S2. Comparison of tantan and TRF annotated tandem repeatsNumber of intersections are counted using bedtools (https://bedtools.readthedocs.io/en/latest/). Each repeat unit was counted separately. Table S3. Phased repeat length and near-by SNPs.SNP genotype on the short and long alleles were shown in each dataset. Repeat copy number changes on both alleles were genotyped using tandem-genotypes. (A) ATXN3 repeat and (B, C) GLS repeat. SNP rs numbers are from dbSNP (https://www.ncbi.nlm.nih.gov/snp/). Table S4. Polymorphic tandem repeat and near-by GWAS signals.Highly polymorphic exonic triplet repeats (IQR ≥ 5) with near GWAS signals (< 100bp). GWAS signals were from a GWAS catalog [22].
Additional file 2. Figure S1. Variation of tandem repeat length (copy number) in long reads from 21 individuals. x-axis: copy number change relative to the human reference (hg38). y-axis: read number. Three different repeat types ((A) exonic CAG, (B) exonic GGC and (C) intronic AAAAT) are shown. Disease repeats: 12 CAG repeats, 14 GGC repeats, and 7 intronic AAAAT repeats. Other repeats: exonic CAG: n = 1840, exonic GGC: n = 3073, intronic AAAAT: n = 19,744. For each repeat type, we show ten sets of “other repeats” for comparison. Each set of “other repeats” is a random selection of the same number of repeats as the number of disease repeats. Figure S2. Repeat length mean and spread of each triplet repeat type. There are 10 kinds of triplet repeat, AAC, CAC, CCT, CTT, GAT, GTA, GTC, TAA CAG and GGC. The numbers of repeats are; 650 (AAC), 632 (CAC), 1862 (CCT), 737 (CTT), 430 (GAT), 47 (GTA), 73 (GTC), 682 (TAA), 1839 (CAG) and 2907 (GGC). The variation of the repeat length in 21 individuals are shown. x-axis: interquartile range (IQR), y-axis: mean repeat length (bp). In merged boxplots, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S3. Variability of exonic repeats. Number of exonic tandem repeats (A) and IQR (B) of each unit are shown. Shorter-unit repeats have more variation. Dots represent outliers. Boxplot ranges are the 25th and 75th percentiles. Lines in boxes are median. Most of the IQRs from repeats whose length are more than six are zero. Figure S4. Comparison of tantan and TRF annotated tandem repeats (A) There are more tantan annotated tandem-repeats than TRF-annotated repeats, however, the distribution of the number of the loci has similar tendency. x-axis: length of repeat unit, y-axis: number of loci. (B) Proportions of triplet repeat sequences are similar between tantan and TRF annotation. Figure S5. Variability of CAG repeats using TRF-annotated repeats.Variation (IQR) and length of repeats with disease-associated sequences. Coding CAG repeats (A), and non-coding exonic CAG repeats (B). x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S6. Variability of GGC repeats using TRF-annotated repeats.Variation (IQR) and length of repeats with disease-associated sequences. Coding GGC repeats (A), and non-coding exonic GGC repeats (B). x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S7. Variability of AAAAT repeats using TRF-annotated repeats.Variation (IQR) and length of repeats with disease-associated intronic AAAAT sequences. x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S8. Repeat length correlates with near-by SNPs. (A) Distribution of tandem repeat length (copy number) in combined long reads of GLS and ATXN3 disease-associated repeats from 21 individuals. x-axis: copy number change relative to the human reference (hg38). y-axis: read count. (B) Three GWAS reported SNPs were near the GLS and ATXN3 repeats. Repeat lengths of each genotype were compared using unpaired t test. P values are shown. Figure S9. Gaucher disease severity modifying SNPs correlate with repeat length.(A) Distribution of tandem repeat length (copy number) in combined long reads of CLN8 from 21 individuals. Note that there is bimodal distribution, with peaks around zero and − 13 copy number changes. x-axis: copy number change relative to the human reference (hg38). y-axis: read count. (B) One GWAS reported SNP rs11986414 is inside and another SNP rs4875960 is near this repeat. In both SNPs, genotype A tends to have larger repeat length.
Acknowledgements
Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Web resources
tantan (http://cbrc3.cbrc.jp/~martin/tantan/). tandem-genotypes (https://github.com/mcfrith/tandem-genotypes). lamassemble (https://gitlab.com/mcfrith/lamassemble/blob/master/lamassemble). bedtools (https://bedtools.readthedocs.io/en/latest/).
Extended data
IQR data for all the repeat loci except homopolymers.
Abbreviations
- GWAS
Genome-wide association studies
- SNP
Single nucleotide polymorphism
- IQR
Interquartile range
- FECD
Fuchs endothelial corneal dystrophy
Authors’ contributions
SM, MCF, and NM contributed to the conception of the work and acquisition/analysis/interpretation of the data. All authors have read and approved the manuscript.
Funding
This work was supported by AMED under the grant numbers JP20ek0109486, JP20dm0107090, JP20ek0109301, JP20ek0109348, JP20kk0205012 (to N. Matsumoto); JSPS KAKENHI under the grant numbers JP17H01539 (to N. Matsumoto) and JP19K07977 and 16H06279 (PAGS) (to S. Mitsuhashi); intramural grants of NCNP from the Ministry of Health, Labor, and Welfare (30–6 and 30–7) (to N. Matsumoto); and the Takeda Science Foundation (to N. Matsumoto). The funding body had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
PromethION WGS sequence data is available from DDBJ (DRA009852). Other public data were downloaded from NCBI or Human PanGenome Project (https://github.com/human-pangenomics/hpgp-data) under accession numbers described in Table S1. SNP identifiers were used under accession numbers from dbSNP (https://www.ncbi.nlm.nih.gov/snp/).
Ethics approval and consent to participate
All genomic DNA were examined after obtaining written informed consent. Experimental protocols were approved by institutional review board of Yokohama City University under the number of A19080001.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Satomi Mitsuhashi, Email: satomits.gfd@mri.tmd.ac.jp.
Naomichi Matsumoto, Email: naomat@yokohama-cu.ac.jp.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-020-00853-3.
References
- 1.Tang H, Kirkness EF, Lippert C, Biggs WH, Fabani M, Guzman E, Ramakrishnan S, Lavrenko V, Kakaradov B, Hou C, et al. Profiling of short-tandem-repeat disease alleles in 12,632 human whole genomes. Am J Hum Genet. 2017;101:700–715. doi: 10.1016/j.ajhg.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitsuhashi S, Matsumoto N. Long-read sequencing for rare human genetic diseases. J Hum Genet. 2020;65:11–19. doi: 10.1038/s10038-019-0671-8. [DOI] [PubMed] [Google Scholar]
- 3.Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, Koike H, Hashiguchi A, Takashima H, Sugiyama H, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. 2019;51:1215–1221. doi: 10.1038/s41588-019-0459-y. [DOI] [PubMed] [Google Scholar]
- 4.Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, Toyoshima Y, Kakita A, Takahashi H, Suzuki Y, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50:581–590. doi: 10.1038/s41588-018-0067-2. [DOI] [PubMed] [Google Scholar]
- 5.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs’ endothelial corneal dystrophy. Investig Ophthalmol Vis Sci. 2014;55:33–42. doi: 10.1167/iovs.13-12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS ONE. 2012;7:e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gymrek M. A genomic view of short tandem repeats. Curr Opin Genet Dev. 2017;44:9–16. doi: 10.1016/j.gde.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 9.Wenger AM, Peluso P, Rowell WJ, Chang PC, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–345. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- 12.Amiel J, Trochet D, Clement-Ziza M, Munnich A, Lyonnet S. Polyalanine expansions in human. Hum Mol Genet. 2004;13(Spec No 2):R235–243. doi: 10.1093/hmg/ddh251. [DOI] [PubMed] [Google Scholar]
- 13.Adegbuyiro A, Sedighi F, Pilkington AW, Groover S, Legleiter J. Proteins containing expanded polyglutamine tracts and neurodegenerative disease. Biochemistry. 2017;56:1199–1217. doi: 10.1021/acs.biochem.6b00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti S, Suppa A, et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat Commun. 2019;10:4920. doi: 10.1038/s41467-019-12671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Florian RT, Kraft F, Leitao E, Kaya S, Klebe S, Magnin E, van Rootselaar AF, Buratti J, Kuhnel T, Schroder C, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat Commun. 2019;10:4919. doi: 10.1038/s41467-019-12763-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeetong P, Pongpanich M, Srichomthong C, Assawapitaksakul A, Shotelersuk V, Tantirukdham N, Chunharas C, Suphapeetiporn K, Shotelersuk V. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain. 2019;142:3360–3366. doi: 10.1093/brain/awz267. [DOI] [PubMed] [Google Scholar]
- 17.Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitsuhashi S, Frith MC, Mizuguchi T, Miyatake S, Toyota T, Adachi H, Oma Y, Kino Y, Mitsuhashi H, Matsumoto N. Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 2019;20:58. doi: 10.1186/s13059-019-1667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, Armstrong J, Tigyi K, Maurer N, Koren S, et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol. 2020;38:1044–1053. doi: 10.1038/s41587-020-0503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Coster W, De Rijk P, De Roeck A, De Pooter T, D'Hert S, Strazisar M, Sleegers K, Van Broeckhoven C. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019;29:1178–1187. doi: 10.1101/gr.244939.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frith MC. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 2011;39:e23. doi: 10.1093/nar/gkq1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitsuhashi S, Ohori S, Katoh K, Frith MC, Matsumoto N. A pipeline for complete characterization of complex germline rearrangements from long DNA reads. Genome Med. 2020;12(1). [DOI] [PMC free article] [PubMed]
- 24.Frith MC, Mitsuhashi S, Katoh K. lamassemble: multiple alignment and consensus sequence of long reads. Methods Mol Biol. 2021;2231:135–145. doi: 10.1007/978-1-0716-1036-7_9. [DOI] [PubMed] [Google Scholar]
- 25.Ohshima K, Kang S, Wells RD. CTG triplet repeats from human hereditary diseases are dominant genetic expansion products in Escherichia coli. J Biol Chem. 1996;271:1853–1856. doi: 10.1074/jbc.271.4.1853. [DOI] [PubMed] [Google Scholar]
- 26.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wojczynski MK, Li M, Bielak LF, Kerr KF, Reiner AP, Wong ND, Yanek LR, Qu L, White CC, Lange LA, et al. Genetics of coronary artery calcification among African Americans, a meta-analysis. BMC Med Genet. 2013;14:75. doi: 10.1186/1471-2350-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. 2018;97(1268–1283):e1266. doi: 10.1016/j.neuron.2018.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167(1415–1429):e1419. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang CK, Stein PB, Liu J, Wang Z, Yang R, Cho JH, Gregersen PK, Aerts JM, Zhao H, Pastores GM, Mistry PK. Genome-wide association study of N370S homozygous Gaucher disease reveals the candidacy of CLN8 gene as a genetic modifier contributing to extreme phenotypic variation. Am J Hematol. 2012;87:377–383. doi: 10.1002/ajh.23118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toriumi K, Oma Y, Kino Y, Futai E, Sasagawa N, Ishiura S. Expression of polyalanine stretches induces mitochondrial dysfunction. J Neurosci Res. 2008;86:1529–1537. doi: 10.1002/jnr.21619. [DOI] [PubMed] [Google Scholar]
- 32.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome. Ann N Y Acad Sci. 2015;1338:58–70. doi: 10.1111/nyas.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JK, Conrad A, Epping E, Mathews K, Magnotta V, Dawson JD, Nopoulos P. Effect of trinucleotide repeats in the Huntington’s gene on intelligence. EBioMedicine. 2018;31:47–53. doi: 10.1016/j.ebiom.2018.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM. Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol. 2014;71:1529–1534. doi: 10.1001/jamaneurol.2014.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bilgin Sonay T, Carvalho T, Robinson MD, Greminger MP, Krutzen M, Comas D, Highnam G, Mittelman D, Sharp A, Marques-Bonet T, Wagner A. Tandem repeat variation in human and great ape populations and its impact on gene expression divergence. Genome Res. 2015;25:1591–1599. doi: 10.1101/gr.190868.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fotsing SF, Margoliash J, Wang C, Saini S, Yanicky R, Shleizer-Burko S, Goren A, Gymrek M. The impact of short tandem repeat variation on gene expression. Nat Genet. 2019;51:1652–1659. doi: 10.1038/s41588-019-0521-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breschel TS, McInnis MG, Margolis RL, Sirugo G, Corneliussen B, Simpson SG, McMahon FJ, MacKinnon DF, Xu JF, Pleasant N, et al. A novel, heritable, expanding CTG repeat in an intron of the SEF2–1 gene on chromosome 18q21.1. Hum Mol Genet. 1997;6:1855–1863. doi: 10.1093/hmg/6.11.1855. [DOI] [PubMed] [Google Scholar]
- 38.Baratz KH, Tosakulwong N, Ryu E, Brown WL, Branham K, Chen W, Tran KD, Schmid-Kubista KE, Heckenlively JR, Swaroop A, et al. E2–2 protein and Fuchs’s corneal dystrophy. N Engl J Med. 2010;363:1016–1024. doi: 10.1056/NEJMoa1007064. [DOI] [PubMed] [Google Scholar]
- 39.Quilez J, Guilmatre A, Garg P, Highnam G, Gymrek M, Erlich Y, Joshi RS, Mittelman D, Sharp AJ. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 2016;44:3750–3762. doi: 10.1093/nar/gkw219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JM, Kim KH, Shin A, Chao MJ, Abu Elneel K, Gillis T, Mysore JS, Kaye JA, Zahed H, Kratter IH, et al. Sequence-level analysis of the major European huntington disease haplotype. Am J Hum Genet. 2015;97:435–444. doi: 10.1016/j.ajhg.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doi H, Okubo M, Fukai R, Fujita A, Mitsuhashi S, Takahashi K, Kunii M, Tada M, Fukuda H, Mizuguchi T, et al. Reply to “GGC repeat expansion of NOTCH2NLC is rare in European leukoencephalopathy”. Ann Neurol. 2020. [DOI] [PubMed]
- 43.Rumping L, Jans JJ, van Hasselt PM. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N Engl J Med. 2019;381:1185. doi: 10.1056/NEJMc1907427. [DOI] [PubMed] [Google Scholar]
- 44.Whillier S, Garcia B, Chapman BE, Kuchel PW, Raftos JE. Glutamine and alpha-ketoglutarate as glutamate sources for glutathione synthesis in human erythrocytes. FEBS J. 2011;278:3152–3163. doi: 10.1111/j.1742-4658.2011.08241.x. [DOI] [PubMed] [Google Scholar]
- 45.Ellory JC, Preston RL, Osotimehin B, Young JD. Transport of amino acids for glutathione biosynthesis in human and dog red cells. Biomed Biochim Acta. 1983;42:S48–52. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table S1. Data sets used in this study.Detection rates are the number of tandem repeats whose length is predicted from at least one DNA read. *3,312,291 loci. Table S2. Comparison of tantan and TRF annotated tandem repeatsNumber of intersections are counted using bedtools (https://bedtools.readthedocs.io/en/latest/). Each repeat unit was counted separately. Table S3. Phased repeat length and near-by SNPs.SNP genotype on the short and long alleles were shown in each dataset. Repeat copy number changes on both alleles were genotyped using tandem-genotypes. (A) ATXN3 repeat and (B, C) GLS repeat. SNP rs numbers are from dbSNP (https://www.ncbi.nlm.nih.gov/snp/). Table S4. Polymorphic tandem repeat and near-by GWAS signals.Highly polymorphic exonic triplet repeats (IQR ≥ 5) with near GWAS signals (< 100bp). GWAS signals were from a GWAS catalog [22].
Additional file 2. Figure S1. Variation of tandem repeat length (copy number) in long reads from 21 individuals. x-axis: copy number change relative to the human reference (hg38). y-axis: read number. Three different repeat types ((A) exonic CAG, (B) exonic GGC and (C) intronic AAAAT) are shown. Disease repeats: 12 CAG repeats, 14 GGC repeats, and 7 intronic AAAAT repeats. Other repeats: exonic CAG: n = 1840, exonic GGC: n = 3073, intronic AAAAT: n = 19,744. For each repeat type, we show ten sets of “other repeats” for comparison. Each set of “other repeats” is a random selection of the same number of repeats as the number of disease repeats. Figure S2. Repeat length mean and spread of each triplet repeat type. There are 10 kinds of triplet repeat, AAC, CAC, CCT, CTT, GAT, GTA, GTC, TAA CAG and GGC. The numbers of repeats are; 650 (AAC), 632 (CAC), 1862 (CCT), 737 (CTT), 430 (GAT), 47 (GTA), 73 (GTC), 682 (TAA), 1839 (CAG) and 2907 (GGC). The variation of the repeat length in 21 individuals are shown. x-axis: interquartile range (IQR), y-axis: mean repeat length (bp). In merged boxplots, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S3. Variability of exonic repeats. Number of exonic tandem repeats (A) and IQR (B) of each unit are shown. Shorter-unit repeats have more variation. Dots represent outliers. Boxplot ranges are the 25th and 75th percentiles. Lines in boxes are median. Most of the IQRs from repeats whose length are more than six are zero. Figure S4. Comparison of tantan and TRF annotated tandem repeats (A) There are more tantan annotated tandem-repeats than TRF-annotated repeats, however, the distribution of the number of the loci has similar tendency. x-axis: length of repeat unit, y-axis: number of loci. (B) Proportions of triplet repeat sequences are similar between tantan and TRF annotation. Figure S5. Variability of CAG repeats using TRF-annotated repeats.Variation (IQR) and length of repeats with disease-associated sequences. Coding CAG repeats (A), and non-coding exonic CAG repeats (B). x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S6. Variability of GGC repeats using TRF-annotated repeats.Variation (IQR) and length of repeats with disease-associated sequences. Coding GGC repeats (A), and non-coding exonic GGC repeats (B). x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S7. Variability of AAAAT repeats using TRF-annotated repeats.Variation (IQR) and length of repeats with disease-associated intronic AAAAT sequences. x-axis: IQR, y-axis: mean repeat length (bp). n provides the numbers of repeat loci. In merged boxplots on the right and upper, ranges are the 25th and 75th percentiles, dots are outliers and lines in boxes are median. Figure S8. Repeat length correlates with near-by SNPs. (A) Distribution of tandem repeat length (copy number) in combined long reads of GLS and ATXN3 disease-associated repeats from 21 individuals. x-axis: copy number change relative to the human reference (hg38). y-axis: read count. (B) Three GWAS reported SNPs were near the GLS and ATXN3 repeats. Repeat lengths of each genotype were compared using unpaired t test. P values are shown. Figure S9. Gaucher disease severity modifying SNPs correlate with repeat length.(A) Distribution of tandem repeat length (copy number) in combined long reads of CLN8 from 21 individuals. Note that there is bimodal distribution, with peaks around zero and − 13 copy number changes. x-axis: copy number change relative to the human reference (hg38). y-axis: read count. (B) One GWAS reported SNP rs11986414 is inside and another SNP rs4875960 is near this repeat. In both SNPs, genotype A tends to have larger repeat length.
Data Availability Statement
PromethION WGS sequence data is available from DDBJ (DRA009852). Other public data were downloaded from NCBI or Human PanGenome Project (https://github.com/human-pangenomics/hpgp-data) under accession numbers described in Table S1. SNP identifiers were used under accession numbers from dbSNP (https://www.ncbi.nlm.nih.gov/snp/).