

SUMMARY
Glucocorticoids (GC) are the mainstay treatment option for inflammatory conditions. Despite the broad usage of GC, the mechanisms by which GC exerts its effects remain elusive. Here, utilizing murine autoimmune and allergic inflammation models, we report that Foxp3+ regulatory T (Treg) cells are irreplaceable GC target cells in vivo. Dexamethasone (Dex) administered in the absence of Treg cells s completely lost its ability to control inflammation, and the lack of glucocorticoid receptor in Treg cells alone resulted in the loss of therapeutic ability of Dex. Mechanistically, Dex induced miR-342–3p specifically in Treg cells and miR-342–3p directly targeted the mTORC2 component, Rictor. Altering miRNA-342–3p or Rictor expression in Treg cells dysregulated metabolic programming in Treg cells, controlling their regulatory functions in vivo. Our results uncover a previously unknown contribution of Treg cells during glucocorticoid-mediated treatment of inflammation and the underlying mechanisms operated via the Dex-miR-342-Rictor axis.
In Brief
Glucocorticoids are the first line treatment option for acute and chronic inflammatory conditions, yet the underlying mechanisms remain largely elusive. Kim et al demonstrate that Foxp3+ regulatory T cells are key mediators of glucocorticoid-induced treatment by miR-342-dependent metabolic control in Treg cells.
Graphical Abstract
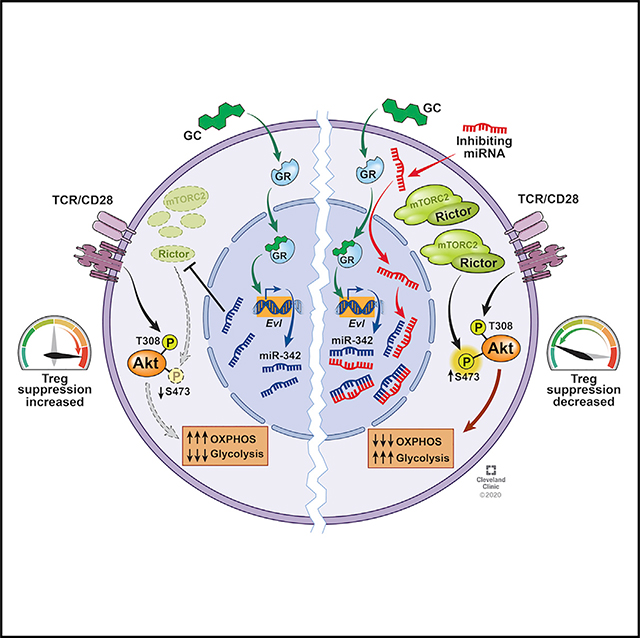
INTRODUCTION
Glucocorticoids (GC) are a class of cholesterol-derived corticosteroid hormones playing highly diverse roles in homeostasis, development, metabolism, and immunity (Barnes, 2011; Cain and Cidlowski, 2017). Due to potent immunosuppressive and anti-inflammatory properties, synthetic GC such as dexamethasone (Dex) serves as the frontline treatment option for various inflammatory conditions including multiple sclerosis and asthma (Alangari, 2014; Barnes et al., 1997). GC exerts its functions by binding the cytosolic glucocorticoid receptor (GR) expressed in almost every nucleated cell (Necela and Cidlowski, 2004; Oakley and Cidlowski, 2013). The cytosolic GR, which remains inactive, undergoes conformational change upon ligand binding, translocates into the nucleus, where it binds the glucocorticoid-responsive DNA elements, and acts as transcriptional activators or repressors (Barnes, 2006; Necela and Cidlowski, 2004). Despite the long history of GC discovery and broad applications to attenuate inflammatory responses, the precise cellular mechanisms underlying GC-induced immune modulation are not entirely understood.
The GR is ubiquitously expressed. Yet, there is emerging evidence that the target cells mediating the GC effects differ depending on the types or tissues of inflammation (Cain and Cidlowski, 2017; Franco et al., 2019). Utilizing cell-type-specific GR-deficient animal models, it has been shown that conventional T cells are the primary GC target cells during Dex-mediated treatment of autoimmune inflammation in the central nervous system (CNS) (Wüst et al., 2008). On the other hand, myeloid and airway epithelial cells have been shown to be the GC-responsive cells during contact hypersensitivity and airway inflammation, respectively (Klaßen et al., 2017; Tuckermann et al., 2007). In addition to the complexity behind the cell-type-specific GC actions, the ‘effector’ mechanisms underlying GC-induced immune modulation are thought to be diverse. GC induces apoptotic cell death in inflammatory cells such as airway infiltrating eosinophils (Meagher et al., 1996; Ohta and Yamashita, 1999; Schleimer and Bochner, 1994), although some cell types express resistance against GC-induced apoptosis (Gruver-Yates and Cidlowski, 2013; Liles et al., 1995).
Foxp3-expressing regulatory T (Treg) cells are the central regulator of immunity and tolerance, and defects in Treg cell generation or function result in systemic autoimmune inflammation (Bennett et al., 2001; Bluestone and Tang, 2018; Josefowicz et al., 2012). GC is known to modulate Treg cell homeostasis (Cari et al., 2019; Ugor et al., 2018). More specifically, GC can amplify interleukin-2 (IL-2)-dependent Treg cell expansion and enhance their suppressive activity to control autoimmune inflammation and graft-versus-host diseases (Chen et al., 2006; Xie et al., 2009). GC may upregulate Foxp3 mRNA expression in asthmatic patients (Karagiannidis et al., 2004) and promote the generation of antigen-specific induced Treg cells via GC-induced leucine zipper (GILZ) protein (Bereshchenko et al., 2014; Hamdi et al., 2007). However, discrepant findings have also been reported (Olsen et al., 2015; Sbiera et al., 2011). Therefore, the precise roles of GC in Treg cell biology and the underlying mechanisms remain elusive.
In the present study, we report that Treg cells are necessary for GC to control inflammatory diseases, autoimmune neuroinflammation, and allergic airway inflammation. Dex administered completely lost its treatment effects when Treg cells were absent. Likewise, inflammation in Treg cell-specific GR-deficient mice was not alleviated by Dex administration, suggesting a direct involvement of GC signaling in Treg cells during the treatment. Mechanistically, from transcriptome analysis we uncovered miRNA-342–3p induced by Dex specifically in Treg cells. miR-342–3p targeted mTORC2 component, Rictor. Dex-induced miR-342–3p played a key role in regulating metabolic profiles of Treg cells in response to Dex stimulation. Modulating miR-342 and Rictor expression directly controlled Treg cell suppressive function in vivo. Here, we report that Treg cells are indispensable direct target cells of Dex to control both autoimmune and allergic inflammation and that miR-342 and Rictor-dependent metabolic control in Treg cells plays a key role during glucocorticoid-mediated treatment.
RESULTS
Dexamethasone Fails to Attenuate Inflammation without Treg Cells
Systemic GC is the mainstay treatment option for inflammatory disorders. Experimental autoimmune encephalomyelitis (EAE) is a mouse model of autoimmune neuroinflammation induced by myelin oligodendrocyte glycoprotein (MOG) peptide immunization. Dexamethasone (Dex) administered at the onset of the disease effectively attenuated the development of clinical signs (Figure S1A). The accumulation of CD4+ T cells expressing inflammatory cytokines in the CNS tissues was significantly reduced by the treatment (Figures S1B and S1C). Anti-inflammatory effects of Dex were also examined in murine model of allergic airway inflammation induced by cockroach antigens (CA), in which lung-infiltrating CD4+ T cells secreting T helper-2 (Th2) cell type cytokines drive the inflammation (Figure S1D) (Jang et al., 2017). Dex systemically delivered during CA challenge diminished inflammatory cell infiltration in the airway and lung tissues (Figures S1E and S1G). CD4+ T cells expressing inflammatory cytokines in the lung tissues were also reduced following the treatment (Figure S1F). Therefore, Dex attenuates Th17- and Th2 cell-associated inflammatory responses.
Treg cells play an essential role in regulating immune responses during autoimmune and allergic inflammation (Dominguez-Villar and Hafler, 2018; Noval Rivas and Chatila, 2016). As the effect of GCon Treg cells in inflammation remains unclear, we sought to directly test the role of Treg cells during Dex treatment of inflammation. EAE was induced in Foxp3DTR mice that express the diphtheria toxin receptor (DTR) under the Foxp3 promoter, and the mice were treated with Dex at the disease onset in the presence or absence of Treg cells (DTx or vehicle injected). Treg cell depletion following DTx treatment was validated in every experiment (data not shown). Mice rapidly succumbed to death following endogenous Treg cell depletion at the EAE onset (Figure 1A; Kim et al., 2019). However, Dex administration was unable to rescue mice from lethal disease when Treg cells were absent (Figure 1A). The accumulation of total CD4+ T cells and of T cells expressing proinflammatory cytokines in the CNS was greatly reduced by Dex treatment, and the reduction was lost when Treg cells were absent (Figures 1B and 1C). Histopathologic examination further validated Treg-cell-dependent Dex treatment effects (Figure 1D). CNS expression of inflammatory cytokine mRNA, Ifng, and Il17, and the key transcription factors, Tbx21 and Rorc, followed similar expression patterns (Figure 1E). Treg-cell-dependent Dex effects were similarly observed in allergic airway inflammation. Dex treatment during CA challenge failed to diminish eosinophil and CD4+ T cell infiltration in the bronchoalveolar lavage (BAL) and lung tissues following Treg cell depletion (Figures 1F and 1G; Figure S2A). Likewise, Dex administration failed to curtail inflammatory CD4+ T cell accumulation in the lung when Treg cells were depleted (Figure 1G; Figure S2B). Secretion of IL-4 and IL-13 in the BAL fluid and the expression of the Muc5a and Muc5b genes was reduced by Dex, but the reduction was only seen when Treg cells were present (Figure 1H; Figure S2C). Lung inflammation and airway resistance showed similar Treg-cell-dependent treatment effects (Figures 1I and 1J). Treg-cell-dependent treatment by Dex was also found following intranasal Dex administration, a route commonly used in asthmatic patients, suggesting that both systemically and locally administered Dex requires Treg cells for its anti-inflammatory effects (Figure 1K; Figure S2D).
Figure 1. Dex-Induced Treatment Requires Treg Cells.

(A–E) Foxp3DTR mice were induced for EAE and injected with Dex daily on days 9–11 and/or with DTx on day 7. (A) The mice were daily scored for clinical diseases and survival.
(B) CD4+ T cells infiltrating the CNS tissues were enumerated on day 14.
(C) Intracellular IFN-γ+ and IL-17+ CD4+ T cells in the CNS were measured on day 14.
(D) H&E staining of the spinal cords (day 14 post immunization, original magnification x100). Scale bar, 10 μm. (E) qPCR analysis of the Ifng, Il17, Rorc, and Tbx21 expression in the CNS. Veh (n = 4–9), Dex (n = 4–13), DTx (n = 4–9), and DTx + Dex (n = 4–9) from two to three independent experiments.
(F–K) Foxp3DTR mice were injected i.p. with CA in alum adjuvant on days 0 and 7. Starting at day 14, mice were intranasally challenged for 4 consecutive days with CA. DTx was injected 1 day before and on the day of first CA challenge.
(F–J) Vehicle or Dex was intraperitoneally injected on the first and third days of CA challenge (days 14 and 16). Mice were sacrificed 24 h after the last challenge. Total numbers of eosinophils and CD4+ T cells in the BAL (F) and of cytokine expressing CD4+ T cells in the lung (G) were calculated by flow cyometry analysis. (H) IL-4 and IL-13 secretion in the BALF was determined using a CBA assay. (I) H&E staining and histology score of the lung tissues are shown (original magnification × 4). Scale bar, 250 μm. (J) Airway resistance was measured by flexivent experiments.
(K) Dex (0.25 mg/kg) was administered intranasally on the first and third days of CA challenge. Eosinophils and CD4+ T cells in the BAL were enumerated. Veh (n = 4–13), Dex (n = 4–14), DTx (n = 4), and DTx + Dex (n = 4–18) from two to three independent experiments. Each symbol represents an individually tested animal. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, as determined by Mann-Whitney or Kruskal-Wallis nonparametric test. Please also see Figure S2.
Dex therapeutically given to mice with ongoing EAE rapidly lowered the clinical signs (Figure 2A). Dex-induced attenuation also required the presence of Treg cells, as Treg cell depletion abrogated the treatment effects (Figure 2A). The inability of Dex to reverse EAE without Treg cells was not due to severity of the disease, because similar Treg cell dependency was also seen during mild EAE induced by suboptimal MOG peptide immunization (Figure 2C). Immunizing mice with low dose of MOG peptide induced delayed and mild disease (peaked at the clinical score of ~2), and Dex treatment further lowered the disease severity (Figure 2C). Mild EAE turned into a lethal, albeit slightly delayed, EAE when Treg cells were depleted, and Dex treatment had no impact on rescuing mice from the disease (Figures 2C and 2E). Lethality was rescued by adoptive transfer of thymus derived regulatory (tTreg) or in vitro generated regulatory (iTreg) cells (Figures 2B, 2C, and 2D), and Dex treatment further diminished the disease severity (Figures 2B and 2D). Likewise, iTreg cells transferred reduced inflammatory CD4+ T cell infiltration and lung inflammation in endogenous Treg-cell-depleted mice treated with Dex (Figure S3). Collectively, these results demonstrate that the presence of Treg cells is necessary for Dex to control autoimmune and allergic inflammation in vivo.
Figure 2. Treg Cells Transferred Restore Dex-Induced Treatment in Treg-Depleted Mice.

(A) Foxp3DTR mice were induced for EAE. Dex treatment started at the average score of 2 (i.e., days 13–15, arrows). n = 5–7 for each group.
(B) Foxp3DTR mice (n = 5–6) induced for EAE and treated with DTx or with DTx + Dex as above were transferred with flow cytometry sorted tTreg cells (1×106 cells per recipient).
(C–E) Foxp3DTR mice were immunized with MOG 35–55 (150 μg for C or 300 μg for D), treated with Dex for 3 days (arrows). Mice received 2×106 Foxp3+ flow cytometry sorted iTreg cells together with DTx on day 7. The mice were daily monitored and scored for clinical diseases and survival. n = 3–13 for each group. **p < 0.01; ***p < 0.001, as determined by Mann-Whitney or Kruskal-Wallis nonparametric test. Please also see Figure S3.
Dex Directly Acts on GR Expressed in Treg Cells for its Functions
GC mediates its immunomodulatory effects through cytosolic receptors, the glucocorticoid receptors (GR, encoded by the Nr3c1 gene) that are ubiquitously expressed (Kadmiel and Cidlowski, 2013; Oakley and Cidlowski, 2013). The loss of treatment effects of Dex without Treg cells suggested that Treg cells may have been the primary target cells of GC in vivo, although conventional T cells are also known as potential Dex targets capable of mediating its anti-inflammatory effects (Engler et al., 2017; Wüst et al., 2008). To directly test whether GC signaling (i.e., GR expression) in Treg cells is necessary for Dex treatment effects, we utilized Treg-cell-specific Nr3c1−/− (Foxp3-cre x Nr3c1fl/fl, Nr3c1ΔTreg) mice. Treg-cell-specific GR deletion was validated by genomic DNA PCR (data not shown). Dex-induced dual specificity phosphatase 1 (Dusp1) gene expression was lost in Treg cells from Nr3c1ΔTreg but not from control Nr3c1WT mice, whereas conventional T cells from both types of mice upregulated Dusp1 expression following Dex stimulation (data not shown). T cell development in the thymus was not affected by the GR deficiency in Treg cells (Figure S4A), and peripheral CD4+ and Treg cells were comparable between the groups (Figure S4B). Both conventional CD4+ and Treg cells displayed similar surface phenotypes including Nrp1, Tim3, ICOS, GITR, PD1, and CD25, as well as in vivo turnover (Figure S4C; data not shown). Cellular expression of inflammatory cytokines and lineage transcription factors was also comparable between the groups (Figure S4D). Therefore, the lack of endogenous GC signaling in Treg cells appears to have little impact on immune homeostasis at steady state. Both Nr3c1ΔTreg and Nr3c1WT mice developed comparable EAE (Figure 3A, left). Following Dex treatment, EAE was significantly attenuated in Nr3c1WT mice, whereas the disease in Nr3c1ΔTreg mice remained unchanged (Figure 3A, right). Consistent with the disease severity, CD4+ T cell infiltration in the CNS was significantly greater in Dex treated Nr3c1ΔTreg mice (Figure 3B). However, Treg cell accumulation was comparable (Figure 3B), suggesting that the inability of Dex to control inflammation cannot be attributed to Treg cell recruitment. Treg cell expression of surface phenotypes was similar between the groups (Figure 3C). CNS infiltrating CD4+ T cells from Dex-treated Nr3c1WT mice secreted less IFN-γ and more IL-10 following recall stimulation compared to those from Dex-treated Nr3c1ΔTreg mice (Figure 3D). The amount of serum cytokines (IL-17, IFN-γ, and TNF-α) was found greater in Dex-treated Nr3c1ΔTreg mice (Figure 3E). The expression of proinflammatory cytokines in the CNS tissues measured by qPCR analysis also supported the results (Figure 3F). Similar results were observed in allergic inflammation model. Dex treatment significantly diminished inflammatory cell recruitment in the BAL cells in Nr3c1WT but not in Nr3c1ΔTreg mice (Figure 3G), and lung-infiltrating CD4+ T cells expressing inflammatory cytokines followed the same pattern (Figure 3H). Histopathologic examination of the lung tissues showed key involvement of GC signaling in Treg cells during Dex-mediated treatment of lung inflammation (Figure 3I). Lastly, lung infiltration of Treg cells was reduced by Dex treatment in Nr3c1WT but not in Nr3c1ΔTreg mice, although expression of Treg-cell-associated markers and lineage transcription factors remained comparable between the groups (Figure 3J; Figure S5). Therefore, direct Dex signaling via GR in Treg cells is essential for Dex to control inflammation.
Figure 3. Dex Directly Acts on GR Expressed in Treg Cells for its Functions.

(A–F) Treg cell-specific Nr3c1−/− and control mice (n = 7–11) were induced for EAE and treated with vehicle or Dex on days 9–11 (arrows).
(A) Clinical score was daily monitored.
(B) Total number of CNS-infiltrating CD4+ T cells and Foxp3+ Treg cells was enumerated on day 14 post induction. (C) The mean fluorescence intensity (MFI) of ICOS, CD25, CD44, and CTLA4 on CNS infiltrating Foxp3+ Treg cells was determined by flow cytometry.
(D) CNS cells were isolated and restimulated with MOG peptide for 48 h. IL-10 and IFN-γ secretion in the culture supernatant was determined by ELISA.
(E) Serum IL17, TNF-α, and IFN-γ was determined by CBA assay (day 14 post EAE induction).
(F) Ifng and Il17 mRNA expression in the CNS was determined by qPCR on day 14 post immunization. Data shown are from the mean ± SD or individually tested mouse of three independent experiments.
(G–J) Treg-cell-specific Nr3c1−/− and control mice were sensitized with CA in alum adjuvant and intranasally challenged as above. Dex (0.25 mg/kg) was administered intranasally on the first and third days of allergen challenge. Total numbers of eosinophils and CD4+ T cells in the BAL (G) and of cytokine expressing CD4 T cells in the lung (H) were determined by flow cytometry analysis. (I) H&E staining and histology score of the lung tissues (original magnification x10). Scale bar, 100 μm. (J) The total number and frequency of lung infiltrating Foxp3+ Treg cells, and the mean fluorescence intensity of Foxp3 and CD25 of Treg cells were determined by flow cytometry. Each symbol represents an individually tested animal. Veh (n = 6–8) and Dex (n = 7–9) from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, as determined by Mann-Whitney or Kruskal-Wallis nonparametric test. Please also see Figure S4.
Differential Gene Signatures Are Induced by Dex in Treg Cells
To gain closer insights into the molecular mechanisms underlying Treg-cell-dependent Dex effects, we performed an RNA-seq experiment comparing gene expression between Treg and CD4+ T effector cells isolated from the inflamed lung tissues of mice induced for allergic inflammation and treated with Dex or vehicle. In particular, we compared genes differentially expressed by Dex over vehicle treatment in each population (Treg versus Teff cells, after normalization by vehicle group, p < 0.001) and the total 1752 genes were identified (Figure S6). These genes were reanalyzed using repetitive normalization by vehicle treatment in each cell type, and 150 genes were identified based on the expression pattern by Dex treatment (|FC|>2, p < 0.001, Figure S6). Differentially expressed genes were equally distributed based on the functions and location (Figure 4A). We particularly focused on the genes whose expression is elevated by Dex treatment only in Treg cells. They included Ccr9, Lrrc32, Esam, Ly6c1, Napb, Rragd, and Evl (Figure 4B; Figure S7). Treg-cell-specific upregulation by Dex was validated by qPCR analysis. Since Treg-cell-dependent Dex treatment effects were found in both allergic and autoimmune inflammation models, the validation was performed using Treg and effector cells isolated from inflamed tissues from EAE (Figure 4C) and from allergic inflammation (Figure 4D). In support of the RNA-seq data, Dex treatment significantly elevated the expression of the selected genes in Treg cells, while the same treatment lowered the expression in effector T cells (Figures 4C and 4D). Of note, Dex-induced downregulation in effector T cells was dependent on Treg cells or on Treg cell GR expression, as Treg cell depletion or GR deficiency in Treg cells abrogated the downregulation (Figures 4C and 4D). Glucocorticoid-induced leucine-zipper (Gilz), GR (Gra), dual specificity phosphatase 1 (Dusp1), and FK506 binding protein 5 (Fkbp5) are canonical Dex-inducible genes (Ayroldi and Riccardi, 2009; Kelly et al., 2012; Tchen et al., 2010), and they were equally induced by Dex treatment in both Treg and effector cells (Figure 4E). Moreover, in Nr3c1ΔTreg mice treated with Dex, the canonical gene expression was lost only in Treg but not in effector cells, validating Treg cell-specific loss of GR signaling (Figure 4E). Unlike Treg-cell-dependent genes shown above, Treg cell deletion had no impact on the expression of the canonical Dex-induced genes in effector T cells (Figure 4E). Therefore, these results demonstrate groups of genes controlled by Dex treatment via different mechanisms between Treg and effector cells.
Figure 4. Differentially Expressed Genes between Treg Cells and T Effector following Dex Treatment.

(A, B, and D) Foxp3GFP or Foxp3DTR mice were induced for allergic inflammation, treated with vehicle or Dex as described above. At sacrifice, Foxp3+ Treg and effector CD4+ T cells were flow cytometry sorted from the lung tissues.
(A and B) RNA-seq analysis was performed (n = 3 each group). Based on the function and location, gene sets were marked for each category.
(B) Heatmap shows expression of genes (|fold change| > 2, p < 0.001 in Treg cells versus effector cells).
(C–E) RNA was isolated from the sorted cells and evaluated for gene expression by qPCR. Expression of the indicated genes was normalized to the housekeeping Gapdh gene, and relative quantification (RQ) value was calculated.
(C and E) Foxp3DTR or Treg-cell-specific Nr3c1−/− mice induced for EAE and treated with Dex or DTx + Dex were sacrificed as above. Treg cells and effector CD4+ T cells were flow cytometry sorted from the CNS tissues, and relative expression of the indicated genes was determined by qPCR. Data are the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01, as determined by Kruskal-Wallis nonparametric test. Please also see Figure S5.
Dex Induces microRNA-342 Specifically in Treg Cells
Ena-vasodilator stimulated phosphoprotein (EVL) is an actin-associated protein involved in various processes dependent on cytoskeleton remodeling and cell polarity (Trichet et al., 2008) and is one of the Dex-induced genes in Treg cells (Figures 4C and 4D). Notably, the third intron of the Evl gene encodes a highly conserved microRNA-342 (miR-342) (Figure 5A) (Grady et al., 2008; Li et al., 2018), and the expression of intronic miR-342 mirrors that of the host gene, Evl (De Marchis et al., 2009; Grady et al., 2008). Indeed, miR-342–3p expression was significantly enhanced by Dex treatment in Treg cells in vivo, whereas the expression was reduced in effector T cells by the treatment (Figure 5B). GR expression of Treg cells was necessary for miR-342–3p induction by Dex (Figure 5B). Searching for its target molecules, we found that the Rictor, a mTORC2 complex molecule, was a potential target of miR-342–3p based on the sequence homology (Figure S8). Indeed, Rictor expression was inversely regulated by Dex treatment in Treg and effector cells via a GR-dependent manner (Figure 5C). Increased expression of the miR-342–3p and downregulation of the Rictor were also observed in Treg cells isolated from inflamed lung tissues, and effector T cells displayed a reversed expression pattern (Figure 5D). Therefore, Rictor expression appears to be controlled by miR-342–3p induced by Dex stimulation.
Figure 5. Dex Induces miRNA-342–3p Specifically in Induced Treg Cells.

(A) Schematic representation of the Evl gene and the location of the conserved miRNA-342.
(B and C) Foxp3DTR or Treg-cell-specific Nr3c1−/− mice induced for EAE and treated with Dex were sacrificed day 14 post induction as in Figure 4. qPCR analysis of miR-342–3p and Rictor expression in the Treg and effector cells isolated from the CNS.
(D) Foxp3DTR mice induced for allergic inflammation were treated with vehicle or Dex as described in Figure 1. At sacrifice, Treg and effector CD4 T cells flow cytometry sorted from the lung were evaluated for gene expression by qPCR.
(E and F) Relative expression of the indicated miRNAs in naive or in vitro differentiated CD4+ T cells with or without Dex.
(G) miR-342–3p expression in the thymus-derived tTreg cells. tTreg cells isolated from Foxp3GFP mice were activated with or without Dex. miR-342–3p and Rictor expression was determined by qPCR analysis.
(H) Immuno blot analysis for Rictor, Raptor, and β-actin in lysates from iTreg cells treated with or without Dex.
(I) Eosinophils and B cells isolated from the BAL and mediastinal lymph node of mice with allergic inflammation, respectively, were evaluated for miR-342–3p expression by qPCR.
(J) Expression of miR-342–3p and miR-16 in the spinal cord and brain tissues was determined (day 14 post EAE induction). Data are the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01, as determined by Mann-Whitney or Kruskal-Wallis nonparametric test. Please also see Figure S6.
We next determined whether Dex-induced miR-342–3p expression is Treg cell-specific. miR-342–3p expression was pronounced in in vitro differentiated Treg (iTreg) cells compared to other conventional effector T cell subsets differentiated in vitro (Figure 5E). Other miRs known to be expressed in Treg cells, including miR-146, miR-155, miR-15b, and miR-16, were highly expressed in iTreg cells (Figure 5E; Figure S9A). However, miR-342–3p was the only miR elevated by Dex treatment in Treg cells among those tested (Figure 5F; Figure S9B). Similar expression pattern was also observed in thymus-derived tTreg cells (Figure 5G). Dex stimulation further enhanced miR-342–3p expression, while the Rictor expression was downregulated (Figure 5G), suggesting that both subsets of Treg cells (thymus derived and induced) similarly respond to Dex. The expression of Rptor, a mTORC1 complex molecule, remained unchanged by Dex treatment, suggesting a mTORC2-specific effect (Figure S9C). Of note, Gilz expression was comparably induced by Dex treatment in all tested effector subsets (Figure S9C). Immunoblot analysis further validated diminished Rictor but not Raptor expression in iTreg cells following Dex stimulation (Figure 5H). Dex stimulation did not induce miR-342–3p expression in other immune cells such as BAL eosinophils or lymph node B cells from the mice induced for allergic inflammation, suggesting Dex-induced miR-342–3p expression in Treg lineage cells (Figure 5I). Elevated miR-342–3p expression by Dex in Treg cells also occurred in vivo. Dex treatment increased miR-342–3p expression in the CNS tissues of EAE mice, and Treg cell depletion abrogated the expression, while miR-16 expression remained comparable by Dex treatment (Figure 5J). Collectively, these results demonstrate that Dex induces miR-342–3p expression specifically in iTreg cells.
miRNA-342 Controls Dex-Mediated Treg Cell Functions
We next examined whether miR-342–3p plays a role in Treg cell functions during Dex-mediated control of inflammation. iTreg cells were generated in the presence or absence of Dex. miR-342–3p expression was elevated in iTreg cells compared to Th0 cells, and further enhanced by Dex treatment (Figure 6A). Dex treatment increased Foxp3 mRNA expression (Figure 6A), although Foxp3 expression based on GFP expression remained comparable between the groups (Figure 6B). Inhibiting miR-342 expression using miR-342–3p specific antisense oligonucleotide inhibitors abolished miR-342–3p expression in iTreg cells as well as in tTreg cells even with Dex stimulation (Figure 6A; data not shown). miR-342 inhibition slightly but significantly diminished the Foxp3 mRNA expression and Dex treatment was unable to restore the expression (Figure 6A). Yet, Foxp3 protein expression remained unchanged by miR-342 inhibition (Figure 6B). miR-342 inhibition drastically reversed Rictor expression, supporting that the Rictor is a direct target of miR-342–3p, whereas the Rptor expression remained unchanged in all tested conditions (Figure 6A). Evl expression was further enhanced by Dex treatment regardless of miR-342 inhibition, validating that miR-342–3p is a downstream molecule of the Evl (Figure 6A). To test the role of miR-342–3p in Treg cells in vivo, iTreg cells treated with miR-342-specific or control inhibitors in the presence or absence of Dex were adoptively transferred into Foxp3DTR mice induced for EAE and treated with DTx at the onset. As shown above, endogenous Treg cell depletion at EAE onset resulted in lethal disease and exogenous Treg cell transfer rescued mice from the lethality (Figure 6C). Control inhibitor treated iTreg cell transfer rescued mice from death, and Dex stimulation of iTreg cells further lowered the disease severity (Figure 6C). By contrast, miR-342 inhibitor treated iTreg recipients developed severe (but not lethal) EAE compared to control iTreg cell recipients, and more importantly, Dex stimulation of these iTreg cells had no impact on attenuating the disease severity, suggesting that miR-342–3p expression in Treg cells appears to be critical for Dex-mediated Treg suppressive function in vivo (Figure 6C).
Figure 6. miR-342–3p Controls Treg Functions and Metabolism.

(A and B) Naive CD4+ T cells isolated from Foxp3GFP mice were nucleofected with control (NC) or miR-342–3p specific antisense oligonucleotide inhibitor and then activated in vitro under Th0 or iTreg cell conditions with or without Dex for 72 h.
(A) Foxp3, miR-342–3p, Rictor, Rptor, and Evl expression were determined by qPCR analysis.
(B) Foxp3 (GFP) expression was determined by flow cytometry analysis.
(C) Foxp3DTR mice were induced for EAE and treated with DTx day 7 post induction. 2×106 control iTreg cells or miR-342–3p inhibitor treated iTreg cells as above were transferred on day 7. Dex treatment was performed on days 9 through 11. The mice were daily monitored and scored for clinical diseases. N.C (n = 5), Dex (n = 7), miR-342–3p inh (n = 8), and miR-342–3p inh + Dex (n = 8) from two to three independent experiments.
(D) Expression and phosphorylation of the indicated proteins in control or miR-342–3p inhibitor treated iTreg cells with or without Dex were determined by immuno blot analysis.
(E–G) Naive CD4+ T cells nucleofected with the indicated inhibitors or si-Rictor were stimulated under iTreg condition with or without Dex for 72 h. (E) OCR and EACR measurement. Quantitation of basal respiration and glycolysis are shown. n = 5 each group. (F) qPCR analysis of Rictor expression. (G) qPCR analysis of Hk2 and Hif1a expression. Data are the mean ± SD of two to three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, as determined by Kruskal-Wallis nonparametric test. Please also see Figure S7.
miR-342 Controls Treg Cell Metabolism
Rictor is a key adaptor protein of the mTORC2 complexes (Guertin et al., 2006; Oh and Jacinto, 2011). Unlike Raptor, Treg-cell-specific Rictor deletion does not result in gross abnormalities, although it has been implicated that Rictor may play a role in Treg maintenance (Chapman and Chi, 2014; Zeng et al., 2013). It has been recently reported that mTORC2 inactivation or Rictor deficiency may improve Treg cell function (Charbonnier et al., 2019). Since Dex-induced miR-342–3p in iTreg cells targets Rictor and miR-342 directly controls Treg functions in vivo, we asked whether miR-342-dependent inhibition of Rictor has impact on the mTOR pathway and on the metabolic profile of Treg cells during Dex stimulation. Dex treatment significantly downregulated Rictor protein expression, and miR-342 inhibition completely reversed the downregulation even with Dex treatment (Figure 6D; Figure S10A). Rictor is involved in phosphorylating Akt at Ser 473 site (Dibble et al., 2009; Sarbassov et al., 2005). Indeed, Akt Ser473 phosphorylation was diminished by Dex treatment, while it was enhanced by miR-342 inhibition (Figure 6D; Figure S10A). Akt Thr308 phosphorylation induced by PDK1 was not affected by Dex and/or miR-342 inhibition (Figure 6D; Figure S10A). Fully activated Akt phosphorylates FoxO1 (Pan et al., 2017), and indeed enhanced FoxO1 transcription factor phosphorylation was observed following miR-342 inhibition (Figure 6D; Figure S10A). Dex treatment inhibited S6K activity, whereas miR-342 inhibition enhanced S6 phosphorylation regardless of Dex (Figure 6D; Figure S10A).
Treg cells utilize fatty acid oxidation and oxidative phosphorylation (OXPHOS) to meet their bioenergetic needs, and glycolysis is under tight control (Gerriets et al., 2015; Howie et al., 2017; Michalek et al., 2011). Foxp3 supports OXPHOS and suppresses glycolysis (Angelin et al., 2017). Enforcing glycolysis in Treg cells by upregulating glucose transporter Glut1 activates mTORC1 pathway and reduces Foxp3 expression as well as the suppressive function (Gerriets et al., 2016). Based on miR-342-dependent regulation of Rictor and other metabolic regulators, we determined metabolic profiles of Treg cells stimulated with Dex in the presence or absence of miR-342 inhibition. Basal oxygen consumption rate (OCR), an indication of oxidative phosphorylation, was elevated by Dex treatment, whereas miR-342 inhibition lowered the OCR regardless of Dex stimulation (Figure 6E; Figure S10B). Conversely, Dex treatment reduced extracellular acidification rate (ECAR), a measurement of overall glycolytic flux (Figure 6E; Figure S10B). miR-342 inhibition reprogrammed Treg cells to glycolytic pathways and Dex treatment had little impact on glycolytic pathways induced by miR-342 inhibition (Figure 6E; Figure S10B). In support, the expression of hexokinase 2 (Hk2), a key mediator of aerobic glycolysis (Wolf et al., 2011), and hypoxia inducible factor 1a (Hif1a), a key factor upregulating many enzymes in glycolytic pathway (Mathupala et al., 2001), was decreased by Dex, and miR-342 inhibition reversed the Dex-induced reduction (Figure 6G; Figure S10B). To examine if miR-342 inhibitor induced alteration in Treg cell metabolism is attributed to Rictor, the cells were cotreated with si-Rictor. si-Rictor treatment abolished Rictor expression even with miR-342 inhibitor treatment (Figure 6F). These Treg cells restored OXPHOS and suppressed glycolysis (Figure 6E). Hk2 and Hif1a expression further corroborated the findings (Figure 6G). When tTreg cells were treated with Dex, miR-342 inhibitor, and si-Rictor, we observed similar metabolic profiles (Figure S11). Thus, Dex controls the metabolic profiles in both tTreg and iTreg cells via the miR-342-Rictor pathways. Dex had no impact on OXPHOS and glycolysis in tTreg cells isolated from Nr3c1ΔTreg mice, suggesting that Dex induces metabolic programming via GC signaling (Figure S12). miR-342 inhibition abolished Dex-induced OXPHOS and glycolysis regardless of GR expression (Figure S12).
To further validate that miR-342–3p controls Treg cell function, we examined if miR-342–3p overexpression in Treg cells enhances their suppressive function. miR-342–3p overexpression significantly reduced the Rictor expression in Treg cells even without Dex treatment (Figure 7A). In vivo, miR-342 mimic transfected iTreg cells effectively attenuated EAE comparable to Dex treated WT iTreg cell recipients, even without Dex treatment (Figure 7B), suggesting that miR-342 is capable of enhancing Treg cell suppressive function independently of Dex. If Dex-mediated Rictor downregulation via miR-342–3p in Treg cells strengthens suppressive functions of Treg cells by supporting OXPHOS and by suppressing glycolytic pathways, it is then possible that enforced Rictor expression may abrogate such effects. To test this possibility, we overexpressed Rictor in Treg cells. miR-342–3p expression remained unchanged by Rictor overexpression, although Hk2 and Hif1a mRNA expression was substantially upregulated, suggesting altered metabolic program by Rictor overexpression (Figure 7C). In vivo, Rictor overexpressing iTreg cell recipients with EAE did not respond to Dex treatment, and the disease severity remained unchanged, while control iTreg cell recipients displayed attenuated disease following Dex treatment (Figure 7D). Therefore, these results demonstrate that enforced Rictor expression in Treg cells negates Dex treatment effects in vivo, further supporting the key contribution of the miR-342–3p-Rictor axis to Treg cell functions during Dex treatment.
Figure 7. Dex Controls Treg Functions via the miR-342–3p-Rictor Axis.

(A and B) Naive CD4+ T cells isolated from Foxp3GFP mice were nucleofected with control (NC) or miR-342–3p mimic, and then activated in vitro under iTreg conditions with or without Dex for 72 h.
(A) miR-342–3p and Rictor expression were determined by qPCR analysis.
(B) Foxp3DTR mice induced for EAE and treated with DTx day 7 post induction were transferred with 2×106 control or miR-342–3p mimic treated iTreg cells and daily treated with Dex on days 9–11 (arrows). The mice were daily monitored and scored for clinical diseases. n = 5–9 each group.
(C and D) Naive CD4+ T cells isolated from Foxp3GFP mice were nucleofected with control or Rictor expression vectors and subsequently polarized under iTreg condition with or without Dex for 72 h.
(C) qPCR analysis of miR-342–3p, Rictor, Hk2, and Hif1a expression.
(D) Foxp3DTR mice induced for EAE and treated with DTx day 7 post induction were transferred with 2×106 control or Rictor overexpressing iTreg cells. Dex treatment was performed on days 9 through 11 as above (arrows). The mice were daily monitored and scored for clinical diseases. n = 6–10 each group. Data are the mean ± SD of two to three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001, as determined by Mann-Whitney or Kruskal-Wallis nonparametric test.
DISCUSSION
In this study, we report that Treg cells were necessary for Dex to control autoimmune and allergic inflammation. Treg cell depletion itself induces severe inflammatory responses from conventional T cells, raising the possibility that the ineffective Dex effect could result from heightened Th cell responses (Arvey et al., 2014). However, we found that the lack of GR only in Treg cells impaired Dex effects in vivo, supporting a direct role for Treg cells in response to Dex stimulation. Mechanistically, Dex induced miR-342–3p in Treg cells, and the mTORC2 complex adaptor molecule, Rictor, was the direct target of miR-342–3p. Inhibiting miR-342 expression in Treg cells reversed Dex-mediated Treg-cell-dependent inhibition of the inflammation in vivo. miR-342–3p-mediated Rictor downregulation enforced Dex-induced OXPHOS metabolic profiles in Treg cells. Inhibiting miR-342 expression reversed Rictor expression, caused metabolic reprogramming to glycolytic pathways, and impaired Treg cell suppressive functions, during which Dex administration was unable to reverse impaired Treg cell functions. Likewise, miR-342 overexpression enhanced Treg cell functions even without Dex treatment, while Rictor overexpression in Treg cells abrogated Dex-mediated inhibition of inflammation in vivo. Collectively, these results uncover a previously unknown mechanism by which Treg cells mediate Dex-induced inhibition of inflammatory responses via a miR-342- and Rictor-dependent mechanism.
GC including Dex activates cytosolic GR expressed in almost every cell type (Oakley and Cidlowski, 2013). Previous studies demonstrate that the in vivo GC target cells differ depending on the tissues or the types of inflammatory responses. Lck-cre-induced GR deletion, targeting the exon 3 encoding the first zinc finger DNA binding domain (Tronche et al., 1999), uncovered that T cells are the primary GC-responding cells in Th1 and Th17 cell-mediated autoimmune inflammation (Engler et al., 2017; Wüst et al., 2008), whereas T cell expression of the GR seems dispensable during GC-mediated treatment of contact hypersensitivity (Tuckermann et al., 2007). In the present study, we utilized GR-floxed mice targeting the exon 3, independently developed by Ashwell and colleagues (Mittelstadt et al., 2012), to delete GR expression in Treg cells and found that Treg cells are necessary for GC to control both Th1 and Th17 cell-associated autoimmune and Th2 cell-associated allergic airway inflammation. Of note, Wüst et al. (2008) ruled out that Treg cells play an anti-inflammatory role of Dex in EAE based on the finding that Dex treatment diminishes peripheral Treg cell frequency in control but not in Lck-cre Nr3c1fl/fl mice (Wüst et al., 2008). We found comparable Treg cell accumulation in the CNS in Dex-treated Nr3c1ΔTreg mice. Since Lck-cre-induced deletion of the GR in all T cells including Treg cells could have masked the contribution of Treg cells in EAE, we would argue that Treg-cell-specific Nr3c1 deletion is the better approach to precisely test the contribution of Treg cells during Dex-induced treatment. Alternatively, different Dex doses used may account for the discrepancy in these studies (100mg/kg versus 5mg/kg). A study from Klaßen et al. reported that airway epithelial cells are the major GC target cells and that Nr3c1 deletion in total T cells had no impact on GC-mediated inhibition of allergic inflammation (Klaßen et al., 2017). Again, the reason behind the discrepancy is not clear. We used 2.5mg/kg of Dex in cockroach antigen-induced inflammation, while Klaben et al. used 10mg/kg of Dex in OVA-induced allergic inflammation. Although the dose of GC used may play a role in GC action on different target cells, we found that increasing Dex dose to 10mg/kg in our model still required the presence of Treg cells for Dex to control cockroach antigen-induced airway inflammation (Nguyen and Min, unpublished results). Alternatively, allergens with protease activity, such as cockroach antigens, are known to enhance inflammatory responses via epithelial activation (Kale et al., 2017), potentially altering GC responsiveness. The nature of allergens may dictate the target cells through which Dex mediates anti-inflammatory roles.
GR-deficient Treg cells are unable to control experimental T cell-induced colitic inflammation, although GR expression (i.e., GC signal) plays little role in Treg cell homeostasis and survival at steady state (Rocamora-Reverte et al., 2019). IFN-γ production in GR deficient Treg cells was noticed, suggesting that endogenous GC signal may prevent Treg cells from acquiring Th1 cell-like phenotypes (Rocamora-Reverte et al., 2019). In agreement with this report, we found that the lack of GR in Treg cells does not alter immune homeostasis in the primary and secondary lymphoid tissues at steady state. Likewise, EAE severity remained comparable between Treg-cell-specific Nr3c1 deficient and control mice without Dex administration, suggesting that endogenous GC signaling in Treg cells does not appear to affect Treg cell’s ability to control inflammation in the CNS tissues. This is in contrast with a previous study that Lck-cre induced Nr3c1 deletion results in early onset of EAE (Wüst et al., 2008). Therefore, endogenous GC signaling may have distinct impact on conventional T versus Treg cells. Further investigation is needed to better understand Dex effects in different cell types.
The facts that miR-342–3p expression is induced by Dex stimulation only in Treg but not in other effector lineage cells and that miR-342–3p plays an instrumental role during Dex-mediated control of inflammation raise several questions. First, how is miR-342 expression specifically regulated in Treg cells? miR-342 expression is controlled by its host gene, Evl, encoding cytoskeletal effector protein implicated in the motility and adhesion (Estin et al., 2017). In vitro generated Foxp3+ Treg cells express higher Evl compared to other lineage effector subsets including Th17 lineage cells, suggesting that TGF-β may not be involved. However, we cannot exclude the possibility that TGF-β signaling in the absence of inflammatory factors such as IL-6 may be important for the Evl expression. Alternatively, Foxp3 expression itself may be a factor driving the expression of miR-342–3p. In fact, Dex enhances miR-342–3p and Foxp3 gene expression in developing iTreg cells (Kim and Min, unpublished results). The mechanisms behind miR-342 and Treg cell development are currently being examined. Second, how does miR-342 control Treg cell function? Based on the sequence homology, we found that Rictor expression was directly controlled by miR-342–3p. Metabolic profiles, particularly the mTOR pathways, of Treg cells have extensively been studied (Kishore et al., 2017; Zeng and Chi, 2015). Unlike Rptor, deletion of the Rictor in Treg cells does not alter Treg cell homeostasis, and Rictor-deficient Treg cells express normal Treg cell gene signatures and mitochondrial activity (Zeng et al., 2013). However, our study demonstrates that Rictor expression in Treg cells was critical especially in maintaining OXPHOS metabolic programming and in limiting glycolytic pathway activation during Dex treatment, for neutralizing miR-342–3p expression with miR-342-specific antisense inhibitors and overexpressing miR-342–3p using miR mimics abolishes and bolsters Treg cell suppressive functions in vivo, respectively. It is worth noting that the cells in which miR-342 expression was inhibited or Rictor was overexpressed become GC refractory. The condition, referred to as steroid resistance, is often associated with severe inflammatory conditions. It will thus be important to examine the causal relationship between GC resistance and miR-342–3p expression (or metabolic programming) of Treg cells. Third, is Rictor expression controlled by miR-342–3p the sole mechanism underlying GC treatment? Since a single miRNA could target multiple genes (Creighton et al., 2012), there may be additional factors that are controlled by miR-342 and are involved in Treg-cell-dependent control of inflammatory responses. We are currently investigating this question.
The fact that Treg cells are necessary in responding to therapeutically administered GC is a compelling yet puzzling finding, especially given the ubiquitous expression of the GC receptor in every cell type. The molecular basis underlying the Treg-cell-specific GC responses will require future investigation. It is estimated that there are more than 1000 GR binding sites, i.e., glucocorticoid responsive element (GRE), in the genome (Vockley et al., 2016). It is particularly worth noting that GR itself is a Foxp3 binding protein (Rudra et al., 2012). Therefore, depending on the cell types, different GR signaling complexes including DNA binding proteins and other transcription modulators may be formed, driving cell-type-specific regulation (Franco et al., 2019). In support of this notion, we found a cohort of genes that is commonly induced or that is specifically altered in Treg cells in response to Dex stimulation. It will therefore be important to compare the DNA binding landscape of GR complexes between different cell types, especially lymphocyte subsets.
In summary, the current study identifies a previously unappreciated mechanism by which Treg cells play a key role during GC-induced treatment of inflammation. The study uncovers miR-342–3p as a potential regulator of metabolic programming in Treg cells in response to GC stimulation. Further investigation may enable us to understand how GC modulates inflammation and possibly to identify therapeutic targets to treat severe inflammatory conditions including steroid-resistant inflammation by targeting the miR-342-Rictor axis in Treg cells.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Booki Min (minb@ccf.org or booki.min@northwestern.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the RNA-seq reported in this paper is GEO: GSE136969.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6 (B6), C57BL/6 Foxp3DTR, Foxp3Cre/YFP mice, and Nr3c1fl/fl mice were purchased from the Jackson Laboratory (Bar Harbor, ME). C57BL/6 Foxp3GFP mice were obtained from Dr. Yasmine Belkaid (NIH) (Bettelli et al., 2006). Six to twelve-week old male and female (sex- and age-matched) mice were used for the experiments. All of the mice were maintained in a specific pathogen-free facility located at the Lerner Research Institute. All experiments were performed in accordance with the guidelines of the Lerner Research Institute Animal Care and Use Committee.
METHODS DETAILS
EAE induction
EAE was induced by injecting mice subcutaneously into the flanks with 200 μL of emulsion containing 300 μg or 150 μg MOG35–55 peptide (BioSynthesis, Lewisville, TX) and 5mg/mL Mycobacterium tuberculosis strain H37Ra (Difco, Detroit, MI). In addition, the mice received 200 ng pertussis toxin (Sigma, St. Louis, MO) intraperitoneally injected on days 0 and 2 after immunization. Vehicle or Dex (5mg/kg, Sigma) were intraperitoneally injected on days 9–11 after immunization. Clinical signs of EAE were assessed daily according to the standard 5-point scale. For depletion of Treg cells in Foxp3DTR mice, diphtheria toxin (DTx, Sigma) was injected intraperitoneally (1 μg) on day 7 after immunization. Spinal cords were fixed in 4% paraformaldehyde, and paraffin embedded, and sections were stained with hematoxylin and eosin (H&E).
Allergic Airway inflammation
Mice were injected intraperitoneally on days 0 and 7 with 5 μg of Cockroach antigen (CA, Greer Laboratory, Lenoir, NC) mixed in 100 μL of aluminum hydroxide adjuvant (Alum, Sigma). Starting on day 14, the mice were intranasally challenged for 4 consecutive days with 5 μg CA in PBS. Vehicle or Dex (2.5 mg/kg) were intraperitoneally injected on the first and third days of allergen challenge (day 14 and day 16). Mice were sacrificed 24 h after the last Ag challenge. In some experiments, Treg cells were depleted by intraperitoneal injection of 1 μg of DTx. Lung tissues were prepared from paraffin-embedded blocks and stained with H&E. Lung mechanics was measured using the FlexiVent ventilator (FlexiVent, Scireq). Lung resistance was measured in response to increasing doses of inhaled methacholine, as previously described (Nguyen et al., 2019).
Antibodies and Flow cytometry
At sacrifice, BALF cells, lung, and CNS tissue were collected and prepared as previously described (Kim et al., 2019; Nguyen et al., 2019). Briefly, the cells were stained with anti-CD4 (RM4–5, BioLegend), anti-Ly6G (1A8, BD Biosciences), and anti-Siglec-F (1RNM44N, eBioscience) mAbs. For intracellular staining, cells were stained with anti-IL-4 (11B11, eBioscience), anti-IL-13 (eBio13A, eBioscience), anti-IL-17 (TC11–18H10, BD Bioscience), anti-IFN-γ (XMG1.2, BD Bioscience), anti-IL-5 (TRFK5, BD Biosciences), anti-IL-10 (JES5–16E3, BD Biosciences) and anti-IL-2 (JES6–5H4, BD Biosciences) mAbs. Cells were acquired using a FACSLSRII or FACSFortessa X-20 (BD Biosciences) and analyzed using a FlowJo software (Tree Star Inc.). For intracellular staining, cells were harvested separately and stimulated ex vivo with PMA (10ng/mL, Millipore-Sigma) and ionomycin (1 μM, Millipore-Sigma) for 4 h in the presence of 2 μM monensin (Calbiochem) during the last 2 h of stimulation. Cells were immediately fixed with 4% paraformaldehyde, permeabilized, and stained with fluorescence-conjugated mAbs.
Nucleofection
Nucleofection was performed with Mouse T Cell Nucleofector Kit and Nucleofector device (Amaxa, Koelin, Germany). First, 1 × 106 naive CD4+ T cells were resuspended in 100 μL nucleofector solution. 100 pM oligonucleotides (including miR-342–3p inhibitor and control-NC5; Integrated DNA Technologies, San Jose, CA) were added into the solution and mixed gently. Then the mixtures were gently transferred to electroporation cuvettes and placed in the Nucleofector device. Cells were nucleofected in the X-001 program. Finally, transfected cells were transferred to a 24-well plate with 1.5 mL prepared Mouse T Cell Nucleofector Medium in each plate and incubated in a humidified 37°C/5% CO2 incubator for 2 h. Then, transfected cells were activated under the indicated polarizing conditions and treated with Dex (50nM).
In vitro T cell differentiation and adoptive transfer
NaiveCD4+Foxp3–CD44low T cells from lymph nodes of Foxp3GFP mice were sorted by cell sorting and treated with immobilizedanti-CD3 (2C11) and soluble anti-CD28 (37.51) mAbs in the presence of recombinant cytokines and antibodies to promote effector T cell differentiation. For Th1 and Th2 cells, IL-12 (10ng/mL)/anti-IL-4 (10 μg/mL) and IL-4 (1000U/mL)/anti-IFN-γ (10 μg/mL) were used, respectively. For Th17 cells, IL-6 (10ng/mL)/TGF-β (2.5ng/mL) and anti-IL-4/anti-IFN-γ (10 μg/mL) were used. For iTreg cells, IL-2 (100U/mL) and 5 ng/mLTGF-β (2.5 ng/mL) were used. After 3 days, iTreg cells (CD4+ Foxp3+) cells were cell sorted using a FACSAria (BD, San Jose,CA) prior to transfer. Cytokines and antibodies were purchased from Peprotech (Rocky Hill, NJ) and eBioscience (San Diego, CA), respectively.
Cytokine analysis by a cytometric bead array
The serum and BALF were collected from mice induced for autoimmune and allergic inflammation, respectively. The cytokines and IL-13 secretion was quantified using a cytometric bead array (CBA) kit (BD Biosciences) and an enhanced sensitivity flex set (BD Biosciences), respectively, according to the manufacturer’s instructions. The data were analyzed using the CBA software, and the standard curve for each cytokine was generated using the mixed cytokine standard.
ELISA
CNS cells isolated from mice with ongoing EAE were in vitro stimulated with 20 μg/mL MOG35–55 peptide for 48 h. ELISA kits were used to detect the levels of IL-10 and IFN-γ according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). Five repeated wells were set for each sample and standard product, and the optical density was measured at 492 nm using a microplate reader (Thermo Fisher Scientific).
Quantitative RT-PCR
RNA was isolated from inflamed lung, CNS or FACS sorted cells using a GeneJet RNA isolation kit (Thermo Fisher Scientific) or Quick-RNA Microprep kit (Zymo Research, Irvine, CA). cDNA was synthesized using Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Promega, Madison, WI). For miRNA detection, cDNA was prepared using the miRscript cDNA synthesis kit (QIAGEN, Hilden, Germany), according to manufacturer’s protocols. qPCR reactions were set up with the miScript SYBR Green PCR Kit. qPCR analysis was performed using a QuantStudio 3 Real-Time PCR System (Applied Biosystems, Waltham, MA) using TaqMan reagents (Applied Biosystems) or SYBR green master mixes (Applied Biosystems). The gene expression level of miRNAs was normalized to that of snoRNA U61 and of mRNAs to that of Gapdh.
Immuno blot analysis
Cells were lysed in lysis buffer (Cell Signaling Technologies). Protein separation was performed on4%polyacrylamide gel and electro blotted on nitrocellulose membrane (Whatman Inc., Florham Park, NJ.). The membrane was incubated for 60 min in5%skimmed milk powder in PBS with 0.1% Tween (PBS-T), and were then incubated overnight at 4°C with the appropriate primary antibody (see star method, Cell Signaling Technologies, Danvers, MA). On the next day, the membrane was washed with PBS-T, and incubated with the appropriate peroxidase secondary antibody (Cell Signaling Technologies) for 1 h at room temperature. Bands were then detected using an enhanced chemiluminescent HRP substrate (supersignal west femto maximum sensitivity substrate; Fisher Scientific) and visualized after exposure onto HyBlot CL Autoradiography Film (Denville Scientific) and processing through an Ecomax X-ray film processor.
Metabolic assay
Cellular metabolism was measured using an XF24 cellular flux analyzer instrument from Seahorse Bioscience (North Billerica, MA, USA). Transfected cells were prepared as described above and adhered to the plate with Cell-Tak (1 μg/well, Corning). OXPHOS was measured using a Mitostress test kit (Agilent Technologies, Wilmington, DE) according to the manufacturer’s instructions. Oligomycin (1 μM), FCCP (2 μM) and rotenone/antimycin A (0.5 μM) were successively injected while measuring OCR. Basal respiration was defined as baseline OCR. Maximal respiration was defined as OCR after FCCP treatment. ATP production was calculated as the difference between basal respiration and OCR after oligomycin treatment, and spare capacity was calculated as the difference between maximal and basal respiration. Extracellular acidification rate (ECAR) was measured using the XFe24 Extracellular Flux Analyzer and the glycolysis stress test kit (Seahorse Bioscience). Glucose (1mM), oligomycin (2 μM) and 2-desoxy-D-glucose (0.5 μM) were then successively added while measuring ECAR. Glycolytic capacity was defined as ECAR after glucose and oligomycin injection.
RNA-seq
RNA was isolated using the RNeasy Micro kit (QIAGEN). All subsequent sample quality assessment was performed on a Fragment Analzyer electrophoresis system (Agilent). Total RNA was normalized prior to oligo-dT capture and cDNA synthesis with SMART-Seq v4 (Takara). The resulting cDNA was quantified using a Qubit 3.0 fluorometer (Life Technologies). Libraries were generated using the Nextera XT DNA Library Prep kit (Illumina). Medium depth sequencing (25 million reads per sample) was performed with a HiSeq 2500 (Illumina) on a Rapid Run flow cell using a 100 base pairs, Paired End run. Raw data files and processed files have been deposited in the Gene Expression Omnibus under accession number GSE136969.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined by the Mann-Whitney (2-tailed) or Kruskal-Wallis test using the Prism 7 software (GraphPad) as indicated in the legends to the figures. A p value < 0.05 was considered statistically significant. EAE scores between groups were analyzed as disease burden per individual day with one-way-ANOVA and post hoc test as indicated. Sample sizes for each experiment are included in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD4 PE-Cy7 (clone RM4-5) | Biolegend | Cat # 100528 |
| anti-mouse IFNγ PE-CF594 (clone XMG1.2) | BD Biosciences | Cat # 562303 |
| anti-mouse IL17 PE (clone TC11-18H10) | BD Biosciences | Cat # 559502 |
| anti-mouse CD44 PE (clone IM7) | Thermo Fisher | Cat # 12-0441-83 |
| anti-mouse Ly-6G, PE (clone 1A8) | BD Biosciences | Cat # 551461 |
| anti-mouse Siglec-F, APC (clone 1RNM44N) | eBioscience | Cat # 50-1702-82 |
| anti-mouse IL-4, APC (clone 11B11) | BD Biosciences | Cat # 557739 |
| anti-mouse IL-13, PE (clone eBio13A) | eBioscience | Cat # 12-7133-82 |
| anti-mouse IL-17, FITC (clone TC11-18H10) | BD Biosciences | Cat # 560220 |
| anti-mouse IL-5, PE (clone TRFK5) | BD Biosciences | Cat # 554395 |
| anti-mouse IL-10, APC (clone JES5-16E3) | BD Biosciences | Cat # 554468 |
| anti-mouse IL-2, FITC (clone JES6-5H4) | BD Biosciences | Cat # 554427 |
| anti-mouse CD62L, PE (clone MEL-14) | BD Biosciences | Cat #553151 |
| anti-mouse CD8, APC (clone 53-6.7) | BD Biosciences | Cat #553035 |
| anti-mouse CD4, BV421 (clone RM4-5) | Biolegend | Cat # 100563 |
| anti-mouse CD25, PE-cy5 (clone PC61.5) | eBioscience | Cat # 15-0251-82 |
| anti-mouse ICOS, APC (clone C398.4A) | BD Pharmingen | Cat # 565882 |
| anti-mouse Nrp1, PE (clone 3E12) | Biolegend | Cat # 145204 |
| anti-mouse CD44, BV510 (clone IM7) | BD Biosciences | Cat # 563114 |
| anti-mouse GITR, PE-cy7 (clone DTA-1) | BD Biosciences | Cat # 558140 |
| anti-mouse PD1, APC (clone J43) | Invitrogen | Cat # 17-9985-82 |
| anti-mouse CTLA4, PE-cy7 (clone UC10-4B9) | Biolegend | Cat # 106314 |
| anti-mouse Foxp3, FITC (clone FJK-16s) | Invitrogen | Cat # 11-5773-82 |
| anti-mouse Tbet, APC (clone eBio4B10) | eBioscience | Cat # 50-5825-82 |
| anti-mouse Gata3, PE (clone TWAJ) | eBioscience | Cat # 12-9966-42 |
| anti-mouse RORγ, PE-TR (clone B20) | Invitrogen | Cat # 61-6981-82 |
| anti-mouse Ki67, PE-cy7 (clone SolA15) | Invitrogen | Cat # 25-5698-82 |
| anti-Rictor (clone 53A2) | CST | Cat # 2114 |
| anti-Raptor (24C12) | CST | Cat # 2280 |
| anti-Beta actin | CST | Cat # 4967 |
| anti-pAKT (S473) (clone D9E) | CST | Cat # 4060 |
| anti-pAKT (T308) (clone C31E5E) | CST | Cat # 2965 |
| anti-AKT (clone C67E7) | CST | Cat # 4691 |
| anti-pFoxo1a (S256) | CST | Cat # 9461 |
| anti-Foxo1a (clone C29H4) | CST | Cat # 2880 |
| anti-pS6 (clone D57.2.2E) | CST | Cat # 4858 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cockroach antigens | Greer Laboratory | Item# B26 |
| Aluminum hydroxide (Alum) | MilliporeSigma | Cat # 239186 |
| Anti-mouse CD3 mAb (clone 2C11) | Envigo Bioproducts | N/A |
| Transforming growth factor β (TGF-β) | PeproTech | Cat# 100-21 |
| Diphtheria toxin (DTx) | Sigma-Aldrich | Cat # D0564 |
| Pertussis toxin (PTx) | Sigma-Aldrich | Cat # P2980 |
| Dexamethasone-Water Soluble (Dex) | Sigma-Aldrich | Cat # D2915 |
| MOG (35–55) | Biosynthesis | Cat # 12668-01 |
| PMA (phorbol 12-myristate 13-acetate) | Sigma-Aldrich | Cat # P1585 |
| Ionomycin | Sigma-Aldrich | Cat # I9657 |
| TRIzol | Invitrogen | Cat # 15596026 |
| Power SYBR Green PCR Master Mix | Thermo Fisher | Cat # 4367659 |
| Recombinant DNA | ||
| Myc_Rictor | Addgene | Plasmid #11367 |
| Critical Commercial Assays | ||
| Mouse IL-10 DuoSet ELISA | R&D systems | Cat # DY417-05 |
| Mouse IFN-gamma DuoSet ELISA | R&D systems | Cat # DY485-05 |
| miScript II RT Kit | Qiagen | Cat # 218161 |
| miScript SYBR Green PCR Kit | Qiagen | Cat # 218073 |
| Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Cytokine Kit | BD Biosciences | Cat # 560485 |
| CBA mouse IL-13 Enhanced Sensitivity Flex Set | BD Biosciences | Cat # 562273 |
| Quick-RNA™ Miniprep Kit | Zymo research | Cat # R1051 |
| Mouse T cell nucleofector kit | Amaxa | Cat # VPA-1006 |
| MojoSort™ Mouse CD4 Naïve T Cell Isolation Kit | Biolegend | Cat # 480040 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat # 34095 |
| Mouse: GAPDH FAM/MGB probe | Thermo Fisher | ID# 4352661 |
| Mouse: Foxp3 FAM/MGB probe | Thermo Fisher | ID# Mm00475162_m1 |
| Mouse: IFNg FAM/MGB probe | Thermo Fisher | ID# Mm01168134_m1 |
| Mouse: IL17 FAM/MGB probe | Thermo Fisher | ID# Mm00439618_m1 |
| Mouse: Rragd FAM/MGB probe | Thermo Fisher | ID# Mm00546741_m1 |
| Mouse: Galnt9 FAM/MGB probe | Thermo Fisher | ID# Mm01196716_m1 |
| Mouse: Esam FAM/MGB probe | Thermo Fisher | ID# Mm00518378_m1 |
| Mouse: Evl FAM/MGB probe | Thermo Fisher | ID# Mm00468405_m1 |
| Mouse: Nabp FAM/MGB probe | Thermo Fisher | ID# Mm00658947_m1 |
| Mouse: Rictor FAM/MGB probe | Thermo Fisher | ID# Mm01307318_m1 |
| Mouse: Raptor FAM/MGB probe | Thermo Fisher | ID# Mm01242613_m1 |
| Mouse: Dusp1 FAM/MGB probe | Thermo Fisher | ID# Mm00457274_g1 |
| Mouse: Fkbp5 FAM/MGB probe | Thermo Fisher | ID# Mm00487406_m1 |
| Mouse: Muc5ac FAM/MGB probe | Thermo Fisher | ID# Mm01276718_m1 |
| Mouse: Muc5b FAM/MGB probe | Thermo Fisher | ID# Mm00466391_m1 |
| Mouse: Hk2 FAM/MGB probe | Thermo Fisher | ID# Mm00443385_m1 |
| Mouse: Hif1a FAM/MGB probe | Thermo Fisher | ID# Mm00468869_m1 |
| Mm_miR-15b_2 miScript Primer Assay | Qiagen | ID# MS00011235 |
| Mm_miR-16_2 miScript Primer Assay | Qiagen | ID# MS00037366 |
| Mm_miR-146_1 miScript Primer Assay | Qiagen | ID# MS00001638 |
| Mm_miR-155_1 miScript Primer Assay | Qiagen | ID# MS00001701 |
| Mm_miR-342_1 miScript Primer Assay | Qiagen | ID# MS00002184 |
| 5 nmol IDT miRNA Inhibitor_NC5; mG/ZEN/mCmGmAmCmUmAmUmAmCmGmCmGmCmAmAmUmAmUmGmG/3ZEN/ | IDT | Ref # 210223534 |
| 5 nmol IDT miRNA Inhibitor_mmu-miR342-3p; mA/ZEN/mCmGmGmGmUmGmCmGmAmUmUmUmCmUmGmUmGmUmGmAmG/3ZEN/ | IDT | Ref # 210223535 |
| mirVan miRNA Mimic, Negative Control #1 | Thermo Fisher | Cat # 4464058 |
| mirVana miR-342 Mimic | Thermo Fisher | ID#MC12328 |
| Accell Mouse Rictor siRNA | Horizon Discovery | Cat # A06459813-0005 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent | Cat # 103020-100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat # 103015-100 |
| Deposited Data | ||
| RNAseq | This paper | GEO: GSE136969 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice (B6) | Jackson Laboratory | JAX: 000664 |
| B6 Foxp3GFP (B6 background) | Generated by V. Kuchroo and Provided by Y. Belkaid (Bettelli et al., 2006) | N/A |
| B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J (B6 background) | Jackson Laboratory | JAX: 016958 |
| B6.Cg-Nr3c1tm1.1Jda/J (B6 background) | Jackson Laboratory | JAX: 021021 |
| B6.129(Cg)-Foxp3tm4(YFP/icre)Ayr/J (B6 background) | Jackson Laboratory | JAX: 016959 |
| Oligonucleotides | ||
| GAPDH forward: 5’- ATGCCTGCTTCACCACCTTCT-3’ | IDT | N/A |
| GAPDH reverse: 5’- CATGGCCTTCCGTGTTCCTA-3’ | IDT | N/A |
| common GR forward: 5’-AAAGAGCTAGGAAAAGCCATTGTC-3’ | IDT | N/A |
| GRα reverse: 5’-TCAGCTAACATCTCTGGGAATTCA-3’ | IDT | N/A |
| Gilz forward: 5’- AATGCGGCCACGGATG -3’ | IDT | N/A |
| Gilz reverse: 5’- GGACTTCACGTTTCAGTGGACA -3’ | IDT | N/A |
| Software and Algorithms | ||
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Flowjo | TreeSta | https://www.flowjo.com/solutions/flowjo/downloads |
| Seahorse Wave | Seahorse Wave | |
Highlights.
Without Foxp3+ Treg cells, glucocorticoid loses its therapeutic effects
Glucocorticoid induces miR-342–3p expression specifically in Treg cells
miR-342–3p targets Rictor and regulates the metabolic programming in Treg cells
miR-342–3p and Rictor expression controls Treg cell function in vivo
ACKNOWLEDGMENTS
The authors thank Ms. Jennifer Powers for cell sorting and Brian Richardson for RNA-seq data preparation. This work was supported by the National Institutes of Health grants R01-AI125247 and R01-AI147498 (B.M.) and by the National Multiple Sclerosis Society Research Grants RG1411–02051 and RG1806–31374 (B.M.). This work was also supported in part by Cancer Prevention & Research Institute of Texas grant RP170307 (J-S.L.), by Congressionally Directed Medical Research Program grant CA160616 (J-S.L.), and by National Institutes of Health grants HL103453, HL081064, HL60917, and HL109250 (K.A. and S.C.E).
Footnotes
DECLARATION OF INTERESTS
Authors declare that they have no competing interests.
REFERENCES
- Alangari AA (2014). Corticosteroids in the treatment of acute asthma. Ann. Thorac. Med. 9, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, et al. (2017). ). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–1293e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, and Rudensky AY (2014). Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 15, 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, and Riccardi C (2009). Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 23, 3649–3658. [DOI] [PubMed] [Google Scholar]
- Barnes PJ (2006). Corticosteroid effects on cell signalling. Eur. Respir. J. 27, 413–426. [DOI] [PubMed] [Google Scholar]
- Barnes PJ (2011). Glucocorticosteroids: current and future directions. Br. J. Pharmacol. 163, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes D, Hughes RA, Morris RW, Wade-Jones O, Brown P, Britton T, Francis DA, Perkin GD, Rudge P, Swash M, et al. (1997). Randomised trial of oral and intravenous methylprednisolone in acute relapses of multiple sclerosis. Lancet 349, 902–906. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, and Ochs HD (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27, 20–21. [DOI] [PubMed] [Google Scholar]
- Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, Sorcini D, Venanzi A, Di Sante M, and Riccardi C (2014). GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-β signaling. Cell Rep. 7, 464–475. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. [DOI] [PubMed] [Google Scholar]
- Bluestone JA, and Tang Q (2018). Treg cells-the next frontier of cell therapy. Science 362, 154–155. [DOI] [PubMed] [Google Scholar]
- Cain DW, and Cidlowski JA (2017). Immune regulation by glucocorticoids. Nat. Rev. Immunol. 17, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cari L, De Rosa F, Nocentini G, and Riccardi C (2019). Context-Dependent Effect of Glucocorticoids on the Proliferation, Differentiation, and Apoptosis of Regulatory T Cells: A Review of the Empirical Evidence and Clinical Applications. Int. J. Mol. Sci 20, 10.3390/ijms20051142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, and Chi H (2014). mTOR signaling, Tregs and immune modulation. Immunotherapy 6, 1295–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, Garcia-Lloret MI, Chen K, Ozen A, Carmeliet P, et al. (2019). Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat. Immunol. 20, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, and Howard OM (2006). Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur. J. Immunol. 36, 2139–2149. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Hernandez-Herrera A, Jacobsen A, Levine DA, Mankoo P, Schultz N, Du Y, Zhang Y, Larsson E, Sheridan R, et al. ; Cancer Genome Atlas Research Network (2012). Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS ONE 7, e34546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchis ML, Ballarino M, Salvatori B, Puzzolo MC, Bozzoni I, and Fatica A (2009). A new molecular network comprising PU.1, interferon regulatory factor proteins and miR-342 stimulates ATRA-mediated granulocytic differentiation of acute promyelocytic leukemia cells. Leukemia 23, 856–862. [DOI] [PubMed] [Google Scholar]
- Dibble CC, Asara JM, and Manning BD (2009). Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 29, 5657–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Villar M, and Hafler DA (2018). Regulatory T cells in autoimmune disease. Nat. Immunol. 19, 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler JB, Kursawe N, Solano ME, Patas K, Wehrmann S, Heckmann N, Lühder F, Reichardt HM, Arck PC, Gold SM, and Friese MA (2017). Glucocorticoid receptor in T cells mediates protection from autoimmunity in pregnancy. Proc. Natl. Acad. Sci. USA 114, E181–E190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estin ML, Thompson SB, Traxinger B, Fisher MH, Friedman RS, and Jacobelli J (2017). Ena/VASP proteins regulate activated T-cell trafficking by promoting diapedesis during transendothelial migration. Proc. Natl. Acad. Sci. USA 114, E2901–E2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco LM, Gadkari M, Howe KN, Sun J, Kardava L, Kumar P, Kumari S, Hu Z, Fraser IDC, Moir S, et al. (2019). Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med. 216, 384–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest. 125, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, et al. (2016). Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 17, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz AM, et al. (2008). Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene 27, 3880–3888. [DOI] [PubMed] [Google Scholar]
- Gruver-Yates AL, and Cidlowski JA (2013). Tissue-specific actions of glucocorticoids on apoptosis: a double-edged sword. Cells 2, 202–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, and Sabatini DM (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 11, 859–871. [DOI] [PubMed] [Google Scholar]
- Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, Lemoine FM, Zou W, and Emilie D (2007). Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood 110, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, Huang H, Roberts DJ, Thomas B, Hester SS, et al. (2017). Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2, e89160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang E, Nguyen QT, Kim S, Kim D, Le THN, Keslar K, Dvorina N, Aronica MA, and Min B (2017). Lung-Infiltrating Foxp3+ Regulatory T Cells Are Quantitatively and Qualitatively Different during Eosinophilic and Neutrophilic Allergic Airway Inflammation but Essential To Control the Inflammation. J. Immunol. 199, 3943–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel M, and Cidlowski JA (2013). Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 34, 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale SL, Agrawal K, Gaur SN, and Arora N (2017). Cockroach protease allergen induces allergic airway inflammation via epithelial cell activation. Sci. Rep. 7, 42341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Rückert B, Mantel PY, Menz G, Akdis CA, Blaser K, and Schmidt-Weber CB (2004). Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J. Allergy Clin. Immunol. 114, 1425–1433. [DOI] [PubMed] [Google Scholar]
- Kelly MM, King EM, Rider CF, Gwozd C, Holden NS, Eddleston J, Zuraw B, Leigh R, O’Byrne PM, and Newton R (2012). Corticosteroid-induced gene expression in allergen-challenged asthmatic subjects taking inhaled budesonide. Br. J. Pharmacol. 165, 1737–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Le HT, Nguyen QT, Kim S, Lee J, and Min B (2019). Cutting Edge: IL-27 Attenuates Autoimmune Neuroinflammation via Regulatory T Cell/Lag3-Dependent but IL-10-Independent Mechanisms In Vivo. J. Immunol. 202, 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, Ward EJ, Colamatteo A, Jangani M, Baragetti A, et al. (2017). Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 47, 875–889e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaßen C, Karabinskaya A, Dejager L, Vettorazzi S, Van Moorleghem J, Lühder F, Meijsing SH, Tuckermann JP, Bohnenberger H, Libert C, and Reichardt HM (2017). Airway Epithelial Cells Are Crucial Targets of Glucocorticoids in a Mouse Model of Allergic Asthma. J. Immunol. 199, 48–61. [DOI] [PubMed] [Google Scholar]
- Li Z, Wong KY, Chan GC, Chng WJ, and Chim CS (2018). Epigenetic silencing of EVL/miR-342 in multiple myeloma. Transl. Res. 192, 46–53. [DOI] [PubMed] [Google Scholar]
- Liles WC, Dale DC, and Klebanoff SJ (1995). Glucocorticoids inhibit apoptosis of human neutrophils. Blood 86, 3181–3188. [PubMed] [Google Scholar]
- Mathupala SP, Rempel A, and Pedersen PL (2001). Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 276, 43407–43412. [DOI] [PubMed] [Google Scholar]
- Meagher LC, Cousin JM, Seckl JR, and Haslett C (1996). Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J. Immunol. 156, 4422–4428. [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelstadt PR, Monteiro JP, and Ashwell JD (2012). Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J. Clin. Invest. 122, 2384–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necela BM, and Cidlowski JA (2004). Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc. Am. Thorac. Soc. 1, 239–246. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Jang E, Le HT, Kim S, Kim D, Dvorina N, Aronica MA, Baldwin WM 3rd, Asosingh K, Comhair S, and Min B (2019). IL-27 targets Foxp3+ Tregs to mediate anti-inflammatory functions during experimental allergic airway inflammation. JCI Insight 4, 10.1172/jci.insight.123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noval Rivas M, and Chatila TA (2016). Regulatory T cells in allergic diseases. J. Allergy Clin. Immunol. 138, 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, and Cidlowski JA (2013). The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 132, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh WJ, and Jacinto E (2011). mTOR complex 2 signaling and functions. Cell Cycle 10, 2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, and Yamashita N (1999). Apoptosis of eosinophils and lymphocytes in allergic inflammation. J. Allergy Clin. Immunol. 104, 14–21. [DOI] [PubMed] [Google Scholar]
- Olsen PC, Kitoko JZ, Ferreira TP, de-Azevedo CT, Arantes AC, and Martins MA (2015). Glucocorticoids decrease Treg cell numbers in lungs of allergic mice. Eur. J. Pharmacol. 747, 52–58. [DOI] [PubMed] [Google Scholar]
- Pan CW, Jin X, Zhao Y, Pan Y, Yang J, Karnes RJ, Zhang J, Wang L, and Huang H (2017). AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. EMBO J. 36, 995–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocamora-Reverte L, Tuzlak S, von Raffay L, Tisch M, Fiegl H, Drach M, Reichardt HM, Villunger A, Tischner D, and Wiegers GJ (2019). Glucocorticoid Receptor-Deficient Foxp3+ Regulatory T Cells Fail to Control Experimental Inflammatory Bowel Disease. Front. Immunol. 10, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, and Rudensky AY (2012). Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 13, 1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Sbiera S, Dexneit T, Reichardt SD, Michel KD, van den Brandt J, Schmull S, Kraus L, Beyer M, Mlynski R, Wortmann S, et al. (2011). Influence of short-term glucocorticoid therapy on regulatory T cells in vivo. PLoS ONE 6, e24345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleimer RP, and Bochner BS (1994). The effects of glucocorticoids on human eosinophils. J. Allergy Clin. Immunol. 94, 1202–1213. [DOI] [PubMed] [Google Scholar]
- Tchen CR, Martins JR, Paktiawal N, Perelli R, Saklatvala J, and Clark AR (2010). Glucocorticoid regulation of mouse and human dual specificity phosphatase 1 (DUSP1) genes: unusual cis-acting elements and unexpected evolutionary divergence. J. Biol. Chem. 285, 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trichet L, Sykes C, and Plastino J (2008). Relaxing the actin cytoskeleton for adhesion and movement with Ena/VASP. J. Cell Biol. 181, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schütz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103. [DOI] [PubMed] [Google Scholar]
- Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, Clausen BE, Stride B, Förster I, Habenicht AJ, et al. (2007). Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Invest. 117, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugor E, Prenek L, Pap R, Berta G, Ernszt D, Najbauer J, Németh P, Boldizsár F, and Berki T (2018). Glucocorticoid hormone treatment enhances the cytokine production of regulatory T cells by upregulation of Foxp3 expression. Immunobiology 223, 422–431. [DOI] [PubMed] [Google Scholar]
- Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, and Reddy TE (2016). Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, and Guha A (2011). Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 208, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüst S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, Lühder F, and Reichardt HM (2008). Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J. Immunol. 180, 8434–8443. [DOI] [PubMed] [Google Scholar]
- Xie Y, Wu M, Song R, Ma J, Shi Y, Qin W, and Jin Y (2009). A glucocorticoid amplifies IL-2-induced selective expansion of CD4(+)CD25(+) FOXP3(+) regulatory T cells in vivo and suppresses graft-versus-host disease after allogeneic lymphocyte transplantation. Acta Biochim. Biophys. Sin. (Shanghai) 41, 781–791. [DOI] [PubMed] [Google Scholar]
- Zeng H, and Chi H (2015). Metabolic control of regulatory T cell development and function. Trends Immunol. 36, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Yang K, Cloer C, Neale G, Vogel P, and Chi H (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq reported in this paper is GEO: GSE136969.